 Cheng-Guang Yang
Cheng-Guang Yang Tao Chen2†
Tao Chen2† Wen-Teng Si
Wen-Teng Si Hong-Can Ren
Hong-Can Ren
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 26 February 2025
Sec. Experimental Pharmacology and Drug Discovery
Volume 16 - 2025 | https://doi.org/10.3389/fphar.2025.1521068
This article is part of the Research TopicNew Analytical Technology and Method in Systems PharmacologyView all 5 articles
Introduction: The cytochrome P450 enzyme 3A4 (CYP3A4) mediates numerous drug-drug interactions (DDIs) by inducing the metabolism of co-administered drugs, which can result in reduced therapeutic efficacy or increased toxicity. This study developed and validated a Physiologically Based Pharmacokinetic (PBPK) model to predict CYP3A4 induction-mediated DDIs, focusing on the early stages of clinical drug development.
Methods: The PBPK model for rifampicin, a potent CYP3A4 inducer, was developed and validated using human pharmacokinetic data. Subsequently, PBPK models for ‘victim’ drugs were constructed and validated. The PBPK-DDI model’s predictive performance was assessed by comparing predicted area under the curve (AUC) and maximum concentration (Cmax) ratioswith empirical data, using both the 0.5 to 2-fold criterion and theGuest criteria.
Results: The rifampicin PBPK model accurately simulated human pharmacokinetic profiles. The PBPK-DDI model demonstrated high predictive accuracy for AUC ratios, with 89% of predictions within the 0.5 to 2-fold criterion and 79% meeting the Guest criteria. For Cmax ratios, an impressive 93% of predictions were within the acceptable range. The model significantly outperformed the static model, particularly in estimating DDI risks associated with CYP3A4 induction.
Discussion: The PBPK-DDI model is a reliable tool for predicting CYP3A4 induction-mediated DDIs. Its high predictive accuracy, confirmed by adherence to evaluation standards, affirms its reliability for drug development and clinical pharmacology. Future refinements may further enhance its predictive value.
In the realm of pharmacology and drug development, interactions between concurrently administered drugs have emerged as a critical focus due to their profound impact on patient safety and therapeutic efficacy (Prueksaritanont et al., 2013). Among these, interactions mediated by the CYP3A4 are of particular concern, given its central role in metabolizing a broad spectrum of therapeutic agents. The CYP3A4-mediated potential to either induce or inhibit the metabolism of concurrently administered drugs can profoundly modify their pharmacokinetic profiles, which may consequently elevate the risk of adverse events due to drug-drug interactions. This sometimes can lead to the withdrawal of the medication from the market (Smith and Schmid, 2006).
The induction of CYP3A4 can accelerate the clearance of co-administered drugs, potentially leading to suboptimal therapeutic effects (Skolnick et al., 1976). Additionally, this induction may result in an increased exposure to toxic metabolites, thereby raising safety concerns due to the potential toxicity (Gramec Skledar et al., 2016; Murphy et al., 2005). Therefore, the accurate prediction of CYP3A4-mediated interactions is essential for ensuring the safe and effective use of new molecular entities in drug development.
The PBPK model is particularly relevant in scenarios such as dose optimization for individualized therapy, prediction of drug-drug interactions, and evaluation of the impact of disease states on drug disposition (Chang et al., 2023; Rowland Yeo et al., 2024). It is also instrumental in extrapolating preclinical data to human conditions and in guiding clinical trial design by simulating various dosing regimens and schedules (Seo et al., 2022; Statelovaet al., 2023). PBPK models have shown promise as predictive tools for DDIs (Yamashita et al., 2013). While there have been numerous individual case reports attesting to the predictive capabilities of PBPK models, particularly for CYP3A4 induction-related DDIs, a systematic assessment of these models across a broader spectrum of DDIs is lacking. Addressing this gap is essential for refining predictive methodologies and enhancing our understanding of the models’ implications for drug development and clinical practice.
Building on our previous work, which successfully developed and validated a PBPK-DDI model for CYP3A4 inhibition-mediated DDIs (Jiang et al., 2023; Ren et al., 2021), the current study addresses the need for robust predictive methodologies for CYP3A4 induction-mediated DDIs, especially during the early clinical stages of drug development. Our approach involves developing a PBPK-DDI model that integrates a comprehensive dataset of substrates for PBPK model development of substrates and their documented interaction outcomes with strong CYP3A4 inducer (rifampicin). This PBPK-DDI model prioritizes the use of empirical data from in vitro and clinical studies, employing mechanistic predictions only for parameters that are difficult to measure experimentally. By comparing the model’s predictions with documented DDIs, we aim to validate its efficacy in anticipating the clinical impact of CYP3A4-mediated interactions, thereby contributing to a more accurate and reliable prediction tool for drug developers and clinicians.
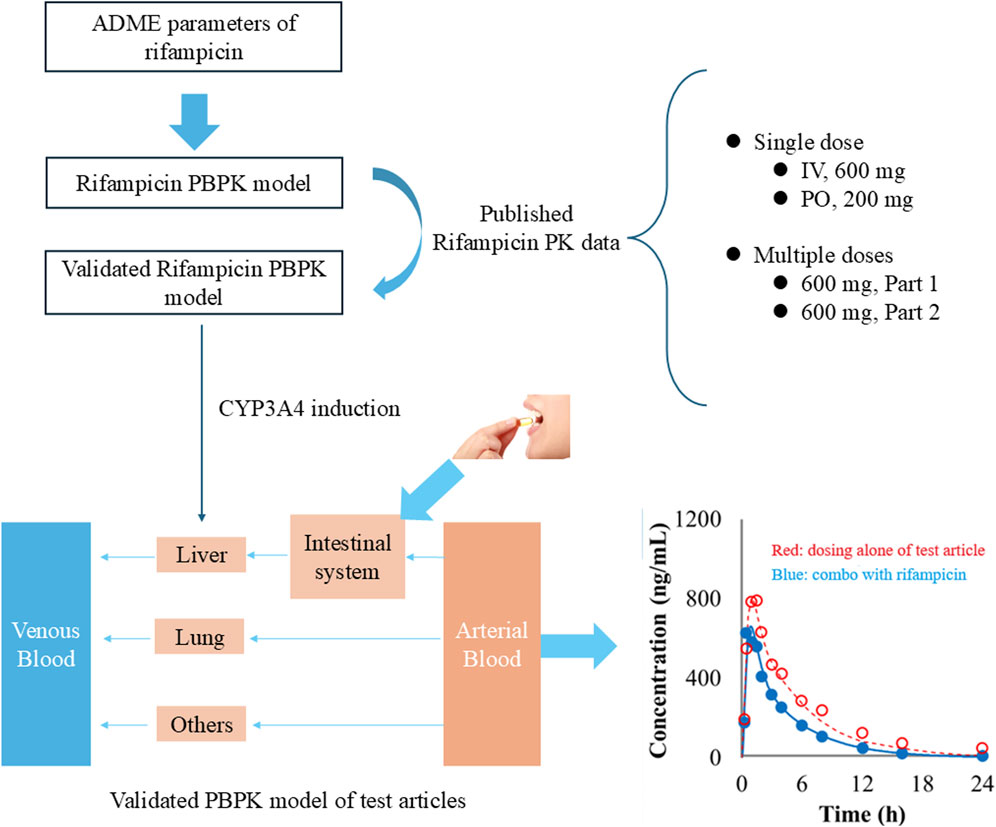
The study employs a comprehensive workflow to model and anticipate DDIs driven by CYP3A4 induction. The process begins with the development of a PBPK model for the inducing agent, rifampicin, which is meticulously validated against empirical human PK data. Following this, a PBPK model for the substrate drug is meticulously crafted, utilizing human PK and mass balance data to ensure model accuracy. In the subsequent phase, a PBPK-DDI model is meticulously formulated, as illustrated in Figure 1, to project the interaction profile between rifampicin and the substrate drug. The predictive accuracy of this model is rigorously evaluated by comparing its predictive accuracy with actual DDI data. Finally, to benchmark the performance, a traditional static model is applied to predict DDIs, and its predictions are critically compared with those from the PBPK model, thereby facilitating an assessment of the comparative efficacy and applicability of these methodologies.
