
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 05 January 2021
Sec. Drug Metabolism and Transport
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.593518
This article is part of the Research Topic Pharmacology and Metabolism of Phytochemicals View all 14 articles
 Jinhui Wang1†Feifei Chen2†Hui Jiang2Jia Xu2Deru Meng2Peiwu Geng2Dapeng Dai3Jingbo Hu1Yunfang Zhou2Quan Zhou2*Shuanghu Wang2*
Jinhui Wang1†Feifei Chen2†Hui Jiang2Jia Xu2Deru Meng2Peiwu Geng2Dapeng Dai3Jingbo Hu1Yunfang Zhou2Quan Zhou2*Shuanghu Wang2*Poziotinib is an orally active, irreversible, pan-HER tyrosine kinase inhibitor used to treat non-small cell lung cancer, breast cancer, and gastric cancer. Poziotinib is currently under clinical investigation, and understanding its drug-drug interactions is extremely important for its future development and clinical application. The cocktail method is most suitable for evaluating the activity of cytochrome P450 enzymes (CYPs). As poziotinib is partially metabolized by CYPs, cocktail probes are used to study the interaction between drugs metabolized by each CYP subtype. Midazolam, bupropion, dextromethorphan, tolbutamide, chlorzoxazone, phenacetin, and their metabolites were used to examine the effects of poziotinib on the activity of cyp1a2, 2b1, 2d1, 2c11, 2e1, and 3a1/2, respectively. The in vitro experiment was carried out by using rat liver microsomes (RLMs), whereas the in vivo experiment involved the comparison of the pharmacokinetic parameters of the probes after co-administration with poziotinib to rats to those of control rats treated with only probes. UPLC-MS/MS was used to detect the probes and their metabolites in rat plasma and rat liver microsomes. The in vitro results revealed that the half-maximal inhibitory concentration values of bupropion and tolbutamide in RLMs were 8.79 and 20.17 μM, respectively, indicating that poziotinib showed varying degrees of inhibition toward cyp2b1 and cyp2c11. Poziotinib was a competitive inhibitor of cyp2b1 and cyp2c11, with Ki values of 16.18 and 17.66 μM, respectively. No time- or concentration-dependence of inhibition by poziotinib was observed toward cyp2b1 and cyp2c11 in RLMs. Additionally, no obvious inhibitory effects were observed on the activity of cyp1a2, cyp2d1, cyp2e1, and cyp3a1/2. In vivo analysis revealed that bupropion, tolbutamide, phenacetin, and chlorzoxazone showed significantly different pharmacokinetic parameters after administration (p < 0.05); there was no significant difference in the pharmacokinetic parameters of dextromethorphan and midazolam. These results show that poziotinib inhibited cyp2b1 and cyp2c11, but induced cyp1a2 and cyp2e1 in rats. Thus, poziotinib inhibited cyp2b1 and cyp2c11 activity in rats, suggesting the possibility of interactions between poziotinib and these CYP substrates and the need for caution when combining them in clinical settings.
Drug-drug interaction (DDI) occurs during the co-administration of two or more drugs and may increase toxicity or weaken the therapeutic effect of the drugs, leading to the occurrence of serious clinical complications such as adverse drug reactions or treatment failure (Lee et al., 2015; Chen Z. H. et al., 2016). DDI is believed to be closely linked to the inhibition or induction of the activity of cytochrome P450 (CYP) enzymes because CYP enzymes play major roles in the metabolism of various drugs (Polasek et al., 2011; Wang et al., 2019). As multi-drug combination therapy is a common therapeutic strategy, the risk of CYP inhibition or induction is greater for drugs with a narrow therapeutic window, with even a small change in concentration causing toxicity or therapeutic failure of the drug (Guidoni et al., 2016). Therefore, it is necessary to understand potential DDIs in the early stages of drug development.
CYP is a superfamily of oxygenases with mixed functions that are involved in the metabolism of endogenous and exogenous substances and are abundantly present in the liver and intestines (Wang et al., 2014; Yin et al., 2016). CYPs are responsible for ∼75% of human drug metabolism and play an important role in the elimination of commonly used drugs (Carrão et al., 2018; Jo et al., 2018). Among the many CYPs, CYP3A, CYP1A, CYP2B, CYP2C, and CYP2D play important roles in drug metabolism. CYP3A4 is the most important isoenzyme subfamily comprising 40% of all CYPs and metabolizing ∼50% of drugs such as immunosuppressants, antibiotics, statins, and herbal drugs (Loue and Tod, 2014; Wang S. H. et al., 2015; Liu et al., 2017; Wang et al., 2017; Lu et al., 2018).
CYP2C is the second most abundant isoenzyme subfamily. Its main isoenzymes are CYP2C9 and CYP2C19 can metabolize ∼25% of all drugs, of which CYP2C9 metabolizes 16% of all drugs, e.g., oral anticoagulants, non-steroidal anti-inflammatory drugs, various fatty acids, and steroid hormones (Cheng et al., 2013; Dai et al., 2014; Sun et al., 2014; Zhang et al., 2018). CYP1A2 comprises ∼13% of all CYPs and metabolizes several commonly used drugs such as theophylline, tacrine, and even some important endogenous compounds (Ragia et al., 2014; Wang X. S. et al., 2015). CYP2D6 (∼2% of all CYPs) metabolizes ∼20% of all marketed drugs (Tracy et al., 2016). CYP2E1 (∼10% of all CYPs) plays an important role in the metabolism of alcohol, organic solvents, and carcinogens (Ye et al., 2015; Xu et al., 2017; Jin et al., 2018). Unlike other CYPs, CYP2B6 does not play a major role in drug metabolism, but is known to metabolize bupropion and methadone and is believed to increase the risk of breast cancer (Justenhoven et al., 2014; Yuce-Artun et al., 2014; Grangeon et al., 2017).
The cocktail method, involving the simultaneous use of several probe substrates, has been used to investigate DDIs and examine changes in CYP activity in drug metabolism (Spaggiari et al., 2016). The FDA recommends the study of DDIs for seven main isoenzymes—CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. Although the CYP2E1 substrate, chlorzoxazone, is not commonly used as a cocktail probe because both chlorzoxazone and its metabolites are mainly detected in the negative ion mode of electrospray ionization (ESI), many studies have included it as a component of their cocktail approach (De Andrés et al., 2014; Tanaka et al., 2014).
Poziotinib (HM781-36B) is a new, orally active, irreversible pan-HER tyrosine kinase inhibitor (Kim et al., 2013). Poziotinib is currently undergoing clinical trials for various indications such as non-small cell lung cancer, breast cancer, and gastric cancer. Common adverse reactions caused by poziotinib treatment include rash, diarrhea, and itching (He et al., 2019; Kim T. Y. et al., 2019). In addition, in recent years, studies have found that poziotinib has a therapeutic effect on lung cancer with EGFR and HER2 exon 20 mutations (Koga et al., 2018). Analyses of its metabolites by LC-MS/MS reveal that its two main metabolites, M1 (dihydroxylation) and M2 (demethylation), are mainly produced by CYP3A4 and CYP2D6 activity (Kim et al., 2013). This evidence shows that poziotinib is mainly metabolized by the liver; however, the effects of poziotinib on CYP450 isoenzymes have not been reported.
