
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 07 April 2020
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00441
This article is part of the Research TopicTargeting the PD-1/PD-L1 Cancer Immune Evasion Axis: Challenges and Emerging Strategies, Volume IView all 18 articles
 Luisa Chocarro de Erauso1Miren Zuazo1Hugo Arasanz1,2Ana Bocanegra1Carlos Hernandez1Gonzalo Fernandez1,2Maria Jesus Garcia-Granda1Ester Blanco1Ruth Vera2Grazyna Kochan1*David Escors1*
Luisa Chocarro de Erauso1Miren Zuazo1Hugo Arasanz1,2Ana Bocanegra1Carlos Hernandez1Gonzalo Fernandez1,2Maria Jesus Garcia-Granda1Ester Blanco1Ruth Vera2Grazyna Kochan1*David Escors1*Cancer immunotherapies targeting immune checkpoints such as programmed cell-death protein 1 (PD-1) and its ligand programmed cell-death 1 ligand 1 (PD-L1), are revolutionizing cancer treatment and transforming the practice of medical oncology. However, despite all the recent successes of this type of immunotherapies, most patients are still refractory and present either intrinsic resistance or acquired resistance. Either way, this is a major clinical problem and one of the most significant challenges in oncology. Therefore, the identification of biomarkers to predict clinical responses or for patient stratification by probability of response has become a clinical necessity. However, the mechanisms leading to PD-L1/PD-1 blockade resistance are still poorly understood. A deeper understanding of the basic mechanisms underlying resistance to cancer immunotherapies will provide insight for further development of novel strategies designed to overcome resistance and treatment failure. Here we discuss some of the major molecular mechanisms of resistance to PD-L1/PD-1 immune checkpoint blockade and argue whether tumor intrinsic or extrinsic factors constitute main determinants of response and resistance.
Cancer immunotherapies aim at stimulating the immune system of patients to reactivate its anti-oncogenic activities (Escors, 2014). The most successful anti-cancer immunotherapies are currently those based on immune checkpoint blockade with antibodies (ICIs). Under normal physiologic conditions, immune checkpoints function as regulators of excessive inflammation following T-cell activation, and mechanisms to prevent auto-reactive responses. Unfortunately many cancer cells exploit these T-cell inhibitory mechanisms by up-regulating the expression of immune checkpoint molecules that will bind their ligands on activated T cells leading to their inactivation. It is thought that ICI therapies act primarily on the reactivation of T lymphocytes to exert cytotoxic activities over cancer cells. The emergence of ICI therapies over the last decade has transformed to the core cancer treatments, as they show good efficacies, and less toxicity than conventional chemotherapy or targeted therapies. However, for most cancer types only a subset of all patients effectively respond to these therapies, which is a major clinical, economic, and ethical problem (Topalian et al., 2011; Nishino et al., 2017; Prasad et al., 2017; Kamada et al., 2019; Martins et al., 2019). It is often said that ICI therapies have revolutionized oncology, although their efficacy is still limited. But, what do we mean when we claim that ICI therapies have caused a revolution?
Before the success stories of ipilimumab (Hodi et al., 2008), and before the publication of the results from the first clinical trials of PD-L1/PD-1 blockers (Brahmer et al., 2012; Topalian et al., 2012), immunotherapies were not seriously considered as viable therapeutic options by most oncologists and pharmaceutical companies. Most of their efforts were directed towards the development of small molecule inhibitors for targeted therapies, or novel chemotherapies. And even though targeted therapies showed good efficacies, they were largely limited to patients with tumors harboring the targeted mutations. So, what did ICI treatments truly change? The truly astonishing result is that with only a single drug, objective responses were obtained in a very large number of cancer types largely independent of their ontogeny. Moreover, these drugs are not even directed towards the cancer cell. For example, the anti-PD-1 antibody pembrolizumab has achieved objective responses in cancers as different as melanoma, lung cancer, head and neck, urothelial, gastric cancer, mesothelioma, and Hodgkin lymphoma, among others.
The inhibitory co-receptors that modulate the activation of T cells are generally associated with the T-lymphocyte receptor (TCR) complex at the immunological synapse. These molecules constitute major control points and serve as targets to enhance antitumor immune responses. Some examples expressed in T cells are programmed cell-death protein 1 (PD-1), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), cytotoxic T-lymphocyte antigen 4 (CTLA-4), or lymphocyte-activation gene 3 (LAG-3) (Saito et al., 2010; Chen and Flies, 2013; Esensten et al., 2016; Schildberg et al., 2016; Lichtenegger et al., 2018). Several ICI antibodies targeting CTLA-4 or the PD-L1/PD-1 axis are approved for use by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for treatment of different cancer types. These antibodies have demonstrated clinical efficacy, with durable clinical responses. Due the success of blockade strategies of CTLA-4 and PD-1 pathways, several antibodies targeting other immune checkpoints are now at different stages of development. Moreover, several combination strategies with ICIs are under evaluation in clinical trials, emerging as new opportunities to enhance anti-tumor immunity (Table 1) (Pardoll, 2015).

Table 1 Clinical trials targeting the PD-L1/PD-1 axis and combinations.
Since 2012, antibodies blocking PD-1/PD-L1 interactions are demonstrating very promising results (Brahmer et al., 2012; Topalian et al., 2012), demonstrating their efficacies and safety. Truly, these results have no precedent in the history of cancer treatments due to their wide range of activities and the durability of responses. To date, six immune checkpoint inhibitors blocking the PD-L1/PD-1 axis are approved by the FDA and the EMA: three PD-1 inhibitors (nivolumab, pembrolizumab, and cemiplimab), and three PD-L1 inhibitors (atezolizumab, durvalumab, and avelumab). Most of them have also been approved by the Chinese National Medical Products Administration (NMPA), and by the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan. Additionally, the NMPA has recently approved the use of four more PD-1 inhibitors (toripalimab, tislelizumab sintilimab, and camrelizumab) in China. These drugs are indicated for the treatment of several cancer types such us melanoma, non–small cell lung cancer (NSCLC), renal cell carcinoma, head and neck squamous cell carcinoma, urothelial carcinoma, microsatellite instability–high colorectal cancer and metastatic cutaneous squamous cell carcinoma.
However, despite these successes the majority of patients in many cancer types do not truly benefit from PD-L1/PD-1 blockade therapies and show resistance, either intrinsic resistance when the treatment directly fails, or acquired resistance where a proportion of responders will also develop resistance. Other patients show some response in the form of stable disease, or acceleration of disease in the form of hyperprogression (Zuazo et al., 2018). Still, the specific mechanisms of resistance and response remain to be elucidated. Therefore, the understanding of the basic mechanistic pathways of resistance and the identification of predictive biomarkers of response have become a clinical necessity. Here, we review the current knowledge on resistance to PD-L1/PD-1 blockade therapies and discuss whether tumor intrinsic or extrinsic factors are the main determinants of response and resistance.
PD-1 (CD279) is a type 1 transmembrane glycoprotein from the B7-CD28 immunoglobulin superfamily discovered in 1992 for which Prof Honjo received the Nobel Prize (Ishida et al., 1992). This protein is encoded by Pdcd1 gene on the human chromosome 2, and it is composed of a short signal sequence, an extracellular IgV-like domain, a stalk region, a transmembrane domain, and an intracellular cytoplasmatic tail containing the two tyrosine-based signaling motifs; the immunoreceptor tyrosine-based inhibitory motif (ITIM) and the immunoreceptor tyrosine-based switch motif (ITSM) (Figure 1). These two motifs contribute to the inhibitory functions of PD-1. PD-1 has two main ligands, PD-L1 (B7-H1, CD274) and PD-L2 (B7-DC, CD273) (Dong et al., 1999; Freeman et al., 2000; Latchman et al., 2001; Tseng et al., 2001) (16–19). PD-L1 is a type I transmembrane protein encoded by the Cd274 gene on the human chromosome 9 discovered in 1999 as an additional member of the B7 family. PD-L1 is composed of a signal sequence, an IgV-like domain, an IgC-like domain, a transmembrane domain, and a highly conserved short intracellular region with intracellular signal transduction capacities (Pascolutti et al., 2016; Gato-Canas et al., 2017; Escors et al., 2018) (Figure 1). The intracellular domain presents three highly conserved sequence motifs, two of which are required for regulating interferon-mediated cytotoxicity (RMLDVEKC and DTSSK) (Gato-Canas et al., 2017; Escors et al., 2018). PD-L2 is a type I transmembrane protein encoded by the Pdcd1lg2 gene was discovered in 2001 (Latchman et al., 2001; Tseng et al., 2001) and exhibits a similar molecular oganization than PD-L1.
