 Ziyang Jiang
Ziyang Jiang Zhihan Gu1
Zhihan Gu1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 November 2024
Sec. Thoracic Oncology
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1447678
This article is part of the Research Topic Treatment Response and Resistance to Targeted Therapies for NSCLC View all 16 articles
The clinical application of small molecule tyrosine kinase inhibitors (TKIs) has significantly improved the quality of life and prognosis of patients with non-small cell lung cancer (NSCLC) carrying driver genes. However, resistance to TKI treatment is inevitable. Bypass signal activation is one of the important reasons for TKI resistance. Although TKI drugs inhibit downstream signaling pathways of driver genes, key signaling pathways within tumor cells can still be persistently activated through bypass routes such as MET gene amplification, EGFR gene amplification, and AXL activation. This continuous activation maintains tumor cell growth and proliferation, leading to TKI resistance. The fundamental strategy to treat TKI resistance mediated by bypass activation involves simultaneously inhibiting the activated bypass signals and the original driver gene signaling pathways. Some clinical trials based on this combined treatment approach have yielded promising preliminary results, offering more treatment options for NSCLC patients with TKI resistance. Additionally, early identification of resistance mechanisms through liquid biopsy, personalized targeted therapy against these mechanisms, and preemptive targeting of drug-tolerant persistent cells may provide NSCLC patients with more sustained and effective treatment.
Lung cancer is one of the leading causes of cancer-related deaths worldwide. According to the 2022 Global Cancer Research Report, nearly 20 million people were newly diagnosed with cancer globally in 2022, with approximately 8 million deaths. Primary lung cancer is the most common among all newly diagnosed cancer cases, with about 2.5 million new cases, accounting for 12.4% of global cancer cases (1). In the United States, thanks to advancements in cancer screening, diagnosis, and treatment technologies, the cancer mortality rate decreased annually by approximately 1.2% from 2016 to 2020, with a particularly noticeable decline in lung cancer mortality (2). The quality of life and prognostic outcomes of lung cancer patients have been greatly improved by the use of TKIs drugs.
Based on histological types, lung cancer can be classified into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), with NSCLC accounting for 80%-85% of all lung cancers (3–5). Current research indicates that more than two-thirds of NSCLC cases carry tumor driver genes (6, 7), including epidermal growth factor receptor (EGFR) gene (10%-15% in Western populations and 30%-50% in East Asian populations) (8, 9), Kirsten rat sarcoma viral oncogene (KRAS) (15%-20%) (10), anaplastic lymphoma kinase (EML4-ALK) (3%-7%) (10, 11), and c-ros oncogene 1 (ROS1) (1%-2%) (12) etc. The U.S. Food and Drug Administration (FDA) has approved various tyrosine kinase inhibitors (TKIs) for clinical treatment of NSCLC with these driver genes, such as first-generation EGFR TKIs [Gefitinib (13, 14) and Erlotinib (15)], second-generation EGFR TKIs [Afatinib (16) and Dacomitinib (17)], third-generation EGFR TKIs [Osimertinib (18–20)], first-generation ALK TKIs [Crizotinib (21–23)], second-generation ALK TKIs [Ceritinib (24–26), Alectinib (27), and Brigatinib (28)], and third-generation ALK TKIs [Lorlatinib (29, 30)] (see Table 1). The application of TKI drugs has significantly improved the prognosis of NSCLC patients with driver genes. Unfortunately, most patients develop resistance to TKI drugs after 1 to 2 years of treatment, causing tumor progression and making it difficult for patients to achieve long-term benefits from TKI therapy (31–33).
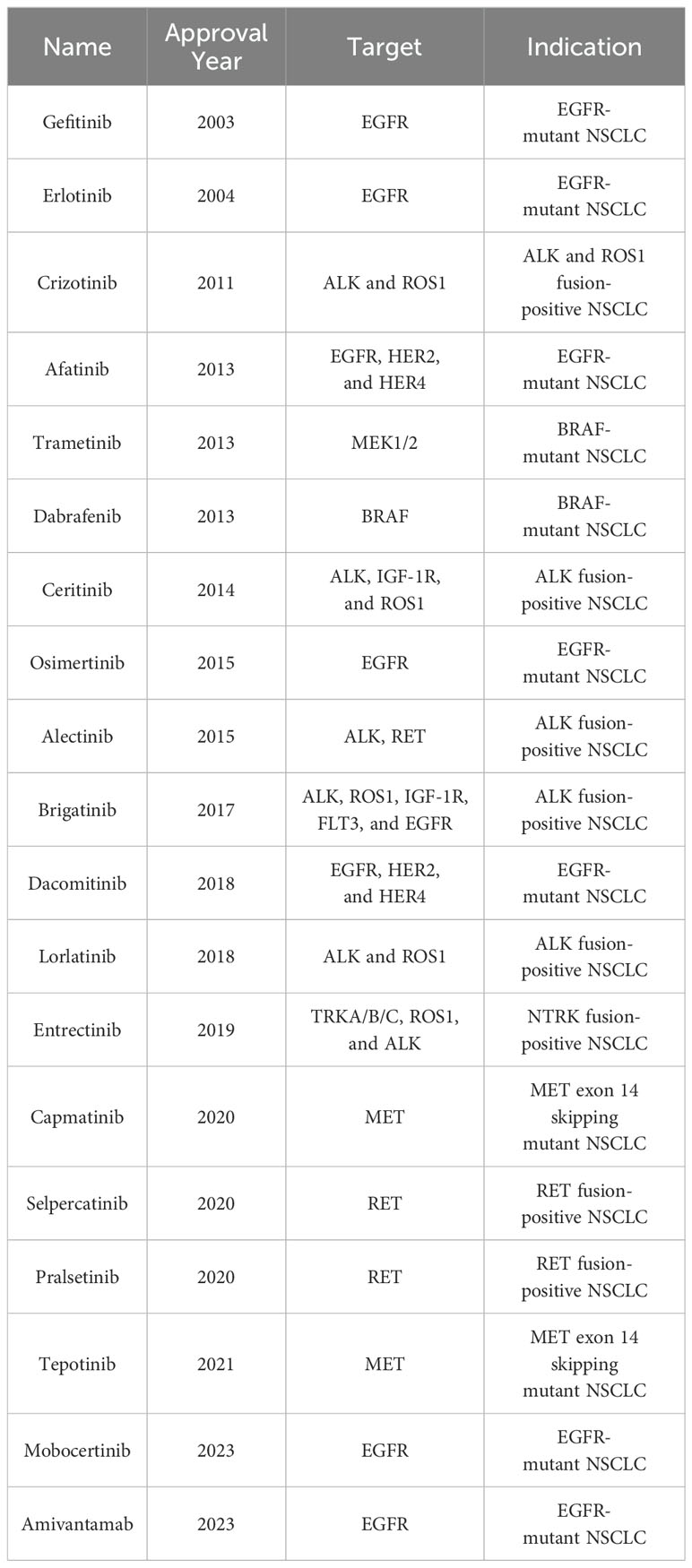
Table 1. FDA-approved kinase inhibitors for advanced NSCLC treatment.
The mechanisms of TKI resistance in NSCLC mainly include primary resistance and acquired resistance (31, 34, 35). Studies have shown that 4%-10% of NSCLC patients exhibit primary resistance to TKIs, which may be due to the presence of other non-sensitive mutations in the target gene, although the exact molecular mechanisms are not fully understood (31). Over 90% of TKI resistance is acquired resistance, which includes mechanisms such as secondary mutations in the target gene, bypass activation, and histological transformation (31, 34). In patients with EGFR mutations, approximately 60% of TKI resistance is caused by secondary mutations in the EGFR gene, while about 20% is due to bypass activation (31, 34, 35). For patients with ALK gene fusions, 28%-50% of TKI resistance is due to gene mutations in the ALK kinase domain, and 40%-50% of patients develop resistance to ALK TKIs through bypass activation (31, 34, 35). Secondary mutations in the target gene can impede the binding of TKI drugs to the kinase by altering the conformation of the tyrosine kinase and/or changing the affinity between the kinase and ATP, resulting in the loss of the drug’s cytotoxic effect on tumor cells (36, 37). New generations of TKI drugs are typically designed to target secondary mutation sites that confer resistance to previous generations, allowing patients with secondary mutations to continue benefiting from treatment. For example, in EGFR-mutant NSCLC patients, the EGFR T790M mutation is a significant cause of resistance to first- and second-generation EGFR TKIs (38, 39). Third-generation EGFR TKIs, such as Osimertinib, are designed to target the EGFR T790M mutation, significantly improving the prognosis of NSCLC patients with this mutation (19). In bypass activation-mediated TKI resistance, tumor cells evade the cytotoxic effects of TKI drugs through continuous activation of bypass signaling pathways. The mechanisms of bypass activation-mediated resistance are complex, currently there is no standard clinical treatment for bypass activation resistance. Therefore, this review will summarize the research progress on bypass activation mechanisms in NSCLC TKI resistance, aiming to provide new insights and clues for future research on NSCLC TKI resistance mechanisms and the development of combination treatment strategies that can overcome or reverse resistance.
Tumor cells maintain proliferation and differentiation through a coordinated network of intracellular signaling pathways. When the primary signaling pathways in tumor cells are inhibited by TKI drugs, the tumor cells can reactivate key downstream effectors required for cell survival and proliferation through parallel signaling pathways of other receptor tyrosine kinases (RTKs). This allows the tumor to bypass the inhibition of the driver gene by the TKI drug and continue to survive and grow (34, 40). This mechanism is known as bypass activation. In 2007, Engelman et al. (41) first described this resistance mechanism. They found Mesenchymal to Epithelial Transition Factor(MET) gene amplification in about 20% of patients with EGFR TKI resistance. MET gene amplification bypasses the EGFR signaling pathway by mediating HER3 phosphorylation, thereby reactivating the PI3K/AKT signaling pathway (41).
Theoretically, the presence of each driver gene in NSCLC is mutually exclusive, but under TKI drug treatment, driver genes often operate in a complementary manner to sustain cell survival and growth. MET gene amplification, which mediates resistance to EGFR TKIs, is one such example. Additionally, in EML4-ALK fusion NSCLC, approximately 40% of Crizotinib-resistant cases exhibit activation of the EGFR signaling pathway (42–44). In these cases, EGFR pathway activation is often due to EGFR gene amplification rather than EGFR gene mutation.
Besides MET gene amplification and EGFR pathway activation, other bypass activation pathways can mediate NSCLC TKI resistance. These include AXL activation (45), insulin-like growth factor receptor (IGF-1R) activation (46), and human epidermal growth factor receptor 2 (HER2) amplification (47). Common bypass activation pathways in NSCLC TKI resistance are summarized in Table 2.
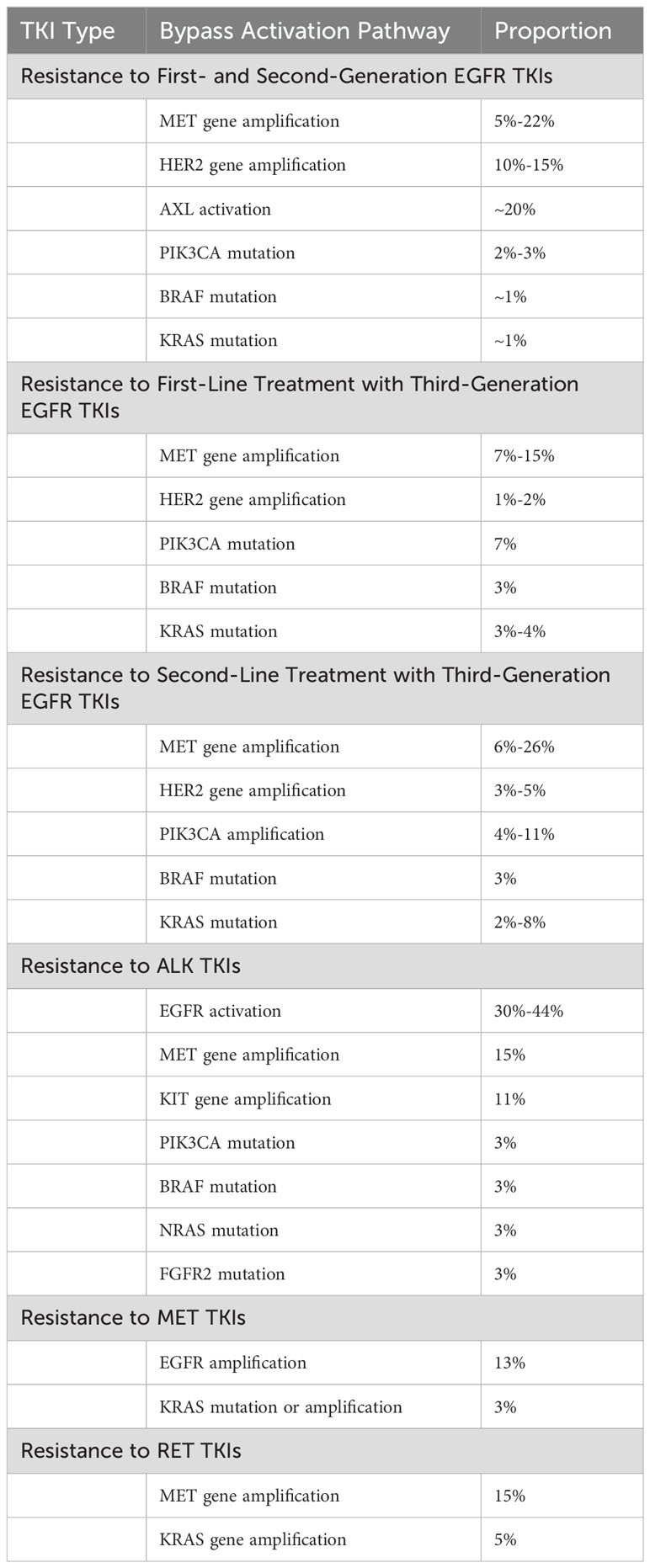
Table 2. Common bypass activation pathways in NSCLC TKI resistance.
