 Rafael Fernades Costa1†
Rafael Fernades Costa1† Larissa Córdova Turones2†Keilah Valéria Naves Cavalcante3
Larissa Córdova Turones2†Keilah Valéria Naves Cavalcante3 Ismael Aureliano Rosa Júnior1,4
Ismael Aureliano Rosa Júnior1,4 Carlos Henrique Xavier5Lucimar Pinheiro Rosseto1Hamilton Barbosa Napolitano1,6Patrícia Ferreira da Silva Castro1,7Marcos Luiz Ferreira Neto8Gustavo Mota Galvão9Ricardo Menegatti9
Carlos Henrique Xavier5Lucimar Pinheiro Rosseto1Hamilton Barbosa Napolitano1,6Patrícia Ferreira da Silva Castro1,7Marcos Luiz Ferreira Neto8Gustavo Mota Galvão9Ricardo Menegatti9 Gustavo Rodrigues Pedrino3Elson Alves Costa2José Luis Rodrigues Martins1
Gustavo Rodrigues Pedrino3Elson Alves Costa2José Luis Rodrigues Martins1 James Oluwagbamigbe Fajemiroye2*
James Oluwagbamigbe Fajemiroye2*- 1Universitary Center of Anápolis, UniEvangélica, Anápolis, Brazil
- 2Laboratory of Pharmacology of Natural and Synthetic Products, Institute of Biological Sciences, Federal University of Goiás, Goiânia, Brazil
- 3Center for Neuroscience and Cardiovascular Research, Department of Physiology, Institute of Biological Sciences, Federal University of Goiás, Goiânia, Brazil
- 4Institute of Science, Technology and Quality (ICTQ), Anápolis, Brazil
- 5Systems Neurobiology Laboratory, Department of Physiology, Institute of Biological Sciences, Federal University of Goiás, Goiânia, Brazil
- 6Theoretical and Structural Chemistry Group, Universidade Estadual de Goiás, Anápolis, Brazil
- 7Goiás State University, Itumbiara, Brazil
- 8Laboratory of Electrophysiology and Cardiovascular Physiology, Departament of Physiology, Institute of Biomedical Science, Federal University of Uberlândia, Uberlândia, Brazil
- 9Laboratory of Medicinal Pharmaceutical Chemistry, Faculty of Pharmacy, Federal University of Goiás, Goiânia, Brazil
Low quality of life and life-threatening conditions often demand pharmacological screening of lead compounds. A spectrum of pharmacological activities has been attributed to pyrazole analogs. The substitution, replacement, or removal of functional groups on a pyrazole ring appears consistent with diverse molecular interactions, efficacy, and potency of these analogs. This mini-review explores cytotoxic, cytoprotective, antinociceptive, anti-inflammatory, and antidepressant activities of some pyrazole analogs to advance structure-related pharmacological profiles and rational design of new analogs. Numerous interactions of these derivatives at their targets could impact future research considerations and prospects while offering opportunities for optimizing therapeutic activity with fewer adverse effects.
Introduction
Medicinal chemistry is engaged in the research area to ameliorate new derivatives (Zhao, 2007). With wide application in medicine and industry (Al-Omar, 2010), the pyrazole ring is important in rational drug development. As a privileged structure present in different classes of drugs, the pyrazole moiety has inspired new classes of drug development (Faria et al., 2017; Patil, 2020; Yet, 2018). The pyrazole moiety is a nitrogen-containing heterocyclic core with diverse targets and effects (Figure 1) (Faisal et al., 2019; Ran et al., 2019; Taher et al., 2019; Badavath and Jayaprakash, 2020). The series of available pyrazole analogs could provide clues to structural activity relationship (SAR) and predict potential therapeutic or adverse effects.
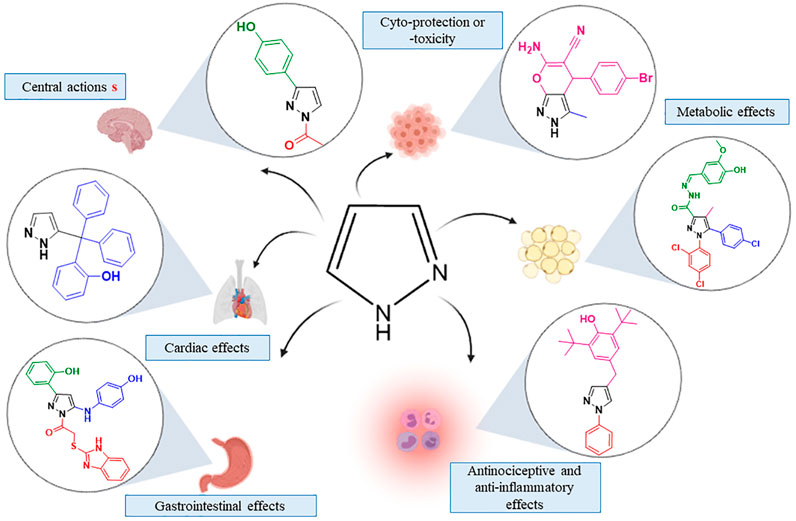
FIGURE 1. Effects evoked by pyrazole analogs in different cells target.
The substitutions, additions, removal, or fusion of different functional groups in the pyrazole ring are key to the synthesis of lead compounds that are effective against emerging and complex diseases (Faisal et al., 2019; Aziz et al., 2020; Ramsay et al., 2018; Benek et al., 2020; He et al., 2016). Pharmacological characterization of these analogs will benefit from our comprehension of the functional group modifications on the central pyrazole ring (Figure 2). The standard drugs containing the pyrazole ring [pyrazofurin (anticancer), crizotinib (cytoprotective), celecoxib and lonazolac (anti-inflammatory), difenamizole (analgesic), rimonabant (anti-obesity), sildenafil (vasodilator), and fezolamide (antidepressant)] (Hill and Whelan, 1980; Kameyama et al., 1981; Clemett and Goa, 2000; Samat et al., 2008; Straube, 2012; Dopp et al., 2013; Mitou et al., 2015; Karrouchi et al., 2018) provide ample opportunity for continuous research and analysis of new analogs.
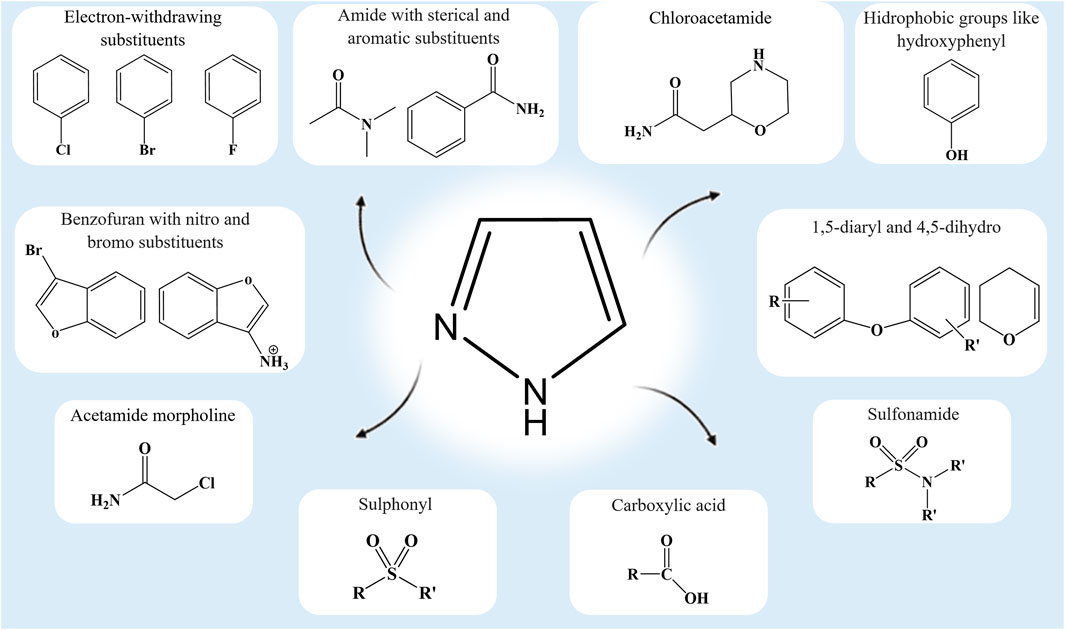
FIGURE 2. Functional groups that confer some distinctive interactions and activities on pyrazole analogs.
