 Marina Marsan1*†
Marina Marsan1*†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 07 March 2025
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2025 | https://doi.org/10.3389/fped.2025.1549504
This article is part of the Research Topic Advances in molecular genetics of Marfan syndrome and related disorders View all articles
Background: Marfan Syndrome (MS) is a connective tissue disorder, an autosomal dominant condition mostly caused by variants in the FBN1 gene, which encodes for fibrillin-1 protein. Anomalies in the gene lead to a wide variety of clinical manifestations, including disorders of the cardiac, ocular and musculoskeletal system. We present a case of a child belonging to a Sardinian family of four generations, with a novel variant found in the FBN1 gene.
Objective: To include this novel missense FBN1 variant into genetic counselling for Marfan Syndrome and to discuss its genotypic-phenotypic correlation.
Methods: Firstly, the proband was diagnosed with Marfan Syndrome using 2020 Revised Ghent Criteria, and she then underwent genetic testing using Next Generation sequencing.
Results: The NGS revealed a novel heterozygous missense variant (c.2348A>G) in the FBN1 gene, in exon 20. This genetic variant caused a missense substitution of a serine residue with an arginine residue in the position 783 of Fibrillin-1 protein. The variant was then evaluated in the other family members, and was eventually only found in symptomatic individuals, regardless of the severity of their phenotype, demonstrating the segregation with MS; furthermore, it showed complete penetrance with the disease.
Conclusions: Our results suggest that this variant is responsible for MS and it therefore should be included in genetic diagnoses and counselling discussion.
Marfan syndrome (MS) is one of the most common inherited disorders of connective tissue. It has an estimated incidence of 1 in 3,000–5,000 individuals and a prevalence of 1 in 10,000–20,000 individuals without geographic, ethnic or gender predilection (1). In 1896 Antoine-Bernard-Jean Marfan described the first case. The involvement of the aorta in the syndrome was recognized in 1943 (2), and Hollister et al. demonstrated the fibrillin-1-related pathogenesis in 1990 (3).
Inheritance is predominantly autosomal dominant with complete penetrance and considerable clinical variability. A “de novo” variant can be found in 25% of cases, mostly (90%) associated with mutation of FBN1 gene on chromosome 15q21.1, which encodes protein fibrillin-1 (4, 5). This protein is one of the main components of elastic matrix microfibrils, and it is especially present at the cardiovascular and musculoskeletal level (6). Fewer cases are due to mutations of other genes, such as TGFBR1 or TGFBR2.
Point mutations are the most common: 60% missense and 10% nonsense (7, 8). Less frequent mutations consist of small insertions, deletions or duplications (10%–15%) or various classes of splicing errors (10%–15%). Finally, few larger rearrangements have been reported (9), while whole gene deletions are rare (10).
Although specific genotypes do not seem to be related to specific phenotypes (7, 9, 11), variants in exons 24–32 of the FBN1 gene seem to be characterized by the most severe prognosis: the clinical onset is immediately after birth and death usually occurs within the second year of life (7, 12). Moreover, TGFBR2 mutations seem to be associated with worse cardiovascular involvement and minor ocular involvement (13). Finally, as well as Marfan Syndrome, other fibrillinopathies might carry variants in the FBN1 gene, and a differential diagnosis should frequently be considered (7, 8).
Our case report focuses on a novel missense variant located in exon 20 of FBN1 gene which was found in a family of four generations; inheritance was autosomal dominant and had complete penetrance, whereas phenotype varied significantly among family members.
We present the case of a family (see Figure 1), located in Sardinia, whose members showed Marfan-related clinical manifestations, which varied both in phenotype as well in different levels of organ involvement.
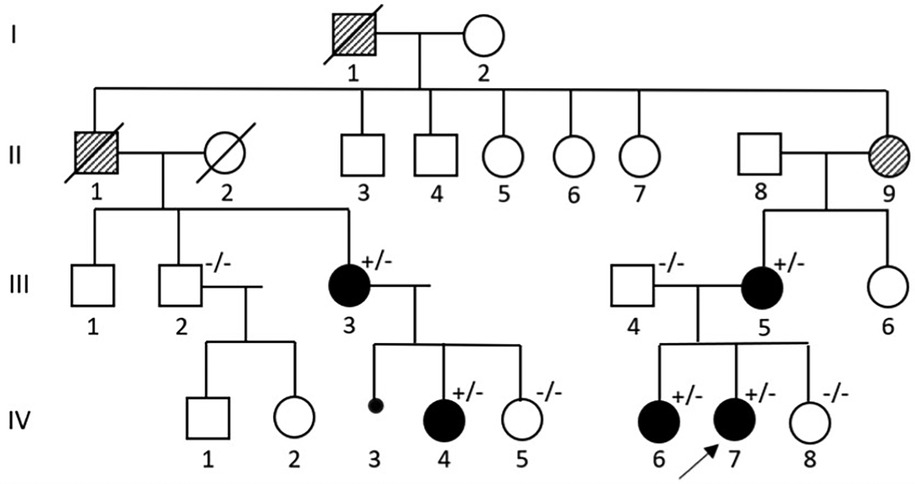
Figure 1. Pedigree of a Sardinian family with Marfan syndrome. The proband is indicated with a black arrow. (+/−): variant carrier and symptomatic. (−/−): not variant carrier and asymptomatic.
