 Jaeyong Jung
Jaeyong Jung Samantha Gokhale
Samantha Gokhale Ping Xie
Ping Xie- 1Department of Cell Biology and Neuroscience, Rutgers University, Piscataway, NJ, United States
- 2Graduate Program in Cellular and Molecular Pharmacology, Rutgers University, Piscataway, NJ, United States
- 3Rutgers Cancer Institute of New Jersey, New Brunswick, NJ, United States
Mitochondria, the organelle critical for cell survival and metabolism, are exploited by cancer cells and provide an important therapeutic target in cancers. Mitochondria dynamically undergo fission and fusion to maintain their diverse functions. Proteins controlling mitochondrial fission and fusion have been recognized as essential regulators of mitochondrial functions, mitochondrial quality control, and cell survival. In a recent proteomic study, we identified the key mitochondrial fission factor, MFF, as a new interacting protein of TRAF3, a known tumor suppressor of multiple myeloma and other B cell malignancies. This interaction recruits the majority of cytoplasmic TRAF3 to mitochondria, allowing TRAF3 to regulate mitochondrial morphology, mitochondrial functions, and mitochondria-dependent apoptosis in resting B lymphocytes. Interestingly, recent transcriptomic, metabolic and lipidomic studies have revealed that TRAF3 also vitally regulates multiple metabolic pathways in B cells, including phospholipid metabolism, glucose metabolism, and ribonucleotide metabolism. Thus, TRAF3 emerges as a novel regulator of mitochondrial physiology and metabolic pathways in B lymphocytes and B cell malignancies. Here we review current knowledge in this area and discuss relevant clinical implications.
Introduction
Tumor necrosis factor receptor-associated factor 3 (TRAF3), a cytoplasmic adaptor protein of the TRAF family, regulates the signal transduction pathways of a wide variety of immune receptors, including the TNF-R superfamily, lymphocyte antigen receptors, pattern recognition receptors (PRRs), and cytokine receptors (1–4). Through its scaffolding function and E3 ubiquitin ligase activity, TRAF3 differentially modulates a plethora of downstream signal transduction cascades, such as the activation of nuclear factor-κBs (NF-κB1 and NF-κB2), mitogen-activated protein kinases (MAPKs), and interferon-regulatory factors (IRFs), among others (1–4). Such regulatory function of TRAF3 exhibits an interesting dependence on the specific receptor engaged as well as the cellular context (1–4).
TRAF3 is ubiquitously expressed in various immune and non-immune cell types of mammals (1, 2, 5). Mice made genetically deficient in Traf3 exhibit global defects and die by 10 days after birth (6). Subsequent studies of conditional Traf3 knockout and cell type-specific Traf3 transgenic mice revealed that TRAF3 plays critical and diverse roles in adaptive immunity and innate immunity as well as the homeostasis and stress responses of many tissues (1–4). Consequently, aberrant function of TRAF3 leads to a broad array of serious diseases in mouse models, including cancers, autoimmune diseases, inflammatory diseases, and infectious diseases (1–4, 7, 8).
Reinforcing the evidence obtained from mouse models, somatic mutations (such as homozygous deletions and inactivating mutations) of the TRAF3 gene in humans were first identified in multiple myeloma (MM) and then other B cell malignancies (1, 3, 9–11). Somatic alterations of the TRAF3 gene are also present in other human cancers (3). The first germline mutation of TRAF3, an autosomal loss-of-expression mutation (R338W), was initially reported in a patient with a history of herpes simplex virus-1 (HSV-1) encephalitis (12) and also detected in another patient with recurrent Mycobacterium abscessus infection (13). Interestingly, heterozygous germline mutations in TRAF3 (premature stop codon mutations) were recently identified in 9 patients from five unrelated families, causing an immune dysregulation syndrome characterized by recurrent bacterial infection, autoimmunity, systemic inflammation, B cell lymphoproliferation, and hypergammaglobulinemia (14). Furthermore, genome-wide association studies (GWAS) and targeted analyses demonstrated that common genetic variants of TRAF3, which reduce TRAF3 expression, are associated with an increased risk of B cell malignancies, systemic lupus erythematosus, hypergammaglobulinemia, and recurrent bacterial infection in a wider population (14). Taken together, the above evidence highlights the importance of TRAF3 in the immune system, and particularly in B lymphocytes.
TRAF3 in B cell biology and B cell malignancies
In normal B lymphocytes, TRAF3 is a critical regulator of mature B cell survival, B cell activation, and plasma cell differentiation and maturation (15–23). These important physiological functions of TRAF3 in B lymphocytes are achieved through its negative regulatory roles in the signal transduction pathways of multiple receptors that are central to B cell biology, including B cell antigen receptor (BCR), the co-stimulatory receptor CD40, receptors of the principle B cell survival factor BAFF, Toll-like receptors (TLRs), and IL-6 receptor (15–23). Specific deletion of the Traf3 gene from B lymphocytes in mice leads to prolonged B cell survival, constitutive NF-κB2 activation, augmented BCR signaling, elevated T-dependent and T-independent antibody responses, enhanced TLR responses, and increased IL-6-induced plasma cell differentiation, which culminate in autoimmunity and B lymphomagenesis (15–24). Interestingly, transgenic overexpression of TRAF3 in B cells also renders B cells exhibiting enhanced reactions (also termed hyperreaction) to antigens and TLRs, resulting in autoimmunity and chronic inflammation (25). Thus, an appropriate level of TRAF3 proteins is required for normal B cell survival and functionality.
Consistent with the importance of TRAF3 in B cell biology, deletions and inactivating mutations of the TRAF3 gene were first reported in human multiple myeloma (MM) (9, 10), a malignancy derived from plasma cells. According to the study by Keats et al., the deletion frequency of TRAF3 is 15.8% in 158 analyzed MM patients (9). The high frequency of TRAF3 deletions and inactivating mutations was verified in a larger cohort of patient study by Walker et al., which reported 16.7% genetic alterations of TRAF3 in 463 examined MM patients, including 13% of deep deletions, 3.26% of mutations, and 0.43% of truncations of the TRAF3 gene (26). Beyond genetic alterations, the Epstein-Barr virus (EBV)-encoded oncoprotein latent membrane protein 1 (LMP1) sequesters TRAF3 in B lymphocytes and can render EBV-infected wild type B cells functionally TRAF3-deficient, which may also contribute to the pathological mechanisms of B cell oncogenesis (27–29).