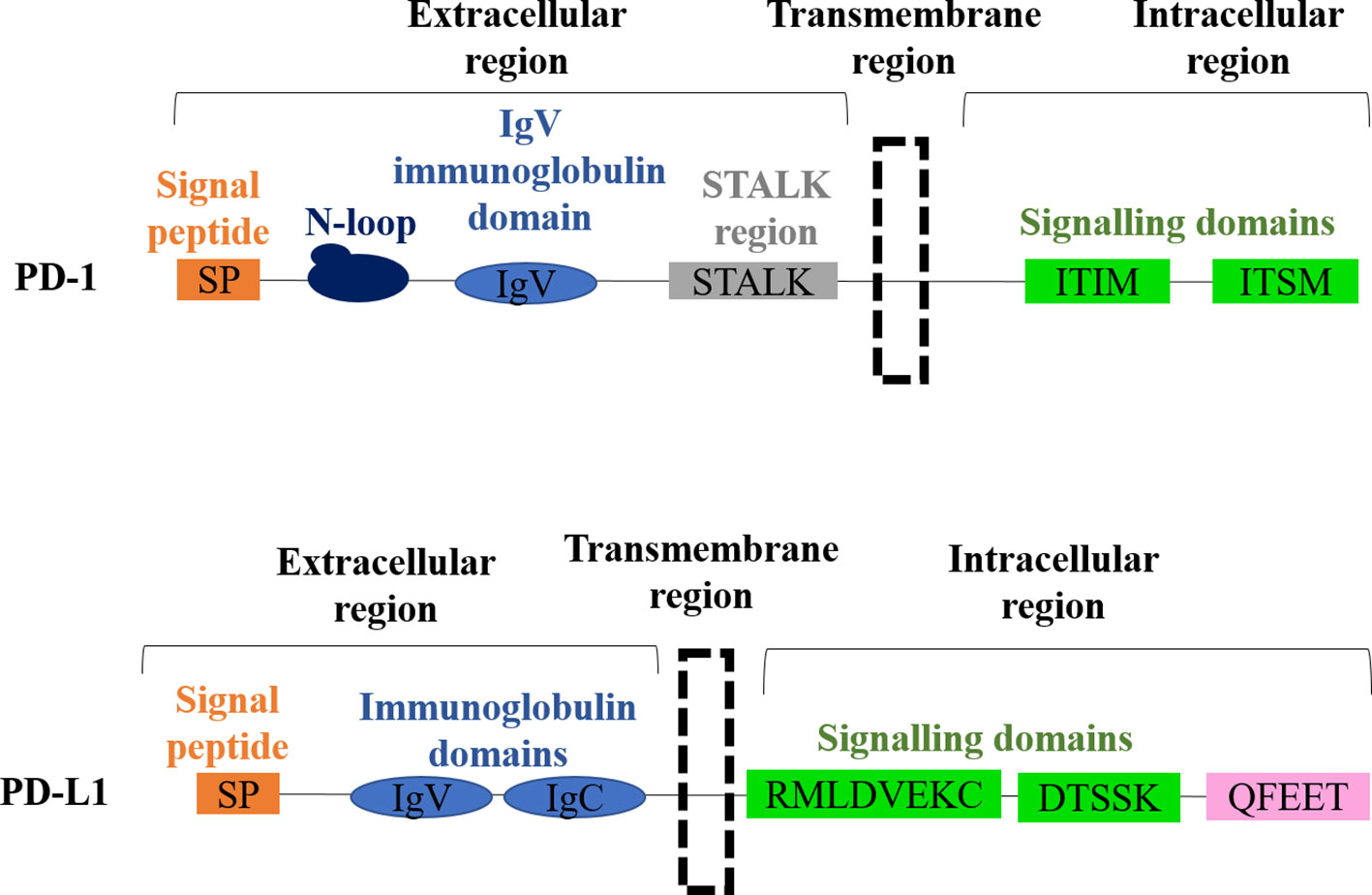
Figure 1 Molecular structures of PD-1 and PD-L1. The domain organization of PD-1 is shown on top, with each domain indicated. The domain organization of PD-L1 is shown below, with each domain indicated.
After engagement with PD-L1, PD-1 inhibits T cell functions through direct and indirect pathways (Arasanz et al., 2017) (Figure 2). Direct pathways are dependent on the recruitment of SHP-1 and SHP-2 phosphatases phosphatases to PD-1 ITIM and ITISM motifs following their tyrosine phosphorylation by Lck (Plas et al., 1996; Chemnitz et al., 2004; Sheppard et al., 2004; Hui et al., 2017). SHP phosphatases inhibit ZAP70 and PI3K activities by dephosphorylation, and thus ending the TCR-CD28 signal transduction and its downstream dependent intracellular pathways (ERK and PKCθ). PD-1 also inhibits T cell activities through indirect pathways. After engaged with PD-L1, PD-1 leads to increased expression of CBL E3 ubiquitin ligases, which ubiquitylate components of the TCR leading to its internalization and degradation (Karwacz et al., 2011; Karwacz et al., 2012; Liechtenstein et al., 2014). Also, an indirect pathway of PD-1-dependent inhibition of TCR signal transduction is caused when PD-L1 engages to PD-1 by inhibiting the transcription of CK2 through an unclear mechanism, resulting in de-phosphorylated PTEN that will in turn de-phosphorylate PI3K and terminating in this way downstream pathways (Patsoukis et al., 2013; Arasanz et al., 2017).
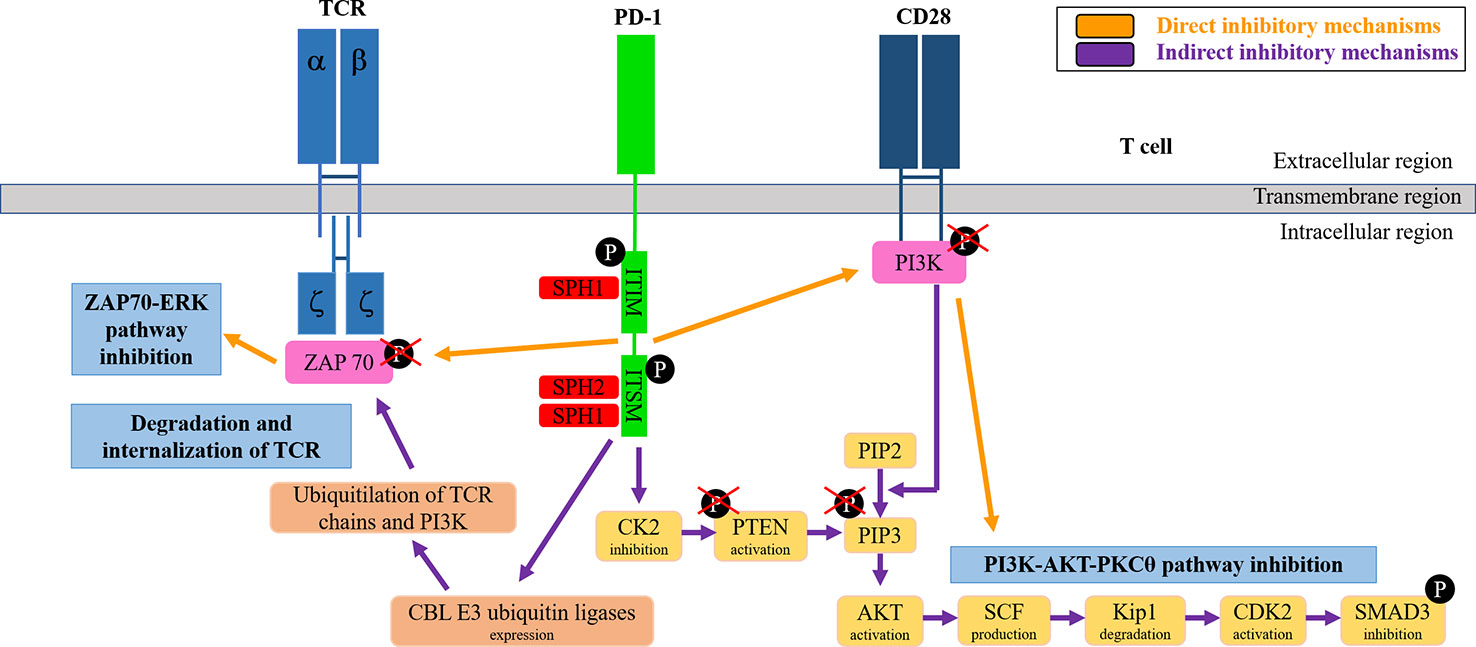
Figure 2 PD-1 signaling pathways in T cells. The figure schematically summarizes the direct and indirect T cell inhibitory signaling mechanisms as indicated.
In physiological conditions PD-L1/PD-1 interactions keep T cell tolerance toward autoantigens (Latchman et al., 2004). Conversely, in pathological conditions these inhibitory receptors lead to regulation of T-cell effector functions in autoimmunity and infection (Barber et al., 2006; Sharpe et al., 2007). Tumor survival can depend on the PD-L1/PD-1 pathway to attenuate immunogenicity and facilitate resistance to anti-apoptotic stimuli (Hirano et al., 2005; Azuma et al., 2008; Keir et al., 2008; Gato-Canas et al., 2017; Escors et al., 2018). PD-L1 is overexpressed in many tumor types to evade the immune attack and its expression generally (but not always) correlates with progression (Gato-Canas et al., 2017; Escors et al., 2018; Bocanegra et al., 2019; Kattan et al., 2019). PD-1 is expressed in T lymphocytes and interferes with their activation when bound with their ligands PD-L1, inhibiting the effector phase and thus dampening the ability of these T cells to kill cancer cells (Keir et al., 2008; Gato-Canas et al., 2017; Zuazo et al., 2019).
PD-L1/PD-1 blockade immunotherapy demonstrates longer duration of responses, and it is better tolerated than traditional therapies. However, despite the recent successes, a large number of patients do not respond to the therapy. This fact indicates intrinsic (or primary) resistance. In addition, a percentage of responder patients end up progressing through mechanisms of acquired resistance. Primary and acquired resistances are important barriers in terms of benefit to the patient (Pitt et al., 2016; Restifo et al., 2016; Sharma et al., 2017; O’Donnell et al., 2019).
Some of the patients treated with PD-L1/PD-1 immunotherapy show hyperprogressive disease, characterized by an unexpected drastic acceleration in tumor growth after the initiation of the therapy with fatal consequences (Champiat et al., 2017; Kato et al., 2017; Saada-Bouzid et al., 2017; Champiat et al., 2018; Ferrara et al., 2018; Zuazo et al., 2018; Kim et al., 2019). Moreover, a certain percentage of responder patients show an apparent progression of neoplastic lesions caused by massive tumor infiltration by immune cells. This response has been termed pseudoprogression, and it is a confounding factor for evaluation of responses by standard techniques such as computerized tomography (Onesti et al., 2019). These variety of atypical responses have prompted the development of immune-related response criteria (irRC) to better characterize the distinct types of responses associated to immunotherapies (Wolchok et al., 2009), in contrast to conventional evaluation criteria by Response Evaluation Criteria in Solid Tumors (RECIST). Nonetheless, the techniques and biomarkers currently integrated in clinical practice are not sufficient to identify responses. A deeper understanding of the mechanisms leading to resistance to PD-L1/PD-1 blockade is required.
In addition, every patient is unique as a result of genetic and clinical backgrounds. Hence, the mechanisms leading to clinical response or resistance are highly complex and might differ not only according to tumor type but also to patient-specific factors. Therefore, the contribution of tumor-cell intrinsic and patient-specific extrinsic factors needs to be elucidated. In the context of immunotherapies, it is unclear which ones are the main determinants of response and resistance.
A number of intrinsic characteristics of the patients are prognostic markers. In principle, we will disregard these general characteristics and focus on more specific factors contributing to immunoresistance. Without any doubt, tumor-intrinsic factors definitely contribute to response or progression in immune checkpoint blockade (Sharma et al., 2017; Chowell et al., 2018; Kalbasi and Ribas, 2020).