IV-7 was the first patient to come to our attention. At 8 years of age, she presented with mild scoliosis, bilateral “pes cavus”, aortic root diameter at the upper limits and bicuspid aortic valve with mild insufficiency. At clinical examination she had an arm span-height ratio of 0.98, joint laxity with a Beighton score >5, no major facial deformities with the exception of anteverted auricles, and a 2/6 mesosystolic heart murmur. In the following years she had a worsening of the lumbar scoliosis, that required a corrective orthopedic surgery, an increase in the arm span-height ratio, positive thumb and wrist signs, elastic skin, hallux valgus, while the aortic ectasia paralleled the general growth of the patient (z-score being 2.99 in 2019, 3.34 in 2020, 3, 7 in 2022); she was therefore started on beta-blockers and angiotensin receptor blockers (ARBs) to minimize aortic dilatation.
Her older sister, IV-6, presented with a progressing dorso-lumbar scoliosis with a 65° Cobb angle (see Figure 2), which was diagnosed when she was 10 years old. She then developed an aortic root ectasia at the upper limits, which is being currently treated with anti-hypertensive medication, mitral valve with irregular, redundant thickened and prolapsed flaps, with slight insufficiency and tricuspid valve with mild-moderate secondary insufficiency. Physical examination pointed out an arm span-height ratio of 1.05, arachnodactyly, reduced elbow extension, positive wrist and thumb signs, flat foot, cutaneous striae at the hips, geographic tongue, systolic murmur, reduced vesicular murmur on the right side of the chest because of the scoliosis. Meeting the Revised Ghent criteria (8 total points) she was diagnosed with Marfan' syndrome.
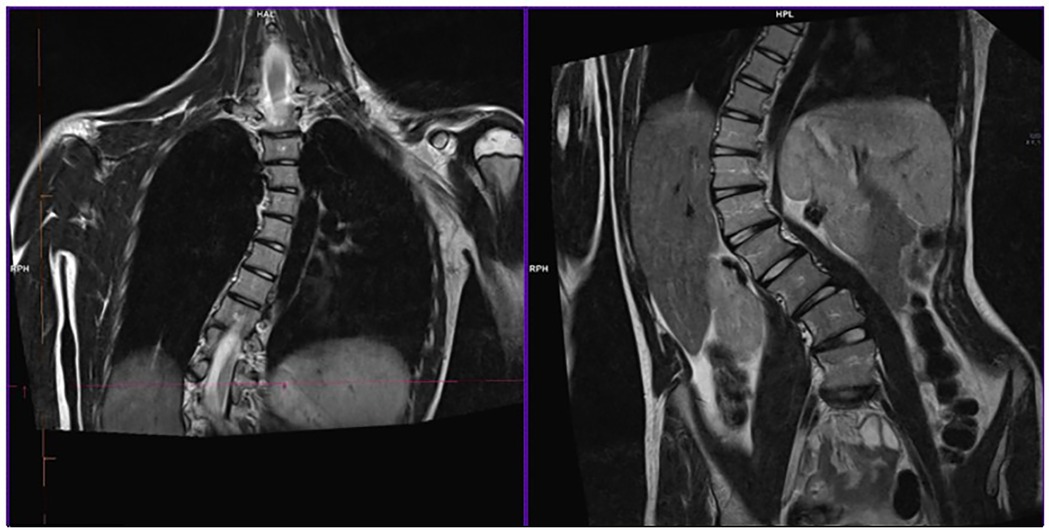
Figure 2. RMI of IV-6's dorsal spine tract before surgery. The image shows dorso-lumbar scoliosis with a 65° Cobb angle.
