 Qingyang Lei
Qingyang Lei Dan Wang
Dan Wang Kai Sun
Kai Sun Liping Wang
Liping Wang Yi Zhang
Yi Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 21 July 2020
Sec. Molecular and Cellular Oncology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00672
In cancer-immunity cycle, the immune checkpoint PD1 and its ligand PDL1 act as accomplices to help tumors resist to immunity-induced apoptosis and promote tumor progression. Immunotherapy targeting PD1/PDL1 axis can effectively block its pro-tumor activity. Anti-PD1/PDL1 therapy has achieved great success in the past decade. However, only a subset of patients showed clinical responses. Most of the patients can not benefit from anti-PD1/PDL1 therapy. Furthermore, a large group of responders would develop acquired resistance after initial responses. Therefore, understanding the mechanisms of resistance is necessary for improving anti-PD1/PDL1 efficacy. Currently, researchers have identified primary resistance mechanisms which include insufficient tumor immunogenicity, disfunction of MHCs, irreversible T cell exhaustion, primary resistance to IFN-γ signaling, and immunosuppressive microenvironment. Some oncogenic signaling pathways also contribute to the primary resistance. Under the pressure applied by anti-PD1/PDL1 therapy, tumors experience immunoediting and preserve beneficial mutations, upregulate the compensatory inhibitory signaling and induce re-exhaustion of T cells, all of which may attenuate the durability of the therapy. Here we explore the underlying mechanisms in detail, review biomarkers that help identifying responders among patients and discuss the strategies that may relieve the anti-PD1/PDL1 resistance.
Cancer immunotherapy has achieved great clinical advances over the past few years, especially in the fields of adoptive cell transfer therapy and the application of immune checkpoint blockade (ICB). Among those developing and inspiring immunotherapies, ICB targeting programmed cell death 1/programmed cell death ligand 1 (PD1/PDL1) axis is famous for the Nobel Prize in 2018 and has been approved for use in different solid tumors.
Antigen-specific T cells play pivotal roles in the process of eliminating tumor cells. Tumor cells trigger the cancer-immunity cycle once they release tumor antigens (Chen and Mellman, 2013). Antigen-specific T cells initially undergo recognitions of tumor antigens presented by major histocompatibility complexes (MHCs) on antigen presenting cells (APCs), and subsequently experience priming and activation. After activation and proliferation, T cells travel to the particular sites following chemokine concentration gradient. For an optimal activation, except that costimulatory signal transductions are required between T cells and APCs, immune checkpoints like PD1 and its ligand PDL1 are also needed to avert the excessive activation of T cells (Chen and Mellman, 2013; Maimela et al., 2018). However, researchers have found that tumors accommodate themselves to resisting the apoptosis induced by immune system in multiple ways. One tactic tumors use to escape immune rejection is to take advantage of immune checkpoints (Pardoll, 2015).
Upon confronting the same antigen on MHCs, T cells release IFN-γ to enhance efficiency of tumor killing. The release of IFN-γ from CD8+ T cells upregulate the expression of PDL1 on tumor cells and stromal cells (Ribas, 2015; Garcia-Diaz et al., 2017). Meanwhile, TCR signaling upregulates the expression of PD1 on T cell surface which binds to PDL1 to exert negative regulatory effects and blunt the antitumor function of T cells (Agata et al., 1996; Ribas and Wolchok, 2018; Akinleye and Rasool, 2019). Immunotherapy against the interaction between PD1 and PDL1 reinvigorates T cells that were inactive because of the PD1/PDL1 signaling inhibition. It has been proved that PD1/PDL1 blockade significantly enhances antitumor effects in different solid tumors including melanoma, non–small cell lung cancer (NSCLC), renal cell carcinoma, head and neck squamous cell carcinoma, urothelial carcinoma, microsatellite instability–high colorectal cancer etc., and the list is still growing (Ansell et al., 2015; Motzer et al., 2015; Robert et al., 2015; Chow et al., 2016; Kaufman et al., 2016; Bellmunt et al., 2017; El-Khoueiry et al., 2017; Overman et al., 2017; Powles et al., 2017; Sharma et al., 2017b; Fuchs et al., 2018; Akinleye and Rasool, 2019; Chung et al., 2019; Figure 1).
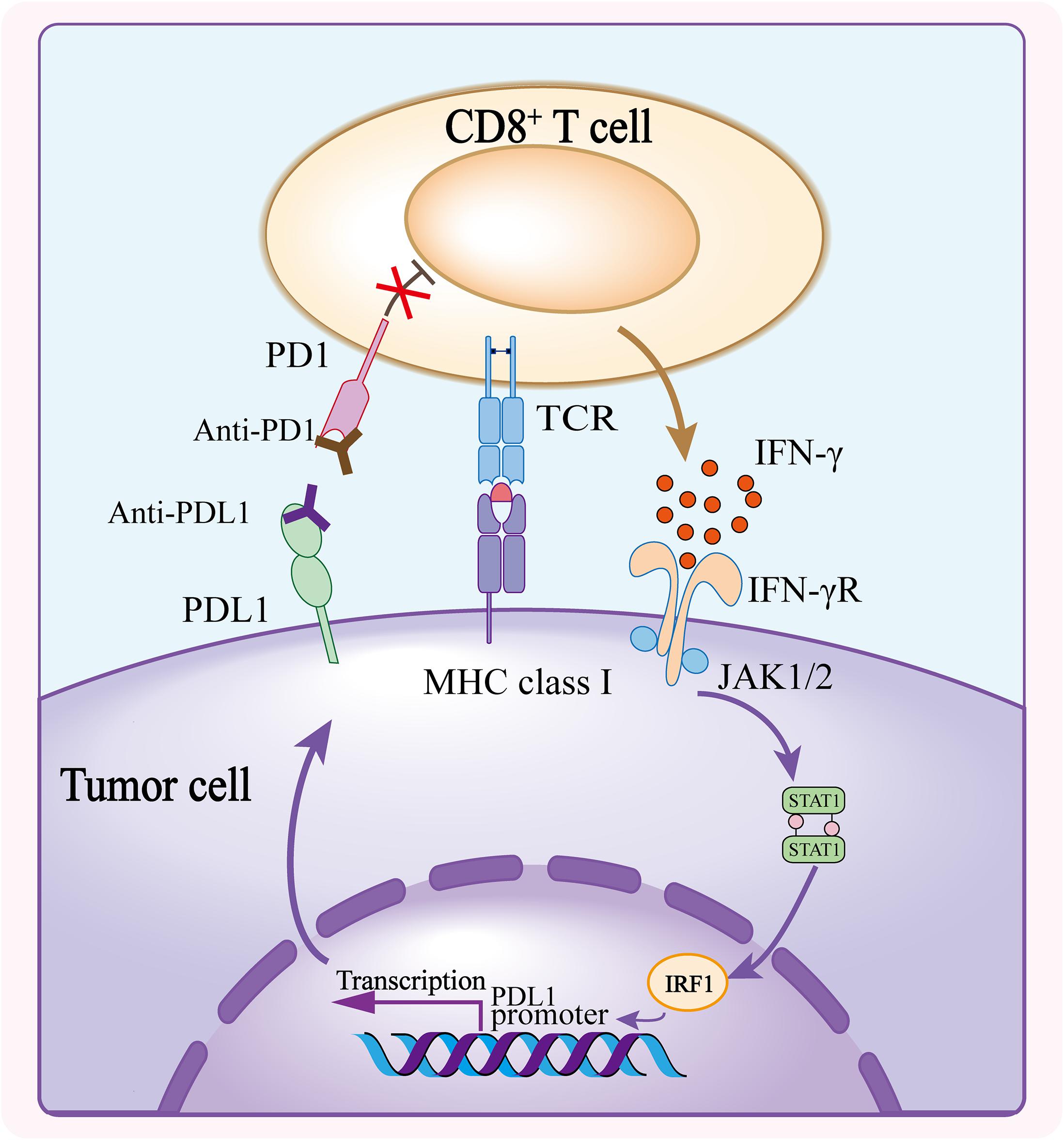
Figure 1. Mechanism of PD1/PDL1 blockade. The CD8+ T cell activates upon recognizing the tumor antigen presented on MHC class I and releases IFN-γ to bind to IFN-γ receptor, and consequently induces the expression of PDL1 on tumor cells. PDL1 conjugates the elevated PD1 on T cell surface, triggering inhibitory effect of PD1/PDL1 axis. Anti-PD1 or anti-PDL1 antibody blocks the interaction of PD1 and PDL1, and abolishes the inhibition of CD8+ T cell thus enhancing the antitumor activity.
However, clinical data of anti-PD1/PDL1 therapy showed limited response rates. Previous investigations revealed that a large group of patients suffered primary resistance and did not respond to PD1/PDL1 blockade, and some of the responders developed acquired resistance after initial responses as well (Sharma et al., 2017a). More disturbingly, the underlying mechanisms largely remain unknown.
In this review, we will focus on documented mechanisms of primary and acquired resistance of anti-PD1/PDL1 therapy and summarize the biomarkers that can potentially discriminate responders from non-responders. Then we will discuss the development of strategies for enhancing the antitumor efficacy and overcoming the resistance of anti-PD1/PDL1 therapy.
Despite the fact that the immunotherapy against cancer PD1/PDL1 axis is becoming more feasible, the limited efficacy of anti-PD1/PDL1 therapy was observed in patients with different solid tumors. Anti-PD1/PDL1 therapy works through strengthening the functions of preexisting CD8+ T cells (Tumeh et al., 2014; Tang et al., 2016). Nonetheless, in anti-PD1/PDL1 therapy, tumors can escape tumor rejection by shaping a hostile tumor microenvironment (TME) to impede the antitumor efficacy of T cells. This may occur due to insufficient antigen immunogenicity, disfunction of antigen presentation, irreversible T cell exhaustion, resistance of IFN-γ signaling, and immunosuppressive TME (Figure 2).