The MET proto-oncogene is located on human chromosome 7q21-q31, spanning approximately 125 kb and containing 21 exons and 20 introns (48). The protein encoded by the MET gene, c-MET, is a transmembrane receptor tyrosine kinase composed of an extracellular SEMA (Semaphorin) domain, PSI (Plexin Semaphoring Integrin) domain, and IPT (Immunoglobulin Plexins Transcription) domain, a transmembrane domain, and an intracellular juxtamembrane (JM) domain, tyrosine kinase domain, and a carboxy-terminal region. This heterodimer has self-phosphorylation capability (49). Hepatocyte Growth Factor (HGF) is currently the only known high-affinity ligand for c-MET. When the HGF ligand binds to the SEMA domain of c-MET, the intracellular tyrosine residues of c-MET undergo homodimerization and phosphorylation, thereby activating c-MET. This activation subsequently triggers downstream signaling pathways such as RAS/ERK/MAPK, PI3K/AKT, Wnt/β-catenin, and STAT, promoting cell growth, proliferation, migration, angiogenesis, and epithelial-to-mesenchymal transition (EMT) (50, 51). In NSCLC, the abnormal activation of the c-MET signaling pathway mainly includes MET gene amplification (52), MET exon 14 skipping mutations (53), MET gene rearrangements (54, 55), and c-MET protein overexpression (56, 57).
MET gene amplification refers to an increase in the copy number of the MET gene (52). This increase is often achieved through gene breakage-fusion-bridge mechanisms that replicate the gene region or local area (58). MET gene amplification leads to overexpression of the c-MET protein, thereby activating downstream signaling pathways. Studies (59) have shown that MET gene amplification can often coexist with other tumor driver genes in NSCLC, such as EGFR and KRAS mutations and ALK and ROS1 fusions, indicating that MET gene amplification may not act as an intrinsic driver factor within tumors. In NSCLC, MET gene amplification is considered the most common mechanism of TKI resistance mediated by bypass activation pathways (31, 32, 34). The frequency of MET gene amplification in various gene mutation types of NSCLC TKI resistance is shown in Table 3.
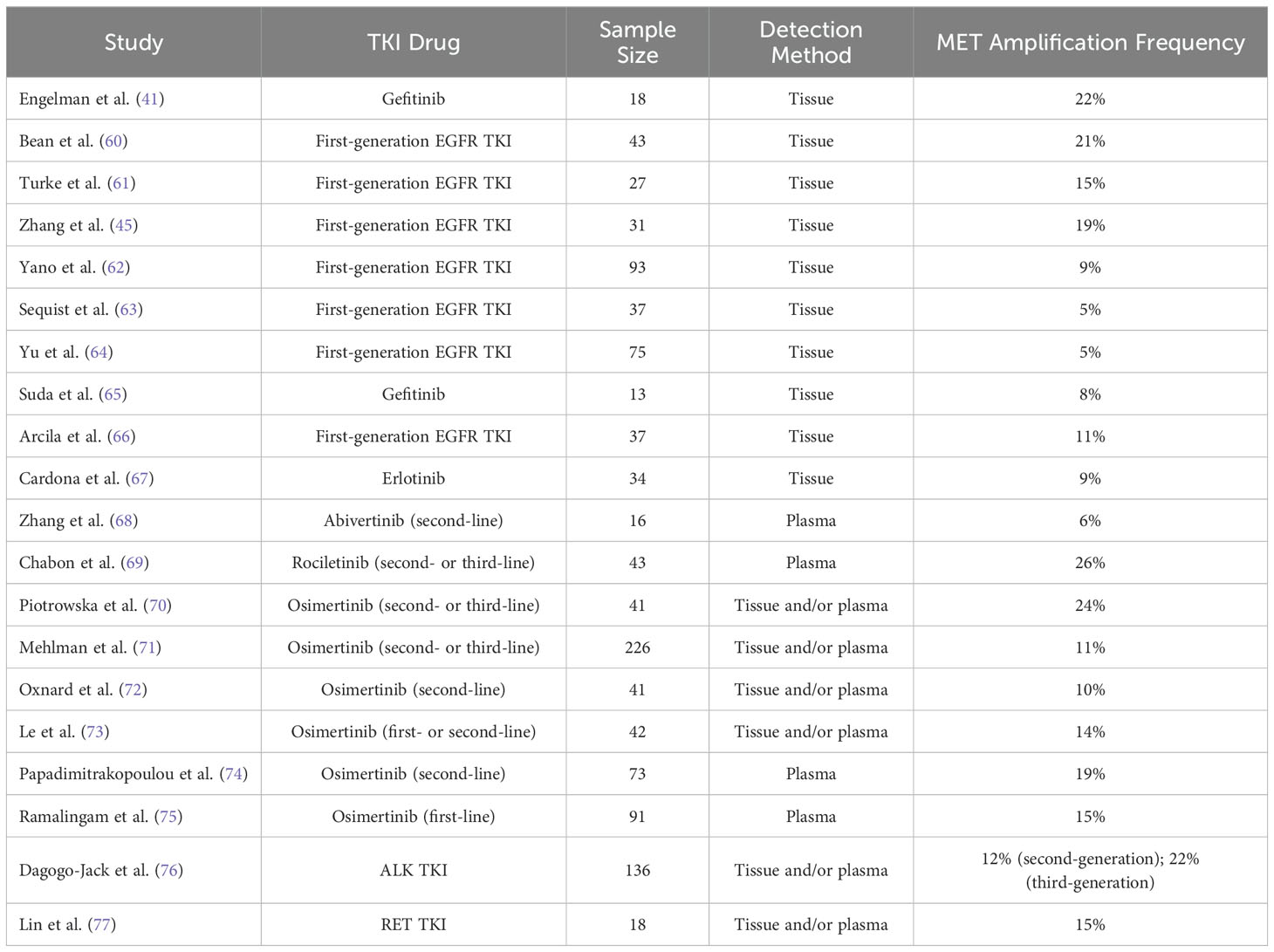
Table 3. Frequency of MET gene amplification in NSCLC TKI resistance.
Research has reported that the occurrence rate of MET gene amplification in first-generation EGFR TKI-resistant patients is approximately 5% to 20% (41, 60–67). In the earliest related reports, among 18 NSCLC patients resistant to the first-generation EGFR TKIs erlotinib or gefitinib, MET gene amplification was found in 4 cases (22%) (41). Similarly, in the study by Bean et al. (60), among 43 NSCLC patients who developed resistance to erlotinib or gefitinib treatment, 9 cases (21%) were found to have MET gene amplification, while among 62 NSCLC patients who did not receive any treatment, only 2 cases (3%) carried MET gene amplification. In another study by Yu et al. (64), involved 155 NSCLC patients resistant to gefitinib or erlotinib, only 5% of the patients had MET gene amplification. In the study by Sequist et al. (63), only 5% of the patients resistant to gefitinib or erlotinib showed MET gene amplification. Nevertheless, MET gene amplification has been considered an important marker for bypass activation mediating resistance to first-generation EGFR TKIs. Meanwhile, the c-MET ligand HGF can also mediate resistance of NSCLC to gefitinib through the activation of the PI3K/AKT signaling pathway via GAB1 (Grb2 associated binder 1) (61). In the study by Yano et al. (62), 61% of NSCLC patients exhibited high expression of HGF after acquiring resistance to EGFR TKIs, with high HGF expression occurring in 29% of patients with primary resistance to EGFR TKIs. Additionally, in the study by Zhong et al. (78), approximately 20% of patients with primary resistance to erlotinib or gefitinib had MET gene amplification, indicating that MET gene amplification may also be one of the causes of primary resistance of NSCLC to first-generation EGFR TKIs.
In third-generation EGFR TKI resistance, when osimertinib is used in cases of prior failure of EGFR TKIs in second-line treatment, regardless of whether T790M mutation existed previously in patients, resistance may occur through MET gene amplification-mediated bypass activation pathways. After resistance to osimertinib, the incidence of MET gene amplification can reach 19% (74). When osimertinib is used as first-line treatment, the incidence of MET gene amplification in osimertinib-resistant samples can reach 15% (75). In the study by Oxnard et al. (72), approximately 10% of NSCLC patients with EGFR mutations resistant to osimertinib had MET gene amplification. Similarly, in the study by Le et al. (73), 14% of NSCLC patients developed MET gene amplification after resistance to osimertinib. In this study, MET gene amplification was the most common molecular mechanism leading to osimertinib resistance besides EGFR secondary mutations. In another study, Chabon et al. (69) explored the mechanism of resistance to another third-generation EGFR TKI, rociletinib, in 43 NSCLC patients by detecting circulating tumor DNA (ctDNA), and found that 26% of patients developed resistance to rociletinib through MET gene amplification, which was the most common molecular mechanism of rociletinib resistance in the study. Moreover, in this study, when EGFR T790M and MET gene amplification coexisted, it could mediate primary resistance of NSCLC patients to rociletinib (69). Additionally, MET gene amplification can also mediate primary resistance of EGFR mutated NSCLC to osimertinib (79). It is worth noting that MET gene amplification can coexist with secondary mutations of driver genes in the same resistant sample. Papadimitrakopoulou et al. (74) showed in their study that in osimertinib-resistant cases mediated by MET amplification, approximately 7% of patients simultaneously had EGFR C797S mutation.
Similar to mediating EGFR TKI resistance, MET gene amplification can mediate resistance of NSCLC patients to ALK TKIs by activating the c-MET signaling pathway. Berger et al. (80) reported a case of MET gene amplification occurring in an ALK fusion NSCLC patient after resistance to the first-generation ALK TKI crizotinib. Additionally, Ueda et al. (81) reported a case of MET gene amplification occurring in an ALK fusion NSCLC patient after resistance to the third-generation ALK TKI lorlatinib. In the study by Dagogo-Jack et al. (76), which included 136 NSCLC patients who had received at least one ALK TKI drug and developed resistance, approximately 15% of patients showed MET gene amplification after resistance. Furthermore, studies have reported that overexpression of the c-MET ligand HGF can also mediate resistance of NSCLC cells to ALK TKIs through the activation of the MET bypass signaling pathway (82, 83).
Additionally, in a study, after resistance to RET inhibitors, 15% of RET fusion NSCLC patients developed MET gene amplification, while only 10% of patients developed resistance through secondary mutations in the RET gene, demonstrating that MET gene amplification is also one of the most common causes of resistance to RET inhibitors (77).
Interestingly, whether in EGFR TKI resistance or ALK TKI resistance, the incidence of MET gene amplification in the latest generation TKI resistance is higher than in the previous generation TKI resistance. For example, in NSCLC patients resistant to first-generation EGFR TKIs, the incidence of MET gene amplification is 5%-22% (41, 60–67), while in patients resistant to third-generation EGFR TKIs, the incidence of MET gene amplification can be as high as 10%-26% (69, 72–75); in NSCLC patients resistant to second-generation ALK TKIs, the incidence of MET gene amplification is approximately 12%, whereas in patients resistant to third-generation ALK TKIs, MET gene amplification can reach 22% (76). New generations of TKI drugs often have higher target selectivity and treatment efficacy, which may be an important reason for the change in TKI resistance patterns, but there is currently no relevant research confirming this hypothesis, future studies can further explore this phenomenon.
In summary, MET gene amplification plays an important role in mediating resistance to EGFR and ALK TKIs, especially as the most important bypass activation resistance pathway in the latest generation TKI resistance. In clinical practice, combination therapy using MET inhibitors and EGFR/ALK TKIs may overcome resistance caused by MET gene amplification.
Under normal circumstances, exon 14 of the MET gene is spliced from both sides of the introns to form mRNA precursor, which mainly encodes the JM domain of MET, containing the Y1003 c-Cbl E3 ubiquitin ligase binding site. Ubiquitination of this site leads to degradation of the c-MET receptor (84). When exon 14 of the MET gene undergoes skipping mutation, the translated c-MET receptor lacks the Y1003 c-Cbl binding site, leading to reduced ubiquitination and slower degradation of the c-MET receptor, thereby sustaining activation of the c-MET signaling pathway (85). The MET gene exon 14 skipping mutation is considered a primary oncogenic driver in NSCLC and does not coexist with other NSCLC driver genes such as EGFR, KRAS, ALK, and ROS1 (86). In a study of 933 non-squamous NSCLC cases, no patients with MET gene exon 14 skipping mutation had mutations in KRAS, EGFR, and ERBB2, or rearrangements in ALK, ROS1, and RET (87). As a primary oncogenic driver, the incidence of MET gene exon 14 skipping mutation in NSCLC is 3%-4% (10, 84, 86, 87).
