 Jeong-In Baek
Jeong-In Baek Ye-Ri Kim2,3†
Ye-Ri Kim2,3† Un-Kyung Kim
Un-Kyung Kim
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 31 March 2023
Sec. Experimental Pharmacology and Drug Discovery
Volume 14 - 2023 | https://doi.org/10.3389/fphar.2023.1176881
This article is part of the Research Topic Advances in Novel Pharmacotherapeutics and Drug Discovery: Computational, Experimental, Translational, and Clinical Models. View all 12 articles
Noise (noise-induced hearing loss), and ototoxic drugs (drug-induced ototoxicity), and aging (age-related hearing loss) are the major environmental factors that lead to acquired sensorineural hearing loss. So far, there have been numerous efforts to develop protective or therapeutic agents for acquired hearing loss by investigating the pathological mechanisms of each types of hearing loss, especially in cochlear hair cells and auditory nerves. Although there is still a lack of information on the underlying mechanisms of redox homeostasis and molecular redox networks in hair cells, an imbalance in mitochondrial reactive oxygen species (ROS) levels that enhance oxidative stress has been suggested as a key pathological factor eventually causing acquired sensorineural hearing loss. Thus, various types of antioxidants have been investigated for their abilities to support auditory cells in maintenance of the hearing function against ototoxic stimuli. In this review, we will discuss the scientific possibility of developing drugs that target particular key elements of the mitochondrial redox network in prevention or treatment of noise- and ototoxic drug-induced hearing loss.
Sensorineural hearing loss (SNHL), the most common type of permanent hearing impairment, is caused by physical and/or functional damage of cochlear hair cells in inner ear or auditory nerves including spiral ganglion neurons. Acquired SNHL, resulting from continuous accumulation of cellular damage due to various environmental stimuli, increases in prevalence with aging: Being identified in approximately 5–8 of 1,000 children, 33% of adults 65–74 years of age, and over 80% of adults 85 years of age (Walling et al., 2012; Baumgartner et al., 2021). This prevalence of acquired SNHL is much higher than congenital SNHL that occurs in two to four of 1,000 newborns. The major environmental factors that lead to acquired SNHL are aging (age-related hearing loss), noise (noise-induced hearing loss), and ototoxic drugs (drug-induced ototoxicity). Over the past few decades, there have been numerous efforts to develop therapeutic agents for each type of acquired hearing loss by investigating the pathological mechanisms of hearing loss at the molecular level. An imbalance in mitochondrial reactive oxygen species (ROS) levels that enhance oxidative stress has been suggested as a key pathological factor that eventually causes hair cell death in all three types of acquired SNHL (Bottger and Schacht, 2013; Fujimoto and Yamasoba, 2014; Kamogashira et al., 2015; Wong and Ryan, 2015). Attempts to decrease ROS levels in order to prevent or slow acquired SNHL led researchers to test the protective or alleviative effects of various antioxidants, such as vitamins, lipoic acids, polyphenols, and other small molecules (Hildebrand et al., 2008; Tavanai and Mohammadkhani, 2017; Ibrahim et al., 2018; Fetoni et al., 2019; Pak et al., 2020). Most of these investigations have been conducted in animal models with only a few studies in humans, and there is still a lack of information on the underlying mechanisms of redox homeostasis and molecular redox networks in hair cells. Due to these limitations, there is currently only one medicine at a clinically applicable level. In 2022, the Food and Drug Administration (FDA) of United States approved the use of sodium thiosulfate, an antioxidant, as a therapeutic agent of cisplatin-induced hearing loss, based on the results of clinical trials (Freyer et al., 2017; Brock et al., 2018a; Brock et al., 2018b). In the clinical test, sodium thiosulfate successfully reduced the incidence of cisplatin-induced ototoxicity by nearly 50% in the hepatoblastoma patients. However, verification of the therapeutic effect was limited to patients under the age of 18 with non-metastic solid cancer, and the effect has also been shown to vary depending on the time intervals between administration of cisplatin and sodium thiosulfate (Hazlitt et al., 2018). It means, the development of otoprotectants that are effective for a wide range of acquired hearing loss is still needed. In this review, we will discuss previous efforts to develop protective or therapeutic agents focusing on oxidative stress-induced mitochondrial damage in hair cells. We will also propose the possibility of developing drugs that target particular elements of the mitochondrial redox network in hearing loss pathology.
The main pathological factors of acquired SNHL, aging, noise exposure, and ototoxic drugs, induce multiple, simultaneous responses in cochlear hair cells, that directly damage macromolecules (nucleic acids, proteins, and lipids), change ion homeostasis, and activate/inhibit intrinsic and extrinsic signaling pathways (Wong and Ryan, 2015; Wu et al., 2020). These responses ultimately produce irreversible hair cell damage when the mechanical or biochemical stimuli overwhelm cellular homeostatic capacity. Disruption of cellular redox homeostasis by either or both increased ROS generation and inhibition of antioxidant defense systems, is known to be mutually influenced by other cellular responses induced by noise, drugs, and aging (Wu et al., 2020) (Figure 1).
