 Jorge E. B. da Rocha1,2‡Houcemeddine Othman1‡Gerrit Botha3‡Laura Cottino1,2‡David Twesigomwe1,2‡Samah Ahmed4‡Britt I. Drögemöller5Faisal M. Fadlelmola4‡Philip Machanick6‡Mamana Mbiyavanga3‡Sumir Panji3‡Galen E. B. Wright7,8Clement Adebamowo9,10‡Mogomotsi Matshaba11,12‡Michéle Ramsay1,2‡Gustave Simo13‡Martin C. Simuunza14‡Caroline T. Tiemessen15Sandra Baldwin16Mathias Chiano17Charles Cox17Annette S. Gross18Pamela Thomas19Francisco-Javier Gamo20Scott Hazelhurst1,21*‡the H3Africa Consortium
Jorge E. B. da Rocha1,2‡Houcemeddine Othman1‡Gerrit Botha3‡Laura Cottino1,2‡David Twesigomwe1,2‡Samah Ahmed4‡Britt I. Drögemöller5Faisal M. Fadlelmola4‡Philip Machanick6‡Mamana Mbiyavanga3‡Sumir Panji3‡Galen E. B. Wright7,8Clement Adebamowo9,10‡Mogomotsi Matshaba11,12‡Michéle Ramsay1,2‡Gustave Simo13‡Martin C. Simuunza14‡Caroline T. Tiemessen15Sandra Baldwin16Mathias Chiano17Charles Cox17Annette S. Gross18Pamela Thomas19Francisco-Javier Gamo20Scott Hazelhurst1,21*‡the H3Africa Consortium- 1Sydney Brenner Institute for Molecular Bioscience (SBIMB), Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
- 2Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
- 3Computational Biology Division and H3ABioNet, Department of Integrative Biomedical Sciences, University of Cape Town, Cape Town, South Africa
- 4Centre for Bioinformatics and Systems Biology, Faculty of Science, University of Khartoum, Khartoum, Sudan
- 5Department of Biochemistry and Medical Genetics, University of Manitoba, Winnipeg, MB, Canada
- 6Department of Computer Science, Rhodes University, Makhanda, South Africa
- 7Neuroscience Research Program, Winnipeg Health Sciences Centre and Max Rady College of Medicine, Kleysen for Advanced Medicine, University of Manitoba, Winnipeg, MB, Canada
- 8Department of Pharmacology and Therapeutics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada
- 9Institute for Human Virology, Abuja, Nigeria
- 10Institute of Human Virology and Greenebaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, MD, United States
- 11Botswana-Baylor Children’s Clinical Center of Excellence, Gaborone, Botswana
- 12Baylor College of Medicine, Houston, TX, United States
- 13Molecular Parasitology and Entomology Unit, Department of Biochemistry, Faculty of Science, University of Dschang, Dschang, Cameroon
- 14Department of Disease Control, School of Veterinary Medicine, University of Zambia, Lusaka, Zambia
- 15Centre for HIV and STIs, National Institute for Communicable Diseases, National Health Laboratory Services and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
- 16Drug Metabolism and Pharmacokinetics, GlaxoSmithKline R&D, Ware, United Kingdom
- 17Human Genetics, GlaxoSmithKline R&D, Stevenage, United Kingdom
- 18Clinical Pharmacology Modelling and Simulation, GlaxoSmithKline R&D, Sydney, NSW, Australia
- 19Data and Computational Sciences, GlaxoSmithKline R&D, Stevenage, United Kingdom
- 20Global Health, GlaxoSmithKline R&D, Tres Cantos, Madrid, Spain
- 21School of Electrical and Information Engineering, University of the Witwatersrand, Johannesburg, South Africa
Introduction: Investigating variation in genes involved in the absorption, distribution, metabolism, and excretion (ADME) of drugs are key to characterizing pharmacogenomic (PGx) relationships. ADME gene variation is relatively well characterized in European and Asian populations, but data from African populations are under-studied—which has implications for drug safety and effective use in Africa.
Results: We identified significant ADME gene variation in African populations using data from 458 high-coverage whole genome sequences, 412 of which are novel, and from previously available African sequences from the 1,000 Genomes Project. ADME variation was not uniform across African populations, particularly within high impact coding variation. Copy number variation was detected in 116 ADME genes, with equal ratios of duplications/deletions. We identified 930 potential high impact coding variants, of which most are discrete to a single African population cluster. Large frequency differences (i.e., >10%) were seen in common high impact variants between clusters. Several novel variants are predicted to have a significant impact on protein structure, but additional functional work is needed to confirm the outcome of these for PGx use. Most variants of known clinical outcome are rare in Africa compared to European populations, potentially reflecting a clinical PGx research bias to European populations.
Discussion: The genetic diversity of ADME genes across sub-Saharan African populations is large. The Southern African population cluster is most distinct from that of far West Africa. PGx strategies based on European variants will be of limited use in African populations. Although established variants are important, PGx must take into account the full range of African variation. This work urges further characterization of variants in African populations including in vitro and in silico studies, and to consider the unique African ADME landscape when developing precision medicine guidelines and tools for African populations.
1 Introduction and Background
Pharmacogenomics (PGx) aims to improve drug safety and efficacy using genomic knowledge for genes involved in drug action (Roden et al., 2019) with a focus on genes that have important roles in drug safety, pharmacokinetics and pharmacodynamics. Genes involved in pharmacokinetics are typically defined by the role they play in the absorption, distribution, metabolism and excretion (ADME) of drug molecules.
Variation in ADME genes play an important role in determining the response to drug treatment in an individual patient. We characterize the extent and impact of variation in these genes in a novel, high-coverage whole genome sequence dataset from a diverse group of Africans.
ADME genes have different functions: 1) phase I metabolizing enzymes, 2) phase II metabolizing enzymes, 3) drug transporters and 4) modifiers. PharmaADME (http://pharmaadme.org) classifies the ADME genes in two classes. The 32 core genes have known biomarkers linked to ADME outcomes. For the 267 extended ADME genes, there is weaker evidence of functional consequences in vitro or in vivo, or they are important for a limited number of drugs only.
1.1 Rationale
Currently the majority of patients studied in drug development programmes are of European or Asian ancestry. The African continent is the cradle of human origin and African populations are characterized by high genetic diversity and complex population structure. Despite this genetic variation, drug efficacy and safety have not been comprehensively studied in the populations of sub-Saharan Africa (SSA) (Radouani et al., 2020). This is of specific relevance to SSA, where high burdens of disease are amplified by non-optimal treatment outcomes.
The particular diversity of ADME genes in SSA has been reported in some studies. Hovelson et al. (Hovelson et al., 2017) and Lakiotaki et al. (Lakiotaki et al., 2017) found that the greatest levels of coding ADME variation per personal haplotype were shown in some African populations sampled in the 1,000 Genomes Project (KGP) data. Examples of the impact of this variation can be seen in CYP2B6 and CYP2D6 variation affecting efavirenz and primaquine respectively. An efavirenz dosage reduction has been recommended for HIV patients in SSA due to the high frequency of functional variants in the CYP2B6 gene that result in a higher risk of adverse drug reactions (Mukonzo, 2014). Potential polymorphisms in the human cytochrome CYP2D6 gene may negatively influence efficacy of primaquine, and significantly affect malaria elimination strategies (Dandara et al., 2014; Awandu et al., 2018). African specific variation in several genes may impact the pharmacokinetics of rosuvastatin, a drug used to treat hypercholesterolemia (Soko et al., 2018). While these studies represent only a fraction of the continent, they serve to highlight the importance of future studies which are aimed at providing a more comprehensive overview of the landscape of ADME variation across Africa.
Therefore it is important to gain a better understanding of the variation that exists in ADME genes, both within and between different SSA populations. This information could be used to inform recommended drug dosage regimens for patients in SSA based on potential pharmacokinetic effects and consequently efficacy and safety. To date, no studies have systematically investigated ADME variation within a diverse set of African populations. We therefore aim to provide valuable information regarding the variation that exists in ADME genes, both within and between different SSA populations. This information could provide insight into drug efficacy and safety for patients in SSA and play a role in ensuring safe and efficacious treatments for the high burden of diseases in populations in SSA.
2 Results
2.1 Description of Samples
Four hundred and fifty eight high coverage whole genome sequences were used in the study as the primary data set (we call this the high coverage African ADME Dataset—HAAD). Most sequences were generated by the Human Health and Heredity in Africa (H3A) consortium (The H3Africa Consortium, 2014; Choudhury et al., 2020) and we also used public data and additional novel data from collaborators—see Table 1 and Figure 1. The population structure of participants in this study is broadly representative of speakers of Niger-Congo languages from West through South Africa. Representation from Nilo-Saharan and Afro-Asiatic populations is sparse. There also are few individuals of Khoe and San heritage, although significant admixture from Khoe and San speakers is found in Bantu-speakers in Southern Africa (Choudhury et al., 2017).

TABLE 1. Sources of high-coverage data sets used to form HAAD: 272 genomes were generated by a supplementary grant from the NIH to the H3A Consortium (Choudhury et al., 2020) for the primary purpose of designing a custom genotyping array; 100 were produced by AWI-Gen; 40 were shared by African collaborators; 15 genomes came from the Southern African Human Genome Program (SAHGP), and 31 genomes were from the Simons Foundation Genome Diversity Project.
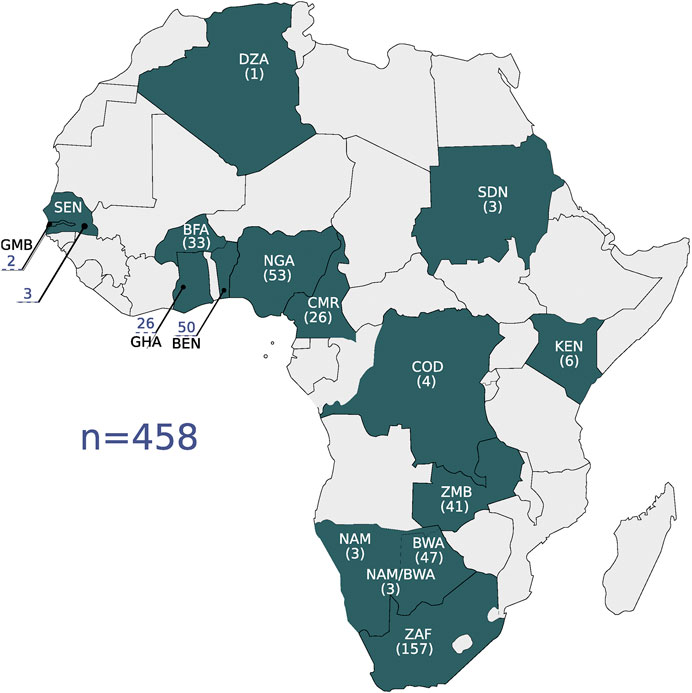
FIGURE 1. The geographic locations of the high coverage WGS data are shown on the map. Countries are referenced by their ISO 3166-1 alpha-3 code: BEN, Benin; BFA, Burkina Faso; BWA, Botswana; CMR, Cameroon; COD, Democratic Republic of the Congo; DZA, Algeria; GHA, Ghana; GMB, Gambia; KEN, Kenya; NAM, Namibia; NGA, Nigeria; SDN, Sudan; SEN, Senegal; ZAF, South Africa; ZMB, Zambia. The number of samples per country is shown in parentheses.
