 Helge Waldum
Helge Waldum Geir Slupphaug
Geir Slupphaug- Department of Clinical and Molecular Medicine, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway
Malignancy manifests itself by deregulated growth and the ability to invade surrounding tissues or metastasize to other organs. These properties are due to genetic and/or epigenetic changes, most often mutations. Many aspects of carcinogenesis are known, but the cell of origin has been insufficiently focused on, which is unfortunate since the regulation of its growth is essential to understand the carcinogenic process and guide treatment. Similarly, the concept of cancer stem cells as cells having the ability to stop proliferation and rest in a state of dormancy and being resistant to cytotoxic drugs before “waking up” and become a highly malignant tumor recurrence, is not fully understood. Some tumors may recur after decades, a phenomenon probably also connected to cancer stem cells. The present review shows that many of these questions are related to the cell of origin as differentiated cells being long-term stimulated to proliferation.
1 Introduction
Malignancy is characterized by deregulated growth and the ability to invade surrounding tissues or metastasize to other organs. Current treatments for malignant tumors include surgery, irradiation, and cytotoxic drugs, often leading to the apparently complete removal of tumors. Unfortunately, many such tumors recur with reduced treatment susceptibility, even after initially effective interventions. Such recurrence often leads to the death of the patient and has been explained by the dormancy of malignant cells that initially survived the cytotoxic treatment (1). The mechanisms governing entry into and emergence from dormancy remain inadequately understood (1). The present review aims to elucidate this phenomenon with a particular emphasis on the cells of origin of cancers.
2 Cell of origin: differentiated versus stem cells
Our points of view are mainly based upon long-term experience in gastroenterology including gastric physiology and pathology. During the late seventies and early eighties, it became evident that the gastric hormone gastrin stimulated the enterochromaffin-like (ECL) cell to proliferation and neoplasia in rodents and humans (2–5), and before the identification of the ECL cell, Azzopardi and Pollock (6) focused on argentaffin (neuroendocrine cell marker) cells in gastric carcinomas. The description of ECL cell carcinoids in rodents after long-term profound acid inhibition (7, 8) made the gastrin hypothesis accepted (9). Curiously, although the use of proton pump inhibitors (PPIs) in clinical doses induced hypergastrinemia and ECL cell hyperplasia (10), it was claimed that in humans, unlike animals, hypergastrinemia did not transform ECL cells into cancer (11). Surprisingly, only our group started to examine gastric cancers for ECL cell markers. Our initial publication revealed ECL cell differentiation mainly in diffuse-type gastric carcinomas (12). A critical comment from Creutzfeldt and Solcia (13) was addressed, demonstrating that the criticism was unfounded (14). Continuing our investigation into the role of ECL cells in gastric carcinogenesis, we collaborated with leading pathologist Julia Polack, validating our initial results through thorough immuno-histochemical methods (15). Employing immunoelectron microscopy to demonstrate neuroendocrine granules (16) and in situ hybridization to detect expression of the gastrin receptor on gastric cancer cells (17–19), we substantiated that the ECL cell was central in gastric carcinogenesis. Moreover, we tracked the progression of an ECL cell carcinoid to a highly malignant cancer (20) and the evident transformation potential of long-term trophic stimulation of differentiated cells, leading to the development of highly malignant tumors. Similarly, the inactivation of the von Hippel-Lindau factor, whether hereditary or sporadic, results in the activation of hypoxia-inducible factor 2α (HIF-2α) (21), which chronically stimulates the erythropoietin (EPO) cell, being the cell of origin of the most prevalent type of renal cancer (clear cell renal cell cancer) (22). Moreover, the progression to pituitary carcinoma after bilateral adrenalectomy may also be seen as a consequence of long-term growth stimulation (23) like the occurrence of postmenopausal estrogen receptor-positive breast cancer after sex steroid hormone treatment (24).
Another interesting aspect lies in understanding why certain cells are more prone to develop into malignancy. For instance, the ECL cell, characterized by its lack of expression of the adherence molecule E-cadherin (25), may be particularly predisposed to malignancy. This is evident in hereditary gastric cancer, where mutations in the CDH1 gene result in the absence of E-cadherin, ultimately leading to gastric cancer (26). The temporal relationship between hypergastrinemia and the development of malignancy was demonstrated by a Spanish family with a missense mutation in the gene encoding the catalytic subunit of the gastric H+/K+-ATPase pump (ATP4A), which transports H+ ions in exchange for K+ ions across the apical membrane of parietal cells. Individuals with this mutation developed tumors of varying malignancy in their twenties and thirties (27). Accordingly, it can be concluded that differentiated cells may progress to malignancy through long-term stimulation of proliferation (20, 27) via phases of hyperplasia to benign neoplasia to malignant tumors in the stomach, but probably also in other organs. This process unfolds over extended periods, often spanning decades. Cells with proliferative capacity and low adherence to neighboring cells like the diffuse neuroendocrine (NE) cells are especially prone. Carcinogenesis is a multistage process, and therefore, cells of varying malignancy may co-exist. During the early stages, cells with low adherence and even low proliferation may nevertheless metastasize. These cells, which in many aspects resemble normal cells, may not readily respond to cytotoxic treatment but gradually accumulate new mutations leading to increased malignancy and subsequently manifest as late metastases. Tumors belonging to the phaeochromocytoma/paraganglioma group show histology close to normal, making it impossible to predict malignancy (28). In fact, only metastases that may occur decades after initial diagnosis (29) can definitively determine whether these tumors were benign or malignant. Unfortunately, there are no studies investigating the expression of E-cadherin or other adherence molecules in neuroendocrine cells except the ECL cell (25), but the fact that they appear intermingled between other epithelial cells must imply that they have low cell-to-cell adhesion. Low cell-to-cell adhesion secondary to changes in the E-cadherin–catenin complex has also been focused on in gastric carcinogenesis (30).
The enterochromaffin (EC) cell in the small intestine (SI) can also develop into neuroendocrine tumors (SI-NETs), but in contrast to ECL cell-derived tumors, the pathogenesis of EC cell SI-NETs is not known. Nevertheless, they probably play a role in the development of SI adenocarcinomas (31). Neuroendocrine tumors (NETs) in the small intestine are the most prevalent tumors in that organ. Interestingly, such tumors may develop in clusters, and they most often lack driver mutations (32), indicating a local carcinogenic factor.
The ability to develop into malignant tumors is not restricted to differentiated cells presently accepted as neuroendocrine but probably also other proliferating cells with low adherence. Moreover, more tumors develop from NE cells than presently recognized. For instance, we have shown that the most prevalent type of kidney cancer, clear cell renal cell cancer (CCRCC), most likely originates from EPO cells (21, 22, 33). Clinically, these tumors exhibit similarities to NETs, including early metastases, low response to cytotoxic drugs, and, parallel to hormonal overstimulation in NETs, erythrocytosis in a subset of patients. They also share positivity for neuron-specific enolase (NSE) (33, 34). NSE is a more specific neuroendocrine marker than ordinarily believed (34). It is probably the most sensitive neuroendocrine marker, which has also contributed to its dubious reputation as non-specific. Hypoxia-inducible factor (HIF) is the most important regulator of erythropoietin release (35). Von Hippel-Lindau (VHL) disease predisposes to CCRCC by stabilization of HIF, thereby leading to increased and continuous stimulation of the EPO cell, both functionally and proliferatively (36). Lack of VHL factor function is also central in CCRCCs in general (37, 38), but the cancers not being a part of inherent VHL disease manifest themselves a few decades later. The prolonged hyperstimulation by HIF causing CCRCC resembles how excessive gastrin hyperstimulation leads to gastric cancer and is another example of cancer developing from a differentiated cell exposed to prolonged hyperstimulation. This improved understanding of the pathogenesis of CCRCC has already resulted in potentially improved treatments such as using HIF inhibitors (36).