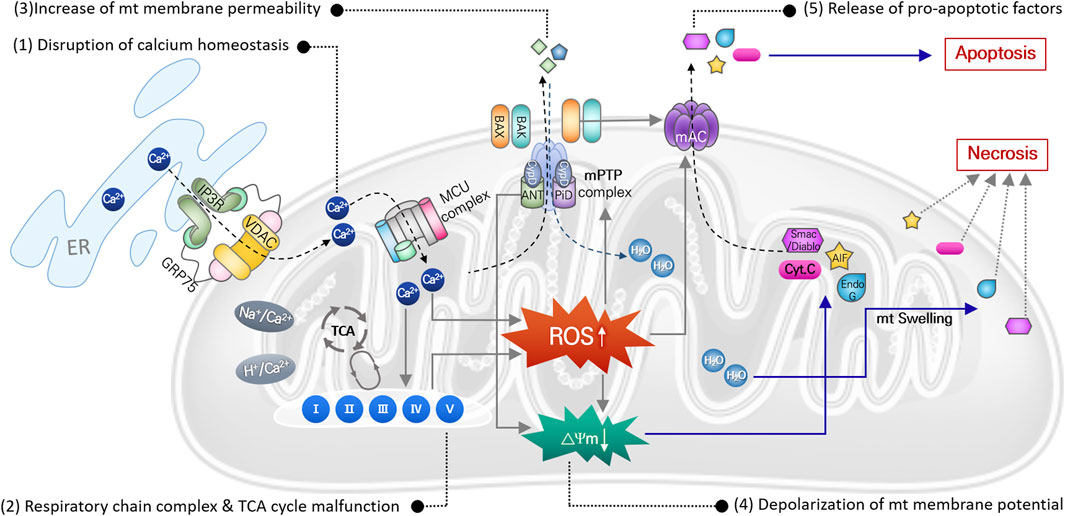
FIGURE 1. Noise, aging and ototoxic drug-induced intracellular signaling pathways leading to hair cell death. It indicates that organelle stress (ER and mitochondria), inflammation, MAPK signaling pathways induced by three environmental stimuli are all mutually interact each other, which finally leads to apoptosis or necroptosis of hair cells. Importantly, increase in mitochondrial ROS is at the center of all the pathways.
Intense noise can directly cause mechanical disruption of the hair cell stereocilia, resulting in their dysfunction in the auditory pathway (Slepecky, 1986; Patuzzi et al., 1989). However, most noise-induced hearing loss is caused by an accumulation of biochemical damage due to prolonged exposure to sound stimuli below the threshold of mechanical damage. Stimulation of the mitochondrial respiratory system by ischemia/reperfusion (Henderson et al., 2006) and release of free Ca2+ from Ca2+ stores, such as endoplasmic reticulum (ER) to cytosol and into mitochondria (Gorlach et al., 2015; Wong and Ryan, 2015), are the major noise-induced intracellular responses that cause biochemical hair cell damage. Importantly, these processes are commonly linked to overgeneration of ROS (Yamashita et al., 2004a). Noise exposure also induces insufficiency of the antioxidant system. Consequently, increased ROS randomly reacts with lipids, nucleic acids, and proteins, resulting in dysfunction of these macromolecules, and subsequently inducing release of pro-apoptotic factors. Cytochrome C promotes caspase 3-mediated apoptosis, and translocation of Endonuclease G (EndoG) and apoptosis-inducing factor (AIF) from mitochondria into the nucleus, triggering apoptotic hair cell death with condensed nuclei (Yamashita et al., 2004b; Wong and Ryan, 2015; Sha and Schacht, 2017). Increased ROS has also been shown to activate the c-Jun N-terminal kinase (JNK) signaling pathway, leading to apoptosis in an animal model (Wang et al., 2007; Wu et al., 2020).
Because oxidative stress has been strongly implicated as a cause of noise-induced hearing loss, various types of antioxidant molecules have been used to protect or recover hair cells. Glutathione, D-methionine, resveratrol, salicylate, ebselen, and coenzymeQ10 administered in animal studies had significant protective effects on noise-induced hair cell damage (Sha and Schacht, 2017; Pak et al., 2020). Specifically, N-acetyl cysteine (NAC), α-lipoic acid, and ebselen have advanced to clinical trials and shown a substantial otoprotective effect. In two randomized clinical studies, a group of textile workers who received NAC had a reduced temporary threshold shift after noise exposure (Fetoni et al., 2009), as well as army members who received NAC for 14 days who had a reduced temporary threshold shift (Lorito et al., 2008; Kopke et al., 2015). Alpha-lipoic acid also had a protective effect on noise-induced hearing threshold shift. When a group of healthy subjects received oral α-lipoic acid 1 h before noise exposure, their temporary threshold shift at 6 kHz was reduced after noise exposure (Campbell et al., 2007). These results suggest a strong possibility that reducing ROS accumulation may effectively prevent noise-induced hearing loss.