This mini-review explores selected activities and structural modifications of some pyrazole analogs. The available data on the concentration of analogs required for 50% inhibition (IC50), equilibrium dissociation constant for the inhibitor (Ki), the dose that elicited 50% of the maximum possible effect (ED50), ulcerogenic index (UI), or selectivity index (SI) are provided to further comparative discussions on their affinity, efficacy, and potency. As the present review is limited to selected biological activities, the author’s considerations, and future perspectives on some representative analogs (see Supplementary Table S1), additional references are listed for the aspects that are out of the present scope. All the chemical structures and figures were designed using either ChemDraw JS program or biorender.com.
Selected Activities
Cytotoxic and Cytoprotective Activities
The cytotoxic effects of some pyrazole analogs against specific cell lines including human breast cancer cells (MDA-MB-231 and MCF-7) appear promising to the development of anticancer drugs (Kamel, 2015). Compound 1 containing a distal pyrazole ring and sulphonyl moiety decreased the viability of MCF7 (IC50 = 39.70 µM). Unlike non-heterocyclic compound 4, the activities of caspase-3 and caspase-7 in MDA-MB-231 were gradually reduced by compounds 1–3. With the data showing the cleavage of caspase-8 and caspase-9 through caspase-3 and caspase-7 activation in the cascade of caspases (Toton et al., 2013), compound 4 is unlikely to elicit a specific effect. Since caspases play different roles in cellular survival, differentiation, and proliferation process, specific caspase inhibition by compounds 1–3 suggests a safety profile (Lehmann et al., 2017). Potent antiproliferative activity (IC50 = 0.26 μM) and mitogen-activated protein kinase inhibition (MEK, IC50 = 91 nM) have been attributed to methyl and ortho-fluorine groups on pyrazole carboxamide of compound 5. This result suggests suppression of abnormal signaling of the MEK-dependent pathways. In the docking assay, the contribution of the 1,3-diphenyl-1H-pyrazole to hydrophobic interactions (Val 82, Ala 95, Val 127, Met 143, Met 143, and Leu 197 of the ATP-binding pocket) and MEK inhibition (Lv et al., 2016) was considered significant. Recently, a thiazole moiety was attached to the pyrazole ring and assayed against different cancer cell lines (Masaret, 2021). Relatively high cytotoxic efficacy of compounds 6–8 (IC50 = 14.32, 11.17, and 10.21, respectively) against MCF-7 supports the antitumor role of the thiazole moiety (Masaret, 2021). However, the specificity of the cytotoxic potential and emerging resistance still need to be extensively evaluated.
As some analogs are considered potent IC50 < 100 nM; mild IC50 > 10,000 nM cytotoxic or noncytotoxic on some cell lines, and others elicit cyto- or neuroprotection. The restoration of redox homeostasis or prevention of oxidative stress, inflammation, glycation, and vascular injury is key to neuroprotection (Hrelia et al., 2020). Chemically diverse pyrazole analogs have shown promising cellular or neuronal protections. The bulky dicyclohexylamide (9), smaller dimethylamide (10), and aminophenyl (11) (pyrazole analogs with sterically hindered substituents on amide) attenuated microglia-mediated neurotoxicity (IC50 = 10–50 µM) better than compound 12 (IC50 = 100 µM) with high susceptibility to electrophilic displacement (McKenzie et al., 2019). This compound contains an allyl group on an electron-deficient amide. Suppression of toxin-induced microglia overactivation offers a therapeutic opportunity capable of halting the progression of reactive oxygen species–induced neurodegenerative disease (Block et al., 2007). The electron-withdrawing substituents such as para-bromo on the aldehyde aromatic ring (13) elicited higher neuroprotective activity than the electron-donating substituent (Gameiro et al., 2017). This activity involves the inhibition of glycogen synthase kinase 3β (GSK3β; IC50 = 3.77 µM) and induction of nuclear factor (erythroid-derived 2)–like 2 (Nrf2; luciferase activity = 3–30 µM). As regulators of cellular responses, both GSK3β and Nrf2 are key neurodegenerative targets (Brandes and Gray, 2020; Toral-Rios et al., 2020).
Analogs with the para-bromophenyl radical attached to the pyrazole ring (14, 15, and 16) target the metabolic enzymes (acetylcholinesterase—AChE, carbonic anhydrase—hCA, and α-glycosidase—α-GlyIs) that are relevant to neurodegenerative disorders. However, the number of phenyl substituents on the pyrazole ring seems to create a specific pattern of metabolic enzyme inhibition. Turkan et al. (2018) attributed potent AChE inhibition to compound 15 with triphenyl substitutions (IC50 = 66.37 nM), α-GlyIs inhibition to analogs with diphenyl substitutions (14 and 16; IC50 = 43.72 and 36.02 nM, respectively), and hCA I and II isoform inhibition (IC50 = 0.93 and 0.75 nM, respectively) to compound 9 with two bromophenyl substitutions. Altogether, the inhibitory effects of these analogs on specific regulators of cellular processes could alter ATP production, functions of organelles, redox reactions, and proliferative, vascular, and inflammatory responses.
Antinociceptive and Anti-inflammatory Activities
As pain is a hallmark of tissue damage and inflammatory processes (Baral et al., 2019), pyrazole analogs with antinociceptive and anti-inflammatory activities are important to analgesic drug development. The pyrazole compounds with fluorine at para (17), meta (18), and ortho (19) positions on the phenyl ring demonstrated an antinociceptive effect in the previous studies (de Oliveira et al., 2017; Florentino et al., 2019). This effect was associated with the activation of the opioid receptor and blockage of the acid-sensing ion channel subtype 1α (ASIC-1α). The para substitution improved the interaction with peripheral opioid receptors, while ortho substitution reduced the ASIC-1α channel’s affinity. The antagonism of transient receptor potential vanilloid subtype 1 (TRPV-1) seems to be optimized by the 3-chlorophenyl and 3-chloro-4-fluorophenyl substitutions (20 and 21, respectively) at the N1 position of the pyrazole C-region. The opioid receptors ASIC-1α and TRPV-1 are important targets of nociceptive modulation. Higher antinociceptive efficacy was reported in compound 21 (ED50 = 57 mg/kg). Like indomethacin (24% of pain inhibition), compound 22 with chloro and trifluoromethyl groups on phenyl and benzofuran template, respectively, elicited a similar 24% of pain inhibition (El Shehry et al., 2019). According to Kenchappa and Bodke (2020), compound 23 (benzofuran pyrazole substituted with nitro and bromo groups) inhibited pain response (60%) better than compounds 24 and 25 with benzofuran carbaldehyde (35 and 50%, respectively). These results support the elevation of antinociceptive efficacy through the addition of electron-withdrawing groups. Meanwhile, different group substitutions on other positions of pyrazole rings could offer additional data on potency and efficacy. The 4-(arylchalcogenyl)-1H-pyrazole analog containing the selanyl or sulfenyl group (26 and 27) elicited a higher nociceptive threshold in formalin, glutamate, and acid-induced abdominal writhing than compound 28 without the organochalcogen group (Oliveira et al., 2020). The modulation of oxidative and inflammatory pathways has been associated with pain inhibition (23%) by analogs with 2,3-di-tert-butylphenol (29) in Freund’s complete adjuvant–induced mechanical hyperalgesia (Galvão et al., 2020). A longer carbon chain (30) and levulinic (31) analogs with 47 and 50% pain inhibition, respectively (Taher et al., 2019), suggests that the length of the aliphatic chain could elicit a subtle change in the antinociceptive effect. In respect of first-line analgesic drugs, the fusion of pyrimidine moiety to the pyrazole backbone (32) similarly increased thermal latency (160%) and reduced abdominal writhing (83%) as compared with tramadol (175%) and aspirin (78%), respectively (Khalifa et al., 2019).
