 Yingpeng Li
Yingpeng Li- 1College of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 2College of Integrative Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
BRD4 inhibitors have demonstrated promising potential in cancer therapy. However, their therapeutic efficacy in breast cancer varies depending on the breast cancer subtype, particularly in the treatment of TNBC. In this study, we designed and synthesized 94 derivatives of 4-(3-(3,5-dimethylisoxazol-4-yl)benzyl)phthalazin-1(2H)-one to evaluate their inhibitory activities against BRD4. Notably, compound DDT26 exhibited the most potent inhibitory effect on BRD4, with an IC50 value of 0.237 ± 0.093 μM. DDT26 demonstrated significant anti-proliferative activity against both TNBC cell lines and MCF-7 cells. Intriguingly, the phthalazinone moiety of DDT26 mimicked the PAPR1 substrate, resulting in DDT26 displaying a moderate inhibitory effect on PARP1 with an IC50 value of 4.289 ± 1.807 μM. Further, DDT26 was shown to modulate the expression of c-MYC and γ-H2AX, induce DNA damage, inhibit cell migration and colony formation, and arrest the cell cycle at the G1 phase in MCF-7 cells. Our findings present potential lead compounds for the development of potent anti-breast cancer agents targeting BRD4.
1 Introduction
In 2022, breast cancer remained a significant health concern, with China and the United States reporting 429,105 and 259,827 new cases, respectively (Giaquinto et al., 2022; Xia et al., 2022). In both nations, breast cancer stands as the predominant cancer among women. Traditional chemotherapeutic agents, including platinum, anthracycline, and paclitaxel, continue to be the primary drugs for adjuvant and neo-adjuvant chemotherapy in breast cancer treatment. However, resistance to these drugs is not uncommon, often leading to a grim prognosis. Advanced patients with systemic recurrence or metastasis frequently require high-dose chemotherapy, which unfortunately can result in severe adverse reactions (Feng et al., 2023; Hussen et al., 2023; Obidiro et al., 2023). Given these challenges, there is a pressing need and significant clinical value in developing novel therapeutic agents with different mechanisms of action as alternatives to traditional chemotherapy.
Bromodomain-containing protein 4 (BRD4) possesses two bromodomains at its N-terminal, enabling it to bind acetylated histones and non-histones, thereby influencing gene transcription, cell cycle regulation, and apoptosis (Cochran et al., 2019). As a transcriptional coactivator, BRD4 has been implicated in the aberrant expression of several oncogenes in breast cancer cells, including c-MYC, p53, FOXM1, and NF-κB (Pérez-Salvia et al., 2017; Pérez-Peña et al., 2018; Yang et al., 2021; Zhou et al., 2022). Furthermore, BRD4 has been shown to upregulate PD-L1, aiding breast cancer cells in evading immune surveillance (Jing et al., 2020). Collectively, these findings position BRD4 as a promising therapeutic target with the potential to revolutionize breast cancer chemotherapy (Sahni and Keri, 2018).
As illustrated in Figure 1, since the introduction of JQ1 by Mitsubishi in 2010 as the first efficient BRD4 inhibitor, both academic and industrial sectors have dedicated significant efforts to develop an array of BRD4 inhibitors for breast cancer treatment, with a particular focus on triple-negative breast cancer (TNBC) (Gajjela and Zhou, 2023). Regrettably, no BRD4 inhibitors have yet cleared clinical trials to gain approval as cancer chemotherapy agents. A few BRD4 inhibitors are currently in phase I/II clinical trial stages for breast cancer treatment, such as OTX-015, ABBV-075 and I-BET762.
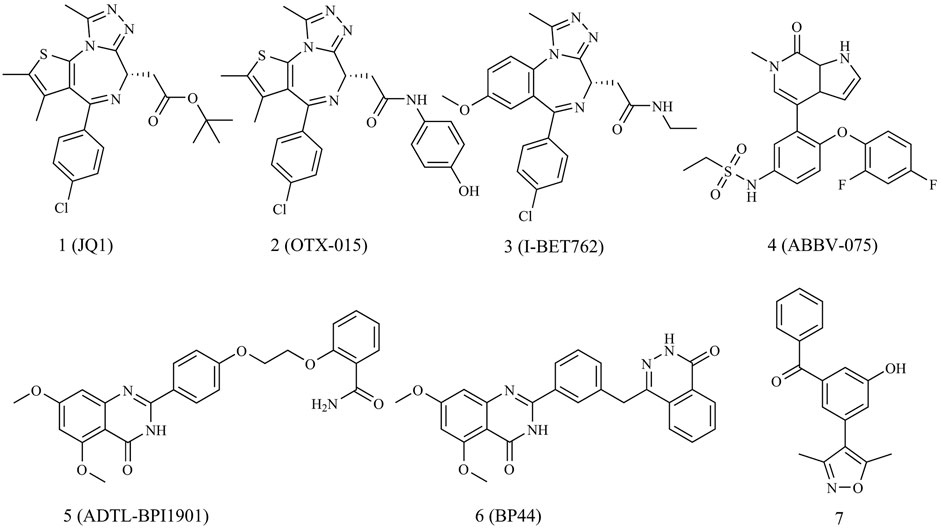
FIGURE 1. The structures of BRD4 inhibitors.
Given the multifaceted roles of BRD4, combinations of BRD4 inhibitors with other drugs have showcased advantages, including potent therapeutic effects, wide safety index and reduced susceptibility to drug resistance in cancer treatments (Jin et al., 2022). Dual-target BRD4 inhibitors have exhibited synergistic effects in breast cancer therapy. For instance, Ouyang’s group reported dual-target inhibitors of PARP1 and BRD4, namely, ADTL-BPI1901 and BP44, which demonstrated excellent antitumor efficacy against TNBC with a high safety profile (Chang et al., 2021; Zhang et al., 2022).
In 2013, Hewings’s group designed and synthesized compound 7, which was proven to be a modest BRD4 inhibitor with an IC50 value of 0.544 μM against the BD1 domain of BRD4 (Hewings et al., 2013). After analyzing the binding mode of compound 7 and BRD4 through molecular docking, we believed that proper structural modification of compound 7 could improve its binding ability with BRD4 to obtain potent BRD4 inhibitors, which was conducive to our next research on anti-breast cancer. The pursuit of BRD4 inhibitors with novel core structures can cover broader chemical space, potentially enhancing drug-like properties and deepening our understanding of structure-activity relationships. In this study, we chose compound 7 as the lead compound to design a series of novel BRD4 inhibitors featuring the 4-(3-(3,5-dimethylisoxazol-4-yl)benzyl)phthalazin-1(2H)-one scaffold. We further investigated their anti-breast cancer efficacies and underlying mechanisms. Our findings offer valuable insights for drug research and development aimed at breast cancer.
2 Results and discussion
2.1 Design
Analysis of the binding modes between compound 7 and BRD4 through molecular docking revealed several key interactions. As shown in Figures 2A, B, the 3,5-dimethylisoxazole moiety, serving as a ε-N-lysine acetylation (KAc) motif mimic, established a hydrogen bond with Asn140 and a water-bridging hydrogen bond with Tyr97. The benzoyl group of compound 7 engaged in hydrophobic interactions with the “WPF shelf”, a hydrophobic region in the binding pocket which was composed of Trp81, Pro82, and Phe83. Interestingly, the hydroxyl group of compound 7 did not interact with BRD4 and was oriented toward the ZA channel, a hydrophobic channel region which was composed of Pro82 to Leu91.