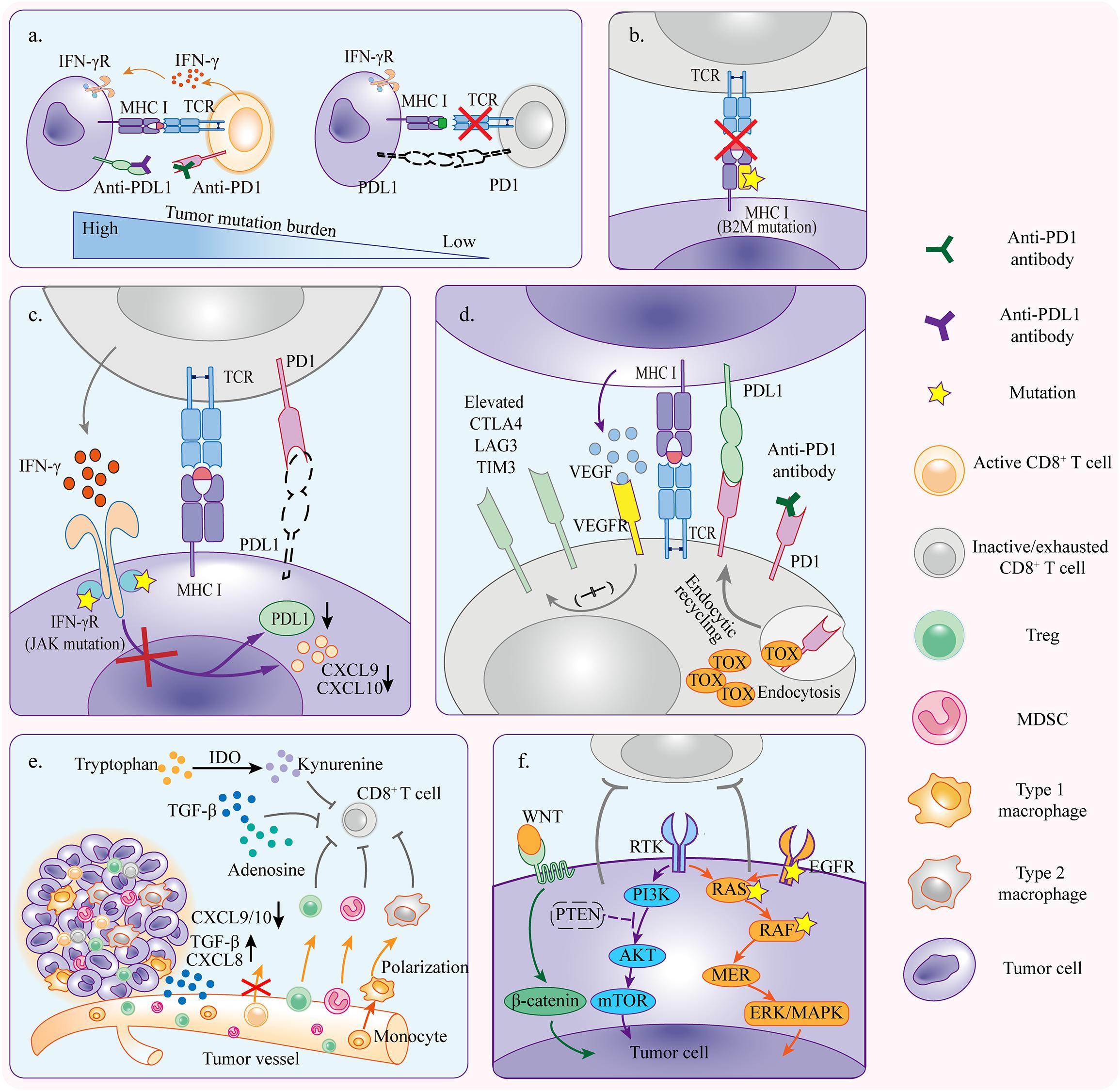
Figure 2. Mechanisms of primary resistance. (a) Tumors with high mutation burden are more likely to respond to anti-PD1/PDL1 therapy because there is a greater possibility of generating immunogenic neoantigens which activate CD8+ T cells and induce tumor rejection reactivity. (b) Tumor cells with primary B2M mutation fail to present tumor antigens and elicit antitumor immunity which is required for tumor cell killing. (c) Tumor cells resistant to IFN-γ signaling because of primary JAK1/2 mutation can not induce PDL1 upregulation but inhibit T cell reactivity in PD1/PDL1 independent pathway. The inactivation of IFN-γ signaling also downregulates CXCL9 and CXCL10 expressions which are necessary for T cell trafficking. (d) Alternative immune checkpoints are upregulated in tumor infiltrating T cells, and thus only blocking PD1/PDL1 axis is not enough to rescue the severely exhausted T cells. Upregulation of VEGFR signaling and TOX expression aggravates the activation of inhibitory signaling. (e) TME contains diverse immunosuppressive cells that influence anti-PD1/PDL1 efficacy through inhibiting T cell reactivity. Cytokines derived from tumors recruit more immunosuppressive cells to TME and promote polarization toward pro-tumor phenotype. (f) Oncogene mutation and abnormal activation contribute to inhibition of antitumor immunity, resulting in primary resistance to anti-PD1/PDL1 therapy.
The antitumor efficacy of PD1/PDL1 blockade depends on the existence of antigen-specific T cell reactivity in TME, which requires potential tumor rejection antigens presented by dendritic cells (DCs) to cross-priming CD8+ T cells and triggering subsequent antitumor activity (Mempel et al., 2004). Theoretically, there are two classes of antigens capable of inducing antitumor immunity; one is formed by non-mutated proteins with incomplete T cell tolerance and another one is formed by mutated proteins which are known as neoantigens (Heemskerk et al., 2013). Deep sequencing technology has helped to identify non-synonymous mutations which are able to generate neoantigens in tumors (Schumacher and Schreiber, 2015). Exome sequencing of two independent cohorts of NSCLC patients who have received anti-PD1 therapy showed that the non-synonymous mutational burden was higher in patients with durable clinical benefit (Rizvi et al., 2015). Further analysis of neoantigen landscape demonstrated that it was the candidate neoantigen burden which reflected the quantity of putative HLA class epitopes, rather than frequency of non-synonymous mutation that related to anti-PD1 response (Rizvi et al., 2015). Coincidentally, tissues derived from melanoma patients who were sensitive to PD1 blockade harbored more non-synonymous single nucleotide variants and were detected with higher putative HLA class I and class II neoepitope loads compared with those from non-responders’ (Hugo et al., 2016). Extrapolating from those evidences, we can speculate that tumors with high mutational burden have higher probability to generate more neoantigens with adequate immunogenicity capable of inducing antigen-specific T cell reactivity. Based on the evidence that the infiltration of CD8+ T cells is higher in tumors with higher mutational burden, neoantigens are thought particularly relevant to antitumor immunity (Brown et al., 2014). Moreover, cancers deficient in mismatch repair genes harbored high numbers of mutations that would generate neoantigens, making them sensitive to anti-PD1 therapy despite of different tissue origins (Le et al., 2017). On the contrary, tumors with low mutational burdens, such as prostate and pancreatic cancer, are more likely not to respond to therapy, due to the lack of immunogenic neoantigens (Schumacher and Schreiber, 2015).
Antigen presentation mainly occurs through MHC class I pathway in TME, and thus tumors can escape T cell killing by inactivating MHC class I complex. B2M is essential for assembly and stabilization of HLA class I complex (Hulpke and Tampé, 2013). Hypothetically, disfunction of HLA class I due to B2M mutation causes antigen-presenting malfunction, resulting in the attenuation of T cell cytotoxicity. Indeed, an investigation of tumor tissues illustrated that B2M mutations were frequently present in CRC with microsatellite instability, indicating the existence of B2M mutation in pre-therapy MSI-H CRC (Yeon Yeon et al., 2019). Further, a more direct evidence revealed that loss of heterozygosity (LOH) at B2M locus happened three folds higher in non-responders compared with responders from a cohort of melanoma patients treated with PD1 blockade (Sade-Feldman et al., 2017). Similar phenomenon was also observed in an anti-CTLA4 treated cohort, proposing B2M mutation as a common mechanism of primary ICB resistance. Collectively, tumors may resist to anti-PD1/PDL1 therapy by disturbing MHC class I function with B2M mutation.
T cell exhaustion has been viewed as a dysfunctional state upon long-lasting existence of tumor antigens and immunosuppressive TME (Wherry and Kurachi, 2015). Considering that PD1/PDL1 blockade works by reinvigorating impaired cytotoxic T cells, mechanisms that engender irreversible T cell exhaustion probably contribute to anti-PD1/PDL1 resistance (McLane et al., 2019; Zhang Z. et al., 2020). Exhausted CD8+ T cells express multiple inhibitory receptors, encompassing PD1, T-cell immunoglobulin and mucin-domain containing 3 (TIM3), lymphocyte-activation gene 3 (LAG3) and cytotoxic T lymphocyte-associated antigen 4 (CTLA4). It is supposed that anti-PD1/PDL1 antibodies can only surmount part of the suppressive signaling in TME, but there are more inhibitory axes that preclude the function of T cells in the TME. Exactly, analysis of tissues from NSCLC patients elucidated that TIM3 was upregulated on T cell surface, resulting in anti-PD1 resistance (Limagne et al., 2019). In addition, high amount of tumor-derived VEGF induced high level of PD1, CTLA4, TIM3, and LAG3 expression which were makers of exhaustion on the surface of CD8+ T cells, contributing to resistance in anti-PD1 treatment (Voron et al., 2015). Importantly, the amount of PD1 expression on T cell surface also interferes the efficacy of anti-PD1 therapy. T cells that express high level of PD1 along with multiple inhibitory receptors from NSCLC patients had poor functional recovery after anti-PD1 therapy (Thommen et al., 2015). Severely exhausted CD8+ T cells that express high level of PD1 do not respond to PD1 blockade and, instead, T cells with intermediate expression level of PD1 is responsive to anti-PD1 therapy (Blackburn et al., 2008; Wherry, 2011). Thymocyte selection-associated high mobility group box gene (TOX) was reported as a positive regulator of PD1, TIM3, TIGIT, and CTLA4 expression in the tumor infiltrating CD8+ T cells, regulating CD8+ T cell exhaustion. The expression level of TOX on tumor infiltrating CD8+ T cells was negatively correlated with anti-PD1 response rates in melanoma and NSCLC patients (Kim et al., 2020). TOX was reported maintaining the PD1 expression on T cell surface by mediating endocytic recycling of PD1, and overexpression of TOX exacerbated exhaustion of CD8+ T cells, which could hinder the CD8+ T cells from responding to anti-PD1 therapy (Khan et al., 2019; Wang et al., 2019).
IFN-γ produced by T cells upon tumor antigen recognition signals through IFN-γ receptor and consequently results in expression of IFN-γ stimulated genes by inducing activation of Janus kinase JAK1/2, and phosphorylation of the signal transducers and activators of transcription (STAT) (Bach et al., 1997; Platanias, 2005). Most of the stimulated genes benefit to antitumor immunity, while others like CD274 (encoding PDL1) lead to inactivation of tumor-specific T cells (Ribas, 2015). Mutations of JAK1/2 disrupt the IFN-γ signaling transduction and lead to paucity of PDL1 expression. Despite high tumor mutational burden (TMB) being often considered as a marker of responsive anti-PD1/PDL1 therapy, studies revealed that the resistance of PD1/PDL1 blockade in some high-mutated tumors was probably attributed to the JAK1/2 mutations. Researchers analyzed samples from melanoma and colon cancer patients who were tested having a high TMB, yet did not respond to PD1 blockade therapy (Shin et al., 2017). They found that those patients had homozygous loss-of-function mutations in JAK1/2, which led to deficiency of PDL1 expression even in the presence of IFN-γ, making it fruitless to block PD1 and PDL1 interaction. Moreover, the JAK1/2 controls expression of chemokines (e.g., CXCL9, CXCL10, and CXCL11) which are potent to attract T cells. Therefore, it was rational that tumors with loss-of-function mutations of JAK1 were indeed short of T-cell infiltration (Shin et al., 2017).
Tumor cells educate surrounding environment to suppress antitumor immunity and support their proliferation, differentiation, expansion, and invasion. Immunosuppressive cells, cytokines and tumor metabolites in the microenvironment restrain antitumor efficacy (Gajewski et al., 2013; Li X. et al., 2019). Regulatory T cells (Tregs) act as negative mediators of antigen-specific T cell function, which gives the privilege to tumors for escaping the antitumor immunity (Tanaka and Sakaguchi, 2017). Tregs suppress activation, proliferation and functions of CD8+ T cells through generating immunosuppressive substances such as IL-10, TGF-β and extracellular adenosine, depriving IL-2 in TME, and constitutively expressing CTLA4 (Tanaka and Sakaguchi, 2017). Increased infiltration of Tregs in tumors is correlated with poor prognosis (Sasada et al., 2003; Curiel et al., 2004; Bates et al., 2006). In vivo studies showed that Tregs which induced high level of PD1 expression in CD8+ T cells were responsible for the primary anti-PD1 resistance (Ngiow et al., 2015).