Subsequent studies revealed that deletions and inactivating mutations of TRAF3 are frequently detected in many other types of mature B cell malignancies, including diffuse large B-cell lymphoma (DLBCL), splenic marginal zone lymphoma (MZL), B-cell chronic lymphocytic leukemia (B-CLL), mantle cell lymphoma (MCL), Waldenström’s macroglobulinemia, and Hodgkin lymphoma (HL) (1, 3, 9–11, 30). This is corroborated by frequent TRAF3 mutations detected in canine non-Hodgkin lymphomas (NHL) (31–34). Similar to Traf3-/- mouse B cells, human B cells with germline TRAF3 mutations that reduce TRAF3 expression also exhibit constitutive NF-κB2 activation and enhanced responses to BCR, BAFF, TLR9, and IL-6 signaling, including increased proliferation and plasma cell formation associated with elevated activation of NF-κB1, ERK, AKT, and STAT3 (14). Reconstitution of TRAF3 expression in TRAF3-deficient human MM cells induces apoptosis, demonstrating a tumor suppressive role of TRAF3 in B cell malignancies (9, 35). Of clinical significance, it is being appreciated that TRAF3 alterations contribute to patient resistance to various therapies of B cell malignancies, including BTK inhibitors, PI3K inhibitors, proteasome inhibitors, HDAC inhibitors, cIAP antagonists, immunotherapy (e.g., rituximab), and chemotherapy (e.g., R-CHOP) (9, 19, 20, 36–39). In this context, a deeper understanding of the molecular mechanisms underlying TRAF3-mediated regulation of B lymphocytes is required to inform better treatment strategies for patients with B cell malignancies involving TRAF3 deletions and other relevant alterations.
TRAF3 is a novel regulator of mitochondrial physiology in B cells
While analyzing the subcellular distribution of TRAF3 in resting B lymphocytes, Liu et al. found that the majority of cytoplasmic TRAF3 is localized at the mitochondria in the absence of stimulation (40). They also noticed that BAFF-induced recruitment and subsequent degradation of TRAF3 mainly affect the proteins localized at the mitochondria in B cells (40). Given the central role of mitochondria in regulating apoptosis (41–43), Liu et al. pursued how TRAF3 is localized at mitochondria and what it does there.
Since TRAF3 does not contain any mitochondrial targeting motif or transmembrane domain, Liu et al. tested if TRAF3 interacts with mitochondrial outer membrane (MOM) proteins by employing a proteomic approach, including biochemical fractionation to isolate mitochondria and affinity purification to pull down mitochondrial TRAF3-interacting proteins followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based sequencing (40). To facilitate affinity purification, they transduced TRAF3-deficient human MM cells with lentiviruses expressing tagged TRAF3 (40). Liu et al. identified the MOM protein MFF (44) as a TRAF3-interacting protein and further verified the TRAF3-MFF interaction by co-immunoprecipitation and GST pull-down assays (40). MFF contains a coiled-coil domain that is known to mediate the interactions of other proteins with TRAFs (45–47). The domain structural analyses revealed that the TRAF-C domain of TRAF3 is required for binding to MFF, which was verified by specific pull-down of in vitro translated MFF by GST-TRAF3 but not by GST-TRAF3ΔTRAF-C (40). Furthermore, TRAF3 inhibits the phosphorylation and ubiquitination of MFF in resting B cells and co-transfected HEK293T cells, whereas overexpression of MFF leads to decreased ubiquitination of TRAF3 (40). Thus, MFF is a novel TRAF3-interacting protein that recruits TRAF3 to the MOM in B cells in the absence of receptor engagement.
The principal function of MFF is to promote mitochondrial fission, thereby contributing to the regulation of mitochondrial number, morphology, function, and quality (44, 48, 49). Consistent with the detected TRAF3-MFF interaction, increased protein levels of mitochondrial TRAF3 are associated with decreased mitochondrial number, altered mitochondrial morphology, reduced mitochondrial respiration, and increased mitochondrial ROS production and membrane permeabilization, which lead to caspase 9-dependent apoptosis in resting wild type B cells (40). Liu et al. and Mambetsariev et al. found that deletion of TRAF3 has the opposite effects on mitochondrial morphology, respiration, ROS production, and mitochondria-dependent apoptosis in resting B cells (40, 50). Interestingly, lentivirus-mediated overexpression of MFF restores mitochondria-dependent apoptosis in TRAF3-deficient human MM cells (40). Corroborating these findings, Rae et al. recently reported that B lymphoblastoid cell lines (BLCLs) derived from patients with germline premature stop codon mutations of TRAF3 display an increased oxygen consumption rate, indicative of elevated mitochondrial respiration, which is accompanied by altered mitochondrial morphology, up-regulated COX II expression and enhanced cytochrome c oxidase activity (14). Reconstitution of TRAF3 expression in patient-derived BLCLs inhibits mitochondrial respiration and the expression of the mitochondrial co-transcriptional regulator PGC1α (14). Moreover, transgenic overexpression of TRAF3 in B cells also promotes NHL development in mice when the anti-apoptotic protein BCL-2 is simultaneously overexpressed (51), suggesting a need for BCL-2-mediated protection of mitochondria in TRAF3-overexpressing B cells. Therefore, TRAF3 is a novel regulator of mitochondrial physiology in normal and malignant B cells.