Lung cancer is another prevalent cancer where NE cells play an important role in carcinogenesis since small cell and large cell pulmonary cancers are regarded as NE tumors (39). In fact, the prevalence of the more benign variant, NETs, localized to the lungs, is secondary only to the gastrointestinal tract. There are many types of NE cells in the lungs either spread in the mucosa (40) or as collections as neuroendocrine bodies (41) often localized to bifurcations of the airways. The latter type plays an important function in the regulation of blood oxygenation (42). The diffusely spread NE cells in the airway surface are also believed to be regulatory, but their precise functions remain to be elucidated. Lung NE cells are the source of the presently called NETs, previously carcinoids (43), whereas the cell of origin of the small and large cell pulmonary carcinomas remains unknown and is not clarified since the early stages of these tumors have not been identified (44). Finally, trans-differentiation may be an alternative for NE cells as the cell of origin for lung NE cancers as well as cancers of the prostate (45).
3 Breast and prostate cancers
Prolonged hormonal stimulation is central in the pathogenesis of breast and prostate cancers, which both probably originate from differentiated cells. Unfortunately, there has been no definitive identification of the specific cells of origin for cancers in these two organs. Both breast and prostate cancers exhibit significant heterogeneity (46, 47). Interestingly, there are similarities regarding cells of origin, whether basal or luminal cells (48, 49). Tumors are also categorized based on their expression of sex hormone receptors, estrogen receptor (ER) (48, 50), or androgen receptor (AR) (49). The central role of estrogen in mammary carcinogenesis is underscored by the large contrast in cancer incidence between the two sexes (51).
Although sex hormones are central in the pathogenesis of both breast and prostate cancers, many of these cancers do not express sex hormone receptors. This could be due to mutations during the carcinogenic process or that the tumor nevertheless developed from a sex hormone receptor-negative cell. Thus, while AR was found to be expressed in virtually all luminal cells, half of the basal cells, and 60% of fibroblasts in the normal prostate, there was a loss of AR particularly in the fibroblasts and the tumor cells during the evolution of cancer (52). Another explanation may be that a receptor-positive cell may itself develop into the tumor, and simultaneously, it may provoke neoplasia in another cell (receptor-negative) type by releasing a signal substance-stimulating secretion and proliferation finally leading to neoplasia in this cell as well. This would be parallel to gastric cancers, where gastrin stimulates the proliferation of ECL cells leading to ECL cell NETs and gastric carcinoma of diffuse type. Concomitantly, the release of signal substances from the ECL cell stimulates the proliferation of stem cells (gastrin receptor negative) causing cancer of intestinal type (53) (Figure 1).
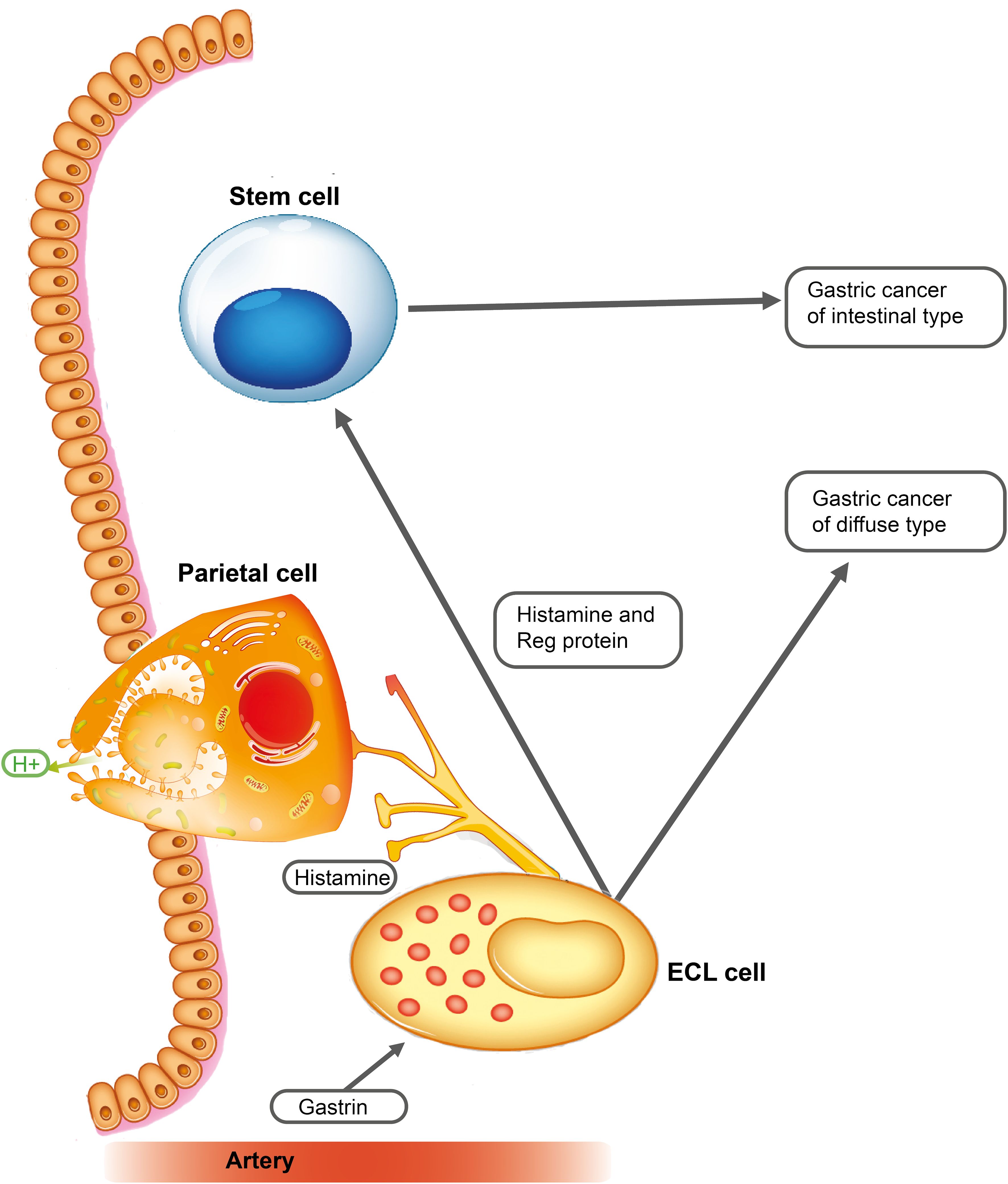
Figure 1 Gastrin stimulates the enterochromaffin-like (ECL) cell to proliferate and to release mediators including regenerating gene (REG) proteins, a family of small proteins with many functions (54), including stimulation of stem cell proliferation (55). Long-term stimulation of these two cells leads to gastric cancers of diffuse and intestinal types [with permission from Waldum et al. (53)].