Myeloid-derived suppressive cells (MDSCs) are a group of immature myeloid cells with suppressive competence in tumor microenvironment. MDSCs consist of two large groups of cells: granulocytic or polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). MDSCs produce immunosuppressive factors including but not limited to ROS, NO, and IL-10, through which can suppress both antigen-specific and non-specific T cell response, and instigate tumor invasion and angiogenesis (Marvel and Gabrilovich, 2015; Veglia et al., 2018). Besides, it is reported that the increased galectin-9+ M-MDSC in peripheral blood of NSCLC patients is involved in resistance of anti-PD1 therapy (Limagne et al., 2019). Thereby, the presence of MDSCs in TME is detrimental for anti-PD1/PDL1 response. As expected, several studies revealed the relationship between MDSCs infiltration and PD1 blockade resistance, and selective depletion of MDSCs could restore the anti-PD1 efficacy (Highfill et al., 2014; De Henau et al., 2016).
Tumor associated macrophages (TAMs) are theoretically divided into two phenotypes: M1 macrophages and M2 macrophages. TAMs, especially those belonging to M2 phenotype, are considered to suppress functions of CTL, recruit immunosuppressive cells and promote tumor progression through secreting inhibitory cytokines and generating other suppressive factors (Yang and Zhang, 2017). Clinical studies identified a correlation between TAMs accumulation and poor clinical outcomes. Therefore, targeting TAMs is expected to induce tumor regression (Yang and Zhang, 2017; Zhou et al., 2020). Presence of TAMs in pancreatic cancer exaggerates immunosuppression within microenvironment and leads to the PD1/PDL1 blockade resistance. Inhibition of colony-stimulating factor 1 receptor (CSF1R) on TAMs can upregulate the expression of PDL1 and increase CD8+ T cell infiltration, which ablates anti-PD1/PDL1 resistance (Zhu et al., 2014).
Cytokines are key modulators in TME mediating recruitment and polarization of immune cells. For example, transforming growth factor beta (TGF-β) plays a multifaceted role in TME. TGF-β promotes tumor progression by inducing epithelial-mesenchymal transition of tumor cells, recruiting immunosuppressive cells like Tregs and MDSCs as well as inhibiting functions of CD8+ T cells (Batlle and Massagué, 2019). Studies found that TGF-β was associated with poor clinical outcomes and limited the response of anti-PDL1 therapy which was attributed to T cell exclusion in urothelial and colorectal cancer (Mariathasan et al., 2018; Tauriello et al., 2018). TGF-β1, the universal isoform of TGF-β, presents in many human cancers and contributes to anti-PD1 resistance (Martin et al., 2020). Tumor-derived chemokines recruit immunosuppressive cells including Tregs, macrophages and MDSCs to the TME to create an immunosuppressive but tumor-supportive environment, interfering the immunotherapy targeting adaptive immune resistance (Roussos et al., 2011). Elevated CXCL8 expression in tumors recruited MDSCs or M2-like TAMs that disrupted the efficacy of anti-PD1 (Highfill et al., 2014; Zhang M. et al., 2020). Furthermore, tumor-derived chemokines not only regulate suppressive cell recruitment, but also prevent T cell infiltration. Epigenetic modulation involves in silencing Th1 type chemokines such as CXCL9 and CXCL10, and thereby impeding anti-PD1/PDL1 efficacy by mediating T cell trafficking to the tumor site (Peng et al., 2015).
Indoleamine 2,3-dioxygenase (IDO) catalyzes tryptophan (TRP) catabolism to generate kynurenine (KYN). In TME, IDO creates an immunosuppressive climate through suppressing the functions of T cells while promoting generation and activation of Tregs and MDSCs (Prendergast et al., 2014). In soft tissue sarcoma treatment, the upregulation of IDO may cause anti-PD1 resistance. After the combination therapy of pembrolizumab and metronomic cyclophosphamide, the increased infiltration of IDO+ M2 failed the therapy because tumor shrinkage was observed in just 3 of the patients with only one experiencing partial response when the total number was 57 and over 40% patients expressed PDL1 in the TME (Toulmonde et al., 2018).
As a consequence of oxygen deprivation, adenosinergic pathway activates in tumors and induces the extracellular adenosine accumulation. CD39 and CD73 are two important enzymes for converting ATP into adenosine (Vijayan et al., 2017). Redundant extracellular adenosine has been identified as an inhibitor of the antitumor activity, which directly binds to A2A receptor on T cell surface or favors polarization of immune cells toward immunosuppressive phenotypes (Cekic et al., 2014; Vijayan et al., 2017). Consistently, preclinical studies demonstrated an enhancement of anti-PD1 efficacy when combining anti-PD1/PDL1 therapy with treatment of A2A receptor antagonist (Beavis et al., 2015; Willingham et al., 2018).
Studies have demonstrated that oncogenic signaling plays a pivotal role in the formation of immunosuppressive TME and promoting tumor immune escape (Spranger and Gajewski, 2016; Yang et al., 2019). The contribution of tumor-intrinsic oncogenic pathways for resistance of PD1/PDL1 blockade should not be overlooked.
EGFR is an important molecular phenotype of NSCLC. Study found that EGFR mutation rates were negatively correlated with PDL1 expression which is necessary for PD1/PDL1 blockade (Ji et al., 2016). A retrospective analysis elucidated that patients with EGFR and ALK mutation had poor response rates when treated with anti-PD1/PDL1 therapy. Intriguingly, patients with EGFR mutation had lower PDL1 expression level than others with ALK rearrangement. The concurrent existence of PDL1 expression and high levels of CD8+ tumor infiltrating lymphocyte (TILs) were only present in a small group of EGFR mutant tumors, suggesting an underlying mechanism of anti-PD1/PDL1 resistance in patients with EGFR or ALK mutation (Gainor et al., 2016).
NSCLC patients with KRAS mutation were reported more responsive to anti-PD1/PDL1 therapy. The mechanism is that KRAS mutation promotes infiltration of CD8+ T cells and increases TMB and tumor immunogenicity (Liu et al., 2020). However, another study revealed that KRAS mutation in tumors can cause anti-PD1 resistance by repressing IRF2 expression which correspondingly promoted the recruitment of MDSC through increasing CXCL3 expression (Liao et al., 2019). Combining PD1 blockade with IRF2 overexpression or CXCR2 (CXCL3 receptor) blockade relieved anti-PD1 resistance (Liao et al., 2019). Moreover, co-mutation in KRAS and STK11/LKB1 contributed to a lower objective response rate in lung adenocarcinoma treated with nivolumab (Skoulidis et al., 2018).
Mitogen-activated protein kinase (MAPK) pathway mediates multiple cellular processes, such as proliferation, apoptosis, and migration. The aberration of MAPK pathway may give rise to carcinogenesis (Dhillon et al., 2007). Alteration of the Ras-MAPK pathway inhibits the recruitment and infiltration of T cells in triple negative breast cancer (Loi et al., 2016). Expectedly, preclinical studies demonstrated intensified efficacy of PD1/PDL1 blockade when combined with MAPK-targeted therapy (Hu-Lieskovan et al., 2015; Hugo et al., 2016; Loi et al., 2016).
Phosphatase and tensin homolog (PTEN) loss results in activation of the phosphatidylinositol-3-kinase (PI3K) pathway and consequently causes tumorigenesis (Keniry and Parsons, 2008). Evidence showed that patients with metastatic uterine leiomyosarcoma which uniquely harbored biallelic PTEN loss were resistant to anti-PD1 therapy (George et al., 2017). PTEN loss in tumor cells leads to inhibition of T cell function by secreting VEGF as well as recruiting immunosuppressive cells. Additionally, elevated expression of immunosuppressive cytokines reduced the infiltration of T cells in tumor sites (Peng et al., 2016).
Wnt/β-catenin signaling is essential for embryonic development and tissue homeostasis (MacDonald et al., 2009). Dysregulation of Wnt/β-catenin signaling pathway was observed promoting multiple carcinogenesis (Zhan et al., 2017). The abnormal activation of WNT/β-catenin signaling pathway was responsible for the impaired naïve T cell priming in melanoma (Spranger et al., 2015). In vivo study revealed that the β-catenin signaling downregulated CCL4 expression, which led to the defective recruitment of DCs to the tumor (Spranger et al., 2015).
Some patients will develop resistance or relapse eventually after the initial response in the treatment with PD1/PDL1 blockade. The host immune system sometimes is an accomplice of therapy resistance. During the course of anti-PD1/PDL1 therapy, tumor cells which enable to escape from antitumor immunity gradually occupied a predominant proportion via cancer immunoediting (O’Donnell et al., 2019). In addition, the activation of PD1/PDL1 independent inhibitory pathways and re-exhaustion of reinvigorated T cells can retard the function of T cells again in the presence of PD1/PDL1 blockade (Figure 3).
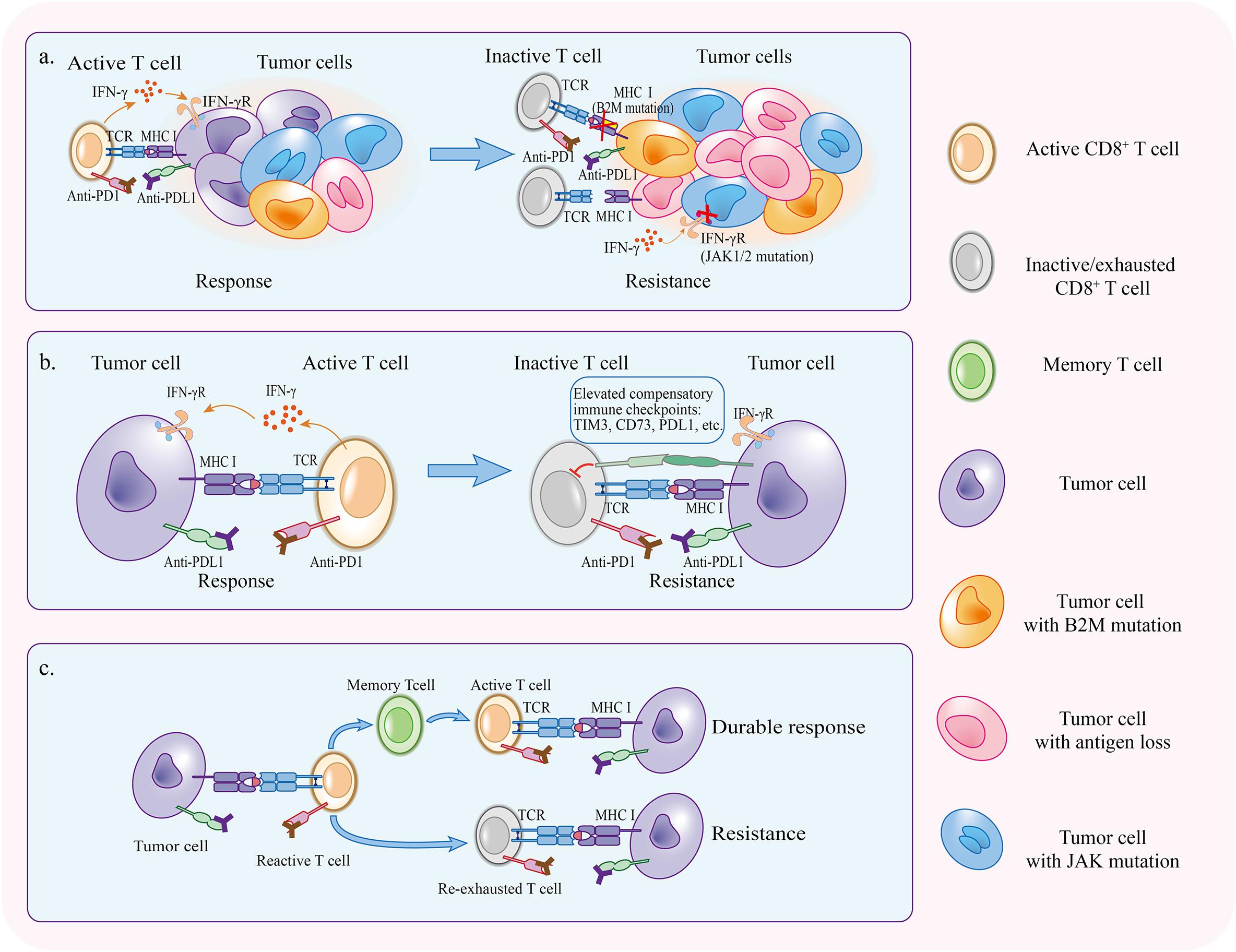
Figure 3. Mechanisms of acquired resistance. (a) Immunoediting under the pressure applied by PD1/PDL1 blockade selectively preserves tumor cells that have superiority to escape antitumor immunity. (b) Compensatory inhibitory signaling is upregulated in the course of therapy, making it difficult for PD1/PDL1 blockade to reinvigorate CD8+ T cells. (c) If tumor specific T cells fail to become memory T cells, therapy response will not persist and T cells will be re-exhausted.