It is noteworthy that BAFF inhibits mitochondrial ROS production and prevents mitochondria-dependent apoptosis in wild type (WT) but not Traf3-/- B cells (40). Upon BAFF stimulation, BAFF-Rs recruit TRAF3 from the MOM to plasma membrane, which would lead to disruption of the TRAF3-MFF interaction and therefore modulate mitochondrial functions in WT B cells (40). Interestingly, the TRAF3-MFF interaction appears to result in decreased phosphorylation and ubiquitination of MFF as well as decreased ubiquitination of TRAF3 (40). Phosphorylation of MFF has been shown to increase the activity of MFF in recruiting Drp1, the GTPase that executes mitochondrial fission, to the MOM, promoting mitochondrial fission (52–55). Ubiquitination of MFF may enhance clearance of damaged mitochondria via mitophagy or may induce the degradation of MFF under non-stressed conditions (56, 57). Detailed mechanisms of how TRAF3 inhibits the phosphorylation and ubiquitination of MFF in resting B cells remain unclear. TRAF3 is known as an E3 ubiquitin ligase. Other E3 ubiquitin ligases that can interact with MFF include Parkin and MARCH5 (56–58). However, only Parkin has been shown to directly catalyze the ubiquitination of MFF (56, 57). Deubiquitinating enzymes of MFF have not been reported yet. It is possible that TRAF3 may catalyze the ubiquitination of Parkin or relevant deubiquitinating enzymes, kinases or phosphatases to indirectly inhibit the ubiquitination and phosphorylation of MFF in resting B cells. Alternatively, the TRAF3-MFF interaction may interfere with the accessibility of MFF by Parkin and kinases or facilitate the recruitment of relevant deubiquitinating enzymes or phosphatases, leading to reduced ubiquitination and phosphorylation of MFF. Such detailed mechanisms await further investigation.
An interesting open question is whether TRAF2, another member of the TRAF family that has overlapping functions with TRAF3 in B cells, can also interact with MFF. The TRAF3-TRAF2 heterotypic interaction is known to bridge the formation of the cIAP1/2-TRAF2-TRAF3-NIK complex in B cells (1). The TRAF3-TRAF2 interaction minimally involves the TRAF-C domain of TRAF3 and the TRAF-N domain and zinc fingers 4 and 5 of TRAF2 (59). It remains unknown whether the TRAF3-MFF interaction interferes with the TRAF3-TRAF2 interaction when TRAF3 proteins are limiting in B cells. Following viral infection, both TRAF3 and TRAF2 are recruited to the mitochondrial antiviral signaling complexes by MAVS (60–63), while in response to ER stress, TRAF2 is translocated to ER via interacting with the ER stress sensor IRE1α (64, 65). In addition, cIAP1/2 also binds to caspases and upon mitochondrial membrane permeabilization, cIAP1/2 binds to Smac that is released from mitochondrial intermembrane space to cytosol (66, 67). However, it is unclear whether and how the TRAF3-MFF, TRAF3-TRAF2, and TRAF2-cIAP1/2 interactions are affected by viral infection, ER stress, and mitochondrial membrane permeabilization in B cells. These unanswered questions await further investigation.
TRAF3 regulates specific metabolic pathways in resting B cells
The metabolic mechanisms underlying TRAF3-mediated regulation of B cells have just begun to be unraveled. To understand the metabolic basis of TRAF3-mediated regulation of B cell survival, we recently exploited multiple “omics” approaches, including metabolomics, lipidomics, and transcriptomics (35). Integrated analyses of these “omics” datasets revealed that TRAF3 regulates specific metabolic pathways in resting B cells, including phospholipid, glucose, and ribonucleotide metabolism (35).
We found that a variety of metabolites, lipids and enzymes regulated by TRAF3 in B cells are clustered in the interconnected phosphatidylcholine (PC) and phosphatidylethanolamine (PE) metabolic pathways (Figure 1A) (35). Enzymes that are regulated by TRAF3 and may contribute to the altered PC and PE metabolism include choline kinase α (Chkα), lysophosphatdiylcholine acyltransferease 1 (Lpcat1), glycerophosphodiester phosphodiesterase3 (Gdpd3), diacylglycerol kinase α (Dgkα), and fatty acid amide hydrolase (Faah), etc. (35, 68). Using stable isotope labeling, we demonstrated that Chkα-driven de novo biosynthesis of PC is remarkably elevated in Traf3-/- mouse B cells and decreased in TRAF3-reconstituted human MM cells containing biallelic TRAF3 deletions (35). Inhibition of Chkα by RSM932A (also named TCD-717) or MN58B substantially reverses the survival phenotype of TRAF3-deficient B cells both in vitro and in vivo (35). Thus, TRAF3-regulated choline metabolism has diagnostic and therapeutic value for B cell malignancies with TRAF3 deletions or relevant alterations (35, 68).
