 Fei Feng
Fei Feng Lin-Jun Wang
Lin-Jun Wang Jian-Chun Li
Jian-Chun Li Ting-Ting Chen
Ting-Ting Chen Liping Liu
Liping Liu- 1The First Clinical Medical School of Lanzhou University, Lanzhou, China
- 2Departments of Emergency Critical Care Medicine, The First Hospital of Lanzhou University, Lanzhou, Gansu, China
Acute respiratory distress syndrome (ARDS) is the most common respiratory disease in ICU. Although there are many treatment and support methods, the mortality rate is still high. The main pathological feature of ARDS is the damage of pulmonary microvascular endothelium and alveolar epithelium caused by inflammatory reaction, which may lead to coagulation system disorder and pulmonary fibrosis. Heparanase (HPA) plays an significant role in inflammation, coagulation, fibrosis. It is reported that HPA degrades a large amount of HS in ARDS, leading to the damage of endothelial glycocalyx and inflammatory factors are released in large quantities. HPA can aggrandize the release of exosomes through syndecan-syntenin-Alix pathway, leading to a series of pathological reactions; at the same time, HPA can cause abnormal expression of autophagy. Therefore, we speculate that HPA promotes the occurrence and development of ARDS through exosomes and autophagy, which leads to a large amount of release of inflammatory factors, coagulation disorder and pulmonary fibrosis. This article mainly describes the mechanism of HPA on ARDS.
1 Introduction
Acute respiratory distress syndrome (ARDS) is an acute respiratory failure characterized by pulmonary edema, progressive dyspnea and refractory hypoxemia caused by increased alveolar capillary permeability (Thompson et al., 2017). The prevalence of ARDS in intensive care units (ICU) in 50 countries is 10.4% (Murray et al., 2019). Although ARDS has been widely studied, its mortality rate is still as high as 34.9%–46% (Zhang et al., 2017). In recent years, many COVID-19 cases have developed into ARDS (Batah and Fabro, 2021).
There are a lot of inducing factors for the occurrence and development of ARDS, including direct lung injury (bacterial and viral pneumonia, inhalation of gastric contents and pulmonary contusion) and indirect extrapulmonary injury (sepsis, severe trauma, blood transfusion, pancreatitis, drug reaction etc.). Once these inducing factors occur, the pathophysiology of ARDS will be manifested in the complex interaction between the inflammatory, coagulation, immune system and the alveolar capillary barrier, which will eventually lead to fiber proliferation (Hughes and Beasley, 2017). It includes the following three aspects: (1) Acute inflammatory response (Simeonovic et al., 2013): increased permeability of alveolar capillaries, injury of alveolar epithelial cells, decreased secretion of pulmonary surfactant, impaired clearance of alveolar fluid, increased expression of adhesion molecules, extravasation of white blood cells and their released products, immune system disorder, and finally tissue destruction; (2) Coagulation disorder (Freeman and Parish, 1998): intravascular coagulation, fibrin deposition and hyperfibrinolysis; (3) Fibrosis: regeneration of alveolar endothelial cells and epithelial type I/II cells, fiber proliferation (Fernandez-Francos et al., 2021). So far, despite the progress in understanding the biology, physiology and pathology of ARDS, there is no effective drug for the treatment of ARDS, and its treatment scheme is mainly limited to supportive therapy (Neff et al., 2003). Therefore, there is a need for pioneering methods to achieve effective treatment of ARDS.
Heparanase (HPA) is the only functional endoglycosidase capable of cleaving heparan sulfate (HS) chains (Vlodavsky et al., 1999). HPA specifically induces the degradation of HS in glycocalyx, thereby aggravating the injury of pulmonary endothelial barrier (Chappell et al., 2008) and the increase of alveolar permeability12, leading to the formation of ARDS (Coulson-Thomas et al., 2015; Chen et al., 2017), affecting the recovery of lung injury (LaRiviere et al., 2020). HPA also has non enzymatic functions (Secchi et al., 2015), such as regulating gene expression, promoting cell adhesion (Nadir, 2014; Sanderson et al., 2017). HPA increased the release of exosomes and caused abnormal expression of autophagy. It has been reported that there is a relationship between the development of ARDS and exosomes, autophagy (Yamada, 2021). Therefore, we searched “ARDS, heparanase, inflammation, immune, coagulation, fibrosis, exosome, autophagy” in electronic databases such as “PubMed” and “Web of Science,” to explore the mechanism of HPA participating in the occurrence and development of ARDS.
2 Heparanase
HPA is an endonuclease-β-D-glucuronidase, the only endoglycosidase to degrade basement membrane extracellular matrix (ECM) and heparan sulfate glycoprotein (Vlodavsky et al., 2007). The gene expression of HPA can be regulated by early growth response genes in tumor cells (de Mestre et al., 2005), inflammatory cytokines and fatty acids in endothelial cells (Chen et al., 2004). The human HPA gene is located on chromosome 4q22 and has two splice variants (Dong et al., 2000). Its cDNA (complementary deoxyribonucleic acid) contains an open reading frame of 1629 base pairs. Cleavage of the N-terminal signal peptide generates a 65kda inactive HPA precursor (Vreys and David, 2007).
According to the effect of HPA on heparin and HS, the enzymes are divided into three categories: HPA I (heparin lyase I), which mainly acts on heparin; HPA II (heparin lyase II), acting on heparin and HS; HPA III (heparin lyase III) mainly acts on HS (Yamada and Sugahara, 1998; Hu et al., 2015). HPA I is known to destroy specific binding points between glucosamine residues (called s-domain), where sulfate groups bind to uronic acid (Bame, 2001; Gingis-Velitski et al., 2004; Reiland et al., 2004). HPA III can cleave bioactive HS fragments by degrading HS (Kato et al., 1998).
The physiological expression of HPA is limited to a few cell and tissue types, such as platelets, immune cells and placenta (Vlodavsky et al., 1992; Goshen et al., 1996; Hulett et al., 1999; Gutter-Kapon et al., 2016; Putz et al., 2017). In adults, HPA may play a role in wound repair, tissue regeneration and immune monitoring. HPA plays a role in cell adhesion, migration and survival (Goldshmidt et al., 2003). HPA can affect the migration of inflammatory cells, inflammatory cell specific cytokine interferon-γ (IFN-γ) and tumor necrosis factor (TNF) can stimulate endothelial cells to produce HPA and enhance the activity of HPA (Bartlett et al., 1995; Edovitsky et al., 2006; Ilan et al., 2006), destroy the cell barrier by degrading HS (Simeonovic et al., 2013), causes a series of inflammatory reactions. Therefore, HPA can cause inflammatory reaction under pathological conditions, leading to a series of reactions in the body.
HPA has both enzymatic and non enzymatic activities. HPA selectively cleaves HS polymers to produce fragments of variable size, usually 10–20 sugar units, indicating that only a limited proportion of glucuronic acid bonds in the HS chain are vulnerable to the enzyme (Goldshmidt et al., 2003). HPA plays a role by releasing growth factors from ECM (Ilan et al., 2006), participates in the degradation and remodeling of extracellular matrix, promotes cell invasion related to inflammation metastasis (Myler and West, 2002). The cleavage of HS seems to be the key for leukocytes to pass through the basement membrane (Finkel, 1999a; Bame, 2001). Its activity is related to inflammatory acidosis (McKenzie et al., 2003). Its enhanced activity can also regulate macrophage activation (cytokine expression induced by c-fos) (Parish, 2006), promotes the recruitment of immune cells to the injury site (remodeling through extracellular matrix) (Shteingauz and Vlodavsky, 2015) and enhances the neuroinflammatory effect of autophagy (Dai et al., 2014). Non enzymatic HPA can induce endothelial cell invasion and migration through PI3K/Akt pathway (Gingis-Velitski et al., 2004; Yuan et al., 2012). Non enzymatic HPA also enhance T cell adhesion, mediated by integrin β (Sotnikov et al., 2004). After the non enzymatic active HPA is activated through PKA (protein kinase A) and PKC (protein kinase C) signaling pathways, the lysosome secretes the active form of HPA (Shafat et al., 2006).
