 Piotr Buda1*†Maciej Chyb2†Anna Smorczewska-Kiljan1Anna Wieteska-Klimczak1Agata Paczesna3Monika Kowalczyk-Domagała3Magdalena Okarska-Napierała4Marta Sobalska-Kwapis2,5Łukasz Grochowalski2Marcin Słomka2,5Aneta Sitek6Janusz Ksia̧żyk1Dominik Strapagiel2,5*
Piotr Buda1*†Maciej Chyb2†Anna Smorczewska-Kiljan1Anna Wieteska-Klimczak1Agata Paczesna3Monika Kowalczyk-Domagała3Magdalena Okarska-Napierała4Marta Sobalska-Kwapis2,5Łukasz Grochowalski2Marcin Słomka2,5Aneta Sitek6Janusz Ksia̧żyk1Dominik Strapagiel2,5*- 1Department of Pediatrics, Nutrition, and Metabolic Diseases, Children's Memorial Health Institute, Warsaw, Poland
- 2Biobank Lab, Department of Molecular Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, Łódź, Poland
- 3Department of Cardiology, The Children's Memorial Health Institute, Warsaw, Poland
- 4Department of Pediatrics With Clinical Decisions Unit, Medical University of Warsaw, Warsaw, Poland
- 5BBMRI.pl Consortium, Wrocław, Poland
- 6Department of Anthropology, Faculty of Biology and Environmental Protection, University of Lodz, Łódź, Poland
Background: Kawasaki disease (KD) is an acute self-limited febrile vasculitis that mainly affects young children. Coronary artery involvement is the most serious complication in children with KD. It is currently the leading cause of acquired cardiac disease in children from developed countries. Literature data indicate a significant role of genetic susceptibility to KD.
Objective: The aim of this study was to perform the first Genome-Wide Association Study (GWAS) in a population of Polish children with KD and identify susceptible genes involved in the pathogenesis of KD.
Materials and Methods: The blood samples of Kawasaki disease patients (n = 119) were collected between 2016 and 2020, isolated and stored at the Department of Pediatrics, Nutrition and Metabolic Diseases, Children's Memorial Health Institute in Warsaw. The control group was based on Polish donors (n = 6,071) registered as the POPULOUS collection at the Biobank Lab of The Department of Molecular Biophysics in University of Lodz. DNA samples were genotyped for 558,231 Single Nucleotide Polymorphisms (SNPs) using the 24 × 1 Infinium HTS Human Core Exome microarrays according to the protocol provided by the manufacturer. In order to discover and verify genetic risk-factors for KD, association analysis was carried out using PLINK 1.9.
Results: Of all 164,395 variants, 5 were shown to occur statistically (padjusted < 0.05) more frequent in Kawasaki disease patients than in controls. Those are: rs12037447 in non-coding sequence (padjusted = 8.329 × 10−4, OR = 8.697, 95% CI; 3.629–20.84) and rs146732504 in KIF25 (padjusted = 0.007354, OR = 11.42, 95% CI; 3.79–34.43), rs151078858 in PTPRJ (padjusted = 0.04513, OR = 8.116, 95% CI; 3.134–21.01), rs55723436 in SPECC1L (padjusted = 0.04596, OR = 5.596, 95% CI; 2.669–11.74), rs6094136 in RPN2 (padjusted = 0.04755, OR = 10.08, 95% CI; 3.385–30.01) genes.
Conclusion: Polymorphisms of genes KIF25, PTRPJ, SPECC1L, RNP2 may be linked with the incidence of Kawasaki disease in Polish children.
Introduction
Kawasaki disease (KD) is an acute self-limited febrile vasculitis that mainly affects young children. It is currently the leading cause of acquired cardiac disease in children from developed countries, associated with an increased risk of coronary heart disease (1). Although KD was first described in 1967, the etiology of the disease remains unknown (2). It is believed that there is a genetic susceptibility to incorrect activation of the immune system, and oligoclonal immune response to bacterial, viral or other unidentified environmental factors, which results in damage to vascular endothelial cells and necrotizing vasculitis (3, 4). The disease is most common in young children, with most cases between 6 months and 5 years old, with predominance in males. The clinical manifestations of KD include high fever, polymorphic rash, swelling and redness of the hands and feet, changes of the lips and oral mucous membranes, cervical lymphadenopathy, and aseptic conjunctivitis. Since the inflammatory process affects all vessels, the clinical manifestation can also involve many other systemic symptoms. The atypical form of KD is increasingly recognized and defined according to the American Society of Cardiology (American Heart Association, AHA) (5). The only clinical symptom found may be fever and abnormalities in additional tests, which can cause diagnostic errors. In infants the diagnosis is particularly challenging and often late, and the disease is more frequently resistant to treatment (5, 6).
Majority of vascular compromise occurs in the coronary arteries. In about 20% of patients, vasculitis leads to coronary artery lesions (dilatation, aneurysm) that are the principal cause of acquired heart disease of children. To-date's literature data indicate a significant role of genetic susceptibility to KD (3, 4). Many genes and chromosomal regions have been identified through genome-wide association studies to have an association with KD. Polymorphisms in ITPKC, ORAI, STIM, CD40, BLK, FCGR2A, and CASP3 were among the most commonly identified as the susceptibility genes for KD (7–10). However, many other new risk loci have been recently found (NMNAT2, HCP5, ZFHX3, NAALADL2, NEBL, TUBA3C) that indicate that KD could be regarded as a multifactorial and polygenic (complex) disorder (11–14). We performed the first genetic studies in a population of Polish children with KD and identified susceptibility genes involved in the pathogenesis of KD that may provide new insights into diagnosis and treatment of this condition.
Materials and Methods
Control Group
The control group is based on polish donors recruited within the research project TESTOPLEK between 2010 and 2012 and is registered as the POPULOUS collection at the Biobank Lab of The Department of Molecular Biophysics, University of Lodz, Poland, which is currently registered in Directory (v. 4.0) of BBMRI-ERIC consortium under bbmri-eric:ID:PL_BLUL:collection:POPULOUS_BLUL registration number (15, 16) (https://directory.bbmri-eric.eu/). Samples were obtained from individuals without chronic or active diseases, based on a statement in the survey, therefore it can be marked as a control group. The control group can be considered homogenous within the same pattern as other populations within European countries, based on carried out mitochondrial DNA and Y chromosome variability studies (17, 18). It consists of 3 109 females and 2 962 males of which age ranged from 20 to 77 years old. This project had the approval from The University of Lodz's Review Board (32/KBBN-UL/I/2018), and all procedures were performed in accordance with the Declaration of Helsinki. A total of 6,071 participants were used as control group in the study. Samples of saliva were collected into Oragene OG-500 DNA collection/storage tubes (DNA Genotek, Kanata, Canada) from each individual. Genomic DNA from saliva samples was manually isolated from 500 μL using the manufacturer's instructions (PrepitL2P, PD-PR-052, DNA Genotek, Kanata, ON, Canada).