Sex hormone-induced breast and prostate cancers may promote tumorigenesis by stimulating the proliferation of their target cells, which by releasing signal substances stimulates the growth of another cell type leading to a hormone receptor-negative cancer. Furthermore, overexpression of the chemokine CXCL12-y in prostate and breast epithelial cells induces a shift from a luminal cell type to a non-luminal cell type (56). Among candidates for signal-producing cells in the prostate is the recently identified PEG10+ NE cell (57). NE cells are found in most prostate cancers. However, among the serum markers for NE cells tested, only chromogranin A (CgA) was positively correlated to the number of CgA-staining cells in the tumors (58). Recently, neuropeptide Y was reported to be increasingly expressed during the tumorigenic process in the prostate (59) and thus is a possible driver of carcinogenesis. In families with hereditary increased risk of prostate cancer, there is also an increased risk of breast, gastric, and renal cancers (60). Like in the gastric oxyntic mucosa, there are NE cells located basally in the glands in the prostate (61), whereas this is debatable in the breast (62, 63). Electron microscopy has revealed intracellular secretory granules in prostate NE cells, distinguishing between open-type cells connected to the gland lumen and closed type cells lacking such connection (61, 64). NE cells in the normal prostate cannot be identified by histochemistry using hematoxylin and eosin but with immune histochemistry using traditional markers like chromogranin, synaptophysin, and NSE. Prostate NE cells do not express the androgen receptor or prostate-specific antigen (PSA), and based upon negativity for Ki67, they are also claimed to be non-proliferative (61, 64). Human ECL cells, which for years were claimed not to divide (65), showed a very slow proliferation in that capturing these scarce cells in the act of proliferation was challenging (66). Nevertheless, the lineage plasticity of prostate carcinomas from adenocarcinomas to neuroendocrine androgen receptor-negative cancers represents a major change in cancers, worsening prognosis, and may be due to epigenetic differences (67).
Distinct NE differentiation is evident in various well-defined conventional-type breast carcinomas, but there is still a controversy about whether true NE neoplasms originating from NE cell precursors exist (63). CgA-positive cells have been described in both normal breast tissue and carcinomas (68, 69). Furthermore, by combining immunohistochemistry with gene expression analysis, the percentage of tumors being positive for NE markers increased (70). Breast NE cell carcinoma phenotypes can also be clear cell carcinoma (71), mucinous carcinoma (72), or small cell carcinoma (73). The distinction between luminal and basal cell breast cancers is not definitive, as there is a lineage plasticity where luminal tumors with low ER change in the direction of basal cells (74). The shared reliance on sex hormones in carcinogenesis as well as the parallel mechanisms involved underscores the significant similarities between breast and prostate cancers.
4 Metastases
Differentiated cells with poor adherence to each other or supportive tissue cells, such as NE cells interspersed among other cells, are probably more prone to lose connections and disseminate, that is, to metastases. Thus, not only neuroendocrine carcinomas but also the more benign variant NETs often show metastases at an asymptomatic stage (75, 76). This pattern is akin to CCRCCs (77), which most likely also are of neuroendocrine origin (21, 22, 33). Melanoma, originating from melanocytes derived from neural crest cells, also demonstrates early metastasis as well as cellular plasticity (78). This underscores the broader pattern that many cancers develop from differentiated cells (79). Additionally, endometriosis, which is treated like a non-neoplastic benign disease, nevertheless, can metastasize (80). The central role of cellular adherence in carcinogenesis is exemplified by hereditary diffuse gastric cancer, resulting from inactivating mutations in the E-cadherin gene (81). Also, breast cancer (82, 83) and prostate cancer (84) exhibit early metastasis. Thus, these two cancers, being among the most prevalent in women and men, may often have metastasized at diagnosis. Breast cancer screening programs may hopefully increase the rate of diagnosis before metastasis (85), and the new improved methods in the diagnosis of prostate cancer (86) may also improve the prognosis of this cancer.
Nonetheless, the prevailing theory of carcinogenesis is that most cancers develop from stem cells (87), characterized by their rapid proliferation and plasticity. Accumulated evidence suggests that this is an oversimplification. For instance, gastric cancer can develop from an ECL cell, a differentiated NE cell leading to diffuse type cancers, or from stem cells, causing the intestinal type of gastric cancer (53). Although multiple stem cell receptors have been identified (88), their roles in carcinogenesis have received less attention, possibly due to their widespread effects, in contrast to the specific hormone receptors that play a central role in carcinogenesis from differentiated cells. Consequently, the knowledge of the cell of origin as a differentiated cell is presently more useful in tailoring cancer treatment.
5 DNA damage resistance in recurrent cancer cells
DNA damage-inducing therapies are fundamental in cancer treatment, as evidenced by the crucial roles of chemotherapy and radiotherapy, both of which operate by directly or indirectly inflicting DNA damage (89). These therapies can alter the DNA nucleotides, induce single-strand (SSBs) or double-strand breaks (DSBs), intercalate between bases, or form crosslinks within DNA or between DNA and proteins (90–94). Additionally, certain compounds induce DNA damage indirectly, e.g., by inhibiting the synthesis of deoxyribonucleotides (95, 96). Cancer cells may develop mechanisms to increase their resistance against DNA-damaging agents, including i) increased efflux or altered metabolism of genotoxic compounds, ii) suppression of apoptosis, or iii) enhanced DNA repair.
Cells harbor elaborate mechanisms to counteract various types of DNA damage, collectively referred to as the DNA damage response (DDR). The DDR is not a rigid pathway but rather a dynamic collection of pathways that are mobilized based on various factors such as type of damage, cell type, chromatin environment, and cell cycle phase (97). Examples of these pathways include base excision repair (BER), which corrects small lesions and certain mismatches (98); ribonucleotide excision repair (RER), which removes mis-incorporated ribonucleotides (99); nucleotide excision repair (NER), which is employed to eliminate bulky and helix-distorting adducts (100); and mismatch repair (MMR), which corrects mismatches generated mainly during replication (101). In the case of DSBs, cells employ either high-fidelity homologous recombination (HR) or the more error-prone non-homologous end-joining (NHEJ) pathway (102, 103). Importantly, the boundaries between these pathways are flexible, and the repair of certain lesions often involves proteins from multiple pathways (104). Additionally, the DDR includes mechanisms to arrest the cell cycle. This is important since DNA-damaging agents primarily exert their cytotoxic effects during the S-phase, and quiescent cells exhibit greater resistance to DNA-damaging drugs (105).
DNA repair plays a crucial but complex role in stem cells. It safeguards normal stem cells and ensures the preservation of genetic integrity as tissues regenerate. Efficient DNA repair mechanisms are particularly prominent in normal stem and progenitor cells. However, as cells differentiate, the tolerance for somatic mutation increases, leading to a reduction in DNA repair activity (106, 107). Enhanced repair of the lesions induced by a specific DNA-damaging agent probably increases resistance to that agent in most cells, including normal and cancer stem cells. However, there are exceptions to this. For example, bone marrow myeloid progenitor cells derived from mice defective in the 3-methyladenine DNA glycosylase Aag exhibit unexpected resistance to alkylation in comparison to progenitor cells from wild-type mice (108). The underlying reason for this is probably that repair intermediates in the BER pathway (AP sites and SSBs) may be more cytotoxic than the original base lesion it left unprocessed. Thus, balanced levels of the factors contributing within that pathway and the availability of redundant repair mechanisms will affect the overall outcome.
The activity of a distinct repair pathway must also be evaluated in relation to the timeframe available for repairing the DNA damage. Even if the expression of many DNA repair proteins is cell cycle regulated and displays elevated levels in rapidly proliferating cells (109), DNA repair processes also take place in quiescent cells. However, the repair pathways employed may differ between these two situations. One example of this is in the repair of DSBs, the most deleterious form of DNA damage. Error-free repair of DSBs via HR depends on the presence of an intact copy of the DNA in a newly replicated sister chromatid and thus cannot occur in post-mitotic quiescent cells. Conversely, error-prone NHEJ operates throughout the cell cycle and is active in quiescent cells, including stem cells (110, 111). This shift in repair mode may potentially contribute to increased genomic instability within dormant cancer cells, thereby promoting cancer recurrence by enhancing drug resistance and metastatic potential.