3 Heparanase promotes ARDS through inflammation and immune disorders
The pathogenesis of ARDS is complex, and the disorder of inflammatory regulation plays an important role (Matthay et al., 2019). In the early stage of ARDS, microvascular endothelial cells are first affected and are less resistant to injury than epithelial cells (Wiener-Kronish et al., 1991; Matthay et al., 2019). Extensive damage of endothelial cell (EC) barrier and inflammatory cascade lead to increased permeability, which is the core pathogenesis of ARDS (Wiener-Kronish et al., 1991; Han and Mallampalli, 2015). Macrophages in the lung secrete a large number of proinflammatory factors, leading to the recruitment of macrophages and activation of effector T cells. These processes further exacerbate the inflammatory response and tissue damage patients with ARDS (Aggarwal et al., 2014; Long et al., 2019; Park et al., 2019), an inflammatory response called “cytokine storm” may lead to multiple organ failure and increase patient mortality (Chappell et al., 2008). With the secretion of inflammatory factors, cytokines increase abnormally in other tissues and organs, interfere with the immune system, cause excessive immunity, and lead to diffuse lung cell injury, pulmonary fibrosis and multiple organ injury (Zhang et al., 2021a). The innate immune response is initially triggered by pulmonary epithelial cells, alveolar macrophages and neutrophils (Rokni et al., 2020). In mucosal immune response, interleukin-22 (IL-22) upregulates mucin, fibrinogen and anti-apoptotic protein; therefore, IL-22 may contribute to the formation of life-threatening edema, and the lungs may be rich in mucin and fibrin, leading to the progress of ARDS (Wu and Yang, 2020). When the immune system is reduced by lymphocytes, it may be one of the mechanisms leading to the development of ARDS (Yazdanpanah et al., 2020). The substances in ARDS may have a binding and signal transduction mode similar to that of lung tissue cells, and may be responsible for the recruitment and activation of corresponding immune cells and acute lung injury (Lee, 2017). Therefore, the occurrence and development of ARDS are related to the aggravation of inflammation and immune disorders.
HPA can affect the migration of inflammatory cells and destroy the cell barrier by degrading HS (Simeonovic et al., 2013). Inflammatory cell specific cytokine IFN-γ and TNF can stimulate endothelial cells to produce HPA and enhance the activity of HPA (Bartlett et al., 1995; Edovitsky et al., 2006; Ilan et al., 2006). Therefore, HPA derived from endothelial cells can also promote the migration of lymphocytes and granulocytes, macrophages and dendritic cells through bone marrow (BM) and ECM under endothelium (Parish et al., 2013). HPA produced by leukocytes can be induced by various cell activation stimuli (de Mestre et al., 2003; Chen et al., 2004; Putz et al., 2017), promoting leukocyte migration (Poon et al., 2014; Digre et al., 2017), cell rolling and adhesion (Lever et al., 2014; Changyaleket et al., 2017), proinflammatory factors are upregulated (Goodall et al., 2014) and activation of innate immune cells (Gutter-Kapon et al., 2016). In colitis, epithelial derived HPA regulates the inflammatory phenotype of macrophages, prevents inflammation from fading, and converts macrophage responses to chronic inflammatory patterns (Lerner et al., 2011). HPA also enhanced macrophage activation in vitro through lipopolysaccharide (LPS) and increased TNF-α, IL-6 and IL-12. Activated macrophages in turn can induce epithelial HPA expression and promote self-sustaining inflammatory circuits through increased secretion of cathepsin-l (Lerner et al., 2011). HPA is involved in the recruitment of pulmonary inflammatory factors in allergic asthma models (Morris et al., 2015). HPA causes the degradation of endothelial glycocalyx in sepsis associated lung injury, which aggregates neutrophils and inflammatory factors (Schmidt et al., 2012), causing a series of inflammatory reactions. Inhibition of HPA activity can prevent endotoxemia related loss of pulmonary endothelial glycocalyx, thus alleviating sepsis induced inflammatory lung injury in mice (Schmidt et al., 2012). In sepsis related intestinal injury, HPA inhibitors prevent the destruction of glycocalyx of intestinal mucosa, inhibit neutrophil infiltration, protect mucosal integrity, and inhibit inflammatory response by inhibiting HPA activity (Chen et al., 2017). Heparin and heparin derived compounds can compete with HS chain to bind HPA and inhibit the activity of HPA (Goldshmidt et al., 2004; Waterman et al., 2007), so they are potent HPA inhibitors (Bar-Ner et al., 1987). It plays an anti-inflammatory role in the treatment of asthma, patients undergoing extracorporeal circulation and cataract surgery (Mousavi et al., 2015).
Therefore, HPA maybe promote the activity of inflammatory cells and immune disorders, increases the aggregation of neutrophils, degrades HS in the glycocalyx of lung endothelium, and causes the permeability of alveolar vascular wall to increase, thus leading to ARDS. But the specific mechanism is still unclear, further discussion is needed.
4 Heparanase promotes ARDS through coagulation
Thrombosis and coagulation disorder maybe the main factors leading to ARDS (Helms et al., 2020). However, the underlying mechanism remains unclear. Tissue factor (TF) can be produced by endothelial cells, smooth muscle cells, neutrophils and monocytes to respond to various stimuli in vivo and in vitro (DelGiudice and White, 2009; Kambas et al., 2012), which is a key link in the process of coagulation in vivo (Rapaport and Rao, 1995; Mackman et al., 2007). Platelets are the key to thrombosis. During blood transfusion, activated platelets can induce acute lung injury (Caudrillier et al., 2012). In recent corona virus disease 2019 (COVID-19) pathology, activated platelets from patients showed the production of TF bearing nets, inducing thrombotic activity of human aortic endothelial cells (HAECs) (Skendros et al., 2020). Experiments have proved that (Zhang et al., 2021b) in the plasma of patients with ARDS, the TF expression of neutrophils is significantly increased and reticular cells are exposed. Thrombin is reported to be necessary for protease activated receptor-1 (PAR-1) to activate platelets (Petzold et al., 2020). Thrombin activated platelets can increase the formation of TF network and subsequent immune thrombosis in patients with ARDS (Zhang et al., 2021b). With the support of immune cells, platelets and coagulation related molecules, immune thrombosis is considered to be a key event in the pathophysiology of ARDS. When sepsis mediated ARDS occurs, neutrophils are activated and then form TF rich neutrophil extracellular traps in pulmonary vessels (the first step), resulting in thrombin production; Platelets are activated by thrombin and then interact with neutrophils to form TF networks (step 2). All these factors lead to a vicious circle leading to a large number of thrombosis (Zhang et al., 2021b). Therefore, the development of ARDS may be closely related to coagulation.
Under normal conditions, HPA activity is limited to placental and skin tissues, as well as blood cells, such as neutrophils, monocytes, mast cells, T lymphocytes and platelets. Among them, platelets have the highest HPA activity and are used as a source of activated HPA (Freeman and Parish, 1998; Freeman et al., 1999). There is evidence that HPA may also affect the coagulation system (Matzner et al., 1985; El-Assal et al., 2001; Koliopanos et al., 2001). HPA may be a co-factor of TF and directly participate in the activation of coagulation factors (Nadir et al., 2010). Increased HPA regulates the expression of coagulation factor TF13 and interacts with TFPI (tissue factor pathway inhibitor) on the cell membrane surface of endothelial cells and tumor cells, resulting in the dissociation of TFPI, thereby increasing the coagulation activity on the cell surface (El-Assal et al., 2001). In addition, HPA can directly enhance TF activity, increase the production of factor Xa, and then activate the coagulation system (Rohloff et al., 2002). Over expression of HPA in human leukemia, glioma and breast cancer cells leads to a significant increase in TF level. HPA may promote angiogenesis by inducing the expression of angiogenesis promoting factor (VEGF) and reducing the expression of angiogenesis inhibitory (thrombospondin) mediators by TF (Zhang et al., 1994; Abe et al., 1999). Discovery that natural anticoagulant heparin can inhibit the spread of cancer in animals (Finkel, 1999b). It can prevent platelets from coagulating around cancer cells, and even if the anticoagulant activity of heparin is exhausted, it can still inhibit tumor metastasis (Eccles, 1999), so heparin inhibits HPA activity by competing with HS to bind heparin or HS binding domain (HBD) (Levy-Adam et al., 2005).
In conclusion, the occurrence of ARDS is related to the disorder of the coagulation system, and HPA leads to the disorder of the coagulation system. Therefore, HPA may cause the occurrence and development of ARDS through the disorder of the coagulation system. However, there is no specific report that HPA can cause ARDS through coagulation disorder, so further study is needed.