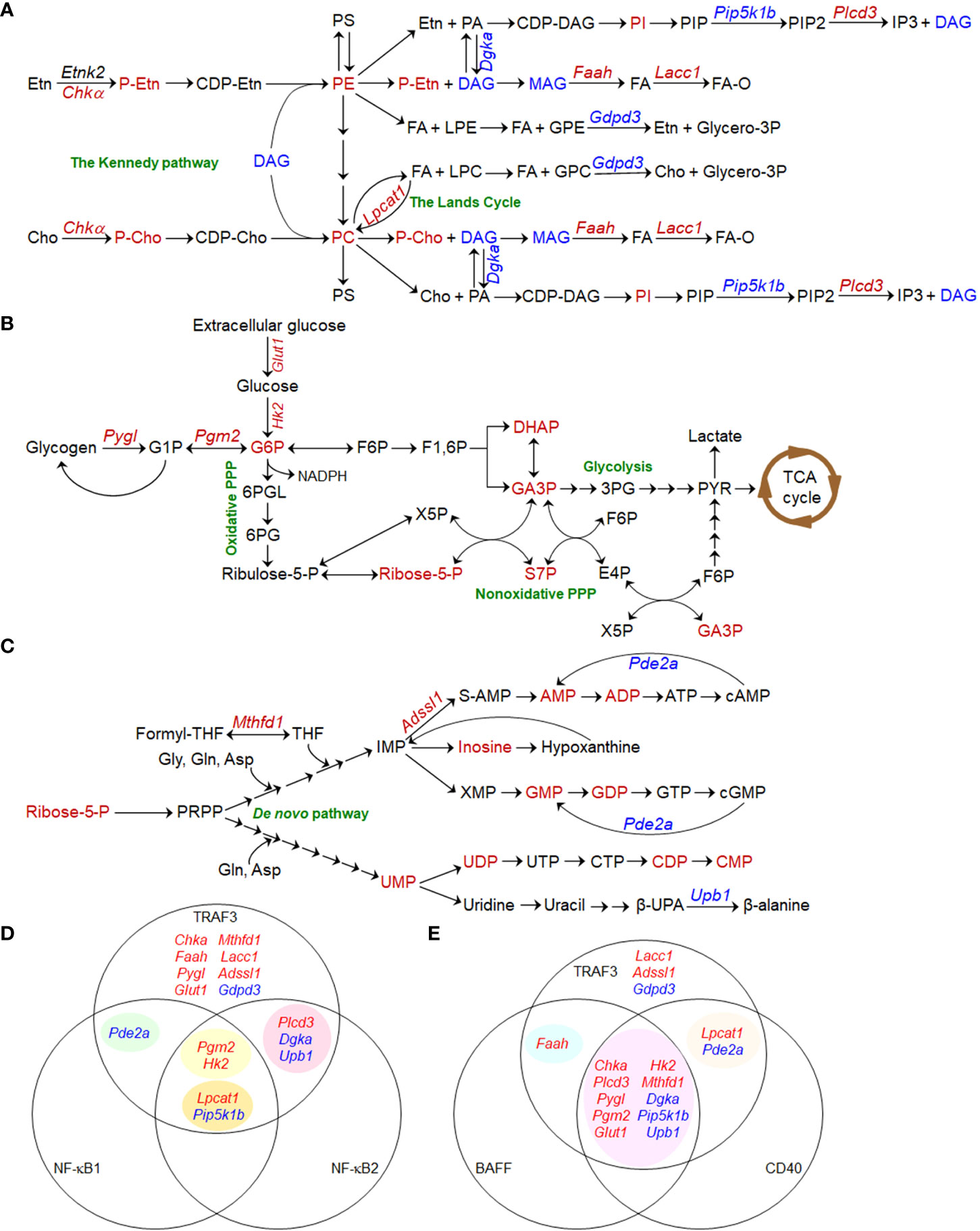
Figure 1 Metabolic pathways regulated by TRAF3 in B lymphocytes. (A–C) Pathway schematics showing TRAF3-mediated metabolic regulation in B cells. Small metabolites, lipids, transporters, and metabolic enzymes that are dysregulated in Traf3-/- B cells are shown in red (for up-regulated) or blue (for down-regulated). Enzymes and transporters are denoted in Italic font in the schematics. (A) The interconnected phosphatidylcholine (PC) and phosphatidylethanolamine (PE) metabolic pathways. (B) Glucose metabolic pathways. (C) Ribonucleotide metabolic pathways. (D, E) Venn diagram of TRAF3-regulated metabolic genes and those regulated by NF-κB1 and NF-κB2 (D) or BAFF and CD40 (E). Metabolic genes regulated by NF-κB1, NF-κB2, BAFF or CD40 are extracted from the published Gene Expression Omnibus (GEO) datasets GSE75761, GSE75762, GSE58972, and GSE62559. (D) TRAF3 regulates the expression of 8 metabolic genes via NF-κB-independent mechanisms. NF-κB2 and NF-κB1 act synergistically to promote Lpcat1 expression and suppress Pip5k1b expression. In contrast, Pgm2 and Hk2 expression are only inhibited by compound deletion of both Relb and cRel, suggesting redundant roles of NF-κB2 and NF-κB1 in up-regulating the expression of these two enzymes. (E) The majority (10/16) of the differentially expressed metabolic genes identified in Traf3-/- B cells are consistently changed in WT B cells following BAFF and CD40 stimulation. TRAF3 also regulates the expression of 3 unique enzymes, which are not affected by BAFF or CD40 stimulation.
We observed significant elevation of 5 glucose metabolic intermediates in resting Traf3-/- B cells, including glucose-6-phosphate (G6P), the convergence point of the glycolytic and pentose phosphate pathways (PPP) (69), and 4 metabolites of nonoxidative PPP (35). Our transcriptomic analysis identified up-regulation of two key enzymes in Traf3-/- B cells: phosphoglucomutase 2 (Pgm2) and glycogen phosphorylase L (Pygl) that is responsible for glycogen breakdown (70, 71). Mambetsariev et al. reported that glucose transporter 1 (Glut1) and hexokinase II (HKII) are also up-regulated in Traf3-/- B cells and that these cells exhibit increased glucose uptake (50). Inhibition of glucose metabolism by the Glut1 inhibitor STF-31 or the glycolysis inhibitor 2-deoxyglucose (2-DG) suppresses B cell survival, while glucose supplementation in serum-free medium is required for long-term survival of Traf3-/- B cells in culture (50). These findings are strengthened by the evidence that B cell-specific deletion of Glut1 leads to substantially decreased numbers of peripheral B cells in mice (72) and that Glut1 expression is necessary to maintain elevated glucose metabolism and to promote cell survival of human MM and B cell acute lymphoblastic leukemia (B-ALL) (73, 74). Therefore, TRAF3 can regulate both glycogen breakdown and glucose uptake to modulate glucose metabolism (Figure 1B), which also affects B cell survival.
In line with elevation of ribose-5-phosphate (Ribose-5-P), a metabolite generated by nonoxidative PPP that serves as the molecular backbone of ribonucleotide biosynthesis (69, 75–77), we detected significantly increased levels of 9 ribonucleotides in resting Traf3-/- B cells (Figure 1C) (35). We also identified up-regulation of two enzymes responsible for ribonucleotide biosynthesis (Mthfd1 and Adssl1) as well as down-regulation of two enzymes involved in ribonucleotide catabolism (Upb1 and Pde2a) in Traf3-/- B cells (35). However, we did not observe significant changes in ribonucleotide triphosphates (35), probably because they are consumed to support elevated transcription and other reactions required for the prolonged B cell survival. MTHFD1, an enzyme crucial for de novo purine biosynthesis, is up-regulated in human MM, NHL, and HL (Oncomine) (78–81). Interestingly, a common polymorphism of MTHFD1 R653Q (MTHFD1 G1958A) in the synthetase domain impairs purine synthesis and the corresponding AA genotype is associated with a decreased risk of human B-ALL and NHL (82, 83), indicating a role of this enzyme in B cell oncogenesis. Taken together, elevated PC and PE synthesis, glucose metabolism, and ribonucleotide synthesis are the metabolic basis mediating the aberrant survival of TRAF3-deficient B cells.