6 Cancer stem cells
Cancer stem cells (CSCs) (112) are a subpopulation of cells within a tumor with the ability to self-renew and generate the diverse cell types found in the tumor. They are often considered responsible for tumor initiation, maintenance, and resistance to therapy (113). The proliferative activity of CSCs varies depending on the type of cancer and the specific characteristics of the CSC population. While some CSCs proliferate rapidly and are sensitive to chemotherapy, others proliferate slowly and are more resistant to such treatments, although there is no experimental evidence that CSCs may undergo cell cycle arrest. Thus, dormant cancer cells and CSCs should be considered separate entities (114). Nevertheless, the link between tumor dormancy and CSCs has long been recognized (115) along with the significance of such cells in driving metastasis (116). Although the concept of CSCs initially emerged from observations in acute leukemia (112), these cells have been mainly attributed to cancers originating in cells needing long-term proliferative hyperstimulation before reaching malignancy. Examples include NE cells (different hormones), EPO cells (HIF) in CCRCC, and the cells of cancer origin in the breast (estrogen), the prostate (androgens), and melanocytes (ultraviolet irradiation). Interestingly, in a recent study comparing the genomes at an early stage of cancer with metastatic lesions, the differences were particularly marked for cancers of the prostate, thyroid, CCRCC, breast, and pancreatic NE tumors (116), all belonging to cancers known to lead to late metastases. Tumor cells may be identified by examining bone marrow or blood for specific markers including nucleic acids (113). Comparing mutations in tumor cells with those in primary tumors, early and late metastases will allow an estimation of the stage at which the different tumor cells separated from each other and thus give an indication of the mechanism behind the occurrence of late metastases (117).
7 Late metastases/dormancy
Late metastases will accordingly be expected to occur in cancers originating in differentiated cells with a slow proliferation and little adherence to neighboring cells (Figure 2).
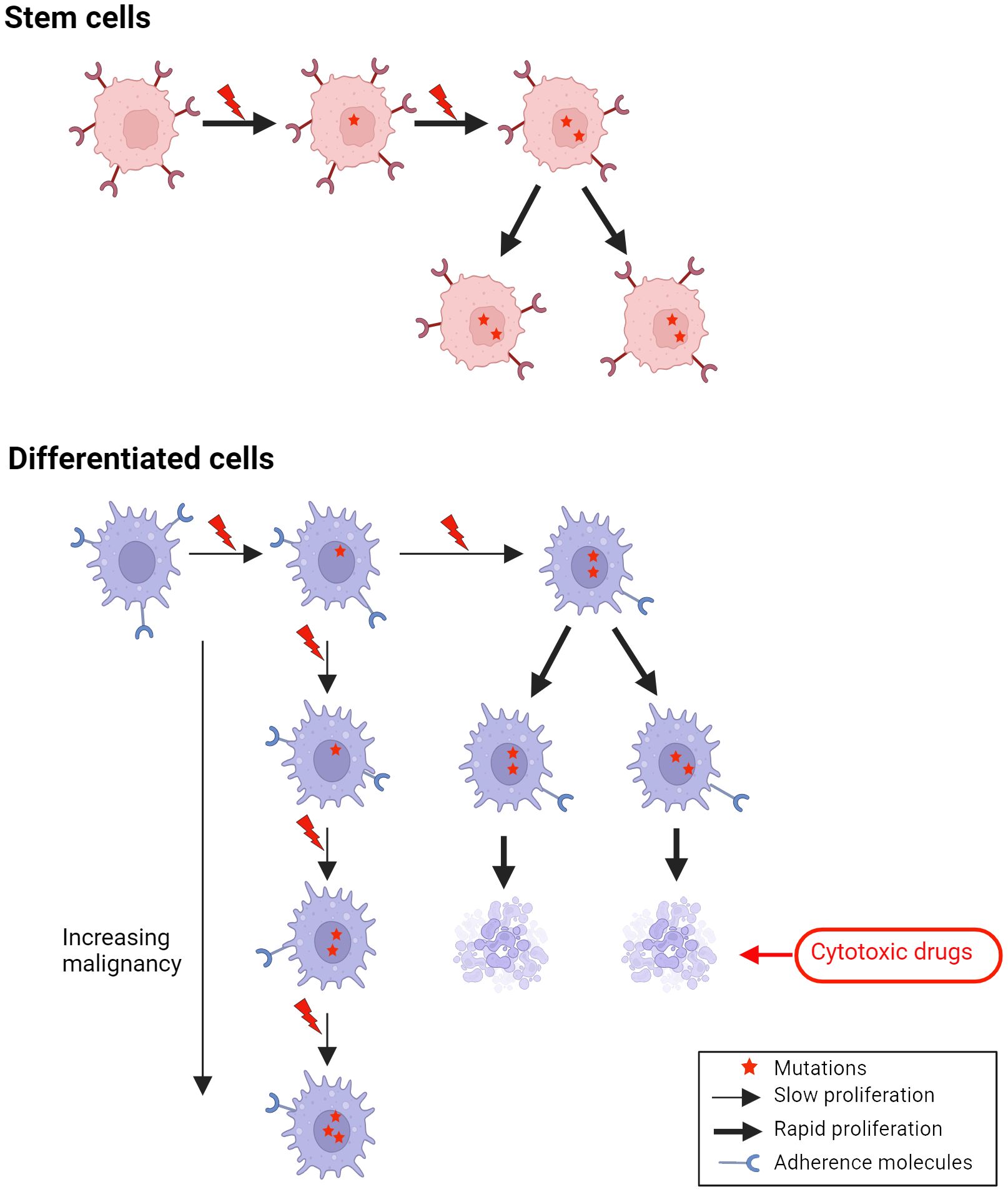
Figure 2 A missense mutation in a gene involved in adherence in differentiated cells, which normally have low adherence, may lead to metastases of cells with rather slow proliferation, which may need years to develop into a clinical tumor (late metastases).
Moreover, long-term stimulation of proliferation of the cell of origin mainly by circulating hormones or paracrine substances is often involved. Another peculiar trait of these cancers is metastases to bones (118–124). Even NE tumors, previously thought rarely to metastasize to bones, now are realized to do that often (125). The recognition of bone metastases in NE tumors may be a consequence of increased survival as well as better diagnostic tools for detecting bone metastases. Tumor cell dormancy seems to occur particularly in the bone marrow where both osteoblasts and osteoclasts play a role (125, 126). Late metastases are connected to tumor cell dormancy. The common traits of cancers showing late metastases are summarized in Table 1. In a mouse study where prostate cancer tissue was injected subcutaneously, cancer cells were found in most organs during the growth of the tumor tissue, but removal of the cancer resulted in a reduction of cancer cells in all tissues except bones where osteoblasts seemed to induce dormancy, possibly through an adhesion kinase (127). The latter study indicates a possible treatment option in patients with bone metastases due to prostate cancer.
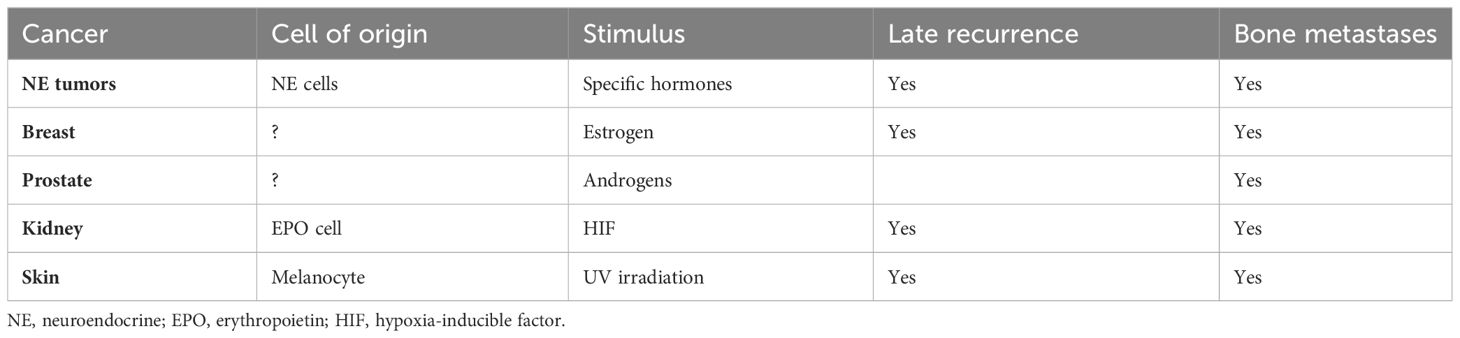
Table 1 Common features in cancers with late metastases.