Case Group
The blood samples of 119 patients, 73 males and 46 females diagnosed with Kawasaki disease were collected between 2016 and 2020, isolated and stored at Department of Pediatrics, Nutrition and Metabolic Diseases, Children's Memorial Health Institute, Warsaw, Poland. Patients' ages ranged from 3 months to 14 years old. All children came from Poland (Mazowieckie Voivodeship), there were no ethnic differences (homogeneous Caucasian population). The diagnosis of Kawasaki disease was established according to the American Heart Association (AHA) criteria (5). Samples can be differentiated into additional phenotypes groups (Table 1). Parents of all patients gave written consent for the participation of their children in the study “Searching for molecular markers related to Kawasaki disease.” This project has the approval from the Bioethics Committee at the Children's Memorial Health Institute Review Board. Genomic DNA was isolated from blood samples mostly using MagCore pipetting station (RBC Bioscience) with the MagCore Genomic DNA Whole Blood Kit (TK Biotech). Some of the samples were isolated manually using Genomic Midi AX kit (A&A Biotechnology).
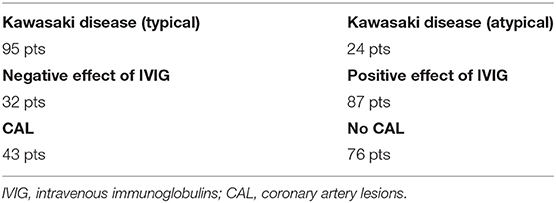
Table 1. Summary of the case group phenotypes.
Quality Control
In order to be qualified for microarray genotyping, samples had to pass quality control. Samples with DNA concentration equal or >50 ng/μl were qualified for microarray genotyping. DNA concentration was measured using broad range Quant-iT™ dsDNA Broad Range Assay Kit (Invitrogen™, Carlsbad, CA, USA). All DNA samples passed quality control in PCR reaction for sex determination (19).
Microarrays Analysis
DNA samples were genotyped using the 24 × 1 Infinium HTS Human Core Exome (Illumina Inc., San Diego, CA, USA) microarrays according to the protocol provided by the manufacturer.
Pre-processing
Raw fluorescence intensities were loaded into GenomeStudio 2.0 with Genotyping Module (Illumina, Inc.) in order to do Quality Control. Six thousand one hundred ninety (6,071 controls, 119 cases) samples with Call Rate above 0.95 were included into further analysis. Variants with incorrect clustering, Multi-allelic SNPs, and polymorphisms located at X and Y chromosomes were excluded from the analysis. 323,583 remaining SNPs where further processed in PLINK 1.9 software (20). One hundred fifty-six thousand three hundred forty-one variants were removed due to minor allele threshold and an additional 2,847 variants were removed due to Hardy–Weinberg exact-test (21). Therefore, statistical analysis was carried out on 164,395 variants. StrandScript was then used to ensure forward strand orientation (22).
Statistical Analysis
Statistical analysis was based on the chi-square statistic with odds ratio and 95% confidence interval for 2 × 2 contingency tables and was carried out in PLINK 1.9 (20, 23). Bonferroni correction was used to counteract the problem of multiple comparisons. Manhattan plot was generated using the Haploview (24).
Prediction of Single Nucleotide Polymorphism Effect
To estimate the effect of polymorphism on protein function, in silico online tools such as PredictSNP2 (25) and SIFT (Sorting Intolerant From Tolerant) was used (26). PredictSNP2 allowed us to use five different prediction tools and compare the results. Prediction is based on tools for scoring the deleteriousness of single nucleotide variants such as: CADD- (Combined Annotation Dependent Depletion) based on a support-vector machine (SVM) classifier; DANN (deleterious Annotation of Genetic Variants using Neural Networks) is based on a deep neutral network classifier; FATHMM (Functional Analysis through Hidden Markov Models) is based on an SVM classifier; FunSeq2 based on a weighted scoring system that combines genetic, epigenetic, and gene expression information; GWAVA (Genome-Wide Annotation of Variants) is based on a random forest classifier. PredictSNP2 score is based on the tools described above (25). To assess the significance of a variant placed in non-coding sequence as a candidate associated with Kawasaki disease, the HaploReg v4.1 tool was used (27).
Results
Comparison of All Allele Frequencies in Kawasaki Disease Patients (n = 119) and Controls (n = 6,071)
Of all 164,395 variants, 5 were shown to occur statistically (padjusted < 0.05) more frequent in Kawasaki disease patients than in controls. Those are: rs12037447 in non-coding sequence (padjusted = 8.329 × 10−4, OR = 8.697, 95% CI; 3.629–20.84) and rs146732504 in KIF25 (padjusted = 0.007354, OR = 11.42, 95% CI; 3.79–34.43), rs151078858 in PTPRJ (padjusted = 0.04513, OR = 8.116, 95% CI; 3.134–21.01), rs55723436 in SPECC1L (padjusted = 0.04596, OR = 5.596, 95% CI; 2.669–11.74), rs6094136 in RPN2 (padjusted = 0.04755, OR = 10.08, 95% CI; 3.385–30.01) genes (Table 2 and Figure 1).