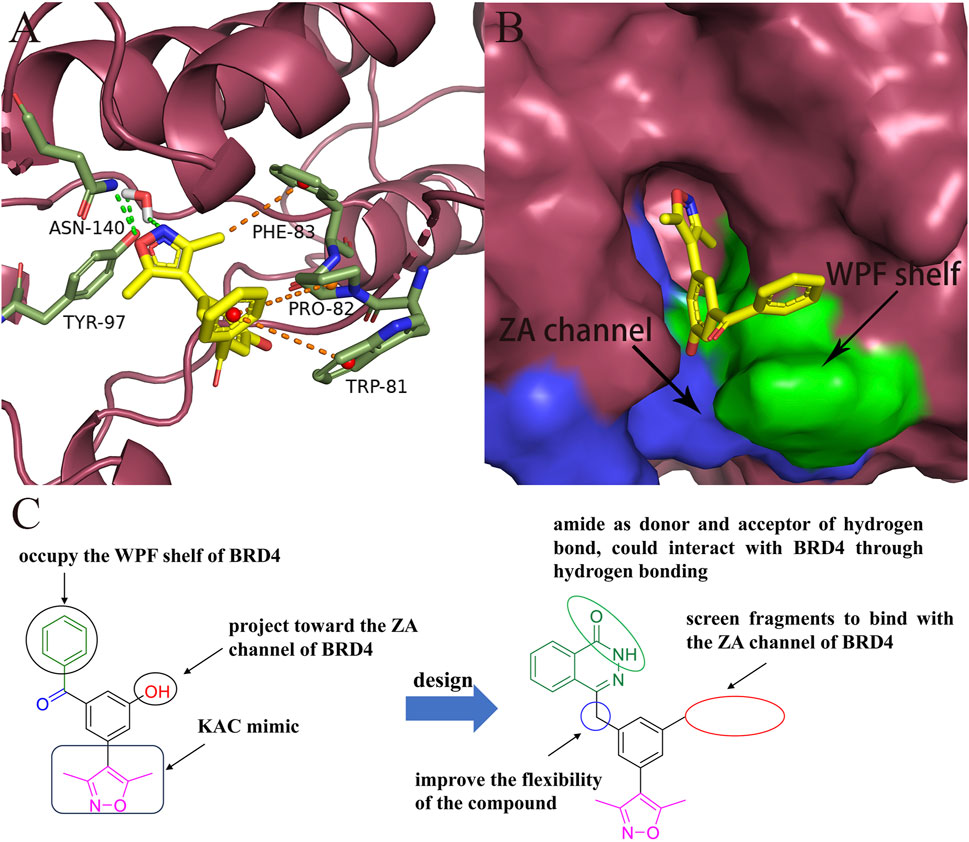
FIGURE 2. Design of novel BRD4 inhibitors based on lead compound 7. (A) Binding mode of compound 7 in the BD1(PDB ID: 4J0S) domain of BRD4. (B) The locations of ZA channel and WPF shelf in BRD4 protein. (C) Design strategy of novel BRD4 inhibitors.
As shown in Figure 2C, to enhance the binding affinity of compound 7 to BRD4, we employed several strategies. First, we replaced the benzene ring in compound 7 with phthalazinone. The lactam moiety of phthalazinone, acting as both hydrogen bond donor and acceptor, could form hydrogen bonds with BRD4. Second, we substituted the carbonyl group of compound 7 with a methylene group. This modification was aimed at increasing the compound’s flexibility, thereby facilitating a more favorable conformation for phthalazinone to bind with BRD4. Finally, we replaced the hydroxyl group of compound 7 with various fragments to interact with ZA channel of BRD4.
2.2 Biological screening
2.2.1 Enzymatic activities against BRD4
The inhibitory effects of all synthesized compounds on BRD4 were assessed at a concentration of 1 μM, and the structure-activity relationship (SAR) will be discussed in detail. As presented in Table 1, the amide compound DDT01 displayed no inhibitory activity against BRD4. This observation prompted us to introduce various substituents on the nitrogen atom to enhance the inhibitory potential. When the methyl group in DDT02 was substituted with cyclopentyl or cyclohexyl, there was a marked improvement in inhibition (as seen in DDT03 and DDT04). Compounds DDT05-DDT07 exhibited weak inhibitory effects, suggesting that the aromatic structure might be detrimental to potency. Modifying the methylene group in the cyclohexane ring of DDT04 with either oxygen or sulfone led to decreased inhibitory effects (as observed in DDT08 and DDT09). Introducing hydroxyl groups into the cyclohexane ring of DDT04 yielded two optical isomers, DDT10 and DDT11. Notably, DDT10 demonstrated a higher inhibitory rate than DDT11, indicating the stereochemistry of the chiral carbon influenced the compound’s potency. Interestingly, both DDT13 and DDT14, derived from the reduction of the carbonyl group in DDT12, exhibited enhanced inhibitory effects on BRD4. However, the stereochemistry of DDT13 and DDT14 did not seem to influence their inhibitory potential. Lastly, most compounds synthesized by substituting cyclohexylamine with either piperidine or piperazine lost their inhibitory effects on BRD4, with the exceptions being DDT15 and DDT16.

TABLE 1. Inhibition rates of compounds DDT01-DDT21 against BRD4.
As depicted in Table 2, compounds synthesized by reversing the amide orientation in DDT05 generally displayed high inhibitory rates against BRD4. The positions of the substituents had a more pronounced effect on inhibitory activity than their electronic properties. Specifically, ortho-substituted derivatives exhibited the most potent inhibitory activity. Upon removal of the carbonyl group’s oxygen in DDT22, a series of benzyl-substituted amine derivatives were synthesized. However, most of these derivatives showed weak inhibitory effects against BRD4, with the exceptions being DDT47-DDT49.
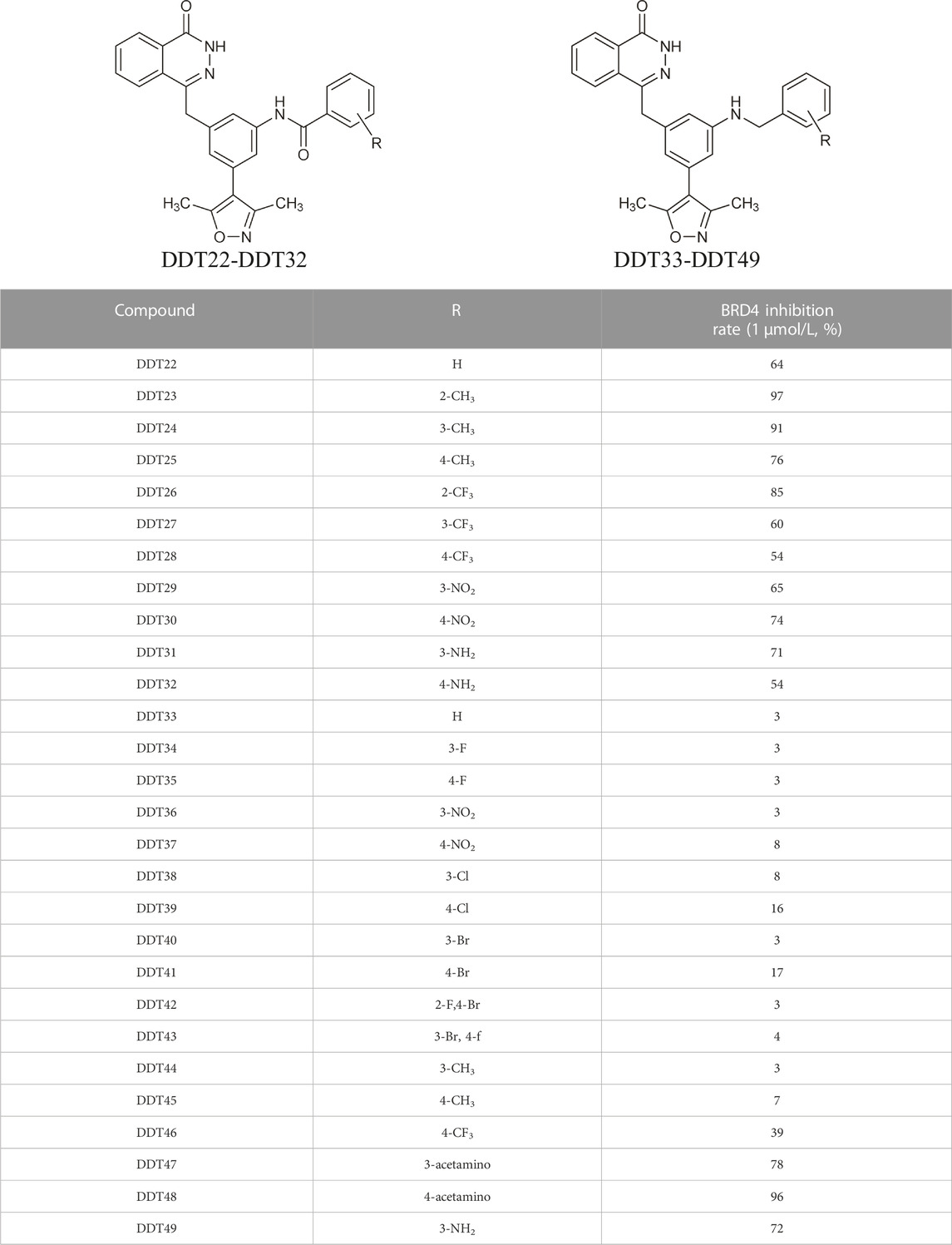
TABLE 2. Inhibition rates of compounds DDT22-DDT49 against BRD4.