BAFF-R or CD40 signaling recruits TRAF3 to receptor complexes at plasma membrane rafts, inducing TRAF3 degradation, NF-κB2 activation, and B cell survival (1). TRAF3 also inhibits NF-κB1 activation induced by CD40 and BCR signaling (18, 19). We thus analyzed the Gene Expression Omnibus (GEO) datasets for relevant metabolic genes in mouse B cells genetically deficient in different subunits of NF-κB in comparison to WT B cells, including Nfkb2-/- (GSE75761 and GSE75762), Rela-/- (GSE58972), cRel-/-, and Relb-/-cRel-/- (GSE62559) B cells, in the absence or presence of stimulation with BAFF, CD40 or CD40 plus IgM (84–86). The results of our analyses revealed that 8 of the 16 TRAF3-regulated metabolic genes (35, 50) are independent of NF-κBs (Figure 1D). Down-regulation of Pde2a is dependent on NF-κB1, while regulation of 3 other genes is dependent on NF-κB2. Interestingly, NF-κB2 and NF-κB1 act synergistically to promote Lpcat1 expression and suppress Pip5k1b expression. In contrast, NF-κB2 and NF-κB1 appear to play redundant roles in up-regulating the expression of Pgm2 and Hk2, which is only inhibited by compound deficiency in both Relb and cRel (84–86). On the other hand, the majority (10/16) of the metabolic genes differentially expressed in Traf3-/- B cells are consistently changed in WT B cells following BAFF and CD40 stimulation (Figure 1E) (84–86). Faah and 2 other regulated genes (Lpcat1 and Pde2a) are only shared with either BAFF or CD40 stimulation, respectively. However, 3 additional genes (Lacc1, Adssl1, and Gdpd3) are uniquely altered by Traf3 deficiency. Thus, although similar to BAFF- or CD40-induced physiological B cell survival, aberrant survival of TRAF3-deficient B cells exhibits certain distinct features in metabolic reprogramming.
Discussion
In summary, recent proteomic and metabolic evidence reveals that TRAF3 regulates mitochondrial physiology and metabolic pathways to control B cell survival (Figure 2). TRAF3 can regulate mitochondrial morphology and function via interacting with MFF (40). Interestingly, TRAF3-mediated metabolic regulation leads to reduced levels of the phospholipids PC and PE (35), which are the most abundant phospholipids of mitochondrial membranes – comprising ∼40% and ∼30% of total mitochondrial phospholipids, respectively (87). Moderate dysregulation of PC and PE has profound effects on mitochondrial physiology and mitochondria-dependent apoptosis (88, 89). Moreover, the expression of anti-apoptotic proteins such as Mcl1 and Bcl-xL, critical regulators of mitochondrial physiology and intrinsic apoptosis (90, 91), is inhibited by TRAF3 via the downstream NIK-NF-κB2 and nuclear CREB pathways (16, 17, 92, 93). Therefore, TRAF3 can regulate mitochondrial physiology and mitochondria-dependent apoptosis in B cells via multi-layered mechanisms. In addition, TRAF3 also regulates the expression of key enzymes responsible for glucose and ribonucleotide metabolism (35, 50), which coordinately provide the metabolic basis to control B cell survival.
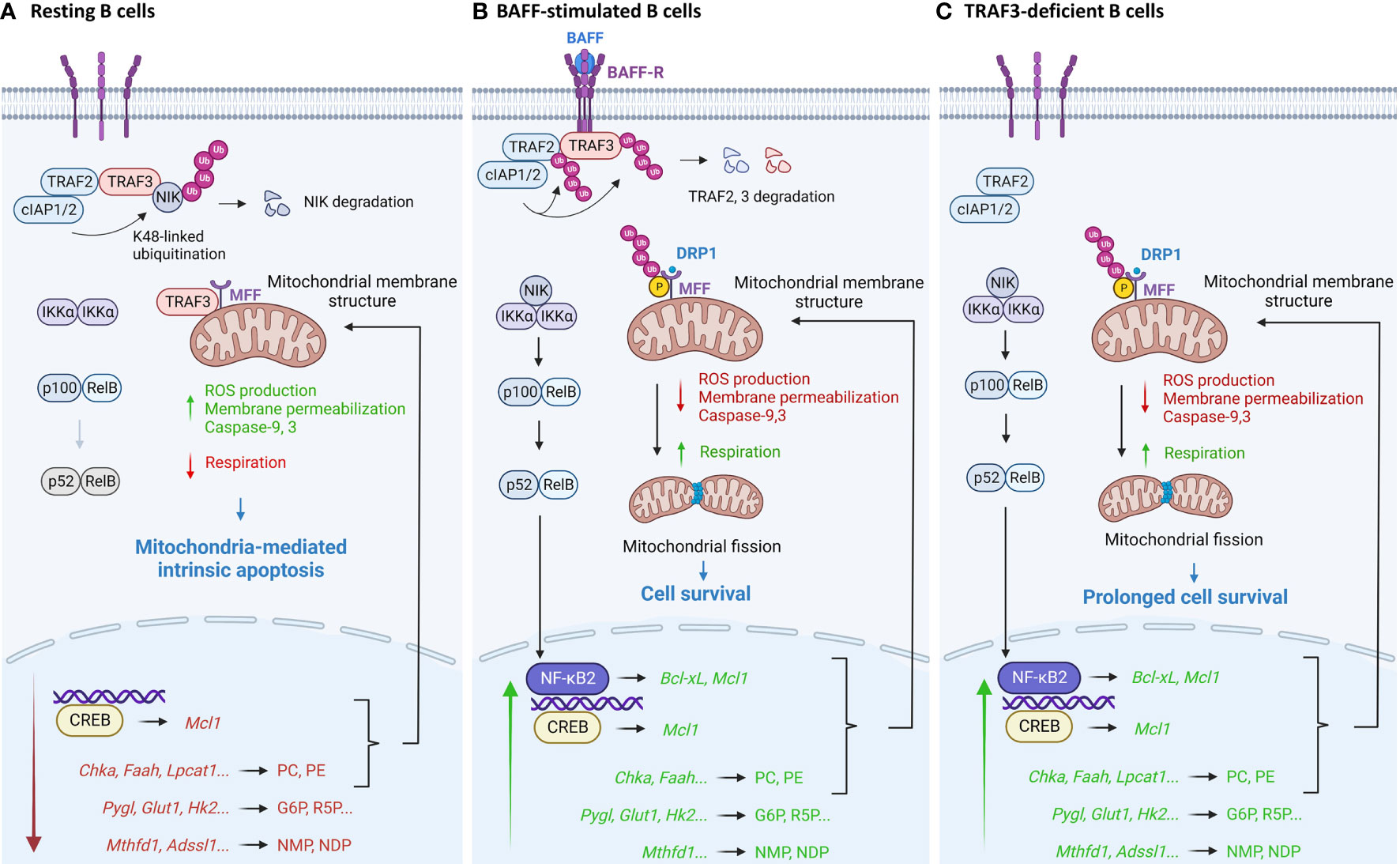
Figure 2 Pathway model depicting TRAF3-mediated regulation of mitochondria-dependent apoptosis in B lymphocytes. (A) In resting B cells, TRAF3 regulates mitochondrial physiology and metabolic pathways to promote mitochondria-dependent apoptosis. TRAF3 controls mitochondrial morphology and function via its direct interaction with MFF to inhibit MFF phosphorylation and ubiquitination, via metabolic reduction of phospholipids PC and PE to alter mitochondrial membrane structure, and via inhibition of the NIK-NF-κB2 and nuclear CREB pathways to down-regulate the expression of anti-apoptotic proteins such as Mcl1 and Bcl-xL. In addition, TRAF3-mediated repression of glucose and ribonucleotide metabolism also coordinately suppresses B cell survival. (B) BAFF stimulation recruits TRAF3 to membrane rafts and subsequently induces TRAF3 degradation, thereby reversing many TRAF3-mediated effects to promote B cell survival. Separation of TRAF3 from MFF at the MOM also results in increased phosphorylation of MFF, leading to enhanced activity of MFF in recruiting Drp1, the GTPase that executes mitochondrial fission, to the MOM, promoting mitochondrial fission. (C) Disruption of all the TRAF3-dependent mechanisms in TRAF3-deficient B cells leads to prolonged survival independent of BAFF, which eventually contributes to B cell malignant transformation. This figure was created with BioRender.com.