5 Heparanase promotes ARDS through fibrosis
Tissue fibrosis is an unregulated wound healing response characterized by gradual accumulation and reduced remodeling of ECM (Rockey et al., 2015a). In organs such as heart, lung, kidney or liver, the accumulation of fibrous tissue can gradually change its normal structure and function, and may cause destructive results (Rockey et al., 2015b; Jun and Lau, 2018). The main pathogenesis of pulmonary fibrosis is that the injury of alveolar epithelial cells activates lung fibroblasts and promotes their transformation into matrix producing myofibroblasts. Replacing normal lung parenchyma with fibrotic tissue will lead to irreversible reduction of oxygen diffusion capacity (Sakai and Tager, 2013). The acute exudative inflammation stage of ARDS is followed by the proliferation stage characterized by the proliferation of alveolar epithelial cells (Matthay et al., 2019), some ARDS survivors will further develop fibroblast proliferation responses, including fibroblast aggregation, deposition of collagen and other pulmonary ECM components (Burnham et al., 2014). In the ARDS animal model, type II alveolar epithelial intracellular stress is regulated by inducing abnormal mucin expression (Hancock et al., 2018), increased endoplasmic reticulum (ER) stress (Lawson et al., 2011), induce local tissue hypoxia (Xi et al., 2017; Burman et al., 2018), impairing the normal repair process and aggravating the fibrotic response (Michalski et al., 2022). It has recently been reported that high levels of heat shock protein 90 (HSP90) play an important role in the development of pulmonary fibrosis (Sontake et al., 2017). HSP90 is a highly expressed molecular chaperone protein that is important for the physiological function of human cells (Wu et al., 2018). HSP90 is critical in the treatment of lung injury (Sibinska et al., 2017; Marinova et al., 2020). HSP90 has a proinflammatory role in the development and progression of ARDS, and inhibition of HSP90 can maintain pulmonary endothelial integrity (Barabutis and Siejka, 2020) and anti-inflammatory effects (Barabutis et al., 2018). HSP90 inhibitors can promote protein ubiquitination and protease degradation, and significantly improve pulmonary fibrosis (Marinova et al., 2020; Colunga Biancatelli et al., 2021). Therefore, pulmonary fibrosis is the final development result of ARDS, but its specific mechanism has not been clarified, so finding the specific mechanism of pulmonary fibrosis is an important target to improve its prognosis.
The involvement of HPA in fibrosis depends on the fact that HPA promotes the release and diffusion of various HS linked molecules, rather than the catalytic activity responsible for cutting HS side chains (Masola et al., 2020). HPA is a key regulator of fibroblast growth factor-basic 2 (FGF-2) and transfroming growth factor-β (TGF-β) activities, and is a major pro-fibrotic factor and inducer of kidney (Masola et al., 2012; Masola et al., 2014). The increase of HPA activity at the renal tubular level can regulate the epithelial to mesenchymal transition (EMT) of proximal renal tubular cells, thus forming a profibrotic environment (Secchi et al., 2017), lack of HPA can prevent the overexpression of TGF- β (Masola et al., 2014), HPA deficient mice did not show TGF-α increased without fibrosis (Gil et al., 2012), HPA regulates TGF-β by releasing syndecan-1, and the upregulation of TGF-β is associated with intestinal fibrosis in vivo (Davids et al., 2010). In non cancerous tissues, HPA expression was negatively correlated with fibrosis stage (Ikeguchi et al., 2003). HPA levels were elevated in the fibrotic livers of thioacetamide treated rats (Goldshmidt et al., 2004; Ohayon et al., 2008). HPA activity in the plasma of patients with mild and severe liver fibrosis increased, while HPA activity in the plasma of patients with cirrhosis decreased to the basal level (Secchi et al., 2017). High mobiliby group box 1 (HMGB1) can activate fibroblasts to myofibroblasts and activate NF through its receptor RAGE-κB and increased HPA expression. Upregulated HPA releases TGF stored in ECM by decomposing HS-β, thus promoting the progress of pulmonary fibrosis (He et al., 2016). At the same time, a related protein also plays a related role in pulmonary fibrosis in ARDS. Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases whose main role is to degradw collagen and ECM components and can also act on cell surface proteins (Zinter et al., 2019). Elevated plasma MMP-3 levels in sepsis patients are associated with endothelial damage and impaired oxygenation in ARDS (Zinter et al., 2019). MMPs not only play an important role in physiological tissue remodeling and wound repair (Page-McCaw et al., 2007; Hadler-Olsen et al., 2011), but also are associated with pathological processes such as rheumatoid arthritis, cancer, liver, kidney, heart and lung fibrosis (Zuo et al., 2002; Green et al., 2003; Heymans et al., 2005; Uchinami et al., 2006; Rosas et al., 2008). MMP-3, MMP-7 and MMP-9 can all promote the formation of pulmonary fibrosis (Craig et al., 2015). At present, the relationship between HPA and MMP is still unclear, and whether they interact with each other in the occurrence and development of ARDS needs further exploration.
In conclusion, ARDS eventually develops into pulmonary fibrosis, and HPA leads to pulmonary fibrosis. Therefore, HPA may lead to the development of ARDS through pulmonary fibrosis. However, there is no specific report on HPA causing ARDS through fibrosis, so further research is needed.
6 Heparanase may affect ARDS through exosomes and autophagy
ARDS is a destructive clinical syndrome characterized by non cardiogenic pulmonary edema, respiratory distress and hypoxemia (Murray et al., 2019). Autophagy is a multi-step dynamic process in cells: damaged or dysfunctional proteins and organelles are separated by double membrane vesicles and fused with lysosomes to form autolysosomes for degradation and recycling (Klionsky et al., 2008; Barth et al., 2010). Under physiological conditions, autophagy is expressed at a low level, which is essential to maintain the stability of the intracellular environment (Sanderson et al., 2017). However, autophagic disorders were observed in different lung diseases, including lung injury (Chang et al., 2015; Chu et al., 2018) and pulmonary fibrosis (Meng et al., 2019). Light chain 3 (LC3) is a major regulator of autophagy formation (Sou et al., 2006). In lung diseases, the penetration of acinetobacter baumannii activates autophagy, which is caused by the Beclin1 dependent AMPK-ERK-mTOR pathway (Liao et al., 2019). MSCs may alleviate LPS induced ALI by downregulating miR-142a-5p to enable pulmonary epithelial cells (PECs) to undergo beclin mediated autophagy (Zhou and You, 2016). The most important cell types involved in the pathogenesis of ARDS are macrophages, epithelial cells and neutrophils (Saidi et al., 2018). During the occurrence of ARDS, the relationship between these cell types is mutual influence and interaction, and one cell type “indicates” that another cell type changes its phenotype, while the other type refutes in turn. In the microenvironment (alveolar lavage fluid BALF), the information transmitted between these cells is assembled into some small goods, such as exosomes, which are released from one cell into BALF and then obtained by another cell to decode signals (Lee, 2016) (Soni et al., 2016). Exosomes are nano double lipid membranes secreted by cells in the secretory body, with a diameter of 30–100nm, which are generated in large cells with multiple vesicles (Jun and Lau, 2018). Bidirectional communication occurs in microenvironment through exosomes and microbubbles (MVs) (Stahl and Raposo, 2019). Exosomes carry nucleic acids, proteins and lipids between different cells in the tumor microenvironment, which affects many ways in biology. Bone marrow mesenchymal stem cells (MSCs) are increasingly used to treat ARDS and sepsis because of their immunomodulatory and regenerative properties (Walter et al., 2014). MSC can also inhibit proinflammatory cytokines secretion, thereby potentially relieving the subsequent cytokine storm (Ye et al., 2020). In fact, preliminary preclinical and clinical results show that bone marrow mesenchymal stem cells can alleviate lung dysfunction in animal lung injury models (Curley et al., 2012), ARDS and patients with neocoronal pneumonia (Zheng et al., 2014; Xiao et al., 2020). Bone marrow derived exosomes are a new, multi-target next-generation biological agent, which may be the key to downregulate cytokine storm and reverse host antiviral defense inhibition characterized by neocoronal pneumonia (Hessvik and Llorente, 2018). The exosomes contain a whole set of chemokines, growth factors, mRNA and microRNAs with anti-inflammatory, regenerative and immune regulatory functions. They are paracrine and endocrine mediators, which endow BMSCs with healing characteristics (De Jong et al., 2014; Yu et al., 2014; Alipoor et al., 2016; Hessvik and Llorente, 2018). The exosomes secreted by bone marrow mesenchymal stem cells (BMSCs) are a new therapy for ARDS, but the mechanism between them and HPA is still unclear.
HPA exists in autophagy and promotes autophagy (Shteingauz et al., 2015). The level of LC3II was found to be decreased in cells and tissues obtained from HPA knockout mice, while the level of LC3-II was found to be increased in transgenic mice overexpressing HPA (Shteingauz et al., 2015). The mechanism of HPA induced autophagy is not completely clear, but it may involve mTOR1 (Dunlop and Tee, 2014). Overexpression of HPA is associated with decreased mTOR1 activity (Shteingauz et al., 2015). Electron microscope analysis of cells overexpressing HPA showed that not only more autophagic vacuoles were found, but also a large number of vesicles were released on the cell surface, which may be exosomes (Thompson et al., 2013; Roucourt et al., 2015). HPA localizes to the surface of exosomes secreted by various cell type (Sanderson et al., 2017; Nawaz et al., 2018). HPA affects the composition of protein and mRNA in exosomes, participates in mediating the secretion and function of exosomes, and enhances the secretion of exosomes (Thompson et al., 2013). HPA stimulates endosomal budding of syntenin and syndecan, and Alix is required to achieve these effects. Therefore, HPA is an activator of the syndecan-syntenin-Alix pathway of exosome biogenesis (Roucourt et al., 2015). In myeloma cells, HPA stimulates the accumulation of syndecan-1 and specific cargoes such as hepatocyte growth factor and VEGF in exosomes (Thompson et al., 2013). Recently, HPA has also been involved in the autophagy process. Exosomes and autophagy are connected through the endolysosomal pathway, and there is a strong interaction between them (Xu et al., 2018). HPA exists in autophagy and promotes autophagy, making HPA overexpressing cells more resistant to stress and chemotherapy. The mechanism of increased autophagy is not fully understood, but may involve decreased mTOR1 activity (Shteingauz et al., 2015; Sanderson et al., 2017).