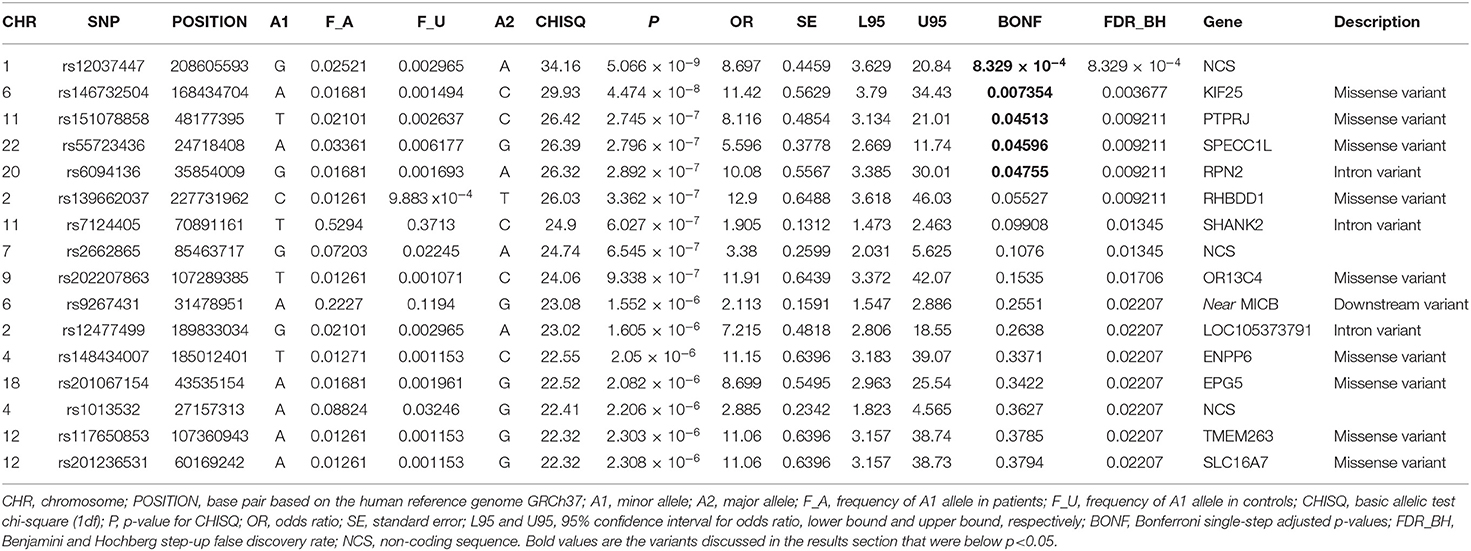
Table 2. Comparison of the frequency of SNPs in patients with KD disease (n = 119) and control groups (n = 6,071).
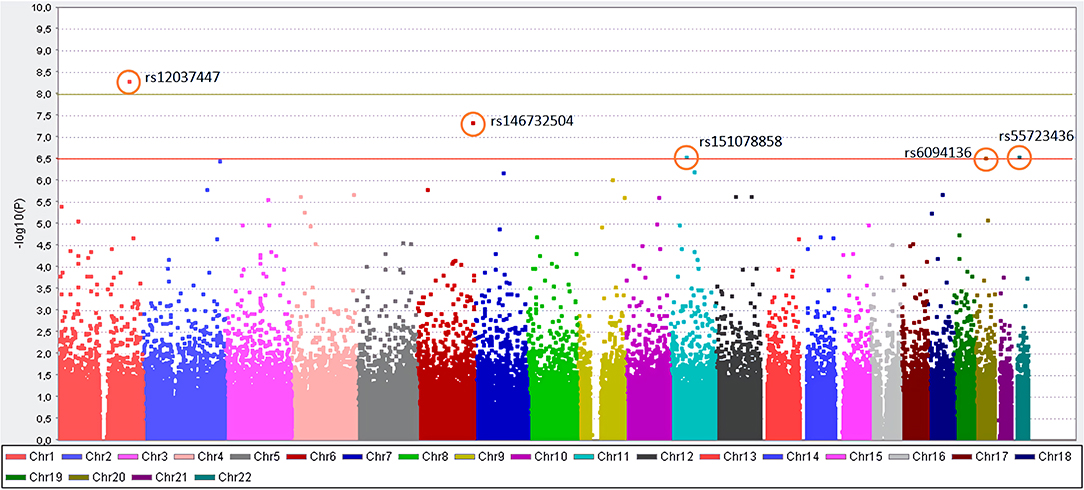
Figure 1. Manhattan plot for the genome-wide association study of patients with KD. The y-axis shows the- log10 of the unadjusted p-values (without Bonferroni correction). On the x-axis, each color represents different chromosomes, and the dots represent SNPs. The red line indicates the association threshold (10−6.5), which represents the corrected p-value = 0.05, while the green line indicates GWAS significant threshold (10−8).
Other variants that did not pass, but were the closest to the established padjusted−value <0.05 were: rs139662037 in RHBDD1 (padjusted = 0.05527, OR = 12.9, 95% CI; 3.618–46.03), rs7124405 in SHANK2 (padjusted = 0.09908, OR = 1.905, 95% CI; 1.473–2.463), rs202207863 in OR13C4 (padjusted = 0.1535, OR = 11.91, 95% CI; 3.372–42.07) genes and rs2662865 in non-coding sequence (padjusted = 0.1076, OR = 3.38, 95% CI; 2.031–5.625) (Table 2). Information about all variants, frequencies, and statistics in analysis is presented in Supplementary Table 1.
Prediction of SNP Effect
The effect of five SNPs that showed to be the most statistically associated with the Kawasaki disease in this study was evaluated, this includes: rs12037447 in non-coding sequence and rs146732504 in KIF25, rs151078858 in PTPRJ, rs55723436 in SPECC1L, rs6094136 in RPN2 genes (Table 3). Rs55723436 is a missense variant G>A in the SPECC1L gene and was predicted as deleterious by all five used tools. Additionally, this result was confirmed by SIFT predictions in one of three possible outcomes of nucleotide change (Supplementary Table 2). A similar situation was observed in the case of rs151078858, a missense variant C>T in the PTPRJ gene, which was predicted as deleterious by four out of five tools use by PredictSNP2, and as unfavorable change by SIFT. On the other hand, rs146732504, a missense variant C>A in the KIF25 gene, and rs6094136 an intron variant A>G in the RPN2 gene, were classified as deleterious only by one of the tools CADD and GAWVA, respectively. However, rs146732504 was marked as deleterious by two out of three SIFT possible predictions, while rs6094136 was not found by this tool. Rs12037447is the only variant that was evaluated as neutral by all PredictSNP2 prediction tools and this is due to the location of this polymorphism. Therefore, to assess its effect, the HaploReg v4.1 tool was used. This SNP is placed in a non-coding sequence, but it appears to have regulatory functions. The results showed that this variant influences 16 regulatory motifs, potentially affecting various functions throughout the genome (Supplementary Table 3).