The above proteomic and metabolic findings have therapeutic implications, suggesting that manipulation of mitochondrial dynamics and inhibition of key metabolic enzymes or transporters offer new perspectives of treatment strategies for B cell malignancies, especially those with TRAF3 deletions or relevant alterations. Indeed, we demonstrated that overexpression of MFF restores the intrinsic apoptosis in TRAF3-deficient human MM cells, supporting a therapeutic potential of drugs that have been developed to target mitochondrial dynamics and are being tested in other disease models, including cell permeable peptidomimetics of MFF, mitochondrial division inhibitor-1 (mdivi-1), dynasore, P110, and 15-oxospiramilactone, etc. (44, 48, 94, 95). We also showed that inhibition of choline metabolism by the Chkα inhibitors TCD-717 or MN58B substantially reduces the expanded B cell compartment in B-Traf3-/- mice and induces apoptosis in TRAF3-deficient human MM cells (35), while Mambetsariev et al. demonstrated that inhibition of glucose metabolism by the Glut1 inhibitor STF-31 or the glycolysis inhibitor 2-DG dampens Traf3-/- B cell survival (50). Moreover, available information suggests that several other TRAF3-regulated metabolic enzymes (e.g., Lpcat1, Pygl, Hk2, and Mthfd1) are also targetable points in cancers (70, 82, 83, 96–104). Thus, all these mitochondria-targeting drugs and pharmacological inhibitors of metabolic enzymes/transporters can be exploited, alone or in combination with current therapies, for the treatment of human B cell malignancies to improve patient outcome.
Increasing evidence indicates that TRAF3 is a tumor suppressor not only in B cell malignancies, but also in a variety of cancers derived from macrophages, osteoblasts, and epithelial cells of different tissues. Examples include histiocytic sarcoma, osteosarcoma, head and neck cancer, bladder cancer, colorectal cancer, breast cancer, liver cancer, and lung cancer (3, 7, 8, 105–112). Loss of TRAF3 also leads to constitutive NF-κB2 activation in macrophages, osteoblasts/osteoclasts, and epithelial cells (7, 107, 111–114). Similar to that observed in B cells, TRAF3 also regulates cell survival and mitochondrial ROS production in macrophages and epithelial carcinoma cells under specific circumstances (109, 115). We detected co-immunoprecipitation of TRAF3 with MFF in transfected HEK293T epithelial cells. Interestingly, Liu et al. recently reported that TRAF3 interacts with the mitochondrial fusion protein mitofusin-1 (MFN1) in ovarian cancer cells, which is enhanced upon TLR4 signaling induced by Selene nanoparticles (116). Furthermore, Zhou et al. previously reported co-immunoprecipitation of TRAF3 with the MOM protein PINK1 in resting primary mouse peritoneal macrophages (117). Therefore, whether and how TRAF3 regulates mitochondria-dependent apoptosis in these cell types via the TRAF3-MFF axis and metabolic mechanisms as described for B cells or via distinct MFN1- and PINK1- dependent mechanisms are significant areas for future exploration. Such knowledge would lay the foundation for testing the therapeutic potential of TRAF3 gene therapy, mitochondria-targeting drugs, and pharmacological inhibitors of metabolic enzymes/transporters in histiocytic sarcoma, osteosarcoma, and epithelial cell-derived cancers of various tissues, especially those with TRAF3 deletions or relevant alterations.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported by the National Institutes of Health grants R01 CA158402 (PX) and R21 AI128264 (PX), the Department of Defense grant W81XWH-13-1-0242 (PX), a Pilot Award of Cancer Institute of New Jersey through Grant Number P30CA072720 from the National Cancer Institute (PX) and a Busch Biomedical Grant (PX).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal (2013) 8:7. doi: 10.1186/1750-2187-8-7
2. Lalani AI, Zhu S, Gokhale S, Jin J, Xie P. TRAF molecules in inflammation and inflammatory diseases. Curr Pharmacol Rep (2018) 4:64–90. doi: 10.1007/s40495-017-0117-y
3. Zhu S, Jin J, Gokhale S, Lu A, Shan H, Feng J, et al. Genetic alterations of TRAF proteins in human cancers. Front Immunol (2018) 9:2111. doi: 10.3389/fimmu.2018.02111
4. Arkee T, Bishop GA. TRAF family molecules in T cells: Multiple receptors and functions. J Leukoc Biol (2020) 107:907–15. doi: 10.1002/JLB.2MR1119-397R
5. Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol (2011) 11:457–68. doi: 10.1038/nri2998
6. Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity (1996) 5:407–15. doi: 10.1016/s1074-7613(00)80497-5
7. Lalani AI, Moore CR, Luo C, Kreider BZ, Liu Y, Morse HC 3rd, et al. Myeloid cell TRAF3 regulates immune responses and inhibits inflammation and tumor development in mice. J Immunol (2015) 194:334–48. doi: 10.4049/jimmunol.1401548
8. Lalani AI, Luo C, Han Y, Xie P. TRAF3: a novel tumor suppressor gene in macrophages. Macrophage (Houst) (2015) 2:e1009. doi: 10.14800/macrophage.1009
9. Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell (2007) 12:131–44. doi: 10.1016/j.ccr.2007.07.003
10. Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell (2007) 12:115–30. doi: 10.1016/j.ccr.2007.07.004
11. Moore CR, Edwards SK, Xie P. Targeting TRAF3 downstream signaling pathways in b cell neoplasms. J Cancer Sci Ther (2015) 7:67–74. doi: 10.4172/1948-5956.1000327
12. Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity (2010) 33:400–11. doi: 10.1016/j.immuni.2010.08.014
13. Liew MF, Lim HF, Liang MC, Lim I, Tan Z, Tan RYM, et al. Negative TRAF3 variant with recurrent mycobacterium abscessus infection and bronchiectasis. Open Forum Infect Dis (2022) 9:ofac379. doi: 10.1093/ofid/ofac379
14. Rae W, Sowerby JM, Verhoeven D, Youssef M, Kotagiri P, Savinykh N, et al. Immunodeficiency, autoimmunity, and increased risk of b cell malignancy in humans with TRAF3 mutations. Sci Immunol (2022) 7:eabn3800. doi: 10.1126/sciimmunol.abn3800
15. Bishop GA, Stunz LL, Hostager BS. TRAF3 as a multifaceted regulator of b lymphocyte survival and activation. Front Immunol (2018) 9:2161. doi: 10.3389/fimmu.2018.02161
16. Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of b cell homeostasis in secondary lymphoid organs. Immunity (2007) 27:253–67. doi: 10.1016/j.immuni.2007.07.012
17. Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to b cells by the BAFF receptor. Immunity (2008) 28:391–401. doi: 10.1016/j.immuni.2008.01.009
18. Chen Z, Krinsky A, Woolaver RA, Wang X, Chen SMY, Popolizio V, et al. TRAF3 acts as a checkpoint of b cell receptor signaling to control antibody class switch recombination and anergy. J Immunol (2020) 205:830–41. doi: 10.4049/jimmunol.2000322
19. Whillock AL, Ybarra TK, Bishop GA. TNF receptor-associated factor 3 restrains b-cell receptor signaling in normal and malignant b cells. J Biol Chem (2021) 296:100465. doi: 10.1016/j.jbc.2021.100465
20. Paiva C, Rowland TA, Sreekantham B, Godbersen C, Best SR, Kaur P, et al. SYK inhibition thwarts the BAFF - b-cell receptor crosstalk and thereby antagonizes mcl-1 in chronic lymphocytic leukemia. Haematologica (2017) 102:1890–900. doi: 10.3324/haematol.2017.170571
21. Fang DF, He K, Wang N, Sang ZH, Qiu X, Xu G, et al. NEDD4 ubiquitinates TRAF3 to promote CD40-mediated AKT activation. Nat Commun (2014) 5:4513. doi: 10.1038/ncomms5513
22. Xie P, Poovassery J, Stunz LL, Smith SM, Schultz ML, Carlin LE, et al. Enhanced toll-like receptor (TLR) responses of TNFR-associated factor 3 (TRAF3)-deficient b lymphocytes. J Leukoc Biol (2011) 90:1149–57. doi: 10.1189/jlb.0111044
23. Lin WW, Yi Z, Stunz LL, Maine CJ, Sherman LA, Bishop GA. The adaptor protein TRAF3 inhibits interleukin-6 receptor signaling in b cells to limit plasma cell development. Sci Signal (2015) 8:ra88. doi: 10.1126/scisignal.aaa5157
24. Moore CR, Liu Y, Shao CS, Covey LR, Morse HC 3rd, Xie P. Specific deletion of TRAF3 in b lymphocytes leads to b lymphoma development in mice. Leukemia (2012) 26:1122–7. doi: 10.1038/leu.2011.309
25. Zapata JM, Llobet D, Krajewska M, Lefebvre S, Kress CL, Reed JC. Lymphocyte-specific TRAF3-transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood (2009) 113:4595–603. doi: 10.1182/blood-2008-07-165456
26. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational spectrum, copy number changes, and outcome: Results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol (2015) 33:3911–20. doi: 10.1200/JCO.2014.59.1503
27. Xie P, Hostager BS, Bishop GA. Requirement for TRAF3 in signaling by LMP1 but not CD40 in b lymphocytes. J Exp Med (2004) 199:661–71. doi: 10.1084/jem.20031255
28. Bangalore-Prakash P, Stunz LL, Mambetsariev N, Whillock AL, Hostager BS, Bishop GA. The oncogenic membrane protein LMP1 sequesters TRAF3 in b-cell lymphoma cells to produce functional TRAF3 deficiency. Blood Adv (2017) 1:2712–23. doi: 10.1182/bloodadvances.2017009670
29. Saha A, Robertson ES. Mechanisms of b-cell oncogenesis induced by Epstein-Barr virus. J Virol (2019) 93:e00238-19. doi: 10.1128/JVI.00238-19
30. Perez-Carretero C, Hernandez-Sanchez M, Gonzalez T, Quijada-Alamo M, Martin-Izquierdo M, Santos-Minguez S, et al. TRAF3 alterations are frequent in del-3'IGH chronic lymphocytic leukemia patients and define a specific subgroup with adverse clinical features. Am J Hematol (2022) 97:903–14. doi: 10.1002/ajh.26578
31. Bushell KR, Kim Y, Chan FC, Ben-Neriah S, Jenks A, Alcaide M, et al. Genetic inactivation of TRAF3 in canine and human b-cell lymphoma. Blood (2015) 125:999–1005. doi: 10.1182/blood-2014-10-602714
32. Smith PAD, Waugh EM, Crichton C, Jarrett RF, Morris JS. The prevalence and characterisation of TRAF3 and POT1 mutations in canine b-cell lymphoma. Vet J (2020) 266:105575. doi: 10.1016/j.tvjl.2020.105575