Table 3 presents a series of sulfonamide derivatives and their inhibitory effects on BRD4. Excluding DDT50, alkyl-substituted sulfonamides did not exhibit inhibitory activity against BRD4. A subset of benzene sulfonamide compounds was then evaluated. While the 3-methyl and 4-methyl substituted compounds demonstrated potent inhibitory effects, the 3,4-dimethyl and 3,5-dimethyl derivatives showed reduced activity (refer to DDT58-DDT61). Introducing larger substituents in place of the 4-methyl group led to decreased inhibitory effects (see DDT62-DTT65). DDT66, derived by substituting the benzene ring of DDT59 with pyridine, showed a marked reduction in inhibitory activity. Among the methoxy-substituted derivatives, the 4-methoxy compound DDT69 exhibited the highest inhibitory rate. Compounds DDT76-DTT78 displayed low inhibitory rates, suggesting that inserting a methylene group between the benzene and sulfonyl groups hindered BRD4 binding. Compounds DDT80-DDT83 and DDT94, with electron-withdrawing groups, showed weak inhibitory activities. Among halogenated derivatives, only the ortho and para chlorine-substituted compounds demonstrated potent inhibitory effects (refer to DDT87 and DDT89). These findings suggested that, for benzene sulfonamide derivatives, electron-donating groups and para substituents enhanced the inhibitory potency against BRD4.
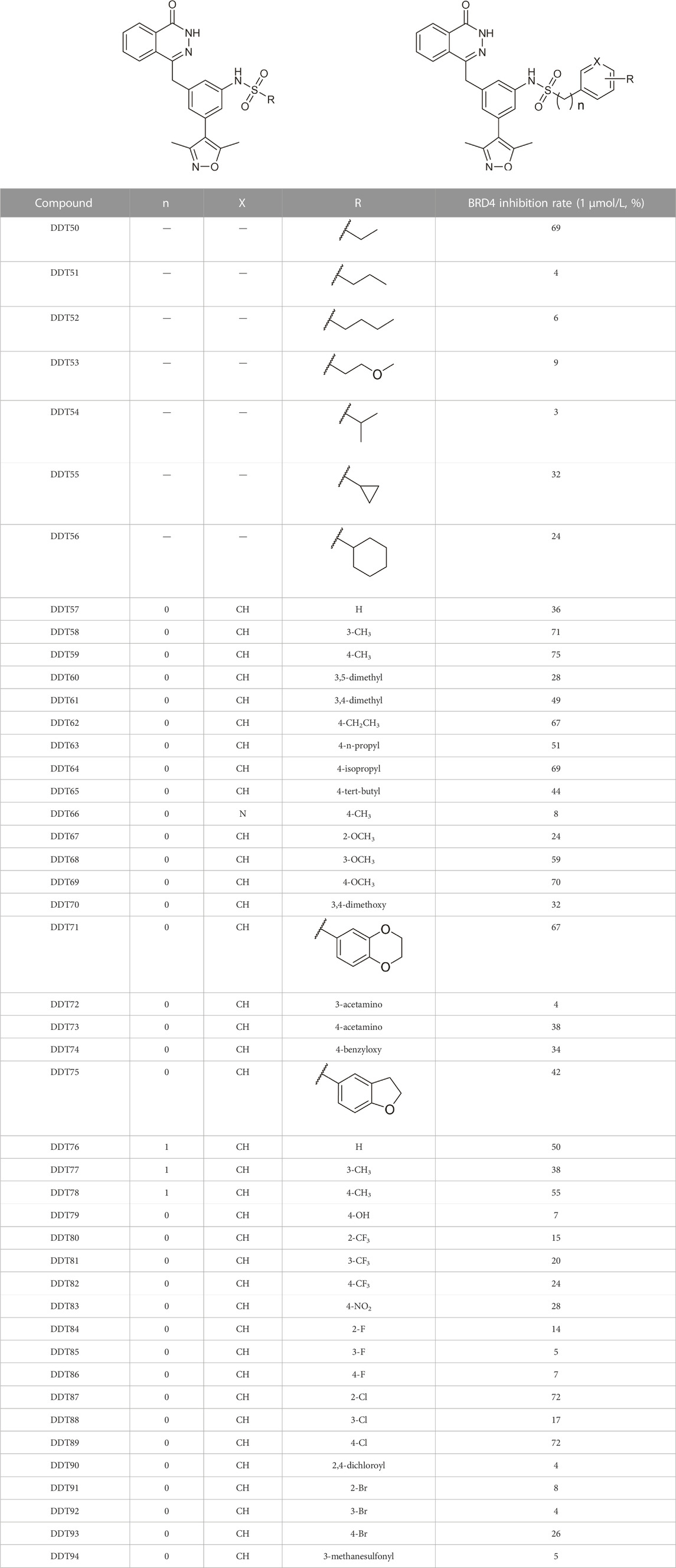
TABLE 3. Inhibition rates of compounds DDT50-DDT94 against BRD4.
We determined the IC50 values of several synthesized compounds for their inhibitory effects on BRD4. As presented in Table 4, among the compounds tested, DDT26 exhibited the most potent inhibitory activity against BRD4, with an IC50 value of 0.237 ± 0.093 μM. In subsequent investigations, we observed that DDT26 could induce DNA damage in MCF-7 cells. This observation led us to hypothesize that DDT26 might interact with targets other than BRD4, potentially interfering with cellular DNA damage repair mechanisms. Notably, the 4-benzylphthalazin-1(2H)-one structure in DDT26 bore resemblance to Olaparib, a known PARP1 inhibitor. Supporting this hypothesis, biochemical assays revealed that DDT26 indeed possessed a modest inhibitory effect on PARP1, with an IC50 value of 4.289 ± 1.807 μM. Interestingly, both DDT14 and DDT49 demonstrated stronger inhibitory activities against PARP1 than DDT26 when tested at a concentration of 0.5 μM.
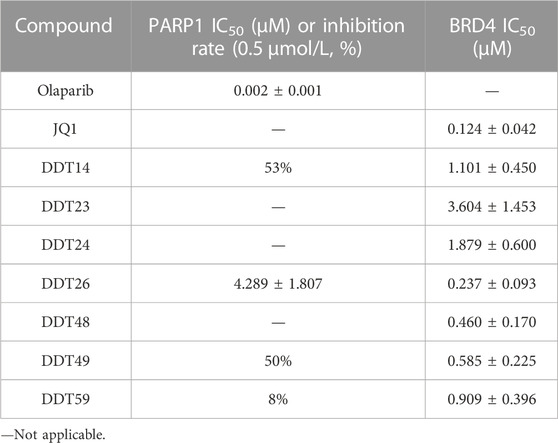
TABLE 4. IC50 values of target compounds against BRD4 and PARP1.
2.2.2 Cytotoxicity assay
Several compounds exhibiting strong inhibitory effects against BRD4 were evaluated for their anti-proliferative activities on three breast cancer cell lines, using JQ1 and Olaparib as positive controls. As detailed in Table 5, JQ1 demonstrated potent anti-proliferative effects on MDA-MB-231 and MDA-MB-468 cells. However, MCF-7 cells appeared to be resistant to JQ1. The anti-proliferative effects of Olaparib on three breast cancer cell lines were modest. Notably, among the synthesized compounds tested, DDT26 displayed robust inhibitory effects on all three breast cancer cell lines, and its inhibitory potency on MCF-7 cells surpassed that of JQ1.
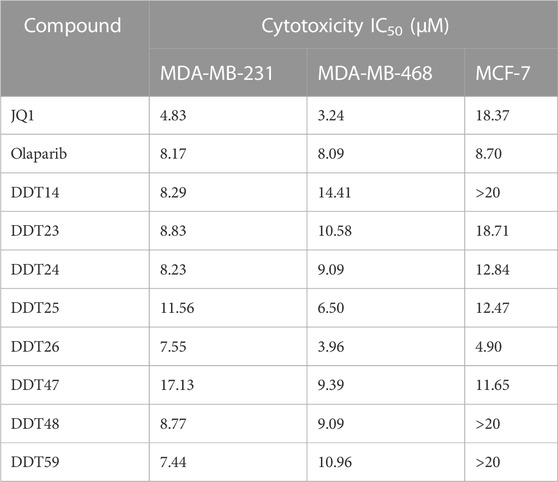
TABLE 5. In vitro anti-proliferative activity of target compounds against breast cancer cell lines.