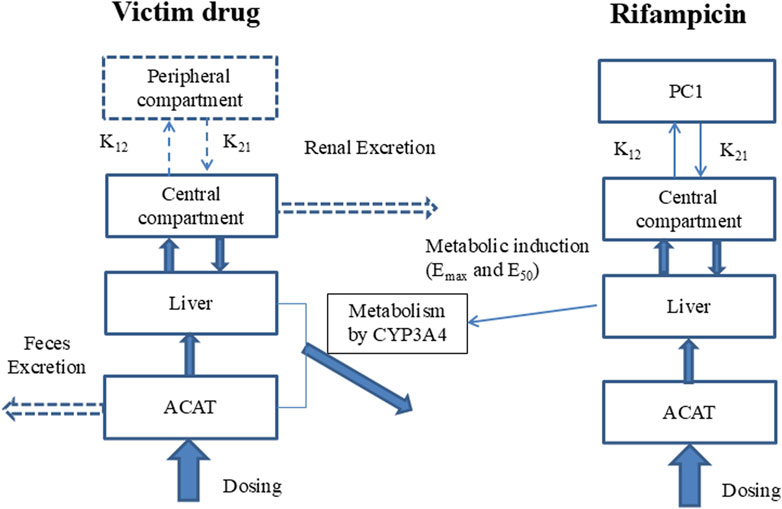
Figure 1. Model of PBPK-DDI. Note: Compartments and arrows with dash lines represent the options in the model. PC1, peripheral compartment 1; PC2, peripheral compartment 2; K12, transfer rate constant from central compartment to peripheral compartment; K21, transfer rate constant from peripheral compartment to peripheral central compartment; Vmax and Km, the constants of Michaelis-Menten reaction kinetics mediated by CYP3A4, which was assumed to be the unique metabolism rout; EC50 and Emax are the induction potency and magnitude to CYP3A4 of rifampicin; ACAT, Advanced Compartmental Absorption & Transit model.
The construction of the rifampicin PBPK model was developed on the integration of core physico-chemical and biopharmaceutical attributes, including aqueous solubility, logarithm of the octanol-water partition coefficient (logD), fraction unbound in plasma (fup), and red blood cell partitioning (Rbp). The human effective permeability (Peff) was meticulously calibrated to correspond with the absorption phase of the observed PK profile, with residual model parameters either prognosticated through the ADMET Predictor module or derived from the default settings within the GastroPlus simulation platform.
In the present investigation, PK parameters pertaining to the compartmental disposition were determined utilizing the PKPlus module of GastroPlus, calculated on the observed plasma concentration-time (C-T) profiles after intravenous administration. The system clearance included 1) the hepatic non-saturated metabolism mediated by CYP3A4 (about 0.0179 L/h/kg), which was defined in the Enzyme Table of GastroPlus using Km and Vmax of CYP3A4; 2) the remained liver metabolism (0.0623 L/h/kg), which was defined as the linear clearance in the Pharmacokinetics of GastroPlus; 3) and the renal clearance (0.0169 L/h/kg), which was defined as the clearance by glomerular filtration (GFR) based on the result of fup·GFR.
The induction potency (EC50) and the magnitude (Emax) of rifampicin’s effect on CYP3A4 were extracted from the published data. The validation of the PBPK model was executed by juxtaposing the simulated plasma C-T profiles of rifampicin against empirical data following a 600 mg intravenous dose, a 400 mg single oral dose, and a 600 mg single and multiple oral doses. A comparative analysis of the PK parameters of rifampicin was also undertaken to substantiate the fidelity of the model.
A total of 28 small molecule drugs approved by the FDA were selected as “victim” drugs for this study. These cases were culled from a previous investigation conducted by our research team, with the exclusion of those lacking reported interactions with rifampicin (Ren et al., 2021). The PBPK models for these victim drugs were constructed with detailed parameters sourced from the literature. Notably, these models incorporated the fraction metabolized (fm) parameter, which represents the proportion of a drug metabolized by CYP3A4. The determination and integration of the fm parameter are detailed in the referenced literature (Jiang et al., 2023).
Each of these PBPK models was rigorously validated using human PK data and mass balance data. The validation process ensured the models’ fidelity to empirical observations. For further scrutiny, the validation datasets and detailed analyses are provided in the Supplementary Material.
The PBPK modelling and simulations of victim drugs and perpetrator (rifampicin), as well as the DDI predictions, were conducted using GastroPlus™ Software (version 9.7; Simulations Plus, Inc., Lancaster, CA, United States). The Cmax and AUC were calculated by the PKPlus module of GastroPlus.
In addition to the PBPK-DDI models, static mechanistic models were employed to predict DDIs, adhering to the guidelines outlined in “In Vitro Drug Interaction Studies: Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions for Industry.” The steady-state DDI predictions, as default in GastroPlus, facilitate the generation of a comprehensive suite of DDI outcomes (Einolf et al., 2014). This includes a tabulation of the predicted plasma AUC ratios in the presence and absence of the perpetrator drug for each specified concentration. The models delineate the individual contributions of the gut and liver to the overall DDI, alongside the projected total change in AUC. The accuracy of these predictions is evaluated by comparing the anticipated AUCR with empirically derived ratios. Furthermore, the performance of the static mechanistic models is juxtaposed with that of the PBPK-DDI model, thereby highlighting the superior predictive capabilities of the PBPK-DDI model.
The predictive accuracy of the model was assessed by comparing the predicted ratios of the AUC and Cmax of the victim drug when co-administered with rifampicin to those observed when the victim drug was administered alone. Specifically, the AUCR and CmaxR were calculated. The predictive performance was deemed acceptable when the model’s projected ratios were within a two-fold deviation of the observed Geometric Mean Ratio (GMR), with the acceptable range defined as 0.5 to 2 times the GMR. Additionally, the methodology reported by Guest was employed to further evaluate the predictive efficacy of model (Guest et al., 2011).