Finally, single-cell genetic analysis of cancer cells is gaining popularity and may improve the identification of cells of origin with greater certainty as well as give information for individual treatment. In a lineage analysis in a mouse model, basal epithelial cells were shown to express great plasticity, and both basal and luminal cells were shown to develop into cancer (128).
8 Conclusion
To understand carcinogenesis, it is important to identify the cell of origin and its growth regulation, which also makes it possible to tailor treatment. It is obvious that differentiated cells secondary to long-term stimulation may develop into cancers. The role of cancer stem cells in carcinogenesis has often been explained by their ability to stop proliferation but at a later stage are highly proliferating. There is, however, a lack of experimental evidence supporting this idea. Conversely, differentiated cells with low adherence occurring spread among other cell types in the normal situation may not need great changes before being able to metastasize at a stage of minimal alterations of other functions, including cell division. Such metastasized slow-growing cells will naturally need a long time to develop into a macroscopic tumor. No common new mutations between the initial tumor and the late metastasis, in contrast, indicate that the tumors developed separately due to a common carcinogenic cause.
Author contributions
HW: Visualization, Writing – original draft, Writing – review & editing. GS: Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Singvogel K, Schittek B. Dormancy of cutaneous melanoma. Cancer Cell Int. (2024) 24:88. doi: 10.1186/s12935-024-03278-5
2. Soga J, Tazawa MD, Ito H. Ultrastructural demonstration of specific secretory granules of Mastomys gastric carcinoids. Acta Med Biol (Niigata). (1969) 17:119–24.
3. Soga J, Tazawa K, Aizawa O, Wada K, Tuto T. Argentaffin cell adenocarcinoma of the stomach: an atypical carcinoid? Cancer. (1971) 28:999–1003. doi: 10.1002/(ISSN)1097-0142
4. Wilander E. Achylia, pernicious anaemia, ECL cells and gastric carcinoids. Virchows Arch A Pathol Anat Histol. (1980) 387:371–3. doi: 10.1007/BF00454839
5. Wilander E, El-Salhy M, Pitkanen P. Histopathology of gastric carcinoids: a survey of 42 cases. Histopathology. (1984) 8:183–93. doi: 10.1111/j.1365-2559.1984.tb02335.x
6. Azzopardi JG, Pollock DJ. Argentaffin and argyrophil cells in gastric carcinoma. J Pathol Bacteriol. (1963) 86:443–51. doi: 10.1002/path.1700860219
7. Havu N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion. (1986) 35 Suppl 1:42–55. doi: 10.1159/000199381
8. Poynter D, Pick CR, Harcourt RA, Selway SA, Ainge G, Harman IW, et al. Association of long lasting unsurmountable histamine H2 blockade and gastric carcinoid tumours in the rat. Gut. (1985) 26:1284–95. doi: 10.1136/gut.26.12.1284
9. Hakanson R, Sundler F. Proposed mechanism of induction of gastric carcinoids: the gastrin hypothesis. Eur J Clin Invest. (1990) 20 Suppl 1:S65–71. doi: 10.1111/j.1365-2362.1990.tb01780.x
10. Lamberts R, Creutzfeldt W, Stockmann F, Jacubaschke U, Maas S, Brunner G. Long-term omeprazole treatment in man: effects on gastric endocrine cell populations. Digestion. (1988) 39:126–35. doi: 10.1159/000199615
11. Solcia E, Rindi G, Silini E, Villani L. Enterochromaffin-like (ECL) cells and their growths: relationships to gastrin, reduced acid secretion and gastritis. Baillieres Clin Gastroenterol. (1993) 7:149–65. doi: 10.1016/0950-3528(93)90035-Q
12. Waldum HL, Haugen OA, Isaksen C, Mecsei R, Sandvik AK. Enterochromaffin-like tumour cells in the diffuse but not the intestinal type of gastric carcinomas. Eur J Gastroenterol Hepatol 1991 3 245-249/Scand J Gastroenterol Suppl. (1991) 180:165–9. doi: 10.3109/00365529109093195
13. Creutzfeldt W, Solcia E. Are diffuse gastric carcinomas neuroendocrine tumours ECL-omas? Eur J Gastroenterol Hepatol. (1991) 3:862–3.
14. Waldum H. Are diffuse gastric carcinomas neuroendocrine tumours ECL-omas? (Reply). Eur J Gastroenterol Hepatol. (1991) 3:863–5.
15. Waldum HL, Aase S, Kvetnoi I, Brenna E, Sandvik AK, Syversen U, et al. Neuroendocrine differentiation in human gastric carcinoma. Cancer. (1998) 83:435–44. doi: 10.1002/(SICI)1097-0142(19980801)83:3<435::AID-CNCR11>3.3.CO;2-9
16. Martinsen TC, Skogaker NE, Fossmark R, Nordrum IS, Sandvik AK, Bendheim MO, et al. Neuroendocrine cells in diffuse gastric carcinomas: an ultrastructural study with immunogold labeling of chromogranin A. Appl Immunohistochem Mol Morphol. (2010) 18:62–8. doi: 10.1097/PAI.0b013e3181b70594
17. Sordal O, Qvigstad G, Nordrum IS, Gustafsson B, Waldum HL. In situ hybridization in human and rodent tissue by the use of a new and simplified method. Appl Immunohistochem Mol Morphol. (2013) 21:185–9. doi: 10.1097/PAI.0b013e31825a0048
18. Sordal O, Qvigstad G, Nordrum IS, Sandvik AK, Gustafsson BI, Waldum H. The PAS positive material in gastric cancer cells of signet ring type is not mucin. Exp Mol Pathol. (2014) 96:274–8. doi: 10.1016/j.yexmp.2014.02.008
19. Mjones P, Nordrum IS, Sordal O, Sagatun L, Fossmark R, Sandvik A, et al. Expression of the cholecystokinin-B receptor in neoplastic gastric cells. Horm Cancer. (2018) 9:40–54. doi: 10.1007/s12672-017-0311-8
20. Qvigstad G, Falkmer S, Westre B, Waldum HL. Clinical and histopathological tumour progression in ECL cell carcinoids (“ECLomas”). Apmis. (1999) 107:1085–92. doi: 10.1111/j.1699-0463.1999.tb01513.x
21. Nguyen CB, Oh E, Bahar P, Vaishampayan UN, Else T, Alva AS. Novel approaches with hif-2α Targeted therapies in metastatic renal cell carcinoma. Cancers (Basel). (2024) 16:601. doi: 10.3390/cancers16030601
22. Mjones PG, Nordrum IS, Qvigstad G, Sordal O, Rian LL, Waldum HL. Expression of erythropoietin and neuroendocrine markers in clear cell renal cell carcinoma. Apmis. (2017) 125:213–22. doi: 10.1111/apm.12654
23. Carlstrom LP, Graffeo CS, Perry A, Stokken JK, Van Gompel JJ. Nelson-Salassa syndrome progressing to pituitary carcinoma: A case report and review of the literature. Cureus. (2019) 11:e5595. doi: 10.7759/cureus.5595
24. Albers FEM, Lou MWC, Dashti SG, Swain CTV, Rinaldi S, Viallon V, et al. Sex-steroid hormones and risk of postmenopausal estrogen receptor-positive breast cancer: A case-cohort analysis. Cancer Causes Control. (2024). doi: 10.1007/s10552-024-01856-6
25. Waldum HL, Ringnes E, Nordbo H, Sordal O, Nordrum IS, Hauso O. The normal neuroendocrine cells of the upper gastrointestinal tract lack E-cadherin. Scand J Gastroenterol. (2014) 49:974–8. doi: 10.3109/00365521.2014.909275
26. Guilford PJ, Hopkins JB, Grady WM, Markowitz SD, Willis J, Lynch H, et al. E-cadherin germline mutations define an inherited cancer syndrome dominated by diffuse gastric cancer. Hum Mutat. (1999) 14:249–55. doi: 10.1002/(ISSN)1098-1004
27. Calvete O, Reyes J, Zuniga S, Paumard-Hernandez B, Fernandez V, Bujanda L, et al. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumour. Hum Mol Genet. (2015) 24:2914–22. doi: 10.1093/hmg/ddv054