The addition of the adamantyl residue to 1,5-diaryl pyrazole (33) elicited a higher anti-inflammatory activity over the antinociceptive effect. The anti-inflammatory effect of pyrazole analogs could be assessed through edema formation (a cardinal sign of inflammation). Compound 33 induced a lower antiedematogenic effect than the reference drug celecoxib (39 and 82% of edema inhibition, respectively). Celecoxib exhibited higher potency of cyclooxygenase (COX)-2 inhibition than compound 33 (IC50 = 0.95 and 2.52 µM, respectively) (Abdelazeem et al., 2020). New azomethine compounds with an electron-withdrawing group like nitrogen (34) or chlorine (35) at the ortho position of phenyl ring with a satisfactory anti-inflammatory effect (ED50 = 0.86 and 0.92 mmol/kg, respectively) elicited weak COX-2 inhibition (IC50 = 38.12 and 32.11) as compared with celecoxib (ED50 = 8.03 mmol/kg; IC50 = 0.34) (Murahari et al., 2019). These results support the importance of electron-withdrawing groups toward the development of highly potent anti-inflammatory analogs. The thiohydantoin derivatives with pyrazole core and methoxy substituents (36–39) induced promising antiedematogenic activity (ED50 = 55–62 μmol/kg) as compared with celecoxib (ED50 = 78 μmol/kg). The methoxy moiety (the electron-donating group) of these analogs confers additional hydrogen bonding and interaction with COX-2 active sites (Lys68, Tyr108, Tyr341, Arg106, and Arg499). This attribute supports a higher range of binding energy among compounds 36–39 (16–20 kcal/mol) as compared with celecoxib (17 kcal/mol) with fewer hydrogen bonding (Ser516 and Tyr371) (Abdellatif et al., 2019). An additional evaluation revealed a better COX-2 inhibition with chloroacetamide (40) and acetamide morpholine (41) compounds (IC50 = 20 and 34 nM, respectively). In comparison with compound 40, the propionamide morpholine (42) analog improved the selectivity index (SI = 5 and 22, respectively). Compounds 40 and 42 significantly inhibited cytosolic (31 and 44%) and microsomal (81 and 74%) prostaglandin E2 synthase. The antiedematogenic activity of these analogs (46 and 44%) appears similar to the analog of celecoxib (42%).
Molecular docking data portraying compound 42 as the most active analog (Hassan et al., 2019) provide clues to the role of the morpholine (heterocyclic nucleus) with electron-withdrawing substituents on the phenyl ring. Additional COX-2 interactions (Gln192 and Phe518) and improved anti-inflammatory effects seem to be related to this chemical modification. Meanwhile, the COX-2 selectivity index (SI = 417) and the anti-inflammatory effect (% edema inhibition = 87%) of compound 43 with amino and methanesulphonyl groups are very close to that of celecoxib (SI = 327; % edema inhibition = 83%). This finding suggests robust aminosulfonyl moiety interaction with Gln178, Arg499, Phe504, and Gln178 amino acid residues of COX-2 (Abdellatif et al., 2020). The growing interest in pyrazole analogs with dual COX/LOX (lipoxygenase) inhibition has widened the scope of developing potent anti-inflammatory drugs. According to Gedawy et al. (2020), analogs with benzotiophenyl and carboxylic acid (44) inhibited COX-2 better than celecoxib (IC50 = 0.01 and 0.70 µM, respectively) with almost similar 5-LOX inhibition as compared to the reference drug licofelone (IC50 = 1.78 and 0.51 µM, respectively). Prodrugs of compound 44 (45 and 46 with % edema inhibition = 57 and 72%, respectively) with dual COX/LOX inhibition elicited a better anti-inflammatory effect than celecoxib (36%). Additional interactions of these analogs with COX-2 (Arg106 and Ser339) and 5-LOX (Leu368, Leu414, Ile415, and Phe421) could inspire additional modification in the subsequent series of pyrazole analogs with the anti-inflammatory potential.
Antidepressant Activity
The level of monoamine is etiologically relevant to several psychiatric diseases such as anxiety and depression (Özdemir et al., 2020). In principle, the inhibition of monoamine oxidase (MAO) provides a good assessment of the antidepressant property of new drug candidates (Fajemiroye et al., 2015). This flavin adenine dinucleotide (FAD)–dependent enzyme exists in two isoforms (MAO-A and MAO-B). The monoamines (serotonin and norepinephrine) often associated with the etiology of depression are principal substrates of MAO-A (Özdemir et al., 2020). Analogs with a positively charged pyrazole moiety at N1 (hydroxy or dihydroxy, phenyl, chloro, methoxy, or dimethoxy groups) showed higher selectivity to MAO-A than MAO-B. The 4,5-dihydro-1H-pyrazoles (47 and 48) showed a considerable affinity with MAO-A amino acid residues (Ala68, Tyr69, Phe208, Tyr407, and Tyr444). The N1-benzenesulfonyl ring at the pyrazoline nucleus seems to mediate additional interactions with the side chain and backbone residues of amino acids at the binding pocket of MAO-A (Tripathi et al., 2018). The analogs with halogen groups in the phenyl ring could also promote hydrophobic interactions with MAO-A. The replacement of a polar 4-hydroxyphenyl substituent in pyrazoline nucleus (47) for a bulky hydrophobic 1-naphthyl substituent (48) significantly reduced the antidepressant effect. The addition of the N1-acetyl group at the pyrazole nucleus in combination with polar substituents (49–53) appears to stabilize FAD bonding and improve efficacy. According to Chimenti et al. (2006), the presence of dimethoxyphenyl on the pyrazole ring lowered the MAO inhibition potency of compound 51 (IC50 = 1.0 × 10–7 M) as compared with hydroxyphenyl radical in compound 52 (IC50 = 8.8 × 10–9 M). The incorporation of functional groups capable of increasing strain energy and facilitating ring-opening could increase the potency drastically. In this manner, the 4,5-dihydro-1H-pyrazole appears to be a better template for the design of MAO-A inhibitor (Chimenti et al., 2004; Tripathi et al., 2016; Upadhyay et al., 2017) than MAO-B inhibitor (Manna et al., 2002). The tautomeric forms of pyrazole by the active site of the enzyme play a key role in the development of antidepressant drugs (Secci et al., 2011). Altogether, many analogs are still without data on potency, efficacy, and MAO-A/MAO-B selectivity index that allow for comparative analysis and determination of the rational template for new pyrazole analogs with the antidepressant property. Although MAO inhibition offers a viable antidepressant mechanism, further screening of these analogs in the models of obsessive-compulsive, panic, anxiety disorder, and post-traumatic stress disorders could expand their potential applications, contraindications, and adverse events in the psychiatry setting.