Aminoglycoside, broad-spectrum antibiotics, and platinum-based anticancer agents such as cisplatin are the most well-known ototoxic drugs that can cause irreversible, bilateral, and high frequency hearing loss (Musial-Bright et al., 2011). Following entry into the hair cells, both aminoglycosides and cisplatin directly bind to hundreds of intracellular proteins such as kinases, transcription factors, and ion channels that potentially cause dell death (Karasawa et al., 2010; Karasawa et al., 2011; Karasawa et al., 2013; Karasawa and Steyger, 2015).
Although they affect multiple cellular signaling pathways by directly interacting with various proteins, the central affected pathological pathway is the accumulation of oxidative stress caused by accelerated ROS generation and inhibition of antioxidant enzyme activities, eventually leading to apoptosis or necrosis of hair cells (Steyger, 2021). Aminoglycosides are known to accelerate both enzymatic and non-enzymatic ROS formation (Priuska and Schacht, 1995), thereby activating the JNK signaling pathway followed by apoptotic hair cell death (Mielke and Herdegen, 2000). In enzymatic ROS formation, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases are considered a primary source of ROS generation. NADPH oxidase is a protein complex composed of several subunits including a catalytic subunit, NOX (Altenhofer et al., 2015; Steyger, 2021). In previous studies, NOX2 expression was abundantly increased in outer hair cells after neomycin administration in rat cochlea, and inhibition of NOX2 significantly reduced neomycin-induced hair cell damage (Qi et al., 2018). Cisplatin also upregulates NOX3 expression and activates the NOX3 signaling pathway, increasing superoxide production in cultured cells and rat cochlea (Banfi et al., 2004; Mukherjea et al., 2008; Mukherjea et al., 2011).
Disruption of the intracellular antioxidant system also contributes to aminoglycoside- or cisplatin-induced oxidative stress. For example, cisplatin can bind directly to sulfhydryl groups within antioxidant enzymes causing enzyme dysfunction, and can also deplete glutathione (GSH) and NADPH that are essential factors for antioxidant enzyme function (Rybak et al., 2007). Cisplatin and kanamycin are known to decrease expression and activity of the primary antioxidant enzymes: Superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione reductase (GR), and catalase (CAT) (Rybak et al., 2000; Rybak et al., 2007). Although there are various molecular factors directly affected by drugs, ultimately they cause an excessive accumulation of ROS, and numerous studies have shown that the subsequent cell death signaling pathways shared with noise-induced hearing loss.
Various types of antioxidants were also tested in drug-induced ototoxicity to examine their ability to prevent hearing loss. The widely-used antioxidants, NAC, sodium thiosulfate, and D-methionine, effectively protected or rescued cisplatin-induced ototoxicity, due to their high affinity for platinum molecules, as well as their antioxidative activity (Wu et al., 2020). In our previous studies, we identified several antioxidants that had protective effects against drug-induced ototoxicity. In mouse cochlear explants treated with amikacin, kanamycin, or cisplatin, pre-treatment with galangin (flavonoid antioxidant), fursultiamine (thiamine disulfide derivative), or berberine chloride (alkaline isoquinoline) reduced intracellular ROS levels in hair cells. These treatments prevented ROS-mediated caspase-3 activation, DNA fragmentation, and apoptosis of hair cells, indicating that an antioxidant can be used to prevent both noise- and drug-induced hearing loss. Sodium thiosulfate (STS) is currently the only clinically approved treatment for cisplatin-induced ototoxicity. As an antioxidant, it is known to plays a role in directly removing ROS and promote antioxidative and anti-apoptotic enzymes by activating Nuclear factor erythroid-related factor 2 (Nrf2) (Zhang et al., 2021). Moreover, it directly binds to cisplatin, inhibiting and deactivating the metabolism of cisplatin by formation of platinum (Pt)-STS complex. Cisplatin is metabolized and activated in the body through the process of hydrolysis. In this process, each of activated cisplatin metabolites has different tumor selectivity, finally causing normal cell damages. Since reducing the level of activated form of cisplatin inhibit cytotoxicity of normal cells, it is suggested that the amelioration of cisplatin-induced cytotoxicity by STS could be explained in terms of the rapid formation of inactive Pt-STS complex (Sooriyaarachchi et al., 2012). Therefore, it is thought that STS can play a role as a direct inhibitor of cisplatin, selectively reducing toxicity to normal cells, rather than as an antioxidant.
Mitochondria, referred to as the center of cellular energy metabolism, are organelles that synthesize ATP through the TCA cycle and oxidative phosphorylation processes. Because this aerobic respiration unavoidably produces ROS, mitochondria are the primary cellular source of ROS (Wei et al., 2001; Islam, 2017). Mitochondria also play critical roles in maintaining cellular homeostasis and contribute to homeostatic regulation of calcium and iron concentrations (Contreras et al., 2010), autophagy (Scherz-Shouval and Elazar, 2007), and cell death (Borutaite, 2010). Thus, mitochondrial damage or dysfunction can directly activate cell death signaling pathways. Noise- or cytotoxic drug-induced intracellular responses, including ischemia/reperfusion, DNA damage, ER stress, etc., increase ROS levels and DNA damage in mitochondria within auditory cells. Even though mitochondrial ROS can be easily scavenged by mitochondrial antioxidant systems under normal physiological conditions, excessive ROS generation or failure to remove mitochondrial ROS can cause oxidative stress leading to mitochondrial dysfunction in multiple ways (Figure 2).