Their mother, III-5, has a mild aortic valve insufficiency and a history of scoliosis treated with back-brace at 15 years old. III-5's cousin, III-3, has a marfanoid habitus, while her daughter, IV-4, of 11 years of age, showed mild ligamentous laxity, arm span-height ratio of 1,03, cutaneous striae on the hips, positive thumb sign, kyphotic appearance, mild scoliosis, bilateral flat foot.
IV-7 and IV-6's clinical signs and symptoms prompted the execution of a NGS gene panel involving Marfan-related genes, which identified a missense variant on gene FBN1. After the NGS test of the index case, familial segregation analysis was performed by Sanger sequencing.
See Figure 3.
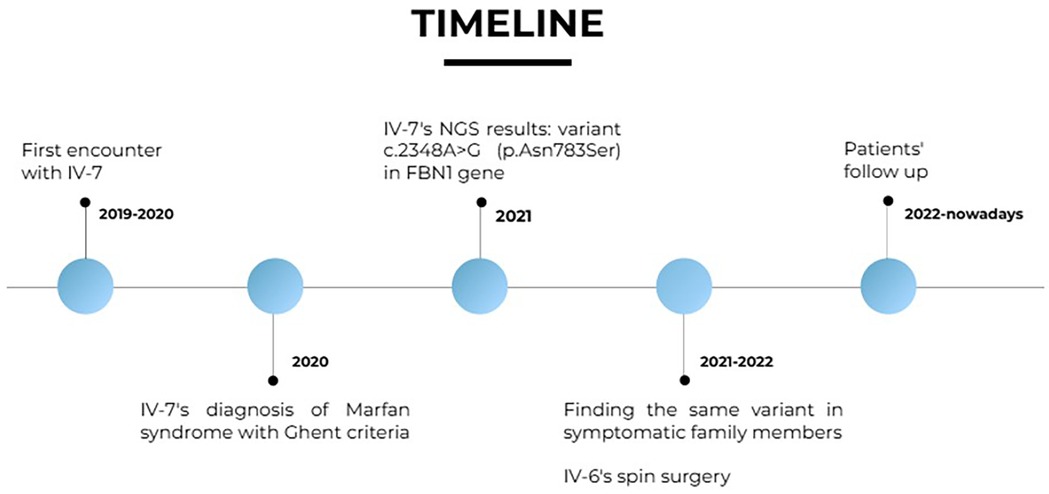
Figure 3. Timeline.
Patients were evaluated in the Pediatric Clinic and Rare Diseases, “Microcitemico Hospital”, Cagliari (Italy). All patients' data were collected from July 2019 to April 2024.
Genetic testing was performed in Medical Genetics Unit, University Hospital of Parma, Parma, Italy. Genomic DNA was extracted from peripheral blood using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany).
Next generation Sequencing of a gene panel of 5 genes (FBN1, TGFBR1, TGFBR2, TGFB2, TGFB3) was carried out on the proband and her parents. The coding exons were captured using the Illumina AmpliSeq focus panel and sequenced using a MiSeq sequencer (Illumina, San Diego, CA, USA) to an average depth of 100 reads per target base. Vcf files were analysed with Base Space Variant Interpreter Illumina software, aligned to a human reference genome (HG19). For variant annotation and prediction, non-synonymous substitutions and SNPs with minor allele frequencies (MAFs) lower than 5% were filtered out. Variant classification followed the ACMG/AMP 2015 guidelines (14).
The function of gene variants and their potential pathogenicity were analyzed referencing gnomAD, dbSNP, OMIM, ClinVar, LOVD, Franklin, REVEL and alphamissense tools.
Candidate variants and regions with low coverage were verified using Sanger sequencing on an Applied Biosystems 3500Dx sequencer (Applied Biosystems, Waltham, MA, USA).
According with ACMG/AMP rules, the assigned class of pathogenicity was Likely Pathogenic, based on the following scores:
• PM1: moderate (Non-truncating non-synonymous variant is located in a mutational hot spot and/or critical and well-established functional domain)
• PP2: supporting (Missense variant in a gene with low rate of benign missense mutations and for which missense mutation is a common mechanism of a disease)
• PM2: moderate (Extremely low frequency in gnomAD population databases; gnomAD there are not population frequencies)
• PP3: supporting (Extremely low frequency in gnomAD population databases)
• PP1: supporting (Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease)
A novel heterozygous nucleotide variant c.2348A>G (p.Asn783Ser) in exon 20 of the FBN1 gene (see Figure 4) was at first detected in the proband and her sister (IV-6, IV-7) and subsequently in the other symptomatic family members (III-3, III-5, IV-4). No other VUS, likely pathogenic and pathogenic variants were found.