Other Activities
The modulation of central and peripheral targets by the pyrazole ring and aforementioned moieties may induce diverse autonomic manifestations. Alteration in the activity of the autonomic nervous system could impact cardiac, vascular, and respiratory functions. The pyrazole-induced endothelium-dependent vascular relaxation, sympathoinhibition, and suppression of bronchial remodeling suggest cardiovascular or respiratory effects (Girodet et al., 2013; Fajemiroye et al., 2014; Basu et al., 2017; Menegatti et al., 2019). Compounds 54 and 55 with antihypertensive properties possess a piperazine ring with carboxylic acid or phenyl substituents, respectively. The introduction of 3,5-bis(trifluoromethyl) backbone with electron-withdrawing substituents in compounds 56 (37-fold Orai1 pore inhibition potency) and 57 (18-fold transient receptor potential canonical type 3 suppression potency) reduced respiratory impairments (Schleifer et al., 2012). Bronchial remodeling suppression with details on the efficacy, potency, selectivity, or mechanism of action has been reported for calcium-activated potassium channel blocker (58) and adenosine receptor subtype 2A inhibitor (59) with chloro or a meta-trifluoromethoxy substituent on the phenyl ring, respectively (Girodet et al., 2013; Basu et al., 2017).
Some of the analogs that share the presence of electron-withdrawing groups in common tend to interact with inflammatory mediators that are relevant to gastrointestinal and metabolic disorders. Some of these analogs inhibit COX enzymes and suppress the synthesis of prostaglandin. Although both COX-1 and COX-2 isoforms are involved in homeostatic functions, the gastrointestinal ulceration of nonsteroidal anti-inflammatory drugs (NSAIDs) has been attributed to COX-1 inhibition and subsequent reduction in gastroprotective prostaglandin (Kanno and Moayyedi, 2020). Hence, pyrazole analogs with little or no effect on prostaglandin synthesis and release remain a viable therapeutic option in patients that are highly susceptible to NSAID-induced ulceration. The anti-inflammatory and antiedematogenic pyrazole analog (60; 40% of edema inhibition) with a very low ulcerogenic index (UI = 60) as compared with phenylbutazone and indomethacin (UI = 275 and 300, respectively) appears promising (Maggio et al., 2001). Unlike compound 60, both phenylbutazone and indomethacin are traditional NSAIDs well known for reducing prostaglandin levels through reversible and nonselective COX inhibition. In the ethanol-induced ulcer model, the benzimidazole–pyrazole hybrids (61–66) with substitutions on both aromatic rings of the pyrazole moiety reduced the ulcer index (UI = 72–83). The ortho-hydroxyl group on the phenyl ring seems to be involved in hydrogen bonding with the proton bomb (H+/K+ ATPase) since the highest binding affinity (–9.8 kcal/mole) reported for compound 64 was lost in compounds 63 and 66 with methoxy group replacement (Noor et al., 2017). The suppression of harmful gastrointestinal effects has been partially attributed to the meta-methyl substitution on the pyrazole ring. The mepirizole analog (67) with methoxy and methyl groups at meta and para-positions on a pyrimidine ring exhibited 93% gastroprotective activity. However, the replacement of the pyrazole ring of mepirizole considerably attenuated this effect (7% inhibition of ulcer formation) (Ikeda et al., 1996). These data indicate that appropriate substitutions on the pyrazole ring could reinforce gastroprotection.
The analogs that suppress pro-inflammatory mediators could also modulate insulin sensitivity and protect against metabolic syndrome. Insulin resistance, oxidative products, and atherogenic dyslipidemia are among metabolic abnormalities underlying diabetes. A molecular hybrid of rimonabant (68) with chloro and methoxy substitutions on the phenyl ring elicited a significant antidiabetic effect (Hernández-Vázquez et al., 2015; Hernández-Vázquez et al., 2017). In a separate study, the water-soluble pyrazole curcumin analog (69) inhibited advanced glycation end products (AGEs) and eliminated excess glucose (Sribalan et al., 2017). The AGEs, oxidative derivatives resulting from diabetic hyperglycemia, are increasingly seen as a potential risk for islet β-cell injury, insulin resistance, and diabetes (Vlassara and Uribarri, 2014). Although, compound 69 seems to inhibit AGEs (IC50 = 56.24 μg/ml) better than the parent curcumin (IC50 = 79.34 μg/ml) and standard drug phloroglucinol (IC50 = 135.73 μg/ml) sufficient data for comparative analysis of these analogs is still lacking. The chronic nature of the metabolic syndrome that warrants prolonged and often indefinite medications (Rochlani et al., 2017) makes the development of new analogs a promising strategy against the burden of adverse effects and patient nonadherence.
Final Considerations and Future Perspective
The hunt for new analogs with desirable pharmacological profiles is a never-ending task in drug discovery programs. With the available literature on pyrazole analogs (Fadaly et al., 2020; Mohamed et al., 2020; Azimi et al., 2021; Faudzi et al., 2021; Verma et al., 2021), it is relatively easy for medicinal chemists to proceed with rational synthesis or modification capable of enhancing biological activities. The substitutions, additions, or removal of functional groups are effective strategies for designing biologically important analogs. The presence of new chemical entities could lead to additional or loss of molecular interactions. Cellular proliferation and metabolic enzymes provide important cytotoxic, cytoprotective, antinociceptive, anti-inflammatory, and antidepressant targets. However, specific contributions of the pyrazole ring and functional groups to these biological activities remain largely unclear.
In light of this review, promising cytotoxic activities of some analogs against breast cancer cell lines (MCF-7 and MDA-MB-231) is expected to stimulate extensive evaluation of their effects on lung cancer (A-549), liver cancer (HepG-2), and brain cancer (HeLa) cell lines (Lv et al., 2016; Lehmann et al., 2017; Turkan et al., 2018; McKenzie et al., 2019; Bennani et al., 2020; Masaret, 2021). As a life-threatening health issue and one of the most lethal diseases known, cancer can be managed by targeting gene expression, certain protein synthesis, functions of organelles, pH, and electrolytes (Johnston and Strobel, 2020). For instance, the cytotoxic hypothesis appears to favor exposure to halogenated analogs capable of increasing halogen influx. This small-sized functional group with high electronegativity could lower cytosolic pH (cellular acidification). According to Johnston and Strobel (2020), increasing acidification and electrolyte imbalance by fluoride could disrupt metabolic processes and induce stress signaling, underpinning cellular toxicity. However, a detailed investigation of the level of analog-induced changes in redox reactions, oxidative stress, inflammation, glycation, and other metabolic processes could facilitate the repositioning of cytotoxic analogs for cytoprotection, and vice versa. Altogether, these effects may impact autonomic and central components vis-à-vis cardiovascular, cardiopulmonary, metabolic, and affective manifestations.