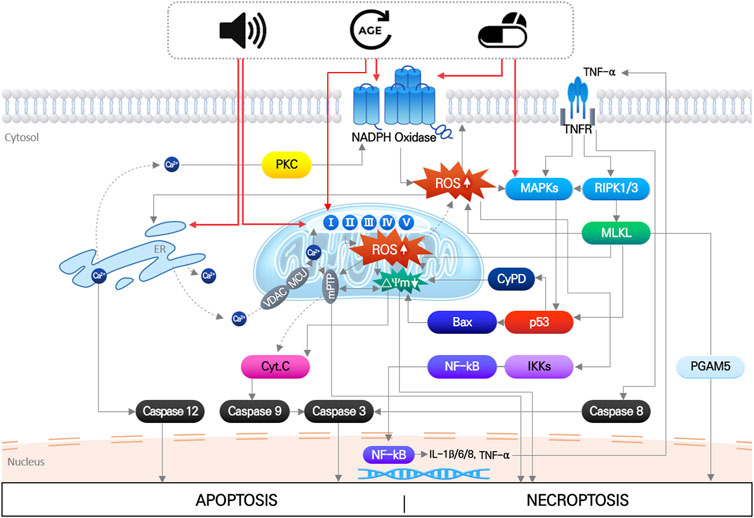
FIGURE 2. Mitochondrial ROS-induced damages causing mitochondrial dysfunction. Direct damages of mitochondrial DNA and antioxidant enzyme, or disruption of ion homeostasis 1), or malfunction of the mitochondrial respiration system 2), can cause increase in mitochondrial ROS. It subsequently leads to increase in mitochondrial membrane potential 3) with depolarization of membrane potential 4), causing osmotic swelling of mitochondria and release of pro-apoptotic factors 5). Finally, cell death signaling pathways are activated by these apoptotic factors.
In mitochondria, free Ca2+ ions are the most likely to trigger ROS elevation. Particularly after intense noise stimulation, Ca2+ is released from the ER and translocated into mitochondria through an ion transporter such as VDAC or MCU. Abnormal Ca2+ influx can dysregulate Ca2+-regulated enzymes that are involved in ROS metabolism, leading to increased mitochondrial ROS levels, free-radical damage, and ultimately to mitochondrial dysfunction (Tretter and Adam-Vizi, 2004; Bottger and Schacht, 2013). Accumulated mitochondrial ROS can directly damage mitochondrial DNA (mtDNA), causing mtDNA mutations. Accumulation of mtDNA is responsible for dysfunction of various mitochondrial proteins, which disrupts metabolic homeostasis and induces mitochondrial dysfunction (Baker and Staecker, 2012). Finally, mitochondrial damage caused by increased mitochondrial ROS can depolarize mitochondrial membrane potential, which increases mitochondrial membrane permeability and initiates apoptosis or necrosis signaling pathways. Increased ROS activates p53 in the cytosol, leading to translocation of Bcl-2 associated X (BAX) from the cytosol to the mitochondrial outer membrane. Mitochondrial ROS-induced activation of the BAX pore and other mitochondrial pore complexes, e.g., the mitochondrial permeability transition pore (mPTP) and mitochondrial apoptosis-induced channel (mAC), contributes to the release of pro-apoptotic factors (cytochrome C, EndoG, and AIF) from the mitochondrial matrix to the cytosol to activate the apoptotic signaling pathway (Dejean et al., 2005; Briston et al., 2017). By contrast, loss of mitochondrial membrane potential activates the mPTP pore complex, allowing H2O and other small molecules to enter the mitochondrial matrix, which induces mitochondrial osmotic swelling and initiates the necrosis signaling pathway (Briston et al., 2017).
Aminoglycosides tend to accumulate in the hair cell mitochondria, and oxidative stress-induced mitochondrial damage and subsequent cell death by antibiotics are also observed consistently in cochlear hair cells (Hobbie et al., 2008). Gentamicin directly inhibits mitochondrial protein synthesis, which triggers opening of the mPTP pore complex that can then release pro-apoptotic factors (Dehne et al., 2002). Cisplatin administration increases mitochondrial ROS, leading to loss of mitochondrial membrane potential and BAX expression, which causes increased cleaved caspase-3 expression and hair cell apoptosis in mouse cochlear explants (Kim et al., 2018).
Because mitochondria act as a pathological link between intracellular oxidative stress and apoptotic cell death, inhibiting mitochondrial oxidative stress or decreasing mitochondrial membrane permeability using mitochondria-targeted molecules, could be an effective approach to alleviating noise- and drug-induced hearing loss. Thus, mitochondria-specific targeting of potent compounds has been considered important to maximize effectiveness of the compounds. Especially Lipophilic cation-based modification of compounds is one of the most successful mitochondria-targeting techniques, due to improved ability to cross polarized mitochondrial membrane. Triphenylphosphonium (TPP), a kind of lipophilic cation, has been conjugated to various potent antioxidants (Wang et al., 2020), and numerous TPP-tagged mitochondria-targeted antioxidants including MitoQ, SkQ1, SkQR1, MitoVitE, and MitoPeroxidase, have been tested to prevent or alleviate acquired hearing loss (Zielonka et al., 2017; Fujimoto and Yamasoba, 2019).