In conclusion, HPA can increase the release of exosomes and promote autophagy. However, the specific mechanism of ARDS is still unclear. Therefore, exploring the specific mechanism between HPA and exosomes, autophagy and ARDS can become an important way to treat ARDS.
7 Summary
With the increase of research on HPA, the influence of HPA in disease is increasing. This review summarizes that HPA can aggravate inflammation, immune system disorder, coagulation dysfunction and tissue fibrosis, which may play a significant role in the occurrence and development of ARDS through inflammation, immune disorder, coagulation disorder and fibrosis. However, the specific mechanism needs to be further explored. At the same time, we speculate that HPA may affect the occurrence and development of ARDS through some signaling pathway of exosomes or autophagy, which needs further study. Therefore, HPA may affects the occurrence and development of ARDS, which may become a new idea to reduce the mortality of ARDS. (Figure 1).
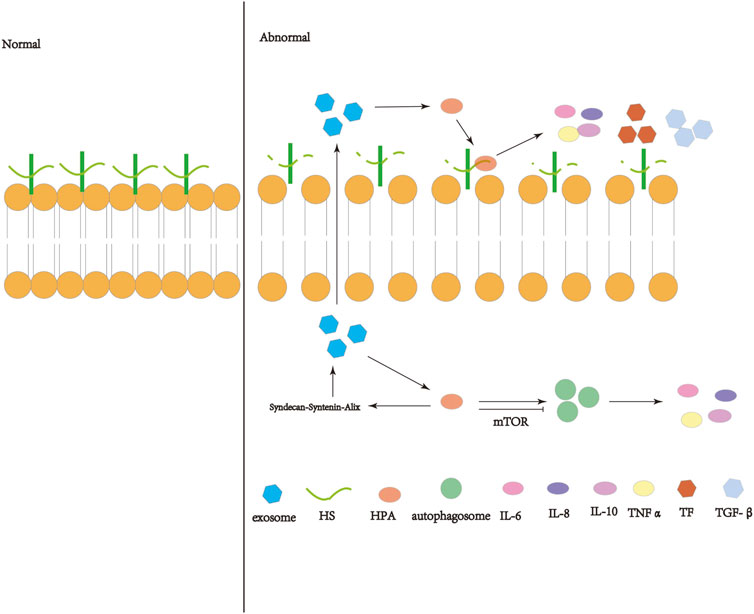
FIGURE 1. Schematic diagram of the operation and action of heparanase (HPA). HPA can interact with HSPG on the cell membrane, shear the heparin sulfate (HS) side chain, destroy the extracellular matrix (ECM) and basement membrane (BM), and release inflammatory immune factors, coagulation factors, etc. Active HPA can enhance autophagy through the mTOR pathway and increase exosome formation through the syndecan-syntenin-Alix pathway, so that exosomes are released into the extracellular space and act on the extracellular HPA.
Author contributions
L-PL proposed the idea, FF designed the article, and wrote the article. L-JW analyzed the feasibility of the article. T-TC collected relevant information and corrected the article. J-CL collected relevant information and corrected the article. All authors contributed to the article and approved the submitted version.
Funding
The present study was funded by the Science and Technology Department of Gansu Province (grant no. 20JR5RA35); Talent Innovation and Entrepreneurship Project of Science and Technology Bureau of Chengguan District, Lanzhou, (grant no. 2020RCCX0030); Lanzhou Science and Technology Development Guiding Plan Project (grant no. 2019-ZD-37); Fund of The First Hospital of Lanzhou University (grant no. Ldyyyn 2018-48); Fund of Donggang Branch, The First Hospital of Lanzhou University (grant no. ldyydgyn-201705); and The Open Topics of the Key Laboratory of Biological Treatment and Regenerative Medicine in Gansu Province (grant no. zdsyskfkt-201702).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abe, K., Shoji, M., Chen, J., Bierhaus, A., Danave, I., Micko, C., et al. (1999). Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc. Natl. Acad. Sci. U. S. A. 96 (15), 8663–8668. doi:10.1073/pnas.96.15.8663
Aggarwal, N. R., King, L. S., and D'Alessio, F. R. (2014). Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell Mol. Physiol. 306 (8), L709–L725. doi:10.1152/ajplung.00341.2013
Alipoor, S. D., Mortaz, E., Garssen, J., Movassaghi, M., Mirsaeidi, M., and Adcock, I. M. (2016). Exosomes and exosomal miRNA in respiratory diseases. Mediat. Inflamm. 2016, 5628404. doi:10.1155/2016/5628404
Bame, K. J. (2001). Heparanases: Endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology 11 (6), 91R–98R. doi:10.1093/glycob/11.6.91r
Bar-Ner, M., Eldor, A., Wasserman, L., Matzner, Y., Cohen, I. R., Fuks, Z., et al. (1987). Inhibition of heparanase-mediated degradation of extracellular matrix heparan sulfate by non-anticoagulant heparin species. Blood 70 (2), 551–557. doi:10.1182/blood.v70.2.551.551
Barabutis, N., Schally, A. V., and Siejka, A. (2018). P53, GHRH, inflammation and cancer. EBioMedicine 37, 557–562. doi:10.1016/j.ebiom.2018.10.034
Barabutis, N., and Siejka, A. (2020). The highly interrelated GHRH, p53, and Hsp90 universe. Cell Biol. Int. 44 (8), 1558–1563. doi:10.1002/cbin.11356
Barth, S., Glick, D., and Macleod, K. F. (2010). Autophagy: Assays and artifacts. J. Pathol. 221 (2), 117–124. doi:10.1002/path.2694
Bartlett, M. R., Underwood, P. A., and Parish, C. R. (1995). Comparative analysis of the ability of leucocytes, endothelial cells and platelets to degrade the subendothelial basement membrane: Evidence for cytokine dependence and detection of a novel sulfatase. Immunol. Cell Biol. 73 (2), 113–124. doi:10.1038/icb.1995.19
Batah, S. S., and Fabro, A. T. (2021). Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir. Med. 176, 106239. doi:10.1016/j.rmed.2020.106239
Burman, A., Kropski, J. A., Calvi, C. L., Serezani, A. P., Pascoalino, B. D., Han, W., et al. (2018). Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight 3 (16), e99543. doi:10.1172/jci.insight.99543
Burnham, E. L., Janssen, W. J., Riches, D. W. H., Moss, M., and Downey, G. P. (2014). The fibroproliferative response in acute respiratory distress syndrome: Mechanisms and clinical significance. Eur. Respir. J. 43 (1), 276–285. doi:10.1183/09031936.00196412
Caudrillier, A., Kessenbrock, K., Gilliss, B. M., Nguyen, J. X., Marques, M. B., Monestier, M., et al. (2012). Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest. 122 (7), 2661–2671. doi:10.1172/JCI61303
Chang, A. L., Ulrich, A., Suliman, H. B., and Piantadosi, C. A. (2015). Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic. Biol. Med. 78, 179–189. doi:10.1016/j.freeradbiomed.2014.10.582
Changyaleket, B., Chong, Z. Z., Dull, R. O., Nanegrungsunk, D., and Xu, H. (2017). Heparanase promotes neuroinflammatory response during subarachnoid hemorrhage in rats. J. Neuroinflammation 14 (1), 137. doi:10.1186/s12974-017-0912-8
Chappell, D., Jacob, M., Rehm, M., Stoeckelhuber, M., Welsch, U., Conzen, P., et al. (2008). Heparinase selectively sheds heparan sulphate from the endothelial glycocalyx. Biol. Chem. 389 (1), 79–82. doi:10.1515/BC.2008.005
Chen, G., Wang, D., Vikramadithyan, R., Yagyu, H., Saxena, U., Pillarisetti, S., et al. (2004). Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry 43 (17), 4971–4977. doi:10.1021/bi0356552
Chen, S., He, Y., Hu, Z., Lu, S., Yin, X., Ma, X., et al. (2017). Heparanase mediates intestinal inflammation and injury in a mouse model of sepsis. J. Histochem Cytochem 65 (4), 241–249. doi:10.1369/0022155417692536
Chu, R., Wang, J., Bi, Y., and Nan, G. (2018). The kinetics of autophagy in the lung following acute spinal cord injury in rats. Spine J. 18 (5), 845–856. doi:10.1016/j.spinee.2018.01.001
Colunga Biancatelli, R. M. L., Solopov, P., Gregory, B., and Catravas, J. D. (2021). The HSP90 inhibitor, AUY-922, protects and repairs human lung microvascular endothelial cells from hydrochloric acid-induced endothelial barrier dysfunction. Cells 10 (6), 1489. doi:10.3390/cells10061489