2.2.3 Molecular docking study
Given the impressive inhibitory performance of DDT26 against both BRD4 and cell proliferation, we undertook a comprehensive analysis of this compound. Through molecular docking, we examined the binding modes of some compounds with BRD4 in Table 4. As shown in Figures 3A, B; Supplementary Table S1, the binding mode of DDT26 with BRD4 was quite similar to that of compound 7. The 3,5-dimethylisoxazole moiety of DDT26 snugly fit into the acetylated lysine binding pocket. The isoxazole’s oxygen formed a hydrogen bond with Asn140, its nitrogen engaged in a water-mediated hydrogen bond with Tyr97, and its methyl group participated in hydrophobic interactions with Pro82. Consistent with our design strategy, the 2-trifluoromethyl benzamide extended into the ZA channel where its benzene and trifluoromethyl groups established T-shaped pi-pi stacking and hydrophobic interactions with Trp81, respectively. Intriguingly, the phthalazinone moiety of DDT26 did not align with the WPF shelf but formed a hydrogen bond with Met149.
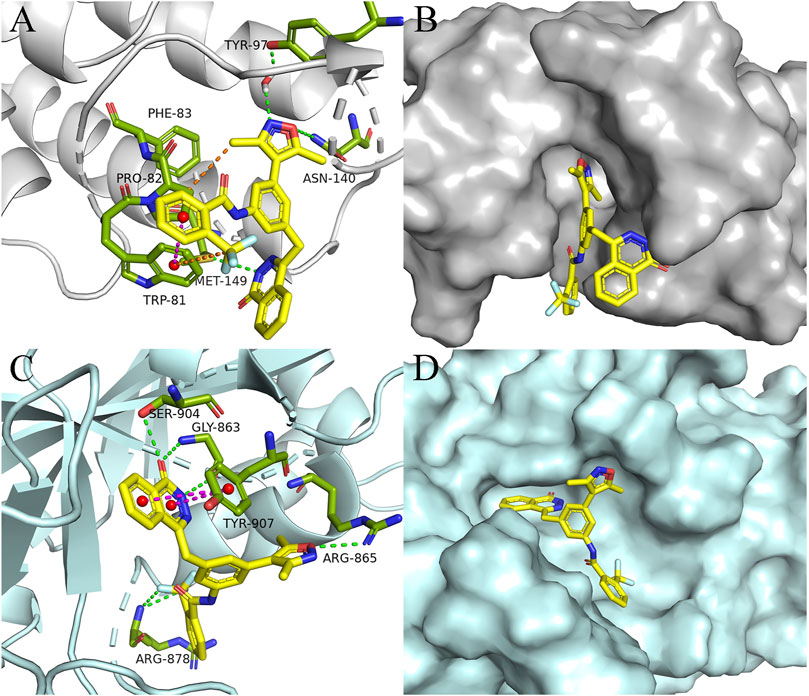
FIGURE 3. Binding mode analysis of DDT26. (A) Binding mode of DDT26 in the BD1(PDB ID: 4J0S) domain of BRD4. (B) Conformation of DDT26 at the binding site of BRD4. (C) Binding mode of DDT26 with PARP1 (PDB ID: 5DS3). (D) Conformation of DDT26 at the binding site of PARP1.
Biochemical experiment showed that DDT26 was a PARP1 inhibitor, which prompted us to analyze the binding mode of DDT26 with PARP1. As shown in Figures 3C, D; Supplementary Table S2, the binding mode of DDT26 with PARP1 closely resembled that of Olaparib. The phthalazinone moiety of DDT26 formed hydrogen bonds with Gly863 and Ser904. Its aromatic ring engaged in face-to-face pi-pi stacking interaction with Tyr907. The isoxazole’s oxygen formed a hydrogen bond with Arg865, and the fluorine of trifluoromethyl group established a hydrogen bond with Arg878.
2.2.4 DDT26 modulated proteins expression in MCF-7 cells
c-Myc, a member of the Myc family of oncogenes, is known to promote cancer cell proliferation, metabolism, and protein synthesis. BRD4, functioning as a transcriptional regulator, plays a role in modulating the expression of c-MYC. As illustrated in Figure 4, Western blot analysis revealed that DDT26 could downregulate the expression of c-MYC in MCF-7 cells in a concentration-dependent manner. γ-H2AX is recognized as a marker for DNA double-strand damage. Western blot results also indicated that DDT26 induced the accumulation of γ-H2AX in MCF-7 cells in a concentration-dependent fashion.
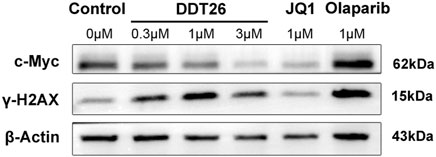
FIGURE 4. The effects of JQ1, Olaparib, and DDT26 on the expression of c-MYC and γ-H2AX analyzed by Western blot.
2.2.5 DDT26 induced G1 phase cell cycle arrest in MCF-7 cells
MCF-7 cells treated with compound DDT26 at concentrations of 0.3, 1, and 3 μΜ for 48 h displayed distinct cell cycle effects. As depicted in Figure 5, the proportions of cells in the G1 phase for the DDT26-treated groups were 53.8%, 59.8%, and 64.5%, respectively. This indicated that DDT26 induced a dose-dependent G1 phase cell cycle arrest in MCF-7 cells. At a concentration of 1 μΜ, the JQ1-treated group exhibited a more pronounced cell cycle arrest compared to the DDT26 group. In contrast, the Olaparib-treated group did not influence the MCF-7 cell cycle. The combination treatment group displayed a slightly reduced cell cycle arrest capability compared to the JQ1-treated group, suggesting that concurrent inhibition of BRD4 and PARP1 did not synergistically enhance cell cycle arrest.
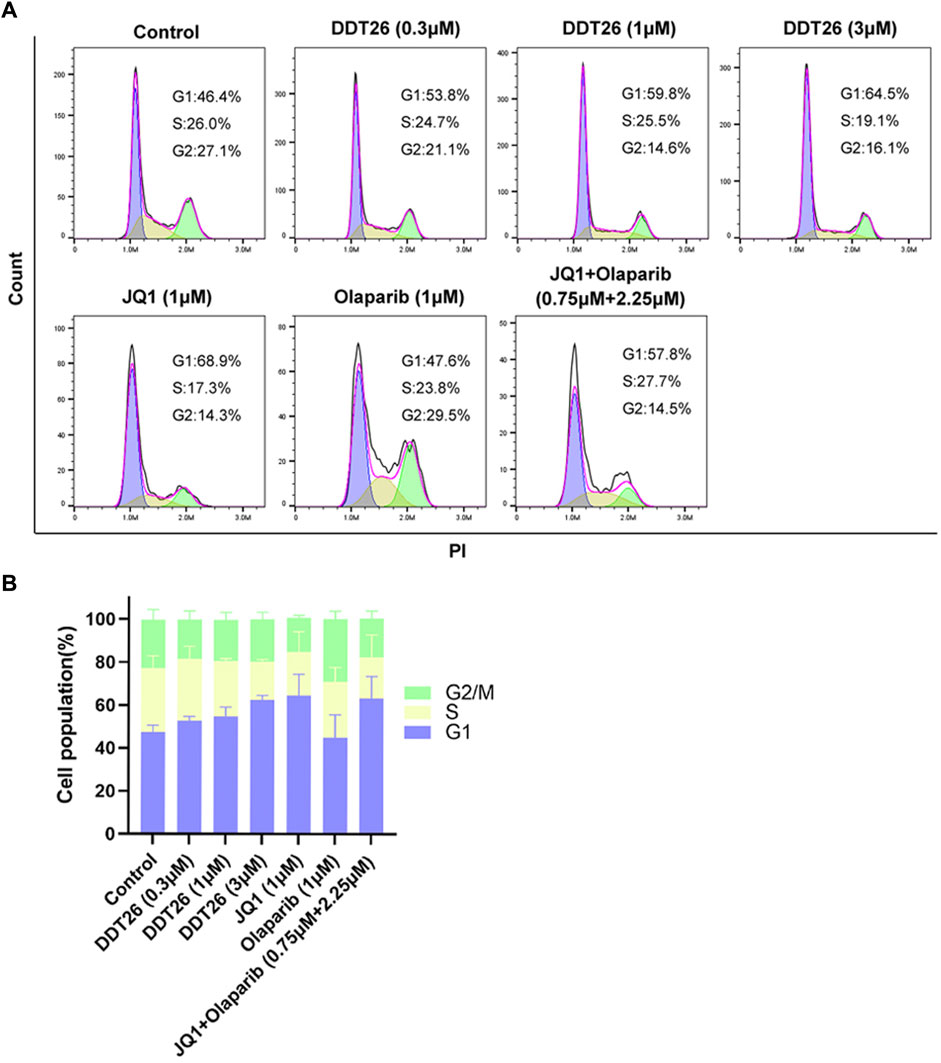
FIGURE 5. DDT26 induced cell cycle of MCF-7 arrest in the G1 phase. (A) Effects of JQ1(1 μM), Olaparib (1 μM), JQ1+Olaparib (0.75 μM + 2.25 μM) and DDT26 (0.3 μM, 1 μM, and 3 μM) on cell cycle progression in MCF-7 cells. (B) The percentage of cells in each population.