Tumor-intrinsic factors that contribute to primary and acquired resistance to PD-L1/PD-1 immunotherapy conform a genetic and signaling landscape that prevents immune cell infiltration in the tumor microenvironment (TME) (Figure 3). Resistance to PD-1 blockade immunotherapy is often associated with insufficient tumor antigenicity, constitutive PD-L1 expression, defects in IFN signal transduction within cancer cells and alterations in the regulation of oncogenic pathways (Escors, 2014; Sharma et al., 2017).
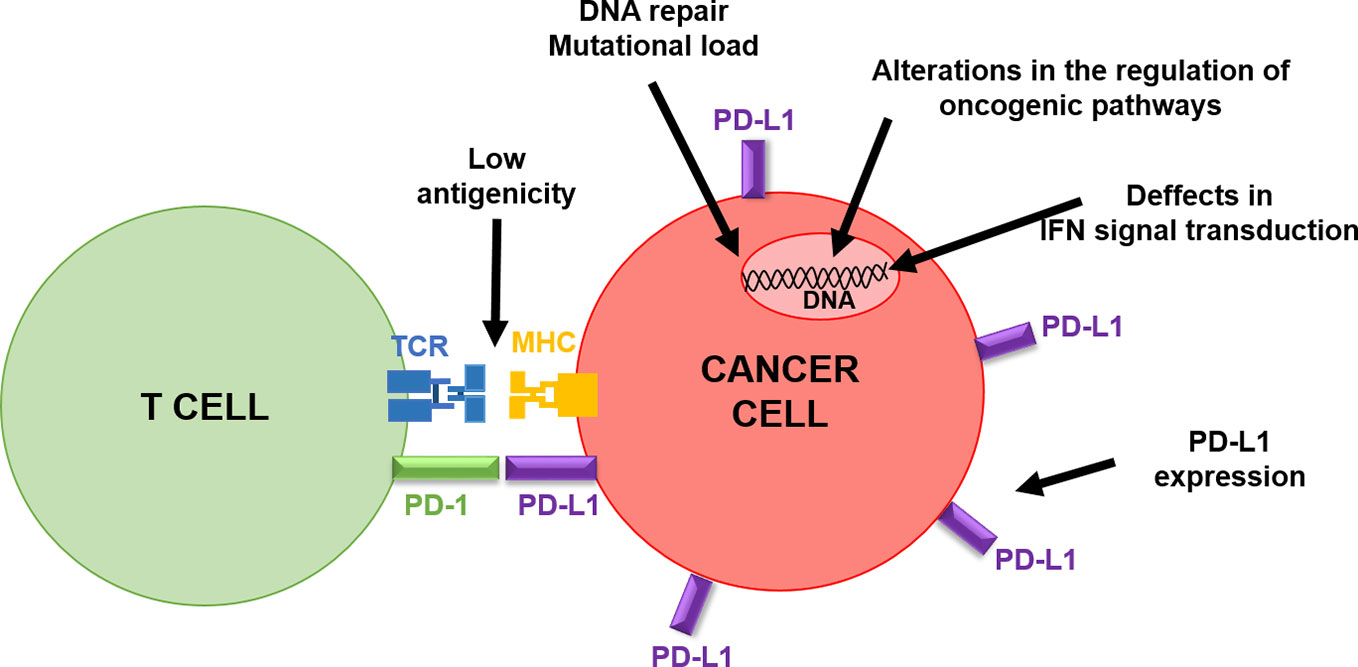
Figure 3 Schematic summary of cancer-intrinsic characteristics influencing clinical responses to PD-L1/PD-1 blockade therapies. The figure depicts the interaction of a T cell with a cancer cell, highlighting cancer cell intrinsic factors that can inactivate T cell activities, as indicated by the arrows.
The loss of tumor antigenicity is a major escape mechanism for many tumor types (Escors, 2014). This is mainly caused by cancer immunoediting, a process by which the immune system exerts a strong and sustained selective pressure over the most immunogenic cancer cell variants (Schreiber et al., 2011). Hence, recognition of tumor-specific antigens by effector T cells is crucial for cancer immunoediting (DuPage et al., 2012). Effector T cells will eliminate the most immunogenic cancer cells and control tumor progression for some time (Restifo et al., 2016; Sharma et al., 2017). However, the less immunogenic cancer cell variants will overgrow and progress. Therefore, tumor immunoediting does constitute a strong mechanism of acquired resistance to immunotherapies. The resulting surviving cancer cells usually show a strong decrease in tumor antigen expression (Matsushita et al., 2012; Escors, 2014), or a down-modulation of molecules involved in antigen presentation such as lack of MHC I or beta-microglobulin expression (Gubin et al., 2014). In this context, ICI therapies will fail simply because no endogenous T cell responses can be raised against these tumors. It has to be noted that immunoediting as a mechanism of immunological escape has been relatively well studied in immunotherapies other than ICIs (Schreiber et al., 2011; Teng et al., 2015; O’Donnell et al., 2019). Therefore, the real extent of the impact of immunoediting over resistance to ICI treatments has not yet been systematically quantified. The detection of less immunogenic variants in samples from patients before the start of immunotherapies may provide the means for adequate patient selection. For instance, characteristics such as genomic instability or epigenetic alterations in pre-existing tumor cell variants, may enable these cancer cells to evade ICI therapies. And these may even facilitate tumor grown, immune evasion, and tumor escape. These escape variants are likely to be naturally selected especially if potent immunostimulatory therapies are applied (Khong and Restifo, 2002). For example, the loss of functional β2 microglobulin from tumor cells, a structural component of the major histocompatibility complex (MHC) 1, confers resistance to tumor-specific CD8 T cells (Restifo et al., 1996). In addition, acquired homeostatic resistance has been described in which tumor cells alter gene expression profiles in response to interactions with the immune system (Pardoll, 2015).
We could include within these mechanisms the adaptive up-regulation of PD-L1 expression as a response to interferons produced during the anti-tumor attack (Garcia-Diaz et al., 2017; Gato-Canas et al., 2017; Escors et al., 2018). Cancer cells with up-regulated PD-L1 would not only inactivate PD-1-expressing T cells, but will also show increased resistance to IFN-mediated apoptosis through reverse signaling by PD-L1 within cancer cells (Gato-Canas et al., 2017; Jalali et al., 2019). It has been known for some time that PD-L1 had intrinsic signaling properties in cancer cells that protected that protected them from a range of apoptotic stimuli, and that its intracellular domain was required for this protection (Azuma et al., 2008). Moreover, PD-L1 was also shown to stimulate cancer cell growth by modulating the activity of AKT/mTOR, autophagy, and glycolysis (Chang et al., 2015; Clark et al., 2016; Gupta et al., 2016). The intracellular part of PD-L1 contains three non-classical signaling motifs; The “RMLD,” “DTSSK,” and “QFEET” motifs (Figure 1). The RMLD sequence is required for the anti-apoptotic activities of PD-L1 through the inhibition of STAT3 expression and alternative phosphorylation. The DTSSK motif has regulatory properties, and when it is removed or mutated, PD-L1 molecules exhibit hyperactivated signaling (Gato-Canas et al., 2017). The QFEET motif has been recently shown to be the docking site for the de-ubiquitinase USP22 (Huang et al., 2019).
Inhibition of STAT3 by PD-L1 intrinsic signaling ensures the abrogation of interferon-mediated apoptosis (Gato-Canas et al., 2017), stimulates the inflammasome pathway in cancer cells (Theivanthiran et al., 2020), and directly inhibits PD-L1-positive T cells (Diskin et al., 2020). PD-L1-regulated inflammasome activation triggers a series of signaling cascades that end up with the recruitment of granulocyte myeloid-derived suppressor cells (MDSC) in the tumor environment. This accumulation of MDSCs contribute to resistance to PD-L1/PD-1 blockade strategies. Therefore, PD-L1 expression by cancer cells regulates several pro-carcinogenic mechanisms that can contribute to resistance: First, PD-L1 as an inhibitor of T cell effector activities; second, PD-L1 as an anti-apoptotic shield; and third, PD-L1 as a recruiter of MDSCs into the tumor microenvironment. In agreement with this, it is not surprising that human carcinomas with inactivating mutations in the DTSSK motif of PD-L1 can be selected by immunoediting (Gato-Canas et al., 2017), as these mutations increase the signaling capacities of PD-L1.
Hence, PD-L1 expression in tumors could be considered a tumor-intrinsic factor of resistance. PD-L1 up-regulation in tumor cells is generally associated with tumor progression, proliferation and invasion, antiapoptotic signaling, and T cell inhibitory activities via engagement with PD-1 (Escors et al., 2018). PD-L1 expression on tumor cells seems to be sufficient for immune evasion and inhibition of CD8 T cell cytotoxicity (Juneja et al., 2017). Therefore, PD-L1 expression is a recognized biomarker for patient stratification in PD-L1/PD-1 blockade immunotherapy. Some immunohistochemistry assays to quantify PD-L1 expression are currently FDA-approved such as Dako 28-8, Dako 22C3, Ventana SP142, and Ventana SP263. However, the systems of detection are not currently standardized, as different immunochemistry assay and scoring system offer different classifications for tumor PD-L1 status (Arasanz et al., 2018; Bocanegra et al., 2019). Additionally, PD-L1 expression can be highly variable and heterogeneous. Some patients with PD-L1-negative tumors may still benefit from anti-PD-L1/PD-1 therapies as PD-L1 is also expressed by many other cell types including myeloid antigen-presenting cells (Karwacz et al., 2011; Motzer et al., 2015; Horn et al., 2017; Bocanegra et al., 2019). Because of these limitations, PD-L1 expression as a predictive biomarker for responses is still under debate. Nevertheless, the application of radioactively-labeled probes specific for PD-L1 and in vivo PET visualization of labeled tumors, and their metastasis is very likely going to solve many of these issues. First, detection of PD-L1 expression levels without the need of obtaining a limited amount of tumor tissue. Second, sensitive detection of “silent” metastases. Third, discrimination of true progression from pseudoprogression, at least for cancers that are PD-L1 positive. So far, several different approaches have been applied in pre-clinical models and in cancer patients. For example, by using PD-L1-specific nanobodies labeled with technetium-99m (Broos et al., 2017), PD-L1-specific cyclic peptides labeled with Gallium (De Silva et al., 2018), and radio-labeled anti-PD-L1 antibodies (Heskamp et al., 2015; Niemeijer et al., 2018).