Coulson-Thomas, V. J., Chang, S. H., Yeh, L. K., Coulson-Thomas, Y. M., Yamaguchi, Y., Esko, J., et al. (2015). Loss of corneal epithelial heparan sulfate leads to corneal degeneration and impaired wound healing. Invest. Ophthalmol. Vis. Sci. 56 (5), 3004–3014. doi:10.1167/iovs.14-15341
Craig, V. J., Zhang, L., Hagood, J. S., and Owen, C. A. (2015). Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 53 (5), 585–600. doi:10.1165/rcmb.2015-0020TR
Curley, G. F., Hayes, M., Ansari, B., Shaw, G., Ryan, A., Barry, F., et al. (2012). Mesenchymal stem cells enhance recovery and repair following ventilator-induced lung injury in the rat. Thorax 67 (6), 496–501. doi:10.1136/thoraxjnl-2011-201059
Dai, T. L., Zhang, C., Peng, F., Niu, X. Y., Hu, L., Zhang, Q., et al. (2014). Depletion of canonical Wnt signaling components has a neuroprotective effect on midbrain dopaminergic neurons in an MPTP-induced mouse model of Parkinson's disease. Exp. Ther. Med. 8 (2), 384–390. doi:10.3892/etm.2014.1745
Davids, J. S., Carothers, A. M., Damas, B. C., and Bertagnolli, M. M. (2010). Chronic cyclooxygenase-2 inhibition promotes myofibroblast-associated intestinal fibrosis. Cancer Prev. Res. (Phila) 3 (3), 348–358. doi:10.1158/1940-6207.CAPR-09-0146
De Jong, O. G., Van Balkom, B. W. M., Schiffelers, R. M., Bouten, C. V. C., and Verhaar, M. C. (2014). Extracellular vesicles: Potential roles in regenerative medicine. Front. Immunol. 5, 608. doi:10.3389/fimmu.2014.00608
de Mestre, A. M., Khachigian, L. M., Santiago, F. S., Staykova, M. A., and Hulett, M. D. (2003). Regulation of inducible heparanase gene transcription in activated T cells by early growth response 1. J. Biol. Chem. 278 (50), 50377–50385. doi:10.1074/jbc.M310154200
de Mestre, A. M., Rao, S., Hornby, J. R., Soe-Htwe, T., Khachigian, L. M., and Hulett, M. D. (2005). Early growth response gene 1 (EGR1) regulates heparanase gene transcription in tumor cells. J. Biol. Chem. 280 (42), 35136–35147. doi:10.1074/jbc.M503414200
DelGiudice, L. A., and White, G. A. (2009). The role of tissue factor and tissue factor pathway inhibitor in health and disease states. J. Vet. Emerg. Crit. Care (San Ant. 19 (1), 23–29. doi:10.1111/j.1476-4431.2008.00380.x
Digre, A., Singh, K., Åbrink, M., Reijmers, R. M., Sandler, S., Vlodavsky, I., et al. (2017). Overexpression of heparanase enhances T lymphocyte activities and intensifies the inflammatory response in a model of murine rheumatoid arthritis. Sci. Rep. 7, 46229. doi:10.1038/srep46229
Dong, J., Kukula, A. K., Toyoshima, M., and Nakajima, M. (2000). Genomic organization and chromosome localization of the newly identified human heparanase gene. Gene 253 (2), 171–178. doi:10.1016/s0378-1119(00)00251-1
Dunlop, E. A., and Tee, A. R. (2014). mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 36, 121–129. doi:10.1016/j.semcdb.2014.08.006
Eccles, S. A. (1999). Heparanase: Breaking down barriers in tumors. Nat. Med. 5 (7), 735–736. doi:10.1038/10455
Edovitsky, E., Lerner, I., Zcharia, E., Peretz, T., Vlodavsky, I., and Elkin, M. (2006). Role of endothelial heparanase in delayed-type hypersensitivity. Blood 107 (9), 3609–3616. doi:10.1182/blood-2005-08-3301
El-Assal, O. N., Yamanoi, A., Ono, T., Kohno, H., and Nagasue, N. (2001). The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin. Cancer Res. 7 (5), 1299–1305.
Fernandez-Francos, S., Eiro, N., González-Galiano, N., and Vizoso, F. J. (2021). Mesenchymal stem cell-based therapy as an alternative to the treatment of acute respiratory distress syndrome: Current evidence and future perspectives. Int. J. Mol. Sci. 22 (15), 7850. doi:10.3390/ijms22157850
Finkel, E. (1999). Does cancer therapy trigger cell suicide? Science 286 (5448), 2256–2258. doi:10.1126/science.286.5448.2256
Finkel, E. (1999). Potential target found for antimetastasis drugs. Science 285 (5424), 33–34. doi:10.1126/science.285.5424.33
Freeman, C., Browne, A. M., and Parish, C. R. (1999). Evidence that platelet and tumour heparanases are similar enzymes. Biochem. J. 342. 361–368. doi:10.1042/bj3420361
Freeman, C., and Parish, C. R. (1998). Human platelet heparanase: Purification, characterization and catalytic activity. Biochem. J. 330, 1341–1350. doi:10.1042/bj3301341
Gil, N., Goldberg, R., Neuman, T., Garsen, M., Zcharia, E., Rubinstein, A. M., et al. (2012). Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes 61 (1), 208–216. doi:10.2337/db11-1024
Gingis-Velitski, S., Zetser, A., Kaplan, V., Ben-Zaken, O., Cohen, E., Levy-Adam, F., et al. (2004). Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J. Biol. Chem. 279 (42), 44084–44092. doi:10.1074/jbc.M402131200
Goldshmidt, O., Yeikilis, R., Mawasi, N., Paizi, M., Gan, N., Ilan, N., et al. (2004). Heparanase expression during normal liver development and following partial hepatectomy. J. Pathol. 203 (1), 594–602. doi:10.1002/path.1554
Goldshmidt, O., Zcharia, E., Cohen, M., Aingorn, H., Cohen, I., Nadav, L., et al. (2003). Heparanase mediates cell adhesion independent of its enzymatic activity. FASEB J. 17 (9), 1015–1025. doi:10.1096/fj.02-0773com
Goodall, K. J., Poon, I. K. H., Phipps, S., and Hulett, M. D. (2014). Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS One 9 (10), e109596. doi:10.1371/journal.pone.0109596
Goshen, R., Hochberg, A. A., Korner, G., Levy, E., Ishai-Michaeli, R., Elkin, M., et al. (1996). Purification and characterization of placental heparanase and its expression by cultured cytotrophoblasts. Mol. Hum. Reprod. 2 (9), 679–684. doi:10.1093/molehr/2.9.679
Green, M. J., Gough, A. K. S., Devlin, J., Smith, J., Astin, P., Taylor, D., et al. (2003). Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatol. Oxf. 42 (1), 83–88. doi:10.1093/rheumatology/keg037
Gutter-Kapon, L., Alishekevitz, D., Shaked, Y., Li, J. P., Aronheim, A., Ilan, N., et al. (2016). Heparanase is required for activation and function of macrophages. Proc. Natl. Acad. Sci. U. S. A. 113 (48), E7808–E7817. doi:10.1073/pnas.1611380113
Hadler-Olsen, E., Fadnes, B., Sylte, I., Uhlin-Hansen, L., and Winberg, J. O. (2011). Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 278 (1), 28–45. doi:10.1111/j.1742-4658.2010.07920.x
Han, S., and Mallampalli, R. K. (2015). Correction: The acute respiratory distress syndrome: From mechanism to translation. J. Immunol. 194 (11), 5569. doi:10.4049/jimmunol.1500741
Hancock, L. A., Hennessy, C. E., Solomon, G. M., Dobrinskikh, E., Estrella, A., Hara, N., et al. (2018). Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 9 (1), 5363. doi:10.1038/s41467-018-07768-9
He, L., Sun, F., Wang, Y., Zhu, J., Fang, J., Zhang, S., et al. (2016). HMGB1 exacerbates bronchiolitis obliterans syndrome via RAGE/NF-κB/HPSE signaling to enhance latent TGF-β release from ECM. Am. J. Transl. Res. 8 (5), 1971–1984.