2.2.6 DDT26 suppressed colony formation in MCF-7 cells
Colony formation assay is an effective method to directly observe the anti-proliferation effect of compounds on cancer cells. As shown in Figure 6, at a concentration of 1 μM, JQ1’s ability to suppress colony formation surpassed that of both DDT26 and Olaparib. DDT26 inhibited colony formation in MCF-7 cells in a dose-dependent manner. The results of colony formation assay showed that DDT26 could effectively inhibit the proliferation of MCF-7 cells at concentrations above 3 μM.
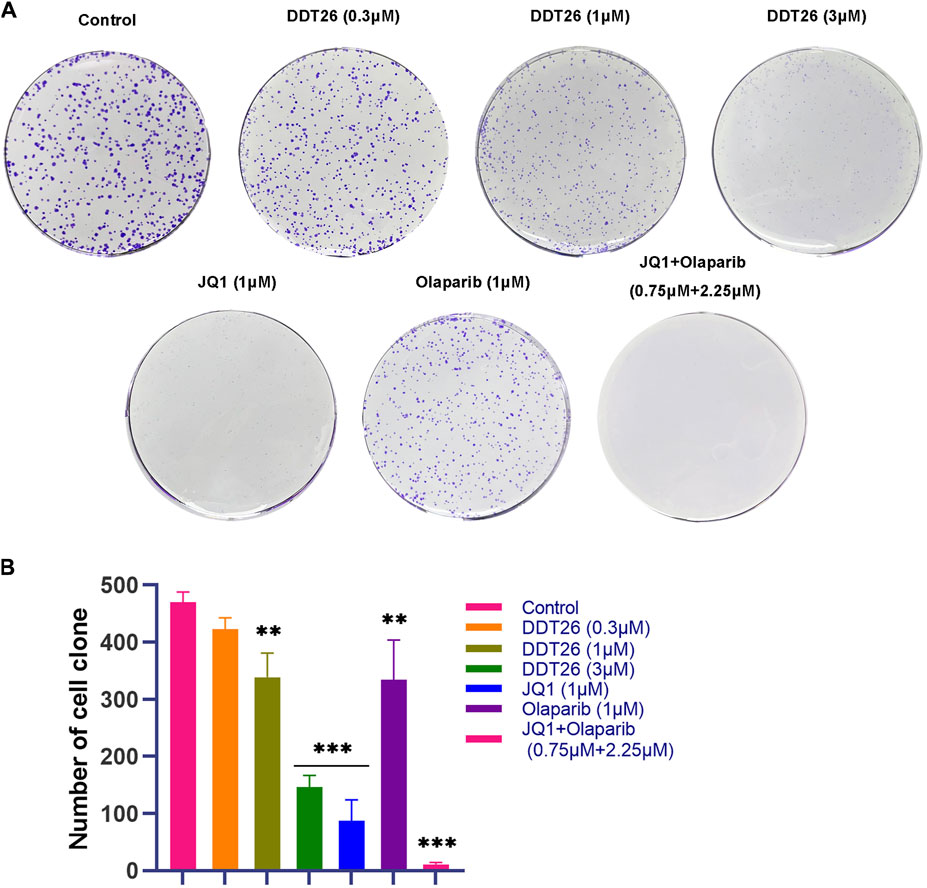
FIGURE 6. DDT26 inhibited colony formation in MCF-7 Cells. (A) Colony formation assay of MCF-7 cells treated with JQ1(1 μM), Olaparib (1 μM), JQ1 + Olaparib (0.75 μM + 2.25 μM) and DDT26 (0.3 μM, 1 μM, and 3 μM), respectively. (B) The number of cell clone in each population.
2.2.7 DDT26 inhibited MCF-7 cells migration
Enhanced migratory capability can lead to the metastasis of cancer cells from the primary site to other tissues via the blood or lymphatic system, culminating in the formation of new tumor lesions. This is a critical indicator of the progression of many cancers. We evaluated the effects of DDT26, JQ1, and Olaparib on the migratory capability of MCF-7 cells using a wound scratch assay. As depicted in Figure 7, the control group demonstrated that MCF-7 cells completely filled the wound area 48 h post-scratching. At a concentration of 1 μM, JQ1 exhibited the most potent inhibitory effect on MCF-7 cell migration. Compared to JQ1, DDT26’s inhibitory effect on MCF-7 cells migration was slightly less pronounced, while Olaparib showed no inhibitory activity. The migration-inhibitory effect of DDT26 on MCF-7 cells was further amplified at a concentration of 3 μM, underscoring DDT26’s concentration-dependent inhibitory action.
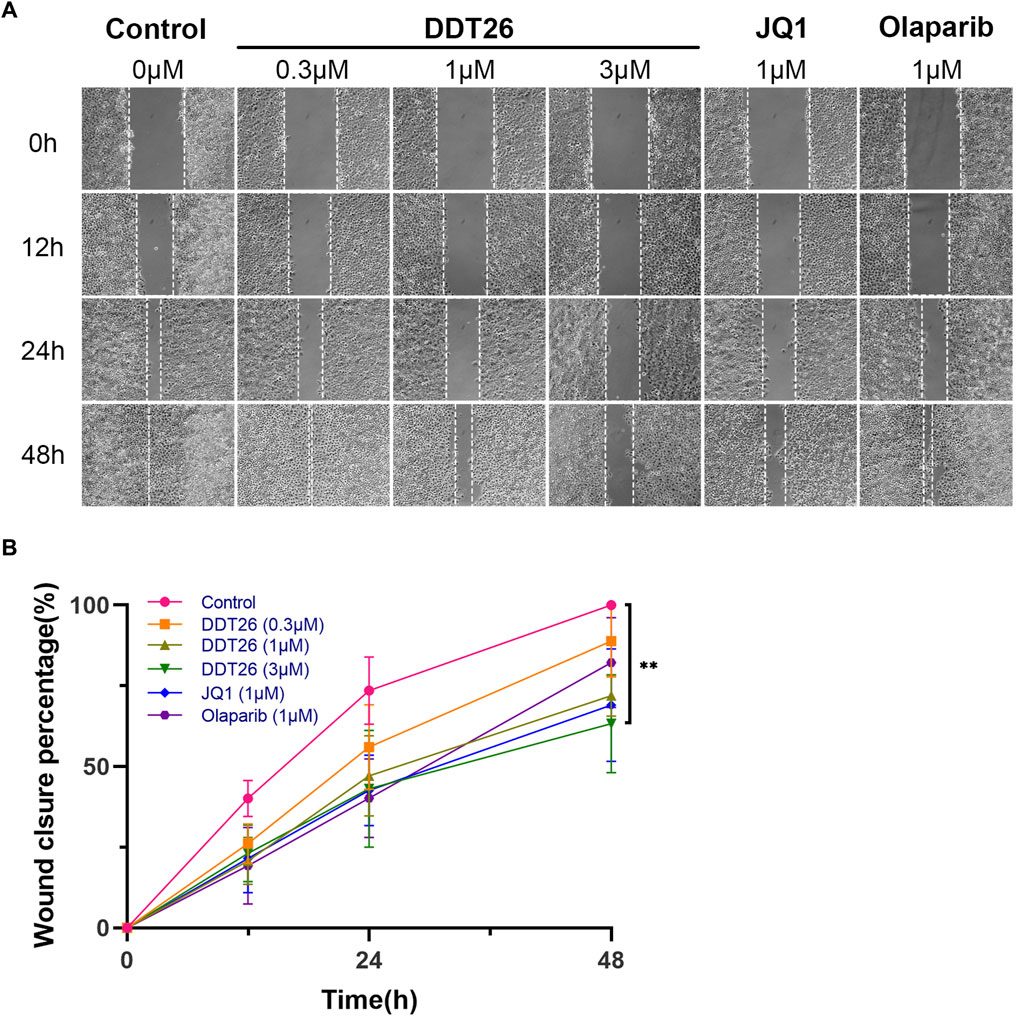
FIGURE 7. DDT26 inhibits the migration of MCF-7 cells in a concentration dependent manner. (A) Effects of DDT26, JQ1 and Olaparib on the migration of MCF-7 cells. (B) Wound closure percentage of DDT26 (0.3 μM, 1 μM, 3 μM), JQ1 (1 μM) and Olaparib (1 μM) in MCF-7 cells.