28. Torres-Costa M, Flores B, Torregrosa N, González M, Ríos A, Febrero B, et al. Malignant prediction in paragangliomas: analysis for clinical risk factors. Langenbecks Arch Surg. (2021) 406:2441–8. doi: 10.1007/s00423-021-02222-9
29. Yew A, Kim W, Chang S, Yang I. Delayed intracranial and bony metastasis of paraganglioma. Surg Neurol Int. (2011) 2:62. doi: 10.4103/2152-7806.81064
30. Kountouras J, Zavos C, Chatzopoulos D, Katsinelos P. New aspects of helicobacter pylori infection involvement in gastric oncogenesis. J Surg Res. (2008) 146:149–58. doi: 10.1016/j.jss.2007.06.011
31. Sei Y, Feng J, Zhao X, Dagur P, McCoy JP, Merchant JL, et al. Tissue- and cell-specific properties of enterochromaffin cells affect the fate of tumorigenesis toward nonendocrine adenocarcinoma of the small intestine. Am J Physiol Gastrointest Liver Physiol. (2023) 324:G177–g89. doi: 10.1152/ajpgi.00205.2022
32. Elias E, Ardalan A, Lindberg M, Reinsbach SE, Muth A, Nilsson O, et al. Independent somatic evolution underlies clustered neuroendocrine tumors in the human small intestine. Nat Commun. (2021) 12:6367. doi: 10.1038/s41467-021-26581-5
33. Waldum H, Mjønes P. Clear cell renal cancer, a tumour with neuroendocrine features originating from the erythropoietin-producing cell. In: Chen J, editor. In Renal Cell Carcinoma - Recent Advances, New Perspectives and Applications [Working Title]. London, England: IntechOpen (2022).
34. Mjønes P, Sagatun L, Nordrum IS, Waldum HL. Neuron-specific enolase as an immunohistochemical marker is better than its reputation. J Histochem Cytochem. (2017) 65:687–703. doi: 10.1369/0022155417733676
35. Semenza GL. Regulation of erythropoiesis by the hypoxia-inducible factor pathway: effects of genetic and pharmacological perturbations. Annu Rev Med. (2023) 74:307–19. doi: 10.1146/annurev-med-042921-102602
36. Suárez C, Vieito M, Valdivia A, González M, Carles J. Selective HIF2A inhibitors in the management of clear cell renal cancer and Von Hippel-Lindau-disease-associated tumors. Med Sci (Basel). (2023) 11:46. doi: 10.3390/medsci11030046
37. Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. (1994) 7:85–90. doi: 10.1038/ng0594-85
38. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. (2010) 463:360–3. doi: 10.1038/nature08672
39. Rekhtman N. Lung neuroendocrine neoplasms: recent progress and persistent challenges. Mod Pathol. (2022) 35:36–50. doi: 10.1038/s41379-021-00943-2
40. Noguchi M, Furukawa KT, Morimoto M. Pulmonary neuroendocrine cells: physiology, tissue homeostasis and disease. Dis Model Mech. (2020) 13. doi: 10.1242/dmm.046920
41. Lauweryns JM, Peuskens JC. Neuro-epithelial bodies (Neuroreceptor or secretory organs)? in human infant bronchial and bronchiolar epithelium. Anat Rec. (1972) 172:471–81. doi: 10.1002/ar.1091720301
42. Cutz E, Jackson A. Neuroepithelial bodies as airway oxygen sensors. Respir Physiol. (1999) 115:201–14. doi: 10.1016/s0034-5687(99)00018-3
43. Miskovic J, Brekalo Z, Vukojevic K, Miskovic HR, Kraljevic D, Todorovic J, et al. Co-expression of TTF-1 and neuroendocrine markers in the human fetal lung and pulmonary neuroendocrine tumors. Acta Histochem. (2015) 117:451–9. doi: 10.1016/j.acthis.2015.02.002
44. Rosen Y. Pulmonary neuroendocrine carcinomas in situ: do they exist? Histopathology. (2022) 80:627–34. doi: 10.1111/his.14596
45. Jing N, Zhang K, Chen X, Liu K, Wang J, Xiao L, et al. ADORA2A-driven proline synthesis triggers epigenetic reprogramming in neuroendocrine prostate and lung cancers. J Clin Invest. (2023) 133. doi: 10.1172/jci168670
46. Guo L, Kong D, Liu J, Zhan L, Luo L, Zheng W, et al. Breast cancer heterogeneity and its implication in personalized precision therapy. Exp Hematol Oncol. (2023) 12:3. doi: 10.1186/s40164-022-00363-1
47. Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. (2015) 163:1011–25. doi: 10.1016/j.cell.2015.10.025
48. Zhou J, Chen Q, Zou Y, Chen H, Qi L, Chen Y. Stem cells and cellular origins of breast cancer: updates in the rationale, controversies, and therapeutic implications. Front Oncol. (2019) 9:820. doi: 10.3389/fonc.2019.00820
49. Xin L. Cells of origin for prostate cancer. Adv Exp Med Biol. (2019) 1210:67–86. doi: 10.1007/978-3-030-32656-2_4
50. Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci U.S.A. (2012) 109:2772–7. doi: 10.1073/pnas.1017626108
51. Silva SN, Gomes BC, André S, Félix A, Rodrigues AS, Rueff J. Male and female breast cancer: the two faces of the same genetic susceptibility coin. Breast Cancer Res Treat. (2021) 188:295–305. doi: 10.1007/s10549-021-06159-x
52. Derderian S, Benidir T, Scarlata E, Altaylouni T, Hamel L, Zouanat FZ, et al. Cell-by-cell quantification of the androgen receptor in benign and Malignant prostate leads to a better understanding of changes linked to cancer initiation and progression. J Pathol Clin Res. (2023) 9:285–301. doi: 10.1002/cjp2.319
53. Waldum HL, Sordal OF, Mjones PG. The enterochromaffin-like [ECL] cell-central in gastric physiology and pathology. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20102444
54. Zhao J, Wang J, Wang H, Lai M. Reg proteins and their roles in inflammation and cancer of the human digestive system. Adv Clin Chem. (2013) 61:153–73. doi: 10.1016/b978-0-12-407680-8.00006-3
55. Kinoshita Y, Ishihara S. Mechanism of gastric mucosal proliferation induced by gastrin. J Gastroenterol Hepatol. (2000) 15 Suppl:D7–11. doi: 10.1046/j.1440-1746.2000.02145.x
56. Jung Y, Kim JK, Lee E, Cackowski FC, Decker AM, Krebsbach PH, et al. CXCL12γ induces human prostate and mammary gland development. Prostate. (2020) 80:1145–56. doi: 10.1002/pros.24043
57. Huang J, Liu D, Li J, Xu J, Dong S, Zhang HA. 12-gene panel in estimating hormone-treatment responses of castration-resistant prostate cancer patients generated using a combined analysis of bulk and single-cell sequencing data. Ann Med. (2023) 55:2260387. doi: 10.1080/07853890.2023.2260387
58. Angelsen A, Syversen U, Haugen OA, Stridsberg M, Mjølnerød OK, Waldum HL. Neuroendocrine differentiation in carcinomas of the prostate: do neuroendocrine serum markers reflect immunohistochemical findings? Prostate. (1997) 30:1–6. doi: 10.1002/(SICI)1097-0045(19970101)30:1<1::AID-PROS1>3.3.CO;2-B
59. Sigorski D, Wesołowski W, Gruszecka A, Gulczyński J, Zieliński P, Misiukiewicz S, et al. Neuropeptide Y and its receptors in prostate cancer: associations with cancer invasiveness and perineural spread. J Cancer Res Clin Oncol. (2023) 149:5803–22. doi: 10.1007/s00432-022-04540-x
60. Grönberg H, Bergh A, Damber JE, Emanuelsson M. Cancer risk in families with hereditary prostate carcinoma. Cancer. (2000) 89:1315–21. doi: 10.1002/(ISSN)1097-0142
61. Butler W, Huang J. Neuroendocrine cells of the prostate: Histology, biological functions, and molecular mechanisms. Precis Clin Med. (2021) 4:25–34. doi: 10.1093/pcmedi/pbab003
62. Nesland JM, Lunde S, Holm R, Johannessen JV. Electron microscopy and immunostaining of the normal breast and its benign lesions. A search for neuroendocrine cells. Histol Histopathol. (1987) 2:73–7.