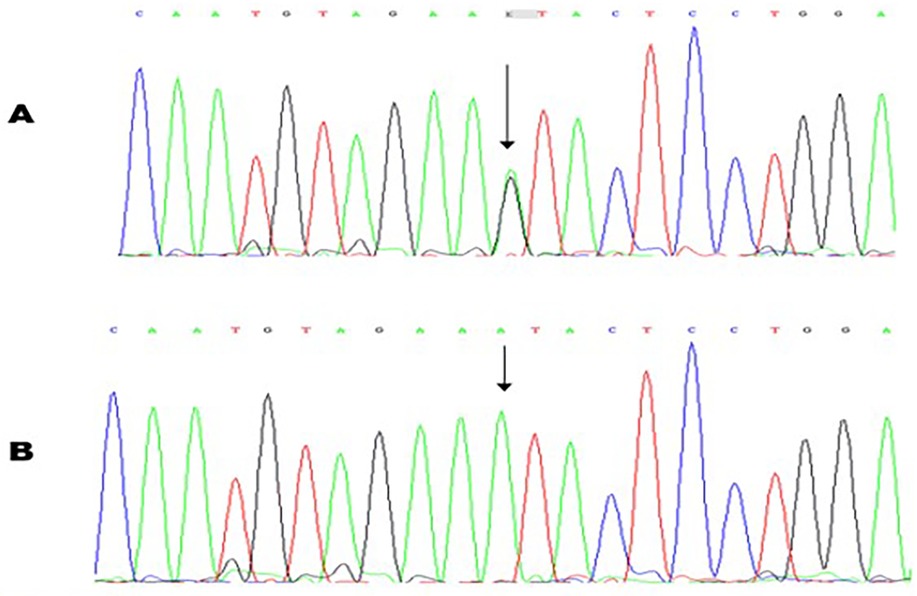
Figure 4. Molecular characterization of a patient with Marfan syndrome. Sequence chromatogram showing the position of the c.234A>G transversion that led to the p.Asn783Ser missense variant. The variant was heterozygous in the proband (A); Sanger sequencing result of an unaffected control (B).
The variant NM_000138.5:c.2348A>G was not described in gnomAD Exomes tool (frequency: 0%), ClinVar and LOVD databases. The c.2348A>G variant was further investigated with the online tools REVEL and alphamissense, resulting in 0.861 and 0.902 scores, respectively; these results strongly support the pathogenicity of the FBN1 genetic variant.
III-2, III-4, IV-5, IV-8 underwent genetic testing as well and do not carry the variant: additionally, they are in good health and they are not showing any symptoms nor signs possibly related to Marfan Syndrome.
Moreover, although II-9 is very likely a carrier of the variant, it was not possible to test her. Finally, thorough family history revealed two other relatives, I-1 and II-1, with “ectopia lentis”, who are thought to be affected because of their clinical history: genetic counselling could not be provided due to their passing.
All of the carriers of the variant have started their follow-up at our Rare Disease Clinic. Up to now, IV-6 has undergone orthopedic scoliosis correction in 2021. IV-7 is now enlisted for the same surgery. Both sisters are medically treated with antihypertensive medication to prevent aortic dilatation. No other treatment has been required.
Marfan Syndrome is a connective tissue disorder. Most of the time it is inherited with an autosomal dominant mechanism, and only 25% of cases are due to sporadic mutations (7, 8, 15). FBN1 gene is mainly involved (4, 5).
Up to now, many variants in the FBN1 gene have been reported regarding this syndrome (7, 16–19); the most commonly found are point mutations, of which 60% are missense and often caused by substitution of cysteine residues, which has been suggested to cause abnormal spliceosome during transcription (7, 20, 21). Nonetheless, many FBN1 mutations could be still unknown.
Nowadays, Revised Ghent Criteria of 2010 are used to diagnose Marfan Syndrome (22, 23), as shown in Table 1.

Table 1. Revised Ghent nosology criteria for Marfan syndrome.
According to these criteria, diagnosis can still be made without a known FBN1 mutation (4). In our study, a clinical diagnosis was made and it was later confirmed by genetic analysis.
As FBN1 gene encodes for one of the main components of elastic matrix microfibrils, connective tissue is mainly involved. However, the phenotypic spectrum is very wide: given the interfamilial and intrafamilial variability, other factors, such as environmental and stochastic effects, are probably involved (7, 8, 21). Indeed, in our case variable phenotypes with different systemic complications were found.
The main symptomatology was found in both our patients and they showed two of the main typical Marfan Syndrome features: scoliosis and aortic root enlargement.