2.2.8 DDT26 induced DNA damage of MCF-7 cells
PARP1 acts as a sensor for DNA single-strand damage, while BRD4 plays a role in regulating the transcription of homologous recombination related proteins. Given the pivotal roles of both PARP1 and BRD4 in DNA damage repair, we employed a comet assay to investigate DDT26’s potential to induce DNA damage in MCF-7 cells. As illustrated in Figure 8, DDT26 induced DNA damage in MCF-7 cells in a dose-dependent manner. At a concentration of 3 μM, DDT26 inflicted more pronounced DNA damage than the combination treatment. These findings suggested that DDT26’s ability to inhibit MCF-7 cell proliferation was intrinsically linked to its DNA damage-inducing properties.
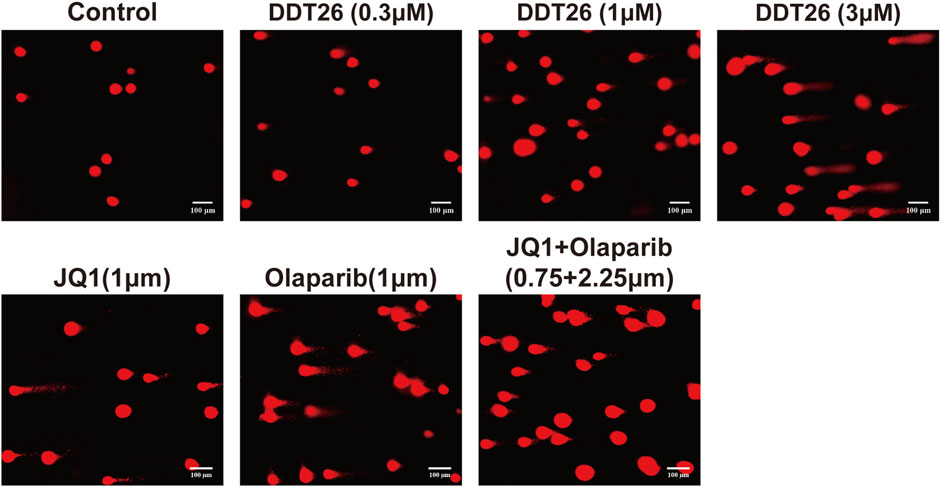
FIGURE 8. Effects of JQ1, Olaparib, JQ1+ Olaparib, DDT26 on the DNA damage of MCF-7 cells.
3 Conclusion
Breast cancer imposes a significant psychological and economic burden on patients. Consequently, the quest for effective targeted therapies for breast cancer remains at the forefront of drug research and development. Given the intricate classifications of breast cancer, not all types are responsive to BRD4 inhibitors. This underscores the need to identify novel BRD4 inhibitors with distinct mechanisms of action to broaden their therapeutic applicability in breast cancer treatment (Sahni and Keri, 2018).
In this study, we designed and synthesized a series of novel 4-(3-(3,5-dimethylisoxazol-4-yl)benzyl)phthalazin-1(2H)-one derivatives as potential BRD4 inhibitors. Among them, DDT26 emerged as the most potent, exhibiting robust inhibitory activity against BRD4 at sub-micromolar concentrations. Furthermore, biochemical analyses revealed that DDT26 also mildly inhibited PARP1, a critical target for treating breast cancers with BRCA1/2 mutations. Molecular docking studies highlighted the phthalazinone moiety in DDT26 as pivotal, facilitating interactions with Met149 of BRD4 and Gly863, Ser904, and Tyr907 of PARP1.
Interestingly, DDT26 demonstrated a more pronounced inhibitory effect on MCF-7 cells proliferation compared to JQ1, even though its direct inhibitory activity against BRD4 was somewhat inferior. We postulate that DDT26’s enhanced cytotoxicity in MCF-7 cells may stem from its dual-targeting capability, simultaneously inhibiting both BRD4 and PARP1 (Sun et al., 2018; Mio et al., 2019). Subsequent investigations confirmed DDT26’s ability to downregulate c-MYC expression dose-dependently, induce γ-H2AX accumulation, inhibit both migration and colony formation, and arrest the cell cycle in the G1 phase in MCF-7 cells. Collectively, these findings position DDT26 as a promising lead compound in the development of novel anti-breast cancer therapeutics.
4 Methods section
4.1 Chemistry
The synthetic pathways for the target compounds are depicted in Figure 9. Starting with commercially available 3,5-dibromobenzaldehyde (compound 8), it was coupled with boric ester (compound 9) to yield intermediate 10 through Suzuki reaction. This was followed by a palladium-catalyzed cyanation of intermediate 10 to produce intermediate 11. A subsequent Horner−Wadsworth−Emmons reaction between intermediate 11 and phosphonate resulted in olefin 12. Refluxing olefin 12 with hydrazine hydrate in ethanol led to the formation of the phthalazinone in intermediate 13. The pivotal intermediate 14 was synthesized by hydrolyzing the nitrile group of intermediate 13 under alkaline conditions. Ultimately, condensing intermediate 14 with the appropriate amines under standard conditions yielded amides DDT01-DDT21.
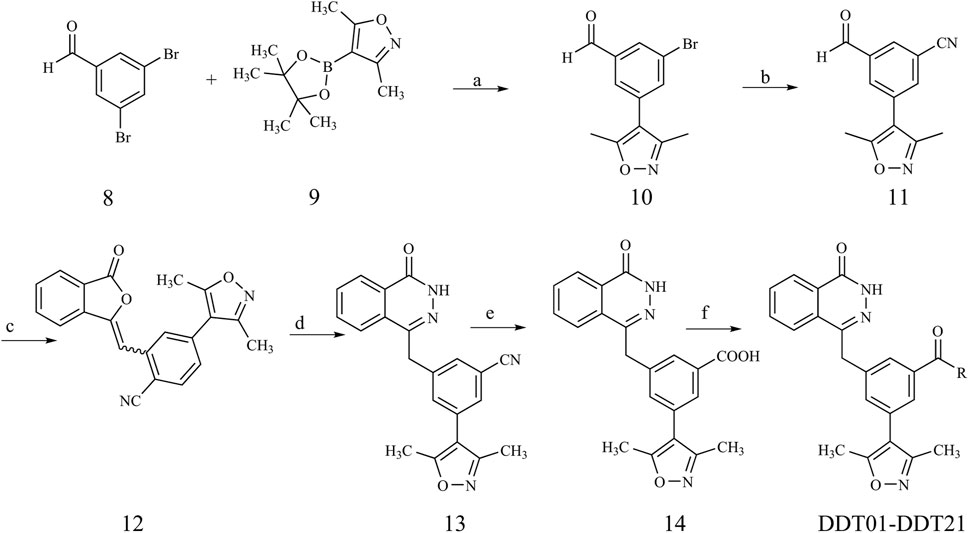
FIGURE 9. Synthetic route of compound DDT01-DDT21. Reagents and conditions: (a) Pd(PPh3)4, K2CO3, N2, dioxane/H2O (3:1), 90°C; (b) Zn(CN)2, Pd(PPh3)4, DMF, N2, 80°C; (c) Dimethyl (3-oxo-1,3-dihydroisobenzofuran-1-yl)phosphonate, Et3N, THF, rt; (d) NH2NH2·H2O, EtOH, reflux; (e) (i) NaOH, MeOH/H2O (1:1), 100°C (ii) HCl (2 N), rt; (f) EDCI, HOBT, DIPEA, DMF, rt.