We previously used a mitochondria-targeted antioxidant, MitoQ, to evaluate the therapeutic potential of mitochondria-specific antioxidants in oxidative stress-induced hair cell damage. In mouse cochlear explants, H2O2 treatment increased mitochondrial ROS leading to loss of mitochondrial membrane potential, which was attributed to decreased expression of the mitochondrial respiratory chain complex I, III, and V. When MitoQ was administered 1 h prior to H2O2 treatment, the mitochondrial oxidative stress responses were almost completely neutralized, thus protecting hair cells from apoptotic cell death (Kim et al., 2019). Other studies on drug-induced ototoxicity also suggested that both MitoQ and SkQR1 significantly reduce aminoglycoside- and cisplatin-induced hearing loss in cultured cells and animal models (Jankauskas et al., 2012; Ojano-Dirain and Antonelli, 2012; Ojano-Dirain et al., 2014; Tate et al., 2017; Dirain et al., 2018).
Extensive scientific evidence strongly suggests mitochondria as a key target to protect auditory cells and maintain hearing function. Although a number of previous studies have shown that general antioxidants can provide protection against drug-induced ototoxicity, there are two significant limitations. First, since the general antioxidants are not highly selective for specific target molecules and can affect multiple signaling pathways simultaneously with low specificity, it is difficult to determine drug efficacy and safety at low concentrations. Second, general antioxidants have the potential to cause unintended side effects when co-administered with other medicines. For instance, antioxidants such as α-lipoic acid can inhibit the death of cancer cells as well as normal cells, thereby inhibiting the anticancer effect. Thus, α-lipoic acid cannot be administered in combination with the anticancer drugs such as cisplatin.
To overcome these limitations, it will be important to discover and develop novel therapeutic agents that interact with specific mitochondrial factors that contribute to mitochondrial redox homeostasis, such as antioxidant enzymes or mitochondrial pore proteins. This will be essential information for developing common drugs that are widely effective in different types of acquired hearing loss.
J-IB and U-KK contributed to conception and design of the study. J-IB and Y-RK wrote the first draft of the manuscript and created the images. K-YL reviewed and edited the manuscript. U-KK organized the whole process and offered constructive criticism. All authors contributed to manuscript revision, read, and approved the submitted version.
This research was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korea government (Ministry of Science and ICT): RS-2023-00212674 (to J-IB), NRF-2020R1A2C2003529 (to K-YL), and NRF-2021R1C1C2006677 (to Y-RK). It was also supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT), No. 2022M3E5F2017487 (to U-KK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Altenhofer, S., Radermacher, K. A., Kleikers, P. W., Wingler, K., and Schmidt, H. H. (2015). Evolution of NADPH oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal 23, 406–427. doi:10.1089/ars.2013.5814
Baker, K., and Staecker, H. (2012). Low dose oxidative stress induces mitochondrial damage in hair cells. Anat. Rec. Hob. 295, 1868–1876. doi:10.1002/ar.22594
Banfi, B., Malgrange, B., Knisz, J., Steger, K., Dubois-Dauphin, M., and Krause, K. H. (2004). NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 279, 46065–46072. doi:10.1074/jbc.M403046200
Baumgartner, J. E., Baumgartner, L. S., Baumgartner, M. E., Moore, E. J., Messina, S. A., Seidman, M. D., et al. (2021). Progenitor cell therapy for acquired pediatric nervous system injury: Traumatic brain injury and acquired sensorineural hearing loss. Stem Cells Transl. Med. 10, 164–180. doi:10.1002/sctm.20-0026
Borutaite, V. (2010). Mitochondria as decision-makers in cell death. Environ. Mol. Mutagen 51, 406–416. doi:10.1002/em.20564
Bottger, E. C., and Schacht, J. (2013). The mitochondrion: A perpetrator of acquired hearing loss. Hear Res. 303, 12–19. doi:10.1016/j.heares.2013.01.006
Briston, T., Roberts, M., Lewis, S., Powney, B., Szabadkai, G., and Duchen, M. R. (2017). Mitochondrial permeability transition pore: Sensitivity to opening and mechanistic dependence on substrate availability. Sci. Rep. 7, 10492. doi:10.1038/s41598-017-10673-8