Since antiproliferative and anti-inflammatory properties could reduce cellular infiltration and modulate gastrointestinal aggressive factors underlying peptic ulcer complications, in hypothesis, some pyrazole analogs could attenuate gastric ulceration, or in association with NSAIDs at subtherapeutic doses without loss of efficacy. The growing number of synthesized analogs without data of biological activity, mechanism of action, and comparative study with standard drugs containing the pyrazole ring (Hill and Whelan, 1980; Kameyama et al., 1981; Clemett and Goa, 2000; Samat et al., 2008; Straube, 2012; Dopp et al., 2013; Mitou et al., 2015; Karrouchi et al., 2018) may yield promising results, if evaluated in the future research.
As the pyrazole moiety offers a central motif to different functional groups (Basu et al., 2017; Noor et al., 2017; Tripathi et al., 2018; Murahari et al., 2019; Kenchappa and Bodke, 2020), its simplified synthetic routes and potential activities are expected to continue to inspire additional chemical modifications (Johnston and Strobel, 2020) toward clinical applications. Emerging analogs with unique physicochemical, pharmacokinetic, and pharmacodynamic properties could be useful scaffolds for future studies. The aforementioned target interactions revealed how structural modification could enrich mechanistic studies. In the future, toxicological studies, reversibility, and selectivity of the effect of these analogs as well as the potential pharmacokinetic and pharmacodynamic interactions with other drugs are expected to predict adverse and therapeutic effects.
Author Contributions
JF contributed to the conception, design, and writing of the work. LT contributed to the writing, revision, discussion, and drafting of the final work. RC, KC, IRJ, LR, HN, PC, MN, and JM contributed to the writing, revision, and discussion. GG contributed to the drafting of chemical structures, analysis of chemical modification, and discussion. RM contributed to the analysis of chemical modification and critical revision for important intellectual content. GP, CX, and EC contributed to the discussion and critical revision.
Funding
The authors appreciate all the institutional supports through Convênio UFG—UniEvangélica, FAPEG, CAPES, and CNPq.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.666725/full#supplementary-material
References
Abdelazeem, A. H., El-Din, A. G. S., El-Saadi, M. T., El-Moghazy, S. M., and Amin, N. H. (2020). Design, Synthesis and Biological Evaluation of Diarylpyrazole/triazole Bearing 1,3,4- Oxadiazole Moiety as Coxs Inhibitors Endowed with Potential Anti-inflammatory and Analgesic Activities. Rese. Jour. Pharm. Technol. 13 (9), 4255–4262. doi:10.5958/0974-360X.2020.00751.9
Abdellatif, K. R. A., Fadaly, W. A. A., Mostafa, Y. A., Zaher, D. M., and Omar, H. A. (2019). Thiohydantoin Derivatives Incorporating a Pyrazole Core: Design, Synthesis and Biological Evaluation as Dual Inhibitors of Topoisomerase-I and Cycloxygenase-2 with Anti-cancer and Anti-inflammatory Activities. Bioorg. Chem. 91, 103132. doi:10.1016/j.bioorg.2019.103132
Abdellatif, K. R. A., Abdelall, E. K. A., Lamie, P. F., Labib, M. B., El-Nahaas, E.-S., and Abdelhakeem, M. M. (2020). New Pyrazole Derivatives Possessing Amino/methanesulphonyl Pharmacophore with Good Gastric Safety Profile: Design, Synthesis, Cyclooxygenase Inhibition, Anti-inflammatory Activity and Histopathological Studies. Bioorg. Chem. 95, 103540. doi:10.1016/j.bioorg.2019.103540
Al-Omar, M. A. (2010). Synthesis and Antimicrobial Activity of New 5-(2-Thienyl)-1,2,4-Triazoles and 5-(2-Thienyl)-1,3,4-Oxadiazoles and Related Derivatives. Molecules 15, 502–514. doi:10.3390/molecules15010502
Azimi, F., Azizian, H., Najafi, M., khodarahmi, G., hassanzadeh, M., Saghaei, L., et al. (2021). Design and Synthesis of Novel Pyrazole-Benzofuran Hybrids: In Vitro α-glucosidase Inhibitory Activity, Kinetic and Molecular Modeling Study. Res. Square. doi:10.21203/rs.3.rs-288227/v1
Aziz, H., Zahoor, A. F., and Ahmad, S. (2020). Pyrazole Bearing Molecules as Bioactive Scaffolds: a Review. J. Chil. Chem. Soc. 65 (1), 4746–4753. doi:10.4067/S0717-97072020000104746
Badavath, V. N., and Jayaprakash, V. (2020). MAO Inhibitory Activity of 4,5-dihydro-1HPyrazole Derivatives: A Platform to Design Novel Antidepressants. Front. Drug Des. Discov. 1, 45. doi:10.2174/9789811421563121100005
Baral, P., Udit, S., and Chiu, I. M. (2019). Pain and Immunity: Implications for Host Defence. Nat. Rev. Immunol. 19, 433–447. doi:10.1038/s41577-019-0147-2
Basu, S., Barawkar, D. A., Ramdas, V., Patel, M., Waman, Y., Panmand, A., et al. (2017). Design and Synthesis of Novel Xanthine Derivatives as Potent and Selective A 2B Adenosine Receptor Antagonists for the Treatment of Chronic Inflammatory Airway Diseases. Eur. J. Med. Chem. 134, 218–229. doi:10.1016/j.ejmech.2017.04.014
Benek, O., Korabecny, J., and Soukup, O. (2020). A Perspective on Multi-Target Drugs for Alzheimer's Disease. Trends Pharmacol. Sci. 41 (7), 434–445. doi:10.1016/j.tips.2020.04.008
Bennani, F. E., Doudach, L., Cherrah, Y., Ramli, Y., Karrouchi, K., Ansar, M. h., et al. (2020). Overview of Recent Developments of Pyrazole Derivatives as an Anticancer Agent in Different Cell Line. Bioorg. Chem. 97, 103470. doi:10.1016/j.bioorg.2019.103470
Block, M. L., Zecca, L., and Hong, J.-S. (2007). Microglia-mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi:10.1038/nrn2038
Brandes, M. S., and Gray, N. E. (2020). NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 12, 175. doi:10.1177/1759091419899782
Chimenti, F., Bolasco, A., Manna, F., Secci, D., Chimenti, P., Befani, O., et al. (2004). Synthesis and Selective Inhibitory Activity of 1-Acetyl-3,5-Diphenyl-4,5-Dihydro-(1h)-Pyrazole Derivatives against Monoamine Oxidase. J. Med. Chem. 47 (8), 2071–2074. doi:10.1021/jm031042b
Chimenti, F., Bolasco, A., Manna, F., Secci, D., Chimenti, P., Granese, A., et al. (2006). Synthesis, Biological Evaluation and 3D-QSAR of 1,3,5-Trisubstituted-4,5- Dihydro-(1h)-Pyrazole Derivatives as Potent and Highly Selective Monoamine Oxidase A Inhibitors. CMC 13 (12), 1411–1428. doi:10.2174/092986706776872907
Clemett, D., and Goa, K. L. (2000). Celecoxib. Drugs 59, 957–980. doi:10.2165/00003495-200059040-00017