It is extremely important to select the appropriate probe substrate for each CYP450 enzyme. Studies reporting the cocktail probe approach have used phenacetin for CYP1A2, bupropion for CYP2B6, tolbutamide for CYP2C9, dextromethorphan for CYP2D6, chlorzoxazone for CYP2E1, and midazolam for CYP3A4 (Palmer et al., 2001; Spaggiari et al., 2014; Giri et al., 2019; Kim H. J. et al., 2019; Chen A. et al., 2016). Therefore, we aimed to investigate the interaction of poziotinib with a cocktail of midazolam, bupropion, dextromethorphan, tolbutamide, chlorzoxazone, and phenacetin and examine their metabolites in rat plasma by UPLC-MS/MS, and compare the effects of poziotinib on cyp1a2, cyp2b1, cyp2c11, cyp2d1, cyp2e1, and cyp3a1/2 in vivo and in vitro. The results of this study will help to understand the potential drug interactions of poziotinib.
Poziotinib (purity > 98%) was purchased from the Beijing Sunflower and Technology Development Co. Ltd. (Beijing, China). Bupropion hydrochloride (purity > 98%) and phenacetin (purity > 99%) were purchased from TCI Chemicals (Shanghai, China). Tolbutamide (purity > 97.5%) and chlorzoxazone (purity > 98%) were purchased from J&K Chemical Ltd. (Shanghai China). α-Naphthoflavone (purity > 98%), Ticlopidine (purity > 98%), sulfaphenazole (purity > 98%), quinidine (purity > 98%), sodium diethyldithiocarbamatre (purity > 97%), ketoconazole (purity > 99%) were purchased from Aladdin. Midazolam injection was obtained from Nhwa Pharma. Co. (Jiangsu, China) and NADPH from Roche (Mannheim, Germany). The internal standard (IS) of diazepam (purity > 98%) was purchased from the Tianjin KingYork Pharmaceutical Co. Ltd. (Tianjin, China). Chromatographic grade methanol and acetonitrile were purchased from Merck Co. Ltd. (Darmstadt, Germany). Rat liver microsomes (RLMs) were prepared in our laboratory (Zhou et al., 2020). All other chemicals were of analytical grade or higher.
Samples were analyzed by a UPLC-MS/MS system equipped with a triple quadrupole mass spectrometer (Waters Corp., Milford, MA, United States) and Acquity UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm) at 40°C. The mobile phase consisted of acetonitrile (A) and 0.1% formic acid (B) with a linear gradient elution at 0.4 ml/min for 3 min. The elution procedure was as follows: 0.0–0.6 min (rapidly rising from 10 to 50% A), 0.6–1 min (increasing from 50 to 80% A), 1–2 min (increasing from 80 to 95% A), 2–2.5 min (maintained at 95% A), and 2.5–2.6 min (reduced to 10% A).
Mass spectrometer contained a triple quadrupole and an ESI source. The optimized parameters of multiple reaction monitoring detection included a source temperature of 150°C and desolvation temperature of 500°C. The MS conditions, chemical structure, and chromatograms for the probe drugs and IS are presented in Table 1 and in Supplementary Figures S1, S2 (Wang et al., 2020). All sample data were acquired with MassLynx 4.1 software (Waters Corp.).
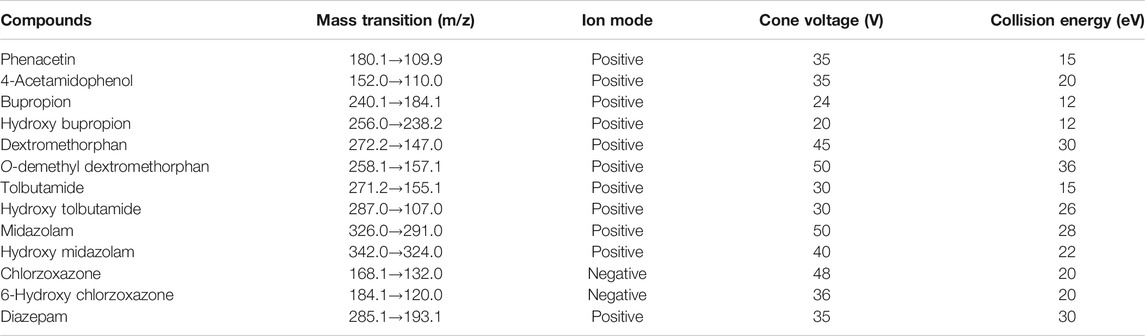
TABLE 1. Mass spectrometric parameters for six substrate probes, their metabolites, and diazepam.
The cocktail method was employed to analyze the effects of poziotinib on cyp1a2, cyp2b1, cyp2c11, cyp2d1, cyp2e1, and cyp3a1/2. First, 100 μM poziotinib was used to test its direct inhibitory effects RLMs. Then half-maximal inhibitory concentration (IC50) of poziotinib was determined with all of the CYPs. Phenacetin, bupropion, tolbutamide, dextromethorphan, chlorzoxazone, and midazolam (40, 20, 100, 10, 40, and 5 μM, respectively) were mixed to form a cocktail of probe substrates for measuring CYP450 activity. To this cocktail, 1 mg/ml of RLMs, 100 mM potassium phosphate buffer (pH 7.4), poziotinib (1, 2.5, 5, 10, 25, 50, or 100 μM) were added to obtain a final volume of 200 μL. After pre-incubation of this mixture for 5 min in a shaking water bath at 37°C, 1 mM NADPH was added to start the reaction. The reactions were allowed to proceed for 30 min, after which 200 μL of acetonitrile and 20 μL of IS were added to terminate the reactions. The mixtures were vortexed for 30 s and centrifuged again at 13,000 rpm for 5 min. Then, 2 μL of the supernatant was injected into the UPLC-MS/MS system for analysis.
To elucidate the mechanism of inhibition of cyp2b1 and 2c11 by poziotinib, we examined the interaction of different concentrations of poziotinib with bupropion and tolbutamide, respectively. Poziotinib was preincubated with 1 mg/ml of RLMs and probe substrate in a 37°C shaking water bath for 5 min, following which NADPH was added and incubated for 30 min. The concentrations of poziotinib were 0, 2, 4, 8 and 16 μM for 2b1, 0, 5, 10 20 and 40 μM for 2c11. The concentrations of bupropion were 5, 10, 20, and 40 μM, and the concentrations of tolbutamide were 25, 50, 100, and 200 μM. Poziotinib was incubated with different concentrations of the probe substrates in triplicate. The reactions were carried out, stopped, and processed as described in 2.3. Lineweaver-Burk plots were graphed.
A mixture containing 1 mg/ml of RLMs, different concentrations of poziotinib (0, 2, 4, 8 and16 μM for 2b1; 0, 5, 10 20 and 40 μM for 2c11), and NADPH was pre-incubated at 37°C for 0, 5, 10, and 20 min; Methanol instead of poziotinib was performed as control. Then 20 μM bupropion or 100 μM tolbutamide was added and incubated at 37°C for 30 min. The reactions were carried out and analyzed in triplicate as described in 2.3.