We supplement some analyses with African datasets from the KGP (we use KGA specifically to refer to the African genomes in KGP). As the KGP datasets are low coverage, not all analyses were performed with the KGA dataset in addition to HAAD.
2.2 Population Structure
A principal component (PC) and structure analysis of our data shows high genome-scale variation and that we have good coverage of African genomic diversity across West, Central and Southern Africa, with less coverage in East Africa. The PC analysis of our data shows a strong correlation to geographical location (Figure 2 and Supplementary Section S1).
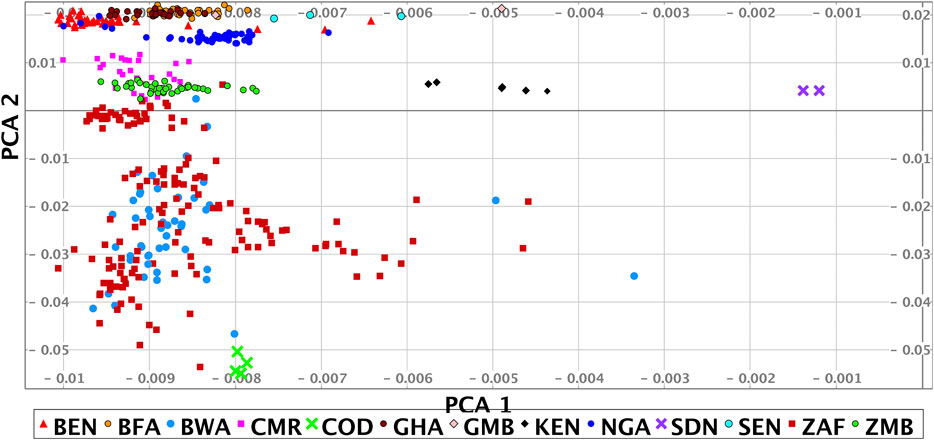
FIGURE 2. Principal component analysis of the HAAD (some outliers are omitted). Abbreviations for sources used are H3A (Human Health and Heredity in Africa Consortium), and SF (Simons Foundation Genome Diversity Project). The countries of origin and source of the samples shown in the PCA are: BEN/H3A, BFA/H3A, BWA/H3A + SF, CMR/H3A, COD/SF, GHA/H3A, GMB/SF, KEN/SF, NGA/H3A + SF, SDN/SF, SEN/SF, ZAF/H3A + Tiemessen Lab + SF + Southern African Human Genome Program, ZMB/H3A. Country codes given in Figure 1.
To explore diversity between different African regions we clustered the studied population together with reference data sets using PC data (see Methods, Table 2). The PC analysis shows that the HAAD samples fall broadly into three groups: West (Ghana, Burkina Faso, Nigeria), South/Central (Cameroon, Zambia, Botswana, South Africa), South (Botswana, South Africa) African populations. The variability in the Southern group primarily arises through differential admixture between Bantu, Khoe and San speakers. There is a Far West group comprising individuals in HAAD and KGA from Gambia, Senegal and Sierra Leone. There are also a few individuals from other African regions. Note that there is significant diversity within countries; and in some cases overlap between countries—e.g. some participants that we label as “South/Central” live to the south of some participants in the “Southern” group.

TABLE 2. Clusters within Africa, including the number of individuals in each cluster. Clusters include both HAAD and 1,000 Genomes African population data.
2.3 Overall Characterization of ADME Variation
Gene-based genetic variation for the core and extended ADME gene categories was assessed for composition and type, including introns, upstream and downstream flanking regions (Figure 3). Comparisons were made between the HAAD dataset and the KGA dataset, which represent samples in the joint called HAAD and African KGP populations respectively (Methods 5.3.1). In ADME core genes, we counted a total of 40,714 and 36,088 variants for HAAD and KGA data respectively while for the extended ADME genes there were 274,798 and 243,022 variants respectively. Intronic variants are most common overall with about the same proportions in both HAAD and KGA datasets of 80 and 77% (for both core and extended genes) respectively. A significant number of variations appear in 3′ untranslated (3′ UTR) and 5′UTR regions. Coding region variants (non-synonymous and synonymous as annotated by VEP v92.0) do not overlap completely between HAAD and KGA groups. For core genes there were 423 coding variants common to both HAAD and KGA datasets, 288 coding variants unique to HAAD, and 252 unique to KGA. For extended genes, there were 17,148 coding variants common to HAAD and KGA, 2,850 unique to HAAD, and 2,318 unique to KGA respectively. Care should be taken in comparing HAAD and KGA data because of the different depth of sequencing.
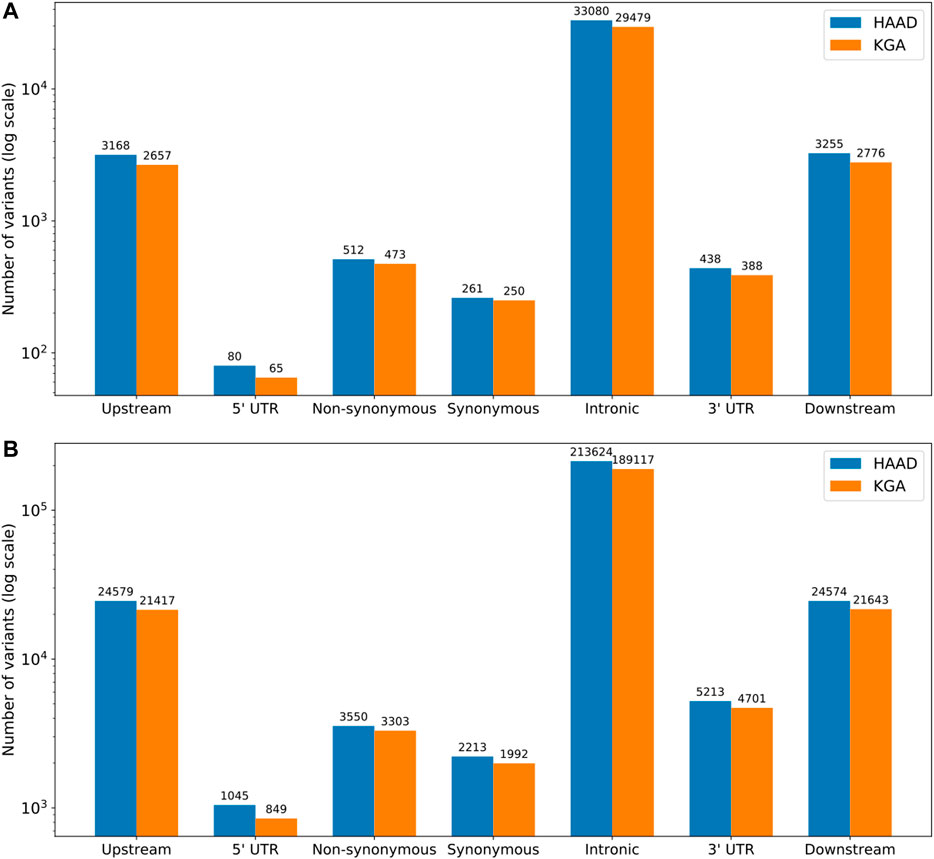
FIGURE 3. Distribution of variant types (as defined by SNPeff annotation) across core (A) and extended (B) ADME gene regions. HAAD (N = 458) represents those samples in the jointly called set from the H3A Consortium data, Simons Foundation, SAHGP and Tiemessen Labs, and KGA (n = 506) represents the African KGP populations from the jointly called set. Upstream and downstream regions are represented by 10 kb flanks from gene start and end respectively.
The importance of using and generating African datasets like ours can be seen in our discovery curves which show the increase in the number of variants found in the core ADME genes as more genomes are included in the study (the results for the extended genes are not shown but are similar). Figure 4A compares our data set to 1,000 Genomes African and European populations. The diversity of African populations compared to European populations is clear and consistent with previous literature (Hovelson et al., 2017). We believe that the increased richness of our data compared to 1,000 Genomes African data is partially due to the fact that our data is high-coverage. This richness is also likely to be driven by the significant numbers of Southern African genomes that have significant Khoe and San ancestry (see (Choudhury et al., 2017) for some discussion) as well some diverse samples from the Simons Foundation. Figure 4B shows the discovery curve for the combined African (HAAD and KGA) dataset. Although the curve has started to plateau, the results show that combining the data sets has value, and that sampling more Africans and more diverse African groups not yet properly captured will reveal considerably more variants.
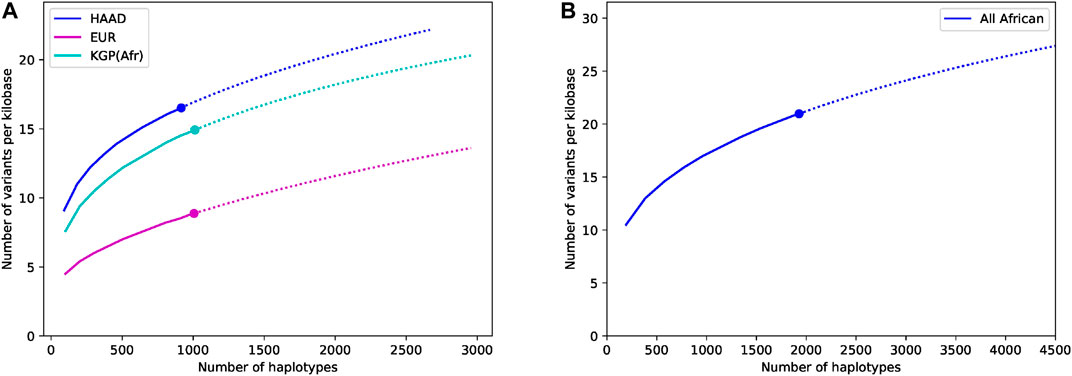
FIGURE 4. Comparative discovery curves of variants in the core ADME genes (including flanks) for the (A) HAAD, the 1,000 Genomes African and European data, and (B) in the combined HAAD and KGA datasets. The results show for a given number of haplotypes the number of variants seen per kilobase. The actual results are shown as a large dot, sub-samples by a solid line, and projections by a dotted line. Sub-samples values are computed averaging over 50 different randomly sampled subsets for intermediate values. Projection is computed using a third order jackknife projection (Burnham and Overton, 1979).