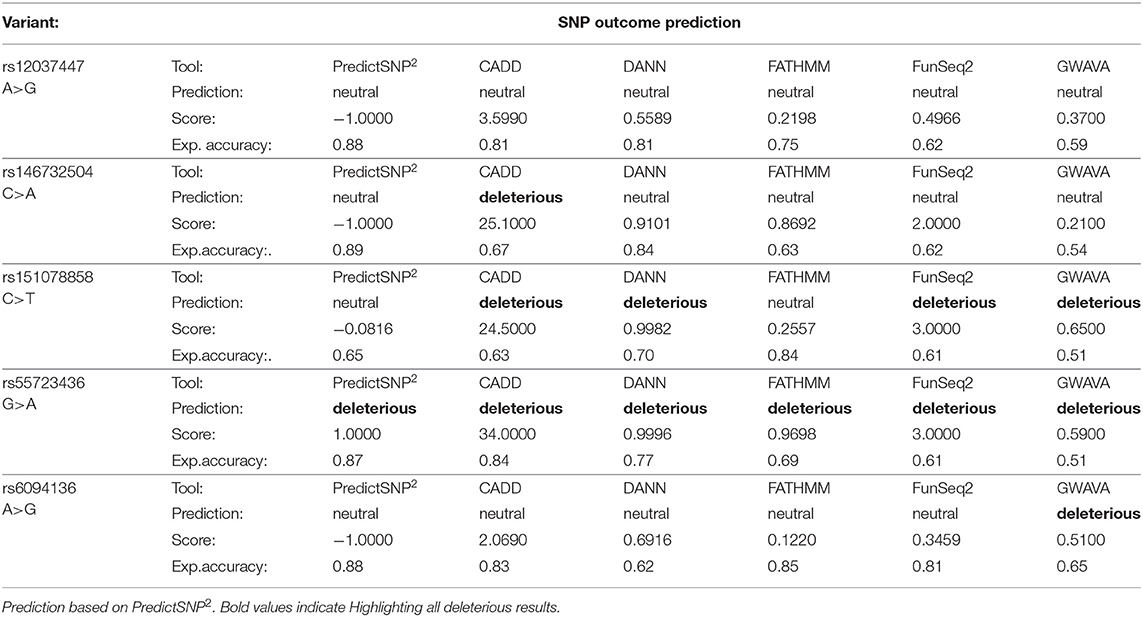
Table 3. Evaluation of SNP effect.
Discussion
We found that five single nucleotide polymorphisms were more commonly found in Polish children with Kawasaki disease than in adults: rs12037447 (non-coding region), rs146732504 (KIF25), rs151078858 (PTRPJ), rs55723436 (SPECC1L), rs6094136 (RNP2). Based on the currently available literature, none of them were previously linked to KD. We discuss below the function of proteins encoded by these genes, susceptibility to a variety of disorders and its potential role in the context of KD.
KIF25
The protein encoded by gene KIF25 (also known as KNSL3) is a member of the kinesin-like protein family involved in the trafficking of vesicles, organelles, and proteins through the cytoskeleton in a microtubule- and ATP-dependent manner (28). This protein is a negative regulator of centrosome separation required to prevent premature centrosome separation during interphase. Intracellular transport is crucial for morphogenesis and functioning of the cell. Kinesins are proteins mainly expressed in neurons, immune cells, and oligodendrocytes. Dysregulation of kinesins has been postulated to aggravate multiple sclerosis disease: lower methylated levels for KIF25 have been measured in these patients in the hippocampi and CD4+ T cells (29–32). In the context of Kawasaki disease, its relation to KIF25 was not previously described. Only Zhang et al. found that specific exosomal miRNA (miR- 671-5p), downregulated in children with KD, is affecting the expression of kinesin family member 1B (33). Another kinesin—Kinesin Superfamily Motor Protein 4 (KIF4)—was found in the muscles of patients with idiopathic inflammatory myopathies and also in activated peripheral blood lymphocytes in vitro (34). Authors concluded that KIF4 is likely to be involved in the cytoskeleton modifications associated with T-cell activation, but further studies are required to elucidate the role of kinesin in inflammatory processes.
The particular function of this gene product has not yet been determined.
PTRPJ (CD148)
Protein Tyrosine Phosphatase Receptor Type J is a protein encoded by gene PTRPJ. It is a member of the protein tyrosine phosphatase (PTP) family, signaling molecules that regulate cell growth, differentiation, mitotic cycle, and oncogenic transformation (35). PTRPJ is found in all hematopoietic lineages. This protein negatively regulates T cell receptor signaling possibly through interfering with the phosphorylation of Phospholipase C Gamma 1 and Linker for Activation of T Cells. PTPRJ/CD148 is a tyrosine phosphatase that has tumor suppressor-like activity due to its reduced expression in some malignant tumors, its regulation by cell density. PTPRJ polymorphisms were found to influence susceptibility to a variety of human cancers, e.g., thyroid, rectal colon cancers, meningiomas (36–39). Marconi et al. showed that biallelic-null mutations in PTPRJ gene impairs spreading on collagen, fibrinogen, and fibronectin; GPVI-mediated aggregation and secretion; and stromal cell-derived factor (SDF)-driven migration on fibronectin, all of which may contribute to thrombocytopenia (40). PTRPJ is also implicated in inflammatory disorders. The activation or enhancement of CD148 expression could lead to modulation of the inflammatory response. CD148 expression is upregulated in chronic inflammatory diseases, such as Crohn's disease and Cogan's syndrome (41, 42). Cogan's syndrome is a rare autoimmune vasculitis, characterized by sensorineural hearing loss, keratitis, and vasculitis. PTRPJ is expressed on the sensory epithelia of the inner ear and on endothelial cells. Antibodies to PTRPJ were identified in patients with Cogan's syndrome. Dave R et al. showed that macrophage-enriched tissues exhibit the highest expression of PTPRJ (43). CD148 is upregulated in response to various toll-like receptor ligands and downregulated by the colony-stimulating factor 1 (CSF1, macrophage colony-stimulating factor, M-CSF). There is an essential role for CSF1 and granulocyte/macrophage colony-stimulating factor (GM-CSF) in the pathogenesis of Kawasaki disease (44, 45). Stock et al. identified GM-CSF as an essential inflammatory cytokine in the development of cardiac inflammation during KD (44). When locally expressed GM-CSF switches on an inflammatory gene profile in resident macrophages of the heart, thus initiating cardiac disease. Another inflammatory pathway linked to PTPRJ was described by Wen et al. (46). It was found that PTPRJ-as1 significantly activated the NF-κB (Nuclear factor kappa-light-chain-enhancer of activated B cells) pathway in microglia under the influence of inflammatory environment and promoted the secretion of inflammatory cytokines: interleukin-6 (IL-6), tumor necrosis factor α (TNF-α) and inducible nitric oxide synthase (iNOS) and nitric oxide (NO) that was involved in inflammatory injury caused by intracerebral hemorrhage. The study by Tian et al. revealed that the expression of caspase-4, mediated by NF-κB signal pathway plays a critical role in KD (47). Human coronary artery endothelial cells (HCAEC) treated with supernatant conditioned by cells from KD patients showed a significant elevation of NF-κB p65 and caspase-4 protein expression vs. HCAEC cells treated with supernatant conditioned by control cells. We hypothesize that understanding the precise mechanism involved in the regulation of inflammation by PTRPJ in KD may be of significance.