Several other approaches based on intrinsic tumor characteristics have been established for patient selection. From these, the tumor mutational burden (TMB) has gained popularity as a potential predictive biomarker associated with response to ICI therapies. TMB provides a quantification of the number of mutations per megabase of genomic DNA within the tumor encoding genome. It is thought that “high” TMB tumors may have increased expression of neoantigens and enhanced immunogenicity (Alexandrov et al., 2013; Yuan et al., 2016). Neoantigen load is associated with response and has some predictive value on long-term clinical benefit of PD-L1/PD-1 blockade therapies. The mutational load before the start of immunotherapies seems to be associated to a higher nonsynonymous mutation burden in tumors, higher neoantigen expression, and mutations within the DNA repair pathways (Gubin et al., 2014; Le et al., 2015; Rizvi et al., 2015; Schumacher and Schreiber, 2015). A reflection of this is exemplified by mismatch repair deficiency in cancers, which predicts response to PD-1 blockade for some tumor types such as colon cancer (Le et al., 2015; Le et al., 2017). Therefore, the FDA approved in 2017 the PD-1 inhibitor pembrolizumab for treatment of progressive mismatch-repair deficient solid tumors, consolidating mismatch repair (MMR) defect as a clinically applicable biomarker.
ICI immunotherapies differ substantially from conventional therapies in which the target is the immune system. Therefore, it is fair to assume that tumor extrinsic factors linked to the immune system will be associated to response or resistance to ICI therapy. So far, a variety of such factors have been associated to resistance. These include irreversible T cell exhaustion, expression of additional immune checkpoint molecules and their ligands (CTLA-4, TIM-3, LAG-3, TIGIT, VISTA, and BTLA), differentiation and expansion of immunosuppressive cell populations, and release of immunosuppressive cytokines and metabolites both systemically and within the TME (IL-10, IL-6, IL-17, IFNγ, CSF-1, tryptophan metabolites, TGF-β, IDO, increased adenosine production) (Figure 4) (Fridman et al., 2017; Sharma et al., 2017; Fares et al., 2019).
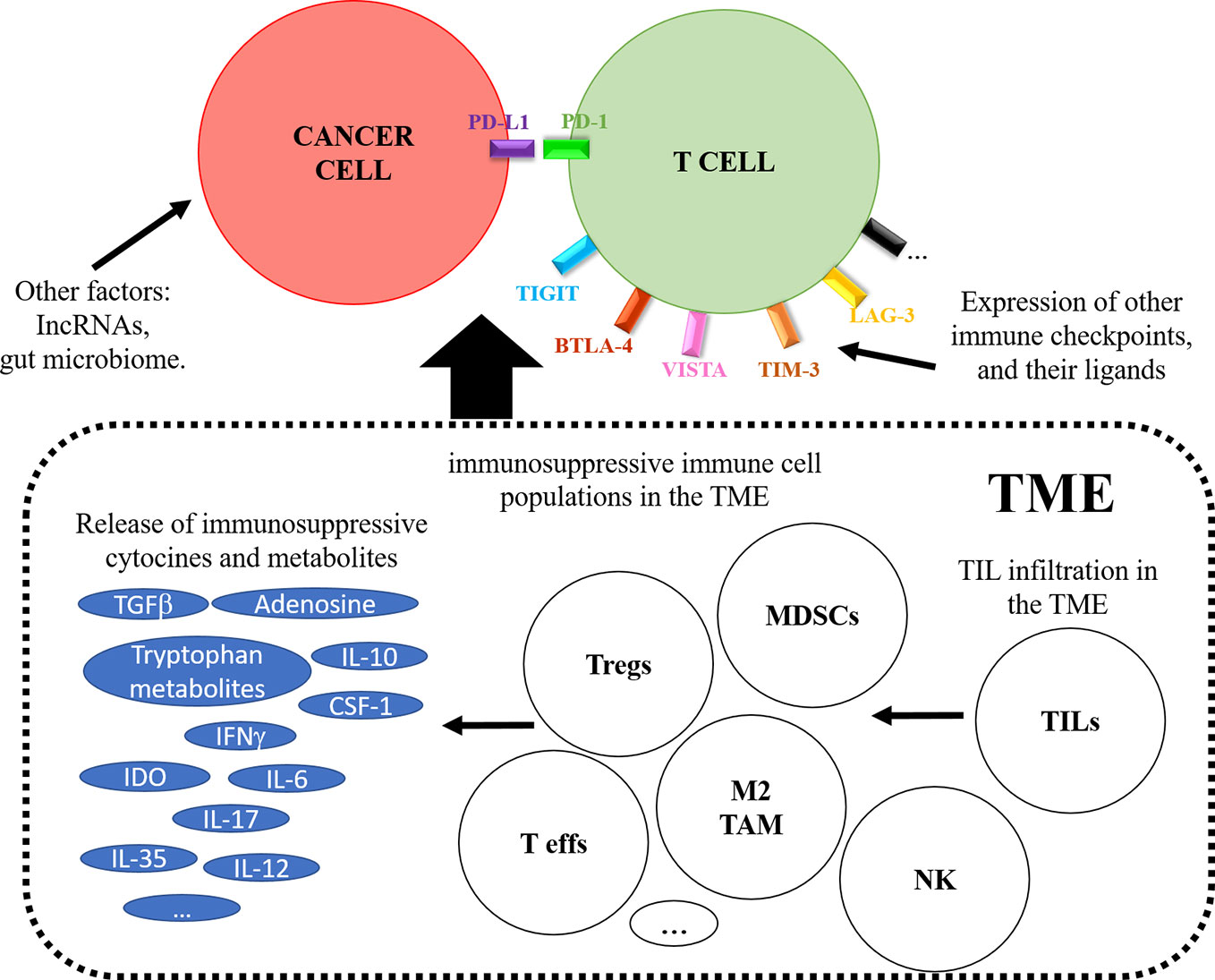
Figure 4 The figure schematically represents tumor-extrinsic mechanisms contributing to response or resistance to PD-L1/PD-1 blockade therapies. The figure depicts on top a T cell interacting with a cancer cell, and the effects caused by the tumor microenvironment (TME) are boxed below. These include the recruitment of immunosuppressive cells as indicated, the expression of immunosuppressive metabolites and the induction of alternative immune checkpoints on the T cell.
One of the oldest prognostic immune biomarkers is the quantification of the type, location, and density of immune cells that infiltrate the TME (O’Donnell et al., 2019). Anti-neoplastic treatments and not only immunotherapies are most efficacious in patients with increased tumor-infiltrating lymphocytes (TILs) in biopsies. This is also true for ICI therapies, and the use of TIL quantification together with PD-L1 tumor positivity is generally associated to good responses (Taube et al., 2012; Bindea et al., 2013). Indeed, there is a positive correlation of TIL infiltration with PD-L1 expression by cancer cells. There are several ways to quantify TIL infiltration, but one of the most successful at least for colon cancer is the so-called “immunoscore” (Galon et al., 2014; Pages et al., 2018; Angell et al., 2020). This biopsy scoring system is a powerful prognostic tool based on the quantification of CD3 and CD8 T lymphocytes on the tumor center and at the tumor invasive margins.
Not surprisingly, TIL infiltration correlates with good prognosis and objective responses to ICI therapies. Oligoclonal TILs are expanded in the tumor tissues of responders to anti-PD-1 blockade. These T cells show enhanced helper T cell type 1 (Th1) cellular immunity (Inoue et al., 2016). Patients can be stratified into four different types according to the characteristics of the TME tumor based on TILs and PD-L1: type I or adaptive immunoresistant (PDL1(+), TIL(+)), type II or immunologically ignorant (PD-L1(-),TIL(-)), type III (PD-L1(+), TIL(-)), and type IV or immune-tolerant (PD-L1(-), TIL(+)) (Teng et al., 2015). This stratification may provide a means for therapy selection. However, other factors contribute to efficacious responses. For instance, the TILs/PD-L1 ratio can be altered according to the expression of oncogene drivers in cancer cells as well as the anatomical location of the neoplastic lesions.
Recent studies demonstrate that ICI therapies do also alter the dynamics and characteristics of systemic immune cell populations. Interestingly, some of these studies highlight the CD28-CD80 costimulation signaling pathway as a major contributor to efficacious responses to ICI (Hui et al., 2017; Zuazo et al., 2019). Indeed, several studies show a key role for IL-12-expressing dendritic cells with cross-presentation capacities for good responses to immunotherapies (Kerkar et al., 2011; Liechtenstein et al., 2014; Goyvaerts et al., 2015; Berraondo et al., 2018; Garris et al., 2018; Etxeberria et al., 2019). These results reinforce the idea that a systemic functional immunity is very likely a required factor for the efficacy of immunotherapies. This was elegantly shown in murine models (Spitzer et al., 2017) as well as in human patients undergoing PD-L1/PD-1 blockade therapies (Kamphorst et al., 2017; Zuazo et al., 2019). A systemic expansion in peripheral blood of a population of CD28+ PD-1+ CD8 T cells was shown in melanoma patients responding to anti-PD-1 therapy (Kamphorst et al., 2017). Patients with non-small cell lung cancer undergoing ICI therapies that presented systemic dysfunctional CD4 T cells that strongly co-expressed PD-1 and LAG-3 failed to respond to therapies (Zuazo et al., 2019). Interestingly, these CD4 T cells did not lose their capacities for multi-cytokine production following in vitro stimulation, albeit with a strong Th17-type of responses. These results suggested that these T cells could not be considered exhausted. However, they showed a degree of proliferative dysfunctionality that was indicative of some type of anergy. Importantly, these patient cohorts were enriched in hyperprogressors, suggesting a key role for T cell dysfunctionality in hyperprogressive disease (Zuazo et al., 2019). These results highlighted the up-regulation of LAG-3 as a major escape mechanism to PD-1/PD-L1 monoblockade strategies. Very similar results were obtained in two other independent studies by Kagamu and collaborators, and Julia and collaborators (Julia et al., 2019; Kagamu et al., 2020). In the study by Zuazo et al. responders had a high percentage of highly differentiated CD27− CD28− memory CD4 T cells before starting immunotherapies, and could be used as a predictive biomarker. Similarly, Kagamu et al. identified this population as CD62Llow CD4 cells, while Julia et al. described this population as central memory CD4 T cells.