2.4 Annotation of High Impact Coding Variants
To annotate ADME genes we used the output of an ADME gene optimized annotation schema. This schema uses five prediction tools, and variants meeting score cutoffs for all five are of the highest confidence for functional impact. We identified 930 high impact variants (HI-vars) (Supplementary Table S8) for 247 ADME genes (from a total of 299 ADME genes) of which 29 are core genes and 218 are extended genes. Of the core genes, seven members of the cytochrome P450 (CYP450) family (CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C19, CYP2A6, CYP2D6) were among those with the highest count of high impact variants. Highest counts of the CYP450 genes were seen in CYP1A1 and CYP2D6 with 12 and 10 HI-vars of respectively. The ATP-Binding Cassette (ABC) transporter gene, ABCB5 showed the highest number of HI-vars overall numbering 20. We also counted three members of ABCC transporter family and three other members of the CYP450 family in the 10 most variable genes.
The 930 HI-vars are mostly rare alleles, with most being singletons or doubletons. There were only 93 variants with a frequency above 1% in the total joint called samples (Figure 5). Overall, the frequency distributions for sub-populations (SA, SC, FW, and WE) are not uniform (we omit discussion of the Khoe and San cluster because of low sample number). Some of the high impact variants tend to show a large disparity in frequency values between some clusters. For example, the CYP27A1 rs114768494 variant (chr2:g.219677301C > T) (28th index in Figure 5) is only present in SC and WE with respective frequencies of 1.1 and 3.7%. Also, variants can exist in all the sub-populations but with significantly different proportions. For instance, the CYP4B1 rs45446505 variant, (chr1:g.47279898C > T) (52nd index) is present at frequencies of 9.5, 2.3, 4.5 and 3.5% for SA, SC, FW, and WE respectively. Another variant: CYP4B1 rs3215983 (ch1:g.47280747_47280747del) (47th index) is common in the SC population with a 10% frequency. This value is at least twice that of other clusters. Frequency differences of ≈10% are observed in common high impact variants.
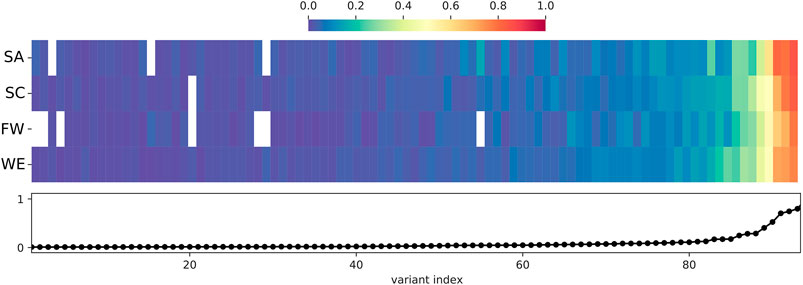
FIGURE 5. Distribution and frequency of HI-vars across sub-populations. Only common HI-vars (Frequency > 1% in the total joint called population dataset) are represented in the figure. Frequency is that of the non-reference (ALT) allele. The frequency per variant is reported in the lower panel of the figure as a line plot (indexed by frequency). The frequency of each of the variants in each sub-population is given by each heat map column, with white indicating 0% frequency. See Table 2 for abbreviations.
The regional overlap of the total HI-vars identified shows the majority of these variants are appear in one population cluster only (Figure 6). There are only ∼100 variants that overlap all African population clusters. These variants appearing in all regions have widely ranging frequencies, with most falling between 1 and 20% for the total African samples assessed. Each population cluster had >110 variants specific to it. Variants that occur only in one cluster are mostly rare, with an average frequency of less than 1% in their own respective cluster. Southern Africans have 20 cluster-specific variants with frequencies above 1% (20 variant)—more than any other cluster. Relatively fewer variants overlap between two clusters alone, with a trend of geographically close clusters sharing more variants than those which are distant.
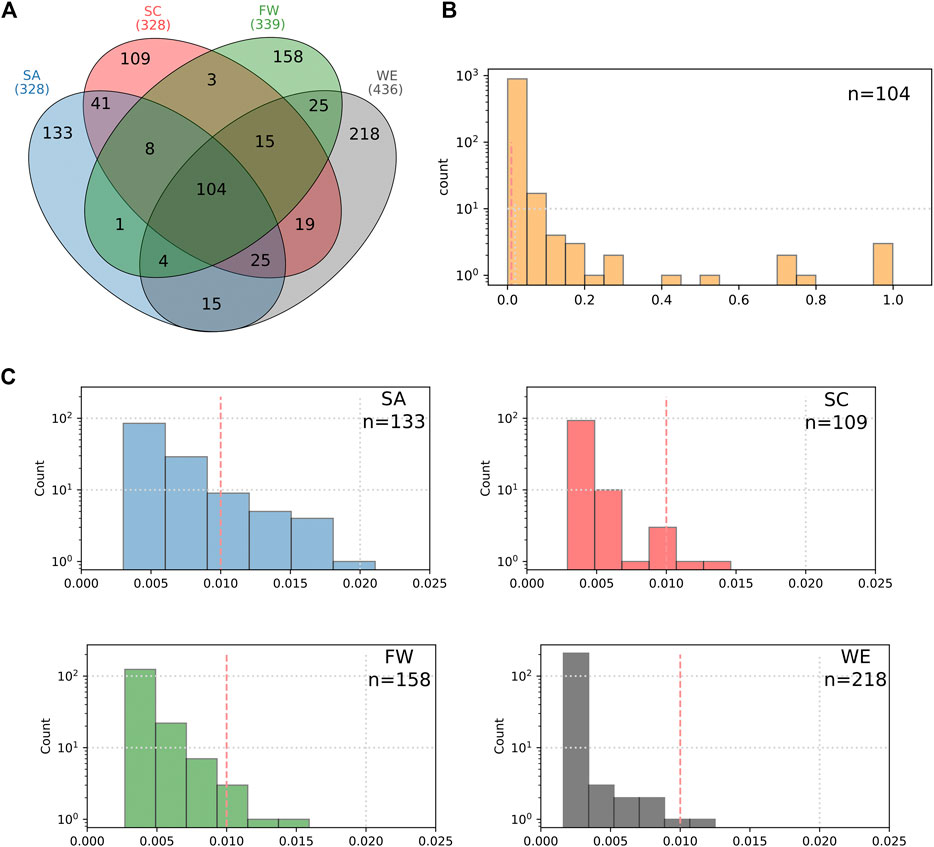
FIGURE 6. Characterization of the HI-vars in clusters SA, SC, FW, and WE. (A) The Venn diagram shows the overlap between the clusters for these variants. Distribution plots of the frequencies of the common variants between the four clusters (B) and for the unique variants found in each sub-population (C).
Fixation index (
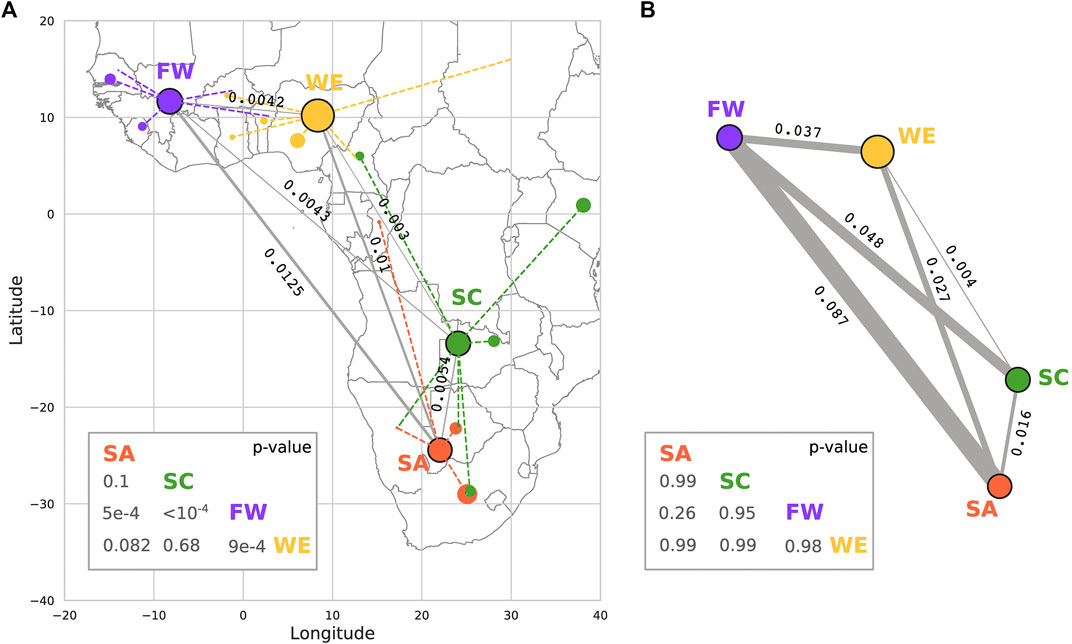
FIGURE 7. Calculation of Fixation index (
In summary, HI-vars are not uniform across African clusters, and that geographical proximity is not a proxy for genetic similarity in ADME genes.
2.5 CNVs
A copy number variant region (CNVR) is determined by aggregating overlapping copy number variants (CNVs) identified in different individuals. A total of 259 CNVRs were identified, consisting of 106 duplications, 106 deletions and 47 mixed CNVRs (i.e. a region that is deleted in some individuals and duplicated in others) (Table 3). Duplications were further separated into biallelic duplications (3 or 4 copies) and multi-allelic duplications (>4 copies). About 54% of CNVRs were unique, while the remaining CNVRs overlapped with one or more of the other CNVRs identified. Of the 299 ADME genes that were analyzed, a total of 116 genes (38.8%) contained at least one CNVR. These include some important core pharmacogenes such as the CYPs, UGTs, and GSTs. Furthermore, the number of CNVs in ADME genes per individual ranged from four to 71, with the majority of individuals (89.9%) harboring between 11 and 30 CNVs.