Scoliosis is usually presented at a young age and has a rapid progression (24), just like in our patients' scenario. Braces and back surgery are usually provided (25). Scoliosis could be accompanied by other musculoskeletal involvements like tall stature, pectum excavatum or carinatum, dolichostenomelia, arachnodactyly, reduced joint mobility at the elbows and digits, and joint laxity (26). Suggestive signs for MS, which were positive in our patients, are Walker-Murdoch (“wrist sign”) and Steinberg (“thumb sign”) (24, 26).
Another important feature in MS is aortic root manifestations (2, 27). They are responsible for the majority of deaths occurring in patients affected by MS (28). In order to prevent aortic dilatation, antihypertensive medications, mainly β-blockers, are used (29, 30); in addition to this, in specific cases, prophylactic aortic root replacement is performed. Other cardiovascular features include aortic dilatation and dissection, coarctation of the aorta, mitral valve prolapse (27, 30, 31). In our scenario, both IV-6 and IV-7 are treated with anti-hypertensive medication to prevent further aortic root dilatation. No cardiac surgery is planned in the near future.
Although “ectopia lentis” (32) usually presents in cysteine missense mutations (21), as noted by Schrijver et al. (33), in our case, it was reported in two dead relatives who, however, could not be tested for gene mutation. In our family, those patients carrying the variant do not present any typical ocular manifestations but, as recommended, periodic ophthalmic examinations are being carried out.
As previously reported, MS is a multisystemic disorder involving organs such as the nervous system (dural ectasia being present in up to 66% of cases), the muscular-skeletal system with various dysmorphisms, and other clinical manifestations (protrusion acetaboli, spontaneous pneumothorax predisposition (34) and sleep disordered breathing (35–37), glaucoma (32), which require frequent follow-up (4, 38).
Although there is not yet a clear understanding regarding the pathogenesis, two main mechanisms have been found using mouse models of MS: the first one being haploinsufficiency, a quantitative defect that leads to a deficiency of fibrillin-1, and the second one being dominant negative mutations, a qualitative defect that generates an aberrant protein with impaired function (6, 7, 39). Both mechanisms seem to play a different, yet crucial, role in the clinical development of the patients carrying the mutations.
Many studies have been brought on regarding the possibility of a correlation between genotype and phenotype, but they showed little support to this hypothesis (8). Nevertheless, some considerations may be made: for example, according to literature, there is a significantly higher risk of severe cardiac involvement, shorter survival and onset of “ectopia lentis” in patients with haploinsufficiency mutations, as opposed to those who carry dominant negative mutations (4). In addition to this, haploinsufficiency tends to lead to a more variable phenotype, according to the percentage of the total proteins involved in the mutation (23, 40–43). As an example, mutations in the exons 24–32 of the gene are associated with the most severe forms of Marfan syndrome, more specifically with early onset of aortic risk (22, 44). In regards to the cardiovascular features, aortic involvement seems to be more frequent when truncating and splicing mutations are found, as well as alterations in cysteine residues (45); moreover, aortic-root dilatation progressed slowly when the mutation occurred in exon 12 or 20. Truncating mutations are also associated with a lower risk of ocular involvement (41), whereas the skeletal findings and joint laxity seem to be more severe (46, 47). Finally, cardiovascular manifestations are mild, at times even absent, when exon 13 or 49 are involved (33, 44). In addition to this, non-sense mutations were never reported in cases of neonatal Marfan syndrome (22), while scoliosis occurred with higher frequency and severity, often requiring surgical intervention, when the C1-C3 or C2-C4 disulfide bonds were disrupted (7).
Early diagnosis and state-of-the-art therapies have increased life expectancy for Marfan patients up to 75 years (48, 49). Our tested patients were relatively young and as a result we managed to intervene to reduce complications or to prevent them at all, greatly improving their quality of life; for example, spine correction surgery in IV-6 has hugely contributed to her well-being, allowing her to stand in an upright position for prolonged periods of time. Moreover, our patients, will undergo periodic follow-up, which is essential to prevent complications (50): ophthalmic examination for glaucoma and cataract, orthopedic checkups in case of scoliosis, annual echocardiography or MRI to monitor aortic root enlargement and mitral prolapse.
Since FBN1 gene mutations have mainly autosomal dominant inheritance with high penetrance, study of the probands families and their descendants is extremely important, even when they do not show any features, as to start a thorough screening process of the most common complications as soon as possible.