The synthetic pathways for target compounds DDT22-DDT94 are illustrated in Figure 10. Intermediate 18 was synthesized following a procedure analogous to that used for intermediate 13. The essential intermediate 19 was derived by reducing the nitro group of intermediate 18 using zinc powder. This intermediate 19 was then reacted with the respective benzoyl chlorides to produce target compounds DDT22-DDT32. Additionally, NaBH3CN-mediated reductive amination reactions between intermediate 19 and the relevant benzaldehydes yielded target compounds DDT33-DDT49. Lastly, reactions between intermediate 19 and sulfonyl chlorides produced target compounds DDT50-DDT94. Detailed synthesis procedures and spectroscopic characterizations for compounds DDT01-DDT94 can be found in the Supplementary Material.
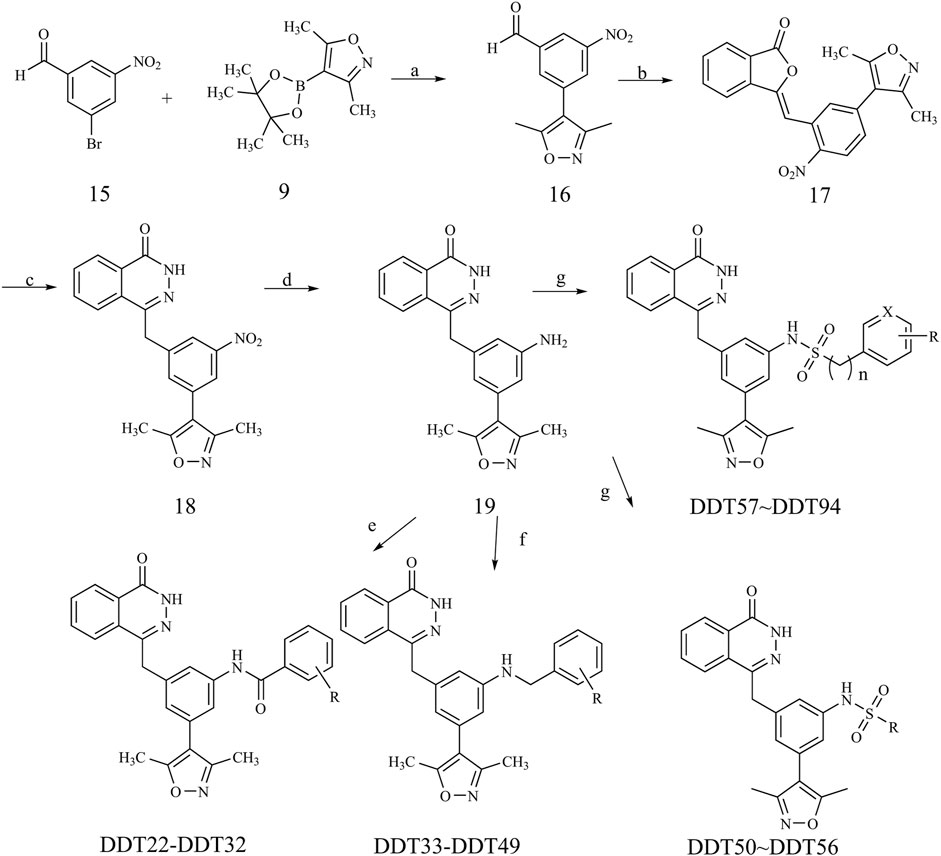
FIGURE 10. Synthetic route of compound DDT22-DDT94. Reagents and conditions: (a) Pd(PPh3)4, K2CO3, N2, dioxane/H2O (3:1), 90°C; (b) Dimethyl (3-oxo-1,3-dihydroisobenzofuran-1-yl)phosphonate, Et3N, THF, rt; (c) NH2NH2·H2O, EtOH, reflux; (d) Zn, NH4Cl, MeOH/H2O (3:1), 60°C; (e) DCM, DIPEA, rt; (f) NaBH3CN, MeOH, 80°C; (g) pyridine/DCM (1:1), rt.
4.2 Molecule docking
Crystal structures of BRD4 (PDB ID: 4J0S) and PARP1 (PDB ID: 5DS3) were chosen for molecular docking. The molecular docking studies were performed by the software Accelrys Discovery Studio (version 2016). The PARP1 protein structure was processed using the Protein Preparation module in this software, with all default parameters. The BRD4 protein structure was processed similarly to PARP1, except that a conserved water molecule (Water 311) was retained. The active sites were defined as spherical regions with a 10 Å diameter, centered on the ligand molecules. The Full Minimization module was employed to minimize the poses of compounds based on the CHARMm forcefield. Docking simulations were conducted using the CDOCKER module, in which twenty random conformations of per compound were generated. The top ten poses of per compound were saved based on CHARMm energy and the poses with the most favorable interactions were chosen for subsequent analysis.
4.3 Determination of inhibition rates and IC50 values against BRD4 and PARP1
The inhibitory effects of target compounds on BRD4 and PARP1 were assessed at Huawei Pharmaceutical Co. Ltd (Jinan, China).
The BRD4 activity assay utilized TR-FRET technology, employing a recombinant BRD4 (BD1 + BD2) and its corresponding ligand. The TR-FRET signal generated in the assay is proportional to the ligand’s binding to the bromodomains. All reactions contained a final DMSO concentration of 1%. All binding reactions were conducted at room temperature. Each 20 µL reaction mixture, in assay buffer, comprised bromodomains, the BET Ligand, and the specified inhibitor amount. For the negative control, 5 µL of assay buffer replaced the BET ligand. After a 120-min incubation with the ligand, the TR-FRET signal was captured using a Tecan Infinite M1000 plate reader. IC50 values were determined through nonlinear regression with a normalized dose-response fit in Prism GraphPad software.
The inhibition of PARP1 enzymatic activity by the tested compounds was gauged using an ELISA in 96-well plates. Each well was pre-coated with histone (20 μg/mL) in 100 μL of PBS buffer and incubated overnight at 4 °C. Subsequently, NAD+ (100 μM), biotinylated NAD+ (25 μM), and slDNA (200 nM) in 30 μL of reaction buffer were added to each well. This was followed by the addition of 5 μL of either the compound at varying concentrations or a solvent control. The reaction was initiated by adding 20 μL of PARP (50 ng/well) and incubating at 30°C for 1 h. Post-reaction, 50 µL of streptavidin-conjugated HRP was introduced, and the assay continued at 30°C for an additional 30 min. Finally, 100 μL of a solution containing H2O2 and luminol in 0.1 M citrate buffer (pH 5.4) was added, and the luminescent signal was recorded using a Molecular Devices SpectraMax M5 microplate reader. The inhibition rate of PARP1 enzymatic activity was determined as: (Lu control −Lu treated/Lu control) ×100%. IC50 values, representing the concentration required for 50% inhibition of PARP1 enzymatic activity, were determined using nonlinear regression with a normalized dose−response fit in Prism GraphPad software.
4.4 Cell culture
MDA-MB-468, MDA-MB-231, and MCF-7 cell lines were sourced from the National Collection of Authenticated Cell Culture, China. Cells were maintained in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin, and incubated at 37°C in a humidified atmosphere containing 5% CO2.
4.5 Cell viability assay
The viability of cells was assessed using the MTT assay. Cells were seeded at a density of 3×103 cells per well in a 96-well plate and treated with varying concentrations (0.05, 0.2, 0.8, 3.2, 6.4, 12.8 µM) of the compounds for 72 h. Post-incubation, 100 µL of MTT solution (0.5 mg/mL) was added to each well and further incubated for 4 h. The resulting formazan crystals were dissolved in 150 µL DMSO, and the absorbance was measured at 570 nm using a microplate reader.
4.6 Western blot analysis
For Western blotting, cells were treated with compound DDT26, JQ1, and Olaparib for a duration of 24 h. Cells were then lysed using RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentrations were determined using the BCA Protein Assay Kit. Equal amounts of protein (20–30 µg) were separated by SDS-polyacrylamide gel electrophoresis and subsequently transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked using 5% milk in PBS prior to incubation with primary antibodies. Primary antibodies against p-H2AX (Cell Signaling Technology™, US), c-MYC, and β-actin (Proteintech, US) were used. After overnight incubation at 4°C, membranes were washed and incubated with HRP-conjugated secondary antibodies for 1 h. Protein bands were visualized using ECL reagents.