Helms, J., Tacquard, C., Severac, F., Leonard-Lorant, I., Ohana, M., Delabranche, X., et al. (2020). High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 46 (6), 1089–1098. doi:10.1007/s00134-020-06062-x
Hessvik, N. P., and Llorente, A. (2018). Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 75 (2), 193–208. doi:10.1007/s00018-017-2595-9
Heymans, S., Lupu, F., Terclavers, S., Vanwetswinkel, B., Herbert, J. M., Baker, A., et al. (2005). Loss or inhibition of uPA or MMP-9 attenuates LV remodeling and dysfunction after acute pressure overload in mice. Am. J. Pathol. 166 (1), 15–25. doi:10.1016/S0002-9440(10)62228-6
Hu, X., Zhang, L., Jin, J., Zhu, W., Xu, Y., Wu, Y., et al. (2015). Heparanase released from mesenchymal stem cells activates integrin beta1/HIF-2alpha/Flk-1 signaling and promotes endothelial cell migration and angiogenesis. Stem Cells 33 (6), 1850–1862. doi:10.1002/stem.1995
Hughes, K. T., and Beasley, M. B. (2017). Pulmonary manifestations of acute lung injury: More than just diffuse alveolar damage. Arch. Pathol. Lab. Med. 141 (7), 916–922. doi:10.5858/arpa.2016-0342-RA
Hulett, M. D., Freeman, C., Hamdorf, B. J., Baker, R. T., Harris, M. J., and Parish, C. R. (1999). Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat. Med. 5 (7), 803–809. doi:10.1038/10525
Ikeguchi, M., Hirooka, Y., and Kaibara, N. (2003). Heparanase gene expression and its correlation with spontaneous apoptosis in hepatocytes of cirrhotic liver and carcinoma. Eur. J. Cancer 39 (1), 86–90. doi:10.1016/s0959-8049(02)00558-0
Ilan, N., Elkin, M., and Vlodavsky, I. (2006). Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell Biol. 38 (12), 2018–2039. doi:10.1016/j.biocel.2006.06.004
Jun, J. I., and Lau, L. F. (2018). Resolution of organ fibrosis. J. Clin. Invest. 128 (1), 97–107. doi:10.1172/JCI93563
Kambas, K., Mitroulis, I., and Ritis, K. (2012). The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front. Immunol. 3, 385. doi:10.3389/fimmu.2012.00385
Kato, M., Wang, H., Kainulainen, V., Fitzgerald, M. L., Ledbetter, S., Ornitz, D. M., et al. (1998). Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat. Med. 4 (6), 691–697. doi:10.1038/nm0698-691
Klionsky, D. J., Abeliovich, H., Agostinis, P., Agrawal, D. K., Aliev, G., Askew, D. S., et al. (2008). Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4 (2), 151–175. doi:10.4161/auto.5338
Koliopanos, A., Friess, H., Kleeff, J., Shi, X., Liao, Q., Pecker, I., et al. (2001). Heparanase expression in primary and metastatic pancreatic cancer. Cancer Res. 61 (12), 4655–4659.
LaRiviere, W. B., Liao, S., McMurtry, S. A., Oshima, K., Han, X., Zhang, F., et al. (2020). Alveolar heparan sulfate shedding impedes recovery from bleomycin-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 318 (6), L1198–L1210. doi:10.1152/ajplung.00063.2020
Lawson, W. E., Cheng, D. S., Degryse, A. L., Tanjore, H., Polosukhin, V. V., Xu, X. C., et al. (2011). Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. U. S. A. 108 (26), 10562–10567. doi:10.1073/pnas.1107559108
Lee, J. W. (2016). Macrophage-derived microvesicles' pathogenic role in acute lung injury. Thorax 71 (11), 975–976. doi:10.1136/thoraxjnl-2016-208895
Lee, K. Y. (2017). Pneumonia, acute respiratory distress syndrome, and early immune-modulator therapy. Int. J. Mol. Sci. 18 (2), 388. doi:10.3390/ijms18020388
Lerner, I., Hermano, E., Zcharia, E., Rodkin, D., Bulvik, R., Doviner, V., et al. (2011). Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J. Clin. Invest. 121 (5), 1709–1721. doi:10.1172/JCI43792
Lever, R., Rose, M. J., McKenzie, E. A., and Page, C. P. (2014). Heparanase induces inflammatory cell recruitment in vivo by promoting adhesion to vascular endothelium. Am. J. Physiol. Cell Physiol. 306 (12), C1184–C1190. doi:10.1152/ajpcell.00269.2013
Levy-Adam, F., Abboud-Jarrous, G., Guerrini, M., Beccati, D., Vlodavsky, I., and Ilan, N. (2005). Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. J. Biol. Chem. 280 (21), 20457–20466. doi:10.1074/jbc.M414546200
Liao, S. X., Sun, P. P., Gu, Y. H., Rao, X. M., Zhang, L. Y., and Ou-Yang, Y. (2019). Autophagy and pulmonary disease. Ther. Adv. Respir. Dis. 13, 1753466619890538. doi:10.1177/1753466619890538
Long, M. E., Gong, K. Q., Eddy, W. E., Volk, J. S., Morrell, E. D., Mikacenic, C., et al. (2019). MEK1 regulates pulmonary macrophage inflammatory responses and resolution of acute lung injury. JCI Insight 4 (23), e132377. doi:10.1172/jci.insight.132377
Mackman, N., Tilley, R. E., and Key, N. S. (2007). Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 27 (8), 1687–1693. doi:10.1161/ATVBAHA.107.141911
Marinova, M., Solopov, P., Dimitropoulou, C., Colunga Biancatelli, R. M. L., and Catravas, J. D. (2020). Post-treatment with a heat shock protein 90 inhibitor prevents chronic lung injury and pulmonary fibrosis, following acute exposure of mice to HCl. Exp. Lung Res. 46 (6), 203–216. doi:10.1080/01902148.2020.1764148
Masola, V., Gambaro, G., and Onisto, M. (2020). Impact of heparanse on organ fibrosis. Adv. Exp. Med. Biol. 1221, 669–684. doi:10.1007/978-3-030-34521-1_27
Masola, V., Gambaro, G., Tibaldi, E., Brunati, A. M., Gastaldello, A., D'Angelo, A., et al. (2012). Heparanase and syndecan-1 interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells. J. Biol. Chem. 287 (2), 1478–1488. doi:10.1074/jbc.M111.279836
Masola, V., Zaza, G., Secchi, M. F., Gambaro, G., Lupo, A., and Onisto, M. (2014). Heparanase is a key player in renal fibrosis by regulating TGF-beta expression and activity. Biochim. Biophys. Acta 1843 (9), 2122–2128. doi:10.1016/j.bbamcr.2014.06.005
Matthay, M. A., Zemans, R. L., Zimmerman, G. A., Arabi, Y. M., Beitler, J. R., Mercat, A., et al. (2019). Acute respiratory distress syndrome. Nat. Rev. Dis. Prim. 5 (1), 18. doi:10.1038/s41572-019-0069-0
Matzner, Y., Bar-Ner, M., Yahalom, J., Ishai-Michaeli, R., Fuks, Z., and Vlodavsky, I. (1985). Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J. Clin. Invest. 76 (4), 1306–1313. doi:10.1172/JCI112104
McKenzie, E., Young, K., Hircock, M., Bennett, J., Bhaman, M., Felix, R., et al. (2003). Biochemical characterization of the active heterodimer form of human heparanase (Hpa1) protein expressed in insect cells. Biochem. J. 373, 423–435. doi:10.1042/BJ20030318
Meng, Y., Pan, M., Zheng, B., Chen, Y., Li, W., Yang, Q., et al. (2019). Autophagy attenuates angiotensin II-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid. Redox Signal 30 (4), 520–541. doi:10.1089/ars.2017.7261
Michalski, J. E., Kurche, J. S., and Schwartz, D. A. (2022). From ARDS to pulmonary fibrosis: The next phase of the COVID-19 pandemic? Transl. Res. 241, 13–24. doi:10.1016/j.trsl.2021.09.001
Morris, A., Wang, B., Waern, I., Venkatasamy, R., Page, C., Schmidt, E. P., et al. (2015). The role of heparanase in pulmonary cell recruitment in response to an allergic but not non-allergic stimulus. PLoS One 10 (6), e0127032. doi:10.1371/journal.pone.0127032
Mousavi, S., Moradi, M., Khorshidahmad, T., and Motamedi, M. (2015). Anti-inflammatory effects of heparin and its derivatives: A systematic review. Adv. Pharmacol. Sci. 2015, 507151. doi:10.1155/2015/507151
Murray, D. D., Itenov, T. S., Sivapalan, P., Eklöf, J. V., Holm, F. S., Schuetz, P., et al. (2019). Biomarkers of acute lung injury the individualized approach: For phenotyping, risk stratification and treatment surveillance. J. Clin. Med. 8 (8), 1163. doi:10.3390/jcm8081163
Myler, H. A., and West, J. L. (2002). Heparanase and platelet factor-4 induce smooth muscle cell proliferation and migration via bFGF release from the ECM. J. Biochem. 131 (6), 913–922. doi:10.1093/oxfordjournals.jbchem.a003182
Nadir, Y., Brenner, B., Fux, L., Shafat, I., Attias, J., and Vlodavsky, I. (2010). Heparanase enhances the generation of activated factor X in the presence of tissue factor and activated factor VII. Haematologica 95 (11), 1927–1934. doi:10.3324/haematol.2010.023713
Nadir, Y. (2014). Heparanase and coagulation-new insights. Rambam Maimonides Med. J. 5 (4), e0031. doi:10.5041/RMMJ.10165