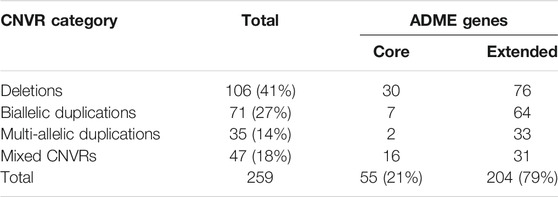
TABLE 3. CNVRs identified in core and extended ADME genes (percentages rounded to closest integer).
2.6 Novel and Highly Differentiated Variants
A novel variant in the context of this study is an SNV that is identified in the high coverage African population datasets, and not present in dbSNP (version 151) (Sherry, 2001) which aggregates variants from various data sources that include the 1,000 Genomes consortium (Altshuler et al., 2010; Auton et al., 2015), GO-ESP (Auer et al., 2016), ExAC consortium (Lek et al., 2016), GnomAD (Karczewski et al., 2019) and TOPMED (Brody et al., 2017).
A total of 343,606 SNVs were called for the ADME genes from the HAAD set of 458 samples, with 12% classified as novel SNPs (Supplementary Table S2). For the 32 core ADME genes, 5,818 novel variants were identified and a further 34,874 novel variants were identified in the 267 extended ADME genes within the HAAD. The majority of these variant types are intronic or intergenic variants (Supplementary Figure S4). Of the novel coding variants, eight were identified as HI-vars in core genes and 88 in extended genes.
The largest number of novel SNVs identified were from populations sampled from the Southern African region (not unexpected as there are no Southern African populations in the KGP). Novel variants in each regional population cluster were characterized according to their effect as summarized in Supplementary Table S3.
We compared the frequencies of ADME variants seen in the HAAD set as well as in at least one of the other large databases including 1,000 Genomes Consortium, ExAC, gnomAD and TOPMED. Any variant with a frequency two-fold more or two-fold less in the HAAD set than in the other datasets was considered as highly differentiated. Approximately 1,957 ADME variants were highly differentiated in the HAAD data compared to 1,000 Genomes consortium, ExAC, gnomAD and TOPMED datasets. Sixteen common variants with Minor Allele Frequency (MAF) ≥1% in eight core genes were more frequent in HAAD than in the KGP including African populations in those datasets. One variant in one of the core genes (rs3017670, SLC22A6) was seen more commonly in the other datasets than in the HAAD data (Supplementary Table S4). In total, 251 core and extended ADME genes harbored highly differentiated variants, with about 80% of them having at least two highly differentiated variants.
We performed a structural analysis of four rare novel HI-vars in the CYP2A13, CFTR, ABCB1, and NAT1 genes, all having a protein structure in the Protein Data Bank (Figure 8). A variant chr19:g.41595975C > G causes a substitution p. Arg123Gly on CYP2A13 (PDB code 2PG5) (Sansen et al., 2007). Mapping this variant on the structure shows a position close to the interaction site belonging to a rigid alpha helix which might affect the binding properties and the local folding integrity.

FIGURE 8. The number of novel high impact variants in ADME protein families. Abbreviation for Protein families: AD, aldehyde dehydrogenase; ANA, arylamine N-acetyltransferase; ATPDABZ, ATP-dependent AMP-binding enzyme; CATA, cation transport ATPase; CYP450, cytochrome P450; DADA, DAMOX/DASOX; DASS, SLC13A/DASS transporter; EPH, epoxide hydrolase; GPER, glutathione peroxidase; GSTA, GST alpha; GSTM, GST Mu; ICAD, iron-containing alcohol dehydrogenase; LAAT, L-type amino acid transporter; NCYP450, NADPH–cytochrome P450 reductase; NHRF, nuclear hormone receptor family; OCTF, organic cation transporter family; PEROX, peroxidase; SDR, short-chain dehydrogenases/reductases; ST2, sulfotransferase 2; SUGT, Sugar transporter; SULF1, sulfotransferase 1; UDPGT, UDP-glycosyltransferase. ABCC, ABCB, ABCA, FMO, ABCG are not abbreviated. On the structures, we showed examples of variants for four proteins belonging to different protein families described in Section 2.6.
For the CFTR gene, a chr7:g.11725069A > G causes a substitution p. Thr1036A which is involved in the interaction of the Lasso domain of the protein serving as a critical interaction segment of CFTR with other proteins (PDB code 6MSM) (Ford, 2017; Zhang et al., 2018). This threonine also appears to form a pseudoproline-like structure in which the side chain OH is hydrogen bonded to its own backbone NH. This may contribute to the bending of the helix in which this residue is found. Mutation to Ala removes this hydrogen bond and may therefore influence the degree of bending of this helix.
A p. H1232Q protein variant in ABCB1 could affect the interactions of this residue with the ATP molecule required for the active transport process (PDB code 6C0V) (Kim and Chen, 2018). In the structure His 1,232 lies in a site to which an ATP is bound approximately 5 Å from the ATP gamma phosphate. Although not in direct contact with the ATP, it could interact with it via a network of hydrogen bonds involving water molecules or, if the histidine is protonated, via an electrostatic interaction with the ATP phosphates. A mutation to Gln could affect both types of interaction with the ATP.
The chr8:g.18079983C > T variant creates a premature stop codon in NAT1 gene (PDB code 2IJA). The variant corresponds to the position p. Q143 which is close to the catalytic site of the protein.
We analyzed the distribution of the novel variants for the HAAD population cluster (Figure 9). The shared variants are generally exclusive for higher index values, which correspond to higher allele frequencies (Figures 9A,B) in their respective cluster for both core (Figure 9C) and extended genes (Figure 9D). Moreover, we noted that the cluster specific variants cover a big portion of the frequency spectrum: most of them are rare (lower limit of the frequency spectrum).
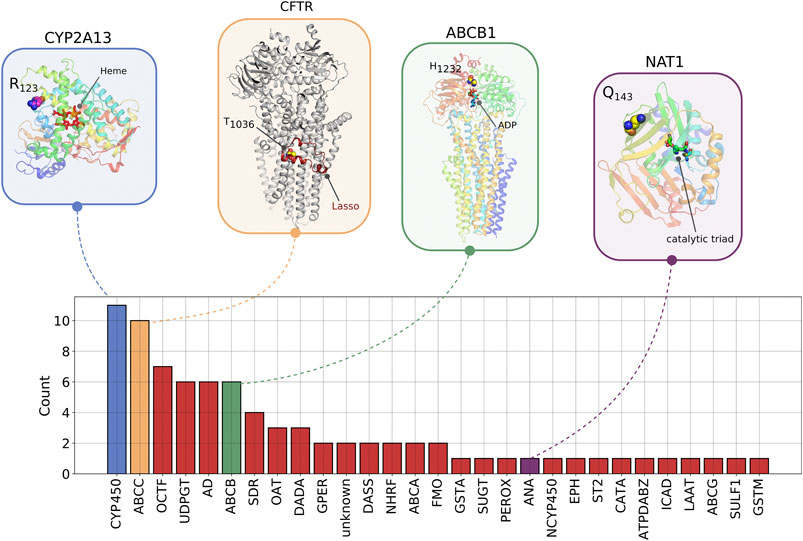
FIGURE 9. Characterization of novel variant distribution across the ADME genes in HAAD. Variants are indexed by descending MAF for core genes (A) and extended genes (B) for the total population of HAAD samples. Circular plots for core (C) and extended genes (D) show the position of the unique variants per each cluster across the index axes represented by points. The index represents the MAF of the variant in the total HAAD dataset. A link is established if two clusters share the same variant. The FW cluster has fewer variants in common with other clusters in this figure due to low sample number used to generate this figure.
2.7 Potential Translational Impact of ADME Pharmacogenomic Variants With Known Clinical Effects
To assess the transferability of variants with known pharmacogenomic effect, we focused on variants with PharmGKB level 1A and 1B clinical annotations. A level 1A annotation denotes a variant-drug combination published as a CPIC guideline or known clinical implementation in a major health system, while a level 1B annotation denotes a variant-drug combination for which a large body of evidence shows an association in the context of altering drug response (Whirl-Carrillo et al., 2012) (Note that the absence of level 1 annotation may be evidence of lack of study of a variant, especially for African-specific variants, rather than evidence against clinical relevance.) There are 21 clinical variants (PharmGKB 1A/B) in total in 11 ADME genes in the entire HAAD set: nine of these variants had AF ≥ 0.05 in HAAD, while 12 are rarer (AF < 0.05); and 7 of the 21 are HI-vars (rs3918290, rs1142345, rs28399504, rs4986893, rs1799853, rs3892097, rs1065852). We compared the frequency of the clinically actionable ADME gene variants in the combined HAAD population with that in the 1,000 Genomes super populations and gnomAD (Table 4). CYP2D6*17 (rs28371706, AF = 0.23) had much higher frequency in HAAD compared to the non-African KGP super populations and the combined gnomAD population. CYP2D6*17 has been associated with decreased CYP2D6 enzymatic activity in African Bantu populations (Masimirembwa et al., 1996). The CYP3A5*3 defining variant (rs776746, splice acceptor) had a much lower frequency in HAAD (AF = 0.17), suggesting differences in the functionality of CYP3A5 in African populations compared to non-African populations where the frequency of this function-obliterating variant is higher.