One limitation of our study is certainly the small number of people examined. Moreover, it was not possible to study all the members of the family. Nonetheless, it is important to highlight the absence of any pathogenic variants in the FBN1 gene in the asymptomatic members of the family, whilst clinical manifestations were present in those who carried said variant, albeit with different levels of severity.
Surely, if the new variant is to be introduced among the already known Marfan syndrome's variants, it will be possible to substantiate our discovery and to further assess the variant's role in its phenotypic presentation.
The existence, in our structure, of the Regional Coordination Centre for Rare Diseases was fundamental. It has made it possible to rapidly extend the genetic exams to adult family members and to follow over time symptomatic patients relatively to age and clinical presentation.
In conclusion, in the present study, we identified a novel heterozygous variant of FBN1 gene, in exon 20: a missense substitution of a serine residue with an arginine residue in the position 783 of Fibrillin-1 protein. Exon 20 is part of FBN1's 5′ region and it's not frequently involved in MS. It has a complete penetrance in our family, as all of the members with this specific genotype showed a pathological—yet different—phenotype. At the same time, all the members without the variant did not present with any Marfan-related symptoms nor signs. This variant had not been reported before neither in literature nor in public databases so we believe that it should be included in genetic diagnoses and counselling discussion of families with MS.
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Comitato Etico Territoriale (CET) della Regione Sardegna. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by- product of routine care or industry. Written informed consent for participation was not required from the participants or the participants' legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
MM: Writing – original draft, Writing – review & editing. BM: Writing – original draft, Writing – review & editing. FM: Data curation, Writing – original draft, Writing – review & editing. MM: Data curation, Writing – original draft, Writing – review & editing. CS: Data curation, Writing – original draft, Writing – review & editing. FL: Data curation, Writing – original draft, Writing – review & editing. DM: Data curation, Writing – original draft, Writing – review & editing. SS: Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MS, Marfan syndrome; NGS, next generation sequencing; RMI, resonance magnetic imaging.
1. Judge DP, Dietz HC. Marfan's syndrome. Lancet. (2005) 366(9501):1965–76. doi: 10.1016/S0140-6736(05)67789-6
2. Graham Stuart A, Williams A. Marfan’s syndrome and the heart. Arch Dis Child. (2007) 92(4):351–6. doi: 10.1136/adc.2006.097469
3. Hollister DW, Godfrey M, Sakai LY, Pyeritz RE. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med. (1990) 323(3):152–9. doi: 10.1056/NEJM199007193230303
4. Dietz H. FBN1-related Marfan syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2001). 1993–2025. PMID: 20301510. (updated February 17, 2022).
5. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene. (2016) 591(1):279–91. doi: 10.1016/j.gene.2016.07.033
6. Coelho SG, Almeida AG. Marfan syndrome revisited: from genetics to clinical practice. Rev Port Cardiol (Engl Ed). (2020) 39(4):215–26. doi: 10.1016/j.repc.2019.09.008.32439107
7. Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De Paepe A, et al. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. (2006) 43(10):769–87. doi: 10.1136/jmg.2005.039669
8. Boileau C, Jondeau G, Mizuguchi T, Matsumoto N. Molecular genetics of Marfan syndrome. Curr Opin Cardiol. (2005) 20(3):194–200. doi: 10.1097/01.hco.0000162398.21972.cd
9. Waldmuller S, Muller M, Warnecke H, Rees W, Schols W, Walterbusch G, et al. Genetic testing in patients with aortic aneurysms/dissections: a novel genotype/phenotype correlation? Eur J Cardiothorac Surg. (2007) 31(6):970–5. doi: 10.1016/j.ejcts.2007.02.027