The concept of immunoediting suggests that the immunity boosted by anti-PD1/PDL1 therapy not only protects the host against tumor progression but also results in selection of tumor subclones that can escape antitumor immunity, which ends up leading to the acquired resistance of anti-PD1/PDL1 therapy (Schreiber et al., 2011; Matsushita et al., 2012; O’Donnell et al., 2019). Antigen loss due to DNA copy-number alterations in tumor cells was observed in the presence of antigen-specific CTLs producing IFN-γ, indicating genetic changes of neoantigens under the pressure of tumor-specific immunity (Takeda K. et al., 2017). Neoantigens generated from non-synonymous mutation play an important role in inducing intratumoral T cell response. Immunoediting that gives rise to neoantigen loss may result in acquired anti-PD1/PDL1 resistance. An analysis of matched pretreatment and posttreatment tumor tissues derived from patient who had developed acquired resistance confirmed that the loss of 7 to 18 putative mutation-associated neoantigens, a consequence of genomic alteration, was detected in tumors with acquired resistance (Anagnostou et al., 2017). Further study found that the eliminated neoantigens were capable of eliciting clonal T cell expansion in autologous T cell culture, suggesting the T-cell-dependent immunoselection occurred under the pressure of PD1/PDL1 blockade (Anagnostou et al., 2017).
Although the B2M mutation has been observed in pre-immunotherapy samples and was partly responsible for primary resistance mechanisms of anti-PD1/PDL1 therapy, more proofs exemplified that tumors including melanoma, CRC and lung cancer acquired B2M mutations in the course of the treatment and were resistant to PD1/PDL1 blockade consequently (acquired resistance) (Zaretsky et al., 2016; Gettinger et al., 2017; Sade-Feldman et al., 2017; Yeon Yeon et al., 2019). Researchers investigated a series of specimen from biopsy of baseline, regressive disease and progressive disease of one melanoma patient who has received sequential treatment of anti-CTLA4 and anti-PD1 antibodies, and samples from anti-PD1 responders and non-responders as well (Sade-Feldman et al., 2017). They found that the LOH and upstream frameshift mutations in B2M were only extensively present during disease progression (Sade-Feldman et al., 2017), suggesting immunoediting of B2M-mutated tumor subclones has already existed earlier in tumor development, or became dominant under selective pressure applied by ICBs. Similar results have also been verified in other studies (Zaretsky et al., 2016; Gettinger et al., 2017; Le et al., 2017; Pereira et al., 2017).
In the setting of patients who developed acquired resistance after continuous anti-PD1/PDL1 therapy, tumors that were no longer sensitive to IFN-γ may survive and grow. Homozygous mutation of JAK1/2 was detected in post-treatment samples collected from melanoma patients who had tumor progression after initial response, suggesting clonal selection and proliferation of JAK-inactive tumor cells after applying the PD1 blockade (Zaretsky et al., 2016).
As was reviewed before, immunotherapy targeting PD1 or PDL1 only blocks one of many immune checkpoints, but there are still chances that tumors would escape immune elimination by activating additional inhibitory signaling or elevating PDL1 expression. A subset of melanoma expressed MHC II and was reported sensitive to anti-PD1 therapy (Johnson et al., 2016). However, another immune checkpoint LAG3 was upregulated on TILs in MHC II+ tumors in the failure of the anti-PD1 treatment after initial response (Johnson et al., 2016, 2018). In preclinical and clinical studies, the upregulation of TIM3 was detected on TILs after treatment, subsequently leading to acquired resistance in PD1/PDL1 blockade (Koyama et al., 2016; Oweida et al., 2018; Limagne et al., 2019). Furthermore, tumors that developed resistance to anti-PD1/PDL1 therapy were found accompanied with upregulation of CD73 expression or higher serum kynurenine/tryptophan ratio, indicating the presence of metabolic alteration in the mechanism of acquired resistance that limits the persistence of anti-PD1/PDL1 efficacy (Reinhardt et al., 2017; Li H. et al., 2019). Additionally, inhibitory signaling derived from suppressive TME may abolish the efficacy of PD1/PDL1 blockade. The reactivation of CD8+ T cells consequently induced the PDL1-NLRP3 inflammasome signaling cascade and eventually led to acquired resistance due to the accumulation of MDSCs (Theivanthiran et al., 2020).
Following effectivity exertion and antigen clearance, a small group of effector T cells transform into memory cells that will be reactivated and expand when reencountering tumor antigens (Harty and Badovinac, 2008). Effector memory T cells were observed predominantly increasing in responders during anti-PD1 therapy (Ribas et al., 2016). If the high level of antigen expression keeps maintained and the exhausted T cells reinvigorated by PD1/PDL1 blockade fail to become memory T cells, the durability of reinvigoration will be limited after the therapy. A study of mouse model of chronic lymphocytic choriomeningitis virus (LCMV) infection found that the exhausted T cells acquired an epigenetic profile which was distinct from memory T cells and became re-exhausted after persistent anti-PDL1 treatment, resulting in relapse which demonstrated the similar viral load compared with control (Pauken et al., 2016).
Despite the impressive success anti-PD1/PDL1 treatment achieved, the primary and acquired resistance still occurs in most of solid tumor patients. Predictive biomarkers are urgently needed to guide the treatment selection, sequentially reducing the economic burden and improving the survival of patients. According to understanding of the underlying mechanisms of PD1/PDL1 blockade, potential biomarkers basically include those which demonstrate immunogenic antigen landscape and preexisting immune context (Ren et al., 2020).
In consideration of the matter that adaptive immunity activation is the basics for PD1/PDL1 blockade, a favorable antigen recognition is required (Tumeh et al., 2014). The antigen recognition may be influenced by the immunogenicity of neoantigens that are closely relevant to TMB and MSI. High TMB and MSI involve in increased total number of neoantigens that may generate more immunogenic neoantigens which induce strong immune response (Schumacher and Schreiber, 2015). Concomitantly, TMB reflecting tumor mutational frequency has been used as an indicator in clinical settings; unresectable solid tumors with MSI-H or dMMR has been approved by FDA as one of the indications of pembrolizumab treatment (Goodman et al., 2017; Cristescu et al., 2018; Marcus et al., 2019). However, biomarkers associated with mutational load are not perfect because the existence of tumor-specific antigen is just one of many necessary conditions for driving the cancer-immunity cycle, and only a tiny part of neoantigens can be presented by MHC and recognized by TCR (Schumacher and Schreiber, 2015; Yi et al., 2018).
The presence of CD8+ T cells in TME is a prerequisite for anti-PD1/PDL1 therapy. Accordingly, the density of TILs is positively correlated with PD1/PDL1 blockade efficacy (Fridman et al., 2012). Measuring TILs by immunohistochemistry (IHC) might be promising but still needs to be standardized. PDL1 expression within tumors has been identified as “canary in a coalmine,” because it proves the existence of T cells (Ribas and Wolchok, 2018). PDL1 expression detected by IHC has been widely used as predicator in multiple cancers (Topalian et al., 2012; Garon et al., 2015; Carbone et al., 2017). However, the reliability of using IHC to detect PDL1 expression within tumor is imperfect (Patel and Kurzrock, 2015). The systemic PDL1 expression in exosome has been explored recently as a potential predictive biomarker for identifying responders (Daassi et al., 2020). Additionally, patients bearing PDL1-positive tumors are not always responsive to anti-PD1/PDL1 therapy. In contrast, PDL1-negative tumors are not meant for anti-PD1/PDL1 resistance (Gibney et al., 2016). Thus, it is not appropriate to regard PDL1 expression as a “yes or no” biomarker. In a pilot study, researchers found that majority of NSCLC patients who were not responsive to anti-PD1 therapy despite with high PDL1 expression were detected having high galectin-3 expression within tumors, suggesting a synergistic use of certain biomarkers may be superior (Capalbo et al., 2019).
Emerging evidence showed that the diversity and composition of the gut microbiota can influence anti-PD1/PDL1 efficiency (Inamura, 2020). Ayelet and colleagues first noticed that the differences among tumor bearing mice rely on distinction of commensal microbiota (Sivan et al., 2015). Oral administration of Bifidobacterium can promote the efficacy of anti-PDL1 antibody in mice (Sivan et al., 2015). In patients, commensal microbial composition was found associated with clinical anti-PD1/PDL1 response rate, and fecal microbiota transplantation from responders into germ-free or antibiotic-treated mice leveraged the restraint of PD1 blockade (Gopalakrishnan et al., 2018b; Matson et al., 2018).
Following development of research, strategies targeting different steps of tumor immune escape have been brought up. Analogous to the “whack a mole” game, unless anti-PD1/PDL1 therapy is combined with other strategies, tumors may pop up at any defenseless step of cancer-immunity cycle. In this case the combination therapies are needed for achieving better clinical outcomes (Song et al., 2018). Currently, strategies mainly focus on enhancing T cell priming, reversing T cell exhaustion, increasing T cell infiltration and improving the immunosuppressive microenvironment (Table 1).