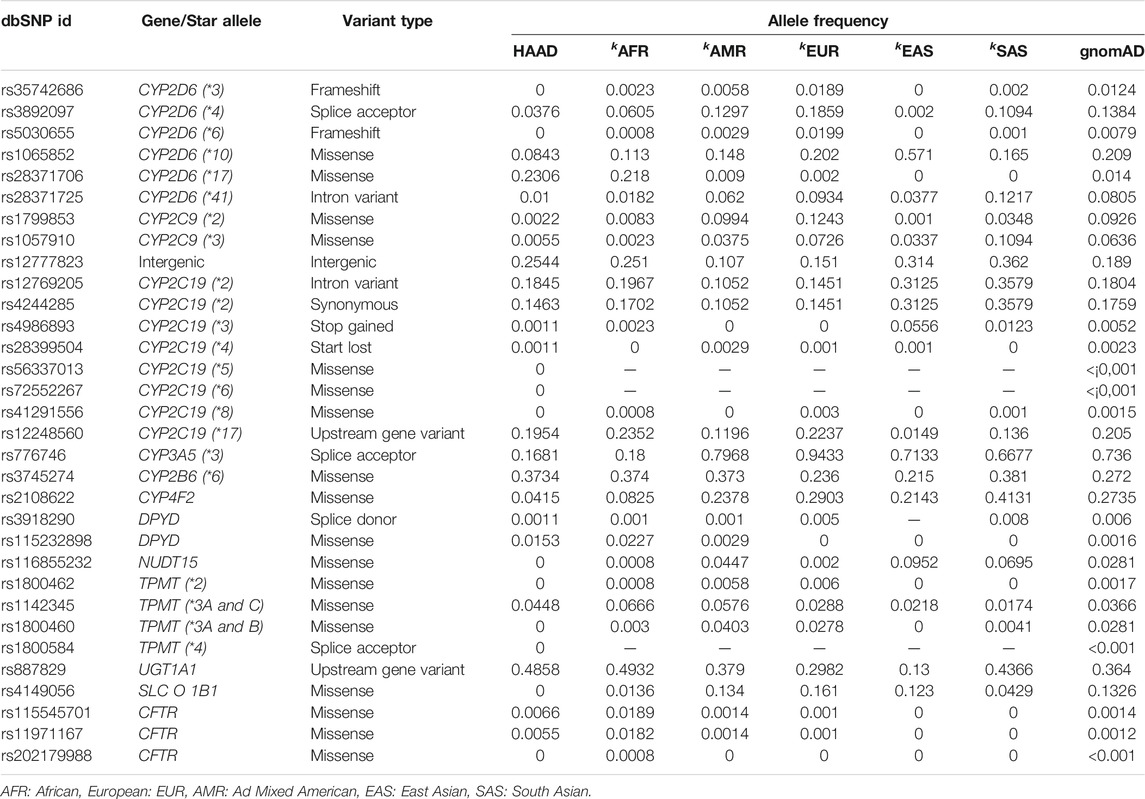
TABLE 4. Allele frequency of the clinically actionable variants (PharmGKB 1A/B) in the combined HAAD dataset compared to the KGP super populationsk as well as gnomAD.
Some clinically actionable ADME gene variants common in the non-African KGP super-populations are rare in the HAAD set. These include the variants SLCO1B1 rs4149056 (SLCO1B1*6), CYP4F2 rs2108622, CYP2D6 rs3892097, CYP2C9 rs1799853, and CYP2C9 rs1057910 (Table 4).
Furthermore, we evaluated the distribution of level 1A/B PharmGKB variants within the African populations (HAAD and KGP) grouped according to the PCA clusters. Variants which show considerable frequency differences among clusters (SA, SC, FW, and WE) include CYP2B6*6 (rs3745274), and CYP2D6*17 (rs28371706) (Figure 10A). The number of level 1A/B PharmGKB variants per individual ranged from 0 to 15 (median of 6, 5, 5, and 6 in SA, SC, FW, and WE respectively) (Figure 10B), with 99.8% of individuals carrying at least one such variants.
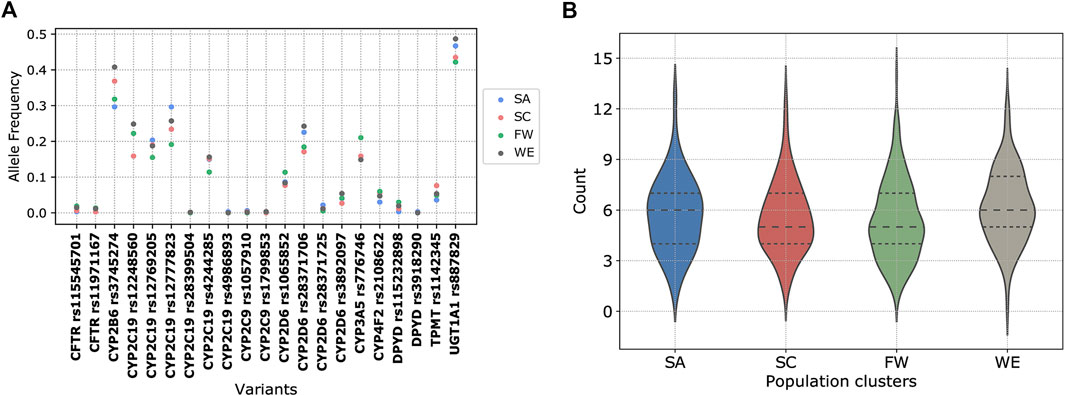
FIGURE 10. Distribution of pharmacogenomic variants with a high level of clinical annotation (PharmGKB level 1A/B). (A) Scatter plot of allele frequency of clinically relevant variants in the PCA clusters. (B) Violin plot of the number of clinically relevant variants carried per individual grouped by the population clusters.
2.8 Coverage of ADME Variants on SNV Genotyping Arrays
To evaluate whether genotyping array chips are suitable for detection of relevant ADME variants in African populations, we compared our whole genome sequencing variants with those captured by current arrays. Table 5 on page five shows the coverage of the variants that we detected in the core ADME genes in the WGS data compared to the Illumina Human Omni 2.5.8 (Omni) and the Illumina Infinium Multi-Ethnic AMR/AFR-8 Kit (MEGA). The Omni is a 2.39 million SNP array commonly used in human GWAS work—previous unpublished work shows that this is one of the best performing arrays on African populations. The MEGA array is 1.43 million SNP array optimized for African and Hispanic American populations (and can be augmented with approx. 200 k user selected SNPs). For different minor allele frequencies of variants we detected (MAF) we show the number of variants that are at least at that threshold, the number of those variants captured by probes by the two arrays, and the percentage of the variants that are captured. As can be seen, even at relatively high frequencies, less than 5% of the variants are captured by the array for core genes, and less than 8% for extended genes. As expected the larger Omni does a better job. However, of the 93 common HI-vars, only 19 (20%) are on the Omni chip whereas 50 (54%) are on the MEGA.
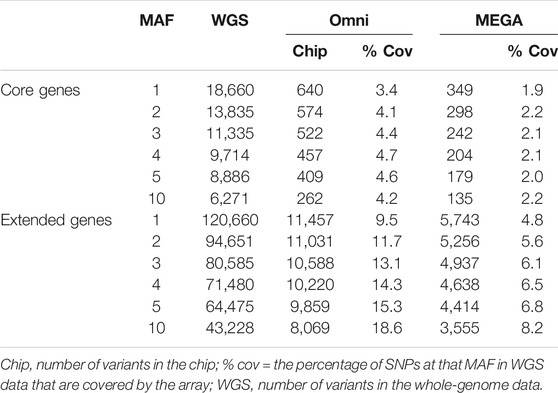
TABLE 5. Variant coverage and overlap for core and extended gene variants detected in HAAD whole genome sequencing datasets vs those captured by the Omni 2.5.8 and the MEGA arrays.
2.9 Other Analysis
We found novel variation in the regulatory regions of the ADME genes and large regions of homozygosity. Supplementary Sections S5, S6 present these analyses.
3 Discussion
Next generation technologies have provided PGx and precision medicine a major increase in their application for disease treatment and drug safety (Claussnitzer et al., 2020). ADME genes have been a focus due to their critical role in pharmacodynamics and pharmacokinetics. Our work presents the first study characterizing the PGx landscape of ADME genes in SSA using high coverage whole-genome sequencing data which has been collected from different sources. The study’s main aim was to assess the variability of ADME genes across Africa and if this could have a significant impact on protein function and other pharmacologic properties and thus the potential impact on drug response.
We focus mainly on four African clusters distinguished geographically and genetically as shown by the PC whole-genome analysis. Overall assessments of structural and regulatory variation were evaluated across the complete dataset, while coding variants were assessed for functional impact. The applicability of known clinical variants and current genotyping technologies was also assessed.
In both novel variant and HI-vars analysis, our study demonstrates a significant level of variability. Most of the variants are rare and are population-specific in accordance with previous studies due mainly to increased population size and to a weak negative selection (Tennessen et al., 2012; Whirl-Carrillo et al., 2012; Nagasaki et al., 2015; Wright et al., 2018). Our high coverage data are adequate to genetically characterize these types of variants at high confidence levels. Evaluations of the false discovery rate of rare variants were previously estimated between 3.6 and 6.3% depending on the platform (Wall et al., 2014). Therefore, a broad extrapolation from our results is that there are between 30 and 60 false positive variants in our HI-vars. In the context of ADME pharmacogenes, although not all variants identified may prove to have functional impact, those that do may have significant consequences in dictating the drug-host response for individuals.
Our
In addition, the important number of cluster-specific novel and high impact rare variants suggest that strategies limited to studies of high-frequency alleles might be considered as an over-generalization to a more complex pharmacogenomic landscape in SSA. In fact, our work highlights a “genetic diversity bottleneck” for precision medicine applications, requiring a balance between variants useful for population-based applications (for a particular cluster of Africans) and between the potential impact posed by variants unique to the individual. Therefore, the complexities of variant interpretation and reporting in PGx testing (Mills et al., 2013) may be exacerbated by the complex African ADME landscape.
While some clinically actionable variants have similar frequencies in European and African populations, our assessment of the top-level clinically validated variants shows a PGx knowledge bias toward European populations, with most variation in African and other global populations still largely uncharacterized in terms of PGx effect. Most are more common in Europeans, though some variants show an opposite trend, such as the CYP3A5*3 rs776746 (less common in Africans compared to Europeans) and CYP2D6*17 rs28371706 (largely African-specific). These enzymes are known to be key metabolisers of a large number of drugs, and these two variants (as they are common) will impact the reliability of using a European based PGx strategy in African populations. Key drugs that may be affected by those variants are codeine (Brousseau et al., 2007), primaquine (Awandu et al., 2018) (CYP2D6), and tacrolimus (Chen and Prasad, 2018) (CYP3A5). We also see an interesting example of SLC O 1B1 rs4149056, which was seen in the KGA populations (albeit rarely), which is not seen in the HAAD samples. This further reiterates the need for additional African sequences, as publicly accessible African genomic data cannot remain represented by the KGP alone.