The development of the rifampicin PBPK model was guided by a comprehensive set of physiological and physicochemical parameters, as detailed in Table 1. Utilizing these parameters, we simulated various dosing regimens to predict the human C-T profiles. The simulated C-T curves were compared with observed data, as depicted in Figure 2. The comparison revealed that the predicted curves closely matched the observed data points, indicating a high degree of fidelity in our model.
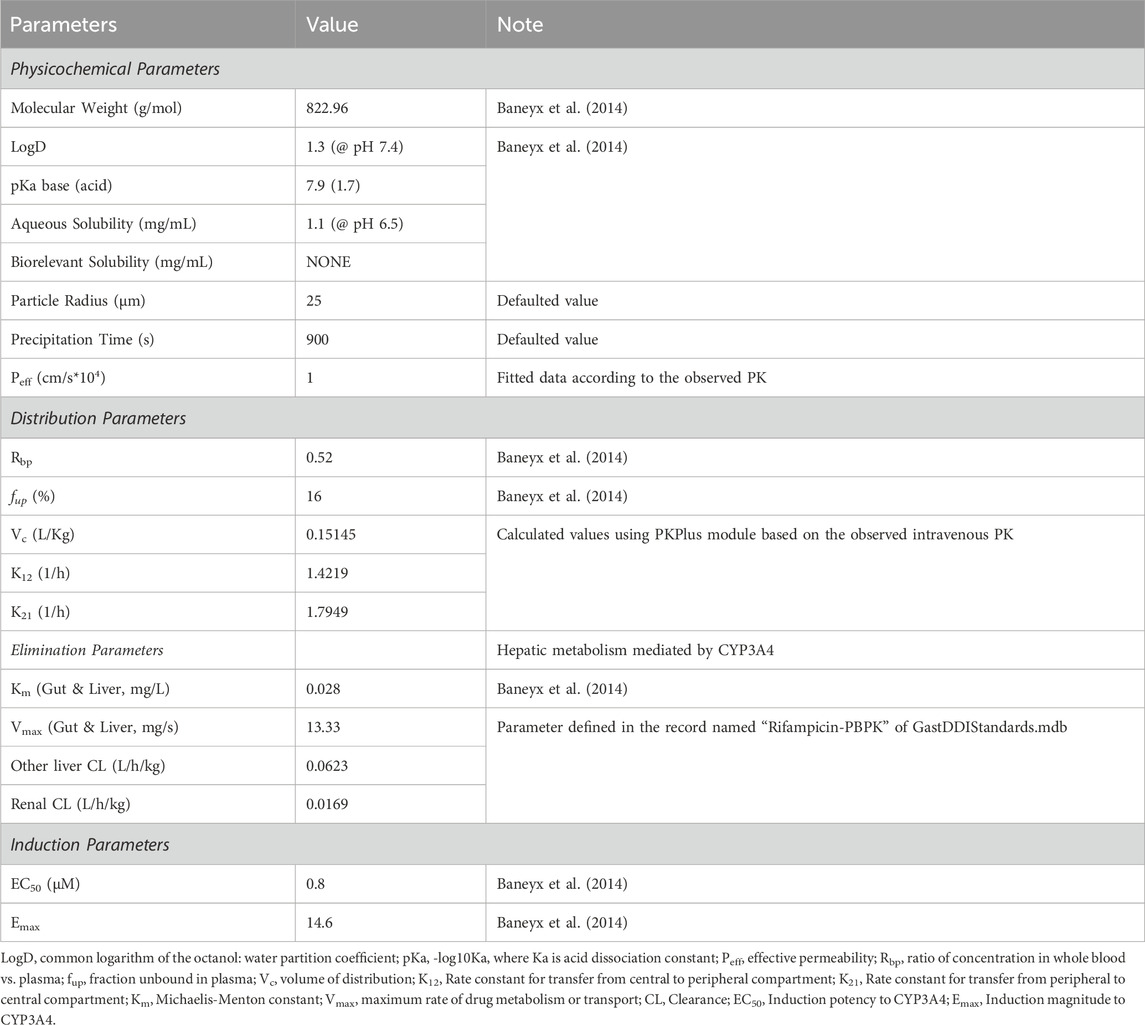
Table 1. Key parameters in rifampicin PBPK model.
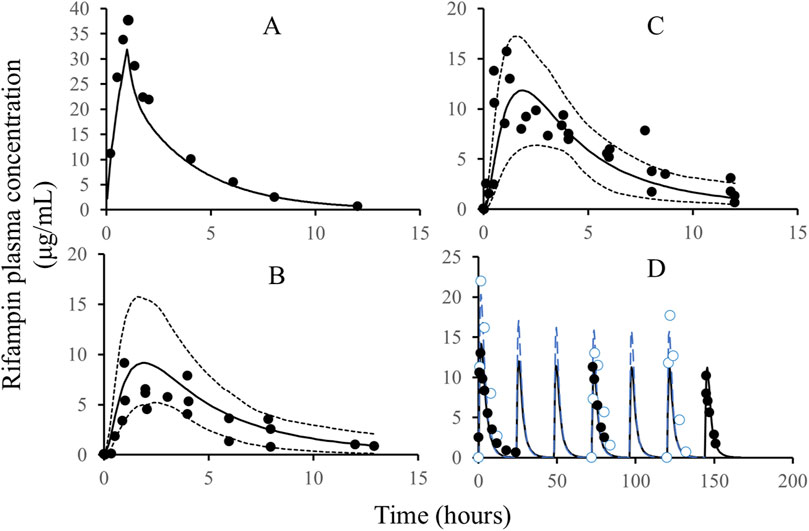
Figure 2. The comparison of simulated rifampicin PK in PBPK model with observed values. Note: Solid line: simulated C-T profiles of rifampicin after IV administration, single dose, 600 mg (A), oral administration, single dose, 200 mg (B), oral administration, single dose, 400 mg (C), and oral administration, QD, 600 mg; dash line: quantile of 5% and 95%; black circles: the observed PK data of rifampicin; blue circles (D): the observed PK data of rifampicin; blue line (D): the simulated C-T profiles of rifampicin. The black and blue C-T profiles in Figure D are from two different publications with different demographic characteristics, such as body weight, height, and age.
To quantitatively assess the accuracy of our model, we calculated the parameters for both the observed and simulated curves using a non-compartmental analysis method. The results of this comparison are presented in Table 2. The predicted error of the parameters was found to be within the range of ±25%, which aligns well with the bioequivalence criteria (>0.8 and <1.25). This level of accuracy confirms the reliability of the rifampicin model and justifies its use for further predictions of DDIs involving CYP3A4 substrates.