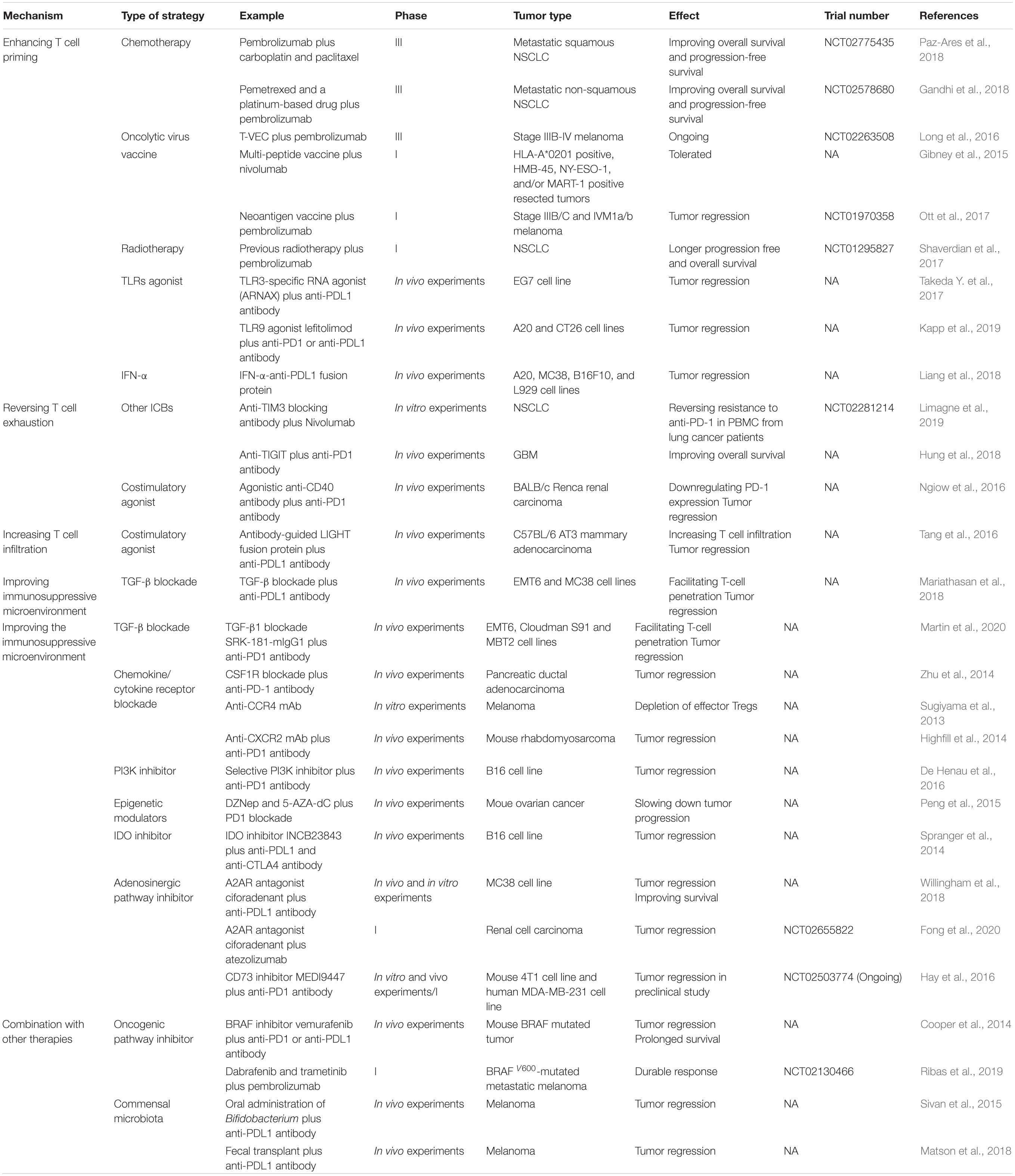
Table 1. Strategies for overcoming anti-PD1/PDL1 resistance.
Chemotherapy can promote tumor antigen release and provide damage-associated signals upon inducing tumor apoptosis, converting DCs to immunostimulatory APCs for CD8+ T cell priming (Grimaldi et al., 2020). For example, decitabine can enhance subsequent antigen recognition through upregulating MAGE-A3 expression in esophageal carcinoma (Shi et al., 2019). Some chemotherapeutic drugs, like gemcitabine and cyclophosphamide, simultaneously deplete MDSC and Tregs while killing tumor cells (Suzuki et al., 2005; van der Most et al., 2009). As expected, studies demonstrated synergistic effects in patients receiving combination of chemotherapy and PD1/PDL1 blockade (Zitvogel et al., 2011; Gandhi et al., 2018; Paz-Ares et al., 2018; Grimaldi et al., 2020).
Clinical studies in melanoma patients displayed improved overall survival when combining anti-PD1 therapy with multi-peptide vaccine which improved T cell priming by increasing antigen presentation (Gibney et al., 2015). Intriguingly, some patients showed complete response in synergistic treatment of personalized tumor neoantigen vaccination and anti-PD1 therapy (Ott et al., 2017). Similar to self vaccination, oncolytic virus therapy also induces tumor antigen release and provides danger signals, which consequently enhances T cell priming and ameliorates anti-PD1/PDL1 resistance (Guo et al., 2014; Long et al., 2016). Radiation therapy upregulates type I interferon production by inducing tumor cell death that activates STING pathway and promotes T cell priming (Twyman-Saint Victor et al., 2015; Darragh et al., 2018). NSCLC patients who receive previous radiotherapy showed a prolonged survival after pembrolizumab treatment (Shaverdian et al., 2017).
Additionally, the regimen of targeting TLRs, such as TLR3, TLR9, promotes DCs maturation and rationally alleviates the anti-PD1/PDL1 resistance (Takeda Y. et al., 2017; Kapp et al., 2019). Since the upregulation of CTLA4 expression has been considered as a negative regulator in mediating T cell priming, the combination therapy of anti-PD1 and anti-CTLA4 antibodies significantly extended the patients’ overall survival (Wolchok et al., 2017). Furthermore, type I IFNs are critical for T cell priming (Deng et al., 2014). In a preclinical study, researchers designed an anti-PDL1-IFN-α fusion protein to specifically deliver IFN-α into tumors. Systemic administration of the fusion protein achieved complete regression in most of the tumor bearing mice (Liang et al., 2018). Collectively, strategies enhancing T cell priming can improve the sensitivity of anti-PD1/PDL1 therapy.
The expressions of coinhibitory immune checkpoint receptors upregulate following T cell activation. Strategies targeting alternative immune checkpoints or amplifying costimulatory signals may contribute to the relief of anti-PD1/PDL1 resistance (Marin-Acevedo et al., 2018). Synergistic therapy of PD1/PDL1 blockade and other ICBs including TIM3 and TIGIT blockade have been proved for harboring superior survival outcomes in several studies (Hung et al., 2018; Limagne et al., 2019). In addition, agonistic antibody targeting CD40 has been identified significantly reversing T cell exhaustion, which was illuminated by reactivated T cells with augmented production of cytokines and capacities of cytotoxicity and proliferation (Ngiow et al., 2016).
Sufficient T cell infiltration is a prerequisite for reinvigorating antitumor immunity through PD1/PDL1 blockade. In a preclinical study, taking tumor bearing mice treated with anti-EGFR-guided LIGHT fusion protein into account, the study illustrated that the activation of lymphotoxin β-receptor induced production of chemokines which led to extensive T cell recruitment. The combination therapy with PDL1 blockade significantly results in tumor regression (Tang et al., 2016). Furthermore, combination therapy of PD1/PDL1 blockade with adoptive cell transfer increases the infiltration and cytotoxicity of T cells, which breaks the restriction of MHC disfunction (Blake et al., 2015).
Immunosuppressive microenvironment inhibits the function of the main force of antitumor immunity i.e., CD8+ T cells, resulting in anti-PD1/PDL1 resistance. Targeting TAMs accumulated in TME can effectively improve response to the therapy (Cassetta and Kitamura, 2018). Combination treatment of CSF1R blockade with anti-PD1 antibody reprogrammed M2 into M1 and promoted tumor regression in tumor bearing mice (Zhu et al., 2014). Immunosuppressive cytokines derived from tumors not only attract immune cell infiltration and drive their polarization into pro-tumor phenotype, but also directly inhibit CD8+ T cell cytotoxicity (Lippitz, 2013). Based on this mechanism, targeting cytokines especially TGF-β promotes T cell penetrating and augments antitumor immunity (Mariathasan et al., 2018), and selective blockade of the most prevalent isoform TGF-β1 can significantly reduce the dose-dependent side effect while enhancing anti-PD1 efficacy (Martin et al., 2020). Moreover, desensitizing chemokine receptors by inhibitors prevents the infiltration of immunosuppressive cells including MDSCs and Tregs, which ultimately increases sensitivity of tumors to PD1/PDL1 blockade (Sugiyama et al., 2013; Highfill et al., 2014). PI3K-γ was reported highly expressed in myeloid cells. Selective inhibition of PI3K-γ shifted myeloid cells to a less suppressive phenotype and increased CD8+ T cell infiltration, hence spurred the efficacy of anti-PD1 therapy (De Henau et al., 2016). Epigenetic silencing of Th1 type chemokines such as CXCL9 and CXCL10, involves in immunosuppression. Epigenetic modulation through DNA methyltransferase and histone deacetylase inhibitors rationally sensitizes tumors to anti-PD1/PDL1 therapy (Peng et al., 2015).
Metabolites play pivotal roles in modulating tumor immunosuppressive context. IDO has been shown to influence the TME by converting tryptophan to kynurenine. Evidence has documented that IDO inhibitors combined with anti-PDL1 and anti-CTLA4 antibodies enhanced proliferation and infiltration of CTLs and IL2 production in preclinical models (Spranger et al., 2014). However, precise biomarker may be needed for selecting responders because there is no significant benefit but side effects that lead to discontinuation of several clinical trials (Günther et al., 2019).
Adenosine is a product of adenosinergic pathway. Accumulation of adenosine in TME abrogates antitumor immunity. Complete tumor elimination has been observed in up to 90% mice treated with antagonist of A2A receptor accompanied by anti-PDL1 or anti-CTLA4 antibody (Willingham et al., 2018). Targeting adenosine production by inhibiting the enzyme CD73 efficaciously facilitated anti-PD1/PDL1 efficiency (Hay et al., 2016; Chen et al., 2019). Recently, the first-in-human study of A2AR antagonist treatment has been conducted in renal cell carcinoma patients with anti-PD1/PDL1 resistance (Fong et al., 2020). Clinical response was observed when treated anti-PD1/PDL1 resistant patients with either A2AR antagonist alone or combining with anti-PDL1 antibody (Fong et al., 2020).
Combination of PD1/PDL1 blockade with other therapies has been widely investigated. Therapies targeting the oncogenic signaling that can influence the TME by mediating cytokine production, bringing benefits to anti-PD1/PDL1 therapy. Take the inhibitors targeting MAPK pathway as an example. In preclinical study, targeting BRAF mutation resulted in an increase in CD8+ T cell infiltration and IFN-γ production (Cooper et al., 2014). Concurrently, triple combination therapy inhibiting BRAF, ERK and PD1 in patients with BRAF V600-mutated metastatic melanoma displayed persistent antitumor response (Ribas et al., 2019). In addition, the influence of composition of gut microbiota in host immunity makes it not only a predicator, but also a target for combination therapy. Strategies including dietary modification, probiotics administration and fecal microbiota transplantation have made progress in preclinical research and still need further investigation in clinical applications (Gopalakrishnan et al., 2018a).
The application of PD1/PDL1 blockade has become a milestone of immunotherapy. Although the anti-PD1/PDL1 therapy has shown impressive efficacy in the treatment of solid tumors, the durable sensitivity only occurred in a small proportion of patients. Some patients who were initially responsive to the therapy eventually developed acquired resistance. Therefore, studying the mechanisms of anti-PD1/PDL1 resistance is critical for figuring out overcoming strategies.
In this review we summarized the mechanisms of primary and acquired resistance of anti-PD1/PDL1 therapy, which contributed to low response rates and limited durability in solid tumors. However, because of the complexity of antitumor immunity, the documented mechanisms are just pieces of the puzzle and varied within each individual, making it intricate for properly selecting patients and designing overcoming strategies. Currently, the efficiency of extensively used biomarkers for identifying responders is not stable, and most of the mechanism-based strategies are still undergoing preclinical studies while some of the clinical trials had been shut down because of severe adverse effects and limited benefits. Thus, picturing a more detailed map of resistant mechanisms, designing proper trials and exploring appropriate biomarkers are necessary for guiding precision therapy and expanding the spectrum of responders in the future.