However, in a recent study, researchers from Memorial Sloan Kettering Cancer Center reported a case of lung adenocarcinoma with EGFR L858R mutation, where MET gene exon 14 skipping mutation (c.2899G>A) occurred after resistance to erlotinib treatment. Subsequent treatment with osimertinib and crizotinib combination therapy led to clinical remission (88). Further research showed that the presence of MET gene exon 14 skipping mutation caused the AKT and ERK downstream signaling pathways of EGFR-mutated lung cancer cells to no longer be inhibited by osimertinib, reducing cell sensitivity to osimertinib by approximately 20% (88). Additionally, Dagogo-Jack et al. (76) also reported a case of lung adenocarcinoma with EML4-ALK fusion after sequential treatment with alectinib and brigatinib, where MET gene exon 14 skipping mutation was detected only in the patient’s plasma cell-free DNA.
MET gene rearrangement refers to fusion rearrangement of the MET gene with other genes, activating the MET signaling pathway. c-MET protein overexpression refers to an increase in c-MET protein expression without MET gene amplification, exon 14 skipping mutation, or rearrangement, resulting in c-MET protein overexpression and activation of the MET signaling pathway. Reports of MET gene rearrangement and c-MET protein overexpression in NSCLC TKI resistance are limited. Dagogo-Jack et al. (76) reported cases of patients resistant to second-generation ALK TKI alectinib and sequential treatment with ceritinib, alectinib, and lorlatinib, all showing ST7-MET gene fusion. In vitro experiments confirmed that this fusion gene mediated resistance of H3122 cells to ALK TKIs, and resistance mediated by rearrangement of ST7-MET gene could be reversed by combination therapy with MET inhibitors and ALK TKIs. Xu et al. (56) reported a case of NSCLC with EGFR L858R mutation, where increased c-MET protein expression was detected after resistance to gefitinib treatment without MET gene amplification or mutation, indicating resistance to gefitinib due to c-MET protein overexpression. The patient responded to gefitinib combined with crizotinib therapy. In summary, although the frequency of MET gene rearrangement and c-MET protein overexpression in NSCLC TKI resistance is low, they are also one of the reasons for mediating resistance through bypass activation pathways.
The EGFR gene is located on the short arm of chromosome 7 (7p12) in humans, spanning approximately 118 kb and comprising 28 exons, with mutations in the tyrosine kinase domain of EGFR primarily occurring in exons 18-21 (89). EGFR is a common receptor tyrosine kinase (RTK) and the first member of the ErbB family. Structurally, EGFR consists of extracellular ligand-binding domains, transmembrane domains, and intracellular tyrosine kinase domains (89). Under normal conditions, EGFR exists as an inactive monomer. Upon binding with ligands such as epidermal growth factor (EGF) or transforming growth factor α (TGFα), the receptor undergoes conformational changes, forming homodimers or heterodimers, thereby activating autophosphorylation of key tyrosine residues in the tyrosine kinase domain. Gene mutations in the EGFR tyrosine kinase domain can also lead to autophosphorylation of tyrosine residues, initiating downstream signaling pathways such as RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, and promoting cell proliferation and growth through cascade reactions (89–91).
In one study, NSCLC patients with ALK fusion showed EGFR gene activation in 44% of tumor samples after developing resistance to ALK TKI crizotinib (43). It has been reported that EGFR bypass activation pathway-mediated resistance to ALK TKIs remains resistant to newer generations of ALK TKIs, but co-administration of EGFR TKIs effectively enhances the cytotoxic effects of ALK TKIs on resistant cells (92). Additionally, overexpression of EGFR ligands EGF and TGFα has been implicated in the mechanism of EGFR bypass activation-mediated resistance (42, 93). EGFR gene mutations and ALK gene fusion are both important driver genes in NSCLC, typically not co-occurring in the same tumor. However, studies have reported that EGFR gene mutations can appear as a means of activating the EGFR bypass signaling pathway in NSCLC patients resistant to ALK TKIs (44, 94). Similarly, the EGFR bypass activation pathway can mediate resistance of ROS1 fusion NSCLC to ROS1 TKIs (95), which can be reversed by co-administration of ROS1 and EGFR inhibitors (96, 97). Study by Lee C et al. (98) revealed that the mutation in EGFR-KDD conferred resistance to 1st and 2nd generation EGFR TKIs, but is sensitive to 3rd generation EGFR TKIs. Furthermore, research by Guo et al. (99) found that approximately 13% of NSCLC patients resistant to MET inhibitors had EGFR gene amplification, suggesting that EGFR bypass signaling pathway activation may be involved in resistance to MET inhibitors.
In summary, EGFR gene activation serves as an important bypass signaling transduction pathway mediating resistance to ALK, ROS1, and MET TKIs. Combination therapy with EGFR TKIs and ALK/ROS1 TKIs may provide an alternative treatment option for patients with resistance mediated by activation of the EGFR signaling pathway.
The AXL gene is located on chromosome 19 (19q13.2) in humans and consists of 20 exons, encoding a protein of 894 amino acids. The AXL protein encoded by the AXL gene is a member of the Tumor-Associated Macrophage (TAM) RTK family, composed of two extracellular immunoglobulin-like repeat sequences, two type III fibronectin repeat sequences, a transmembrane domain, and an intracellular tyrosine kinase domain (100–102). Activation of the AXL receptor can be achieved through ligand binding, AXL overexpression leading to homodimerization, or heterodimerization with other RTKs, with ligand binding being the primary mode of AXL receptor activation (101–103). Growth arrest-specific protein 6 (GAS6) is considered the sole ligand that binds to the extracellular domain of the AXL receptor (101, 102, 104). Upon binding to GAS6, AXL activates downstream signaling pathways such as PI3K/AKT/mTOR, MAPK/ERK, JAK/STAT, and NF-κB through homodimerization and autophosphorylation, thereby promoting activities including cell proliferation, migration, angiogenesis, and mediating cell resistance (101, 102). It is worth noting that unlike other driver genes such as EGFR, ALK, and MET, the expression regulation of AXL is not mediated by gene mutations, fusions, or amplifications (105) but through mechanisms such as transcription factor activation (106–110), microRNA regulation (111–113), or post-transcriptional translation (114–116).
According to a study, approximately 20% of NSCLC patients who developed resistance to first-generation EGFR TKIs erlotinib or gefitinib showed upregulated AXL expression, and about 25% showed upregulated GAS6 expression (45) In preclinical models, AXL has been shown to mediate resistance of NSCLC cells to first-, second-, and even third-generation EGFR TKIs through activation of bypass signaling pathways such as PI3K/AKT, MAPK/ERK, and NF-κB. Restoring sensitivity of NSCLC cells to EGFR TKIs can be achieved by inhibiting AXL gene expression or using AXL inhibitors (116–120). Studies have also reported that AXL can mediate resistance of NSCLC cells to EGFR TKIs by regulating the expression of microRNAs (121). Epithelial-mesenchymal transition (EMT), a biological process involving the transformation of polarized epithelial cells into mesenchymal cells, is also implicated in EGFR TKI resistance (122–124). Activation of AXL often accompanies the occurrence of tumor cell EMT, promoting resistance of NSCLC cells to EGFR TKIs. Inhibiting AXL in resistant cells can reverse the EMT process, overcoming resistance (125). Furthermore, studies have reported that the AXL bypass activation pathway is also involved in mediating resistance of NSCLC cells to first- and second-generation ALK TKIs (126–128). However, currently, there are no studies reporting the role of AXL bypass signaling pathway activation in resistance to third-generation ALK TKIs and ROS1 TKIs.
In conclusion, AXL bypass signaling activation is one of the important reasons for resistance to NSCLC EGFR TKIs and ALK TKIs. Combining AXL inhibitors with EGFR/ALK TKIs may provide a new treatment strategy for patients with TKI resistance mediated by AXL bypass activation.
The IGF-1R gene is located on chromosome 15 (15q26) in humans and spans approximately 315 kb, containing 21 exons (129). The protein encoded by the IGF-1R gene, IGF-1R, is a transmembrane RTK belonging to the insulin receptor (INSR) family, composed of extracellular ligand-binding domains, a transmembrane domain, and intracellular domains near the membrane and tyrosine kinase domains (129, 130). When IGF-1R binds to its ligand, insulin-like growth factor (IGF), receptor structural rearrangements promote transphosphorylation, activating downstream MAPK/ERK, PI3K/AKT, and other cascading signaling pathways, thereby regulating cellular activities such as proliferation, growth, differentiation, and apoptosis (129–131). Recent studies have reported that IGF-1R also mediates resistance to NSCLC TKIs through bypass activation pathways (132–134).
In preclinical models, abnormal activation of the IGF-1R signaling pathway is considered one of the mechanisms by which NSCLC cells develop resistance to first- and second-generation EGFR TKIs via bypass pathways. Co-administration of EGFR TKIs and IGF-1R inhibitors effectively suppresses NSCLC cells resistant through this bypass activation pathway (133–137). For instance, in gefitinib-resistant NSCLC cells A431, the loss of expression of insulin-like growth factor binding proteins (IGFBP) 3 and 4 leads to activation of the IGF-1R/PI3K/AKT pathway. When the EGFR signaling pathway is inhibited, the PI3K/AKT signaling pathway remains activated, allowing NSCLC cells to continue growing and proliferating (132). Additionally, studies have confirmed that abnormal activation of the IGF-1R signaling pathway also contributes to resistance of NSCLC cells to the third-generation EGFR TKI osimertinib (132).
In a study by Lovly et al. (46), immunohistochemical staining of five samples from crizotinib-resistant ALK fusion NSCLC patients revealed activation of IGF-1R in four cases. Preclinical models demonstrated that IGF-1R can mediate crizotinib resistance in ALK fusion NSCLC cells by sustaining activation of the PI3K/AKT signaling pathway. Inhibiting IGF-1R can restore sensitivity of NSCLC cells to crizotinib. Similar conclusions were drawn in the study by Wilson et al. (138) Furthermore, studies have reported IGF-1R-mediated resistance of ALK fusion NSCLC cells to the second-generation ALK TKI ceritinib. Co-administration of ceritinib and IGF-1R inhibitors can reverse resistance in these cells (82). These findings indicate that IGF-1R bypass signaling activation is one of the reasons for resistance of ALK fusion NSCLC to first- and second-generation ALK TKIs. Co-administration of IGF-1R inhibitors with first-generation ALK TKIs may offer further treatment possibilities for patients acquiring resistance through this bypass pathway. However, whether this bypass signaling activation mediates resistance to third-generation ALK TKIs and ROS1 TKIs has not been reported in relevant studies.
The HER2 gene, also known as ERBB2, is located on chromosome 17 (17q21) in humans (139). The HER2 protein encoded by the HER2 gene is a transmembrane glycoprotein of approximately 185 kDa and is the second member of the ErbB family of RTKs (139). The HER3 gene, or ERBB3 gene, is located on the long arm of chromosome 12 (12q13) in humans (140). The HER3 protein encoded by the HER3 gene is the third member of the ErbB family of RTKs (140). Structurally, both HER2 and HER3 consist of extracellular ligand-binding domains, transmembrane domains, and intracellular tyrosine kinase domains (141, 142). HER2 is considered an “orphan receptor” as it lacks known ligands to activate downstream signaling pathways directly, though it consists of tyrosine kinase domain. Instead, HER2 forms heterodimers with other ErbB family members to activate downstream signaling pathways (142, 143). HER3 is considered a “dead kinase.” Although it can bind to multiple ligands, its intrinsic tyrosine kinase activity is impaired. It must form heterodimers with other members of the ErbB family to activate downstream signaling pathways (142). Heterodimers of HER2/HER3 are considered the most potent dimers within the ErbB family due to HER2 being the preferred dimerization partner for all ErbB family members, with HER3 being the preferred partner for HER2 (144, 145). Upon ligand binding, HER3 undergoes conformational changes allowing dimerization, thereby activating downstream PI3K/AKT, MAPK/ERK, and other cascading signaling pathways, regulating cell growth, proliferation, and resistance (142, 145, 146).
In a study, about 12% of NSCLC patients resistant to EGFR TKIs were found to have HER2 amplification, whereas the incidence of HER2 amplification in untreated NSCLC patients was only 1%, suggesting that HER2 amplification may be involved in the resistance process to EGFR TKIs. Further in vitro experiments confirmed that HER2 amplification activated downstream AKT and ERK signaling pathways and continued to activate them even when the EGFR signaling pathway was inhibited, leading to NSCLC cell resistance to EGFR TKIs (47). Additionally, Yu et al. (63) found that approximately 13% of NSCLC patients developed HER2 gene amplification after resistance to gefitinib or erlotinib. Besides mediating resistance to first- and second-generation EGFR TKIs, HER2 gene amplification has also been shown to mediate resistance to third-generation EGFR TKIs through bypass activation pathways (147, 148). When osimertinib was used as second-line treatment, the incidence of HER2 gene amplification in osimertinib-resistant samples was about 3% (74), while it was approximately 2% when osimertinib was used as first-line treatment (75). Studies have also reported that HER2 gene amplification can mediate primary resistance of NSCLC patients to osimertinib. Furthermore, in a study by Zhang et al. (68), it was found that approximately 3% of NSCLC patients developed HER2 gene amplification after resistance to abivertinib, another third-generation EGFR TKI. It is worth noting that the frequency of HER2 gene amplification in resistance to first-generation EGFR TKIs is significantly higher than in resistance to third-generation EGFR TKIs, which is contrary to the frequency of resistance to EGFR TKIs mediated by MET gene amplification. There is currently no relevant research to explain this phenomenon.