The expansion of immunosuppressive immune cell populations systemically or infiltrating the TME also contributes to extrinsic factors of resistance. Regulatory T cells (Tregs) strongly suppress tumor-specific T cell functions and disrupt effector T cell function. The mechanisms of Treg-mediated immune suppression are varied and include direct cell-to-cell contact and secretion of potent immunosuppressive cytokines such us L-10, IL-35 or TGF-β (Viehl et al., 2006; Sakaguchi et al., 2008; Arce et al., 2011). Some of these cytones will differentiate naïve T cells into inducible Tregs especially in the context of antigen presentation from tolerogenic DCs (Arce et al., 2011). It is increasingly clear the negative impact that the expansion of myeloid-derived suppressor cells have not only in immunotherapy, but also in conventional therapies. Although there is some controversy on their ontogeny and nature, MDSCs englobe a collection of myeloid populations with potent immunosuppressive activities. Tumor infiltrating MDSCs promote angiogenesis, tumor cell invasion, and establish distal metastatic niches (Srivastava et al., 2012; Meyer et al., 2013; Liechtenstein et al., 2014; Dufait et al., 2015; Gato-Canas et al., 2015; Ibanez-Vea et al., 2017). A special case of immunosuppressive myeloid cells constitutes tumor associated macrophages (TAMs). Tumor infiltration with TAMs usually correlates with poor prognosis, particularly with M2 macrophages characterized by high production of immunosuppressive cytokines. Therefore, tumor infiltration with M2 macrophages over M1 macrophages has an impact on tumor angiogenesis, invasion, metastasis, and immunosuppression (Chanmee et al., 2014; Gato et al., 2016; Ibanez-Vea et al., 2018). The recruitment of M2 macrophages seems to lead to immunotherapy resistance, and recent reports in murine models of cancer treated with PD-L1/PD-1 blockade therapies link macrophages with hyperprogressive disease by removing therapeutic antibodies through interactions with their Fc receptors (Lo Russo et al., 2019).
Other more subtle mechanisms may also contribute to resistance. In recent years it has been shown that long non-coding RNAs (lncRNAs) constitute systemic regulators of many biological functions including cancer (Schmitt and Chang, 2016). Interestingly, some immune-related lncRNAs regulate immunosuppressive mechanisms leading to immune evasion and resistance to immunotherapy. Some examples include loss of antigen presentation, PD-L1 overexpression, regulation of T-cell exhaustion, and MDSC and Treg differentiation and expansion (Zhou et al., 2019; Zheng et al., 2019).
Finally, recent metagenomic studies have shown that abnormal gut microbiome affects antitumor immunity, influencing on the response to PD-1-based blockade (74, 75). For example, the abundance of Bifidobacterium spp. in the gut microbiome enhances anti-PD-L1 therapy efficacy and improves antitumor immunity by affecting dendritic cells (Sivan et al., 2015). Responders to immunotherapy showed abundant Bifidobacterium longum and adolescentis, Collinesella aerofaciens, Parabacteiodes merdae, and Fecalibacterium spp. on their microbioma, while non-responders had increased abundance of Ruminococcus obeum and Roseburia intestinalis (Gopalakrishnan et al., 2018; Matson et al., 2018). A large presence of Akkermansia muciniphila and A. muciniphila contributes to the immunogenicity of PD-1 blockade, and its abundance was correlated with clinical responses. Fecal microbiota transplantations restore the efficacy of IL-12-dependent anti-PD-1 blockade (Routy et al., 2018). These observations are not restricted to PD-L1/PD-1 blockade, as the presence of Bacteroides spp in the gut microbioma was required for anticancer immunity in anti-CTLA-4 therapy (Vetizou et al., 2015).
It is undisputed that ICI therapies are currently leading the way for the development of efficacious anti-neoplastic treatments. Nevertheless, it is yet unclear which mechanisms are driving resistance to ICI treatments and how to tackle them. The relative contribution of tumor cell intrinsic and extrinsic factors to primary, adaptive, and acquired resistance is currently highly confusing. A deeper understanding of the mechanisms underlying the complex immunological pathways in cancer and the molecular mechanisms underlying the PD-L/PD-1 blockade will provide insight into the subject.
Considering all the current evidence, we propose that performing highly detailed systemic immunological profiling is right now a requirement for any study involving ICIs. Not only to identify potential responders, but also to monitor the “real time” performance of ICI therapies by quantifying the dynamic changes of immune cell populations. An increasing number of clinical studies are addressing this particular issue by quantification of the relative abundance of distinct immunological populations in peripheral blood. Nowadays, flow cytometry panels composed of more than 10 markers are routinely used for immunological profiling without the need of setting up CyTOF technologies. In a recent study published by our group, quantification of the relative proportion of highly differentiated CD27- CD28- CD4 T cells before the start of immunotherapies was sufficient to identify a cohort of NSCLC patients with a high probability of response to PD-L1/PD-1 blockers (Zuazo et al., 2019). More specifically, responder patients had high percentages of central and effector memory CD4 T cells. This analysis relied on a panel of 8 markers to stain T cells from a small blood sample by standard flow cytometry. Importantly, our study was validated by the results from two similar studies which used other alternative T cell markers. The first study correlated the high baseline frequency of central memory CD4 T cells with response to immunotherapy in NSCLC and renal cancer patients using flow cytometry (Julia et al., 2019). In the second study, NSCLC patients with high baseline percentages of CD62Llow effector CD4 T cells quantified by CyTOF had a high chance of responding to PD-L1/PD-1 blockade (Kagamu et al., 2020). The dynamics and behavior of these CD4 T cell subsets were identical to those from highly-differentiated memory CD4 T cells in our study, strongly suggesting that we were all monitoring the same CD4 T cell subsets but with different markers. Cytotoxicity assays performed with peripheral T cells have also been shown to have predictive capabilities for nivolumab efficacy (Iwahori et al., 2019), as well as the quantification of PD-1+ CD8 T cells in peripheral blood after administration of PD-1 blockers (Kamphorst et al., 2017). Therefore, all these studies including our own demonstrate that simple analytical techniques can be effectively applied in clinical practice for defining an immunological profile based on systemic T cell subsets without the need of obtaining a tumor biopsy sample.
In addition, the dynamic changes of the immune populations in peripheral blood provides invaluable clinical information. Changes in T cell compartments have been recently shown by others and us to correlate with progression and even hyperprogression. The study by Kagamu and collaborators showed that a decrease in peripheral CD62Llow CD4 T cells right after therapy correlated with acquired resistance (Kagamu et al., 2020). In our particular NSCLC cohort, a low baseline percentage of memory CD27- CD28- CD4 T cells correlated with intrinsic resistance (Zuazo et al., 2019). Moreover, a sudden increase in highly differentiated CD4 T cells (CD4 THD burst) following the first cycle of immunotherapy was indicative of hyperprogressive disease (Zuazo et al., 2018; Arasanz et al., 2020). The identification of hyperprogressors is also of the outmost importance, as these patients deteriorate very quickly with fatal outcomes. Hence, we propose that the generation of a “systemic immunological file” containing the relative percentages of at least T cell subsets before and after the first cycle of immunotherapies will provide the means to identify patients according to probabilities of response and provide useful information to the clinician.
Considering the most recent evidence, we do think that an “immunological file” on each patient provides information over immediate responses to immunotherapy. However, cancer cells can select several mutations that interfere with the specific molecular pathways stimulated by ICI therapies. For example, mutations in JAK1, JAK2, and beta2-microglobulin in cancer cells abrogate interferon-mediated apoptosis and prevent PD-L1 up-regulation by interferons (Zaretsky et al., 2016; Garcia-Diaz et al., 2017; Sharma et al., 2017; Shin et al., 2017). Some mutations in the DTSSK domain of PD-L1 present in human carcinomas enhance the capacities of PD-L1 to counteract IFN-cytotoxicity by interfering with STAT3 expression and its alternative phosphorylation (Gato-Canas et al., 2017). Moreover, this inhibition of STAT3 has been recently shown to activate the inflammasome in cancer cells leading to the recruitment of granulocytic MDSCs to the tumor and causing acquired resistance to immunotherapy (Theivanthiran et al., 2020). The molecular characterization of cancer cells, particularly focusing on genetic traits and mutations, will identify patients with high risk of acquired resistance. New generation sequencing is currently on the increase in clinical oncology, with panels that cover the major oncogenic and driver mutations. In ICI therapies, it is likely that new panels covering mutations affecting immunological signaling pathways and immune checkpoints will be of relevance in the near future. Currently, this is an expanding research subject that will surely play a key role in the future oncology.
By a better understanding of the key pathways involved in these processes, we will develop treatments to effectively counteract resistance. The identification of truly predictive and prognostic biomarkers of response is currently a top priority in clinical practice. Some therapeutic strategies to overcome resistance could include the modulation of the TME to increase immunogenicity, overcome T-cell exhaustion, enhance tumor infiltration, and modulate epigenetic regulation. The incorporation of the “immunological file” to be included in the clinical profile of each patient could be a practical example. NSCLC patients with dysfunctional CD4 systemic immunity before starting immunotherapies have intrinsic resistance (Julia et al., 2019; Kamada et al., 2019; Zuazo et al., 2019; ; Kagamu et al., 2020). A closer analysis of these patients uncovered a high co-expression of PD-1 and LAG-3 (Zuazo et al., 2019), TIM-3 up-regulation (Julia et al., 2019), or an expansion of Tregs (Kamada et al., 2019). These patients could therefore be selected on the basis of their “systemic immunological profile” for combination therapies with anti-PD-1/anti-LAG-3, anti-PD-1/anti-TIM-3 or anti-PD-1/anti-CTLA4 antibodies. In addition, minimizing immunological escape and the onset of resistance will be likely achieved by combination therapies with targeted therapies. Other combinations such as with chemotherapy, radiotherapy, CAR-T cells, or the application of additional immune checkpoint blockade agents targeting LAG-3, TIM-3, CSF1R, IDO, GITR, or CD134 could be the key to achieve long-lasting clinical responses.
LC conceived the review and wrote the first draft. MZ, HA, AB, CH, GF, MG-G, and EB and RV as contributed author to the final form of the review. GK and DE conceived the review and contributed to the writing of the final version.