QL and YZ conceptualized this review and decided on the content. QL wrote the manuscript. QL and KS prepared the figures and tables. YZ, DW, and LW revised this review. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work.
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos. U1804281, 81771781, 81702810, and 81773060).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
APC, antigen presenting cell; CSF1R, colony-stimulating factor 1 receptor; CTLA4, cytotoxic T lymphocyte-associated antigen 4; DC, dendritic cell; ICB, immune checkpoint blockade; IDO, indoleamine 2,3-dioxygenase; IHC, immunohistochemistry; KYN, kynurenine; LAG3, lymphocyte activation gene 3; LCMV, chronic lymphocytic choriomeningitis virus; LOH, loss of heterozygosity; MAPK, mitogen-activated protein kinase; MDSC, myeloid-derived suppressor cell; MHC, major histocompatibility complexes; NSCLC, non–small cell lung cancer; PD1, programmed cell death 1; PDL1, programmed cell death ligand 1; PI3K, phosphatidylinositol-3-kinase; PTEN, phosphatase and tensin homolog; STAT, signal transducers and activators of transcription; TAM, tumor associated macrophage; TGF-β, transforming growth factor beta; TIL, tumor infiltrating lymphocyte; TIM3, T-cell immunoglobulin and mucin-domain containing 3; TMB, tumor mutation burden; TME, tumor microenvironment; TOX, thymocyte selection-associated high mobility group box gene; Treg, regulatory T cell; TRP, tryptophan.
Agata, Y., Kawasaki, A., Nishimura, H., Ishida, Y., Tsubata, T., Yagita, H., et al. (1996). Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8, 765–772. doi: 10.1093/intimm/8.5.765
Akinleye, A., and Rasool, Z. (2019). Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 12:92. doi: 10.1186/s13045-019-0779-5
Anagnostou, V., Smith, K. N., Forde, P. M., Niknafs, N., Bhattacharya, R., White, J., et al. (2017). Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 7, 264–276. doi: 10.1158/2159-8290.CD-16-0828
Ansell, S. M., Lesokhin, A. M., Borrello, I., Halwani, A., Scott, E. C., Gutierrez, M., et al. (2015). PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 372, 311–319. doi: 10.1056/NEJMoa1411087
Bach, E. A., Aguet, M., and Schreiber, R. D. (1997). The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 15, 563–591. doi: 10.1146/annurev.immunol.15.1.563
Bates, G. J., Fox, S. B., Han, C., Leek, R. D., Garcia, J. F., Harris, A. L., et al. (2006). Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 24, 5373–5380. doi: 10.1200/jco.2006.05.9584
Batlle, E., and Massagué, J. (2019). Transforming growth factor-β signaling in immunity and cancer. Immunity 50, 924–940. doi: 10.1016/j.immuni.2019.03.024
Beavis, P. A., Milenkovski, N., Henderson, M. A., John, L. B., Allard, B., Loi, S., et al. (2015). Adenosine receptor 2A blockade increases the efficacy of anti–PD-1 through Enhanced antitumor T-cell responses. Cancer Immunol. Res. 3:506. doi: 10.1158/2326-6066.CIR-14-0211
Bellmunt, J., de Wit, R., Vaughn, D. J., Fradet, Y., Lee, J. L., Fong, L., et al. (2017). Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 376, 1015–1026. doi: 10.1056/NEJMoa1613683
Blackburn, S. D., Shin, H., Freeman, G. J., and Wherry, E. J. (2008). Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. U.S.A. 105, 15016–15021. doi: 10.1073/pnas.0801497105
Blake, S. J. P., Ching, A. L. H., Kenna, T. J., Galea, R., Large, J., Yagita, H., et al. (2015). Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS One 10:e0119483. doi: 10.1371/journal.pone.0119483
Brown, S. D., Warren, R. L., Gibb, E. A., Martin, S. D., Spinelli, J. J., Nelson, B. H., et al. (2014). Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 24, 743–750. doi: 10.1101/gr.165985.113
Capalbo, C., Scafetta, G., Filetti, M., Marchetti, P., and Bartolazzi, A. (2019). Predictive biomarkers for checkpoint inhibitor-based immunotherapy: the Galectin-3 signature in NSCLCs. Int. J. Mol. Sci. 20:1607. doi: 10.3390/ijms20071607
Carbone, D. P., Reck, M., Paz-Ares, L., Creelan, B., Horn, L., Steins, M., et al. (2017). First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 376, 2415–2426. doi: 10.1056/NEJMoa1613493
Cassetta, L., and Kitamura, T. (2018). Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front. Cell Dev. Biol. 6:38. doi: 10.3389/fcell.2018.00038
Cekic, C., Day, Y.-J., Sag, D., and Linden, J. (2014). Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 74:7250. doi: 10.1158/0008-5472.CAN-13-3583
Chen, D. S., and Mellman, I. (2013). Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10. doi: 10.1016/j.immuni.2013.07.012
Chen, S., Wainwright, D. A., Wu, J. D., Wan, Y., Matei, D. E., Zhang, Y., et al. (2019). CD73: an emerging checkpoint for cancer immunotherapy. Immunotherapy 11, 983–997. doi: 10.2217/imt-2018-0200
Chow, L. Q. M., Haddad, R., Gupta, S., Mahipal, A., Mehra, R., Tahara, M., et al. (2016). Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion cohort. J. Clin. Oncol. 34, 3838–3845. doi: 10.1200/JCO.2016.68.1478
Chung, H. C., Ros, W., Delord, J. P., Perets, R., Italiano, A., Shapira-Frommer, R., et al. (2019). Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 37, 1470–1478. doi: 10.1200/jco.18.01265
Cooper, Z. A., Juneja, V. R., Sage, P. T., Frederick, D. T., Piris, A., Mitra, D., et al. (2014). Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol. Res. 2, 643–654. doi: 10.1158/2326-6066.Cir-13-0215
Cristescu, R., Mogg, R., Ayers, M., Albright, A., Murphy, E., Yearley, J., et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362:6411. doi: 10.1126/science.aar3593
Curiel, T. J., Coukos, G., Zou, L., Alvarez, X., Cheng, P., Mottram, P., et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949. doi: 10.1038/nm1093
Daassi, D., Mahoney, K. M., and Freeman, G. J. (2020). The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 20, 209–215. doi: 10.1038/s41577-019-0264-y
Darragh, L. B., Oweida, A. J., and Karam, S. D. (2018). Overcoming resistance to combination radiation-immunotherapy: a focus on contributing pathways within the tumor microenvironment. Front. Immunol. 9:3154. doi: 10.3389/fimmu.2018.03154
De Henau, O., Rausch, M., Winkler, D., Campesato, L. F., Liu, C., Cymerman, D. H., et al. (2016). Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 539, 443–447. doi: 10.1038/nature20554
Deng, L., Liang, H., Xu, M., Yang, X., Burnette, B., Arina, A., et al. (2014). STING-dependent cytosolic DNA sensing promotes radiation-induced type i interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852. doi: 10.1016/j.immuni.2014.10.019
Dhillon, A. S., Hagan, S., Rath, O., and Kolch, W. (2007). MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290. doi: 10.1038/sj.onc.1210421
El-Khoueiry, A. B., Sangro, B., Yau, T., Crocenzi, T. S., Kudo, M., Hsu, C., et al. (2017). Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502. doi: 10.1016/s0140-6736(17)31046-2
Fong, L., Hotson, A., Powderly, J. D., Sznol, M., Heist, R. S., Choueiri, T. K., et al. (2020). Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 10:40. doi: 10.1158/2159-8290.CD-19-0980
Fridman, W. H., Pages, F., Sautes-Fridman, C., and Galon, J. (2012). The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer 12, 298–306. doi: 10.1038/nrc3245
Fuchs, C. S., Doi, T., Jang, R. W., Muro, K., Satoh, T., Machado, M., et al. (2018). Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 Trial. JAMA Oncol. 4:e180013. doi: 10.1001/jamaoncol.2018.0013
Gainor, J. F., Shaw, A. T., Sequist, L. V., Fu, X., Azzoli, C. G., Piotrowska, Z., et al. (2016). EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin. Cancer Res. 22, 4585–4593. doi: 10.1158/1078-0432.CCR-15-3101
Gajewski, T. F., Schreiber, H., and Fu, Y. X. (2013). Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022. doi: 10.1038/ni.2703
Gandhi, L., Rodriguez-Abreu, D., Gadgeel, S., Esteban, E., Felip, E., De Angelis, F., et al. (2018). Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092. doi: 10.1056/NEJMoa1801005
Garcia-Diaz, A., Shin, D. S., Moreno, B. H., Saco, J., Escuin-Ordinas, H., Rodriguez, G. A., et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201. doi: 10.1016/j.celrep.2017.04.031
Garon, E. B., Rizvi, N. A., Hui, R., Leighl, N., Balmanoukian, A. S., Eder, J. P., et al. (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028. doi: 10.1056/NEJMoa1501824
George, S., Miao, D., Demetri, G. D., Adeegbe, D., Rodig, S. J., Shukla, S., et al. (2017). Loss of PTEN is associated with resistance to anti-pd-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 46, 197–204. doi: 10.1016/j.immuni.2017.02.001
Gettinger, S., Choi, J., Hastings, K., Truini, A., Datar, I., Sowell, R., et al. (2017). Impaired HLA Class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 7, 1420–1435. doi: 10.1158/2159-8290.CD-17-0593
Gibney, G. T., Kudchadkar, R. R., DeConti, R. C., Thebeau, M. S., Czupryn, M. P., Tetteh, L., et al. (2015). Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 21, 712–720. doi: 10.1158/1078-0432.Ccr-14-2468
Gibney, G. T., Weiner, L. M., and Atkins, M. B. (2016). Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 17, e542–e551. doi: 10.1016/s1470-2045(16)30406-5
Goodman, A. M., Kato, S., Bazhenova, L., Patel, S. P., Frampton, G. M., Miller, V., et al. (2017). Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 16, 2598–2608. doi: 10.1158/1535-7163.Mct-17-0386
Gopalakrishnan, V., Helmink, B. A., Spencer, C. N., Reuben, A., and Wargo, J. A. (2018a). The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33, 570–580. doi: 10.1016/j.ccell.2018.03.015
Gopalakrishnan, V., Spencer, C. N., Nezi, L., Reuben, A., Andrews, M. C., Karpinets, T. V., et al. (2018b). Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359:97. doi: 10.1126/science.aan4236
Grimaldi, A., Cammarata, I., Martire, C., Focaccetti, C., Piconese, S., Buccilli, M., et al. (2020). Combination of chemotherapy and PD-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun. Biol. 3:85. doi: 10.1038/s42003-020-0811-x
Günther, J., Däbritz, J., and Wirthgen, E. (2019). Limitations and off-target effects of tryptophan-related IDO inhibitors in cancer treatment. Front. Immunol. 10:1801. doi: 10.3389/fimmu.2019.01801
Guo, Z. S., Liu, Z., and Bartlett, D. L. (2014). Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 4:74. doi: 10.3389/fonc.2014.00074
Harty, J. T., and Badovinac, V. P. (2008). Shaping and reshaping CD8+ T-cell memory. Nat. Rev. Immunol. 8, 107–119. doi: 10.1038/nri2251
Hay, C. M., Sult, E., Huang, Q., Mulgrew, K., Fuhrmann, S. R., McGlinchey, K. A., et al. (2016). Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 5:e1208875. doi: 10.1080/2162402X.2016.1208875
Heemskerk, B., Kvistborg, P., and Schumacher, T. N. M. (2013). The cancer antigenome. Embo J. 32, 194–203. doi: 10.1038/emboj.2012.333
Highfill, S. L., Cui, Y., Giles, A. J., Smith, J. P., Zhang, H., Morse, E., et al. (2014). Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 6:237ra267. doi: 10.1126/scitranslmed.3007974
Hugo, W., Zaretsky, J. M., Sun, L., Song, C., Moreno, B. H., Hu-Lieskovan, S., et al. (2016). Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44. doi: 10.1016/j.cell.2016.02.065
Hu-Lieskovan, S., Mok, S., Homet Moreno, B., Tsoi, J., Robert, L., Goedert, L., et al. (2015). Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 7:279ra241. doi: 10.1126/scitranslmed.aaa4691
Hulpke, S., and Tampé, R. (2013). The MHC I loading complex: a multitasking machinery in adaptive immunity. Trends Biochem. Sci. 38, 412–420. doi: 10.1016/j.tibs.2013.06.003
Hung, A. L., Maxwell, R., Theodros, D., Belcaid, Z., Mathios, D., Luksik, A. S., et al. (2018). TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 7:e1466769. doi: 10.1080/2162402x.2018.1466769
Inamura, K. (2020). Roles of microbiota in response to cancer immunotherapy. Semin. Cancer Biol. 20, 30001–30008. doi: 10.1016/j.semcancer.2019.12.026 [Epub ahead of print].