Brock, P. R., Maibach, R., Childs, M., Rajput, K., Roebuck, D., Sullivan, M. J., et al. (2018a). Sodium thiosulfate for protection from cisplatin-induced hearing loss. N. Engl. J. Med. 378, 2376–2385. doi:10.1056/NEJMoa1801109
Brock, P. R., Maibach, R., and Neuwelt, E. A. (2018b). Sodium thiosulfate and cisplatin-induced hearing loss. N. Engl. J. Med. 379, 1180–1181. doi:10.1056/NEJMc1809501
Campbell, K. C., Meech, R. P., Klemens, J. J., Gerberi, M. T., Dyrstad, S. S., Larsen, D. L., et al. (2007). Prevention of noise- and drug-induced hearing loss with D-methionine. Hear Res. 226, 92–103. doi:10.1016/j.heares.2006.11.012
Contreras, L., Drago, I., Zampese, E., and Pozzan, T. (2010). Mitochondria: The calcium connection. Biochim. Biophys. Acta 1797, 607–618. doi:10.1016/j.bbabio.2010.05.005
Dehne, N., Rauen, U., de Groot, H., and Lautermann, J. (2002). Involvement of the mitochondrial permeability transition in gentamicin ototoxicity. Hear Res. 169, 47–55. doi:10.1016/s0378-5955(02)00338-6
Dejean, L. M., Martinez-Caballero, S., Guo, L., Hughes, C., Teijido, O., Ducret, T., et al. (2005). Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol. Biol. Cell 16, 2424–2432. doi:10.1091/mbc.e04-12-1111
Dirain, C. O., Ng, M., Milne-Davies, B., Joseph, J. K., and Antonelli, P. J. (2018). Evaluation of mitoquinone for protecting against amikacin-induced ototoxicity in Guinea pigs. Otol. Neurotol. 39, 111–118. doi:10.1097/MAO.0000000000001638
Fetoni, A. R., Paciello, F., Rolesi, R., Paludetti, G., and Troiani, D. (2019). Targeting dysregulation of redox homeostasis in noise-induced hearing loss: Oxidative stress and ROS signaling. Free Radic. Biol. Med. 135, 46–59. doi:10.1016/j.freeradbiomed.2019.02.022
Fetoni, A. R., Ralli, M., Sergi, B., Parrilla, C., Troiani, D., and Paludetti, G. (2009). Protective effects of N-acetylcysteine on noise-induced hearing loss in Guinea pigs. Acta Otorhinolaryngol. Ital. 29, 70–75.
Freyer, D. R., Chen, L., Krailo, M. D., Knight, K., Villaluna, D., Bliss, B., et al. (2017). Effects of sodium thiosulfate versus observation on development of cisplatin-induced hearing loss in children with cancer (ACCL0431): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 18, 63–74. doi:10.1016/S1470-2045(16)30625-8
Fujimoto, C., and Yamasoba, T. (2019). Mitochondria-targeted antioxidants for treatment of hearing loss: A systematic review. Antioxidants (Basel) 8, 109. doi:10.3390/antiox8040109
Fujimoto, C., and Yamasoba, T. (2014). Oxidative stresses and mitochondrial dysfunction in age-related hearing loss. Oxid. Med. Cell Longev. 2014, 582849. doi:10.1155/2014/582849
Gorlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: A mutual interplay. Redox Biol. 6, 260–271. doi:10.1016/j.redox.2015.08.010
Hazlitt, R. A., Min, J., and Zuo, J. (2018). Progress in the development of preventative drugs for cisplatin-induced hearing loss. J. Med. Chem. 61, 5512–5524. doi:10.1021/acs.jmedchem.7b01653
Henderson, D., Bielefeld, E. C., Harris, K. C., and Hu, B. H. (2006). The role of oxidative stress in noise-induced hearing loss. Ear Hear 27, 1–19. doi:10.1097/01.aud.0000191942.36672.f3
Hildebrand, M. S., Newton, S. S., Gubbels, S. P., Sheffield, A. M., Kochhar, A., de Silva, M. G., et al. (2008). Advances in molecular and cellular therapies for hearing loss. Mol. Ther. 16, 224–236. doi:10.1038/sj.mt.6300351
Hobbie, S. N., Akshay, S., Kalapala, S. K., Bruell, C. M., Shcherbakov, D., and Bottger, E. C. (2008). Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc. Natl. Acad. Sci. U. S. A. 105, 20888–20893. doi:10.1073/pnas.0811258106
Ibrahim, I., Zeitouni, A., and da Silva, S. D. (2018). Effect of antioxidant vitamins as adjuvant therapy for sudden sensorineural hearing loss: Systematic review study. Audiol. Neurootol 23, 1–7. doi:10.1159/000486274
Islam, M. T. (2017). Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 39, 73–82. doi:10.1080/01616412.2016.1251711
Jankauskas, S. S., Plotnikov, E. Y., Morosanova, M. A., Pevzner, I. B., Zorova, L. D., Skulachev, V. P., et al. (2012). Mitochondria-targeted antioxidant SkQR1 ameliorates gentamycin-induced renal failure and hearing loss. Biochem. (Mosc) 77, 666–670. doi:10.1134/S0006297912060144