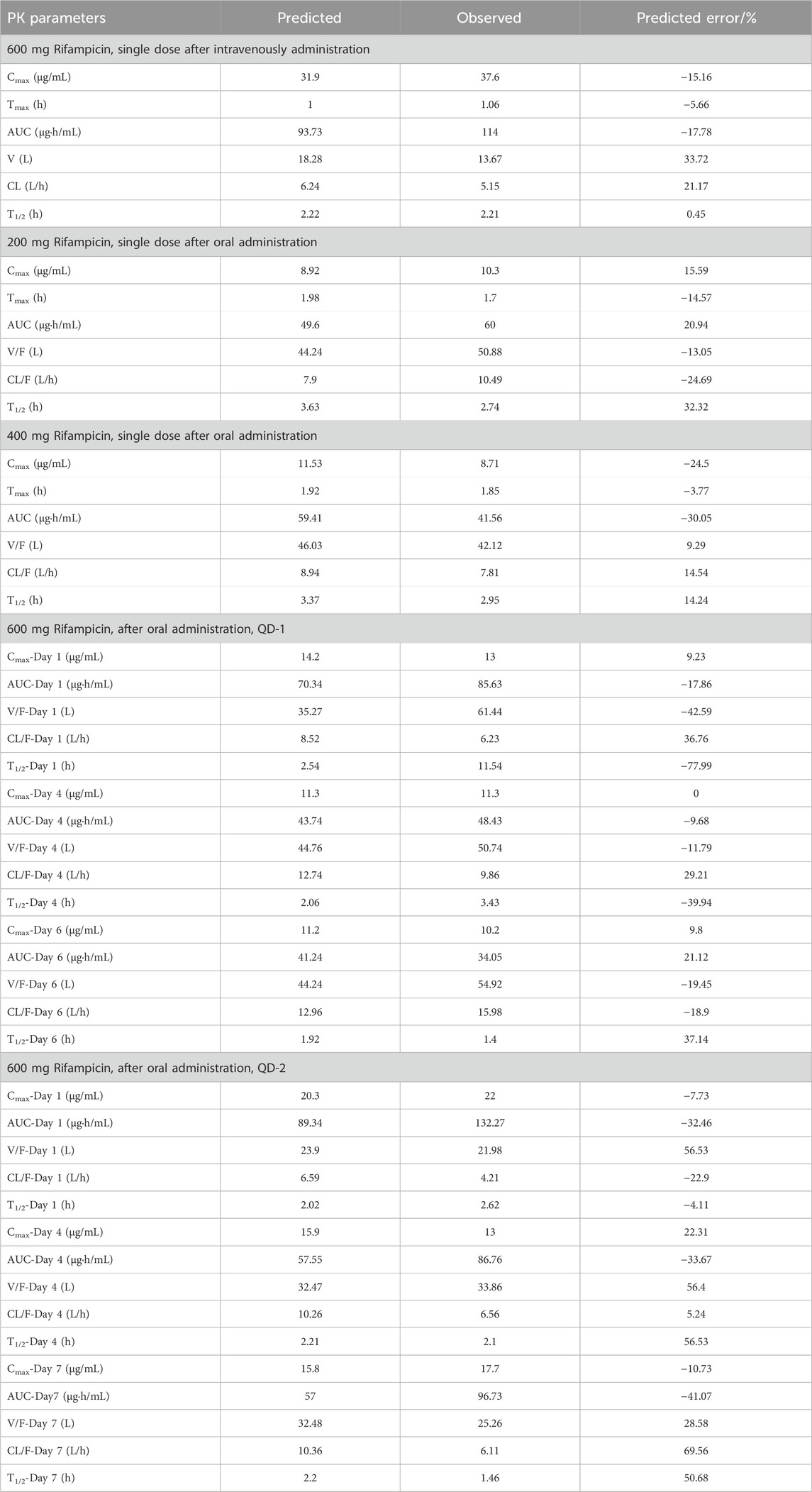
Table 2. The comparison of predicted PK parameters with observed values for rifampicin.
Given the model’s validated predictive capacity, it will be employed in subsequent analyses to predict the interactions between rifampicin and various CYP3A4 substrates, providing a robust foundation for our predictions.
The models for the victim drugs were meticulously constructed and validated using a combination of human data and mass balance data. These models were designed to simulate the disposition of drugs in the human body, accounting for absorption, distribution, metabolism, and excretion processes.
The accuracy of the victim drug models was assessed by comparing the simulated profiles and mass balance data with empirical observations. The results of this validation process demonstrated a high degree of consistency between the simulated and observed data, confirming the predictive power of our models.
For a detailed analysis and a comprehensive set of validation data, readers are directed to the Supplementary Material and our previously published articles (Ren et al., 2021), which lay a foundation for their application in predicting DDIs involving CYP3A4.
Utilizing the validated model parameters for rifampicin and its substrates, coupled with the characterized Emax and EC50 values indicative of rifampicin’s induction potency on CYP3A4, we projected the potential DDIs. The predictive metrics centered on the exposure ratios, quantified by the AUCR and the CmaxR. The predictive performance of our PBPK-DDI model was rigorously assessed by comparing predicted AUCR and CmaxR with empirical data. This comparison is visually represented in Figure 3A, which delineates the alignment between predicted and observed AUC ratios. The analysis revealed a substantial concordance between predicted and observed ratios, with 89% of the predictions falling within the acceptable range according to the 0.5 to 2-fold criterion. When subjected to the Guest criteria, our model demonstrated a commendable alignment, with 79% of the predictions matching the observed data. Furthermore, to provide a more granular analysis, we specifically evaluated the concordance between the predicted and observed Cmax ratios. As depicted in Figure 3B, an impressive 93% of the predicted CmaxR values agreed with the empirical CmaxR values within the 0.5 to 2-fold acceptance criterion. These findings underscore the robustness of our PBPK-DDI model in forecasting the clinical implications of CYP3A4-mediated DDIs induced by rifampin. The high predictive concordance, as evidenced by the adherence to both evaluation standards, affirms the model’s reliability and its potential utility in the realm of drug development and clinical pharmacology.
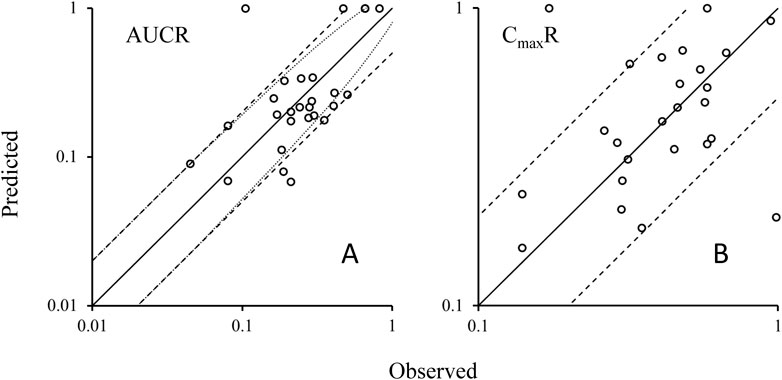
Figure 3. Plots of predicted AUCRs (A) and CmaxR (B) versus the observed values. Note: Solid lines represent the unit line (Y = X). The dash lines are the 0.5 fold and 2 fold lines; The dot lines are the lines using Guest-limit criteria. The circles represent the observed means of AUCR and CmaxR. AUCR, the ratio of area under the concentration-time curve of victim after co-administration with rifampicin over area under the C-T curve of victim with dosing alone; CmaxR, the ratio of victim Cmax after co-administration with rifampicin over victim Cmax with dosing alone.