SPECC1L
The SPECC1L gene encodes a cytoskeletal crosslinking protein named sperm antigen with calponin homology and coiled-coil domains 1 like. It is involved in cytokinesis, spindle organization, actin cytoskeleton reorganization and microtubule stabilization and hence is required for proper cell adhesion and migration (48). Diseases associated with SPECC1L include congenital disorders: oblique facial clefting-1, Opitz GBBB syndrome type II, Teebi hypertelorism syndrome. The potential linkage between SPECC1L and the immune system or inflammation and Kawasaki disease is unknown.
RPN2
This gene encodes ribophorin II (RPN2)—an integral membrane protein found only in the rough endoplasmic reticulum. This protein is part of an N-oligosaccharyltransferase complex that links high mannose oligosaccharides to asparagine residues found in the Asn-X-Ser/Thr consensus motif of nascent polypeptide chains. RPN2 is multifunctional; it has been demonstrated to be a prognostic marker of many cancers (49). RPN2 expression correlates with osteosarcoma, gastric adenocarcinoma and colorectal cancer (50–52). Takahashi et al. showed that RPN2 regulates tumor initiation and metastasis through the stabilization of mutant p53 in breast cancer cells (53). RPN2 is also highly expressed in the CD24+CD44+ cancer stem-like cells of pancreatic cancer (54). Huang L found that RPN2 promotes metastasis of hepatocellular carcinoma cell and inhibits autophagy via STAT3 (signal transducer and activator of transcription-3) and NF-κB pathways (55). The role of NF-κB in KD has been already mentioned. STAT3 is activated by interleukin 6, a pro-inflammatory cytokine that is involved in early innate immune reactivity, and present in the acute phase of KD (12, 56). Wang X et al. showed that microRNA (miR-223-3p) plays a protective role against endothelial injury in KD, by targeting IL6ST and by regulating the STAT3-NF-κB signaling pathway, making it a potential target for the diagnosis and treatment of KD (57). RPN2 is also involved in the binding of prothrombin to the monocyte surface. Fujieda suggested that RPN2 is involved in the pathophysiology of thrombosis in patients with antiphospholipid syndrome (APS) (58). Thrombosis in KD is well-known, but its relation with APS and antiphospholipid antibodies is still debated. It is worth to note that RPN2 could act via another inflammatory—JAK (Janus kinase) pathway. The Janus kinase/signal transducers and activators of transcription (JAK/STAT) axis is an inflammatory-associated pathway that is activated after receptor ligation, it mediates the signaling from cytokine receptors to the nucleus (59). Ni L et al. found that RPN2 targets Janus kinase 1 and promotes osteogenic differentiation of human bone mesenchymal stem cells (60). It is known that JAK1/STAT3 signaling pathway is activated in some systemic vasculitides (Behcet disease) through the activation of Th1/Th17-type cytokines such as IL-2, interferon (IFN-γ), IL-6, IL-17, and IL-23 but its role in KD is not well-understood (61). We speculate that the RPN2/JAK1/STAT3 axis could be a potential therapeutic target in Kawasaki disease because of its known effect on inflammatory reactions.
The most significant variant related to Kawasaki disease according to this study was rs12037447, which is placed in non-coding regulatory sequence and the analysis showed that this variant can potentially alter binding of 16 different regulatory motifs. One of the most interesting findings may be an altered P300 regulatory motif. P300 has been identified as a co-activator of HIF1α (hypoxia-inducible factor 1 alpha) in HIF-1 signaling pathway (GeneID: 2033). On the other hand, HIF-1 produced by macrophages and neutrophils is involved in the regulation of the inflammatory response and the intensification of the innate immune response. Moreover, HIF-1 signaling pathway leads to angiogenesis through activation of the VEGF signaling pathway. It has been previously reported that the serum level of vascular endothelial growth factor A (VEGF-A) in KD patients was correlated with the development of coronary arterial lesions (62). This suggests that this variant may play a role in disease development by influencing the regulatory function of P300. This polymorphism may also affect the enhancer binding protein beta (C/EBPβ). This is interesting because research suggests that C/EBPβ and C/EBPδ activation is associated with intravenous immunoglobulin therapy resistance in KD patients (63). Other altered regulatory motifs do not seem to have a connection to the pathogenesis of KD. Nevertheless, we believe that rs12037447 could be potentially connected with the pathogenesis of KD. Further studies are required to explain the complex effect of this variant.
Interestingly, we did not find the SNPs of known candidate genes for KD to be statistically significant in this analysis: rs361525 (TNF), rs1569723 (CD40), rs17531088 (NAALADL2), rs2078087 (NMNAT2) (Supplementary Table 1) (10, 11, 64, 65). The explanation for that might be the specificity of the GWAS method. Genome Wide Association Studies are a powerful tool to discover novel disease susceptibility genes, but it has its limitations. One of them is the requirement for a larger affected sample group; therefore, by increasing it, we could possibly observe SNPs of known genes in KD to be more statistically significant. Moreover, expanding the study group would allow us to perform more complex analysis, by differentiation of the study group into subgroups by adding secondary phenotypes, for example, information about a patient's resistance for intravenous immunoglobulin therapy and thus make an attempt to identify variants related to this phenomenon (66). Another secondary phenotype may be gender, as research suggests a link to male-specific association of polymorphisms in FCGR2A gene with Kawasaki disease (67). Other well-known SNPs related to the development of KD, such as rs1801274 (FCGR2A) (10), rs2254546 (FAM167A-BLK region) (8), rs2736340 (BLK) (64) were not included in the analysis, because they were filtered out at the beginning of workflow, due to multi-allelism which makes it impossible to predict a risk allele based solely on genotyping data. Despite this, the re-analysis was carried out containing these variants which showed that they did not occur statistically more or less frequently in this study group than in controls (unpublished data).