Nawaz, M., Shah, N., Zanetti, B. R., Maugeri, M., Silvestre, R. N., Fatima, F., et al. (2018). Extracellular vesicles and matrix remodeling enzymes: The emerging roles in extracellular matrix remodeling, progression of diseases and tissue repair. Cells 7 (10), 167. doi:10.3390/cells7100167
Neff, T. A., Stocker, R., Frey, H. R., Stein, S., and Russi, E. W. (2003). Long-term assessment of lung function in survivors of severe ARDS. Chest 123 (3), 845–853. doi:10.1378/chest.123.3.845
Ohayon, O., Mawasi, N., Pevzner, A., Tryvitz, A., Gildor, T., Pines, M., et al. (2008). Halofuginone upregulates the expression of heparanase in thioacetamide-induced liver fibrosis in rats. Lab. Invest. 88 (6), 627–633. doi:10.1038/labinvest.2008.30
Page-McCaw, A., Ewald, A. J., and Werb, Z. (2007). Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8 (3), 221–233. doi:10.1038/nrm2125
Parish, C. R., Freeman, C., Ziolkowski, A. F., He, Y. Q., Sutcliffe, E. L., Zafar, A., et al. (2013). Unexpected new roles for heparanase in Type 1 diabetes and immune gene regulation. Matrix Biol. 32 (5), 228–233. doi:10.1016/j.matbio.2013.02.007
Parish, C. R. (2006). The role of heparan sulphate in inflammation. Nat. Rev. Immunol. 6 (9), 633–643. doi:10.1038/nri1918
Park, I., Kim, M., Choe, K., Song, E., Seo, H., Hwang, Y., et al. (2019). Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur. Respir. J. 53 (3), 1800786. doi:10.1183/13993003.00786-2018
Petzold, T., Thienel, M., Dannenberg, L., Mourikis, P., Helten, C., Ayhan, A., et al. (2020). Response by Petzold et al to Letter Regarding Article, "Rivaroxaban Reduces Arterial Thrombosis by Inhibition of FXa-Driven Platelet Activation via Protease Activated Receptor-1. Circ. Res. 126 (8), e54–e55. doi:10.1161/CIRCRESAHA.120.316786
Poon, I. K., Goodall, K. J., Phipps, S., Chow, J. D. Y., Pagler, E. B., Andrews, D. M., et al. (2014). Mice deficient in heparanase exhibit impaired dendritic cell migration and reduced airway inflammation. Eur. J. Immunol. 44 (4), 1016–1030. doi:10.1002/eji.201343645
Putz, E. M., Mayfosh, A. J., Kos, K., Barkauskas, D. S., Nakamura, K., Town, L., et al. (2017). NK cell heparanase controls tumor invasion and immune surveillance. J. Clin. Invest. 127 (7), 2777–2788. doi:10.1172/JCI92958
Rapaport, S. I., and Rao, L. V. (1995). The tissue factor pathway: How it has become a "prima ballerina. Thromb. Haemost. 74 (1), 007–017. doi:10.1055/s-0038-1642646
Reiland, J., Sanderson, R. D., Waguespack, M., Barker, S. A., Long, R., Carson, D. D., et al. (2004). Heparanase degrades syndecan-1 and perlecan heparan sulfate: Functional implications for tumor cell invasion. J. Biol. Chem. 279 (9), 8047–8055. doi:10.1074/jbc.M304872200
Rockey, D. C., Bell, P. D., and Hill, J. A. (2015). Fibrosis--A common pathway to organ injury and failure. N. Engl. J. Med. 373 (1), 96. doi:10.1056/NEJMc1504848
Rockey, D. C., Bell, P. D., and Hill, J. A. (2015). Fibrosis--a common pathway to organ injury and failure. N. Engl. J. Med. 372 (12), 1138–1149. doi:10.1056/NEJMra1300575
Rohloff, J., Zinke, J., Schoppmeyer, K., Tannapfel, A., Witzigmann, H., Mössner, J., et al. (2002). Heparanase expression is a prognostic indicator for postoperative survival in pancreatic adenocarcinoma. Br. J. Cancer 86 (8), 1270–1275. doi:10.1038/sj.bjc.6600232
Rokni, M., Ghasemi, V., and Tavakoli, Z. (2020). Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: Comparison with SARS and MERS. Rev. Med. Virol. 30 (3), e2107. doi:10.1002/rmv.2107
Rosas, I. O., Richards, T. J., Konishi, K., Zhang, Y., Gibson, K., Lokshin, A. E., et al. (2008). MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 5 (4), e93. doi:10.1371/journal.pmed.0050093
Roucourt, B., Meeussen, S., Bao, J., Zimmermann, P., and David, G. (2015). Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 25 (4), 412–428. doi:10.1038/cr.2015.29
Saidi, H., Bérubé, J., Laraba-Djebari, F., and Hammoudi-Triki, D. (2018). Involvement of alveolar macrophages and neutrophils in acute lung injury after scorpion envenomation: New pharmacological targets. Inflammation 41 (3), 773–783. doi:10.1007/s10753-018-0731-9
Sakai, N., and Tager, A. M. (2013). Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 1832 (7), 911–921. doi:10.1016/j.bbadis.2013.03.001
Sanderson, R. D., Elkin, M., Rapraeger, A. C., Ilan, N., and Vlodavsky, I. (2017). Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. FEBS J. 284 (1), 42–55. doi:10.1111/febs.13932
Schmidt, E. P., Yang, Y., Janssen, W. J., Gandjeva, A., Perez, M. J., Barthel, L., et al. (2012). The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 18 (8), 1217–1223. doi:10.1038/nm.2843
Secchi, M. F., Crescenzi, M., Masola, V., Russo, F. P., Floreani, A., and Onisto, M. (2017). Heparanase and macrophage interplay in the onset of liver fibrosis. Sci. Rep. 7 (1), 14956. doi:10.1038/s41598-017-14946-0
Secchi, M. F., Masola, V., Zaza, G., Lupo, A., Gambaro, G., and Onisto, M. (2015). Recent data concerning heparanase: Focus on fibrosis, inflammation and cancer. Biomol. Concepts 6 (5-6), 415–421. doi:10.1515/bmc-2015-0021
Shafat, I., Vlodavsky, I., and Ilan, N. (2006). Characterization of mechanisms involved in secretion of active heparanase. J. Biol. Chem. 281 (33), 23804–23811. doi:10.1074/jbc.M602762200
Shteingauz, A., Boyango, I., Naroditsky, I., Hammond, E., Gruber, M., Doweck, I., et al. (2015). Heparanase enhances tumor growth and chemoresistance by promoting autophagy. Cancer Res. 75 (18), 3946–3957. doi:10.1158/0008-5472.CAN-15-0037
Shteingauz, N. I. A., and Vlodavsky, I. (2015). Function from within: Autophagy induction by HPSE/heparanasenew possibilities for intervention. Autophagy 11 (12), 2387–2389. doi:10.1080/15548627.2015.1115174
Sibinska, Z., Tian, X., Korfei, M., Kojonazarov, B., Kolb, J. S., Klepetko, W., et al. (2017). Amplified canonical transforming growth factor-beta signalling via heat shock protein 90 in pulmonary fibrosis. Eur. Respir. J. 49 (2), 1501941. doi:10.1183/13993003.01941-2015
Simeonovic, C. J., Ziolkowski, A. F., Wu, Z., Choong, F. J., Freeman, C., and Parish, C. R. (2013). Heparanase and autoimmune diabetes. Front. Immunol. 4, 471. doi:10.3389/fimmu.2013.00471
Skendros, P., Mitsios, A., Chrysanthopoulou, A., Mastellos, D. C., Metallidis, S., Rafailidis, P., et al. (2020). Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 130 (11), 6151–6157. doi:10.1172/JCI141374
Soni, S., Wilson, M. R., O'Dea, K. P., Yoshida, M., Katbeh, U., Woods, S. J., et al. (2016). Alveolar macrophage-derived microvesicles mediate acute lung injury. Thorax 71 (11), 1020–1029. doi:10.1136/thoraxjnl-2015-208032
Sontake, V., Wang, Y., Kasam, R. K., Sinner, D., Reddy, G. B., Naren, A. P., et al. (2017). Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2 (4), e91454. doi:10.1172/jci.insight.91454
Sotnikov, I., Hershkoviz, R., Grabovsky, V., Ilan, N., Cahalon, L., Vlodavsky, I., et al. (2004). Enzymatically quiescent heparanase augments T cell interactions with VCAM-1 and extracellular matrix components under versatile dynamic contexts. J. Immunol. 172 (9), 5185–5193. doi:10.4049/jimmunol.172.9.5185
Sou, Y. S., Tanida, I., Komatsu, M., Ueno, T., and Kominami, E. (2006). Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J. Biol. Chem. 281 (6), 3017–3024. doi:10.1074/jbc.M505888200
Stahl, P. D., and Raposo, G. (2019). Extracellular vesicles: Exosomes and microvesicles, integrators of homeostasis. Physiol. (Bethesda) 34 (3), 169–177. doi:10.1152/physiol.00045.2018
Thompson, B. T., Chambers, R. C., and Liu, K. D. (2017). Acute respiratory distress syndrome. N. Engl. J. Med. 377 (6), 1904–1905. doi:10.1056/NEJMc1711824
Thompson, C. A., Purushothaman, A., Ramani, V. C., Vlodavsky, I., and Sanderson, R. D. (2013). Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J. Biol. Chem. 288 (14), 10093–10099. doi:10.1074/jbc.C112.444562
Uchinami, H., Seki, E., Brenner, D. A., and D'Armiento, J. (2006). Loss of MMP 13 attenuates murine hepatic injury and fibrosis during cholestasis. Hepatology 44 (2), 420–429. doi:10.1002/hep.21268
Vlodavsky, I., Eldor, A., Haimovitz-Friedman, A., Matzner, Y., Ishai-Michaeli, R., Lider, O., et al. (1992). Expression of heparanase by platelets and circulating cells of the immune system: Possible involvement in diapedesis and extravasation. Invasion Metastasis 12 (2), 112–127.