The static model, a widely recognized and straightforward approach for evaluating DDIs as per DDI guidance, was employed for comparative analysis with our PBPK-DDI model. The comparison, detailed in Table 3, reveals significant discrepancies in the predictive accuracy of the Static model, particularly when assessing induction-based DDI risks.
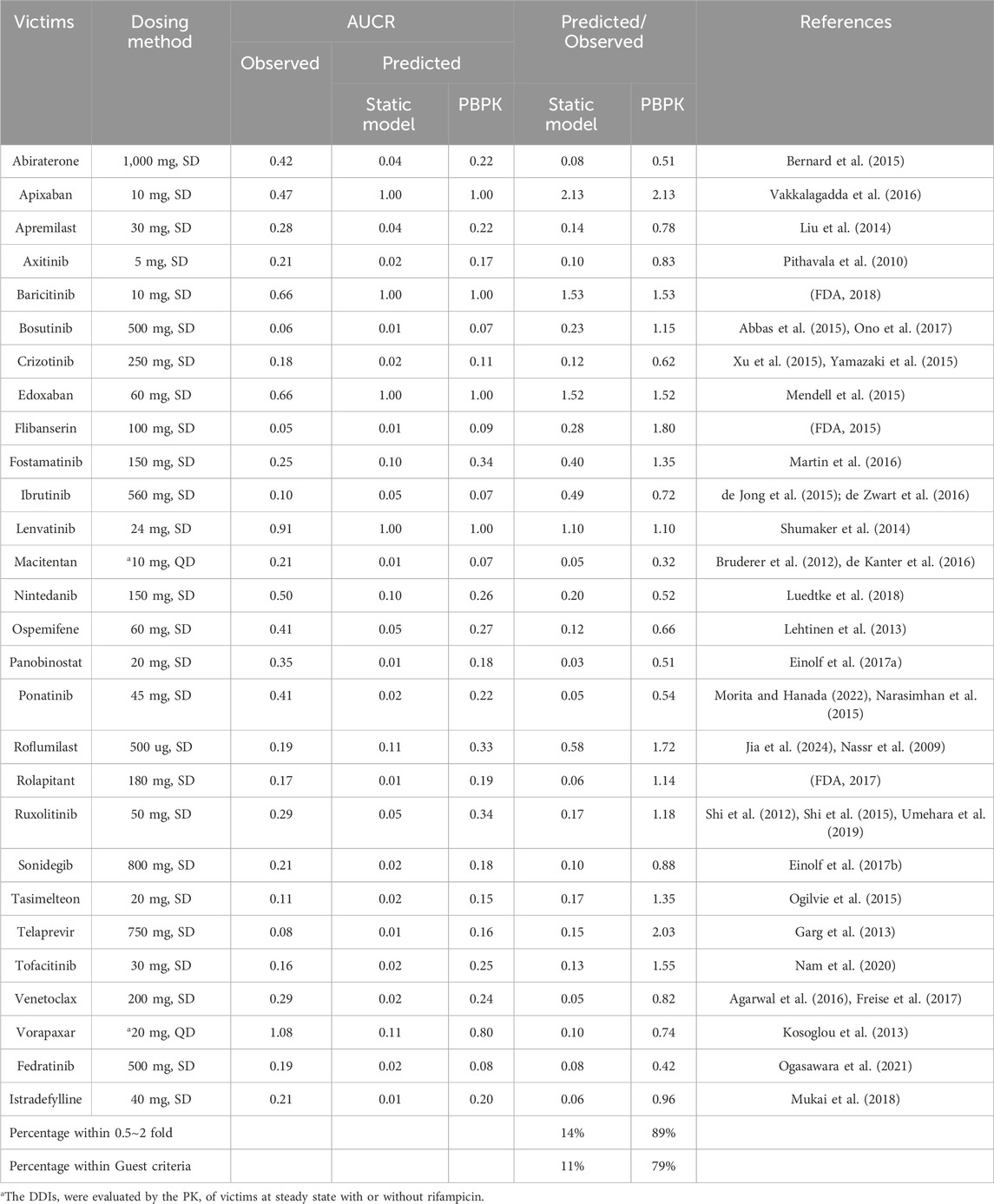
Table 3. The Dosing method of victim drugs and the predictive performance using different approaches.
Upon evaluation, it was observed that the Static model significantly overestimated the DDI risk in 83% of the cases, with only 14% of the predicted AUCR falling within the empirically accepted range of 0.5–2 times the observed AUCR. When applying the more stringent Guest criteria, this predictive accuracy further declined to 11%.
This comparative analysis underscores the Static model’s limitations in accurately forecasting DDIs, particularly when compared to the superior predictive capabilities of the PBPK-DDI model reported in this study. The PBPK-DDI model’s enhanced predictive performance is attributed to its physiologically based, mechanistic approach, which more accurately captures the complex interplay of drug interactions in the human body.
This study presents a comprehensive evaluation of the PBPK model’s predictive capabilities for DDIs mediated by CYP3A4 induction. Through meticulous model development and validation, we have demonstrated the model’s high fidelity in simulating the complex profiles of both rifampicin and its substrates. The comparison with empirical data has confirmed the model’s robustness, with an impressive alignment between predicted and observed parameters, well within the accepted bioequivalence criteria. Furthermore, the predictive performance of our PBPK-DDI model significantly surpassed that of the conventional Static model, particularly in accurately estimating the risks associated with CYP3A4 induction. This superiority is evident in the model’s ability to closely mirror the clinical outcomes, as evidenced by the high percentage of accurate predictions based on both the 0.5 to 2-fold and Guest criteria. The findings underscore the transformative potential of the PBPK-DDI model in enhancing the safety and efficacy assessments of new molecular entities during early clinical development.
Our rifampicin PBPK model, informed by the comprehensive framework within GastroPlus’s full PBPK model, has been strategically simplified to a compartmental model for enhanced practicality and expedited application. This tailored approach, while retaining the physiological essence of the full model, allows for rapid predictions that are crucial in clinical and research settings. Our model parameters, fine-tuned against a spectrum of published literature and aligned with empirical human data, have been optimized to ensure the most accurate predictions, as evidenced by their close match with observed plasma C-T profiles (Acocella, 1978; Loos et al., 1985; Peloquin et al., 1997). This validation not only affirms the model’s reliability but also positions it comparably with other published rifampicin PBPK models, reinforcing the consensus on model parameters that best capture the drug’s behavior (Ono et al., 2017; Yamazaki et al., 2015; de Zwart et al., 2016; Luedtke et al., 2018; Morita and Hanada, 2022; Nassr et al., 2009; Umehara et al., 2019; Freise et al., 2017).
The robustness for the PBPK model of victim drugs is crucial for assessing the DDIs. Our models align with existing scientific literature and highlight the significance of the fm parameter. By incorporating empirically derived in vitro fm values into our models, we prevent the potential overestimation of DDI risk that could occur if we assumed a fm value of 100%. Although this nuanced approach lacks comparative data in this presentation, it is theoretically sound and enhances the precision of DDI predictions, aligning with the current scientific consensus on this topic.