The Oncoimmunology group is supported by: Asociación Española Contra el Cáncer (AECC, PROYE16001ESCO); Instituto de Salud Carlos III, Spain (FIS project grant PI17/02119); Gobierno de Navarra Biomedicine Project grant (BMED 050-2019); TRANSPOCART (Instituto de Salud Carlos III, project: ICI19/00069); “Precipita” Crowdfunding grant (FECYT); Crowdfunding grant from Sociedad Española de Inmunología (SEI); DE is funded by a Miguel Servet Fellowship (ISC III, CP12/03114, Spain); LC is supported by a DESCARTHES project grant (Industry department, Government of Navarre project grant number: 0011-1411-2019-000058); AB and EB are supported by a European Project Horizon 2020-SC1-BHC-2018-2020 project grant; CH is supported by a Roche-funded grant (stop fuga de cerebros); HA is supported by the Clinico Junior 2019 scholarship from AECC; MZ is supported by a scholarship from Universidad Pública de Navarra; and MG.is supported by a scholarship from the Government of Navarre.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We sincerely thank the patients and families that generously agreed to take part in this study. We are thankful as well to the nursing staff of the Medical Oncology Day Care at Hospital Complex of Navarre for their willful collaboration. We also thank the Blood and Tissue Bank of Navarre, Health Department of Navarre, Spain.
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. doi: 10.1038/nature12477
Angell, H. K., Bruni, D., Barrett, J. C., Herbst, R., Galon, J. (2020). The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 26, 332–339. doi: 10.1158/1078-0432.CCR-18-1851
Arasanz, H., Gato-Canas, M., Zuazo, M., Ibanez-Vea, M., Breckpot, K., Kochan, G., et al. (2017). PD1 signal transduction pathways in T cells. Oncotarget 8, 51936–51945. doi: 10.18632/oncotarget.17232
Arasanz, H., Zuazo, M., Vera, R., Kochan, G., Escors, D. (2018). Systemic immunological biomarkers of clinical responses in immune checkpoint blockade therapies. Lung Cancer Manag. 7, LMT07. doi: 10.2217/lmt-2018-0014
Arasanz, H., Zuazo, M., Bocanegra, A., Gato, M., Martinez-Aguillo, M., Morilla, I., et al. (2020). Early detection of hyperprogressive disease in non-small cell lung cancer by monitoring of systemic T cell dynamics. Cancers 12, 344. doi: 10.3390/cancers12020344
Arce, F., Breckpot, K., Stephenson, H., Karwacz, K., Ehrenstein, M. R., Collins, M., et al. (2011). Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 63, 84–95. doi: 10.1002/art.30099
Azuma, T., Yao, S., Zhu, G., Flies, A. S., Flies, S. J., Chen, L. (2008). B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 111, 3635–3643. doi: 10.1182/blood-2007-11-123141
Barber, D. L., Wherry, E. J., Masopust, D., Zhu, B., Allison, J. P., Sharpe, A. H., et al. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. doi: 10.1038/nature04444
Berraondo, P., Etxeberria, I., Ponz-Sarvise, M., Melero, I. (2018). Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. 24, 2716–2718. doi: 10.1158/1078-0432.CCR-18-0381
Bindea, G., Mlecnik, B., Tosolini, M., Kirilovsky, A., Waldner, M., Obenauf, A. C., et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. doi: 10.1016/j.immuni.2013.10.003
Bocanegra, A., Fernandez-Hinojal, G., Zuazo-Ibarra, M., Arasanz, H., Garcia-Granda, M. J., Hernandez, C., et al. (2019). PD-L1 Expression in Systemic Immune Cell Populations as a Potential Predictive Biomarker of Responses to PD-L1/PD-1 Blockade Therapy in Lung Cancer. Int. J. Mol. Sci. 20, pii E1631. doi: 10.3390/ijms20071631
Brahmer, J. R., Tykodi, S. S., Chow, L. Q., Hwu, W. J., Topalian, S. L., Hwu, P., et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl. J. Med. 366, 2455–2465. doi: 10.1056/NEJMoa1200694
Broos, K., Keyaerts, M., Lecocq, Q., Renmans, D., Nguyen, T., Escors, D., et al. (2017). Non-invasive assessment of murine PD-L1 levels in syngeneic tumor models by nuclear imaging with nanobody tracers. Oncotarget 8, 41932–41946. doi: 10.18632/oncotarget.16708
Champiat, S., Dercle, L., Ammari, S., Massard, C., Hollebecque, A., Postel-Vinay, S., et al. (2017). Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 23, 1920–1928. doi: 10.1158/1078-0432.CCR-16-1741
Champiat, S., Ferrara, R., Massard, C., Besse, B., Marabelle, A., Soria, J. C., et al. (2018). Hyperprogressive disease: recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol, 15, 748–762. doi: 10.1038/s41571-018-0111-2
Chang, C. H., Qiu, J., O’Sullivan, D., Buck, M. D., Noguchi, T., Curtis, J. D., et al. (2015). Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. doi: 10.1016/j.cell.2015.08.016
Chanmee, T., Ontong, P., Konno, K., Itano, N. (2014). Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 6, 1670–1690. doi: 10.3390/cancers6031670
Chemnitz, J. M., Parry, R. V., Nichols, K. E., June, C. H., Riley, J. L. (2004). SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173, 945–954. doi: 10.4049/jimmunol.173.2.945
Chen, L., Flies, D. B. (2013). Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242. doi: 10.1038/nri3405
Chowell, D., Morris, L. G. T., Grigg, C. M., Weber, J. K., Samstein, R. M., Makarov, V., et al. (2018). Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359, 582–587. doi: 10.1126/science.aao4572
Clark, C. A., Gupta, H. B., Sareddy, G., Pandeswara, S., Lao, S., Yuan, B., et al. (2016). Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth. Pathog. Autophagy Ovarian Cancer Melanoma Cancer Res. 76, 6964–6974. doi: 10.1158/0008-5472.CAN-16-0258
De Silva, R. A., Kumar, D., Lisok, A., Chatterjee, S., Wharram, B., Venkateswara Rao, K., et al. (2018). Peptide-Based (68)Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol. Pharm. 15, 3946–3952. doi: 10.1021/acs.molpharmaceut.8b00399
Diskin, B., Adam, S., Cassini, M. F., Sanchez, G., Liria, M., Aykut, B., et al. (2020). PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat. Immunol. 21, 442–454. doi: 10.1038/s41590-020-0620-x
Dong, H., Zhu, G., Tamada, K., Chen, L. (1999). B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5, 1365–1369. doi: 10.1038/70932
Dufait, I., Schwarze, J. K., Liechtenstein, T., Leonard, W., Jiang, H., Law, K., et al. (2015). Ex vivo generation of myeloid-derived suppressor cells that model the tumor immunosuppressive environment in colorectal cancer. Oncotarget 6, 12369–12382. doi: 10.18632/oncotarget.3682
DuPage, M., Mazumdar, C., Schmidt, L. M., Cheung, A. F., Jacks, T. (2012). Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409. doi: 10.1038/nature10803
Escors, D., Gato-Canas, M., Zuazo, M., Arasanz, H., Garcia-Granda, M. J., Vera, R., et al. (2018). The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct. Target Ther. 3, 26. doi: 10.1038/s41392-018-0022-9
Escors, D. (2014). Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J. Sci. 2014, pii: 734515. doi: 10.1155/2014/734515
Esensten, J. H., Helou, Y. A., Chopra, G., Weiss, A., Bluestone, J. A. (2016). CD28 Costimulation: From Mechanism to Therapy. Immunity 44, 973–988. doi: 10.1016/j.immuni.2016.04.020
Etxeberria, I., Bolanos, E., Quetglas, J. I., Gros, A., Villanueva, A., Palomero, J., et al. (2019). Intratumor Adoptive Transfer of IL-12 mRNA Transiently Engineered Antitumor CD8(+) T Cells. Cancer Cell 36, 613–629 e7. doi: 10.1016/j.ccell.2019.10.006
Fares, C. M., Van Allen, E. M., Drake, C. G., Allison, J. P., Hu-Lieskovan, S. (2019). Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 39, 147–164. doi: 10.1200/EDBK_240837
Ferrara, R., Mezquita, L., Texier, M., Lahmar, J., Audigier-Valette, C., Tessonnier, L., et al. (2018). Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 4, 1543–1552. doi: 10.1001/jamaoncol.2018.3676
Freeman, G. J., Long, A. J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034. doi: 10.1084/jem.192.7.1027
Fridman, W. H., Zitvogel, L., Sautes-Fridman, C., Kroemer, G. (2017). The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 14, 717–734. doi: 10.1038/nrclinonc.2017.101
Galon, J., Mlecnik, B., Bindea, G., Angell, H. K., Berger, A., Lagorce, C., et al. (2014). Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 232, 199–209. doi: 10.1002/path.4287
Garcia-Diaz, A., Shin, D. S., Moreno, B. H., Saco, J., Escuin-Ordinas, H., Rodriguez, G. A., et al. (2017). Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 19, 1189–1201. doi: 10.1016/j.celrep.2017.04.031
Garris, C. S., Arlauckas, S. P., Kohler, R. H., Trefny, M. P., Garren, S., Piot, C., et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161 e7. doi: 10.1016/j.immuni.2018.09.024
Gato, M., Blanco-Luquin, I., Zudaire, M., de Morentin, X. M., Perez-Valderrama, E., Zabaleta, A., et al. (2016). Drafting the proteome landscape of myeloid-derived suppressor cells. Proteomics 16, 367–378. doi: 10.1002/pmic.201500229
Gato-Canas, M., Martinez de Morentin, X., Blanco-Luquin, I., Fernandez-Irigoyen, J., Zudaire, I., Liechtenstein, T., et al. (2015). A core of kinase-regulated interactomes defines the neoplastic MDSC lineage. Oncotarget 6, 27160–27175. doi: 10.18632/oncotarget.4746
Gato-Canas, M., Zuazo, M., Arasanz, H., Ibanez-Vea, M., Lorenzo, L., Fernandez-Hinojal, G., et al. (2017). PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 20, 1818–1829. doi: 10.1016/j.celrep.2017.07.075
Gopalakrishnan, V., Spencer, C. N., Nezi, L., Reuben, A., Andrews, M. C., Karpinets, T. V., et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 359, 97–103. doi: 10.1126/science.aan4236
Goyvaerts, C., Broos, K., Escors, D., Heirman, C., Raes, G., De Baetselier, P., et al. (2015). The transduction pattern of IL-12-encoding lentiviral vectors shapes the immunological outcome. Eur. J. Immunol. 45, 3351–3361. doi: 10.1002/eji.201545559
Gubin, M. M., Zhang, X., Schuster, H., Caron, E., Ward, J. P., Noguchi, T., et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581. doi: 10.1038/nature13988
Gupta, H. B., Clark, C. A., Yuan, B., Sareddy, G., Pandeswara, S., Padron, A. S., et al. (2016). Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct. Target Ther. 1, pii: 16030. doi: 10.1038/sigtrans.2016.30
Heskamp, S., Hobo, W., Molkenboer-Kuenen, J. D., Olive, D., Oyen, W. J., Dolstra, H., et al. (2015). Noninvasive Imaging of Tumor PD-L1 Expression Using Radiolabeled Anti-PD-L1 Antibodies. Cancer Res. 75, 2928–2936. doi: 10.1158/0008-5472.CAN-14-3477
Hirano, F., Kaneko, K., Tamura, H., Dong, H., Wang, S., Ichikawa, M., et al. (2005). Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 65, 1089–1096.