10. Hilhorst-Hofstee Y, Hamel BC, Verheij JB, Rijlaarsdam ME, Mancini GM, Cobben JM, et al. The clinical spectrum of complete FBN1 allele deletions. Eur J Hum Genet. (2011) 19(3):247–52. doi: 10.1038/ejhg.2010.174
11. Jondeau G, Michel JB, Boileau C. The translational science of Marfan syndrome. Heart. (2011) 97(15):1206–14. doi: 10.1136/hrt.2010.212100
12. Yoon SH, Kong Y. Severe neonatal Marfan syndrome with a novel mutation in the intron of the FBN1 gene: a case report. Medicine (Baltimore). (2021) 100(6):e24301. doi: 10.1097/MD.0000000000024301
13. Attias D, Stheneur C, Roy C, Collod-Béroud G, Detaint D, Faivre L, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. (2009) 120(25):2541–9. doi: 10.1161/CIRCULATIONAHA.109.887042
14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
15. Gray JR, Bridges AB, Faed MJ, Pringle T, Baines P, Dean J, et al. Ascertainment and severity of Marfan syndrome in a Scottish population. J Med Genet. (1994) 31(1):51–4. doi: 10.1136/jmg.31.1.51
16. Ouyang PB, Zhao Y, Peng YQ, Zhang LS, Cao J, Li Y. A novel mutation in FBN1 gene in autosomal dominant Marfan syndrome and macular degeneration in a Chinese consanguineous family. Int J Ophthalmol. (2019) 12(5):725–30. doi: 10.18240/ijo.2019.05.05
17. Meng B, Li H, Yang T, Huang S, Sun X, Yuan H. Identification of a novel FBN1 gene mutation in a Chinese family with Marfan syndrome. Mol Vis. (2011) 17:2421–7.21976953
18. Micheal S, Khan MI, Akhtar F, Weiss MM, Islam F, Ali M, et al. Identification of a novel FBN1 gene mutation in a large Pakistani family with Marfan syndrome. Mol Vis. (2012) 18:1918–26.22876116
19. Yin Y, Liu XH, Li XH, Fan N, Lei DF, Wang Y, et al. A novel FBN1 heterozygous mutation identified in a Chinese family with autosomal dominant Marfan syndrome. Genet Mol Res. (2015) 14(2):4125–32. doi: 10.4238/2015.April.27.27
20. Franken R, Teixido-Tura G, Brion M, Forteza A, Rodriguez-Palomares J, Gutierrez L, et al. Relationship between fibrillin-1 genotype and severity of cardiovascular involvement in Marfan syndrome. Heart. (2017) 103(22):1795–9. doi: 10.1136/heartjnl-2016-310631
21. Loeys B. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med. (2001) 161(20):2447–54. doi: 10.1001/archinte.161.20.2447
22. Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. (2007) 81(3):454–66. doi: 10.1086/520125
23. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. (2010) 47(7):476–85. doi: 10.1136/jmg.2009.072785
24. Demetracopoulos CA, Sponseller PD. Spinal deformities in Marfan syndrome. Orthop Clin North Am. (2007) 38(4):563–72. doi: 10.1016/j.ocl.2007.04.003
25. Foran JRH, Pyeritz RE, Dietz HC, Sponseller PD. Characterization of the symptoms associated with dural ectasia in the Marfan patient. Am J Med Genet. (2005) 134A(1):58–65. doi: 10.1002/ajmg.a.30525
26. Pollock L, Ridout A, Teh J, Nnadi C, Stavroulias D, Pitcher A, et al. The musculoskeletal manifestations of Marfan syndrome: diagnosis, impact, and management. Curr Rheumatol Rep. (2021) 23(11):81. doi: 10.1007/s11926-021-01045-3
27. Détaint D, Faivre L, Collod-Beroud G, Child AH, Loeys BL, Binquet C, et al. Cardiovascular manifestations in men and women carrying a FBN1 mutation. Eur Heart J. (2010) 31(18):2223–9. doi: 10.1093/eurheartj/ehq258
28. Adams JN, Trent RJ. Aortic complications of Marfan’s syndrome. Lancet. (1998) 352(9142):1722–3. doi: 10.1016/S0140-6736(05)79822-6
29. Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan's syndrome. N Engl J Med. (1994) 330(19):1335–41. doi: 10.1056/NEJM199405123301902
30. Bin Mahmood SU, Velasquez CA, Zafar MA, Saeyeldin AA, Brownstein AJ, Ziganshin BA, et al. Medical management of aortic disease in Marfan syndrome. Ann Cardiothorac Surg. (2017) 6(6):654–61. doi: 10.21037/acs.2017.11.09
31. Singh J, Wanjari A. Cardiac complications in Marfan syndrome: a review. Cureus. (2022) 14(9):e29800. doi: 10.7759/cureus.29800
32. Esfandiari H, Ansari S, Mohammad-Rabei H, Mets M. Management strategies of ocular abnormalities in patients with Marfan syndrome: current perspective. J Ophthalmic Vis Res. (2019) 14(1):71. doi: 10.4103/jovr.jovr_29_18
33. Schrijver I, Liu W, Brenn T, Furthmayr H, Francke U. Cysteine substitutions in epidermal growth factor–like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet. (1999) 65(4):1007–20. doi: 10.1086/302582