de Oliveira, L. P., da Silva, D. P. B., Florentino, I. F., Fajemiroye, J. O., de Oliveira, T. S., Marcelino, R. I. d. Á., et al. (2017). New Pyrazole Derivative 5-[1-(4-Fluorophenyl)-1h-Pyrazol-4-Yl]-2h-Tetrazole: Synthesis and Assessment of Some Biological Activities. Chem. Biol. Drug Des. 89 (1), 124–135. doi:10.1111/cbdd.12838
Dopp, J. M., Agapitov, A. V., Sinkey, C. A., Haynes, W. G., and Phillips, B. G. (2013). Sildenafil Increases Sympathetically Mediated Vascular Tone in Humans. Am. J. Hypertens 26 (6), 762–769. doi:10.1093/ajh/hpt018
El Shehry, M. F., Ewies, E. F., and Zayed, E. M. (2019). Synthesis of New Pyrazole Derivatives, Their Anti-inflammatory and Analgesic Activities, and Molecular Docking Studies. Russ. J. Gen. Chem. 89, 492–498. doi:10.1134/S1070363219030216
Fadaly, W. A. A., Elshaier, Y. A. M. M., Hassanein, E. H. M., and Abdellatif, K. R. A. (2020). New 1,2,4-triazole/pyrazole Hybrids Linked to Oxime Moiety as Nitric Oxide Donor Celecoxib Analogs: Synthesis, Cyclooxygenase Inhibition Anti-inflammatory, Ulcerogenicity, Anti-proliferative Activities, Apoptosis, Molecular Modeling and Nitric Oxide Release Studies. Bioorg. Chem. 98, 103752. doi:10.1016/j.bioorg.2020.103752
Faisal, M., Saeed, A., Hussain, S., Dar, P., and Larik, F. A. (2019). Recent Developments in Synthetic Chemistry and Biological Activities of Pyrazole Derivatives. J. Chem. Sci. 131, 70. doi:10.1007/s12039-019-1646-1
Fajemiroye, J. O., Amaral, N. O., da Silva, E. F., Galdino, P. M., de Oliveira, T. S., Ghedini, P. C., et al. (2014). Hypotensive and Antihypertensive Potential of 4-[(1-Phenyl-1h-Pyrazol-4-Yl) Methyl]1-Piperazine Carboxylic Acid Ethyl Ester: A Piperazine Derivative. Life Sci. 112 (1–2), 90–96. doi:10.1016/j.lfs.2014.07.025
Fajemiroye, J. O., Polepally, P. R., Chaurasiya, N. D., Tekwani, B. L., Zjawiony, J. K., and Costa, E. A. (2015). Oleanolic Acid Acrylate Elicits Antidepressant-like Effect Mediated by 5-HT1A Receptor. Sci. Rep. 5, 11582. doi:10.1038/srep11582
Faria, J. V., Vegi, P. F., Miguita, A. G. C., Dos Santos, M. S., Boechat, N., and Bernardino, A. M. R. (2017). Recently Reported Biological Activities of Pyrazole Compounds. Bioorg. Med. Chem. 25 (21), 5891–5903. doi:10.1016/j.bmc.2017.09.035
Faudzi, S. M. M., Leong, S. W., Auwal, F. A., Abas, F., Wai, L. K., Ahmad, S., et al. (2021). In Silico studies, Nitric Oxide, and Cholinesterases Inhibition Activities of Pyrazole and Pyrazoline Analogs of Diarylpentanoids. Arch. Pharm. 354, e2000161. doi:10.1002/ardp.202000161
Florentino, I. F., Silva, D. P. B., Cardoso, C. S., Menegatti, R., de Carvalho, F. S., Lião, L. M., et al. (2019). Antinociceptive Effects of New Pyrazoles Compounds Mediated by the ASIC-1α Channel, TRPV-1 and μMOR Receptors. Biomed. Pharmacother. 115, 108915. doi:10.1016/j.biopha.2019.108915
Galvão, G. M., Florentino, I. F., Sanz, G., Vaz, B. G., Lião, L. M., Sabino, J. R., et al. (2020). Anti-inflammatory and Antinociceptive Activity Profile of a New Lead Compound - LQFM219. Int. Immunopharmacol. 88, 106893. doi:10.1016/j.intimp.2020.106893
Gameiro, I., Michalska, P., Tenti, G., Cores, Á., Buendia, I., Rojo, A. I., et al. (2017). Discovery of the First Dual GSK3β inhibitor/Nrf2 Inducer. A New Multitarget Therapeutic Strategy for Alzheimer's Disease. Sci. Rep. 7, 45701. doi:10.1038/srep45701
Gedawy, E. M., Kassab, A. E., and El Kerdawy, A. M. (2020). Design, Synthesis and Biological Evaluation of Novel Pyrazole Sulfonamide Derivatives as Dual COX-2/5-LOX Inhibitors. Eur. J. Med. Chem. 189, 112066. doi:10.1016/j.ejmech.2020.112066
Girodet, P.-O., Ozier, A., Carvalho, G., Ilina, O., Ousova, O., Gadeau, A.-P., et al. (2013). Ca2+-Activated K+Channel-3.1 Blocker TRAM-34 Attenuates Airway Remodeling and Eosinophilia in a Murine Asthma Model. Am. J. Respir. Cell Mol. Biol. 48 (2), 212–219. doi:10.1165/rcmb.2012-0103O10.1165/rcmb.2012-0103oc
Hassan, G. S., Abdel Rahman, D. E., Abdelmajeed, E. A., Refaey, R. H., Alaraby Salem, M., and Nissan, Y. M. (2019). New Pyrazole Derivatives: Synthesis, Anti-inflammatory Activity, Cycloxygenase Inhibition Assay and Evaluation of mPGES. Eur. J. Med. Chem. 171, 332–342. doi:10.1016/j.ejmech.2019.03.052
He, B., Lu, C., Zheng, G., He, X., Wang, M., Chen, G., et al. (2016). Combination Therapeutics in Complex Diseases. J. Cell Mol. Med. 20 (12), 2231–2240. doi:10.1111/jcmm.12930
Hernández-Vázquez, E., Castañeda-Arriaga, R., Ramírez-Espinosa, J. J., Medina-Campos, O. N., Hernández-Luis, F., Chaverri, J. P., et al. (2015). 1,5-Diarylpyrazole and Vanillin Hybrids: Synthesis, Biological Activity and DFT Studies. Eur. J. Med. Chem. 100, 106–118. doi:10.1016/j.ejmech.2015.06.010
Hernández-Vázquez, E., Ocampo-Montalban, H., Cerón-Romero, L., Cruz, M., Gómez-Zamudio, J., Hiriart-Valencia, G., et al. (2017). Antidiabetic, Antidyslipidemic and Toxicity Profile of ENV-2: A Potent Pyrazole Derivative against Diabetes and Related Diseases. Eur. J. Pharmacol. 803, 159–166. doi:10.1016/j.ejphar.2017.03.036
Hill, B. T., and Whelan, R. D. H. (1980). Antitumour Activity and Cell Kinetic Effects of Pyrazofurin In Vitro. Eur. J. Cancer (1965) 16 (12), 1633–1638. doi:10.1016/0014-2964(80)90039-0
Hrelia, P., Sita, G., Ziche, M., Ristori, E., Marino, A., Cordaro, M., et al. (2020). Common Protective Strategies in Neurodegenerative Disease: Focusing on Risk Factors to Target the Cellular Redox System. Oxid. Med. Cell. Longev. 2020, 1–18. doi:10.1155/2020/8363245
Ikeda, M., Maruyama, K., Nobuhara, Y., Yamada, T., and Okabe, S. (1996). Synthesis and Cytoprotective Antiulcer Activity of 2- or 4-(lH-Pyrazol-1-Yl)pyrimidine Derivatives Related to Mepirizole and Dulcerozine. Chem. Pharm. Bull. 44 (9), 1700–1706. doi:10.1248/cpb.44.1700
Johnston, N. R., and Strobel, S. A. (2020). Principles of Fluoride Toxicity and the Cellular Response: A Review. Arch. Toxicol. 94, 1051–1069. doi:10.1007/s00204-020-02687-5
Kamel, M. (2015). Convenient Synthesis, Characterization, Cytotoxicity and Toxicity of Pyrazole Derivatives. ACSi 62 (1), 136–151. doi:10.17344/acsi.2014.828
Kameyama, T., Nabeshima, T., Yoshida, N., and Yamaguchi, K. (1981). Neurochemical Studies of an Analgesic, 1,3-Diphenyl-5-(2-Dimethylaminopropionamide)-Pyrazole [difenamizole]. Res. Commun. Chem. Pathol. Pharmacol. 31 (1), 31–53.