Ji, M., Liu, Y., Li, Q., Li, X., Ning, Z., Zhao, W., et al. (2016). PD-1/PD-L1 expression in non-small-cell lung cancer and its correlation with EGFR/KRAS mutations. Cancer Biol. Ther. 17, 407–413. doi: 10.1080/15384047.2016.1156256
Johnson, D. B., Estrada, M. V., Salgado, R., Sanchez, V., Doxie, D. B., Opalenik, S. R., et al. (2016). Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 7:10582. doi: 10.1038/ncomms10582
Johnson, D. B., Nixon, M. J., Wang, Y., Wang, D. Y., Castellanos, E., Estrada, M. V., et al. (2018). Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight 3:24. doi: 10.1172/jci.insight.120360
Kapp, K., Volz, B., Oswald, D., Wittig, B., Baumann, M., and Schmidt, M. (2019). Beneficial modulation of the tumor microenvironment and generation of anti-tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology 8:e1659096. doi: 10.1080/2162402X.2019.1659096
Kaufman, H. L., Russell, J., Hamid, O., Bhatia, S., Terheyden, P., D’Angelo, S. P., et al. (2016). Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 17, 1374–1385. doi: 10.1016/s1470-2045(16)30364-3
Keniry, M., and Parsons, R. (2008). The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 27, 5477–5485. doi: 10.1038/onc.2008.248
Khan, O., Giles, J. R., McDonald, S., Manne, S., Ngiow, S. F., Patel, K. P., et al. (2019). TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218. doi: 10.1038/s41586-019-1325-x
Kim, K., Park, S., Park, S. Y., Kim, G., Park, S. M., Cho, J.-W., et al. (2020). Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 12:22. doi: 10.1186/s13073-020-00722-9
Koyama, S., Akbay, E. A., Li, Y. Y., Herter-Sprie, G. S., Buczkowski, K. A., Richards, W. G., et al. (2016). Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7:10501. doi: 10.1038/ncomms10501
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. doi: 10.1126/science.aan6733
Li, H., Bullock, K., Gurjao, C., Braun, D., Shukla, S. A., Bosse, D., et al. (2019). Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat. Commun. 10:4346. doi: 10.1038/s41467-019-12361-9
Li, X., Wenes, M., Romero, P., Huang, S. C., Fendt, S. M., and Ho, P. C. (2019). Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 16, 425–441. doi: 10.1038/s41571-019-0203-7
Liang, Y., Tang, H., Guo, J., Qiu, X., Yang, Z., Ren, Z., et al. (2018). Targeting IFNα to tumor by anti-PD-L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat. Commun. 9:4586. doi: 10.1038/s41467-018-06890-y
Liao, W., Overman, M. J., Boutin, A. T., Shang, X., Zhao, D., Dey, P., et al. (2019). KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 35, 559.e7–572.e7. doi: 10.1016/j.ccell.2019.02.008
Limagne, E., Richard, C., Thibaudin, M., Fumet, J. D., Truntzer, C., Lagrange, A., et al. (2019). Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology 8:e1564505. doi: 10.1080/2162402X.2018.1564505
Lippitz, B. E. (2013). Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 14, e218–e228. doi: 10.1016/S1470-2045(12)70582-X
Liu, C., Zheng, S., Jin, R., Wang, X., Wang, F., Zang, R., et al. (2020). The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 470, 95–105. doi: 10.1016/j.canlet.2019.10.027
Loi, S., Dushyanthen, S., Beavis, P. A., Salgado, R., Denkert, C., Savas, P., et al. (2016). RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 22, 1499–1509. doi: 10.1158/1078-0432.CCR-15-1125
Long, G. V., Dummer, R., Ribas, A., Puzanov, I., VanderWalde, A., Andtbacka, R. H. I., et al. (2016). Efficacy analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec (T-VEC) and pembrolizumab (pembro) for unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 34:9568. doi: 10.1200/JCO.2016.34.15_suppl.9568
MacDonald, B. T., Tamai, K., and He, X. (2009). Wnt/β-Catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. doi: 10.1016/j.devcel.2009.06.016
Maimela, N. R., Liu, S., and Zhang, Y. (2018). Fates of CD8+ T cells in tumor microenvironment. Comput. Struct. Biotechnol. J. 17, 1–13. doi: 10.1016/j.csbj.2018.11.004
Marcus, L., Lemery, S. J., Keegan, P., and Pazdur, R. (2019). FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin. Cancer Res. Clincanres 4070:2018. doi: 10.1158/1078-0432.CCR-18-4070
Mariathasan, S., Turley, S. J., Nickles, D., Castiglioni, A., Yuen, K., Wang, Y., et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. doi: 10.1038/nature25501
Marin-Acevedo, J. A., Dholaria, B., Soyano, A. E., Knutson, K. L., Chumsri, S., and Lou, Y. (2018). Next generation of immune checkpoint therapy in cancer: new developments and challenges. J. Hematol. Oncol. 11:39. doi: 10.1186/s13045-018-0582-8
Martin, C. J., Datta, A., Littlefield, C., Kalra, A., Chapron, C., Wawersik, S., et al. (2020). Selective inhibition of TGFβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 12:eaay8456. doi: 10.1126/scitranslmed.aay8456
Marvel, D., and Gabrilovich, D. I. (2015). Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J. Clin. Invest. 125, 3356–3364. doi: 10.1172/JCI80005
Matson, V., Fessler, J., Bao, R., Chongsuwat, T., Zha, Y., Alegre, M. L., et al. (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108. doi: 10.1126/science.aao3290
Matsushita, H., Vesely, M. D., Koboldt, D. C., Rickert, C. G., Uppaluri, R., Magrini, V. J., et al. (2012). Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404. doi: 10.1038/nature10755
McLane, L. M., Abdel-Hakeem, M. S., and Wherry, E. J. (2019). CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495. doi: 10.1146/annurev-immunol-041015-055318
Mempel, T. R., Henrickson, S. E., and von Andrian, U. H. (2004). T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 427, 154–159. doi: 10.1038/nature02238
Motzer, R. J., Escudier, B., McDermott, D. F., George, S., Hammers, H. J., Srinivas, S., et al. (2015). Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813. doi: 10.1056/NEJMoa1510665
Ngiow, S. F., Young, A., Blake, S. J., Hill, G. R., Yagita, H., Teng, M. W. L., et al. (2016). Agonistic CD40 mAb-Driven IL12 reverses resistance to Anti-PD1 in a T-cell-Rich tumor. Cancer Res. 76, 6266–6277. doi: 10.1158/0008-5472.CAN-16-2141
Ngiow, S. F., Young, A., Jacquelot, N., Yamazaki, T., Enot, D., Zitvogel, L., et al. (2015). A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to Anti-PD1. Cancer Res. 75, 3800–3811. doi: 10.1158/0008-5472.Can-15-1082
O’Donnell, J. S., Teng, M. W. L., and Smyth, M. J. (2019). Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167. doi: 10.1038/s41571-018-0142-8
Ott, P. A., Hu, Z., Keskin, D. B., Shukla, S. A., Sun, J., Bozym, D. J., et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. doi: 10.1038/nature22991
Overman, M. J., McDermott, R., Leach, J. L., Lonardi, S., Lenz, H.-J., Morse, M. A., et al. (2017). Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 18, 1182–1191. doi: 10.1016/s1470-2045(17)30422-9
Oweida, A., Hararah, M. K., Phan, A., Binder, D., Bhatia, S., Lennon, S., et al. (2018). Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-Cell infiltration. Clin. Cancer Res. 24, 5368–5380. doi: 10.1158/1078-0432.CCR-18-1038
Pardoll, D. (2015). Cancer and the immune system: basic concepts and targets for intervention. Semin. Oncol. 42, 523–538. doi: 10.1053/j.seminoncol.2015.05.003
Patel, S. P., and Kurzrock, R. (2015). PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 14, 847–856. doi: 10.1158/1535-7163.Mct-14-0983
Pauken, K. E., Sammons, M. A., Odorizzi, P. M., Manne, S., Godec, J., Khan, O., et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. doi: 10.1126/science.aaf2807
Paz-Ares, L., Luft, A., Vicente, D., Tafreshi, A., Gümüş, M., Mazières, J., et al. (2018). Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. N. Engl. J. Med. 379, 2040–2051. doi: 10.1056/NEJMoa1810865
Peng, D., Kryczek, I., Nagarsheth, N., Zhao, L., Wei, S., Wang, W., et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. doi: 10.1038/nature15520
Peng, W., Chen, J. Q., Liu, C., Malu, S., Creasy, C., Tetzlaff, M. T., et al. (2016). Loss of PTEN promotes resistance to T Cell–mediated immunotherapy. Cancer Discov. 6:202. doi: 10.1158/2159-8290.CD-15-0283
Pereira, C., Gimenez-Xavier, P., Pros, E., Pajares, M. J., Moro, M., Gomez, A., et al. (2017). Genomic profiling of patient-derived xenografts for lung cancer identifies B2M inactivation impairing immunorecognition. Clin. Cancer Res. 23, 3203–3213. doi: 10.1158/1078-0432.Ccr-16-1946-t
Platanias, L. C. (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. doi: 10.1038/nri1604
Powles, T., O’Donnell, P. H., Massard, C., Arkenau, H. T., Friedlander, T. W., Hoimes, C. J., et al. (2017). Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol. 3:e172411. doi: 10.1001/jamaoncol.2017.2411
Prendergast, G. C., Smith, C., Thomas, S., Mandik-Nayak, L., Laury-Kleintop, L., Metz, R., et al. (2014). Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 63, 721–735. doi: 10.1007/s00262-014-1549-4
Reinhardt, J., Landsberg, J., Schmid-Burgk, J. L., Ramis, B. B., Bald, T., Glodde, N., et al. (2017). MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res. 77:4697. doi: 10.1158/0008-5472.CAN-17-0395
Ren, D., Hua, Y., Yu, B., Ye, X., He, Z., Li, C., et al. (2020). Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol. Cancer 19:19. doi: 10.1186/s12943-020-1144-6
Ribas, A. (2015). Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. 5, 915–919. doi: 10.1158/2159-8290.Cd-15-0563
Ribas, A., Lawrence, D., Atkinson, V., Agarwal, S., Miller, W. H., Carlino, M. S., et al. (2019). Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 25, 936–940. doi: 10.1038/s41591-019-0476-5
Ribas, A., Shin, D. S., Zaretsky, J., Frederiksen, J., Cornish, A., Avramis, E., et al. (2016). PD-1 blockade expands intratumoral memory T Cells. Cancer Immunol. Res. 4, 194–203. doi: 10.1158/2326-6066.CIR-15-0210
Ribas, A., and Wolchok, J. D. (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. doi: 10.1126/science.aar4060