Vlodavsky, I., Friedmann, Y., Elkin, M., Aingorn, H., Atzmon, R., Ishai-Michaeli, R., et al. (1999). Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat. Med. 5 (7), 793–802. doi:10.1038/10518
Vlodavsky, I., Ilan, N., Naggi, A., and Casu, B. (2007). Heparanase: Structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr. Pharm. Des. 13 (20), 2057–2073. doi:10.2174/138161207781039742
Vreys, V., and David, G. (2007). Mammalian heparanase: What is the message? J. Cell Mol. Med. 11 (3), 427–452. doi:10.1111/j.1582-4934.2007.00039.x
Walter, J., Ware, L. B., and Matthay, M. A. (2014). Mesenchymal stem cells: Mechanisms of potential therapeutic benefit in ARDS and sepsis. Lancet Respir. Med. 2 (12), 1016–1026. doi:10.1016/S2213-2600(14)70217-6
Waterman, M., Ben-Izhak, O., Eliakim, R., Groisman, G., Vlodavsky, I., and Ilan, N. (2007). Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod. Pathol. 20 (1), 8–14. doi:10.1038/modpathol.3800710
Wiener-Kronish, J. P., Albertine, K. H., and Matthay, M. A. (1991). Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J. Clin. Invest. 88 (3), 864–875. doi:10.1172/JCI115388
Wu, D., and Yang, X. O. (2020). TH17 responses in cytokine storm of COVID-19: An emerging target of JAK2 inhibitor Fedratinib. J. Microbiol. Immunol. Infect. 53 (3), 368–370. doi:10.1016/j.jmii.2020.03.005
Wu, J., Liang, B., Qian, Y., Tang, L., Xing, C., Zhuang, Q., et al. (2018). Down-regulation of CD19 expression inhibits proliferation, adhesion, migration and invasion and promotes apoptosis and the efficacy of chemotherapeutic agents and imatinib in SUP-B15 cells. Cell Biol. Int. 42 (9), 1228–1239. doi:10.1002/cbin.10994
Xi, Y., Kim, T., Brumwell, A. N., Driver, I. H., Wei, Y., Tan, V., et al. (2017). Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 19 (8), 904–914. doi:10.1038/ncb3580
Xiao, K., Hou, F., Huang, X., Li, B., Qian, Z. R., and Xie, L. (2020). Mesenchymal stem cells: Current clinical progress in ARDS and COVID-19. Stem Cell Res. Ther. 11 (1), 305. doi:10.1186/s13287-020-01804-6
Xu, J., Camfield, R., and Gorski, S. M. (2018). The interplay between exosomes and autophagy - partners in crime. J. Cell Sci. 131 (15), jcs215210. doi:10.1242/jcs.215210
Yamada, M. (2021). Extracellular vesicles: Their emerging roles in the pathogenesis of respiratory diseases. Respir. Investig. 59 (3), 302–311. doi:10.1016/j.resinv.2021.02.006
Yamada, S. H., and Sugahara, K. (1998). Structure of oligosaccharides isolated from heparan sulfate heparin and substrate specificities of the degrading enzymes of bacterial origin. Trends Glycosci. Glycotechnol. 10 (52), 95–123. doi:10.4052/tigg.10.95
Yazdanpanah, F., Hamblin, M. R., and Rezaei, N. (2020). The immune system and COVID-19: Friend or foe? Life Sci. 256, 117900. doi:10.1016/j.lfs.2020.117900
Ye, Q., Wang, B., and Mao, J. (2020). The pathogenesis and treatment of the `Cytokine Storm' in COVID-19. J. Infect. 80 (6), 607–613. doi:10.1016/j.jinf.2020.03.037
Yu, B., Zhang, X., and Li, X. (2014). Exosomes derived from mesenchymal stem cells. Int. J. Mol. Sci. 15 (3), 4142–4157. doi:10.3390/ijms15034142
Yuan, L., Hu, J., Luo, Y., Liu, Q., Li, T., Parish, C. R., et al. (2012). Upregulation of heparanase in high-glucose-treated endothelial cells promotes endothelial cell migration and proliferation and correlates with Akt and extracellular-signal-regulated kinase phosphorylation. Mol. Vis. 18, 1684–1695.
Zhang, H., Zhou, Y., Qu, M., Yu, Y., Chen, Z., Zhu, S., et al. (2021). Tissue factor-enriched neutrophil extracellular traps promote immunothrombosis and disease progression in sepsis-induced lung injury. Front. Cell Infect. Microbiol. 11, 677902. doi:10.3389/fcimb.2021.677902
Zhang, R., Wang, Z., Tejera, P., Frank, A. J., Wei, Y., Su, L., et al. (2017). Late-onset moderate to severe acute respiratory distress syndrome is associated with shorter survival and higher mortality: A two-stage association study. Intensive Care Med. 43 (3), 399–407. doi:10.1007/s00134-016-4638-3
Zhang, Y. M., Bachmann, S., Hemmer, C., van Lunzen, J., von Stemm, A., Kern, P., et al. (1994). Vascular origin of Kaposi's sarcoma. Expression of leukocyte adhesion molecule-1, thrombomodulin, and tissue factor. Am. J. Pathol. 144 (1), 51–59.
Zhang, Y., Wang, J., Liu, Y. M., Yang, H., Wu, G. J., and He, X. H. (2021). Analysis of the efficacy and mechanism of action of xuebijing injection on ARDS using meta-analysis and network Pharmacology. Biomed. Res. Int. 2021, 8824059. doi:10.1155/2021/8824059
Zheng, G., Huang, L., Tong, H., Shu, Q., Hu, Y., Ge, M., et al. (2014). Treatment of acute respiratory distress syndrome with allogeneic adipose-derived mesenchymal stem cells: A randomized, placebo-controlled pilot study. Respir. Res. 15, 39. doi:10.1186/1465-9921-15-39
Zhou, Z., and You, Z. (2016). Mesenchymal stem cells alleviate LPS-induced acute lung injury in mice by MiR-142a-5p-controlled pulmonary endothelial cell autophagy. Cell Physiol. Biochem. 38 (1), 258–266. doi:10.1159/000438627
Zinter, M. S., Delucchi, K. L., Kong, M. Y., Orwoll, B. E., Spicer, A. S., Lim, M. J., et al. (2019). Early plasma matrix metalloproteinase profiles. A novel pathway in pediatric acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 199 (2), 181–189. doi:10.1164/rccm.201804-0678OC
Keywords: heparinase, ARDS, exosome, inflammation, coagulation, fibrosis, autophagy
Citation: Feng F, Wang L-J, Li J-C, Chen T-T and Liu L (2023) Role of heparanase in ARDS through autophagy and exosome pathway (review). Front. Pharmacol. 14:1200782. doi: 10.3389/fphar.2023.1200782
Received: 05 April 2023; Accepted: 30 May 2023;
Published: 08 June 2023.
Edited by:
Elena Talero, Sevilla University, SpainReviewed by:
Ayyanar Sivanantham, Boston Medical Center, United StatesPavel Solopov, Old Dominion University, United States
Copyright © 2023 Feng, Wang, Li, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liping Liu, bGl1bGlwaW5nbGR5eUAxMjYuY29t