Male Sprague-Dawley rats were supplied by the Animal Experimental Center of the Wenzhou Medical University (Wenzhou, China). Animals were kept under standard conditions with a 12-h day-night cycle and were provided water and food ad libitum for 2 weeks for acclimation. All of the experimental procedures followed the guidelines for the care and use of laboratory animals and were approved (No. wydw 2019-650) by the Animal Experimental Ethical Inspection of Laboratory Animal Center, Wenzhou Medical University.
Twelve male Sprague-Dawley rats (230 ± 20 g) were randomly divided into treated and control groups, with 6 in each group. Dose of probe substrate mixture (phenacetin, bupropion, midazolam, chlorzoxazone and dextromethorphan) was dissolved in 0.5% CMC-Na solution: 10 mg/kg, Toluamide: 1 mg/kg. After 7 days of continuous oral administration of poziotinib (5 mg/kg, dissolved in 0.5% CMC-Na solution) to the experimental group, the two group rats were treated with an oral gavage of probe substrate mixture. After 0.083, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h following administration of poziotinib, 300 μL blood was collected from the tail vein of each rat, transferred to a heparinized centrifuge tube, and immediately centrifuged at 4,000 rpm for 10 min. The supernatant was mixed with 200 μL of acetonitrile and 20 μL of 0.5 μg/ml diazepam (IS), vortexed for 30 s, centrifuged at 13,000 rpm for 5 min, and the supernatant was injected into the UPLC-MS/MS system for analysis.
Pharmacokinetic parameters were calculated using DAS software (Version 3.2.8). SPSS software (version 16.0; SPSS Inc., Chicago, IL, United States) was used to analyze the significance of differences between the main pharmacokinetic parameters. p < 0.05 was considered statistically significant. GraphPad (version 8.0; GraphPad Software Inc., San Diego, CA, United States) was used to calculate IC50 and plot the plasma concentration-time curves and inhibition plots (type of inhibition, time- and concentration-dependence).
To examine the effects of poziotinib on the six CYPs, different concentrations of poziotinib and the cocktail of all probe substrates were incubated with RLMs to determine the IC50 values of the six probe substrates (Figure 1). The IC50 values of bupropion, tolbutamide, dextromethorphan, phenacetin, chlorzoxazone and midazolam in RLMs were 8.79, 20.17, 61.96, 128.2, 156.3, and 302.7 μM respectively. Poziotinib showed weak inhibition toward cyp2d1 (Figure 2D). In contrast, the inhibitory effects of poziotinib on cyp2b1 and 2c11 were strong (Figures 2B,C). Poziotinib did not show any obvious inhibitory effect on cyp1a2 (Figure 2A), cyp2e1 (Figure 2E) or CYP3a1/2 (Figure 2F). As these results showed that pozitinib could significantly inhibit the activities of cyp2b1 and 2c11 in vitro.
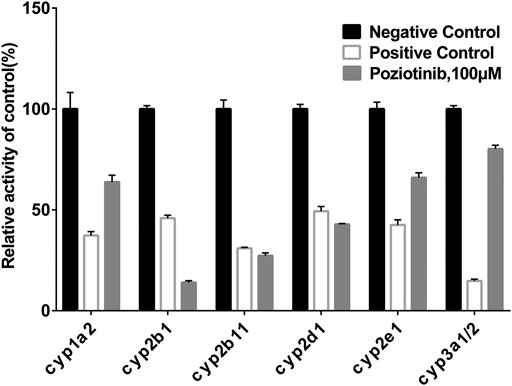
FIGURE 1. Inhibition of poziotinib on rat liver microsome. The concentrations of positive control inhibitors were as follows: 50 μM α-Naphthoflavone for cyp1a2, 50 μM Ticlopidine for cyp2b1, 20 μM sulfaphenazole for cyp2c11, 10 μM quinidine for cyp2d1, 100 μM sodium diethyldithiocarbamatre for cyp2e1, 1 μM ketoconazole for cyp3a1/2. Poziotinib was 100 μM. All data were mean ± SD of triplicate determinations.
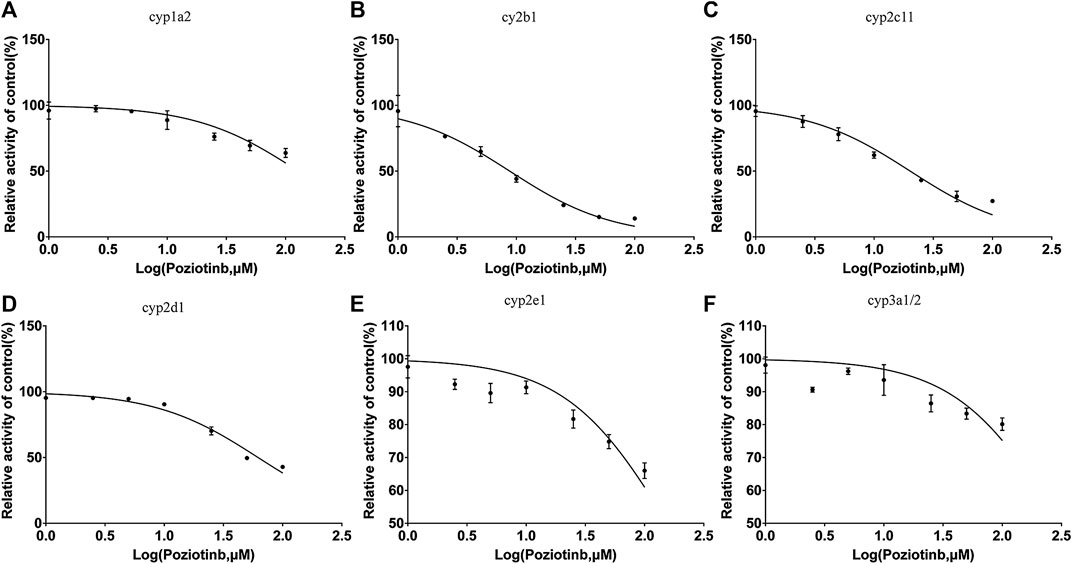
FIGURE 2. Determination of half-maximal inhibitory concentrations (IC50) of poziotinib using (A) 40 μM phenacetin for cyp1a2, (B) 20 μM bupropion for cyp2b1, (C)100 μM tolbutamide for cyp2c11, (D) 10 μM dextromethorphan for cyp2d1, (E) 40 μM chlorzoxazone for cyp2e1, and (F) 5 μM midazolam for cyp3a1/2 in rat liver microsomes. All data were mean ± SD of triplicate determinations.
We investigated the mechanism of the inhibitory effects of poziotinib on cyp2b1 and cyp2c11 activity by constructing Lineweaver-Burk plots (Figures 3A,B) and Michaelis-Menten saturation curves (Figures 4A,B). The Lineweaver–Burk plots show that all the straight lines intersect at a point on the Y axis (Figures 3A,B), indicating that the inhibition of 2b1 and 2c11 by poziotinib was competitive, with Ki values of 16.18 and 17.66 μM, respectively (Figures 3C,D).