The greatest genomic coverage of African populations to date is available in genotyping array format (Gurdasani et al., 2015). These methods are unable to adequately characterize rare ADME variants at high confidence levels compared to high coverage WGS datasets. Moreover, we have also detected a large number of CNVs, and were able to do so robustly with our high coverage sequencing data as compared to other methods (Zhao et al., 2013). The distribution of CNVs and their impact on the ADME landscape in Africans is currently ongoing and will be available in a separate publication. As the state of data availability and type remains in flux, precision medicine approaches in Africa will be limited. In an ideal scenario, high coverage long read WGS will be used for more African samples undergoing clinical trials, as this allows for accurate resolution of haplotypes (including novel haplotypes), and thus, clearer interpretations of their potential impact on drug response.
4 Conclusion
Our work highlights that the ADME landscape in African populations is diverse, and shows the importance of rare variation held within individual population clusters. Therefore, current array-based genotyping technologies have severe limitations to be applied as the high throughput method in precision medicine applications. As sequencing technology becomes more accessible and cheaper, characterization of rare variants would benefit from the ongoing progress. Targeted sequencing and whole-exome sequencing would be better suited for characterizing ADME genes. Moreover, a previous suggestion to consider intra-ethnic genetic characterization in drug-development (Soko et al., 2018) might not be appropriate for SSA due to the important presence of singletons and the subjective assigning of ethnicity for individuals. The “genetic diversity bottleneck” in precision medicine might increase the burden of developing targeted therapies at sub-population levels because of the weak presence of common genetic patterns. However, these patterns might exist at the functional and phenotypic levels which might help to stratify the populations to clusters sharing common pharmacokinetic properties for a given drug. In this context, a proposed plan would integrate genotypic and phenotypic data into predictive models to unveil these patterns (Dandara and Matimba, 2019).
Capacity building efforts for PGx research in Africa is important. Strategies and policies for development of science and technology must ensure a future where Africa can take an active role in harnessing the power of genomic research in addressing its healthcare challenges. Promising positive steps are being taken with the establishment of initiatives such as the Human Heredity and Health in Africa project (http://h3africa.org/) that aims at strengthening research capacity for genomics in Africa.
4.1 Limitations
There are many limitations of our work. The most obvious is the need for significantly more genomic data from Africa. Although, more samples are necessary generally, there is a particular need for more diverse sampling. We focus on SSA, omitting northern Africa completely. We only had limited numbers of samples from Nilo-Saharan and Afroasiatic language speakers as well as speakers of non-Bantu languages in central, southern and eastern Africa (such as San and Khoe speakers). However, with more samples, we expect our conclusions to hold and the additional benefit would be a clearer resolution of the PGx landscape in diverse sub-clusters. Ideally, we would have merged the 1,000 Genomes African data and the HAAD data set and done a combined analysis. However, the bulk of the 1,000 Genomes WGS is low-coverage while the HAAD set is high-coverage which complicates comparative work significantly. As more data becomes available, this challenge will become easier. The discovery curve shown in Figure 4B shows we can expect to find many more variants when they are sequenced. Besides the lack of genomic data, there is very little clinical and drug response data for African populations (despite the effects of groups of excellence across Africa we have cited). Without this it will be difficult to associate the functional effect of variants to the clinical phenotypes. All of this costs money and requires scarce skills. Collaborations like ours, which has brought a diverse group of African scientists together show the potential of what can be done.
4.2 Strengths
Our work investigates novel African datasets and combines these with established African sequences to assess as broad an overview of African ADME variation as possible. This work could lay the foundations for motivation of more PGx related studies in Africans. We applied diverse computational assessment methods to mine the data and retrieve valuable genomic information. This can assist in guiding future research in resource scarce environments.
4.3 Future Work
In our subsequent work, we plan to do linkage disequilibrium analysis and haplotype frequency determination of key ADME genes—an example being CYP2D6, which requires specialized algorithms for accurate haplotyping (Twesigomwe et al., 2020). We are currently in the stage of resequencing CYP2D6 and some other ADME genes with long-read sequencing technology to explore such features, as well as for in-depth analysis of CNVs.
5 Methods
5.1 Data
H3A Consortium set contains 272 samples selected and sequenced for the Human Heredity and Health in Africa (H3Africa) project. Samples cover populations from Benin, Burkina Faso, Botswana, Cameroon, Ghana, Nigeria and Zambia. Samples were shipped to the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine (BCM), Houston, United States, under signed material transfer agreements from each project. Samples were prepared using the TruSeq Nano DNA Library Prep Kits and underwent whole genome sequencing on an Illumina TenX (150 bp) to a minimum depth of coverage of 30×.
AWI-Gen set consists of 100 South Eastern Bantu-Speakers (40× coverage).
Cell Biology Research Unit, Wits set consists of 40 samples from Soweto/Johannesburg South Africa (39 black and one mixed ancestry). Library preparation and sequencing was done at Edinburgh Genomics, Edinburgh, Scotland. Library preparation was done using the TruSeq Nano protocol and high coverage sequencing (∼30×) was done utilizing the Illumina SeqLab workflow system and the Illumina HiSeqX platform.
The SAHGP set is a collection of 15 samples from the Southern African Human Genome Program (Choudhury et al., 2017). Two main Bantu-speaking ethno-linguistic groups were included: The Sotho (Sotho-Tswana speakers; n = 8) and the Xhosa speakers (Nguni language; n = 7 recruited from the Eastern Cape Province). The DNA samples were normalized to ∼60 ng/μl and ∼5 µg DNA was submitted to the Illumina Service Center in San Diego, California, for sequencing on the Illumina HiSeq 2000 instrument (101 bp paired-end reads, ∼314 bp insert size) with a minimum read depth of coverage of 30× (Choudhury et al., 2017).
SF set contains 34 African samples selected from 300 individuals from the Simons Foundation Genome Diversity Project. Samples include populations from Congo, Namibia, Kenya, Senegal, Algeria, Nigeria, Gambia, Sudan and South Africa. Samples were sequenced at an average depth of 43× at Illumina Ltd.; almost all samples were prepared using the same PCR-free library preparation (Mallick et al., 2016).
KGA set consists of 507 African samples from the KGP. These samples include Gambian Mandinka, Mende from Sierra Leone, Yoruba from Ibadan, Nigeria, Esan from Nigeria and Luhya from Webuye, Kenya. Libraries were constructed on either Illumina HiSeq2000 or GAIIX with the use of 101 base pair end reads. Sequencing was done at an average depth of 4× (Auton et al., 2015).
The only phenotype made available to us was sex. In particular, self-identified ethnicity, location in the country, and disease status were not revealed.
5.2 Data Processing
From the BAMs we called gVCFs using HaplotypeCaller in gVCF mode using GATK v4.0.8.1. We combined all the gVCFs into one combined gVCF using GATK’s CombineGVCF (v4.0.8.1). From the combined gVCF we did joint calling using GenotypeGVCFs (v4.1.3.0) and followed GATK’s best practice for variant quality score recalibration for SNPs and INDELs. After applying VQSR we filtered for all the high quality (PASS) sites and used the VCF. The final VCF was used for downstream analysis. All code can be accessed at https://github.com/h3abionet/recalling.
5.3 Population Structure
Population structure was computed using the autosomal data in our samples together with reference data sets in order to ensure a relatively unbiased structure. We included all KGP African data, and two non-African KGP sets (Utah residents (CEPH) with Northern and Western European ancestry—CEU—and Bengali in Bangladesh—BEB) and some chip data from various projects including Khoi-San data (Schlebusch et al., 2012). The CEU and BEB populations were included to identify overall outliers in the African population groups. Prior work of our group has shown that the primary admixture from non-African populations, particularly in Southern Africa, comes from Europe and the Indian subcontinent. The BEB and CEU are good representatives for this study as other European or Indian populations. Only unambiguous, biallelic SNPs (A/C, A/G, C/T, G/T) common in all data sets were used. The data was merged and pruned using PLINK (Chang et al., 2015), leaving 401 k SNPs for analysis. Principal components were computed using PLINK and structure charts were produced using ADMIXTURE (Alexander et al., 2009) (30 independent runs for each value of k) and averaged using CLUMPP (Jakobsson and Rosenberg, 2007). All charts were produced with Genesis (Buchmann and Hazelhurst, 2014).
Population clusters were determined from the PCA values rather than from the project and self-identification labels due to overlapping data. The optimal number of clusters was determined using the method of Solovieff et al. (Solovieff et al., 2010), and clusters determined using k-means clustering with the R MASS package (Venables and Ripley, 2002). In analyses in which population clusters were compared, we only used the samples that appeared in the clusters (e.g., excluding Algerian, San samples). In all other analyses all the data was used. Choudhury et al. (Choudhury et al., 2020) discusses the population structure of the H3A data in more detail.
5.4 ADME Gene Selection
ADME genes as defined by PharmADME (http://pharmaadme.org) (both core and extended definitions) were extracted using current genomic co-ordinates for GRCh37. p13, as obtained through BioMart (Smedley et al., 2009). Gene flanking regions were included in the extraction (10,000 bp upstream from gene start and downstream from gene end).
5.5 Annotation and Functional Prediction
Variants were classified and typed using SnpEff v4.3t (Cingolani et al., 2012) with the GR37Ch base reference for canonical gene transcripts. Variant Effect Predictor (VEP) v92.0 (McLaren et al., 2016) was used for functional prediction based annotation. VEP was configured with dbNSFP v3.0 (Liu et al., 2016), a large database used to retrieve functional prediction scores for coding variants. The annotation analysis is implemented in g_miner workflow (https://github.com/hothman/PGx-Tools/tree/master/workflows/g_miner). An optimized model for functional prediction of pharmacogene variants produced by Zhou et al. (Zhou et al., 2019) was used as the basis for high impact classification of missense variants. The model uses five toolsets (LRT, MutationAssessor, PROVEAN, VEST3, and CADD). Loss of Function variants were classified as high impact if they were present in the canonical transcript of the gene. Singleton or doubleton high impact variants were filtered based on their VCF QUAL scores, using a cutoff of >50. Any variant that did not match such criteria was removed prior to subsequent analyses with bcftools v1.9 (Li, 2011). Three HI-vars were not displayed in Figure 5 due to incorrect reference alleles inducing an erroneous frequency: ALDH3B1 rs11433668, and rs58160034; and ADH1C—rs283413. We have checked these variants in KGP and gnomAD datasets to validate the error.
5.6 Fixation Index (
Differences between African subgroups were calculated by PLINK v1.9 (Chang et al., 2015), using mean, weighted
5.7 CNVs
Discovery and genotyping of CNVs was performed using GenomeSTRiP’s SVPreprocessing and CNVDiscovery (svtoolkit 2.00.1918) pipelines using the default parameters for genomes sequenced at 30–40× coverage (Handsaker et al., 2015).