Following the obtained results, polymorphisms of genes KIF25, PTRPJ, SPECC1L, and RNP2 seemed to be involved in KD in Polish children. They also may explain the incidence of KD in Poland; however geographic distribution of gene polymorphisms in Polish children is unknown. This study's limitation is a relatively small sample size, thus multicenter studies on large sample sizes are needed to further reveal the relationship between the above mentioned genes and KD.
Data Availability Statement
The original contributions generated in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by The Bioethics Committee at the Children's Memorial Health Institute in Warsaw. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
PB: conceptualization, methodology, validation, investigation, formal analysis, writing—original draft preparation, visualization, supervision, and project administration. MC: methodology, validation, formal analysis, resources, software, data curation, writing—original draft preparation, visualization, and supervision. AS-K, AW-K, AP, MK-D, and MO-N: investigation, writing—review, and editing. MS-K: methodology and data curation. ŁG: methodology, writing—review, and editing. MS and AS: data curation. JK: writing—review and editing. DS: methodology, validation, formal analysis, resources, funding acquisition, visualization, supervision, writing—review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financed by Polish Ministry of Science and Higher Education No. DIR/WK/2017/01 and Operational Programme Digital Poland Grant No. POPC.02.03.01-00-0012/17. POPULOUS collection was financed by the Polish POIG Grant 01.01.02-10-005/08.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2021.624798/full#supplementary-material
References
1. Burns JC, Glodé MP. Kawasaki syndrome. Lancet. (2004) 364:533–44. doi: 10.1016/S0140-6736(04)16814-1
2. Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Jpn J Allergol. (1967) 16:178–222.
3. Rowley AH. Kawasaki disease: novel insights into etiology and genetic susceptibility. Annu Rev Med. (2011) 62:69–77. doi: 10.1146/annurev-med-042409-151944
4. Onouchi Y. The genetics of Kawasaki disease. Int J Rheum Dis. (2018) 21:26–30. doi: 10.1111/1756-185X.13218
5. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the American Heart Association. Circulation. (2017) 135:e927–99. doi: 10.1161/CIR.0000000000000484
6. Eleftheriou D, Levin M, Shingadia D, Tulloh R, Klein NJ, Brogan PA. Management of Kawasaki disease. Arch Dis Child. (2014) 99:74–83. doi: 10.1136/archdischild-2012-302841
7. Onouchi Y, Gunji T, Burns JC, Shimizu C, Newburger JW, Yashiro M, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. (2008) 40:35–42. doi: 10.1038/ng.2007.59
8. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
9. Onouchi Y, Ozaki K, Buns JC, Shimizu C, Hamada H, Honda T, et al. Common variants in CASP3 confer susceptibility to Kawasaki disease. Hum Mol Genet. (2010) 19:2898–906. doi: 10.1093/hmg/ddq176
10. Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 13:1241–6. doi: 10.1038/ng.981
11. Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, Kil HR, et al. A genome-wide association analysis identifies NMNAT2 and HCP5 as susceptibility loci for Kawasaki disease. J Hum Genet. (2017) 62:1023–9. doi: 10.1038/jhg.2017.87
12. Burgner D, Davila S, Breunis WB, Ng SB, Li Y, Bonnard C, et al. A genome-wide association study identifies novel and functionally related susceptibility loci for Kawasaki disease. PLoS Genet. (2009) 5:e1000319. doi: 10.1371/journal.pgen.1000319
13. Kuo HC, Li SC, Guo MMH, Huang YH, Yu HR, Huang FC, et al. Genome-wide association study identifies novel susceptibility genes associated with coronary artery aneurysm formation in Kawasaki disease. PLoS ONE. (2016) 11:e0154943. doi: 10.1371/journal.pone.0154943
14. Kuo HC, Chang WC. Genetic polymorphisms in Kawasaki disease. Acta Pharmacologica Sin. (2011) 32:1193–8. doi: 10.1038/aps.2011.93
15. Strapagiel D, Sobalska-kwapis M, Słomka M, Marciniak B. Biobank Lodz—DNA Based Biobank at the University of Lodz. Open J Bioresour. (2016) 25:3–7. doi: 10.5334/ojb.22
16. Dobrowolska S, Michalska-Madej J, Slomka M, Sobalska-Kwapis M, Strapagiel D. BiobankŁódź-population based biobank at the University of Łódź, Poland. Eur J Transl Clin Med. (2019) 2:85–95. doi: 10.31373/ejtcm/109495
17. Jarczak J, Grochowalski Ł, Marciniak B, Lach J, Słomka M, Sobalska-Kwapis M, et al. Mitochondrial DNA variability of the Polish population. Eur J Hum Genet. (2019) 27:1304–14. doi: 10.1038/s41431-019-0381-x
18. Grochowalski Ł, Jarczak J, Urbanowicz M, Słomka M, Szargut M, Borówka P, et al. Y-chromosome genetic analysis of modern polish population. Front Genet. (2020) 11:567309. doi: 10.3389/fgene.2020.567309
19. Strapagiel D, Majewska M, Słomka M, Janik K, Sobalska M, Bartosz G. Method for Determination of Gender. Patent Number: PL 232652 B1 (2019).
20. Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richerdatasets. Giga Sci. (2015) 4:7 doi: 10.1186/s13742-015-0047-8
21. Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet. (2005) 76:887–93. doi: 10.1086/429864
22. Wang J, Samuels DC, Shyr Y, Guo Y. Genetics and population analysis StrandScript: evaluation of Illumina genotyping array design and strand correction. Bioinformatics. (2017) 4:1–3. doi: 10.1093/bioinformatics/btx186
23. Steiß V, Letschert T, Schäfer H, Pahl R. PERMORY-MPI: a program for high-speed parallel permutation testing in genome-wide association studies. Bioinformatics. (2012) 28:1168–9. doi: 10.1093/bioinformatics/bts086
24. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. (2005) 21:263–5. doi: 10.1093/bioinformatics/bth457
25. Bendl J, Musil M, Jan Š, Zendulka J, Damborský J, Brezovský J. PredictSNP2: a unified platform for accurately evaluating SNP effects by exploiting the different characteristics of variants in distinct genomic regions. PLoS Comput Biol. (2016) 12:e1004962. doi: 10.1371/journal.pcbi.1004962
26. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. (2015) 11:1–9. doi: 10.1038/nprot.2015.123
27. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucl Acid Res. (2012) 40:930–4. doi: 10.1093/nar/gkr917
28. Miki H, Setou M, Kaneshiro K, Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA. (2001) 98:7004–11. doi: 10.1073/pnas.111145398
29. Hares K, Kemp K, Rice C, Gray E, Scolding N, Wilkins A. Reduced axonal motor protein expression in non-Lesional grey matter in multiple sclerosis. MultScler J. (2014) 20:812–21. doi: 10.1177/1352458513508836
30. Chomyk AM, Volsko C, Tripathi A, Deckard SA, Trapp BD, Fox RJ, et al. DNA methylation in demyelinated multiple sclerosis hippocampus. Sci Rep. (2017) 7:8696. doi: 10.1038/s41598-017-08623-5
31. Graves MC, Benton M, Lea RA, Boyle M, Tajouri L, Macartney-Coxson D, et al. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. MultScler J. (2014) 20:1033–41. doi: 10.1177/1352458513516529
32. Celarain N, Tomas-Roig J. Aberrant DNA methylation profile exacerbates inflammation and neurodegeneration in multiple sclerosis patients. J Neuroinflamm. (2020) 17:21. doi: 10.1186/s12974-019-1667-1
33. Zhang X, Xin G, Sun D. Serum exosomal miR-328, miR-575, miR-134 and miR-671-5p as potential biomarkers for the diagnosis of kawasaki disease and the prediction of therapeutic outcomes of intravenous immunoglobulin therapy. Exp Ther Med. (2018) 16:2420–32. doi: 10.3892/etm.2018.6458
34. Bernasconi P, Cappelletti C, Navone F, Nessi V, Baggi F, Vernos I, et al. The kinesin superfamily motor protein KIF4 is associated with immune cell activation in idiopathic inflammatory myopathies. J Neuropathol Exp Neurol. (2008) 67:624–32. doi: 10.1097/NEN.0b013e318177e5fd
35. Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, et al. Protein tyrosine phosphatases in the human genome. Cell. (2004) 117:699–711. doi: 10.1016/j.cell.2004.05.018
36. Iuliano R, Le Pera I, Cristofaro C, Baudi F, Arturi F, Pallante PL, et al. The tyrosine phosphatase PTPRJ/DEP-1 genotype affects thyroid carcinogenesis. Oncogene. (2004) 23:8432–8. doi: 10.1038/sj.onc.1207766
37. Ruivenkamp CAL, Van Wezd T, Zanon C, Stassen APM, Vlcek C, Csikós T, et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. (2002) 31:295–300. doi: 10.1038/ng903
38. Petermann A, Haase D, Wetzel A, Balavenkatraman KK, Tenev T, Gührs KH, et al. Loss of the protein-tyrosine phosphatase DEP-1/PTPRJ drives meningioma cell motility. Brain Pathol. (2011) 21:405–18. doi: 10.1111/j.1750-3639.2010.00464.x
39. Mita Y, Yasuda Y, Sakai A, Yamamoto H, Toyooka S, Gunduz M, et al. Missense polymorphisms of PTPRJ and PTPN13 genes affect susceptibility to a variety of human cancers. J Cancer Res Clin Oncol. (2010) 136:249–59. doi: 10.1007/s00432-009-0656-7
40. Marconi C, DI Buduo CA, LeVine K, Barozzi S, Faleschini M, Bozzi V, et al. Loss-of-function mutations in PTPRJ cause a new form of inherited thrombocytopenia. Blood. (2019) 133:1346–57. doi: 10.1182/blood-2018-07-859496
41. Autschbach F, Palou E, Mechtersheimer G, Rohr C, Pirotto F, Gassler N, et al. Expression of the membrane protein tyrosine phosphatase CD148 in human tissues. Tissue Antigens. (1999) 54:485–98. doi: 10.1034/j.1399-0039.1999.540506.x
42. Lunardi C, Bason C, Leandri M, Navone R, Lestani M, Millo E, et al. Autoantibodies to inner ear and endothelial antigens in Cogan's syndrome. Lancet. (2002) 360:915–21. doi: 10.1016/S0140-6736(02)11028-2
43. Dave RK, Dinger ME, Andrew M, Askarian-Amiri M, Hume DA, Kellie S. Regulated expression of PTPRJ/CD148 and an antisense long noncoding RNA in macrophages by proinflammatory stimuli. PLoS ONE. (2013) 8:e68306. doi: 10.1371/journal.pone.0068306
44. Stock AT, Hansen JA, Sleeman MA, McKenzie BS, Wicks IP. GM-CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. (2016) 213:1983–98. doi: 10.1084/jem.20151853
45. Igarashi H, Hatake K, Tomizuka H, Yamada M, Gunji Y, Momoi MY. High serum levels of M-CSF and G-CSF in Kawasaki disease. Br J Haematol. (1999)105:613–5. doi: 10.1046/j.1365-2141.1999.01381.x
46. Wen J, Yang CY, Lu J, Wang XY. Ptprj-As1 mediates inflammatory injury after intracerebral hemorrhage by activating NF-κB pathway. Eur Rev Med Pharmacol Sci. (2018) 22:2817–23. doi: 10.26355/eurrev_201805_14981
47. Tian J, An X, Niu L. Correlation between NF-κB signal pathway-mediated caspase-4 activation and Kawasaki disease. Exp Ther Med. (2017) 13:3333–6. doi: 10.3892/etm.2017.4409
48. Saadi I, Alkuraya FS, Gisselbrecht SS, Goessling W, Cavallesco R, Turbe-Doan A, et al. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am J Hum Genet. (2011) 89:44–55. doi: 10.1016/j.ajhg.2011.05.023
49. Hong F, Li Y, Ni H, Li J. Downregulation of ribophorin II suppresses tumor growth, migration, and invasion of nasopharyngeal carcinoma. Onco Targets Ther. (2018) 11:3485–94. doi: 10.2147/OTT.S158355
50. Fujiwara T, Takahashi RU, Kosaka N, Nezu Y, Kawai A, Ozaki T, et al. RPN2 gene confers osteosarcoma cell malignant phenotypes and determines clinical prognosis. Mol Ther Nucleic Acids. (2014) 3:e189. doi: 10.1038/mtna.2014.35
51. Fujimoto D, Goi T, Hirono Y. Expression of ribophorine II is a promising prognostic factor in human gastric adenocarcinoma. Int J Oncol. (2017) 50:448–56. doi: 10.3892/ijo.2016.3822
52. Li H, Al-Japairai K, Tao Y, Xiang Z. RPN2 promotes colorectal cancer cell proliferation through modulating the glycosylation status of EGFR. Oncotarget. (2017) 8:72633–51. doi: 10.18632/oncotarget.20005
53. Takahashi R, Ochiya T. Small interfering RNA-mediated silencing of the ribophorin II gene: advances in the treatment of malignant breast cancer. In: Filice M, Ruiz-Cabello J, editor. Nucleic Acid Nanotheranostics. Amsterdam: Elsevier (2019). p. 27–41.