In addition to its role in EGFR TKI resistance, activation of the HER2/HER3 signaling pathway can also mediate resistance of NSCLC cells to ALK TKIs (149). Wilson et al. (127) found that treatment of ALK fusion NSCLC cell lines (H3122, H2228, and MGH006) with recombinant NRG1 protein (ligand of HER3) resulted in resistance to the ALK TKI TAE684, which was eliminated by co-administration of TAE684 and the HER2 inhibitor lapatinib, demonstrating that the ligand of HER3, NRG1, can mediate resistance of ALK fusion NSCLC to ALK TKIs through the activation of the HER3/HER2 signaling pathway. Another study also confirmed that the HER2 and HER3 signaling pathways were activated in TAE684-resistant ALK fusion NSCLC cells (150), mediating resistance to TAE684. Additionally, in NSCLC cells resistant to ceritinib, another second-generation ALK TKI, increased expression of HER3 and its ligand neuregulin-1 (NRG1) was observed. The upregulated HER3 protein continued to activate downstream AKT and ERK signaling pathways even when the ALK signaling pathway was inhibited, enabling cells to evade the cytotoxic effects of ceritinib (82). In clinical studies, Minari et al. (149) reported a case of ALK fusion NSCLC patient who developed HER2 gene amplification after treatment with first- and second-generation ALK TKIs. Furthermore, research has found that activation of the HER2 signaling pathway mediates resistance of NSCLC cells with ROS1 fusion to ROS1 TKIs (151).
The RAS/RAF/MAPK/ERK and PI3K/AKT pathways are considered the two most common cascade signaling pathways involved in bypass activation, and mutations or amplifications in key genes in these two pathways can mediate NSCLC TKI resistance (31, 32, 34, 152).
Mutations in genes such as KRAS, NRAS, BRAF, and MAP2K1 have been reported to mediate resistance to EGFR TKIs within the RAS/RAF/MAPK/ERK cascade signaling pathway. Eberlain et al. (153) identified the NRAS E63K mutation involved in resistance to osimertinib in NSCLC cell lines resistant to osimertinib. Both in vitro and in vivo experiments demonstrated that co-administration of osimertinib and the MEK inhibitor selumetinib could reverse the resistance mediated by the NRAS E63K mutation. Ortiz-Cuaran et al. (79) observed the KRAS G12S mutation in patients resistant to osimertinib as second-line treatment. Other KRAS mutations, such as G12D, G13D, Q61R, and Q61, have also been detected in patients resistant to osimertinib (72, 73, 154). KRAS G12D mutation or KARS gene amplification has also been reported in NSCLC patients resistant to MET inhibitors and RET inhibitors (77, 98). Furthermore, approximately 3% of patients resistant to osimertinib carry the BRAF V600E mutation (74, 75). In vitro experiments have confirmed that the BRAF V600E mutation mediates resistance of NSCLC cells to osimertinib, and co-administration of the BRAF inhibitor encorafenib can restore sensitivity of resistant cells to osimertinib (155). Additionally, research has found that the BRAF G469A mutation can mediate acquired resistance of NSCLC cells to osimertinib (156). Moreover, mutations such as MAP2K1 K57N, BRAF G15V, and NRAS A155T have been discovered in NSCLC resistant to second-generation ALK TKIs (157, 158). Thus, key gene mutations in the RAS/RAF/MAPK/ERK cascade signaling pathway play a significant role in NSCLC TKI resistance.
Within the PI3K/AKT cascade signaling pathway, mutations in the PIK3CA gene are the most frequently reported genetic alterations mediating NSCLC TKI resistance. PIK3CA gene mutations have been reported in NSCLC resistant to first-, second-, and third-generation EGFR TKIs (64, 159). In clinical samples of NSCLC resistant to osimertinib as first- or second-line treatment, the incidence of PIK3CA gene mutations can reach 4%-11%, including mutations such as PIK3CA E545K, E542K, R88Q, N345K, and E418K (72–75). The osimertinib resistance mediated by the PIK3CA E545K mutation has been confirmed in vitro (73). Similarly, mutations in the PIK3CA gene have also been found in samples resistant to second-generation ALK TKIs (157). Thus, PIK3CA gene mutations play an important role in mediating NSCLC TKI resistance.
Kim et al. (160) found FGFR1 gene amplification and increased expression of Fibroblast Growth Factor 2 (FGF2) in NSCLC patients resistant to osimertinib, suggesting a potential association between the FGFR1 signaling pathway and acquired resistance to osimertinib. Additionally, FGFR3 gene amplification was observed in NSCLC samples resistant to the ROS TKI crizotinib (151). These studies suggest that aberrant activation of the FGFR signaling pathway may be one of the mechanisms by which NSCLC develops acquired resistance to TKIs through bypass pathways (161, 162).
Furthermore, fusion of driver genes can also activate bypass signaling pathways to mediate acquired resistance of NSCLC to TKIs. Studies have reported that 3%-10% of patients resistant to the third-generation EGFR TKI osimertinib develop resistance through driver gene fusions. Reported fusion genes associated with resistance include MET-UBE2H, FGFR3-TACC3, RET-ERC1, CCDC6-RET, KIF5B-RET, NTRK1-TPM3, NCOA4-RET, GOPC-ROS1, AGK-BRAF, ESYT2–BRAF, PLEKHA7-ALK, and EML4-ALK, among others (70, 72–75, 163–167).
Additionally, KIT gene mutations (KIT D816G) and gene amplifications have been separately reported to mediate resistance to ROS1 TKIs and ALK TKIs (43, 151, 168).
In TKI resistance mediated by bypass pathways, the bypass signaling pathway can remain activated even when the signaling pathway driven by the original driver gene is inhibited. Therefore, simultaneously inhibiting both the bypass signaling pathway and the signaling pathway driven by the original driver gene may overcome TKI resistance mediated by bypass pathways. Currently, numerous phase 1 and phase 2 clinical trials are investigating treatment strategies for NSCLC patients with TKI resistance mediated by bypass pathway activation, and some studies have already achieved promising results (see Table 4).
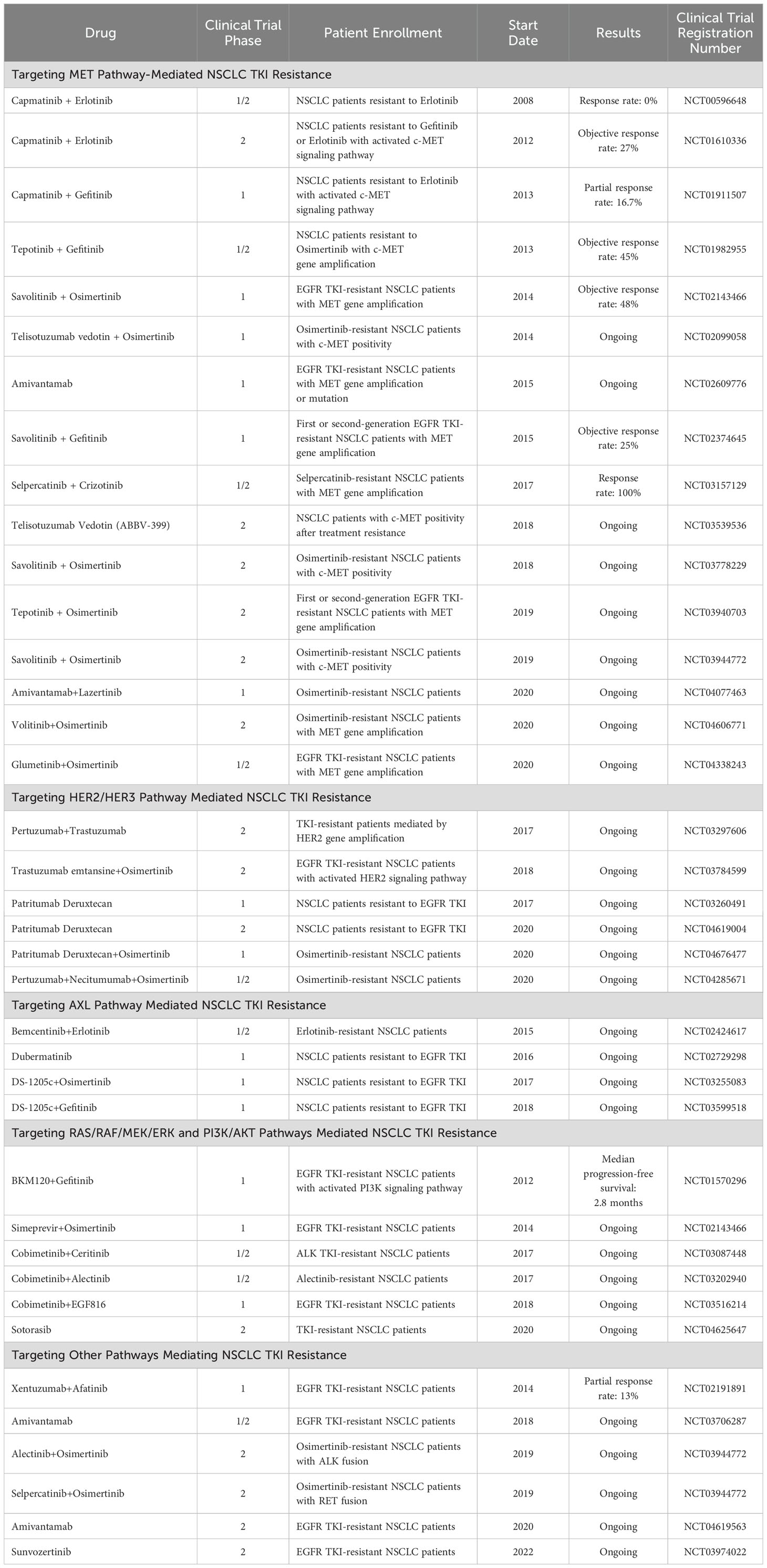
Table 4. Ongoing clinical trials targeting bypass activation pathways.
Currently, over 50 MET-targeted drugs are under research or have entered clinical use, mainly including anti-HGF antibodies, anti-c-MET antibodies, and MET inhibitors, among which MET inhibitors are the most extensively studied and applied c-MET targeted drugs. Presently, there are four FDA-approved small molecule inhibitors targeting c-MET, namely crizotinib (169), cabozantinib (170), capmatinib (171), and tepotinib (172). Among these, crizotinib and cabozantinib are multitargeted drugs (including ALK and ROS1), initially not marketed with c-Met as a therapeutic target, while capmatinib and tepotinib were approved with c-MET as the therapeutic target and are highly selective c-Met inhibitors. Brigatinib also belongs to the new generation of ALK inhibitors and has been approved for use in patients with ALK-positive metastatic NSCLC who have undergone disease progression on crizotinib therapy or who cannot tolerate crizotinib (173). For ROS1 translocation malignancies that have developed resistance to crizotinib, cabozantinib may be effective. Ceritinib has also been shown to be efficacious, but may not be able to overcome the problem of acquired resistance to crizotinib (174). The MET inhibitors glesatinib, savolitinib and the EGFR/MET bispecific antibody amivantamab are also in clinical trials. Worth mentioning is the domestically produced highly selective c-MET inhibitor volitinib (Savolitinib), which has entered the FDA New Drug Application (NDA) stage, representing a promising c-MET inhibitor.
Some preclinical studies have demonstrated that in NSCLC resistant cells mediated by MET gene amplification, the combination of MET inhibitors and EGFR TKIs can reverse the resistance of resistant cells. Simultaneously, in clinical reports, the combination of crizotinib and osimertinib has been proven to be an effective treatment strategy for osimertinib-resistant patients with MET gene amplification. In clinical research, in a phase 1b/2 trial, 47% of EGFR TKI-resistant NSCLC patients with MET gene amplification and 32% of EGFR TKI-resistant patients with MET overexpression showed objective responses to the combination treatment of the MET inhibitor capmatinib and gefitinib (175). In another phase 1b trial of the combination of the MET inhibitor volitinib and gefitinib, 52% of EGFR TKI-resistant NSCLC patients with MET gene amplification exhibited objective responses to the combination therapy (176). In the subsequent INSIGHT study, 67% of EGFR TKI-resistant NSCLC patients with MET gene amplification showed objective therapeutic responses to the combination of the MET inhibitor tepotinib and gefitinib (177). In the phase 1b trial of the TATTON study, 64% of NSCLC patients resistant to first- or second-generation EGFR TKIs and with MET gene amplification demonstrated objective responses to the combination of volitinib and osimertinib, while only 30% of patients resistant to third-generation EGFR TKIs and with MET gene amplification showed objective responses to this combination therapy (178). Based on the results of the TATTON study, the SAVANNAH study and the ORCHARD study are exploring the use of volitinib in combination with osimertinib for the treatment of osimertinib-resistant NSCLC patients with MET gene amplification (NCT03778229 and NCT03944772). The savolitinib + osimertinib combination represents a promising therapy in patients with MET-amplified/overexpressed, EGFRm advanced NSCLC with disease progression on a prior EGFR-TKI (NCT02143466) (179). The combination of osimertinib+chemotherapy in EGFR-Mutated Advanced NSCLC not only helps patients with survival rates and disease progression but also has a higher response rate and longer response duration (180). Additionally, the INSIGHT 2 study is investigating the combination of the MET inhibitor tepotinib with osimertinib for the treatment of EGFR TKI-resistant NSCLC patients with MET amplification (NCT03940703).