Kamogashira, T., Fujimoto, C., and Yamasoba, T. (2015). Reactive oxygen species, apoptosis, and mitochondrial dysfunction in hearing loss. Biomed. Res. Int. 2015, 617207. doi:10.1155/2015/617207
Karasawa, T., Sibrian-Vazquez, M., Strongin, R. M., and Steyger, P. S. (2013). Identification of cisplatin-binding proteins using agarose conjugates of platinum compounds. PLoS One 8, e66220. doi:10.1371/journal.pone.0066220
Karasawa, T., and Steyger, P. S. (2015). An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 237, 219–227. doi:10.1016/j.toxlet.2015.06.012
Karasawa, T., Wang, Q., David, L. L., and Steyger, P. S. (2011). Calreticulin binds to gentamicin and reduces drug-induced ototoxicity. Toxicol. Sci. 124, 378–387. doi:10.1093/toxsci/kfr196
Karasawa, T., Wang, Q., David, L. L., and Steyger, P. S. (2010). CLIMP-63 is a gentamicin-binding protein that is involved in drug-induced cytotoxicity. Cell Death Dis. 1, e102. doi:10.1038/cddis.2010.80
Kim, K. H., Lee, B., Kim, Y. R., Kim, M. A., Ryu, N., Jung, D. J., et al. (2018). Evaluating protective and therapeutic effects of alpha-lipoic acid on cisplatin-induced ototoxicity. Cell Death Dis. 9, 827. doi:10.1038/s41419-018-0888-z
Kim, Y. R., Baek, J. I., Kim, S. H., Kim, M. A., Lee, B., Ryu, N., et al. (2019). Therapeutic potential of the mitochondria-targeted antioxidant MitoQ in mitochondrial-ROS induced sensorineural hearing loss caused by Idh2 deficiency. Redox Biol. 20, 544–555. doi:10.1016/j.redox.2018.11.013
Kopke, R., Slade, M. D., Jackson, R., Hammill, T., Fausti, S., Lonsbury-Martin, B., et al. (2015). Efficacy and safety of N-acetylcysteine in prevention of noise induced hearing loss: A randomized clinical trial. Hear Res. 323, 40–50. doi:10.1016/j.heares.2015.01.002
Lorito, G., Giordano, P., Petruccelli, J., Martini, A., and Hatzopoulos, S. (2008). Different strategies in treating noiseinduced hearing loss with N-acetylcysteine. Med. Sci. Monit. 14, BR159–164.
Mielke, K., and Herdegen, T. (2000). JNK and p38 stresskinases--degenerative effectors of signal-transduction-cascades in the nervous system. Prog. Neurobiol. 61, 45–60. doi:10.1016/s0301-0082(99)00042-8
Mukherjea, D., Jajoo, S., Sheehan, K., Kaur, T., Sheth, S., Bunch, J., et al. (2011). NOX3 NADPH oxidase couples transient receptor potential vanilloid 1 to signal transducer and activator of transcription 1-mediated inflammation and hearing loss. Antioxid. Redox Signal 14, 999–1010. doi:10.1089/ars.2010.3497
Mukherjea, D., Jajoo, S., Whitworth, C., Bunch, J. R., Turner, J. G., Rybak, L. P., et al. (2008). Short interfering RNA against transient receptor potential vanilloid 1 attenuates cisplatin-induced hearing loss in the rat. J. Neurosci. 28, 13056–13065. doi:10.1523/JNEUROSCI.1307-08.2008
Musial-Bright, L., Fengler, R., Henze, G., and Hernaiz Driever, P. (2011). Carboplatin and ototoxicity: Hearing loss rates among survivors of childhood medulloblastoma. Childs Nerv. Syst. 27, 407–413. doi:10.1007/s00381-010-1300-1
Ojano-Dirain, C. P., Antonelli, P. J., and Le Prell, C. G. (2014). Mitochondria-targeted antioxidant MitoQ reduces gentamicin-induced ototoxicity. Otol. Neurotol. 35, 533–539. doi:10.1097/MAO.0000000000000192
Ojano-Dirain, C. P., and Antonelli, P. J. (2012). Prevention of gentamicin-induced apoptosis with the mitochondria-targeted antioxidant mitoquinone. Laryngoscope 122, 2543–2548. doi:10.1002/lary.23593
Pak, J. H., Kim, Y., Yi, J., and Chung, J. W. (2020). Antioxidant therapy against oxidative damage of the inner ear: Protection and preconditioning. Antioxidants (Basel) 9, 1076. doi:10.3390/antiox9111076
Patuzzi, R. B., Yates, G. K., and Johnstone, B. M. (1989). Outer hair cell receptor current and sensorineural hearing loss. Hear Res. 42, 47–72. doi:10.1016/0378-5955(89)90117-2
Priuska, E. M., and Schacht, J. (1995). Formation of free radicals by gentamicin and iron and evidence for an iron/gentamicin complex. Biochem. Pharmacol. 50, 1749–1752. doi:10.1016/0006-2952(95)02160-4
Qi, M., Qiu, Y., Zhou, X., Tian, K., Zhou, K., Sun, F., et al. (2018). Regional up-regulation of NOX2 contributes to the differential vulnerability of outer hair cells to neomycin. Biochem. Biophys. Res. Commun. 500, 110–116. doi:10.1016/j.bbrc.2018.03.141
Rybak, L. P., Husain, K., Morris, C., Whitworth, C., and Somani, S. (2000). Effect of protective agents against cisplatin ototoxicity. Am. J. Otol. 21, 513–520.