In our study, we observed a significant discrepancy in the fm test results for tasimelteon when using microsomal and recombinant enzyme methods. The microsomal method, after multiple repetitions, yielded results ranging from 0% to 15%, while the recombinant enzyme method resulted in a 65% value. The cause of this difference may be attributed to non-enzymatic metabolism in the microsomes; further investigation is underway to pinpoint the exact reason. According to the submission information on tasimelteon published by the FDA (FDA, 2014), CYP3A4 is the primary enzyme involved in the metabolism of tasimelteon. Therefore, in this research, we have adopted the results based on the recombinant enzyme method.
Our current model demonstrates a high degree of predictive accuracy for DDIs, the multifaceted nature of in vivo DDIs suggests that there is still room for refinement. One of the factors that may influence the prediction of rifampicin’s inducing effect is its ability to induce multiple enzymes while also inhibiting transporters, potentially leading to increased drug absorption. This phenomenon is particularly evident with elagolix (FDA, 2020), where co-administration with rifampicin does not reduce but rather increases its exposure, highlighting the significant contribution of transporters over the inhibition of metabolic enzymes. Our model does not currently integrate the impact of transporters, which is why we have not included elagolix as a case study. The examples we have utilized are mostly drugs with minimal transporter involvement, which to some extent, has bolstered the precision of our predictions. Future models could benefit from incorporating the effects of transporters to further enhance the accuracy of DDI predictions.
The PBPK models have effectively predicted DDIs involving rifampicin, highlighting their utility in clinical drug development. The models’ alignment with empirical data confirms their reliability, and their refinement will further enhance predictive precision in future studies.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
C-GY: Writing–review and editing, Formal Analysis, Resources, Writing–original draft. TC: Formal Analysis, Writing–original draft, Methodology, Software. W-TS: Conceptualization, Resources, Writing–review and editing. A-HW: Conceptualization, Writing–review and editing. H-CR: Conceptualization, Writing–review and editing, Data curation, Investigation, Methodology, Supervision. LW: Data curation, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors thank Simulations Plus, Inc. (Lancaster, CA, United States of America) for authorizing the use of the optimization module in this study. The authors thank Dr. Siyuan Le (Translation Medicine department, GenFleet Therapeutics (Shanghai) Inc.) for his proofreading of the manuscript.
TC is an employee of Shanghai PharmoGo Co., Ltd., an authorized distributor of Simulations Plus, the developer of GastroPlus, in China. H-CR and LW were employed by GenFleet Therapeutics (Shanghai) Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1521068/full#supplementary-material
AUCR, the ratio of area under the concentration-time curve of victim after co-administration with rifampicin over area under the concentration-time curve of victim with dosing alone; CmaxR, the ratio of victim Cmax after co-administration with rifampicin over victim Cmax with dosing alone; FDA, Food and Drug Administration; fm, the fraction of metabolism; Fa, cumulative intestinal absorption, %; Vc, distribution volume in central compartment; K12, transfer rate constant from central compartment to peripheral compartment; PC1, peripheral compartment 1; PC2, peripheral compartment 2.
Abbas, R., Boni, J., and Sonnichsen, D. (2015). Effect of rifampin on the pharmacokinetics of bosutinib, a dual Src/Abl tyrosine kinase inhibitor, when administered concomitantly to healthy subjects. Drug Metab. Pers. Ther. 30 (1), 57–63. doi:10.1515/dmdi-2014-0026
Acocella, G. (1978). Clinical pharmacokinetics of rifampicin. Clin. Pharmacokinet. 3 (2), 108–127. doi:10.2165/00003088-197803020-00002
Agarwal, S. K., Hu, B., Chien, D., Wong, S. L., and Salem, A. H. (2016). Evaluation of rifampin's transporter inhibitory and CYP3A inductive effects on the pharmacokinetics of venetoclax, a BCL-2 inhibitor: results of a single- and multiple-dose study. J. Clin. Pharmacol. 56 (11), 1335–1343. doi:10.1002/jcph.730
Baneyx, G., Parrott, N., Meille, C., Iliadis, A., and Lave, T. (2014). Physiologically based pharmacokinetic modeling of CYP3A4 induction by rifampicin in human: influence of time between substrate and inducer administration. Eur. J. Pharm. Sci. 56, 1–15. doi:10.1016/j.ejps.2014.02.002
Bernard, A., Vaccaro, N., Acharya, M., Jiao, J., Monbaliu, J., De Vries, R., et al. (2015). Impact on abiraterone pharmacokinetics and safety: open-label drug-drug interaction studies with ketoconazole and rifampicin. Clin. Pharmacol. Drug Dev. 4 (1), 63–73. doi:10.1002/cpdd.132
Bruderer, S., Aanismaa, P., Homery, M. C., Hausler, S., Landskroner, K., Sidharta, P. N., et al. (2012). Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. AAPS J. 14 (1), 68–78. doi:10.1208/s12248-011-9316-3
Chang, S. Y., Huang, W., Chapron, A., Quinones, A. J. L., Wang, J., Isoherranen, N., et al. (2023). Incorporating uremic solute-mediated inhibition of OAT1/3 improves PBPK prediction of tenofovir renal and systemic disposition in patients with severe kidney disease. Pharm. Res. 40 (11), 2597–2606. doi:10.1007/s11095-023-03594-x
de Jong, J., Skee, D., Murphy, J., Sukbuntherng, J., Hellemans, P., Smit, J., et al. (2015). Effect of CYP3A perpetrators on ibrutinib exposure in healthy participants. Pharmacol. Res. Perspect. 3 (4), e00156. doi:10.1002/prp2.156
de Kanter, R., Sidharta, P. N., Delahaye, S., Gnerre, C., Segrestaa, J., Buchmann, S., et al. (2016). Physiologically-based pharmacokinetic modeling of macitentan: prediction of drug-drug interactions. Clin. Pharmacokinet. 55 (3), 369–380. doi:10.1007/s40262-015-0322-y
de Zwart, L., Snoeys, J., De Jong, J., Sukbuntherng, J., Mannaert, E., and Monshouwer, M. (2016). Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin. Pharmacol. Ther. 100 (5), 548–557. doi:10.1002/cpt.419
Einolf, H. J., Chen, L., Fahmi, O. A., Gibson, C. R., Obach, R. S., Shebley, M., et al. (2014). Evaluation of various static and dynamic modeling methods to predict clinical CYP3A induction using in vitro CYP3A4 mRNA induction data. Clin. Pharmacol. Ther. 95 (2), 179–188. doi:10.1038/clpt.2013.170
Einolf, H. J., Lin, W., Won, C. S., Wang, L., Gu, H., Chun, D. Y., et al. (2017a). Physiologically based pharmacokinetic model predictions of panobinostat (LBH589) as a victim and perpetrator of drug-drug interactions. Drug Metab. Dispos. 45 (12), 1304–1316. doi:10.1124/dmd.117.076851
Einolf, H. J., Zhou, J., Won, C., Wang, L., and Rebello, S. (2017b). A physiologically-based pharmacokinetic modeling approach to predict drug-drug interactions of sonidegib (LDE225) with perpetrators of CYP3A in cancer patients. Drug Metab. Dispos. 45 (4), 361–374. doi:10.1124/dmd.116.073585
FDA (2014). New drug application, Hetlioz® clinical pharmacology and biopharmaceutics reviews. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205677Orig1s000ClinPharmR.pdf (Accessed September 27, 2024).