Currently, sporadic case reports exist regarding the combined use of MET inhibitors with ALK TKIs or RET TKIs, but there are no clinical studies specifically targeting MET inhibitors in combination with ALK TKIs or RET TKIs. Gouji et al. (181) reported a case of an ALK fusion NSCLC patient who developed MET gene amplification after resistance to the second-generation ALK TKI alectinib, achieving a response to treatment with crizotinib; however, the patient died after 7 months of crizotinib treatment. Dagogo-Jack et al (76). reported a case of an ALK fusion NSCLC patient who showed significant MET gene amplification after resistance to alectinib, achieving a response after treatment with crizotinib; however, the tumor recurred after 10 weeks of treatment. Dagogo-Jack et al. (76) also reported another case of an ALK fusion NSCLC patient who developed MET gene amplification after resistance to lorlatinib, achieving tumor control after treatment with lorlatinib in combination with crizotinib, but the tumor progressed again after 3 months of treatment. In the study by Rosen et al. (182), four NSCLC patients treated with the RET inhibitor selpercatinib were found to have MET gene amplification after resistance of selpercatinib treatment, all showing varying degrees of therapeutic response to crizotinib combined with selpercatinib; however, tumors recurred in all patients after 3.5 to 10 months. Larger clinical trials are needed to confirm the efficacy of ALK or RET TKIs in combination with MET inhibitors in NSCLC patients with MET gene amplification who are resistant to ALK or RET TKIs.
Several small molecule inhibitors targeting HER2 or therapeutic antibodies have been FDA-approved, while there are currently no marketed drugs targeting HER3. FDA-approved small molecule inhibitors targeting HER2 include lapatinib (183), afatinib (184), neratinib (185), and dacomitinib (17); therapeutic antibodies targeting HER2 include Herceptin, trastuzumab (186), Phesgo, pertuzumab (187), and Enhertu (trastuzumab deruxtecan) (188).
Currently, multiple trials are investigating the efficacy of HER2/HER3 inhibitors or antibodies in patients with NSCLC TKI resistance driven by HER2/HER3 signaling pathway activation, but standard treatment regimens have not been established. In NSCLC patients with HER2 gene amplification, trastuzumab-DM1 (trastuzumab emtansine) has shown promising efficacy in phase 2 clinical trials (189), and the ongoing TRAEMOS trial is investigating the therapeutic effect of T-DM1 combined with osimertinib in osimertinib-resistant patients with HER2 amplification (NCT03784599). In a study on patients with advanced NSCLC harboring HER-2 mutations, the efficacy and pharmacotoxicity of pyrotinib were analyzed, thereby expanding treatment options for rare gene mutations (NCT03574402) (190). The CAPTUR trial is also investigating the combination of trastuzumab and pertuzumab to overcome resistance mediated by HER2 gene amplification (NCT03297606). Additionally, a phase 1 trial is investigating the therapeutic effect of the HER3-targeting antibody-drug conjugate U3-1402 (patritumab deruxtecan) combined with osimertinib in osimertinib-resistant NSCLC patients (NCT04676477).
Preclinical studies have confirmed that combination therapy targeting AXL can effectively reverse NSCLC TKI resistance mediated by AXL bypass activation. In ongoing clinical trials, three AXL inhibitors (bemcentinib, dubermatinib, and DS-1205c) are being used alone or in combination with EGFR TKIs to treat EGFR TKI-resistant NSCLC patients (NCT02424617, NCT02729298, NCT03255083, NCT03599518). However, unfortunately, these AXL inhibitors have not yet been FDA-approved for clinical use, and there is currently no standard treatment regimen for NSCLC AKI resistance mediated by AXL bypass activation.
Multiple drugs targeting cascade signaling pathways involved in NSCLC TKI resistance are currently being investigated in clinical trials. In studies targeting the PI3K/AKT cascade signaling pathway mediated NSCLC TKI resistance treatment, the PI3K inhibitor BKM120 combined with gefitinib was used to treat EGFR TKI-resistant NSCLC patients with PI3K signaling pathway activation, but unfortunately, the median progression-free survival was only 2.8 months (191). The novel PI3K inhibitor alpelisib was FDA-approved in 2019 for the treatment of PIK3CA-mutated breast cancer, and considering the high incidence of PIK3CA mutations in NSCLC TKI resistance (2%-11%), future clinical trials may use next-generation PI3K inhibitors to explore their efficacy in NSCLC TKI-resistant patients with PIK3CA mutations. Adagrasib is a related molecule that the FDA has also approved for patients with locally advanced or metastatic NSCLC with the KRAS G12C mutation who have received at least one prior systemic therapy (192).
In the treatment of NSCLC TKI resistance mediated by the RAF/RAS/MEK/ERK cascade signaling pathway, the MEK inhibitors selumetinib (190, 192), trametinib (193), and cobimetinib (194) have been FDA-approved. The KRAS inhibitor sotorasib (AMG 510) (195) has also received FDA breakthrough therapy designation and real-time oncology review qualification for the treatment of NSCLC patients with KRAS G12C mutations. A phase I study enrolling 137 patients with advanced KRAS-mutant cancers showed that treatment with the covalent KRAS G12C inhibitor divarasib (GDC-6036) had an ORR of 53% and a median PFS of 13.1 months in 60 patients with NSCLC (196). There are few promising treatments for non-G12C KRAS-mutant NSCLC. A phase III trial combining the oral MEK inhibitor selumetinib with docetaxel did not find a beneficial MEK inhibitor treatment strategy (197). Ongoing clinical trials include trametinib in combination with ceritinib for the treatment of ALK TKI-resistant NSCLC patients (NCT03087448), cobimetinib in combination with alectinib for the treatment of alectinib-resistant NSCLC patients (NCT03202940), selumetinib in combination with osimertinib for the treatment of EGFR TKI-resistant NSCLC patients (NCT02143466), and sotorasib for the treatment of TKI-resistant NSCLC patients (NCT04625647).
RET gene fusion is one mechanism of NSCLC TKI bypass activation resistance, and currently, the FDA has approved two RET inhibitors, pralsetinib (BLU-667) (198) and selpercatinib (LOXO-292) (199), for the treatment of RET fusion-positive NSCLC patients. In clinical reports, two NSCLC patients resistant to osimertinib due to RET gene fusion achieved clinical remission after receiving pralsetinib in combination with osimertinib (70). Additionally, a clinical trial is investigating selpercatinib in combination with osimertinib for the treatment of osimertinib-resistant patients with RET gene fusion (NCT03944772).
Combination chemotherapy presented good results in patients with advanced NSCLC in which EGFR mutations were found. A non-blind randomized trial enrolling 557 patients with advanced NSCLC carrying EGFR mutations found that the addition of platinum and pemetrexed chemotherapy to ositinib improved PFS (180). For primary patients with locally advanced or metastatic NSCLC harboring an EGFR mutation (exon 19 deletion or L858R mutation), treatment with amivantamab + lazertinib resulted in a better PFS improvement than ositinib alone (200).
Furthermore, clinical trials are underway for the combination of alectinib with osimertinib for the treatment of osimertinib-resistant NSCLC patients with ALK fusion (NCT03944772), IGF-1R therapeutic antibody in combination with afatinib for the treatment of EGFR TKI-resistant NSCLC patients (NCT02191891), and FGFR inhibitors for the treatment of EGFR TKI-resistant NSCLC patients (NCT03706287, NCT04619563). Oral inhibitors have been shown to be particularly effective and less toxic for the initial treatment of patients with advanced NSCLC who have concomitant driver mutations and who are older or in poorer health. This approach is particularly useful in patients with EGFR mutations, BRAF mutations, ALK fusion oncogenes, or ROS1 translocations, subject to ongoing discovery of other targetable mutations.
The use of TKI drugs has significantly improved the prognosis of NSCLC patients carrying driver genes. However, the emergence of resistance poses new challenges to further extending patient survival, and a deeper understanding and study of the molecular mechanisms of TKI resistance will be the foundation for overcoming TKI resistance. In NSCLC, the molecular mechanisms of TKI resistance include secondary mutations of driver genes, bypass signal activation, and histological transformation. Secondary mutations of driver genes can be overcome by developing new generations of TKI drugs, while resistance mechanisms caused by bypass signal activation are more complex. In TKI resistance caused by bypass signal activation, although TKI drugs inhibit downstream signaling pathways of driver genes, RTKs can continuously activate key signaling pathways within tumor cells through alternative pathways, thereby sustaining tumor cell growth and proliferation, leading to resistance to TKI drugs. Inhibiting activated bypass signals and the original driver gene signaling pathway is the basic concept for treating bypass activation-mediated TKI resistance. Based on this concept of combination therapy, several clinical trials are currently underway to treat patients resistant to bypass signal activation, although all trials are in early stages, some have yielded promising preliminary results, which will provide more treatment options for NSCLC TKI-resistant patients.
In recent years, based on next-generation sequencing of circulating tumor DNA (ctDNA) detection, dynamic monitoring of molecular genetic changes occurring during TKI treatment of tumor patients through tumor rebiopsy can timely discover changes in bypass signals leading to resistance, such as MET gene amplification, HER2 gene amplification, KRAS gene mutation, PIK3CA gene mutation, etc. (69, 180, 201–203). The FDA has approved a number of ctDNA tests for identifying patients with positive EGFR mutations (204, 205), and one of these tests uses NGS to identify abnormalities in 55 additional genes (205). Although tissue availability is limited, somatic gene alteration testing prior to treatment of advanced NSCLC remains recommended. In a trial of adult patients with advanced NSCLC that used blood liquid biopsies to assess circulating tumor DNA to determine whether patients with ROS1-positive NSCLC were treated with the entrectinib, the assay resulted in an 81% objective response rate to entrectinib, which is consistent with the results of a previous study using a tissue-based assay to identify ROS1 fusions (206). The use of liquid biopsies to assess other molecular abnormalities may become more common as more data become available.
Through this approach, on one hand, more precise individualized tumor treatment plans can be formulated for patients, and on the other hand, effective treatment can be given to patients in a timely manner to prevent further occurrence and development of resistance. The ongoing ELIOS trial is such a study, in which NSCLC patients receiving osimertinib as first-line treatment will obtain tumor biopsy samples and plasma samples before treatment, during treatment, and after treatment resistance, and compare and analyze the genetic variations of tissue samples and plasma samples through next-generation sequencing technology to reveal the role of liquid biopsy technology in resistance monitoring (NCT03239340). Meanwhile, the complementary ORCHARD clinical trial aims to study the treatment plan for NSCLC patients receiving osimertinib as first-line treatment after acquired resistance. In this innovative trial platform, resistant patients will be allocated to corresponding treatment cohorts based on their different tumor molecular characteristics and different resistance mechanisms, including cohorts of wolinib combined with osimertinib, gefitinib combined with osimertinib, alectinib combined with osimertinib, and selpercatinib combined with osimertinib, etc. (NCT03944772). Integrating the results of these two trials will provide more precise, effective, and personalized treatment for NSCLC patients.
In addition, the concept of “Drug-Tolerant Persister (DTP) cells” has been proposed, refreshing researchers’ understanding of the mechanisms of tumor resistance (207). A small proportion of tumor cells (0.3%-5%) can survive by adopting a reversible drug-tolerant state when facing TKI treatment, and under the continuous action of drugs, eventually acquire permanent resistance mechanisms based on genetic changes (207). Turke et al.’s (61) study found that a small number of cells (<1%) with MET gene amplification could be detected in samples from NSCLC patients before using EGFR TKIs, while a large number of cells with MET amplification were present after EGFR TKI resistance, indicating that the small fraction of cells with MET gene amplification before treatment ultimately led to resistance. In Taniguchi et al.’s (208) study, DTP cells obtained after treating PC-9 cells with osimertinib for 9 days showed significantly increased expression of AXL, and resistance was significantly higher than that of sensitive cells, while combined use of osimertinib and AXL inhibitors could prevent the generation of DTP cells. Wang et al.’s (209) study also found that although NSCLC cells with low expression of AXL were more sensitive to osimertinib, there still existed a small group of cells activated by IGF-1R signaling that were resistant to osimertinib treatment, while the use of IGF-1R inhibitors could prevent the generation of DTP cells. These studies suggest that early targeting of DTP cells may delay the occurrence of NSCLC TKI resistance, thereby increasing patient survival. In an ongoing clinical trial, researchers are adding c-MET inhibitors (APL-101) for combination therapy in NSCLC patients receiving first-line osimertinib treatment between 8 and 12 weeks before the occurrence of resistance, to observe whether this treatment regimen can delay the onset of resistance in patients (NCT04743505).