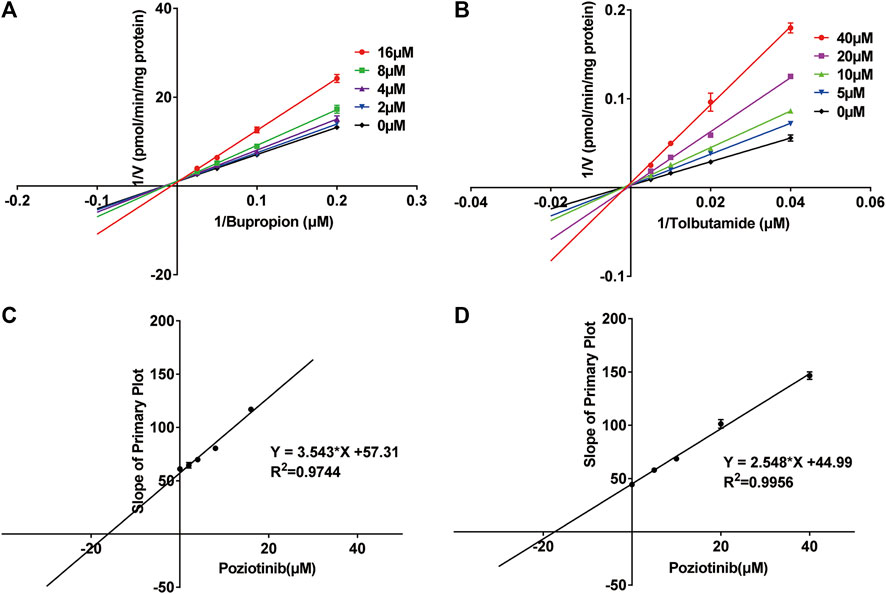
FIGURE 3. Primary Lineweaver–Burk plots of poziotinib (0, 2, 4, 8, 16 μM for bupropion, and 0, 5, 10, 20, 40 μM for tolbutamide) inhibited (A) bupropion (5, 10, 20 and 40 μM) and (B) tolbutamide (25, 50, 100 and 200 μM); Secondary plots for determination of Ki of (C) bupropion and (D) tolbutamide. All data were mean ± SD of triplicate determinations.
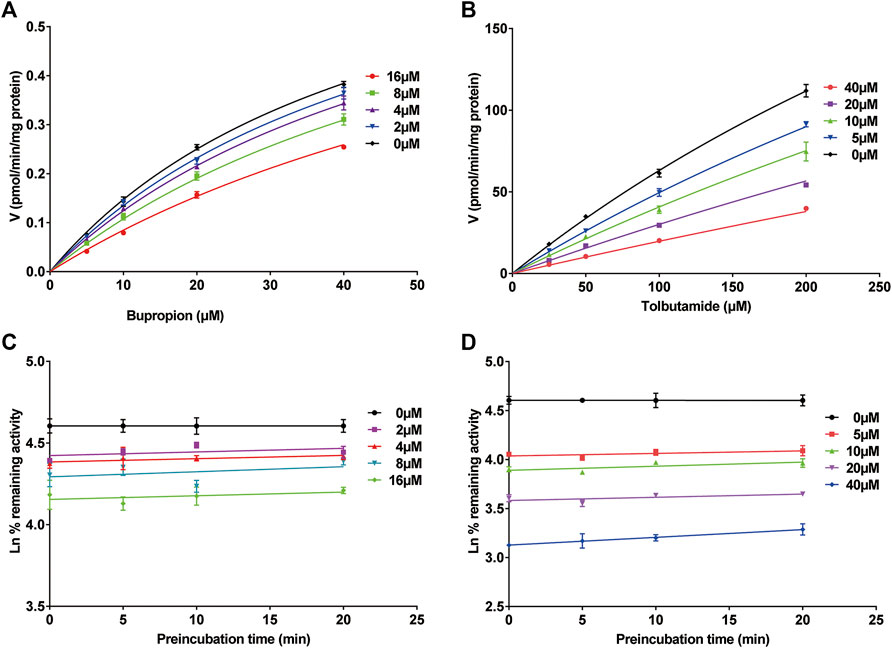
FIGURE 4. Michaelis-Menten saturation curves of (A) bupropion and (B) tolbutamide. Time- and concentration-dependent inhibition of poziotinib on rat liver microsome. Incubation reaction mixtures were pre-incubated for 0, 5, 10 and 20 min without or with poziotinib at 2, 4, 8 and 16 µM for bupropion (C) or 5, 10, 20, 40 µM for tolbutamide (D). All data were mean ± SD of triplicate determinations.
The inhibition of cyp2b1 and 2c11 by poziotinib was reversible, and poziotinib had no time- or concentration-dependent inhibitory effects on cyp2b1 and cyp2c11 (Figures 4C,D).
The pharmacokinetic parameters of phenacetin were used to describe the inhibitory effects of poziotinib on cyp1a2 activity. The area under the plasma concentration-time curve of phenacetin (Figure 5A) in the treated group was lower than that of the control group. The main pharmacokinetic parameters of phenacetin in the treated group were significantly different (p < 0.05) from those of the control group (Table 2). Compared with the control group, the AUC (0–t), AUC (0–∞), and Cmax values of the treated group were significantly reduced by 56.08, 55.81, and 48.06%, respectively. In addition, t1/2, Vz/F, and CLz/F values were significantly increased by 45.45, 203.82, and 116.85%, respectively. The higher AUC and Cmax values and lower Tmax values of the control group indicated that poziotinib induces cyp1a2 activity, thus accelerating the metabolism of phenacetin and reducing its plasma drug concentration in rats.
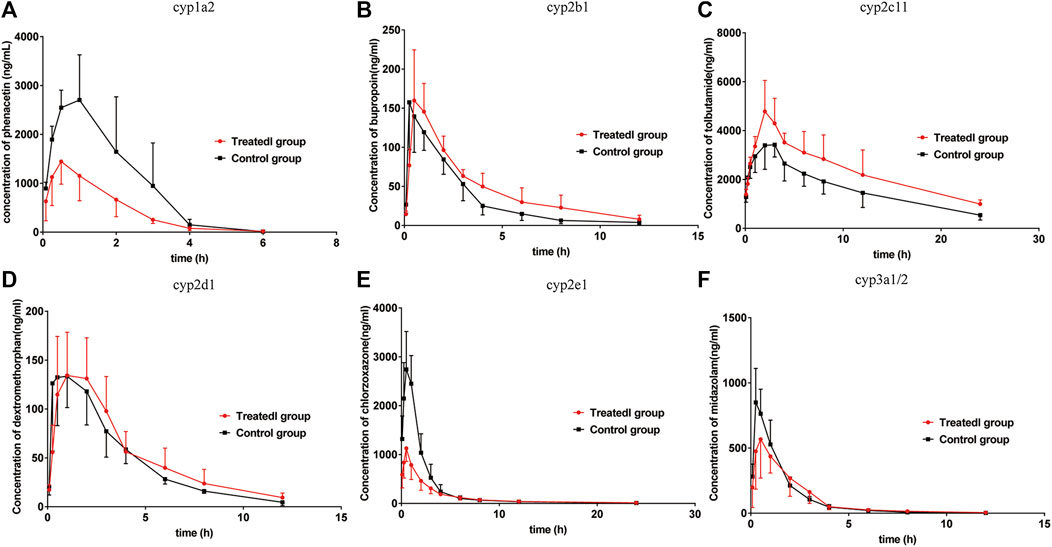
FIGURE 5. Mean plasma concentration-time curves of (A) phenacetin (CYP1A2), (B) bupropion (cyp2b1), (C) tolbutamide (cyp2c11), (D) dextromethorphan (cyp2d1), (E) chlorzoxazone (cyp2e1), and (F) midazolam (cyp3a1/2) in rats (mean ± SD, n = 6).