Kang, J. M., Kwon, S. O., Ann, J., Blumberg, P. M., Ha, H., Yoo, Y. D., et al. (2020). Discovery of 1-(1h-Indazol-4-Yl)-3-((1-Phenyl-1h-Pyrazol-5-Yl)methyl) Ureas as Potent and Thermoneutral TRPV1 Antagonists. Bioorg. Med. Chem. Lett. 30 (23), 127548. doi:10.1016/j.bmcl.2020.127548
Kanno, T., and Moayyedi, P. (2020). Who Needs Gastroprotection in 2020? Curr. Treat. Options. Gastrol. 18, 557–573. doi:10.1007/s11938-020-00316-9
Karrouchi, K., Radi, S., Ramli, Y., Taoufik, J., Mabkhot, Y., Al-aizari, F., et al. (2018). Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 23 (1), 134. doi:10.3390/molecules23010134
Kenchappa, R., and Bodke, Y. D. (2020). Synthesis, Analgesic and Anti-inflammatory Activity of Benzofuran Pyrazole Heterocycles. Chem. Data Collec. 28, 100453. doi:10.1016/j.cdc.2020.100453
Khalifa, N. M., Fahmy, H. H., Nossier, E. S., Amr, A. E., and Herqash, R. N. (2019). Novel Synthesis of Pyrazole-Containing Thiophene, 2-alkyloxypyridine and Thieno[2,3-D]pyrimidine Scaffolds as Analgesic Agents. Bull. Chem. Soc. Ethiop. 33 (3), 505–515. doi:10.4314/bcse.v33i3.11
Lehmann, T. P., Kujawski, J., Kruk, J., Czaja, K., Bernard, M. K., and Jagodzinski, P. P. (2017). Cell-specific Cytotoxic Effect of Pyrazole Derivatives on Breast Cancer Cell Lines MCF7 and MDA-MB-231. J. Physiol. Pharmacol. 68 (2), 201–207.
Lv, X.-H., Ren, Z.-L., Zhou, B.-G., Li, Q.-S., Chu, M.-J., Liu, D.-H., et al. (2016). Discovery of N-(benzyloxy)-1,3-diphenyl-1H-pyrazole-4-carboxamide Derivatives as Potential Antiproliferative Agents by Inhibiting MEK. Bioorg. Med. Chem. 24 (19), 4652–4659. doi:10.1016/j.bmc.2016.08.002
Maggio, B., Daidone, G., Raffa, D., Plescia, S., Mantione, L., Cutuli, V. M. C., et al. (2001). Synthesis and Pharmacological Study of Ethyl 1-methyl-5-(substituted 3,4-Dihydro-4-Oxoquinazolin-3-Yl)-1h-Pyrazole-4-Acetates. Eur. J. Med. Chem. 36 (9), 737–742. doi:10.1016/s0223-5234(01)01259-4
Manna, F., Chimenti, F., Bolasco, A., Secci, D., Bizzarri, B., Befani, O., et al. (2002). Inhibition of Amine Oxidases Activity by 1-Acetyl-3,5-Diphenyl-4,5-Dihydro-(1h)-Pyrazole Derivatives. Bioorg. Med. Chem. Lett. 12 (24), 3629–3633. doi:10.1016/s0960-894x(02)00699-6
Masaret, G. S. (2021). A New Approach for the Synthesis and Biological Activities of Novel Thiazolyl‐Pyrazole Derivatives. ChemistrySelect 6, 974–982. doi:10.1002/slct.202004304
McKenzie, J. A., Barghash, R. F., Alsaggaf, A. T., Kulkarni, O., Boudreau, K., Menard, F., et al. (2019). Synthesis and Evaluation of Novel Pyrazole Ethandiamide Compounds as Inhibitors of Human THP-1 Monocytic Cell Neurotoxicity. Cells 8 (7), 655. doi:10.3390/cells8070655
Menegatti, R., Carvalho, F. S., Lião, L. M., Villavicencio, B., Verli, H., Mourão, A. A., et al. (2019). Novel Choline Analog 2-(4-((1-Phenyl-1h-Pyrazol-4-Yl)methyl)piperazin-1-Yl)ethan-1-Ol Produces Sympathoinhibition, Hypotension, and Antihypertensive Effects. Naunyn-schmiedeberg's Arch. Pharmacol. 392, 1071–1083. doi:10.1007/s00210-019-01649-8
Mitou, G., Frentzel, J., Desquesnes, A., Le Gonidec, S., AlSaati, T., Beau, I., et al. (2015). Targeting Autophagy Enhances the Anti-tumoral Action of Crizotinib in ALK-Positive Anaplastic Large Cell Lymphoma. Oncotarget 6 (30), 30149–30164. doi:10.18632/oncotarget.4999
Mohamed, H. H. M., Hussien, A. B. W. E. M., and Saeed, A. E. M. (2020). In Silico evaluation and Docking Studies of Pyrazole Analogs as Potential Autophagy Modulators against Pancreatic Cancer Cell Line MIA PaCa-2. Eur. J. Chem. 11 (3), 187–193. doi:10.5155/eurjchem.11.3.187-193.1976
Murahari, M., Mahajan, V., Neeladri, S., Kumar, M. S., and Mayur, Y. C. (2019). Ligand Based Design and Synthesis of Pyrazole Based Derivatives as Selective COX-2 Inhibitors. Bioorg. Chem. 86, 583–597. doi:10.1016/j.bioorg.2019.02.031
Noor, A., Qazi, N. G., Nadeem, H., Khan, A.-u., Paracha, R. Z., Ali, F., et al. (2017). Synthesis, Characterization, Anti-ulcer Action and Molecular Docking Evaluation of Novel Benzimidazole-Pyrazole Hybrids. Chem. Cent. J. 11 (1). doi:10.1186/s13065-017-0314-0
Oliveira, D. H., Sousa, F. S. S., Birmann, P. T., Alves, D., Jacob, R. G., and Savegnago, L. (2020). Antinociceptive and Anti-inflammatory Effects of 4-(arylchalcogenyl)-1h-Pyrazoles Containing Selenium or Sulfur. Pharmacol. Rep. 72, 36–46. doi:10.1007/s43440-019-00001-4
Özdemir, Z., Alagöz, M. A., Uslu, H., Karakurt, A., Erikci, A., Ucar, G., et al. (2020). Synthesis, Molecular Modelling and Biological Activity of Some Pyridazinone Derivatives as Selective Human Monoamine Oxidase-B Inhibitors. Pharmacol. Rep. 72, 692–704. doi:10.1007/s43440-020-00070-10.1007/s43440-020-00070-w
Patil, S. B. (2020). Medicinal Siganificance of Pyrazole Analogues: A Review. J. Pharm. Sci. Res. 12 (3), 402–404.