FDA (2015). New drug application, ADDYI®, clinical pharmacology and biopharmaceutics reviews. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/022526Orig1s000ClinPharmR.pdf (Accessed September 27, 2024).
FDA (2017). New drug application, VARUBI®, clinical pharmacology and biopharmaceutics reviews. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208399Orig1s000ClinPharmR.pdf (Accessed September 27, 2024).
FDA (2018). New drug application, Olumiant®, clinical pharmacology and biopharmaceutics reviews. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/207924Orig1s000ClinPharmR.pdf (Accessed September 27, 2024).
FDA (2020). New drug application, Oriahnn®, clinical pharmacology and biopharmaceutics reviews. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213388Orig1s000MultidisciplineR.pdf (Accessed September 27, 2024).
Freise, K. J., Shebley, M., and Salem, A. H. (2017). Quantitative prediction of the effect of CYP3A inhibitors and inducers on venetoclax pharmacokinetics using a physiologically based pharmacokinetic model. J. Clin. Pharmacol. 57 (6), 796–804. doi:10.1002/jcph.858
Garg, V., Chandorkar, G., Yang, Y., Adda, N., McNair, L., Alves, K., et al. (2013). The effect of CYP3A inhibitors and inducers on the pharmacokinetics of telaprevir in healthy volunteers. Br. J. Clin. Pharmacol. 75 (2), 431–439. doi:10.1111/j.1365-2125.2012.04345.x
Gramec Skledar, D., Tomasic, T., Carino, A., Distrutti, E., Fiorucci, S., and Peterlin Masic, L. (2016). New brominated flame retardants and their metabolites as activators of the pregnane X receptor. Toxicol. Lett. 259, 116–123. doi:10.1016/j.toxlet.2016.08.005
Guest, E. J., Aarons, L., Houston, J. B., Rostami-Hodjegan, A., and Galetin, A. (2011). Critique of the two-fold measure of prediction success for ratios: application for the assessment of drug-drug interactions. Drug Metab. Dispos. 39 (2), 170–173. doi:10.1124/dmd.110.036103
Jia, G., Ren, C., Wang, H., and Fan, C. (2024). Prediction of drug-drug interactions between roflumilast and CYP3A4/1A2 perpetrators using a physiologically-based pharmacokinetic (PBPK) approach. BMC Pharmacol. Toxicol. 25 (1), 4. doi:10.1186/s40360-023-00726-2
Jiang, P., Chen, T., Chu, L. F., Xu, R. P., Gao, J. T., Wang, L., et al. (2023). Enhancing drug-drug interaction prediction by integrating physiologically-based pharmacokinetic model with fraction metabolized by CYP3A4. Expert Opin. Drug Metab. Toxicol. 19 (10), 721–731. doi:10.1080/17425255.2023.2263358
Kosoglou, T., Statkevich, P., Kumar, B., Xuan, F., Schiller, J. E., Johnson-Levonas, A. O., et al. (2013). The effect of multiple doses of ketoconazole or rifampin on the single- and multiple-dose pharmacokinetics of vorapaxar. J. Clin. Pharmacol. 53 (5), 540–549. doi:10.1002/jcph.20
Lehtinen, T., Tolonen, A., Turpeinen, M., Uusitalo, J., Vuorinen, J., Lammintausta, R., et al. (2013). Effects of cytochrome P450 inhibitors and inducers on the metabolism and pharmacokinetics of ospemifene. Biopharm. Drug Dispos. 34 (7), 387–395. doi:10.1002/bdd.1853
Liu, Y., Zhou, S., Wan, Y., Wu, A., and Palmisano, M. (2014). The impact of co-administration of ketoconazole and rifampicin on the pharmacokinetics of apremilast in healthy volunteers. Br. J. Clin. Pharmacol. 78 (5), 1050–1057. doi:10.1111/bcp.12448
Loos, U., Musch, E., Jensen, J. C., Mikus, G., Schwabe, H. K., and Eichelbaum, M. (1985). Pharmacokinetics of oral and intravenous rifampicin during chronic administration. Klin. Wochenschr 63 (23), 1205–1211. doi:10.1007/BF01733779
Luedtke, D., Marzin, K., Jungnik, A., von Wangenheim, U., and Dallinger, C. (2018). Effects of ketoconazole and rifampicin on the pharmacokinetics of nintedanib in healthy subjects. Eur. J. Drug Metab. Pharmacokinet. 43 (5), 533–541. doi:10.1007/s13318-018-0467-9
Martin, P., Gillen, M., Millson, D., Oliver, S., Brealey, C., Grossbard, E. B., et al. (2016). Effects of CYP3A4 inhibitors ketoconazole and verapamil and the CYP3A4 inducer rifampicin on the pharmacokinetic parameters of fostamatinib: results from in vitro and phase I clinical studies. Drugs R. D. 16 (1), 81–92. doi:10.1007/s40268-015-0118-4
Mendell, J., Chen, S., He, L., Desai, M., and Parasramupria, D. A. (2015). The effect of rifampin on the pharmacokinetics of edoxaban in healthy adults. Clin. Drug Investig. 35 (7), 447–453. doi:10.1007/s40261-015-0298-2
Morita, T. O., and Hanada, K. (2022). Physiologically based pharmacokinetic modeling of ponatinib to describe drug-drug interactions in patients with cancer. Cancer Chemother. Pharmacol. 90 (4), 315–323. doi:10.1007/s00280-022-04466-8
Mukai, M., Uchimura, T., Zhang, X., Greene, D., Vergeire, M., and Cantillon, M. (2018). Effects of rifampin on the pharmacokinetics of a single dose of istradefylline in healthy subjects. J. Clin. Pharmacol. 58 (2), 193–201. doi:10.1002/jcph.1003
Murphy, P. A., Kern, S. E., Stanczyk, F. Z., and Westhoff, C. L. (2005). Interaction of St. John's Wort with oral contraceptives: effects on the pharmacokinetics of norethindrone and ethinyl estradiol, ovarian activity and breakthrough bleeding. Contraception 71 (6), 402–408. doi:10.1016/j.contraception.2004.11.004
Nam, S. H., Oh, J. S., Hong, S., Shim, T. S., Lee, C. K., Yoo, B., et al. (2020). Early discontinuation of tofacitinib in patients with rheumatoid arthritis co-treated with rifampin for latent tuberculosis. Jt. Bone Spine 87 (5), 475–479. doi:10.1016/j.jbspin.2020.04.010
Narasimhan, N. I., Dorer, D. J., Davis, J., Turner, C. D., and Sonnichsen, D. (2015). Evaluation of the effect of multiple doses of rifampin on the pharmacokinetics and safety of ponatinib in healthy subjects. Clin. Pharmacol. Drug Dev. 4 (5), 354–360. doi:10.1002/cpdd.182