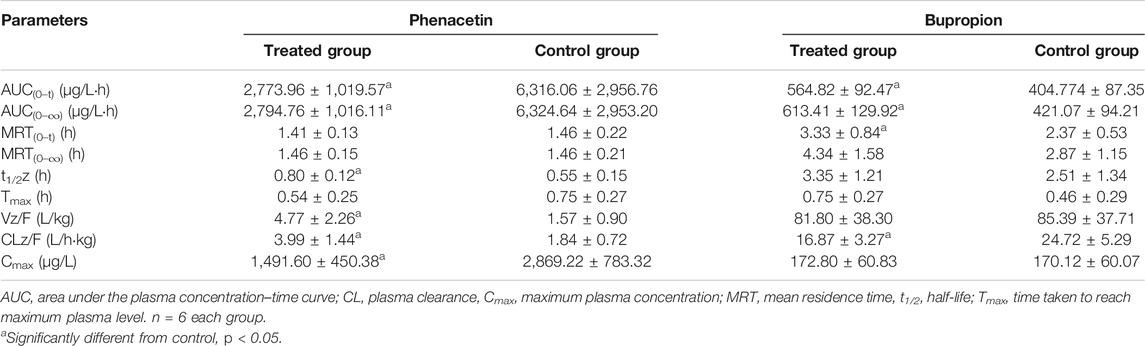
TABLE 2. Primary pharmacokinetic parameters after oral administration of phenacetin and bupropion in rats.
The mean plasma concentration-time curves of bupropion in the treated and control groups show that the AUC of the treated group was greater than that of the control group (Figure 5B). The AUC(0–t), AUC(0–∞), MRT(0–t), and Tmax values in the treated group were 1.39, 1.46, 1.41, and 1.63-fold higher than those of the control group, respectively, whereas the CLz/F and Vz/F values were 1.47 and 1.04-fold lower than those of the control group, respectively (Table 2). Thus, poziotinib reduced the metabolism of bupropion in rats, leading to its accumulation, which indicates that poziotinib had an inhibitory effect on cyp2b1 activity in rats.
The AUC of tolbutamide in the treated group was higher than that of the control group (Figure 5C). AUC (0–t) and AUC (0–∞) values were significantly higher in the treated group than in the control group (p < 0.05), increasing by 45.87 and 51.60%, respectively, whereas CLz/F was significantly lower than control group by 50% (Table 3). Thus, we concluded that poziotinib had a significantly inhibitory effect on cyp2c11 activity in rats, leading to the accumulation of tolbutamide in rats.
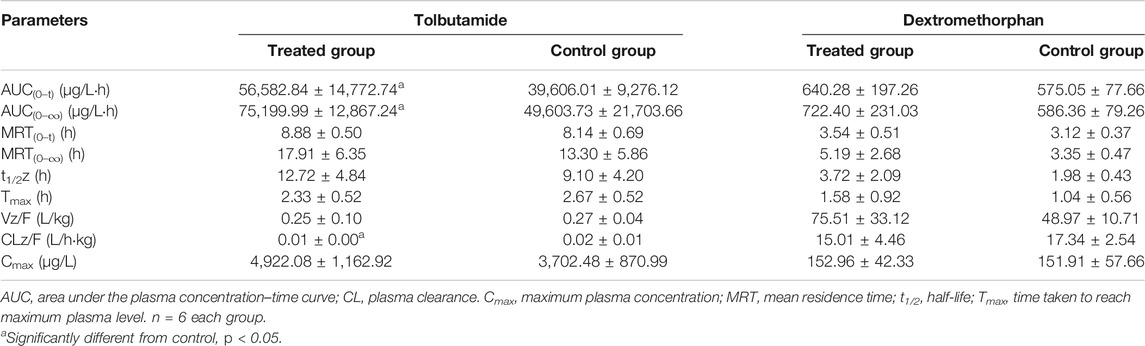
TABLE 3. Primary pharmacokinetic parameters after oral administration of tolbutamide and dextromethorphan in rats.
There was no obvious difference in the average plasma concentration-time curves of dextromethorphan in the treated and control groups (Figure 5D). There were no significant differences in the main pharmacokinetic parameters of dextromethorphan such as AUC, Cmax, Tmax, t1/2, MRT, and CLz/F values between the two groups of rats (Table 3). In summary, poziotinib had no obvious inhibitory effect on cyp2d1 activity in rats.
The AUC of chlorzoxazone in the treated group was lower than that of the control group (Figure 5E). The AUC(0–t), AUC(0–∞), and Cmax values of the treated group were significantly reduced to 1.95, 1.94 and 2.51-fold that of the control group, respectively. The MRT (0–t), MRT (0–∞), Vz/F, and CLz/F of the treated group were 1.60, 1.56, 1.75, and 1.96-fold higher than those of the control group (Table 4). We concluded that poziotinib induced cyp2e1 activity and accelerated the metabolism of chlorzoxazone in rats.
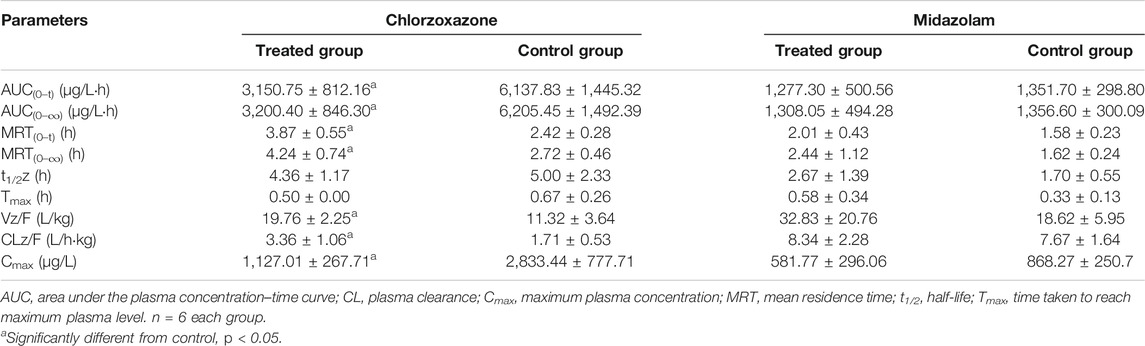
TABLE 4. Primary pharmacokinetic parameters after oral administration of chlorzoxazone and midazolam in rats.
The AUC of midazolam was lower in the treated group than in the control group (Figure 5F). The AUC and Cmax values of the control group were higher than those of the treated group, whereas the CLz/F value of the treated group was higher than that of the control group (Table 4), but these differences were not significant. Therefore, we concluded that poziotinib had no significant inhibitory effect on cyp3a1/2 activity in rats.
This study is the first to examine the induction/inhibition effects of poziotinib on CYP enzyme activity in RLMs by using six substrate probes and investigate the interactions of poziotinib with the probe substrates by using the cocktail method with RLMs as well as in rats.