63. Rakha E, Tan PH. Head to head: do neuroendocrine tumours in the breast truly exist? Histopathology. (2022) 81:2–14. doi: 10.1111/his.14627
64. Huang J, Wu C, di Sant’Agnese PA, Yao JL, Cheng L, Na Y. Function and molecular mechanisms of neuroendocrine cells in prostate cancer. Anal Quant Cytol Histol. (2007) 29:128–38.
65. Barrett P, Hobbs RC, Coates PJ, Risdon RA, Wright NA, Hall PA. Endocrine cells of the human gastrointestinal tract have no proliferative capacity. Histochem J. (1995) 27:482–6. doi: 10.1007/BF02388805
66. Waldum HL, Brenna E. Non-proliferative capacity of endocrine cells of the human gastro-intestinal tract. Histochem J. (1996) 28:397–8. doi: 10.1007/bf02331403
67. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. (2016) 22:298–305. doi: 10.1038/nm.4045
68. Bussolati G, Gugliotta P, Sapino A, Eusebi V, Lloyd RV. Chromogranin-reactive endocrine cells in argyrophilic carcinomas (“carcinoids”) and normal tissue of the breast. Am J Pathol. (1985) 120:186–92.
69. Bofin AM, Qvigstad G, Waldum C, Waldum HL. Neuroendocrine differentiation in carcinoma of the breast. Tyramide signal amplification discloses chromogranin A-positive tumour cells in more breast tumours than previously realized. Apmis. (2002) 110:658–64. doi: 10.1034/j.1600-0463.2002.1100910.x
70. Annaratone L, Medico E, Rangel N, Castellano I, Marchiò C, Sapino A, et al. Search for neuro-endocrine markers (chromogranin A, synaptophysin and VGF) in breast cancers. An integrated approach using immunohistochemistry and gene expression profiling. Endocr Pathol. (2014) 25:219–28. doi: 10.1007/s12022-013-9277-4
71. Shirley SE, Escoffery CT, Titus IP, Williams EE, West AB. Clear cell carcinoma of the breast with immunohistochemical evidence of divergent differentiation. Ann Diagn Pathol. (2002) 6:250–6. doi: 10.1053/adpa.2002.35399
72. Ohashi R, Sakatani T, Matsubara M, Watarai Y, Yanagihara K, Yamashita K, et al. Mucinous carcinoma of the breast: a comparative study on cytohistological findings associated with neuroendocrine differentiation. Cytopathology. (2016) 27:193–200. doi: 10.1111/cyt.12298
73. Jiang Y, Gao Z, Wang Y, Xu W. Primary breast small cell carcinoma with merkel cell features: A case report and literature review. J Breast Cancer. (2023) 26:302–7. doi: 10.4048/jbc.2023.26.e24
74. Mohamed GA, Mahmood S, Ognjenovic NB, Lee MK, Wilkins OM, Christensen BC, et al. Lineage plasticity enables low-ER luminal tumors to evolve and gain basal-like traits. Breast Cancer Res. (2023) 25:23. doi: 10.1186/s13058-023-01621-8
75. Shen H, Yu Z, Zhao J, Li XZ, Pan WS. Early diagnosis and treatment of gastrointestinal neuroendocrine tumors. Oncol Lett. (2016) 12:3385–92. doi: 10.3892/ol.2016.5062
76. Riihimäki M, Hemminki A, Sundquist K, Sundquist J, Hemminki K. The epidemiology of metastases in neuroendocrine tumors. Int J Cancer. (2016) 139:2679–86. doi: 10.1002/ijc.30400
77. Apanovich N, Matveev A, Ivanova N, Burdennyy A, Apanovich P, Pronina I, et al. Prediction of distant metastases in patients with kidney cancer based on gene expression and methylation analysis. Diagnost (Basel). (2023) 13. doi: 10.3390/diagnostics13132289
78. Castro-Pérez E, Rodríguez CI, Mikheil D, Siddique S, McCarthy A, Newton MA, et al. Melanoma progression inhibits pluripotency and differentiation of melanoma-derived iPSCs produces cells with neural-like mixed dysplastic phenotype. Stem Cell Rep. (2019) 13:177–92. doi: 10.1016/j.stemcr.2019.05.018
79. Mills JC, Sansom OJ. Reserve stem cells: Differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal. (2015) 8:re8. doi: 10.1126/scisignal.aaa7540
80. Starzinski-Powitz A, Zeitvogel A, Schreiner A, Baumann R. In search of pathogenic mechanisms in endometriosis: the challenge for molecular cell biology. Curr Mol Med. (2001) 1:655–64. doi: 10.2174/1566524013363168
81. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature. (1998) 392:402–5. doi: 10.1038/32918
82. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature. (2016) 540:588–92. doi: 10.1038/nature20609
83. Hosseini H, Obradović MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. (2016) 540:552–8. doi: 10.1038/nature20785
84. Gorin MA, Verdone JE, van der Toom E, Bivalacqua TJ, Allaf ME, Pienta KJ. Circulating tumour cells as biomarkers of prostate, bladder, and kidney cancer. Nat Rev Urol. (2017) 14:90–7. doi: 10.1038/nrurol.2016.224
85. Monticciolo DL, Hendrick RE, Helvie MA. Outcomes of breast cancer screening strategies based on cancer intervention and surveillance modeling network estimates. Radiology. (2024) 310:e232658. doi: 10.1148/radiol.232658
86. Zhao R, Ke M, Lv J, Liu S, Liu Y, Zhang J, et al. First-in-human study of Psma-targeting agent, [(18)F]Alf-P16-093: dosimetry and initial evaluation in prostate cancer patients. Eur J Nucl Med Mol Imaging. (2024). doi: 10.1007/s00259-024-06596-y
87. Sell S. On the stem cell origin of cancer. Am J Pathol. (2010) 176:2584–494. doi: 10.2353/ajpath.2010.091064
88. Doze VA, Perez DM. GPCRs in stem cell function. Prog Mol Biol Transl Sci. (2013) 115:175–216. doi: 10.1016/b978-0-12-394587-7.00005-1
89. Reuvers TGA, Kanaar R, Nonnekens J. DNA damage-inducing anticancer therapies: from global to precision damage. Cancers (Basel). (2020) 12. doi: 10.3390/cancers12082098
90. Drabløs F, Feyzi E, Aas PA, Vaagbø CB, Kavli B, Bratlie MS, et al. Alkylation damage in DNA and rna–repair mechanisms and medical significance. DNA Repair (Amst). (2004) 3:1389–407. doi: 10.1016/j.dnarep.2004.05.004
91. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. (2007) 7:573–84. doi: 10.1038/nrc2167
92. Ferguson LR, Denny WA. Genotoxicity of non-covalent interactions: DNA intercalators. Mutat Res. (2007) 623:14–23. doi: 10.1016/j.mrfmmm.2007.03.014
93. Melia E, Parsons JL. DNA damage and repair dependencies of ionising radiation modalities. Biosci Rep. (2023) 43. doi: 10.1042/bsr20222586
94. Sharma NK, Bahot A, Sekar G, Bansode M, Khunteta K, Sonar PV, et al. Understanding cancer’s defense against topoisomerase-active drugs: A comprehensive review. Cancers (Basel). (2024) 16. doi: 10.3390/cancers16040680