Members of the Human Heredity and Health in Africa Consortium
Jorge da Rocha, Sydney Brenner Institute for Molecular Bioscience, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, and Division of Human Genetics, National Health Laboratory Service, and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa; Houcemeddine Othman, Sydney Brenner Institute for Molecular Bioscience, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa; Gerrit Botha, Computational Biology Division and H3ABioNet, Department of Integrative Biomedical Sciences, University of Cape Town, South Africa; Laura Cottino, Sydney Brenner Institute for Molecular Bioscience, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, and Division of Human Genetics, National Health Laboratory Service, and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa; David Twesigomwe, Sydney Brenner Institute for Molecular Bioscience, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, and Division of Human Genetics, National Health Laboratory Service, and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa; Samah Ahmed, Centre for Bioinformatics and Systems Biology, Faculty of Science, University of Khartoum, Sudan; Faisal M. Fadlelmola, Centre for Bioinformatics and Systems Biology, Faculty of Science, University of Khartoum, Sudan; Philip Machanick, Department of Computer Science, Rhodes University, Makhanda, South Africa; Mamana Mbiyavanga, Computational Biology Division and H3ABioNet, Department of Integrative Biomedical Sciences, University of Cape Town, South Africa; Sumir Panji, Computational Biology Division and H3ABioNet, Department of Integrative Biomedical Sciences, University of Cape Town, South Africa; Clement Adebamowo, Institute for Human Virology, Abuja, Nigeria, and Institute of Human Virology and Greenebaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, MD, United States; Mogomotsi Matshaba, Botswana-Baylor Children’s Clinical Center of Excellence, Gaborone, Botswanam, and Baylor College of Medicine, Houston, TX, United States; Michéle Ramsay, Sydney Brenner Institute for Molecular Bioscience, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa, and Division of Human Genetics, National Health Laboratory Service, and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa; Gustave Simo, Molecular Parasitology and Entomology Unit, Department of Biochemistry, Faculty of Science, University of Dschang, Dschang, Cameroon; Martin C. Simuunza, Department of Disease Control, School of Veterinary Medicine, University of Zambia, Lusaka, Zambia; Scott Hazelhurst, School of Electrical and Information Engineering, University of the Witwatersrand, Johannesburg, South Africa.
Members of the H3A/GSK ADME Collaboration
Britt I. Drögemöller, Department of Biochemistry and Medical Genetics, University of Manitoba, Winnipeg, MB, Canada; Galen E. B. Wright, Neuroscience Research Program, Kleysen Institute for Advanced Medicine, Winnipeg Health Sciences Centre and Max Rady College of Medicine, University of Manitoba, and Department of Pharmacology and Therapeutics, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada; Caroline T. Tiemessen, Centre for HIV and STIs, National Institute for Communicable Diseases, National Health Laboratory Services and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg South Africa; Sandra Baldwin, Drug Metabolism and Pharmacokinetics, GlaxoSmithKline R&D, Ware, UK; Mathias Chiano, Human Genetics, GlaxoSmithKline R&D, Stevenage, UK; Charles Cox, Human Genetics, GlaxoSmithKline R&D, Stevenage, UK; Annette S. Gross, Clinical Pharmacology Modelling and Simulation, GlaxoSmithKline R&D, Sydney, NSW, Australia; Pamela Thomas, Data and Computational Sciences, GlaxoSmithKline R&D, Stevenage, UK; Francisco-Javier Gamo, Global Health, GlaxoSmithKline R&D, Madrid, Spain.
Data Availability Statement
The Simons Foundation and 1000 Genomes data are publicly available. The HAAD data set is controlled access data due to the need to protect participants, and is available from European Genome-Phenome Archive (https://ega-archive.org/) on application to the relevant Data Access Committee (EGADs EGAD00001003791, EGAD00001006418, EGAD00001004220, EGAD00001004448, EGAD00001004505, EGAD00001004533, EGAD00001004557, EGAD00001004393. The EGAD for the CBRL data is pending).
Ethics Statement
No new data was generated specifically for this project—this is secondary analysis of data that had been generated and studied for other purposes. The H3A AWI-Gen Study (H3A data from Ghana, Burkina Faso and South Africa) was approved by the Human Research Ethics Committee (Medical) of the University of the Witwatersrand (Wits) (protocol numbers M121029 and M170880), and each contributing Centre obtained additional local ethics approval, as required. The H3A Benin study was approved by the Comité d’éthique de la recherche, Université de Montréal. The H3A CAfGEN study (Botswana) was approved by the IRB of the Ministry of Health of the Republic of Botswana (PPPME-13/18/1). The H3A TrypanoGEN Study (Cameroon component) was approved by the Comité National D’Ethique de la Recherche pour la Santé Humaine of the Republic of Cameroon (No 2013/11/364/L/CNERSH/SP). The H3A ACCME Study (Nigeria) was approved by the National Health Research Ethics Committee of Nigeria (NHREC/01/01/2007-29/11/2016). The H3A TrypanoGEN Study (Zambian component) was approved by the Biomedical Research Ethics Committee of the University of Zambia (FWA00000338). The data from the Cell Biology Research Lab, NICD/Wits was generated by a study approved by the Wits Human Research Ethics (Medical) Committee (protocol number M140926).
Author Contributions
JdR led the writing of the paper with assistance from HO. GB was responsible for the joint calling of the data and QC. JdR, HO, GB, SP, MM, SH contributed to the genomic data analysis. LC was primarily responsible for copy number variation analysis. DT and SA studied the transferability of ADME pharmacogenomic variants. PM provided the analysis of the regulatory regions. BD, DT, FMF, PM, MM, SP, GW, and SH all contributed to writing. SB, MC, CC, ASG, GS, CA, MoM, MR, GS, MS, CT, and PT provided the critical analysis of the paper and contributed to writing. SH proposed the project, coordinated the work and co-led it with FJG. All authors contributed to writing the manuscript, and read and approved the final manuscript. This paper describes the views of the authors in their personal capacities and does not necessarily represent the official views of the funders.
Funding
This work was primarily funded through a grant by GlaxoSmithKline Research and Development Ltd. to the Wits Health Consortium. The AWI-Gen Collaborative Center is funded by the NIH/NHGRI (Grant U54HG006938) as part of the H3Africa Consortium. MR is a South African Research Chair in Genomics and Bioinformatics of African Populations hosted by the University of the Witwatersrand, funded by the Department of Science and Technology, and administered by National Research Foundation of South Africa (NRF). The TrypanoGEN project was funded by the Wellcome Trust, study number 099310/Z/12/Z. The Collaborative African Genetics Network (CAfGEN) is funded by the NIH/NHGRI (grant CAfGEN 1U54AI110398). The whole genome sequencing of the H3A Data was supported by a grant from the National Human Genome Research Institute, National Institutes of Health (NIH/NHGRI) U54HG003273. Cell Biology Research Lab component: This work is based on the research supported by grants awards from the Strategic Health Innovation Partnerships (SHIP) Unit of the South African Medical Research Council, a grantee of the Bill and Melinda Gates Foundation, and the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa (84177). JdR and DT were partially funded by the SA National Research Foundation(SFH170626244782; SFH210111581508). GB, SA, FMF, MM, SP and part of the computational infrastructure used are supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U24HG006941. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
GlaxoSmithKline provided funding for this project. The authors SB, MC, CC, ASG, PT and FJG are all employees of GlaxoSmithKline and participated in the analysis and writing of the paper. GlaxoSmithKline had no additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely responsibility of the authors and does not necessarily represent the official views of GlaxoSmithKline. The remaining authors declare no conflicts of interest.
Acknowledgments
Matt Hall’s enthusiasm and guidance was critical in launching this project. We thank the generous collaboration of colleagues in the various H3A groups who contributed to this work. This study would not have been possible without the generosity of the participants. We acknowledge the sterling contributions of our field workers, phlebotomists, laboratory scientists, administrators, data personnel and all other staff who contributed to the data and sample collections, processing, storage, and shipping.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.634016/full#supplementary-material
References
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19 (9), 1655–1664. doi:10.1101/gr.094052.109
Altshuler, D. L., Durbin, R. M., Abecasis, G. R., Bentley, D. R., Chakravarti, A., Clark, A. G., et al. (2010). A map of human genome variation from population-scale sequencing. Nature 467 (7319), 1061–1073. doi:10.1038/nature09534
Auer, P. L., Reiner, A. P., Wang, G., Kang, H. M., Abecasis, G. R., Altshuler, D., et al. (2016). Guidelines for large-scale sequence-based complex trait association studies: lessons learned from the NHLBI exome sequencing project. Am. J. Hum. Genet. 99 (4), 791–801. doi:10.1016/j.ajhg.2016.08.012
Auton, A., Abecasis, G. R., Altshuler, D. M., Durbin, R. M., Bentley, D. R., Chakravarti, A., et al. (2015). A global reference for human genetic variation. Nature 526 (7571), 68–74. doi:10.1038/nature15393
Awandu, S., Raman, J., Makhanthisa, T. I., Kruger, P., Frean, J., Bousema, T., et al. (2018). Understanding human genetic factors influencing primaquine safety and efficacy to guide primaquine roll-out in a pre-elimination setting in southern Africa. Malar. J. 17, 120. doi:10.1186/s12936-018-2271-z
Brody, J. A., Morrison, A. C., Bis, J. C., O’Connell, J. R., Brown, M. R., Huffman, J. E., et al. (2017). Analysis commons, a team approach to discovery in a big-data environment for genetic epidemiology. Nat. Genet. 49 (11), 1560–1563. doi:10.1038/ng.3968
Brousseau, D. C., McCarver, D. G., Drendel, A. L., Divakaran, K., and Panepinto, J. A. (2007). The effect of CYP2D6 polymorphisms on the response to pain treatment for pediatric sickle cell pain crisis. J. Pediatr. 150 (6), 623–626. doi:10.1016/j.jpeds.2007.01.049
Buchmann, R., and Hazelhurst, S. (2014). Genesis manual. Johannesburg, South Africa: University of the Witwatersrand.