The results of in vitro incubation with RLMs showed that IC50 (bupropion) > IC50 (tolbutamide) > IC50 (dextromethorphan) > IC50 (phenacetin) > IC50 (chlorzoxazone) > IC50 (midazolam). According to literature, IC50 < 1 μM is considered to be strong inhibition, 1 μM < IC50 < 10 μM moderate inhibition, and IC50 > 10 μM weak inhibition (Jin et al., 2015). Our results show that poziotinib has moderate and weak inhibitory effects on CYP2B1 (IC50 = 8.79 μM) and CYP2C11 (IC50 = 20.17 μM), respectively, whereas its inhibitory effect on the other four CYPs was not obvious (IC50 of cyp1a2, cyp2d1, cyp2e1 and cyp3a1/2 were 128.2, 61.96, 156.3, and 302.7 μM, respectively). A more detailed investigation of the inhibitory effect of poziotinib on cyp2b1 and 2c11 revealed that the inhibition of both cyp2b1 and 2c11 by poziotinib was competitive, with Ki values of 16.18 and 17.66 μM, respectively.
As the approved EGFR inhibitor, poziotinib is a quinazoline derivative, same as afatinib, erlotinib, gefitinib, dacomitinib. The above quinazoline derivatives are all metabolized by CYP 2D6 and CYP 3A4, however, they have different impacts on CYPs activity. Afatinib has no inhibition or induction on cytochrome P450 (CYP) enzymes (Dungo et al., 2013). Erlotinib may inhibit the activity of cyp2b1 and cyp3a1/2 in rats (Ma et al., 2015), and inhibit warfarin metabolism, a CYP2C9 substrate, in human (Hiraide et al., 2019). Gefitinib not only inhibit CYP2C9, but also CYP2D6 (Prommer, 2004; Hiraide et al., 2019). A similar, dacomitinib is a potent inhibitor of CY2D6 (Shirley, 2018). In our study, poziotinib inhibits cyp2b1 and cyp2c11 in RLM and in rats with competitive model, and induces cyp1a2 and cyp2e1 activities in rats.
This competitive inhibition indicates that the inhibitor, poziotinib, and the enzyme substrate compete at the binding site, thereby interfering with the binding of the substrate to the enzyme and reducing enzymatic activity. In addition, we did not observe any time- and concentration-dependent inhibitory effects of poziotinib on cyp2b1 and 2c11 in RLMs, which implies that the activity of these enzymes did not change over time during the incubation period. These results indicate that cyp2b1 and cyp2c11 were reversibly inhibited by poziotinib. Therefore, attention should be paid to the combined use of poziotinib and drugs to look for possible interactions of poziotinib with drugs that are metabolized by cyp2b1 or cyp2c11 to prevent the occurrence of serious adverse reactions.
We compared pharmacokinetic parameters of the six CYP probe substrates as a result of their interaction with poziotinib in rats. For cyp2b1 and bupropion, the AUC(0–t) and CLz/F values of the treated group were 1.40 and 0.68-fold that of the control group values, respectively. Similarly, for cyp2c11 and tolbutamide, the AUC(0–t) and CLz/F values of the treated group were 1.43 and 0.5-fold that of the control group values, respectively. These results indicate that the activity of cyp2b1 and 2c11 was inhibited by poziotinib, which resulted in increased systemic exposure of bupropion and tolbutamide in rats and slower clearance rates. This was consistent with the results of the in vitro incubation with RLMs, confirming the inhibitory effect of poziotinib on the activity of cyp2b1 and 2c11.
This inhibition could be attributed to the direct binding of poziotinib to the substrate binding site in the enzymes, leading to a decrease in the metabolic activity of these two enzymes. Another study had postulated that long-term use of poziotinib may affect the expression of metabolic enzymes (Wang et al., 2018). In addition, we found that poziotinib induced cyp1a2 and cyp2e1 activity with the two probe substrates, phenacetin and chlorzoxazone, respectively, because the AUC and Cmax values in the treated groups were significantly lower than those in the respective control groups, whereas CLz/F values were much higher than those in the control groups. Thus, poziotinib accelerated the elimination rates and reduced the plasma concentrations of phenacetin and chlorzoxazone in rats. However, our in vitro results did not reveal any inhibitory effect of poziotinib on cyp1a2 and 2e1. This could imply that the observed induction in rats may be caused by gene regulation, which increased protein synthesis and accelerated the metabolism of the probe substrates; however, further research is needed to confirm this hypothesis (Yan, 2012). Poziotinib had no inhibitory effect on the enzymatic activity of cyp2d1 and cyp3a1/2 in vivo or in vitro.
In conclusion, this study shows that poziotinib competitively and reversibly inhibited the metabolism of cyp2b1 and cyp2c11 substrates to varying degrees without any time- or concentration dependence. Poziotinib had no obvious inhibitory effect on the other four CYPs. The results of this study will help in further study of poziotinib in humans and draw attention to its potential DDIs when combined with other drugs metabolized by cyp2b1 and cyp2c11 to enable clinically effective and safe use of poziotinib.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Animal Experimental Ethical Inspection of Laboratory Animal Center, Wenzhou Medical University.
SW and QZ contributed to the conception and design of the study; FC, HJ, JX, PG, and JW performed experiments; DD performed the statistical analysis; FC and JW wrote the first draft of the manuscript; SW and JH wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version. FC is accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
This work was supported by grants funded by Public Welfare Technology Research Funding Project of Zhejiang (LGD19H090001 and LGD20H060001), High-level talent training project of Lishui (2018RC18), Medical and Health Research Project of Zhejiang province (2017KY731 and 2019ZYY10), City-level public welfare technology application research project of Lishui (2017GYX15) Ningbo Public Welfare Science and Technology Project (2019C50072), Scientific Research Fund of Zhejiang Provincial Education Department (Y201940895), and CAMS Innovation Fund for Medical Sciences (2018-I2M-1-002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2020.593518/full#supplementary-material.