54. Zhu J, He J, Liu Y, Simeone DM, Lubman DM. Identification of glycoprotein markers for pancreatic cancer CD24 +CD44 + stem-like cells using nano-LC-MS/MS and tissue microarray. J Proteome Res. (2012) 11:2272–81. doi: 10.1021/pr201059g
55. Huang L, Jian Z, Gao Y, Zhou P, Zhang G, Jiang B, et al. RPN2 promotes metastasis of hepatocellular carcinoma cell and inhibits autophagy via STAT3 and NF-κB pathways. Aging (Albany NY). (2019) 11:6674–90. doi: 10.18632/aging.102167
56. Ueno Y, Takano N, Kanegane H, Yokoi T, Yachie A, Miyawaki T, et al. The acute phase nature of interleukin 6: studies in Kawasaki disease and other febrile illnesses. Clin Exp Immunol. (1989) 76:337–42.
57. Wang X, Ding YY, Chen Y, Xu QQ, Qian HG, Qian GW, et al. MiR-223-3p alleviates vascular endothelial injury by targeting IL6ST in Kawasaki disease. Front Pediatr. (2019) 7:288. doi: 10.3389/fped.2019.00288
58. Fujieda Y, Amengual O, Matsumoto M, Kuroki K, Takahashi H, Kono M, et al. Ribophorin ii is involved in the tissue factor expression mediated by phosphatidylserine dependent antiprothrombin antibody on monocytes. Rheumatol (United Kingdom). (2016) 55:1117–26. doi: 10.1093/rheumatology/kew005
59. Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. (2004) 18:189–218. doi: 10.1038/sj.leu.2403241
60. Ni L, Yu J, Gui X, Lu Z, Wang X, Guo H, et al. Overexpression of RPN2 promotes osteogenic differentiation of hBMSCs through the JAK/STAT3 pathway. FEBS Open Bio. (2020) 10:158–67. doi: 10.1002/2211-5463.12766
61. Nanke Y, Yago T, Kotake S. The role of Th17 cells in the pathogenesis of Behcet's disease. J Clin Med. (2017) 6:74. doi: 10.3390/jcm6070074
62. Jing Y, Ding M, Fu J, Xiao Y, Chen X, Zhang Q. Neutrophil extracellular trap from Kawasaki disease alter the biologic responses of PBMC. Biosci Rep. (2020) 40:BSR20200928. doi: 10.1042/BSR20200928
63. Inoue T, Miyashita M, Murakami S, Igarashi A, Motomura K, Abe J, et al. IL-1β and IL-17A are involved in IVIG resistance through activation of C/EBPβ and δ in a coronary artery model of Kawasaki disease. Allergy. (2020) 75:2102–5. doi: 10.1111/all.14281
64. Lee YC, Kuo HC, Chang JS, Chang LY, Huang LM, Chen MR, et al. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat Genet. (2012) 44:522–5. doi: 10.1038/ng.2227
65. Ferdosian F, Dastgheib SA, Hosseini-jangjou SH, Nafei Z, Lookzadeh MH, Noorishadkam M, et al. Association of TNF- α rs1800629, CASP3 rs72689236 and FCGR2A rs1801274 polymorphisms with susceptibility to Kawasaki disease: a comprehensive meta-analysis. Fetal Pediatr Pathol. (2019) 30:1–17. doi: 10.1080/15513815.2019.1707917
66. Kuo HC, Wong HSC, Chang WP, Chen BK, Wu MS, Yang KD, et al. Prediction for intravenous immunoglobulin resistance by using weighted genetic risk score identified from genome-wide association study in Kawasaki disease. Circ Cardiovasc Genet. (2017) 10:1–7. doi: 10.1161/CIRCGENETICS.116.001625
Keywords: Kawasaki disease, genome-wide association study, susceptibility gene, KIF25, PTPRJ, SPECC1L, RNP2
Citation: Buda P, Chyb M, Smorczewska-Kiljan A, Wieteska-Klimczak A, Paczesna A, Kowalczyk-Domagała M, Okarska-Napierała M, Sobalska-Kwapis M, Grochowalski Ł, Słomka M, Sitek A, Ksia̧żyk J and Strapagiel D (2021) Association Between rs12037447, rs146732504, rs151078858, rs55723436, and rs6094136 Polymorphisms and Kawasaki Disease in the Population of Polish Children. Front. Pediatr. 9:624798. doi: 10.3389/fped.2021.624798
Received: 01 November 2020; Accepted: 27 January 2021;
Published: 22 February 2021.
Edited by:
Kuender D. Yang, Mackay Memorial Hospital, TaiwanReviewed by:
Lovro Lamot, University of Zagreb, CroatiaKlaus Tenbrock, RWTH Aachen University, Germany
Copyright © 2021 Buda, Chyb, Smorczewska-Kiljan, Wieteska-Klimczak, Paczesna, Kowalczyk-Domagała, Okarska-Napierała, Sobalska-Kwapis, Grochowalski, Słomka, Sitek, Ksia̧żyk and Strapagiel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piotr Buda, p.buda@ipczd.pl; Dominik Strapagiel, dominik.strapagiel@biol.uni.lodz.pl
†These authors have contributed equally to this work