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. doi: 10.1126/science.aaa1348
Robert, C., Schachter, J., Long, G. V., Arance, A., Grob, J. J., Mortier, L., et al. (2015). Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532. doi: 10.1056/NEJMoa1503093
Roussos, E. T., Condeelis, J. S., and Patsialou, A. (2011). Chemotaxis in cancer. Nat. Rev. Cancer 11, 573–587. doi: 10.1038/nrc3078
Sade-Feldman, M., Jiao, Y. J., Chen, J. H., Rooney, M. S., Barzily-Rokni, M., Eliane, J. P., et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 8:1136. doi: 10.1038/s41467-017-01062-w
Sasada, T., Kimura, M., Yoshida, Y., Kanai, M., and Takabayashi, A. (2003). CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer 98, 1089–1099. doi: 10.1002/cncr.11618
Schreiber, R. D., Old, L. J., and Smyth, M. J. (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570. doi: 10.1126/science.1203486
Schumacher, T. N., and Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. doi: 10.1126/science.aaa4971
Sharma, P., Hu-Lieskovan, S., Wargo, J. A., and Ribas, A. (2017a). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. doi: 10.1016/j.cell.2017.01.017
Sharma, P., Retz, M., Siefker-Radtke, A., Baron, A., Necchi, A., Bedke, J., et al. (2017b). Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 18, 312–322. doi: 10.1016/s1470-2045(17)30065-7
Shaverdian, N., Lisberg, A. E., Bornazyan, K., Veruttipong, D., Goldman, J. W., Formenti, S. C., et al. (2017). Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 18, 895–903. doi: 10.1016/S1470-2045(17)30380-7
Shi, X., Chen, X., Fang, B., Ping, Y., Qin, G., Yue, D., et al. (2019). Decitabine enhances tumor recognition by T cells through upregulating the MAGE-A3 expression in esophageal carcinoma. Biomed. Pharmacother. 112:108632. doi: 10.1016/j.biopha.2019.108632
Shin, D. S., Zaretsky, J. M., Escuin-Ordinas, H., Garcia-Diaz, A., Hu-Lieskovan, S., Kalbasi, A., et al. (2017). Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7, 188–201. doi: 10.1158/2159-8290.CD-16-1223
Sivan, A., Corrales, L., Hubert, N., Williams, J. B., Aquino-Michaels, K., Earley, Z. M., et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. doi: 10.1126/science.aac4255
Skoulidis, F., Goldberg, M. E., Greenawalt, D. M., Hellmann, M. D., Awad, M. M., Gainor, J. F., et al. (2018). STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835. doi: 10.1158/2159-8290.CD-18-0099
Song, M., Chen, X., Wang, L., and Zhang, Y. (2018). Future of anti-PD-1/PD-L1 applications: combinations with other therapeutic regimens. Chin. J. Cancer Res. 30, 157–172. doi: 10.21147/j.issn.1000-9604.2018.02.01
Spranger, S., Bao, R., and Gajewski, T. F. (2015). Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. doi: 10.1038/nature14404
Spranger, S., and Gajewski, T. F. (2016). Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 5:e1086862. doi: 10.1080/2162402x.2015.1086862
Spranger, S., Koblish, H. K., Horton, B., Scherle, P. A., Newton, R., and Gajewski, T. F. (2014). Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J. Immunother. Cancer 2:3. doi: 10.1186/2051-1426-2-3
Sugiyama, D., Nishikawa, H., Maeda, Y., Nishioka, M., Tanemura, A., Katayama, I., et al. (2013). Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. U.S.A. 110, 17945–17950. doi: 10.1073/pnas.1316796110
Suzuki, E., Kapoor, V., Jassar, A. S., Kaiser, L. R., and Albelda, S. M. (2005). Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 11, 6713–6721. doi: 10.1158/1078-0432.Ccr-05-0883
Takeda, K., Nakayama, M., Hayakawa, Y., Kojima, Y., Ikeda, H., Imai, N., et al. (2017). IFN-γ is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat. Commun. 8:14607. doi: 10.1038/ncomms14607
Takeda, Y., Kataoka, K., Yamagishi, J., Ogawa, S., Seya, T., and Matsumoto, M. (2017). A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. 19, 1874–1887. doi: 10.1016/j.celrep.2017.05.015
Tanaka, A., and Sakaguchi, S. (2017). Regulatory T cells in cancer immunotherapy. Cell Res. 27, 109–118. doi: 10.1038/cr.2016.151
Tang, H., Wang, Y., Chlewicki, L. K., Zhang, Y., Guo, J., Liang, W., et al. (2016). Facilitating T Cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 29, 285–296. doi: 10.1016/j.ccell.2016.02.004
Tauriello, D. V. F., Palomo-Ponce, S., Stork, D., Berenguer-Llergo, A., Badia-Ramentol, J., Iglesias, M., et al. (2018). TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543. doi: 10.1038/nature25492
Theivanthiran, B., Evans, K. S., DeVito, N. C., Plebanek, M. P., Sturdivant, M., Wachsmuth, L. P., et al. (2020). A tumor-intrinsic PD-L1-NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Invest. 130:133055. doi: 10.1172/jci133055
Thommen, D. S., Schreiner, J., Müller, P., Herzig, P., Roller, A., Belousov, A., et al. (2015). Progression of lung cancer is associated with increased dysfunction of T Cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol. Res. 3, 1344–1355. doi: 10.1158/2326-6066.CIR-15-0097
Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott, D. F., et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454. doi: 10.1056/NEJMoa1200690
Toulmonde, M., Penel, N., Adam, J., Chevreau, C., Blay, J.-Y., Le Cesne, A., et al. (2018). Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: a phase 2 clinical trial. JAMA Oncol. 4, 93–97. doi: 10.1001/jamaoncol.2017.1617
Tumeh, P. C., Harview, C. L., Yearley, J. H., Shintaku, I. P., Taylor, E. J., Robert, L., et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. doi: 10.1038/nature13954
Twyman-Saint Victor, C., Rech, A. J., Maity, A., Rengan, R., Pauken, K. E., Stelekati, E., et al. (2015). Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377. doi: 10.1038/nature14292
van der Most, R. G., Currie, A. J., Mahendran, S., Prosser, A., Darabi, A., Robinson, B. W., et al. (2009). Tumor eradication after cyclophosphamide depends on concurrent depletion of regulatory T cells: a role for cycling TNFR2-expressing effector-suppressor T cells in limiting effective chemotherapy. Cancer Immunol. Immunother. 58, 1219–1228. doi: 10.1007/s00262-008-0628-9
Veglia, F., Perego, M., and Gabrilovich, D. (2018). Myeloid-derived suppressor cells coming of age. Nat. Immunol. 19, 108–119. doi: 10.1038/s41590-017-0022-x
Vijayan, D., Young, A., Teng, M. W. L., and Smyth, M. J. (2017). Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 17, 709–724. doi: 10.1038/nrc.2017.86
Voron, T., Colussi, O., Marcheteau, E., Pernot, S., Nizard, M., Pointet, A. L., et al. (2015). VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 212, 139–148. doi: 10.1084/jem.20140559
Wang, X., He, Q., Shen, H., Xia, A., Tian, W., Yu, W., et al. (2019). TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hematol. Oncol. 71, 731–741. doi: 10.1016/j.jhep.2019.05.015
Wherry, E. J., and Kurachi, M. (2015). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499. doi: 10.1038/nri3862
Willingham, S. B., Ho, P. Y., Hotson, A., Hill, C., Piccione, E. C., Hsieh, J., et al. (2018). A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to anti-PD-(L)1 and Anti-CTLA-4 in preclinical models. Cancer Immunol. Res. 6, 1136–1149. doi: 10.1158/2326-6066.CIR-18-0056
Wolchok, J. D., Chiarion-Sileni, V., Gonzalez, R., Rutkowski, P., Grob, J. J., Cowey, C. L., et al. (2017). Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 377, 1345–1356. doi: 10.1056/NEJMoa1709684
Yang, L., Li, A., Lei, Q., and Zhang, Y. (2019). Tumor-intrinsic signaling pathways: key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 12:125. doi: 10.1186/s13045-019-0804-8
Yang, L., and Zhang, Y. (2017). Tumor-associated macrophages: from basic research to clinical application. J. Hematol. Oncol. 10:58. doi: 10.1186/s13045-017-0430-2
Yeon Yeon, S., Jung, S. H., Jo, Y. S., Choi, E. J., Kim, M. S., Chung, Y. J., et al. (2019). Immune checkpoint blockade resistance-related B2M hotspot mutations in microsatellite-unstable colorectal carcinoma. Pathol. Res. Pract. 215, 209–214. doi: 10.1016/j.prp.2018.11.014
Yi, M., Qin, S., Zhao, W., Yu, S., Chu, Q., and Wu, K. (2018). The role of neoantigen in immune checkpoint blockade therapy. Exp. Hematol. Oncol. 7, 28–28. doi: 10.1186/s40164-018-0120-y
Zaretsky, J. M., Garcia-Diaz, A., Shin, D. S., Escuin-Ordinas, H., Hugo, W., Hu-Lieskovan, S., et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829. doi: 10.1056/NEJMoa1604958
Zhan, T., Rindtorff, N., and Boutros, M. (2017). Wnt signaling in cancer. Oncogene 36, 1461–1473. doi: 10.1038/onc.2016.304
Zhang, M., Huang, L., Ding, G., Huang, H., Cao, G., Sun, X., et al. (2020). Interferon gamma inhibits CXCL8-CXCR2 axis mediated tumor-associated macrophages tumor trafficking and enhances anti-PD1 efficacy in pancreatic cancer. J. Immunother. Cancer 8:e000308. doi: 10.1136/jitc-2019-000308
Zhang, Z., Liu, S., Zhang, B., Qiao, L., Zhang, Y., and Zhang, Y. (2020). T cell dysfunction and exhaustion in cancer. Front. Cell Dev. Biol. 8:17. doi: 10.3389/fcell.2020.00017
Zhou, J., Tang, Z., Gao, S., Li, C., Feng, Y., and Zhou, X. (2020). Tumor-associated macrophages: recent insights and therapies. Front. Oncol. 10:188. doi: 10.3389/fonc.2020.00188
Zhu, Y., Knolhoff, B. L., Meyer, M. A., Nywening, T. M., West, B. L., Luo, J., et al. (2014). CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069. doi: 10.1158/0008-5472.CAN-13-3723
Keywords: cancer, immunotherapy, PD1, PDL1, resistance, mechanism
Citation: Lei Q, Wang D, Sun K, Wang L and Zhang Y (2020) Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 8:672. doi: 10.3389/fcell.2020.00672
Received: 05 April 2020; Accepted: 02 July 2020;
Published: 21 July 2020.
Edited by:
Sergio Roa, Cima Universidad de Navarra, SpainReviewed by:
Lidia Avalle, University of Turin, ItalyCopyright © 2020 Lei, Wang, Sun, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhang, eWl6aGFuZ0B6enUuZWR1LmNu; Liping Wang, d2xwQHp6dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.