Carrão, D. B., de Oliveira, A. R., and Magalhães, I. R. (2018). Challenges of probe cocktail approach for human drug-drug interaction assays. Bioanalysis 10, 1969–1972. doi:10.4155/bio-2018-0247
Chen, A., Zhou, X., Tang, S., Liu, M., and Wang, X. (2016). Evaluation of the inhibition potential of plumbagin against cytochrome P450 using LC-MS/MS and cocktail approach. Sci. Rep. 6, 28482. doi:10.1038/srep28482
Chen, Z. H., Zhang, S. X., Long, N., Lin, L. S., Chen, T., Zhang, F. P., et al. (2016). An improved substrate cocktail for assessing direct inhibition and time-dependent inhibition of multiple cytochrome P450s. Acta Pharmacol. Sin. 37, 708–718. doi:10.1038/aps.2016.10
Cheng, Y., Huang, Y., Tian, Y., Xu, L., Liu, G. Q., and Zahng, Z. J. (2013). Assessment of the effects of Radix bupleuri and vinegar-baked Radix bupleuri on cytochrome 450 activity by a six-drug cocktail approach. Chin. J. Nat. Med. 11, 302–308. doi:10.1016/s1875-5364(13)60033-3
Dai, D. P., Wang, S. H., Geng, P. W., Hu, G. X., and Cai, J. P. (2014). In vitro assessment of 36 CYP2C11 allelic isoforms found in the Chinese population on the metabolism of glimepiride. Basic Clin. Pharmacol. Toxicol. 114, 305–310. doi:10.1111/bcpt.12159
De Andrés, F., Sosa-Macías, M., and Llerena, A. (2014). A rapid and simple LC-MS/MS method for the simultaneous evaluation of CYP1A2, CYP2C11, CYP2C19, CYP2D6 and CYP3A1/2 hydroxylation capacity. Bioanalysis 6, 683–696. doi:10.4155/bio.14.20
Dungo, R. T., and Keating, G. M. (2013). Afatinib: first global approval. Drugs 73, 1503–1515. doi:10.1007/s40265-013-0111-6
Giri, P., Patel, H., and Srinivas, N. R. (2019). Use of cocktail probe drugs for indexing cytochrome P450 enzymes in clinical pharmacology studies - review of case studies. Drug Metabol. Lett. 13, 3–18. doi:10.2174/1872312812666181119154734
Grangeon, A., Gravel, S., Gaudette, F., Turgeon, J., and Michaud, V. (2017). Highly sensitive LC-MS/MS methods for the determination of seven human CYP450 activities using small oral doses of probe-drugs in human. J. Chromatogr. B 1040, 144–158. doi:10.1016/j.jchromb.2016.12.006
Guidoni, C. M., Camargo, H. P., Obreli-Neto, P. R., Girotto, E., and Pereira, L. R. (2016). Study of warfarin utilization in hospitalized patients: analysis of possible drug interactions. Int. J. Clin. Pharm. 38, 1048–1051. doi:10.1007/s11096-016-0336-z
He, Y., Chen, F., Wang, B., Fan, Q., Li, H., Hua, A., et al. (2019). Dete pharmacokinetic study. Lat. Am. J. Pharm. 38, 310–315.
Hiraide, M., Minowa, Y., Nakano, Y., Suzuki, K., Shiga, T., Nishio, M., et al. (2019). Drug interactions between tyrosine kinase inhibitors (gefitinib and erlotinib) and warfarin: assessment of international normalized ratio elevation characteristics and in vitro CYP2C9 activity. J. Oncol. Pharm. Pract. 25, 1599–1607. doi:10.1177/1078155218801061
Jin, C., He, X., Zhang, F., He, L., Chen, J., Wang, L., et al. (2015). Inhibitory mechanisms of celastrol on human liver cytochrome P450 1A2, 2C19, 2D6, 2E1 and 3A1/2. Xenobiotica 45, 571–577. doi:10.3109/00498254.2014.1003113
Jin, S. E., Ha, H., Seo, C. S., Shin, H. K., and Jeong, S. J. (2018). Expression of hepatic cytochrome P450s in rats administered with guibi-tang, a traditional herbal formula. Phcog. Mag. 13, S822‒s827. doi:10.4103/pm.pm_107_17
Jo, J. J., Jo, J. H., Kim, S., Lee, J. M., and Lee, S. (2018). Development of a simultaneous LC-MS/MS method to predict in vivo drug-drug interaction in mice. Arch Pharm. Res. (Seoul) 41, 450–458. doi:10.1007/s12272-018-1012-8
Justenhoven, C., Pentimalli, D., Rabstein, S., Harth, V., Lotz, A., Pesch, B., et al. (2014). CYP2B1*6 is associated with increased breast cancer risk. Int. J. Canc. 134, 426–430. doi:10.1002/ijc.28356
Kim, E., Kim, H., Suh, K., Kwon, S., Lee, G., Park, N. H., et al. (2013). Metabolite identification of a new tyrosine kinase inhibitor, HM781-36B, and a pharmacokinetic study by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 27, 1183–1195. doi:10.1002/rcm.6559
Kim, H. J., Lee, H., Ji, H. K., Lee, T., and Liu, K. H. (2019). Screening of ten cytochrome P450 enzyme activities with 12 probe substrates in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Biopharm Drug Dispos. 40, 101–111. doi:10.1002/bdd.2174
Kim, T. Y., Han, H. S., Lee, K. W., Zang, D. Y., Rha, S. Y., Park, Y. I., et al. (2019). A phase I/II study of poziotinib combined with paclitaxel and trastuzumab in patients with HER2-positive advanced gastric cancer. Gastric Cancer 22, 1206–1214. doi:10.1007/s10120-019-00958-4
Koga, T., Kobayashi, Y., Tomizawa, K., Suda, K., Kosaka, T., Sesumi, Y., et al. (2018). Activity of a novel HER2 inhibitor, poziotinib, for HER2 exon 20 mutations in lung cancer and mechanism of acquired resistance: an in vitro study. Lung Cancer 126, 72–79. doi:10.1016/j.lungcan.2018.10.019
Lee, B., Ji, H. K., Lee, T., and Liu, K. H. (2015). Simultaneous screening of activities of five cytochrome P450 and four uridine 5'-diphospho-glucuronosyltransferase enzymes in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Drug Metab. Dispos. 43, 1137–1146. doi:10.1124/dmd.114.063016
Liu, L., Sun, S., Rui, H., and Li, X. (2017). In vitro inhibitory effects of dihydromyricetin on human liver cytochrome P450 enzymes. Pharm. Biol. 55, 1868–1874. doi:10.1080/13880209.2017.1339284
Loue, C., and Tod, M. (2014). Reliability and extension of quantitative prediction of CYP3A1/2-mediated drug interactions based on clinical data. AAPS J. 16, 1309–1320. doi:10.1208/s12248-014-9663-y
Lu, Y. Y., Du, Z. Y., Li, Y., Wang, J. L., Zhao, M. B., Jiang, Y., et al. (2018). Effects of Baoyuan decoction, a traditional Chinese medicine formula, on the activities and mRNA expression of seven CYP isozymes in rats. J. Ethnopharmacol. 225, 327–335. doi:10.1016/j.jep.2018.07.023
Ma, J., Wang, S., Zhang, M., Zhang, Q., Zhou, Y., Lin, C., et al. (2015). Simultaneous determination of bupropion, metroprolol, midazolam, phenacetin, omeprazole and tolbutamide in rat plasma by UPLC-MS/MS and its application to cytochrome P450 activity study in rats. Biomed. Chromatogr. 29, 1203–1212. doi:10.1002/bmc.3409