4.7 Cell cycle analysis
The cell cycle distribution was assessed using the DNA Content Quantitation Assay Kit (Solarbio, China). MCF-7 cells were seeded in six-well plates and exposed to different concentrations of DDT26, JQ1, Olaparib, or their combinations for 48 h. After treatment, cells were harvested, fixed in ice-cold 70% ethanol overnight at 4°C, and subsequently washed with PBS. Cells were then treated with 100 μL RNase at 37°C for 30 min and stained with 400 μL PI for 30 min at 4°C in the dark. The cell cycle distribution was analyzed using an Accuri C6 Plus flow cytometer (BD Bioscience, CA), and data interpretation was performed with FlowJo software.
4.8 Clonogenic assay
MCF-7 cells were seeded in 6-well plates at a density of 500 cells/well and allowed to grow for 8 days to facilitate colony formation. After incubation, cells were washed with PBS, fixed in methanol for 15 min, and stained with 1% crystal violet for an hour. Colonies were then imaged and counted.
4.9 Cell migration assay
MCF-7 cells were seeded in 6-well plates at a density of 5×105 cells/well and incubated overnight. A scratch was made in the cell monolayer using a 10 µL pipette tip. After creating the scratch, cells were washed with PBS and treated with DDT26, JQ1, or Olaparib. Images were captured at 0, 12, 24, and 48-h intervals.
4.10 Comet assay
The comet assay was performed under alkaline conditions using the Comet Assay Kit (Beyotime, China) as per the manufacturer’s instructions. Briefly, after treating cells with DDT26, JQ1, Olaparib, or their combinations for 24 h, they were harvested and mixed with 0.7% low melting point agarose at a 1:7 (v/v) ratio. This mixture was spread onto microscope slides, which were then submerged in lysis solution for an hour. Electrophoresis was conducted in a horizontal apparatus at 25 V and 300 mA. Post-electrophoresis, slides were stained with PI for DNA visualization, and images were captured using a fluorescence inverted microscope.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
YL: Writing–review and editing. XC: Writing–original draft, Methodology. YY: Methodology, Data curation, Writing–review and editing. HL: Investigation. HF: Data curation, Writing–review and editing. XF: Formal Analysis, Writing–review and editing. YD: Methodology, Writing–review and editing. JG: Writing–original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Tianjin Science and Technology Plan Project (21JCQNJC01740) and National Natural Science Foundation of China (82074030).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2023.1289003/full#supplementary-material
References
Chang, X., Sun, D., Shi, D., Wang, G., Chen, Y., Zhang, K., et al. (2021). Design, synthesis, and biological evaluation of quinazolin-4(3H)-one derivatives co-targeting poly(ADP-ribose) polymerase-1 and bromodomain containing protein 4 for breast cancer therapy. Acta Pharm. Sin. B 11, 156–180. doi:10.1016/j.apsb.2020.06.003
Cochran, A. G., Conery, A. R., and Sims, R. J. (2019). Bromodomains: a new target class for drug development. Nat. Rev. Drug Discov. 18, 609–628. doi:10.1038/s41573-019-0030-7
Feng, G., Zhou, X., Chen, J., Li, D., and Chen, L. (2023). Platinum drugs-related safety profile: the latest five-year analysis from FDA adverse event reporting system data. Front. Oncol. 12, 1012093. doi:10.3389/fonc.2022.1012093
Gajjela, B. K., and Zhou, M.-M. (2023). Bromodomain inhibitors and therapeutic applications. Curr. Opin. Chem. Biol. 75, 102323. doi:10.1016/j.cbpa.2023.102323
Giaquinto, A. N., Sung, H., Miller, K. D., Kramer, J. L., Newman, L. A., Minihan, A., et al. (2022). Breast cancer statistics, 2022. CA Cancer J. Clin. 72, 524–541. doi:10.3322/caac.21754
Hewings, D. S., Fedorov, O., Filippakopoulos, P., Martin, S., Picaud, S., Tumber, A., et al. (2013). Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. J. Med. Chem. 56, 3217–3227. doi:10.1021/jm301588r
Hussen, N. H., Hasan, A. H., Muhammed, G. O., Yassin, A. Y., Salih, R. R., Esmail, P. A., et al. (2023). Anthracycline in medicinal chemistry: mechanism of cardiotoxicity, preventive and treatment strategies. Curr. Org. Chem. 27, 363–377. doi:10.2174/1385272827666230423144150
Jin, W. K., Tan, H. D., Wu, J. H., He, G., and Liu, B. (2022). Dual-target inhibitors of bromodomain-containing protein 4 (BRD4) in cancer therapy: current situation and future directions. Drug Discov. Today 27, 246–256. doi:10.1016/j.drudis.2021.08.007
Jing, X., Shao, S., Zhang, Y., Luo, A., Zhao, L., Zhang, L., et al. (2020). BRD4 inhibition suppresses PD-L1 expression in triple-negative breast cancer. Exp. Cell Res. 392, 112034. doi:10.1016/j.yexcr.2020.112034
Mio, C., Gerratana, L., Bolis, M., Caponnetto, F., Zanello, A., Barbina, M., et al. (2019). BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 144, 755–766. doi:10.1002/ijc.31898
Obidiro, O., Battogtokh, G., and Akala, E. O. (2023). Triple negative breast cancer treatment options and limitations: future outlook. Pharmaceutics 15, 1796. doi:10.3390/pharmaceutics15071796
Pérez-Peña, J., Győrffy, B., Amir, E., Pandiella, A., and Ocaña, A. (2018). Epigenetic modulation of FOXM1-gene interacting network by BET inhibitors in breast cancer. Breast Cancer Res. Treat. 172, 725–732. doi:10.1007/s10549-018-4965-x
Pérez-Salvia, M., Simó-Riudalbas, L., Llinàs-Arias, P., Roa, L., Setien, F., Soler, M., et al. (2017). Bromodomain inhibition shows antitumoral activity in mice and human luminal breast cancer. Oncotarget 8, 51621–51629. doi:10.18632/oncotarget.18255
Sahni, J. M., and Keri, R. A. (2018). Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacol. Res. 129, 156–176. doi:10.1016/j.phrs.2017.11.015
Sun, C., Yin, J., Fang, Y., Chen, J., Jeong, K. J., Chen, X., et al. (2018). BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 33, 401–416. doi:10.1016/j.ccell.2018.01.019
Xia, C. F., Dong, X. S., Li, H., Cao, M. M., Sun, D. A. Q., He, S. Y., et al. (2022). Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin. Med. J. Engl. 135, 584–590. doi:10.1097/cm9.0000000000002108
Yang, G.-J., Song, Y.-Q., Wang, W., Han, Q.-B., Ma, D.-L., and Leung, C.-H. (2021). An optimized BRD4 inhibitor effectively eliminates NF-κB-driven triple-negative breast cancer cells. Bioorg Chem. 114, 105158. doi:10.1016/j.bioorg.2021.105158
Zhang, J., Yang, C., Tang, P., Chen, J., Zhang, D., Li, Y., et al. (2022). Discovery of 4-hydroxyquinazoline derivatives as small molecular BET/PARP1 inhibitors that induce defective homologous recombination and lead to synthetic lethality for triple-negative breast cancer therapy. J. Med. Chem. 65, 6803–6825. doi:10.1021/acs.jmedchem.2c00135
Keywords: BRD4, PARP1, BRD4 inhibitor, breast cancer, DNA damage
Citation: Li Y, Chu X, Yin Y, Li H, Fu H, Feng X, Deng Y and Ge J (2023) Design, synthesis, and evaluation of 4-(3-(3,5-dimethylisoxazol-4-yl)benzyl)phthalazin-1(2H)-one derivatives: potent BRD4 inhibitors with anti-breast cancer activity. Front. Pharmacol. 14:1289003. doi: 10.3389/fphar.2023.1289003
Received: 05 September 2023; Accepted: 13 November 2023;
Published: 30 November 2023.
Edited by:
Leilei Fu, Southwest Jiaotong University, ChinaReviewed by:
Berna Dogan, Istanbul Technical University, TürkiyeRadhika Amaradhi, University of Texas at San Antonio, United States
Copyright © 2023 Li, Chu, Yin, Li, Fu, Feng, Deng and Ge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Ge, gejun87@126.com
†These authors have contributed equally to this work and share first authorship