34. Dyhdalo K, Farver C. Pulmonary histologic changes in Marfan syndrome. Am J Clin Pathol. (2011) 136(6):857–63. doi: 10.1309/AJCP79SNDHGKQFIN
35. Kohler M, Blair E, Risby P, Nickol AH, Wordsworth P, Forfar C, et al. The prevalence of obstructive sleep apnoea and its association with aortic dilatation in Marfan’s syndrome. Thorax. (2009) 64(2):162–6. doi: 10.1136/thx.2008.102756
36. Velvin G, Bathen T, Rand-Hendriksen S, Geirdal AØ. Systematic review of chronic pain in persons with Marfan syndrome: systematic review, Marfan syndrome, chronic pain, associated factors. Clin Genet. (2016) 89(6):647–58. doi: 10.1111/cge.12699
37. Goland S, Barakat M, Khatri N, Elkayam U. Pregnancy in Marfan syndrome: maternal and fetal risk and recommendations for patient assessment and management. Cardiol Rev. (2009) 17(6):253–62. doi: 10.1097/CRD.0b013e3181bb83d3
38. Black C, Withers AP, Gray JR, Bridges AB, Craig A, Baty DU, et al. Correlation of a recurrent FBN1 mutation (R122C) with an atypical familial Marfan syndrome phenotype. Hum Mutat. (1998) 11(S1):S198–200. doi: 10.1002/humu.1380110164
39. Finkbohner R, Johnston D, Crawford ES, Coselli J, Milewicz DM. Marfan syndrome: long-term survival and complications after aortic aneurysm repair. Circulation. (1995) 91(3):728–33. doi: 10.1161/01.cir.91.3.728
40. Robinson PN, Booms P, Katzke S, Ladewig M, Neumann L, Palz M, et al. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum Mutat. (2002) 20(3):153–61. doi: 10.1002/humu.10113
41. Nijbroek G, Sood S, McIntosh I, Francomano CA, Bull E, Pereira L, et al. Fifteen novel FBN1 mutations causing Marfan syndrome detected by heteroduplex analysis of genomic amplicons. Am J Hum Genet. (1995) 57(1):8–21. PMID: 7611299.7611299
42. Zhao F, Pan X, Zhao K, Zhao C. Two novel mutations of fibrillin-1 gene correlate with different phenotypes of Marfan syndrome in Chinese families. Mol Vis. (2013) 19:751–8. PMID: 23592911.23592911
43. Gentilini D, Oliveri A, Fazia T, Pini A, Marelli S, Bernardinelli L, et al. NGS Analysis in Marfan syndrome spectrum: combination of rare and common genetic variants to improve genotype-phenotype correlation analysis. Brodehl A, editor. PLoS One. (2019) 14(9):e0222506. doi: 10.1371/journal.pone.0222506
44. Baudhuin LM, Kotzer KE, Lagerstedt SA. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet Med. (2015) 17(3):177–87. doi: 10.1038/gim.2014.91
45. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536(7616):285–91. doi: 10.1038/nature19057
46. Schrijver I, Liu W, Odom R, Brenn T, Oefner P, Furthmayr H, et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet. (2002) 71(2):223–37. doi: 10.1086/341581
47. Loeys B, De Backer J, Van Acker P, Wettinck K, Pals G, Nuytinck L, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat. (2004) 24(2):140–6. doi: 10.1002/humu.20070
48. Landis BJ, Veldtman GR, Ware SM. Genotype–phenotype correlations in Marfan syndrome. Heart. (2017) 103(22):1750–2. doi: 10.1136/heartjnl-2017-311513
49. Faivre L, Collod-Beroud G, Child A, Callewaert B, Loeys BL, Binquet C, et al. Contribution of molecular analyses in diagnosing Marfan syndrome and type I fibrillinopathies: an international study of 1009 probands. J Med Genet. (2008) 45(6):384–90. doi: 10.1136/jmg.2007.056382
Keywords: case report, FBN1, Marfan syndrome, novel variant, pediatrics
Citation: Marsan M, Brutti M, Meloni F, Marica M, Soddu C, Lai F, Martorana D and Savasta S (2025) A novel missense variant of FBN1 gene in a Sardinian family with Marfan syndrome: a case report. Front. Pediatr. 13:1549504. doi: 10.3389/fped.2025.1549504
Received: 21 December 2024; Accepted: 21 February 2025;
Published: 7 March 2025.
Edited by:
Georgia Damoraki, National and Kapodistrian University of Athens, GreeceReviewed by:
Marcos Edgar Herkenhoff, University of São Paulo, BrazilCopyright: © 2025 Marsan, Brutti, Meloni, Marica, Soddu, Lai, Martorana and Savasta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Marsan, marsan.spec.pediatria@gmail.com; Mattia Brutti, mattiabrutti@libero.it
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.