95. Tsesmetzis N, Paulin CBJ, Rudd SG, Herold N. Nucleobase and nucleoside analogues: resistance and re-sensitisation at the level of pharmacokinetics, pharmacodynamics and metabolism. Cancers (Basel). (2018) 10. doi: 10.3390/cancers10070240
96. Gonen N, Assaraf YG. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Update. (2012) 15:183–210. doi: 10.1016/j.drup.2012.07.002
97. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. (2010) 40:179–204. doi: 10.1016/j.molcel.2010.09.019
98. Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. (2013) 5:a012583. doi: 10.1101/cshperspect.a012583
99. Kellner V, Luke B. Molecular and physiological consequences of faulty eukaryotic ribonucleotide excision repair. EMBO J. (2020) 39:e102309. doi: 10.15252/embj.2019102309
100. Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst). (2015) 36:13–8. doi: 10.1016/j.dnarep.2015.09.003
101. Liu D, Keijzers G, Rasmussen LJ. DNA mismatch repair and its many roles in eukaryotic cells. Mutat Res Rev Mutat Res. (2017) 773:174–87. doi: 10.1016/j.mrrev.2017.07.001
102. Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. (2018) 293:10524–35. doi: 10.1074/jbc.TM118.000372
103. Rulten SL, Grundy GJ. Non-homologous end joining: common interaction sites and exchange of multiple factors in the DNA repair process. Bioessays. (2017) 39. doi: 10.1002/bies.201600209
104. Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FM. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer. (2015) 15:166–80. doi: 10.1038/nrc3891
105. Stewart DJ, Chiritescu G, Dahrouge S, Banerjee S, Tomiak EM. Chemotherapy dose–response relationships in non-small cell lung cancer and implied resistance mechanisms. Cancer Treat Rev. (2007) 33:101–37. doi: 10.1016/j.ctrv.2006.12.002
106. Frosina G. The bright and the dark sides of DNA repair in stem cells. J BioMed Biotechnol. (2010) 2010:845396. doi: 10.1155/2010/845396
107. Maynard S, Swistowska AM, Lee JW, Liu Y, Liu ST, Da Cruz AB, et al. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells. (2008) 26:2266–74. doi: 10.1634/stemcells.2007-1041
108. Roth RB, Samson LD. 3-methyladenine DNA glycosylase-deficient Aag null mice display unexpected bone marrow alkylation resistance. Cancer Res. (2002) 62:656–60.
109. Mjelle R, Hegre SA, Aas PA, Slupphaug G, Drabløs F, Saetrom P, et al. Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair (Amst). (2015) 30:53–67. doi: 10.1016/j.dnarep.2015.03.007
110. Mladenov E, Mladenova V, Stuschke M, Iliakis G. New facets of DNA double strand break repair: radiation dose as key determinant of Hr versus C-Nhej engagement. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241914956
111. Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. (2010) 7:174–85. doi: 10.1016/j.stem.2010.06.014
112. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. (1994) 367:645–8. doi: 10.1038/367645a0
113. Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. (2012) 44:2144–51. doi: 10.1016/j.biocel.2012.08.022
114. Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer. (2020) 20:398–411. doi: 10.1038/s41568-020-0263-0
115. Kleffel S, Schatton T. Tumor dormancy and cancer stem cells: two sides of the same coin? Adv Exp Med Biol. (2013) 734:145–79. doi: 10.1007/978-1-4614-1445-2_8
116. Hedley BD, Chambers AF. Tumor dormancy and metastasis. Adv Cancer Res. (2009) 102:67–101. doi: 10.1016/S0065-230X(09)02003-X
117. Martínez-Jiménez F, Movasati A, Brunner SR, Nguyen L, Priestley P, Cuppen E, et al. Pan-cancer whole-genome comparison of primary and metastatic solid tumours. Nature. (2023) 618:333–41. doi: 10.1038/s41586-023-06054-z
118. Coleman RE, Rubens RD. The clinical course of bone metastases from breast cancer. Br J Cancer. (1987) 55:61–6. doi: 10.1038/bjc.1987.13
119. Akhtari M, Mansuri J, Newman KA, Guise TM, Seth P. Biology of breast cancer bone metastasis. Cancer Biol Ther. (2008) 7:3–9. doi: 10.4161/cbt.7.1.5163
120. Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem. (2004) 91:718–29. doi: 10.1002/jcb.10662
121. Archer Goode E, Wang N, Munkley J. Prostate cancer bone metastases biology and clinical management (Review). Oncol Lett. (2023) 25:163. doi: 10.3892/ol.2023.13749
122. Bianchi M, Sun M, Jeldres C, Shariat SF, Trinh QD, Briganti A, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol. (2012) 23:973–80. doi: 10.1093/annonc/mdr362
123. Santini D, Procopio G, Porta C, Ibrahim T, Barni S, Mazzara C, et al. Natural history of Malignant bone disease in renal cancer: final results of an Italian bone metastasis survey. PloS One. (2013) 8:e83026. doi: 10.1371/journal.pone.0083026
124. Matsugaki A, Kimura Y, Watanabe R, Nakamura F, Takehana R, Nakano T. Impaired alignment of bone matrix microstructure associated with disorganized osteoblast arrangement in Malignant melanoma metastasis. Biomolecules. (2021) 11. doi: 10.3390/biom11020131
125. Altieri B, Di Dato C, Martini C, Sciammarella C, Di Sarno A, Colao A, et al. Bone metastases in neuroendocrine neoplasms: from pathogenesis to clinical management. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11091332
126. Dai R, Liu M, Xiang X, Xi Z, Xu H. Osteoblasts and osteoclasts: an important switch of tumour cell dormancy during bone metastasis. J Exp Clin Cancer Res. (2022) 41:316. doi: 10.1186/s13046-022-02520-0
127. Liu R, Su S, Xing J, Liu K, Zhao Y, Stangis M, et al. Tumor removal limits prostate cancer cell dissemination in bone and osteoblasts induce cancer cell dormancy through focal adhesion kinase. J Exp Clin Cancer Res. (2023) 42:264. doi: 10.1186/s13046-023-02849-0
Keywords: bone metastases, cancer cell of origin, cell adherence, differentiated versus stem cell, direct versus indirect stimulation, dormancy, late metastases
Citation: Waldum H and Slupphaug G (2024) Correctly identifying the cells of origin is essential for tailoring treatment and understanding the emergence of cancer stem cells and late metastases. Front. Oncol. 14:1369907. doi: 10.3389/fonc.2024.1369907
Received: 26 January 2024; Accepted: 25 March 2024;
Published: 10 April 2024.
Edited by:
Daitoku Sakamuro, Augusta University, United StatesReviewed by:
Luis Fernando Méndez López, Autonomous University of Nuevo León, MexicoCopyright © 2024 Waldum and Slupphaug. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Helge Waldum, helge.waldum@ntnu.no