In summary, by utilizing liquid biopsy techniques to identify resistance mechanisms early, implementing individualized targeted therapies against these mechanisms at an early stage, and preemptively targeting drug-tolerant persister (DTP) cells, NSCLC patients may achieve more durable and effective treatments, leading to further extensions in survival time.
ZJ: Writing – original draft, Writing – review & editing, Data curation, Investigation, Resources, Visualization. ZG: Writing – original draft, Methodology. XY: Writing – review & editing, Data curation. TC: Writing – review & editing, Funding acquisition, Resources. BL: Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding: This research was funded by the Sichuan Provincial Natural Science Foundation Youth Fund Project under grant number NO. 23NSFSC4251. Funder Role: Sichuan Provincial Natural Science Foundation Youth Fund Project: Provided financial support for the data collection. The funder had no role in the study design, data analysis, or manuscript preparation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
2. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics. CA Cancer J Clin. (2024) 74:203. doi: 10.3322/caac.21820
3. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. (2018) 553:446–54. doi: 10.1038/nature25183
4. Gridelli C, Rossi A, Carbone DP, Guarize J, Karachaliou N, Mok T, et al. Non-small-cell lung cancer. Nat Rev Dis Primers. (2015) 1:15009. doi: 10.1038/nrdp.2015.9
5. Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. (2021) 7(1):3. doi: 10.1038/s41572-020-00235-0
6. Chen J, Yang H, Teo ASM, Amer LB, Sherbaf FG, Tan CQ, et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat Genet. (2020) 52(2):177–86. doi: 10.1038/s41588-019-0569-6
7. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. (2012) 150(6):1121–34. doi: 10.1016/j.cell.2012.08.024
8. Han B, Tjulandin S, Hagiwara K, Normanno N, Wulandari L, Laktionov K, et al. EGFR mutation prevalence in Asia-Pacific and Russian patients with advanced NSCLC of adenocarcinoma and non-adenocarcinoma histology: The IGNITE study. Lung Cancer. (2017) 113:37–44. doi: 10.1016/j.lungcan.2017.08.021
9. Graham RP, Treece AL, Lindeman NI, Vasalos P, Shan M, Jennings LJ, et al. Worldwide frequency of commonly detected EGFR mutations. Arch Pathol Lab Med. (2018) 142(2):163–7. doi: 10.5858/arpa.2016-0579-CP
10. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543–50. doi: 10.1038/nature13385
11. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. (2007) 448:561–6. doi: 10.1038/nature05945
12. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. (2012) 30(8):863–70. doi: 10.1200/JCO.2011.35.6345
13. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. (2010) 362(25):2380–8. doi: 10.1056/NEJMoa0909530
14. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. (2004) 350:2129–39. doi: 10.1056/NEJMoa040938
15. Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinib in lung cancer - molecular and clinical predictors of outcome. N Engl J Med. (2005) 353(2):133–44. doi: 10.1056/NEJMoa050736
16. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. (2014) 15:213–22. doi: 10.1016/S1470-2045(13)70604-1
17. Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. (2017) 18:1454–66. doi: 10.1016/S1470-2045(17)30608-3
18. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
19. Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. (2017) 376:629–40. doi: 10.1056/NEJMoa1612674
20. Wu YL, Tsuboi M, He J, John T, Grohe C, Majem M, et al. Osimertinib in resected EGFR-mutated non-small-cell lung cancer. N Engl J Med. (2020) 383:1711–23. doi: 10.1056/NEJMoa2027071
21. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. (2014) 371:2167–77. doi: 10.1056/NEJMoa1408440
22. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. (2014) 371:1963–71. doi: 10.1056/NEJMoa1406766
23. Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. (2013) 368:2385–94. doi: 10.1056/NEJMoa1214886
24. Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. (2014) 370:1189–97. doi: 10.1056/NEJMoa1311107
25. Soria JC, Tan DSW, Chiari R, Wu YL, Paz-Ares L, Wolf J, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet. (2017) 389:917–29. doi: 10.1016/S0140-6736(17)30123-X
26. Shaw AT, Kim TM, Crino L, Gridelli C, Kiura K, Liu G, et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. (2017) 18:874–86. doi: 10.1016/S1470-2045(17)30339-X
27. Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. (2017) 377:829–38. doi: 10.1056/NEJMoa1704795
28. Camidge DR, Kim HR, Ahn MJ, Yang JC, Han JY, Lee JS, et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N Engl J Med. (2018) 379:2027–39. doi: 10.1056/NEJMoa1810171
29. Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. (2018) 19:1654–67. doi: 10.1016/S1470-2045(18)30649-1
30. Shaw AT, Bauer TM, de Marinis F, Felip E, Goto Y, Liu G, et al. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N Engl J Med. (2020) 383:2018–29. doi: 10.1056/NEJMoa2027187
31. Lin JJ, Shaw AT. Resisting resistance: targeted therapies in lung cancer. Trends Cancer. (2016) 2:350–64. doi: 10.1016/j.trecan.2016.05.010
32. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. (2019) 121:725–37. doi: 10.1038/s41416-019-0573-8
33. Yoda S, Lin JJ, Lawrence MS, Burke BJ, Friboulet L, Langenbucher A, et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discovery. (2018) 8:714–29. doi: 10.1158/2159-8290.CD-17-1256
34. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. (2017) 17:637–58. doi: 10.1038/nrc.2017.84
35. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. (2014) 11:473–81. doi: 10.1038/nrclinonc.2014.104
36. Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. (2018) 17:48. doi: 10.1186/s12943-018-0804-2
37. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U.S.A. (2008) 105:2070–5. doi: 10.1073/pnas.0709662105
38. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. (2005) 352:786–92. doi: 10.1056/NEJMoa044238
39. Kobayashi Y, Fujino T, Nishino M, Koga T, Chiba M, Sesumi Y, et al. EGFR T790M and C797S mutations as mechanisms of acquired resistance to dacomitinib. J Thorac Oncol. (2018) 13:727–31. doi: 10.1016/j.jtho.2018.01.009
40. Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, et al. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discovery. (2015) 5:960–71. doi: 10.1158/2159-8290.CD-15-0063
41. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. (2007) 316:1039–43. doi: 10.1126/science.1141478
42. Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. (2011) 71:6051–60. doi: 10.1158/0008-5472.CAN-11-1340
43. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. (2012) 4:120ra117. doi: 10.1126/scitranslmed.3003316
44. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. (2012) 18:1472–82. doi: 10.1158/1078-0432.CCR-11-2906
45. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. (2012) 44:852–60. doi: 10.1038/ng.2330
46. Lovly CM, McDonald NT, Chen H, Ortiz-Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. (2014) 20:1027–34. doi: 10.1038/nm.3667
47. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discovery. (2012) 2:922–33. doi: 10.1158/2159-8290.CD-12-0108
48. Dean M, Park M, Le Beau MM, Robins TS, Diaz MO, Rowley JD, et al. The human met oncogene is related to the tyrosine kinase oncogenes. Nature. (1985) 318:385–8. doi: 10.1038/318385a0
49. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. (2010) 11:834–48. doi: 10.1038/nrm3012
50. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol. (2017) 12:15–26. doi: 10.1016/j.jtho.2016.10.014
51. Kim ES, Salgia R. MET pathway as a therapeutic target. J Thorac Oncol. (2009) 4:444–7. doi: 10.1097/JTO.0b013e31819d6f91
52. Kawakami H, Okamoto I, Okamoto W, Tanizaki J, Nakagawa K, Nishio K. Targeting MET amplification as a new oncogenic driver. Cancers (Basel). (2014) 6:1540–52. doi: 10.3390/cancers6031540
53. Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. (2005) 65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650
54. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. (2014) 5:4846. doi: 10.1038/ncomms5846
55. Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci U.S.A. (1991) 88:4892–6. doi: 10.1073/pnas.88.11.4892
56. Xu Y, Fan Y. Responses to crizotinib can occur in c-MET overexpressing nonsmall cell lung cancer after developing EGFR-TKI resistance. Cancer Biol Ther. (2019) 20:145–9. doi: 10.1080/15384047.2018.1523851
57. Feng Y, Minca EC, Lanigan C, Liu A, Zhang W, Yin L, et al. High MET receptor expression but not gene amplification in ALK 2p23 rearrangement positive non-small-cell lung cancer. J Thorac Oncol. (2014) 9:646–53. doi: 10.1097/JTO.0000000000000145
58. Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, Smith DI, et al. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. (2002) 1:89–97. doi: 10.1016/S1535-6108(02)00017-X
59. Schildhaus HU, Schultheis AM, Ruschoff J, Binot E, Merkelbach-Bruse S, Fassunke J, et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin Cancer Res. (2015) 21:907–15. doi: 10.1158/1078-0432.CCR-14-0450
60. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U.S.A. (2007) 104:20932–7.
61. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. (2010) 17:77–88. doi: 10.1016/j.ccr.2009.11.022
62. Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, Yatabe Y, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. (2011) 6:2011–7. doi: 10.1097/JTO.0b013e31823ab0dd
63. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. (2011) 3:75ra26. doi: 10.1126/scitranslmed.3002003
64. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. (2013) 19:2240–7. doi: 10.1158/1078-0432.CCR-12-2246
65. Suda K, Murakami I, Katayama T, Tomizawa K, Osada H, Sekido Y, et al. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res. (2010) 16:5489–98. doi: 10.1158/1078-0432.CCR-10-1371
66. Arcila ME, Oxnard GR, Nafa K, Solomon SB, Zakowski MF, Kris MG, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. (2011) 17:1169–80. doi: 10.1158/1078-0432.CCR-10-2277
67. Cardona AF, Arrieta O, Zapata MI, Rojas L, Wills B, Reguart N, et al. Acquired resistance to erlotinib in EGFR mutation-positive lung adenocarcinoma among hispanics (CLICaP). Target Oncol. (2017) 12:513–23. doi: 10.1007/s11523-017-0497-2
68. Zhang YC, Chen ZH, Zhang XC, Xu CR, Yan HH, Xie Z, et al. Analysis of resistance mechanisms to abivertinib, a third-generation EGFR tyrosine kinase inhibitor, in patients with EGFR T790M-positive non-small cell lung cancer from a phase I trial. EBioMedicine. (2019) 43:180–7. doi: 10.1016/j.ebiom.2019.04.030
69. Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. (2016) 7:11815. doi: 10.1038/ncomms11815
70. Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discovery. (2018) 8:1529–39. doi: 10.1158/2159-8290.CD-18-1022
71. Mehlman C, Cadranel J, Rousseau-Bussac G, Lacave R, Pujals A, Girard N, et al. Resistance mechanisms to osimertinib in EGFR-mutated advanced non-small-cell lung cancer: A multicentric retrospective French study. Lung Cancer. (2019) 137:149–56. doi: 10.1016/j.lungcan.2019.09.019
72. Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. (2018) 4:1527–34. doi: 10.1001/jamaoncol.2018.2969
73. Le X, Puri S, Negrao MV, Nilsson MB, Robichaux J, Boyle T, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin Cancer Res. (2018) 24:6195–203. doi: 10.1158/1078-0432.CCR-18-1542
74. Papadimitrakopoulou VA, Wu YL, Han JY, Ahn M-J, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. (2018) 29:VIII741. doi: 10.1093/annonc/mdy424.064
75. Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann Oncol. (2018) 29:VIII740. doi: 10.1093/annonc/mdy424.063
76. Dagogo-Jack I, Yoda S, Lennerz JK, Langenbucher A, Lin JJ, Rooney MM, et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin Cancer Res. (2020) 26:2535–45. doi: 10.1158/1078-0432.CCR-19-3906
77. Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann Oncol. (2020) 31:1725–33. doi: 10.1016/j.annonc.2020.09.015
78. Zhong J, Li L, Wang Z, Bai H, Gai F, Duan J, et al. Potential resistance mechanisms revealed by targeted sequencing from lung adenocarcinoma patients with primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). J Thorac Oncol. (2017) 12:1766–78. doi: 10.1016/j.jtho.2017.07.032
79. Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. (2016) 22:4837–47. doi: 10.1158/1078-0432.CCR-15-1915
80. Berger LA, Janning M, Velthaus JL, Ben-Batalla I, Schatz S, Falk M, et al. Identification of a high-level MET amplification in CTCs and cfTNA of an ALK-positive NSCLC patient developing evasive resistance to crizotinib. J Thorac Oncol. (2018) 13:e243–6. doi: 10.1016/j.jtho.2018.08.2025
81. Ueda S, Shukuya T, Hayashi T, Suzuki M, Kondo A, Arai Y, et al. Transformation from adenocarcinoma to squamous cell lung carcinoma with MET amplification after lorlatinib resistance: A case report. Thorac Cancer. (2021) 12(5):715–719. doi: 10.1111/1759-7714.13829
82. Isozaki H, Ichihara E, Takigawa N, Ohashi K, Ochi N, Yasugi M, et al. Non-small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases. Cancer Res. (2016) 76:1506–16. doi: 10.1158/0008-5472.CAN-15-1010
83. Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res. (2012) 18:3592–602. doi: 10.1158/1078-0432.CCR-11-2972
84. Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. (2009) 4:5–11. doi: 10.1097/JTO.0b013e3181913e0e
85. Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. (2006) 66:283–9. doi: 10.1158/0008-5472.CAN-05-2749
86. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discovery. (2015) 5:850–9. doi: 10.1158/2159-8290.CD-15-0285
87. Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J Clin Oncol. (2016) 34:721–30. doi: 10.1200/JCO.2015.63.4600
88. Suzawa K, Offin M, Schoenfeld AJ, Plodkowski AJ, Odintsov I, Lu D, et al. Acquired MET exon 14 alteration drives secondary resistance to epidermal growth factor receptor tyrosine kinase inhibitor in EGFR-mutated lung cancer. JCO Precis Oncol. (2019) 3:PO.19.00011. doi: 10.1200/PO.19.00011
89. Sabbah DA, Hajjo R, Sweidan K. Review on epidermal growth factor receptor (EGFR) structure, signaling pathways, interactions, and recent updates of EGFR inhibitors. Curr Top Med Chem. (2020) 20:815–34. doi: 10.2174/1568026620666200303123102
90. Purba ER, Saita EI, Maruyama IN. Activation of the EGF receptor by ligand binding and oncogenic mutations: the "Rotation model. Cells. (2017) 6. doi: 10.20944/preprints201705.0212.v1
91. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. (2010) 10:760–74. doi: 10.1038/nrc2947
92. Miyawaki M, Yasuda H, Tani T, Hamamoto J, Arai D, Ishioka K, et al. Overcoming EGFR bypass signal-induced acquired resistance to ALK tyrosine kinase inhibitors in ALK-translocated lung cancer. Mol Cancer Res. (2017) 15:106–14. doi: 10.1158/1541-7786.MCR-16-0211
93. Tani T, Yasuda H, Hamamoto J, Kuroda A, Arai D, Ishioka K, et al. Activation of EGFR bypass signaling by TGFalpha overexpression induces acquired resistance to alectinib in ALK-translocated lung cancer cells. Mol Cancer Ther. (2016) 15:162–71. doi: 10.1158/1535-7163.MCT-15-0084
94. Miyanaga A, Shimizu K, Noro R, Seike M, Kitamura K, Kosaihira S, et al. Activity of EGFR-tyrosine kinase and ALK inhibitors for EML4-ALK-rearranged non-small-cell lung cancer harbored coexisting EGFR mutation. BMC Cancer. (2013) 13:262. doi: 10.1186/1471-2407-13-262
95. Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PloS One. (2013) 8:e82236. doi: 10.1371/journal.pone.0082236
96. Song A, Kim TM, Kim DW, Kim S, Keam B, Lee SH, et al. Molecular changes associated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer. Clin Cancer Res. (2015) 21:2379–87. doi: 10.1158/1078-0432.CCR-14-1350
97. Vaishnavi A, Schubert L, Rix U, Kim TM, Kim S, Im SW, et al. EGFR mediates responses to small-molecule drugs targeting oncogenic fusion kinases. Cancer Res. (2017) 77:3551–63. doi: 10.1158/0008-5472.CAN-17-0109
98. Lee C, Kim M, Kim DW, Chang J, Chow A, Delasos L, et al. Acquired resistance mechanism of EGFR kinase domain duplication to EGFR TKIs in non-small cell lung cancer. Cancer Res Treat. (2022) 54:140–9. doi: 10.4143/crt.2021.385
99. Guo R, Offin M, Brannon AR, Neubauer A, Kitch B, Prokop C, et al. MET exon 14-altered lung cancers and MET inhibitor resistance. Clin Cancer Res. (2021) 27:799–806. doi: 10.1158/1078-0432.CCR-20-2861
100. O'Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. (1991) 11:5016–31. doi: 10.1128/mcb.11.10.5016-5031
101. Antony J, Huang RY. AXL-driven EMT state as a targetable conduit in cancer. Cancer Res. (2017) 77:3725–32. doi: 10.1158/0008-5472.CAN-17-0392
102. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. (2019) 18:153. doi: 10.1186/s12943-019-1090-3
103. Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. (2013) 6:ra66. doi: 10.1126/scisignal.2004155
104. Varnum BC, Young C, Elliott G, Garcia A, Bartley TD, Fridell YW, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature. (1995) 373:623–6. doi: 10.1038/373623a0
105. Scaltriti M, Elkabets M, Baselga J. Molecular pathways: AXL, a membrane receptor mediator of resistance to therapy. Clin Cancer Res. (2016) 22:1313–7. doi: 10.1158/1078-0432.CCR-15-1458
106. Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Luthar N, et al. AXL mediates resistance to cetuximab therapy. Cancer Res. (2014) 74:5152–64. doi: 10.1158/0008-5472.CAN-14-0294
107. Mudduluru G, Allgayer H. The human receptor tyrosine kinase Axl gene–promoter characterization and regulation of constitutive expression by Sp1, Sp3 and CpG methylation. Biosci Rep. (2008) 28:161–76. doi: 10.1042/BSR20080046
108. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. (2011) 30:1229–40. doi: 10.1038/onc.2010.504
109. Sayan AE, Stanford R, Vickery R, Grigorenko E, Diesch J, Kulbicki K, et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene. (2012) 31:1493–503. doi: 10.1038/onc.2011.336
110. Badarni M, Prasad M, Balaban N, Zorea J, Yegodayev KM, Joshua BZ, et al. Repression of AXL expression by AP-1/JNK blockage overcomes resistance to PI3Ka therapy. JCI Insight. (2019) 5(8):e125341. doi: 10.1172/jci.insight.125341
111. Mudduluru G, Ceppi P, Kumarswamy R, Scagliotti GV, Papotti M, Allgayer H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene. (2011) 30:2888–99. doi: 10.1038/onc.2011.13
112. Mackiewicz M, Huppi K, Pitt JJ, Dorsey TH, Ambs S, Caplen NJ. Identification of the receptor tyrosine kinase AXL in breast cancer as a target for the human miR-34a microRNA. Breast Cancer Res Treat. (2011) 130:663–79. doi: 10.1007/s10549-011-1690-0
113. Wang Y, Ou Z, Sun Y, Yeh S, Wang X, Long J, et al. Androgen receptor promotes melanoma metastasis via altering the miRNA-539-3p/USP13/MITF/AXL signals. Oncogene. (2017) 36:1644–54. doi: 10.1038/onc.2016.330
114. Valverde P. Effects of Gas6 and hydrogen peroxide in Axl ubiquitination and downregulation. Biochem Biophys Res Commun. (2005) 333:180–5. doi: 10.1016/j.bbrc.2005.05.086
115. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. (2014) 507:508–12. doi: 10.1038/nature12998
116. Bae SY, Hong JY, Lee HJ, Park HJ, Lee SK. Targeting the degradation of AXL receptor tyrosine kinase to overcome resistance in gefitinib-resistant non-small cell lung cancer. Oncotarget. (2015) 6:10146–60. doi: 10.18632/oncotarget.v6i12
117. Namba K, Shien K, Takahashi Y, Torigoe H, Sato H, Yoshioka T, et al. Activation of AXL as a preclinical acquired resistance mechanism against osimertinib treatment in EGFR-mutant non-small cell lung cancer cells. Mol Cancer Res. (2019) 17:499–507. doi: 10.1158/1541-7786.MCR-18-0628
118. Liu YN, Tsai MF, Wu SG, Chang TH, Tsai TH, Gow CH, et al. Acquired resistance to EGFR tyrosine kinase inhibitors is mediated by the reactivation of STC2/JUN/AXL signaling in lung cancer. Int J Cancer. (2019) 145:1609–24. doi: 10.1002/ijc.v145.6
119. Okura N, Nishioka N, Yamada T, Taniguchi H, Tanimura K, Katayama Y, et al. ONO-7475, a novel AXL inhibitor, suppresses the adaptive resistance to initial EGFR-TKI treatment in EGFR-mutated non-small cell lung cancer. Clin Cancer Res. (2020) 26:2244–56. doi: 10.1158/1078-0432.CCR-19-2321
120. Kim D, Bach DH, Fan YH, Luu TT, Hong JY, Park HJ, et al. AXL degradation in combination with EGFR-TKI can delay and overcome acquired resistance in human non-small cell lung cancer cells. Cell Death Dis. (2019) 10:361. doi: 10.1038/s41419-019-1601-6
121. Wang Y, Xia H, Zhuang Z, Miao L, Chen X, Cai H. Axl-altered microRNAs regulate tumorigenicity and gefitinib resistance in lung cancer. Cell Death Dis. (2014) 5:e1227. doi: 10.1038/cddis.2014.186
122. Tulchinsky E, Demidov O, Kriajevska M, Barlev NA, Imyanitov E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim Biophys Acta Rev Cancer. (2019) 1871:29–39. doi: 10.1016/j.bbcan.2018.10.003
123. Zhu X, Chen L, Liu L, Niu X. EMT-mediated acquired EGFR-TKI resistance in NSCLC: mechanisms and strategies. Front Oncol. (2019) 9:1044. doi: 10.3389/fonc.2019.01044
124. Yochum ZA, Cades J, Wang H, Chatterjee S, Simons BW, O'Brien JP, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene. (2019) 38:656–70. doi: 10.1038/s41388-018-0482-y
125. Wu F, Li J, Jang C, Xiong J. The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int J Clin Exp Pathol. (2014) 7:6653–61.
126. Kim HR, Kim WS, Choi YJ, Choi CM, Rho JK, Lee JC. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol Oncol. (2013) 7:1093–102. doi: 10.1016/j.molonc.2013.08.001
127. Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. (2015) 27:397–408. doi: 10.1016/j.ccell.2015.02.005
128. Gower A, Hsu WH, Hsu ST, Wang Y, Giaccone G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol Oncol. (2016) 10:601–9. doi: 10.1016/j.molonc.2015.11.007
129. Klammt J, Kiess W, Pfaffle R. IGF1R mutations as cause of SGA. Best Pract Res Clin Endocrinol Metab. (2011) 25:191–206. doi: 10.1016/j.beem.2010.09.012
130. Ekyalongo RC, Yee D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis Oncol. (2017) 1. doi: 10.1038/s41698-017-0017-y
131. Forbes BE, Blyth AJ, Wit JM. Disorders of IGFs and IGF-1R signaling pathways. Mol Cell Endocrinol. (2020) 518:111035. doi: 10.1016/j.mce.2020.111035
132. Manabe T, Yasuda H, Terai H, Kagiwada H, Hamamoto J, Ebisudani T, et al. IGF2 autocrine-mediated IGF1R activation is a clinically relevant mechanism of osimertinib resistance in lung cancer. Mol Cancer Res. (2020) 18:549–59. doi: 10.1158/1541-7786.MCR-19-0956
133. Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. (2008) 118:2609–19. doi: 10.1172/JCI34588
134. Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The insulin-like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild-type epidermal growth factor receptor. Int J Cancer. (2014) 135:1002–6. doi: 10.1002/ijc.v135.40
135. Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. (2013) 73:834–43. doi: 10.1158/0008-5472.CAN-12-2066
136. Lee Y, Wang Y, James M, Jeong JH, You M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol Carcinog. (2016) 55:991–1001. doi: 10.1002/mc.22342
137. Peled N, Wynes MW, Ikeda N, Ohira T, Yoshida K, Qian J, et al. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell Oncol (Dordr). (2013) 36:277–88. doi: 10.1007/s13402-013-0133-9
138. Wilson C, Nimick M, Nehoff H, Ashton JC. ALK and IGF-1R as independent targets in crizotinib resistant lung cancer. Sci Rep. (2017) 7:13955. doi: 10.1038/s41598-017-14289-w
139. Rubin I, Yarden Y. The basic biology of HER2. Ann Oncol. (2001) 12 Suppl 1:S3–8. doi: 10.1093/annonc/12.suppl_1.S3
140. Mishra R, Patel H, Alanazi S, Yuan L, Garrett JT. HER3 signaling and targeted therapy in cancer. Oncol Rev. (2018) 12:355. doi: 10.4081/oncol.2018.355
141. Hsu JL, Hung MC. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. (2016) 35:575–88. doi: 10.1007/s10555-016-9649-6
142. Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. (2006) 7:505–16. doi: 10.1038/nrm1962
143. Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. (2003) 11:495–505. doi: 10.1016/S1097-2765(03)00048-0
144. Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. (1997) 16:1647–55. doi: 10.1093/emboj/16.7.1647
145. Engelman JA, Cantley LC. The role of the ErbB family members in non-small cell lung cancers sensitive to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res. (2006) 12:4372s–6s. doi: 10.1158/1078-0432.CCR-06-0795
146. Dey N, Williams C, Leyland-Jones B, De P. A critical role for HER3 in HER2-amplified and non-amplified breast cancers: function of a kinase-dead RTK. Am J Transl Res. (2015) 7:733–50.
147. Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. (2015) 26:2073–8. doi: 10.1093/annonc/mdv319
148. Wang M, Zhang D, Wang G, Xia J, Chen S, Li S, et al. HER2 amplification as a potential mechanism of acquired resistance to afatinib in an advanced non-small-cell lung cancer patient. Lung Cancer. (2021) 151:106–7. doi: 10.1016/j.lungcan.2020.11.004
149. Minari R, Gnetti L, Lagrasta CA, Squadrilli A, Bordi P, Azzoni C, et al. Emergence of a HER2-amplified clone during disease progression in an ALK-rearranged NSCLC patient treated with ALK-inhibitors: a case report. Transl Lung Cancer Res. (2020) 9:787–92. doi: 10.21037/tlcr.2020.04.03
150. Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res. (2012) 18:6219–26. doi: 10.1158/1078-0432.CCR-12-0392
151. McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin Cancer Res. (2018) 24:3334–47. doi: 10.1158/1078-0432.CCR-17-2452
152. Meador CB, Hata AN. Acquired resistance to targeted therapies in NSCLC: Updates and evolving insights. Pharmacol Ther. (2020) 210:107522. doi: 10.1016/j.pharmthera.2020.107522
153. Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, et al. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. (2015) 75:2489–500. doi: 10.1158/0008-5472.CAN-14-3167
154. Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I, et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. (2017) 77:2078–89. doi: 10.1158/0008-5472.CAN-16-2359
155. Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Yang J, et al. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. (2017) 12:567–72. doi: 10.1016/j.jtho.2016.11.2231
156. La Monica S, Minari R, Cretella D, Bonelli M, Fumarola C, Cavazzoni A, et al. Acquired BRAF G469A mutation as a resistance mechanism to first-line osimertinib treatment in NSCLC cell lines harboring an EGFR exon 19 deletion. Target Oncol. (2019) 14:619–26. doi: 10.1007/s11523-019-00669-x
157. Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discovery. (2016) 6:1118–33. doi: 10.1158/2159-8290.CD-16-0596
158. Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. (2014) 346:1480–6. doi: 10.1126/science.1254721
159. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. (2013) 31:1070–80. doi: 10.1200/JCO.2012.43.3912
160. Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B, et al. Mechanisms of acquired resistance to AZD9291: A mutation-selective, irreversible EGFR inhibitor. J Thorac Oncol. (2015) 10:1736–44. doi: 10.1097/JTO.0000000000000688
161. Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, et al. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. (2013) 11:759–67. doi: 10.1158/1541-7786.MCR-12-0652
162. Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P, et al. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PloS One. (2010) 5:e14117. doi: 10.1371/journal.pone.0014117
163. Ou SI, Horn L, Cruz M, Vafai D, Lovly CM, Spradlin A, et al. Emergence of FGFR3-TACC3 fusions as a potential by-pass resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR mutated NSCLC patients. Lung Cancer. (2017) 111:61–4. doi: 10.1016/j.lungcan.2017.07.006
164. Zeng L, Yang N, Zhang Y. GOPC-ROS1 rearrangement as an acquired resistance mechanism to osimertinib and responding to crizotinib combined treatments in lung adenocarcinoma. J Thorac Oncol. (2018) 13:e114–6. doi: 10.1016/j.jtho.2018.02.005
165. Schrock AB, Zhu VW, Hsieh WS, Madison R, Creelan B, Silberberg J, et al. Receptor tyrosine kinase fusions and BRAF kinase fusions are rare but actionable resistance mechanisms to EGFR tyrosine kinase inhibitors. J Thorac Oncol. (2018) 13:1312–23. doi: 10.1016/j.jtho.2018.05.027
166. Offin M, Somwar R, Rekhtman N, Benayed R, Chang JC, Plodkowski A, et al. Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis Oncol. (2018) 2:PO.18.00126. doi: 10.1200/PO.18.00126
167. Klempner SJ, Bazhenova LA, Braiteh FS, Nikolinakos PG, Gowen K, Cervantes CM, et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer. (2015) 89:357–9. doi: 10.1016/j.lungcan.2015.06.021
168. Dziadziuszko R, Le AT, Wrona A, Jassem J, Camidge DR, Varella-Garcia M, et al. An activating KIT mutation induces crizotinib resistance in ROS1-positive lung cancer. J Thorac Oncol. (2016) 11:1273–81. doi: 10.1016/j.jtho.2016.04.001
169. Drilon A, Clark JW, Weiss J, Ou SI, Camidge DR, Solomon BJ, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med. (2020) 26:47–51. doi: 10.1038/s41591-019-0716-8
170. Neal JW, Dahlberg SE, Wakelee HA, Aisner SC, Bowden M, Huang Y, et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): a randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol. (2016) 17:1661–71. doi: 10.1016/S1470-2045(16)30561-7
171. Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N Engl J Med. (2020) 383:944–57. doi: 10.1056/NEJMoa2002787
172. Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC, et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med. (2020) 383:931–43. doi: 10.1056/NEJMoa2004407
173. Brigatinib tablets . United States Prescribing Information. US National Library of Medicine. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/208772s012lbl.pdf (Accessed September 29, 2021).
174. Chong CR, Bahcall M, Capelletti M, Kosaka T, Ercan D, Sim T, et al. Identification of existing drugs that effectively target NTRK1 and ROS1 rearrangements in lung cancer. Clin Cancer Res. (2017) 23:204–13. doi: 10.1158/1078-0432.CCR-15-1601
175. Wu YL, Zhang L, Kim DW, Lee DH, Yang JC-H, Ahn M-J, et al. Phase ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J Clin Oncol. (2018) 36:3101–9. doi: 10.1200/JCO.2018.77.7326
176. Yang JJ, Fang J, Shu YQ, Chang JH, Chen GY, He JX, et al. A phase Ib study of the highly selective MET-TKI savolitinib plus gefitinib in patients with EGFR-mutated, MET-amplified advanced non-small-cell lung cancer. Invest New Drugs. (2020) 39(2):477–87. doi: 10.1007/s10637-020-01010-4
177. Wu YL, Cheng Y, Zhou J, Lu S, Zhang Y, Zhao J, et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir Med. (2020) 8:1132–43. doi: 10.1016/S2213-2600(20)30154-5
178. Sequist LV, Han JY, Ahn MJ, Cho BC, Yu H, Kim SW, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. (2020) 21:373–86. doi: 10.1016/S1470-2045(19)30785-5
179. Hartmaier RJ, Markovets AA, Ahn MJ, Sequist LV, Han JY, Cho BC, et al. Osimertinib + Savolitinib to overcome acquired MET-mediated resistance in epidermal growth factor receptor-mutated, MET-amplified non-small cell lung cancer: TATTON. Cancer Discovery. (2023) 13:98–113. doi: 10.1158/2159-8290.CD-22-0586
180. Planchard D, Jänne PA, Cheng Y, Yang JC, Yanagitani N, Kim SW, et al. Osimertinib with or without chemotherapy in EGFR-mutated advanced NSCLC. N Engl J Med. (2023) 389:1935–48. doi: 10.1056/NEJMoa2306434
181. Gouji T, Takashi S, Mitsuhiro T, Yukito I. Crizotinib can overcome acquired resistance to CH5424802: is amplification of the MET gene a key factor? J Thorac Oncol. (2014) 9:e27–28. doi: 10.1097/JTO.0000000000000113
182. Rosen EY, Johnson ML, Clifford SE, Somwar R, Kherani JF, Son J, et al. Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining selpercatinib with crizotinib. Clin Cancer Res. (2021) 27:34–42. doi: 10.1158/1078-0432.CCR-20-2278
183. Ross HJ, Blumenschein GR Jr., Aisner J, Damjanov N, Dowlati A, Garst J, et al. Randomized phase II multicenter trial of two schedules of lapatinib as first- or second-line monotherapy in patients with advanced or metastatic non-small cell lung cancer. Clin Cancer Res. (2010) 16:1938–49. doi: 10.1158/1078-0432.CCR-08-3328
184. Soria JC, Felip E, Cobo M, Lu S, Syrigos K, Lee KH, et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. (2015) 16:897–907. doi: 10.1016/S1470-2045(15)00006-6
185. Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. (2010) 28:3076–83. doi: 10.1200/JCO.2009.27.9414
186. Gatzemeier U, Groth G, Butts C, Van Zandwijk N, Shepherd F, Ardizzoni A, et al. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann Oncol. (2004) 15:19–27. doi: 10.1093/annonc/mdh031
187. Herbst RS, Davies AM, Natale RB, Dang TP, Schiller JH, Garland LL, et al. Efficacy and safety of single-agent pertuzumab, a human epidermal receptor dimerization inhibitor, in patients with non small cell lung cancer. Clin Cancer Res. (2007) 13:6175–81. doi: 10.1158/1078-0432.CCR-07-0460
188. Tsurutani J, Iwata H, Krop I, Jänne PA, Doi T, Takahashi S, et al. Targeting HER2 with trastuzumab deruxtecan: A dose-expansion, phase I study in multiple advanced solid tumors. Cancer Discovery. (2020) 10:688–701. doi: 10.1158/2159-8290.CD-19-1014
189. Peters S, Stahel R, Bubendorf L, Bonomi P, Villegas A, Kowalski DM, et al. Trastuzumab emtansine (T-DM1) in patients with previously treated HER2-overexpressing metastatic non-small cell lung cancer: efficacy, safety, and biomarkers. Clin Cancer Res. (2019) 25:64–72. doi: 10.1158/1078-0432.CCR-18-1590
190. Liu SM, Tu HY, Wei XW, Yan HH, Dong XR, Cui JW, et al. First-line pyrotinib in advanced HER2-mutant non-small-cell lung cancer: a patient-centric phase 2 trial. Nat Med. (2023) 29:2079–86. doi: 10.1038/s41591-023-02461-x
191. Tan DS-W, Lim KH, Tai WM, Ahmad A, Pan S, Ng QS, et al, et al. A phase Ib safety and tolerability study of a pan class I PI3K inhibitor buparlisib (BKM120) and gefitinib in EGFR TKI-resistant NSCLC. J Clin Oncol. (2013) 31:8107. doi: 10.1200/jco.2013.31.15_suppl.8107
192. KRAZATI (adagrasib) tablets, for oral use . US Food and Drug Administration. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216340s001lbl.pdf (Accessed December 13, 2022).
193. Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. (2017) 18:1307–16. doi: 10.1016/S1470-2045(17)30679-4
194. Hellmann MD, Kim TW, Lee CB, Goh BC, Miller WH Jr, Oh DY, et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol. (2019) 30:1134–42. doi: 10.1093/annonc/mdz113
195. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
196. Sacher A, LoRusso P, Patel MR, Miller WH Jr, Garralda E, Forster MD, et al. Single-agent divarasib (GDC-6036) in solid tumors with a KRAS G12C mutation. N Engl J Med. (2023) 389:710–21. doi: 10.1056/NEJMoa2303810
197. Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. (2017) 317:1844–53. doi: 10.1001/jama.2017.3438
198. Belli C, Penault-Llorca F, Ladanyi M, Normanno N, Scoazec JY, Lacroix L, et al. ESMO recommendations on the standard methods to detect RET fusions and mutations in daily practice and clinical research. Ann Oncol. (2021) 32:337–50. doi: 10.1016/j.annonc.2020.11.021
199. Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, et al. Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N Engl J Med. (2020) 383:813–24. doi: 10.1056/NEJMoa2005653
200. Cho BC, Lu S, Felip E, Spira AI, Girard N, Lee JS, et al. Amivantamab plus lazertinib in previously untreated EGFR-mutated advanced NSCLC. N Engl J Med. (2024). doi: 10.1056/NEJMoa2403614
201. Yeung KT, More S, Woodward B, Velculescu V, Husain H. Circulating tumor DNA for mutation detection and identification of mechanisms of resistance in non-small cell lung cancer. Mol Diagn Ther. (2017) 21:375–84. doi: 10.1007/s40291-017-0260-5
202. Phallen J, Leal A, Woodward BD, Forde PM, Naidoo J, Marrone KA, et al. Early noninvasive detection of response to targeted therapy in non-small cell lung cancer. Cancer Res. (2019) 79:1204–13. doi: 10.1158/0008-5472.CAN-18-1082
203. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. (2013) 497:108–12. doi: 10.1038/nature12065
204. US Food and Drug Administration (FDA). FDA Premarket Approval: Cobas EGFR MutationTest v2, Cobas DNA and cfDNA Sample Preparation Kit . Available online at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P120019s019 (Accessed August 14, 2020).
205. US Food and Drug Administration (FDA). FDA Premarket Approval: Next-generation sequencing oncology panel, somatic or germline variant detection system . Available online at: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=p200010 (Accessed August 14, 2020).
206. Peters S, Gadgeel SM, Mok T, Nadal E, Kilickap S, Swalduz A, et al. Entrectinib in ROS1-positive advanced non-small cell lung cancer: the phase 2/3 BFAST trial. Nat Med. (2024) 30:1923–1932. doi: 10.1016/j.annonc.2023.10.117
207. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. (2010) 141:69–80. doi: 10.1016/j.cell.2010.02.027
208. Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. (2019) 10:259. doi: 10.1038/s41467-018-08074-0
Keywords: tyrosine kinase inhibitors, NSCLC, bypass activation resistance, targeted therapy, gene ontology
Citation: Jiang Z, Gu Z, Yu X, Cheng T and Liu B (2024) Research progress on the role of bypass activation mechanisms in resistance to tyrosine kinase inhibitors in non-small cell lung cancer. Front. Oncol. 14:1447678. doi: 10.3389/fonc.2024.1447678
Received: 12 June 2024; Accepted: 25 September 2024;
Published: 08 November 2024.
Edited by:
Gao Zhang, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Wenyu Zhai, Sun Yat-Sen University Cancer Center, ChinaCopyright © 2024 Jiang, Gu, Yu, Cheng and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bofu Liu, ODM3NzE4MDc3QHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.