Ramsay, R. R., Popovic‐Nikolic, M. R., Nikolic, K., Uliassi, E., and Bolognesi, M. L. (2018). A Perspective on Multi‐target Drug Discovery and Design for Complex Diseases. Clin. Transl. Med. 7 (1), 3. doi:10.1186/s40169-017-0181-2
Ran, F., Liu, Y., Zhang, D., Liu, M., and Zhao, G. (2019). Discovery of Novel Pyrazole Derivatives as Potential Anticancer Agents in MCL. Bioorg. Med. Chem. Lett. 29 (9), 1060–1064. doi:10.1016/j.bmcl.2019.03.005
Rochlani, Y., Pothineni, N. V., Kovelamudi, S., and Mehta, J. L. (2017). Metabolic Syndrome: Pathophysiology, Management, and Modulation by Natural Compounds. Ther. Adv. Cardiovasc. Dis. 11 (8), 215–225. doi:10.1177/1753944717711379
Samat, A., Tomlinson, B., Taheri, S., and Thomas, G. (2008). Rimonabant for the Treatment of Obesity. Recent Pat. Cardiovasc. Drug Discov. 3 (3), 187–193. doi:10.2174/157489008786264014
Schleifer, H., Doleschal, B., Lichtenegger, M., Oppenrieder, R., Derler, I., Frischauf, I., et al. (2012). Novel Pyrazole Compounds for Pharmacological Discrimination between Receptor‐operated and Store‐operated Ca 2+ Entry Pathways. Br. J. Pharmacol. 167, 1712–1722. doi:10.1111/j.1476-5381.2012.02126.x
Secci, D., Bolasco, A., Chimenti, P., and Carradori, S. (2011). The State of the Art of Pyrazole Derivatives as Monoamine Oxidase Inhibitors and Antidepressant/anticonvulsant Agents. CMC 18 (33), 5114–5144. doi:10.2174/092986711797636090
Sribalan, R., Shakambari, G., Banuppriya, G., Varalakshmi, P., Subramanian, E. R., Sudhakar, S., et al. (2017). Synthesis of a Water-Soluble Pyrazole Curcumin Derivative:In VitroandIn VivoAGE Inhibitory Activity and its Mechanism. ChemistrySelect 2 (3), 1122–1128. doi:10.1002/slct.201601740
Straube, S. (2012). Anti-inflammatory and Antipyretic Analgesics and Drugs Used in Gout. Side Effects Drugs Annu., 181–193. doi:10.1016/b978-0-444-59499-0.00009-x
Taher, A. T., Mostafa Sarg, M. T., El-Sayed Ali, N. R., and Hilmy Elnagdi, N. (2019). Design, Synthesis, Modeling Studies and Biological Screening of Novel Pyrazole Derivatives as Potential Analgesic and Anti-inflammatory Agents. Bioorg. Chem. 89, 103023. doi:10.1016/j.bioorg.2019.103023
Toral-Rios, D., Pichardo-Rojas, P. S., Alonso-Vanegas, M., and Campos-Peña, V. (2020). GSK3β and Tau Protein in Alzheimer's Disease and Epilepsy. Front. Cell Neurosci. 14, 19. doi:10.3389/fncel.2020.00019
Toton, E., Ignatowicz, E., Bernard, M. K., Kujawski, J., and Rybczynska, M. (2013). Evaluation of Apoptotic Activity of New Condensed Pyrazole Derivatives. J. Physiol. Pharmacol. 64, 115–123.
Tripathi, A. C., Upadhyay, S., Paliwal, S., and Saraf, S. K. (2016). An Expeditious One-Pot Microwave Facilitated versus Conventional Syntheses: In Vivo Biological Screening and Molecular Docking Studies of Some 3,5-Disubstituted-4,5-Dihydro-(1h)-Pyrazole Derivatives. Med. Chem. Res. 25, 390–406. doi:10.1007/s00044-015-1489-3
Tripathi, A. C., Upadhyay, S., Paliwal, S., and Saraf, S. K. (2018). Derivatives of 4,5-dihydro (1H) Pyrazoles as Possible MAO-A Inhibitors in Depression and Anxiety Disorders: Synthesis, Biological Evaluation and Molecular Modeling Studies. Med. Chem. Res. 27, 1485–1503. doi:10.1007/s00044-018-2167-z
Turkan, F., Cetin, A., Taslimi, P., and Gulçin, İ. (2018). Some Pyrazoles Derivatives: Potent Carbonic Anhydrase, α-glycosidase, and Cholinesterase Enzymes Inhibitors. Arch. Pharm. Chem. Life Sci. 351 (10), e1800200. doi:10.1002/ardp.201800200
Upadhyay, S., Tripathi, A. C., Paliwal, S., and Saraf, S. K. (2017). Facile One-Pot Synthesis Methodology for Nitrogen-Containing Heterocyclic Derivatives of 3,5-disubstituted 4,5-Dihydro-1h-Pyrazole, Their Biological Evaluation and Molecular Docking Studies. Pharm. Chem. J. 51, 564–575. doi:10.1007/s11094-017-1655-7
Verma, R., Verma, S. K., Rakesh, K. P., Girish, Y. R., Ashrafizadeh, M., Sharath Kumar, K. S., et al. (2021). Pyrazole-based Analogs as Potential Antibacterial Agents against Methicillin-Resistance staphylococcus Aureus (MRSA) and its SAR Elucidation. Eur. J. Med. Chem. 212, 113134. doi:10.1016/j.ejmech.2020.113134
Vlassara, H., and Uribarri, J. (2014). Advanced Glycation End Products (AGE) and Diabetes: Cause, Effect, or Both? Curr. Diab Rep. 14 (1), 453. doi:10.1007/s11892-013-0453-1
Yet, L. (2018). “Privileged Structures in Drug Discovery: Medicinal Chemistry and Synthesis,” in Methods and Principles in Medicinal Chemistry. London; Hoboken, NJ: John Wiley & Sons. doi:10.1002/9781118686263
Keywords: heterocyclic ring, functional groups, derivatives, interactions, activities
Citation: Costa RF, Turones LC, Cavalcante KVN, Rosa Júnior IA, Xavier CH, Rosseto LP, Napolitano HB, Castro PFdS, Neto MLF, Galvão GM, Menegatti R, Pedrino GR, Costa EA, Martins JLR and Fajemiroye JO (2021) Heterocyclic Compounds: Pharmacology of Pyrazole Analogs From Rational Structural Considerations. Front. Pharmacol. 12:666725. doi: 10.3389/fphar.2021.666725
Received: 10 February 2021; Accepted: 23 April 2021;
Published: 10 May 2021.
Edited by:
Syed Nasir Abbas Bukhari, Al Jouf University, Saudi ArabiaReviewed by:
Satyendra Mishra, Institute of Advanced Research (IAR), IndiaPierfausto Seneci, Institute of Advanced Research (IAR), India
Copyright © 2021 Costa, Turones, Cavalcante, Rosa Júnior, Xavier, Rosseto, Napolitano, Castro, Neto, Galvão, Menegatti, Pedrino, Costa, Martins and Fajemiroye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James Oluwagbamigbe Fajemiroye, amFtZXNmYWplbWlyb3llQHVmZy5icg==
†These authors have contributed equally to this work and share first authorship