Palmer, J. L., Scott, R. J., Gibson, A., Dickins, M., and Pleasance, S. (2001). An interaction between the cytochrome P450 probe substrates chlorzoxazone (CYP2E1) and midazolam (CYP3A). Br. J. Clin. Pharmacol. 52, 555–561. doi:10.1046/j.0306-5251.2001.01479.x
Polasek, T. M., Lin, F. P., Miners, J. O., and Doogue, M. P. (2011). Perpetrators of pharmacokinetic drug-drug interactions arising from altered cytochrome P450 activity: a criteria-based assessment. Br. J. Clin. Pharmacol. 71, 727–736. doi:10.1111/j.1365-2125.2011.03903.x
Prommer, E. (2004). Gefitinib: a new agent in palliative care. Am. J. Hosp. Palliat. Care 21, 222–227. doi:10.1177/104990910402100313
Ragia, G., Giannakopoulou, E., Karaglani, M., Karantza, I. M., Tavridou, A., and Manolopoulos, V. G. (2014). Frequency of CYP450 enzyme gene polymorphisms in the Greek population: review of the literature, original findings and clinical significance. Drug Metabol. Drug Interact. 29, 235–248. doi:10.1515/dmdi-2014-0006
Shirley, M. (2018). Dacomitinib: first global approval. Drugs 78, 1947–1953. doi:10.1007/s40265-018-1028-x
Spaggiari, D., Daali, Y., and Rudaz, S. (2016). An extensive cocktail approach for rapid risk assessment of in vitro CYP450 direct reversible inhibition by xenobiotic exposure. Toxicol. Appl. Pharmacol. 302, 41–51. doi:10.1016/j.taap.2016.04.013
Spaggiari, D., Geiser, L., Daali, Y., and Rudaz, S. (2014). A cocktail approach for assessing the in vitro activity of human cytochrome P450s: an overview of current methodologies. J. Pharmaceut. Biomed. Anal. 101, 221–237. doi:10.1016/j.jpba.2014.03.018
Sun, M., Tang, Y., Ding, T., Liu, M., and Wang, X. (2014). Inhibitory effects of celastrol on rat liver cytochrome P450 1A2, 2C11, 2D6, 2E1 and 3A2 activity. Fitoterapia 92, 1–8. doi:10.1016/j.fitote.2013.10.004
Tanaka, S., Uchida, S., Inui, N., Takeuchi, K., Watanabe, H., and Namiki, N. (2014). Simultaneous LC-MS/MS analysis of the plasma concentratsions of a cocktail of 5 cytochrome P450 substrate drugs and their metabolites. Biol. Pharm. Bull. 37, 18–25. doi:10.1248/bpb.b13-00401
Tracy, T. S., Chaudhry, A. S., Prasad, B., Thummel, K. E., Schuetz, E. G., Zhong, X. B., et al. (2016). Interindividual variability in cytochrome P450-mediated drug metabolism. Drug Metab. Dispos. 44, 343–351. doi:10.1124/dmd.115.067900
Wang, B., Yang, S., Hu, J., and Li, Y. (2014). Multifaceted interaction of the traditional Chinese medicinal herb Schisandra chinensis with cytochrome P450-mediated drug metabolism in rats. J. Ethnopharmacol. 155, 1473–1482. doi:10.1016/j.jep.2014.07.026
Wang, S. H., Dong, Y. W., Su, K., Zhang, J., Wang, L. Y., Han, A. Y., et al. (2017). Effect of codeine on CYP450 isoform activity of rats. Pharm. Biol. 55, 1223–1227. doi:10.1080/13880209.2017.1297466
Wang, S. H., Wang, Z. Y., Chen, D. X., Chen, M. C., Lin, Y. Y., Liu, Z. Z., et al. (2015). Effect of acute paraquat poisoning on CYP450 isoforms activity in rats by cocktail method. Int. J. Clin. Exp. Med. 8, 19100–19106.
Wang, X. S., Hu, X. C., Chen, G. L., Yuan, X., Yang, R. N., Liang, S., et al. (2015). Effects of vitexin on the pharmacokinetics and mRNA expression of CYP isozymes in rats. Phytother Res. 29, 366–372. doi:10.1002/ptr.5260
Wang, Y., Jin, Y., Yun, X., Wang, M., Dai, Y., and Xia, Y. (2018). Co-administration with simvastatin or lovastatin alters the pharmacokinetic profile of sinomenine in rats through cytochrome P450-mediated pathways. Life Sci. 209, 228–235. doi:10.1016/j.lfs.2018.08.012
Wang, Y., Wang, C., Wang, S., Zhou, Q., Dai, D., Shi, J., et al. (2020). Cytochrome P450-based drug-drug interactions of vonoprazan in vitro and in vivo. Front. Pharmacol. 11, 53. doi:10.3389/fphar.2020.00053
Wang, Z., Sun, W., Lin, Z. F., Sun, R., Huang, C. K., Ye, W. J., et al. (2019). A UHPLC-MS/MS method coupled with liquid-liquid extraction for the quantitation of phenacetin, omeprazole, metoprolol, midazolam and their metabolites in rat plasma and its application to the study of four CYP450 activities. J. Pharmaceut. Biomed. Anal. 163, 204–210. doi:10.1016/j.jpba.2018.10.012
Xu, J., Ma, H. Y., Liang, S., Sun, M., Karin, G., Koyama, Y., et al. (2017). The role of human cytochrome P450 2E1 in liver inflammation and fibrosis. Hepatol. Commun. 1, 1043–1057. doi:10.1002/hep4.1115
Yan, W. (2012). In vitro assessment of CYP Inhibition, time-dependent inhibition in human liver mcrosomes and CYP Induction in human hepatocyte. Dissertation/Master’s thesis. Shanghai, China: Fudan University
Ye, X. H., Song, L., Peng, L., Bu, Z., Yan, S. X., Feng, J., et al. (2015). Association between the CYP2E1 polymorphisms and lung cancer risk: a meta-analysis. Mol. Genet. Genom. 290, 545–558. doi:10.1007/s00438-014-0941-2
Yin, S., Cheng, Y., Li, T., Dong, M., Zhao, H., and Liu, G. (2016). Effects of notoginsenoside R1 on CYP1A2, CYP2C11, CYP2D1, and CYP3A1/2 activities in rats by cocktail probe drugs. Pharm. Biol. 54, 231–236. doi:10.3109/13880209.2015.1029051
Yuce-Artun, N., Kose, G., and Suzen, H. S. (2014). Allele and genotype frequencies of CYP2B1 in a Turkish population. Mol. Biol. Rep. 41, 3891–3896. doi:10.1007/s11033-014-3256-9
Zhang, D., Wu, G. D., Zhang, Y. D., Xu, J. P., Zhen, H. T., and Li, M. H. (2018). Effects of Digeda-4 decoction on the CYP450 activities in rats using a cocktail method by HPLC. BioMed Res. Int. 2018, 1–9. doi:10.1155/2018/1415082
Keywords: pozioitinib, cancer, drug-drug interactions, inhibitor, cytochrome P450, cocktail method, pharmacokinetics
Citation: Wang J, Chen F, Jiang H, Xu J, Meng D, Geng P, Dai D, Hu J, Zhou Y, Zhou Q and Wang S (2021) Inhibition and Induction by Poziotinib of Different Rat Cytochrome P450 Enzymes In Vivo and in an In Vitro Cocktail Method. Front. Pharmacol. 11:593518. doi: 10.3389/fphar.2020.593518
Received: 10 August 2020; Accepted: 14 October 2020;
Published: 05 January 2021.
Edited by:
Jun Lu, Auckland University of Technology, New ZealandReviewed by:
Petr Pavek, Charles University, CzechiaCopyright © 2021 Wang, Chen, Jiang, Xu, Meng, Geng, Dai, Hu, Zhou, Zhou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Zhou, zhouquan1991@lsu.edu.cn; Shuanghu Wang, wangshuanghu@lsu.edu.cn
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.