Rybak, L. P., Whitworth, C. A., Mukherjea, D., and Ramkumar, V. (2007). Mechanisms of cisplatin-induced ototoxicity and prevention. Hear Res. 226, 157–167. doi:10.1016/j.heares.2006.09.015
Scherz-Shouval, R., and Elazar, Z. (2007). ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17, 422–427. doi:10.1016/j.tcb.2007.07.009
Sha, S. H., and Schacht, J. (2017). Emerging therapeutic interventions against noise-induced hearing loss. Expert Opin. Investig. Drugs 26, 85–96. doi:10.1080/13543784.2017.1269171
Slepecky, N. (1986). Overview of mechanical damage to the inner ear: Noise as a tool to probe cochlear function. Hear Res. 22, 307–321. doi:10.1016/0378-5955(86)90107-3
Sooriyaarachchi, M., Narendran, A., and Gailer, J. (2012). The effect of sodium thiosulfate on the metabolism of cis-platin in human plasma in vitro. Metallomics 4, 960–967. doi:10.1039/c2mt20076g
Steyger, P. S. (2021). Mechanisms of aminoglycoside- and cisplatin-induced ototoxicity. Am. J. Audiol. 30, 887–900. doi:10.1044/2021_AJA-21-00006
Tate, A. D., Antonelli, P. J., Hannabass, K. R., and Dirain, C. O. (2017). Mitochondria-targeted antioxidant mitoquinone reduces cisplatin-induced ototoxicity in Guinea pigs. Otolaryngol. Head. Neck Surg. 156, 543–548. doi:10.1177/0194599816678381
Tavanai, E., and Mohammadkhani, G. (2017). Role of antioxidants in prevention of age-related hearing loss: A review of literature. Eur. Arch. Otorhinolaryngol. 274, 1821–1834. doi:10.1007/s00405-016-4378-6
Tretter, L., and Adam-Vizi, V. (2004). Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci. 24, 7771–7778. doi:10.1523/JNEUROSCI.1842-04.2004
Walling, A. D., and Dickson, G. M. (2012). Hearing loss in older adults. Am. Fam. Physician 85, 1150–1156.
Wang, J., Ruel, J., Ladrech, S., Bonny, C., van de Water, T. R., and Puel, J. L. (2007). Inhibition of the c-Jun N-terminal kinase-mediated mitochondrial cell death pathway restores auditory function in sound-exposed animals. Mol. Pharmacol. 71, 654–666. doi:10.1124/mol.106.028936
Wang, J. Y., Li, J. Q., Xiao, Y. M., Fu, B., and Qin, Z. H. (2020). Triphenylphosphonium (TPP)-Based antioxidants: A new perspective on antioxidant design. ChemMedChem 15, 404–410. doi:10.1002/cmdc.201900695
Wei, Y. H., Lu, C. Y., Wei, C. Y., Ma, Y. S., and Lee, H. C. (2001). Oxidative stress in human aging and mitochondrial disease-consequences of defective mitochondrial respiration and impaired antioxidant enzyme system. Chin. J. Physiol. 44, 1–11.
Wong, A. C., and Ryan, A. F. (2015). Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci. 7, 58. doi:10.3389/fnagi.2015.00058
Wu, J., Ye, J., Kong, W., Zhang, S., and Zheng, Y. (2020). Programmed cell death pathways in hearing loss: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 53, e12915. doi:10.1111/cpr.12915
Yamashita, D., Jiang, H. Y., Schacht, J., and Miller, J. M. (2004a). Delayed production of free radicals following noise exposure. Brain Res. 1019, 201–209. doi:10.1016/j.brainres.2004.05.104
Yamashita, D., Miller, J. M., Jiang, H. Y., Minami, S. B., and Schacht, J. (2004b). AIF and EndoG in noise-induced hearing loss. Neuroreport 15, 6452–2722.
Zhang, M. Y., Dugbartey, G. J., Juriasingani, S., and Sener, A. (2021). Hydrogen sulfide metabolite, sodium thiosulfate: Clinical applications and underlying molecular mechanisms. Int. J. Mol. Sci. 22, 6452. doi:10.3390/ijms22126452
Keywords: acquired hearing loss, noise, ototoxic drugs, ROS, mitochondria, drug development
Citation: Baek J-I, Kim Y-R, Lee K-Y and Kim U-K (2023) Mitochondrial redox system: A key target of antioxidant therapy to prevent acquired sensorineural hearing loss. Front. Pharmacol. 14:1176881. doi: 10.3389/fphar.2023.1176881
Received: 01 March 2023; Accepted: 22 March 2023;
Published: 31 March 2023.
Edited by:
Francisco Torrens, University of Valencia, SpainReviewed by:
Amit P. Bhavsar, University of Alberta, CanadaCopyright © 2023 Baek, Kim, Lee and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Un-Kyung Kim, a2ltdWtAa251LmFjLmty
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.