Burnham, K. P., and Overton, W. S. (1979). Robust estimation of population size when capture probabilities vary among animals. Ecology 60 (5), 927–936. doi:10.2307/1936861
Busby, G., Band, G., Si Le, Q., Jallow, M., Bougama, E., Mangano, V., et al. (2016). Admixture into and within sub-saharan africa. eLife 5, e15266. doi:10.1101/038406
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4 (1), 1–16. doi:10.1186/s13742-015-0047-8
Chen, L., and Prasad, G. V. R. (2018). CYP3A5 polymorphisms in renal transplant recipients: influence on tacrolimus treatment. Pharmgenomics Pers Med. 11, 23–33. doi:10.2147/pgpm.s107710
Choudhury, A., Aron, S., Botigué, L., Sengupta, D., Botha, G., Bensellak, T., et al. (2020). High depth African genomes inform human migration and health. Under embargo.
Choudhury, A., Ramsay, M., Hazelhurst, S., Aron, S., Bardien, S., Botha, G., et al. (2017). Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans. Nat. Commun. 8 (1), 2062. doi:10.1038/s41467-017-00663-9
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6 (2), 80–92. doi:10.4161/fly.19695
Claussnitzer, M., Cho, J. H., Collins, R., Cox, N. J., Dermitzakis, E. T., Hurles, M. E., et al. (2020). A brief history of human disease genetics. Nature 577 (7789), 179–189. doi:10.1038/s41586-019-1879-7
Dandara, C., and Matimba, A. (2019). “A glimpse into pharmacogenomics in africa,” in The genetics of african populations in health and disease. Editor M. E. Ibrahim, and C. N. Rotimi (Cambridge, United Kingdom: Cambridge University Press), 128–167. doi:10.1017/9781139680295.007
Dandara, C., Swart, M., Mpeta, B., Wonkam, A., and Masimirembwa, C. (2014). Cytochrome P450 pharmacogenetics in African populations: implications for public health. Expert Opin. Drug Metab. Toxicol. 10 (6), 769–785. doi:10.1517/17425255.2014.894020
Ford, B. (2017). CFTR structure: lassoing cystic fibrosis. Nat. Struct. Mol. Biol. 24 (1), 13–14. doi:10.1038/nsmb.3353
Gurdasani, D., Carstensen, T., Tekola-Ayele, F., Pagani, L., Tachmazidou, I., Hatzikotoulas, K., et al. (2015). The african genome variation project shapes medical genetics in africa. Nature 517 (7534), 327–332. doi:10.1038/nature13997
Handsaker, R. E., Van Doren, V., Berman, J. R., Genovese, G., Kashin, S., Boettger, L. M., et al. (2015). Large multiallelic copy number variations in humans. Nat. Genet. 47 (3), 296–303. doi:10.1038/ng.3200
Hovelson, D. H., Xue, Z., Zawistowski, M., Ehm, M. G., Harris, E. C., Stocker, S. L., et al. (2017). Characterization of {ADME} gene variation in 21 populations by exome sequencing. Pharmacogenet. and genomics 27 (3), 89–100. doi:10.1097/FPC.0000000000000260
Jakobsson, M., and Rosenberg, N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23 (14), 1801–1806. doi:10.1093/bioinformatics/btm233
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Available at: https://www.biorxiv.org/content/10.1101/531210v2.
Kim, Y., and Chen, J. (2018). Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 359 (6378), 915–919. doi:10.1126/science.aar7389
Lakiotaki, K., Kanterakis, A., Kartsaki, E., Katsila, T., Patrinos, G. P., and Potamias, G. (2017). Exploring public genomics data for population pharmacogenomics. PLoS One 12 (8), e0182138. doi:10.1371/journal.pone.0182138
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536 (7616), 285–291. doi:10.1038/nature19057
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27 (21), 2987–2993. doi:10.1093/bioinformatics/btr509
Liu, X., Wu, C., Li, C., and Boerwinkle, E. (2016). dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37 (3), 235–241. doi:10.1002/humu.22932
Mallick, S., Li, H., Lipson, M., Mathieson, I., Gymrek, M., Racimo, F., et al. (2016). The Simons genome diversity project: 300 genomes from 142 diverse populations. Nature 538 (7624), 201–206. doi:10.1038/nature18964
Masimirembwa, C., Persson, I., Bertilsson, L., Hasler, J., and Ingelman-Sundberg, M. (1996). A novel mutant variant of the CYP2D6 gene (CYP2D617) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br. J. Clin. Pharmacol. 42 (6), 713–719. doi:10.1046/j.1365-2125.1996.00489.x
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17 (1), 122. doi:10.1186/s13059-016-0974-4
Mills, R., Voora, D., Peyser, B., and Haga, S. B. (2013). Delivering pharmacogenetic testing in a primary care setting. Pharmgenomics Pers Med. 6, 105–112. doi:10.2147/pgpm.s50598
Mukonzo, J. K. (2014). The challenge of paediatric efavirenz dosing : implications and way forward for the sub-Saharan Africa. AIDS 28 (13), 1855–1857. doi:10.1097/QAD.0000000000000372
Nagasaki, M., Yasuda, J., Katsuoka, F., Nariai, N., Kojima, K., Kawai, Y., et al. (2015). Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat. Commun. 6, 8018. doi:10.1038/ncomms9018
Radouani, F., Zass, L., Hamdi, Y., Rocha, J. D., Sallam, R., Abdelhak, S., et al. (2020). A review of clinical pharmacogenetics studies in African populations. Per Med. 17 (2), 155–179. doi:10.2217/pme-2019-0110
Roden, D. M., Mcleod, H. L., Relling, M. V., Williams, M. S., Mensah, G. A., Peterson, J. F., et al. (2019). Pharmacogenomics. Lancet 394, 521–532. doi:10.1016/S0140-6736(19)31276-0
Sansen, S., Hsu, M. H., Stout, C. D., and Johnson, E. F. (2007). Structural insight into the altered substrate specificity of human cytochrome P450 2A6 mutants. Arch. Biochem. Biophys. 464 (2), 197–206. doi:10.1016/j.abb.2007.04.028
Schlebusch, C. M., Skoglund, P., Gattepaille, L. M., Hernandez, D., Jay, F., Soodyall, H., et al. (2012). Genomic variation in seven khoe-san groups reveals adaptation and complex african history. Science 338 (6105), 374–379. doi:10.1126/science.1227721
Sherry, S. T. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29 (1), 308–311. doi:10.1093/nar/29.1.308
Smedley, D., Haider, S., Ballester, B., Holland, R., London, D., Thorisson, G., et al. (2009). BioMart–biological queries made easy. BMC Genomics 10, 22. doi:10.1186/1471-2164-10-22
Soko, N. D., Masimirembwa, C., and Dandara, C. (2018). Rosuvastatin pharmacogenetics in African populations. Pharmacogenomics 19 (18), 1373–1375. doi:10.2217/pgs-2018-0168
Solovieff, N., Hartley, S. W., Baldwin, C. T., Perls, T. T., Steinberg, M. H., and Sebastiani, P. (2010). Clustering by genetic ancestry using genome-wide SNP data. BMC Genet. 11 (1), 108. doi:10.1186/1471-2156-11-108
Tennessen, J. A., Bigham, A. W., O’Connor, T. D., Fu, W., Kenny, E. E., Gravel, S., et al. (2012). Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337 (6090), 64–69. doi:10.1126/science.1219240
The H3Africa Consortium (2014). Enabling the genomic revolution in Africa. Science 344, 1346–1348. doi:10.1126/science.1251546
Twesigomwe, D., Wright, G. E. B., Drogemuller, B. I., da Rocha, J., Lombard, Z., and Hazelhurst, S. (2020). A systematic comparison of pharmacogene star allele calling bioinformatics algorithms: a focus on CYP2D6 genotyping. NPJ Genom. Med. 5 (1), 30. doi:10.1038/s41525-020-0135-2
Venables, W. N., and Ripley, B. D. (2002). Modern applied statistics with S. 4th Edn. New York, NY: Springer.
Wall, J. D., Tang, L. F., Zerbe, B., Kvale, M. N., Kwok, P. Y., Schaefer, C., et al. (2014). Estimating genotype error rates from high-coverage next-generation sequence data. Genome Res. 24 (11), 1734–1739. doi:10.1101/gr.168393.113
Whirl-Carrillo, M., McDonagh, E. M., Hebert, J. M., Gong, L., Sangkuhl, K., Thorn, C. F., et al. (2012). Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92 (4), 414–417. doi:10.1038/clpt.2012.96
Wright, G. E. B., Carleton, B., Hayden, M. R., and Ross, C. J. D. (2018). The global spectrum of protein-coding pharmacogenomic diversity. Pharmacogenomics J. 18 (1), 187–195. doi:10.1038/tpj.2016.77
Zhang, Z., Liu, F., and Chen, J. (2018). Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 115 (50), 12757–12762. doi:10.1073/pnas.1815287115
Zhao, M., Wang, Q., Wang, Q., Jia, P., and Zhao, Z. (2013). Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics 14 (Suppl. 11), S1. doi:10.1186/1471-2105-14-s11-s1
Keywords: ADME, genetic diversity, Africa, pharmacogenomics, CNV
Citation: da Rocha JEB, Othman H, Botha G, Cottino L, Twesigomwe D, Ahmed S, Drögemöller BI, Fadlelmola FM, Machanick P, Mbiyavanga M, Panji S, Wright GEB, Adebamowo C, Matshaba M, Ramsay M, Simo G, Simuunza MC, Tiemessen CT, Baldwin S, Chiano M, Cox C, Gross AS, Thomas P, Gamo F-J and Hazelhurst S (2021) The Extent and Impact of Variation in ADME Genes in Sub-Saharan African Populations. Front. Pharmacol. 12:634016. doi: 10.3389/fphar.2021.634016
Received: 26 November 2020; Accepted: 10 February 2021;
Published: 28 April 2021.
Edited by:
Volker Martin Lauschke, Karolinska Institutet (KI), SwedenReviewed by:
Sonja Pavlovic, University of Belgrade, SerbiaUmamaheswaran Gurusamy, University of California San Francisco, United States
Copyright © 2021 da Rocha, Othman, Botha, Cottino, Twesigomwe, Ahmed, Drögemöller, Fadlelmola, Machanick, Mbiyavanga, Panji, Wright, Adebamowo, Matshaba, Ramsay, Simo, Simuunza, Tiemessen, Baldwin, Chiano, Cox, Gross, Thomas, Gamo and Hazelhurst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott Hazelhurst, scott.hazelhurst@wits.ac.za
‡Members of the Human Heredity and Health in Africa Consortium. Authors not marked with ‡are members of the H3A/GSK ADME Collaboration but not of the H3Africa Consortium.