Hodi, F. S., Oble, D. A., Drappatz, J., Velazquez, E. F., Ramaiya, N., Ramakrishna, N., et al. (2008). CTLA-4 blockade with ipilimumab induces significant clinical benefit in a female with melanoma metastases to the CNS. Nat. Clin. Pract. Oncol. 5, 557–561. doi: 10.1038/ncponc1183
Horn, L., Spigel, D. R., Vokes, E. E., Holgado, E., Ready, N., Steins, M., et al. (2017). Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 35, 3924–3933. doi: 10.1200/JCO.2017.74.3062
Huang, X., Zhang, Q., Lou, Y., Wang, J., Zhao, X., Wang, L., et al. (2019). USP22 Deubiquitinates CD274 to Suppress Anticancer Immunity. Cancer Immunol. Res. 7, 1580–1590. doi: 10.1158/2326-6066.CIR-18-0910
Hui, E., Cheung, J., Zhu, J., Su, X., Taylor, M. J., Wallweber, H. A., et al. (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 355, 1428–1433. doi: 10.1126/science.aaf1292
Ibanez-Vea, M., Zuazo, M., Gato, M., Arasanz, H., Fernandez-Hinojal, G., Escors, D., et al. (2017). Myeloid-Derived Suppressor Cells in the Tumor Microenvironment: Current Knowledge and Future Perspectives. Arch. Immunol. Ther. Exp. (Warsz), 66, 113–123. doi: 10.1007/s00005-017-0492-4
Ibanez-Vea, M., Huang, H., Martinez de Morentin, X., Perez, E., Gato, M., Zuazo, M., et al. (2018). Characterization of Macrophage Endogenous S-Nitrosoproteome Using a Cysteine-Specific Phosphonate Adaptable Tag in Combination with TiO2 Chromatography. J. Proteome Res. 17, 1172–1182. doi: 10.1021/acs.jproteome.7b00812
Inoue, H., Park, J. H., Kiyotani, K., Zewde, M., Miyashita, A., Jinnin, M., et al. (2016). Intratumoral expression levels of PD-L1, GZMA, and HLA-A along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma. Oncoimmunology 5, e1204507. doi: 10.1080/2162402X.2016.1204507
Ishida, Y., Agata, Y., Shibahara, K., Honjo, T. (1992). Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x
Iwahori, K., Shintani, Y., Funaki, S., Yamamoto, Y., Matsumoto, M., Yoshida, T., et al. (2019). Peripheral T cell cytotoxicity predicts T cell function in the tumor microenvironment. Sci. Rep. 9, 2636. doi: 10.1038/s41598-019-39345-5
Jalali, S., Price-Troska, T., Bothun, C., Villasboas, J., Kim, H. J., Yang, Z. Z., et al. (2019). Reverse signaling via PD-L1 supports malignant cell growth and survival in classical Hodgkin lymphoma. Blood Cancer J. 9, 22. doi: 10.1038/s41408-019-0185-9
Julia, E. P., Mando, P., Rizzo, M. M., Cueto, G. R., Tsou, F., Luca, R., et al. (2019). Peripheral changes in immune cell populations and soluble mediators after anti-PD-1 therapy in non-small cell lung cancer and renal cell carcinoma patients. Cancer Immunol. Immunother. CII 68, 1585–1596. doi: 10.1007/s00262-019-02391-z
Juneja, V. R., McGuire, K. A., Manguso, R. T., LaFleur, M. W., Collins, N., Haining, W. N., et al. (2017). PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 214, 895–904. doi: 10.1084/jem.20160801
Kagamu, H., Kitano, S., Yamaguchi, O., Yoshimura, K., Horimoto, K., Kitazawa, M., et al. (2020). CD4(+) T-cell Immunity in the Peripheral Blood Correlates with Response to Anti-PD-1 Therapy. Cancer Immunol. Res. 8, 334–344. doi: 10.1158/2326-6066.CIR-19-0574
Kalbasi, A., Ribas, A. (2020). Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39. doi: 10.1038/s41577-019-0218-4
Kamada, T., Togashi, Y., Tay, C., Ha, D., Sasaki, A., Nakamura, Y., et al. (2019). PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. United States America 116, 9999–10008. doi: 10.1073/pnas.1822001116
Kamphorst, A. O., Pillai, R. N., Yang, S., Nasti, T. H., Akondy, R. S., Wieland, A., et al. (2017). Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. United States America 114, 4993–4998. doi: 10.1073/pnas.1705327114
Karwacz, K., Bricogne, C., Macdonald, D., Arce, F., Bennett, C. L., Collins, M., et al. (2011). PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8(+) T cells. EMBO Mol. Med. 3, 581–592. doi: 10.1002/emmm.201100165
Karwacz, K., Arce, F., Bricogne, C., Kochan, G., Escors, D. (2012). PD-L1 co-stimulation, ligand-induced TCR down-modulation and anti-tumor immunotherapy. Oncoimmunology 1, 86–88. doi: 10.4161/onci.1.1.17824
Kato, S., Goodman, A., Walavalkar, V., Barkauskas, D. A., Sharabi, A., Kurzrock, R. (2017). Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 23, 4242–4250. doi: 10.1158/1078-0432.CCR-16-3133
Kattan, J., Kattan, C., Farhat, F., Assi, T. (2019). Overcoming the resistance to BRAF inhibitor by the double BRAF and MEK inhibitions in advanced melanoma: a case report. Anticancer Drugs 30, 1052–1054. doi: 10.1097/CAD.0000000000000827
Keir, M. E., Butte, M. J., Freeman, G. J., Sharpe, A. H. (2008). PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704. doi: 10.1146/annurev.immunol.26.021607.090331
Kerkar, S. P., Goldszmid, R. S., Muranski, P., Chinnasamy, D., Yu, Z., Reger, R. N., et al. (2011). IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J. Clin. Invest. 121, 4746–4757. doi: 10.1172/JCI58814
Khong, H. T., Restifo, N. P. (2002). Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 3, 999–1005. doi: 10.1038/ni1102-999
Kim, J. Y., Lee, K. H., Kang, J., Borcoman, E., Saada-Bouzid, E., Kronbichler, A., et al. (2019). Hyperprogressive Disease during Anti-PD-1 (PDCD1) / PD-L1 (CD274) Therapy: A Systematic Review and Meta-Analysis. Cancers 11, 1699. doi: 10.3390/cancers11111699
Latchman, Y., Wood, C. R., Chernova, T., Chaudhary, D., Borde, M., Chernova, I., et al. (2001). PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268. doi: 10.1038/85330
Latchman, Y. E., Liang, S. C., Wu, Y., Chernova, T., Sobel, R. A., Klemm, M., et al. (2004). PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. United States America 101, 10691–10696. doi: 10.1073/pnas.0307252101
Le, D. T., Uram, J. N., Wang, H., Bartlett, B. R., Kemberling, H., Eyring, A. D., et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl. J. Med. 372, 2509–2520. doi: 10.1056/NEJMoa1500596
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., et al. (2017). , Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. doi: 10.1126/science.aan6733
Lichtenegger, F. S., Rothe, M., Schnorfeil, F. M., Deiser, K., Krupka, C., Augsberger, C., et al. (2018). Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front. Immunol. 9, 385. doi: 10.3389/fimmu.2018.00385
Liechtenstein, T., Perez-Janices, N., Blanco-Luquin, I., Schwarze, J., Dufait, I., Lanna, A., et al. (2014). Anti-melanoma vaccines engineered to simultaneously modulate cytokine priming and silence PD-L1 characterized using ex vivo myeloid-derived suppressor cells as a readout of therapeutic efficacy. Oncoimmunology 3, e29178. doi: 10.4161/21624011.2014.945378
Liechtenstein, T., Perez-Janices, N., Gato, M., Caliendo, F., Kochan, G., Blanco-Luquin, I., et al. (2014). A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget 5, 7843–7857. doi: 10.18632/oncotarget.2279
Lo Russo, G., Moro, M., Sommariva, M., Cancila, V., Boeri, M., Centonze, G., et al. (2019). Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin. Cancer Res. 25, 989–999. doi: 10.1158/1078-0432.CCR-18-1390
Martins, F., Sofiya, L., Sykiotis, G. P., Lamine, F., Maillard, M., Fraga, M., et al. (2019). Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 16, 563–580. doi: 10.1038/s41571-019-0218-0
Matson, V., Fessler, J., Bao, R., Chongsuwat, T., Zha, Y., Alegre, M. L., et al. (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108. doi: 10.1126/science.aao3290
Matsushita, H., Vesely, M. D., Koboldt, D. C., Rickert, C. G., Uppaluri, R., Magrini, V. J., et al. (2012). Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404. doi: 10.1038/nature10755
Meyer, C., Cagnon, L., Costa-Nunes, C. M., Baumgaertner, P., Montandon, N., Leyvraz, L., et al. (2013). Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. CII. 63, 247–257. doi: 10.1007/s00262-013-1508-5
Motzer, R. J., Rini, B. I., McDermott, D. F., Redman, B. G., Kuzel, T. M., Harrison, M. R., et al. (2015). Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. 33, 1430–1437. doi: 10.1200/JCO.2014.59.0703
Niemeijer, A. N., Leung, D., Huisman, M. C., Bahce, I., Hoekstra, O. S., van Dongen, G., et al. (2018). Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 9, 4664. doi: 10.1038/s41467-018-07131-y
Nishino, M., Ramaiya, N. H., Hatabu, H., Hodi, F. S. (2017). Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 14, 655–668. doi: 10.1038/nrclinonc.2017.88
O’Donnell, J. S., Teng, M. W. L., Smyth, M. J. (2019). Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167. doi: 10.1038/s41571-018-0142-8