Nassr, N., Huennemeyer, A., Herzog, R., von Richter, O., Hermann, R., Koch, M., et al. (2009). Effects of rifampicin on the pharmacokinetics of roflumilast and roflumilast N-oxide in healthy subjects. Br. J. Clin. Pharmacol. 68 (4), 580–587. doi:10.1111/j.1365-2125.2009.03478.x
Ogasawara, K., Kam, J., Thomas, M., Liu, L., Liu, M., Xue, Y., et al. (2021). Effects of strong and moderate CYP3A4 inducers on the pharmacokinetics of fedratinib in healthy adult participants. Cancer Chemother. Pharmacol. 88 (3), 369–377. doi:10.1007/s00280-021-04292-4
Ogilvie, B. W., Torres, R., Dressman, M. A., Kramer, W. G., and Baroldi, P. (2015). Clinical assessment of drug-drug interactions of tasimelteon, a novel dual melatonin receptor agonist. J. Clin. Pharmacol. 55 (9), 1004–1011. doi:10.1002/jcph.507
Ono, C., Hsyu, P. H., Abbas, R., Loi, C. M., and Yamazaki, S. (2017). Application of physiologically based pharmacokinetic modeling to the understanding of bosutinib pharmacokinetics: prediction of drug-drug and drug-disease interactions. Drug Metab. Dispos. 45 (4), 390–398. doi:10.1124/dmd.116.074450
Peloquin, C. A., Jaresko, G. S., Yong, C. L., Keung, A. C., Bulpitt, A. E., and Jelliffe, R. W. (1997). Population pharmacokinetic modeling of isoniazid, rifampin, and pyrazinamide. Antimicrob. Agents Chemother. 41 (12), 2670–2679. doi:10.1128/AAC.41.12.2670
Pithavala, Y. K., Tortorici, M., Toh, M., Garrett, M., Hee, B., Kuruganti, U., et al. (2010). Effect of rifampin on the pharmacokinetics of Axitinib (AG-013736) in Japanese and Caucasian healthy volunteers. Cancer Chemother. Pharmacol. 65 (3), 563–570. doi:10.1007/s00280-009-1065-y
Prueksaritanont, T., Chu, X., Gibson, C., Cui, D., Yee, K. L., Ballard, J., et al. (2013). Drug-drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 15 (3), 629–645. doi:10.1208/s12248-013-9470-x
Ren, H. C., Sai, Y., Chen, T., Zhang, C., Tang, L., and Yang, C. G. (2021). Predicting the drug-drug interaction mediated by CYP3A4 inhibition: method development and performance evaluation. AAPS J. 24 (1), 12. doi:10.1208/s12248-021-00659-w
Rowland Yeo, K., Gil Berglund, E., and Chen, Y. (2024). Dose optimization informed by PBPK modeling: state-of-the art and future. Clin. Pharmacol. Ther. 116 (3), 563–576. doi:10.1002/cpt.3289
Seo, S. W., Han, D. G., Choi, E., Park, T., Byun, J. H., Cho, H. J., et al. (2022). Development and application of a physiologically based pharmacokinetic model for entrectinib in rats and scale-up to humans: route-dependent gut wall metabolism. Biomed. Pharmacother. 146, 112520. doi:10.1016/j.biopha.2021.112520
Shi, J. G., Chen, X., Emm, T., Scherle, P. A., McGee, R. F., Lo, Y., et al. (2012). The effect of CYP3A4 inhibition or induction on the pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in healthy volunteers. J. Clin. Pharmacol. 52 (6), 809–818. doi:10.1177/0091270011405663
Shi, J. G., Fraczkiewicz, G., Williams, W. V., and Yeleswaram, S. (2015). Predicting drug-drug interactions involving multiple mechanisms using physiologically based pharmacokinetic modeling: a case study with ruxolitinib. Clin. Pharmacol. Ther. 97 (2), 177–185. doi:10.1002/cpt.30
Shumaker, R. C., Aluri, J., Fan, J., Martinez, G., Thompson, G. A., and Ren, M. (2014). Effect of rifampicin on the pharmacokinetics of lenvatinib in healthy adults. Clin. Drug Investig. 34 (9), 651–659. doi:10.1007/s40261-014-0217-y
Skolnick, J. L., Stoler, B. S., Katz, D. B., and Anderson, W. H. (1976). Rifampin, oral contraceptives, and pregnancy. JAMA 236 (12), 1382. doi:10.1001/jama.1976.03270130044027
Smith, D. A., and Schmid, E. F. (2006). Drug withdrawals and the lessons within. Curr. Opin. Drug Discov. Devel 9 (1), 38–46.
Statelova, M., Holm, R., Fotaki, N., Reppas, C., and Vertzoni, M. (2023). Usefulness of the beagle model in the evaluation of paracetamol and ibuprofen exposure after oral administration to pediatric populations: an exploratory study. Mol. Pharm. 20 (6), 2836–2852. doi:10.1021/acs.molpharmaceut.2c00926
Umehara, K., Huth, F., Jin, Y., Schiller, H., Aslanis, V., Heimbach, T., et al. (2019). Drug-drug interaction (DDI) assessments of ruxolitinib, a dual substrate of CYP3A4 and CYP2C9, using a verified physiologically based pharmacokinetic (PBPK) model to support regulatory submissions. Drug Metab. Pers. Ther. 34 (2). doi:10.1515/dmpt-2018-0042
Vakkalagadda, B., Frost, C., Byon, W., Boyd, R. A., Wang, J., Zhang, D., et al. (2016). Effect of rifampin on the pharmacokinetics of apixaban, an oral direct inhibitor of factor xa. Am. J. Cardiovasc Drugs 16 (2), 119–127. doi:10.1007/s40256-015-0157-9
Xu, H., O'Gorman, M., Tan, W., Brega, N., and Bello, A. (2015). The effects of ketoconazole and rifampin on the single-dose pharmacokinetics of crizotinib in healthy subjects. Eur. J. Clin. Pharmacol. 71 (12), 1441–1449. doi:10.1007/s00228-015-1945-5
Yamashita, F., Sasa, Y., Yoshida, S., Hisaka, A., Asai, Y., Kitano, H., et al. (2013). Modeling of rifampicin-induced CYP3A4 activation dynamics for the prediction of clinical drug-drug interactions from in vitro data. PLoS One 8 (9), e70330. doi:10.1371/journal.pone.0070330
Keywords: drug interactions, rifampicin, CYP3A enzyme, PBPK, pharmacokinetics
Citation: Yang C-G, Chen T, Si W-T, Wang A-H, Ren H-C and Wang L (2025) High-performance PBPK model for predicting CYP3A4 induction-mediated drug interactions: a refined and validated approach. Front. Pharmacol. 16:1521068. doi: 10.3389/fphar.2025.1521068
Received: 01 November 2024; Accepted: 06 February 2025;
Published: 26 February 2025.
Edited by:
Huilin Ma, Bristol Myers Squibb, United StatesReviewed by:
Jianchang Qian, Wenzhou Medical University, ChinaCopyright © 2025 Yang, Chen, Si, Wang, Ren and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Teng Si, c3d0ZW5nNTE2QDE2My5jb20=; An-Hai Wang, d2FoMDYxMDAzQDE2My5jb20=; Hong-Can Ren, cmVuaG9uZ2NhbkBnbWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.