Onesti, C. E., Freres, P., Jerusalem, G. (2019). Atypical patterns of response to immune checkpoint inhibitors: interpreting pseudoprogression and hyperprogression in decision making for patients’ treatment. J. Thorac. Dis. 11, 35–38. doi: 10.21037/jtd.2018.12.47
Pages, F., Mlecnik, B., Marliot, F., Bindea, G., Ou, F. S., Bifulco, C., et al. (2018). International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391, 2128–2139. doi: 10.1016/S0140-6736(18)30789-X
Pardoll, D. (2015). Cancer and the Immune System: Basic Concepts and Targets for Intervention. Semin. Oncol. 42, 523–538. doi: 10.1053/j.seminoncol.2015.05.003
Pascolutti, R., Sun, X., Kao, J., Maute, R. L., Ring, A. M., Bowman, G. R., et al. (2016). Structure and Dynamics of PD-L1 and an Ultra-High-Affinity PD-1 Receptor Mutant. Structure 24, 1719–1728. doi: 10.1016/j.str.2016.06.026
Patsoukis, N., Li, L., Sari, D., Petkova, V., Boussiotis, V. A. (2013). PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol. Cell Biol. 33, 3091–3098. doi: 10.1128/MCB.00319-13
Pitt, J. M., Marabelle, A., Eggermont, A., Soria, J. C., Kroemer, G., Zitvogel, L. (2016). Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 27, 1482–1492. doi: 10.1093/annonc/mdw168
Plas, D. R., Johnson, R., Pingel, J. T., Matthews, R. J., Dalton, M., Roy, G., et al. (1996). Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science 272, 1173–1176. doi: 10.1126/science.272.5265.1173
Prasad, V., De Jesus, K., Mailankody, S. (2017). The high price of anticancer drugs: origins, implications, barriers, solutions. Nat. Rev. Clin. Oncol. 14, 381–390. doi: 10.1038/nrclinonc.2017.31
Restifo, N. P., Marincola, F. M., Kawakami, Y., Taubenberger, J., Yannelli, J. R., Rosenberg, S. A. (1996). Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 88, 100–108. doi: 10.1093/jnci/88.2.100
Restifo, N. P., Smyth, M. J., Snyder, A. (2016). Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 16, 121–126. doi: 10.1038/nrc.2016.2
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. doi: 10.1126/science.aaa1348
Routy, B., Le Chatelier, E., Derosa, L., Duong, C. P. M., Alou, M. T., Daillere, R., et al. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97. doi: 10.1126/science.aan3706
Saada-Bouzid, E., Defaucheux, C., Karabajakian, A., Coloma, V. P., Servois, V., Paoletti, X., et al. (2017). Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 28, 1605–1611. doi: 10.1093/annonc/mdx178
Saito, T., Yokosuka, T., Hashimoto-Tane, A. (2010). Dynamic regulation of T cell activation and co-stimulation through TCR-microclusters. FEBS Lett. 584, 4865–4871. doi: 10.1016/j.febslet.2010.11.036
Sakaguchi, S., Yamaguchi, T., Nomura, T., Ono, M. (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. doi: 10.1016/j.cell.2008.05.009
Schildberg, F. A., Klein, S. R., Freeman, G. J., Sharpe, A. H. (2016). Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 44, 955–972. doi: 10.1016/j.immuni.2016.05.002
Schmitt, A. M., Chang, H. Y. (2016). Long Noncoding RNAs in Cancer Pathways. Cancer Cell 29, 452–463. doi: 10.1016/j.ccell.2016.03.010
Schreiber, R. D., Old, L. J., Smyth, M. J. (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570. doi: 10.1126/science.1203486
Schumacher, T. N., Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. doi: 10.1126/science.aaa4971
Sharma, P., Hu-Lieskovan, S., Wargo, J. A., Ribas, A. (2017). Primary. Adapt. Acquir. Resist. Cancer Immunother. Cell 168, 707–723. doi: 10.1016/j.cell.2017.01.017
Sharpe, A. H., Wherry, E. J., Ahmed, R., Freeman, G. J. (2007). The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 8, 239–245. doi: 10.1038/ni1443
Sheppard, K. A., Fitz, L. J., Lee, J. M., Benander, C., George, J. A., Wooters, J., et al. (2004). PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 574, 37–41. doi: 10.1016/j.febslet.2004.07.083
Shin, D. S., Zaretsky, J. M., Escuin-Ordinas, H., Garcia-Diaz, A., Hu-Lieskovan, S., Kalbasi, A., et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discovery 7, 188–201. doi: 10.1158/2159-8290.CD-16-1223
Sivan, A., Corrales, L., Hubert, N., Williams, J. B., Aquino-Michaels, K., Earley, Z. M., et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. doi: 10.1126/science.aac4255
Spitzer, M. H., Carmi, Y., Reticker-Flynn, N. E., Kwek, S. S., Madhireddy, D., Martins, M. M., et al. (2017). Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502 e15. doi: 10.1016/j.cell.2016.12.022
Srivastava, M. K., Zhu, L., Harris-White, M., Kar, U. K., Huang, M., Johnson, M. F., et al. (2012). Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PloS One 7, e40677. doi: 10.1371/annotation/5c756e7d-6e97-416f-836a-dced97cf46af
Taube, J. M., Anders, R. A., Young, G. D., Xu, H., Sharma, R., McMiller, T. L., et al. (2012). Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 4, 127ra37. doi: 10.1126/scitranslmed.3003689
Teng, M. W., Galon, J., Fridman, W. H., Smyth, M. J. (2015). From mice to humans: developments in cancer immunoediting. J. Clin. Invest. 125, 3338–3346. doi: 10.1172/JCI80004
Teng, M. W., Ngiow, S. F., Ribas, A., Smyth, M. J. (2015). Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 75, 2139–2145. doi: 10.1158/0008-5472.CAN-15-0255
Theivanthiran, B., Evans, K. S., DeVito, N. C., Plebanek, M. P., Sturdivant, M., Wachsmuth, L. P., et al. (2020). A tumor-intrinsic PD-L1-NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Invest. pii: 133055. doi: 10.1172/JCI133055
Topalian, S. L., Weiner, G. J., Pardoll, D. M. (2011). Cancer immunotherapy comes of age. J. Clin. Oncol. 29, 4828–4836. doi: 10.1200/JCO.2011.38.0899
Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott, D. F., et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl. J. Med. 366, 2443–2454. doi: 10.1056/NEJMoa1200690
Tseng, S. Y., Otsuji, M., Gorski, K., Huang, X., Slansky, J. E., Pai, S. I., et al. (2001). B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193, 839–846. doi: 10.1084/jem.193.7.839
Vetizou, M., Pitt, J. M., Daillere, R., Lepage, P., Waldschmitt, N., Flament, C., et al. (2015). Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084. doi: 10.1126/science.aad1329
Viehl, C. T., Moore, T. T., Liyanage, U. K., Frey, D. M., Ehlers, J. P., Eberlein, T. J., et al. (2006). Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Ann. Surg. Oncol. 13, 1252–1258. doi: 10.1245/s10434-006-9015-y
Wolchok, J. D., Hoos, A., O’Day, S., Weber, J. S., Hamid, O., Lebbe, C., et al. (2009). Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin. Cancer Res. 15, 7412–7420. doi: 10.1158/1078-0432.CCR-09-1624
Yuan, J., Hegde, P. S., Clynes, R., Foukas, P. G., Harari, A., Kleen, T. O., et al. (2016). Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J. Immunother. Cancer 4, 3. doi: 10.1186/s40425-016-0107-3
Zaretsky, J. M., Garcia-Diaz, A., Shin, D. S., Escuin-Ordinas, H., Hugo, W., Hu-Lieskovan, S., et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl. J. Med. 375, 819–829. doi: 10.1056/NEJMoa1604958
Zheng, Y., Tian, X., Wang, T., Xia, X., Cao, F., Tian, J., et al. (2019). Long noncoding RNA Pvt1 regulates the immunosuppression activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Mol. Cancer 18, 61. doi: 10.1186/s12943-019-0978-2
Zhou, Y., Zhu, Y., Xie, Y., Ma, X. (2019). The Role of Long Non-coding RNAs in Immunotherapy Resistance. Front. Oncol. 9, 1292. doi: 10.3389/fonc.2019.01292
Zuazo, M., Arasanz, H., Fernandez-Hinojal, G., Gato-Canas, M., Hernandez-Marin, B., Martinez-Aguillo, M., et al. (2018). Highly differentiated CD4 T cells Unequivocally Identify Primary Resistance and Risk of Hyperprogression to PD-L1/PD-1 Immune Checkpoint Blockade in Lung Cancer. bioRxiv, 320176. doi: 10.1101/320176
Keywords: immune checkpoint blockade, programmed cell-death protein 1, programmed cell-death 1 ligand 1, immunotherapy, tumor-intrinsic resistance, tumor-extrinsic resistance, biomarkers
Citation: Chocarro de Erauso L, Zuazo M, Arasanz H, Bocanegra A, Hernandez C, Fernandez G, Garcia-Granda MJ, Blanco E, Vera R, Kochan G and Escors D (2020) Resistance to PD-L1/PD-1 Blockade Immunotherapy. A Tumor-Intrinsic or Tumor-Extrinsic Phenomenon? Front. Pharmacol. 11:441. doi: 10.3389/fphar.2020.00441
Received: 20 January 2020; Accepted: 20 March 2020;
Published: 07 April 2020.
Edited by:
Jie Xu, Fudan University, ChinaCopyright © 2020 Chocarro de Erauso, Zuazo, Arasanz, Bocanegra, Hernandez, Fernandez, Garcia-Granda, Blanco, Vera, Kochan and Escors. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grazyna Kochan, grazyna.kochan@navarra.es; David Escors, descorsm@navarra.es
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.