33. Avery AC. The genetic and molecular basis for canine models of human leukemia and lymphoma. Front Oncol (2020) 10:23. doi: 10.3389/fonc.2020.00023
34. Giannuzzi D, Marconato L, Fanelli A, Licenziato L, De Maria R, Rinaldi A, et al. The genomic landscape of canine diffuse large b-cell lymphoma identifies distinct subtypes with clinical and therapeutic implications. Lab Anim (NY) (2022) 51:191–202. doi: 10.1038/s41684-022-00998-x
35. Gokhale S, Lu W, Zhu S, Liu Y, Hart RP, Rabinowitz JD, et al. Elevated choline kinase alpha-mediated choline metabolism supports the prolonged survival of TRAF3-deficient b lymphocytes. J Immunol (2020) 204:459–71. doi: 10.4049/jimmunol.1900658
36. Ondrisova L, Mraz M. Genetic and non-genetic mechanisms of resistance to BCR signaling inhibitors in b cell malignancies. Front Oncol (2020) 10:591577. doi: 10.3389/fonc.2020.591577
37. Wang H, Zhang W, Yang J, Zhou K. The resistance mechanisms and treatment strategies of BTK inhibitors in b-cell lymphoma. Hematol Oncol (2021) 39:605–15. doi: 10.1002/hon.2933
38. Roue G, Sola B. Management of drug resistance in mantle cell lymphoma. Cancers (2020) 12:1565. doi: 10.3390/cancers12061565
39. Zhou L, Zhang Y, Meads MB, Dai Y, Ning Y, Hu X, et al. IAP and HDAC inhibitors interact synergistically in myeloma cells through noncanonical NF-kappaB- and caspase-8-dependent mechanisms. Blood Adv (2021) 5:3776–88. doi: 10.1182/bloodadvances.2020003597
40. Liu Y, Gokhale S, Jung J, Zhu S, Luo C, Saha D, et al. Mitochondrial fission factor is a novel interacting protein of the critical b cell survival regulator TRAF3 in b lymphocytes. Front Immunol (2021) 12:670338. doi: 10.3389/fimmu.2021.670338
41. Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet (2009) 43:95–118. doi: 10.1146/annurev-genet-102108-134850
42. Caroppi P, Sinibaldi F, Fiorucci L, Santucci R. Apoptosis and human diseases: mitochondrion damage and lethal role of released cytochrome c as proapoptotic protein. Curr Med Chem (2009) 16:4058–65. doi: 10.2174/092986709789378206
43. Burke PJ. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer (2017) 3:857–70. doi: 10.1016/j.trecan.2017.10.006
44. Yu R, Lendahl U, Nister M, Zhao J. Regulation of mammalian mitochondrial dynamics: Opportunities and challenges. Front Endocrinol (2020) 11:374. doi: 10.3389/fendo.2020.00374
45. Lee SY, Choi Y. TRAF-interacting protein (TRIP): a novel component of the tumor necrosis factor receptor (TNFR)- and CD30-TRAF signaling complexes that inhibits TRAF2-mediated NF-kappaB activation. J Exp Med (1997) 185:1275–85. doi: 10.1084/jem.185.7.1275
46. Gamper C, van Eyndhoven WG, Schweiger E, Mossbacher M, Koo B, Lederman S. TRAF-3 interacts with p62 nucleoporin, a component of the nuclear pore central plug that binds classical NLS-containing import complexes. Mol Immunol (2000) 37:73–84. doi: 10.1016/s0161-5890(00)00015-8
47. Dadgostar H, Doyle SE, Shahangian A, Garcia DE, Cheng G. T3JAM, a novel protein that specifically interacts with TRAF3 and promotes the activation of JNK(1). FEBS Lett (2003) 553:403–7. doi: 10.1016/s0014-5793(03)01072-x
48. El-Hattab AW, Suleiman J, Almannai M, Scaglia F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases. Mol Genet Metab (2018) 125:315–21. doi: 10.1016/j.ymgme.2018.10.003
49. Serasinghe MN, Chipuk JE. Mitochondrial fission in human diseases. Handb Exp Pharmacol (2017) 240:159–88. doi: 10.1007/164_2016_38
50. Mambetsariev N, Lin WW, Wallis AM, Stunz LL, Bishop GA. TRAF3 deficiency promotes metabolic reprogramming in b cells. Sci Rep (2016) 6:35349. doi: 10.1038/srep35349
51. Perez-Chacon G, Adrados M, Vallejo-Cremades MT, Lefebvre S, Reed JC, Zapata JM. Dysregulated TRAF3 and BCL2 expression promotes multiple classes of mature non-hodgkin b cell lymphoma in mice. Front Immunol (2019) 9:3114. doi: 10.3389/fimmu.2018.03114
52. Ducommun S, Deak M, Sumpton D, Ford RJ, Nunez Galindo A, Kussmann M, et al. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal (2015) 27:978–88. doi: 10.1016/j.cellsig.2015.02.008
53. Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science (2016) 351:275–81. doi: 10.1126/science.aab4138
54. Zong Y, Zhang CS, Li M, Wang W, Wang Z, Hawley SA, et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res (2019) 29:460–73. doi: 10.1038/s41422-019-0163-6
55. Seabright AP, Fine NHF, Barlow JP, Lord SO, Musa I, Gray A, et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-parkin independent manner. FASEB J (2020) 34:6284–301. doi: 10.1096/fj.201903051R
56. Gao J, Qin S, Jiang C. Parkin-induced ubiquitination of mff promotes its association with p62/SQSTM1 during mitochondrial depolarization. Acta Biochim Biophys Sin (Shanghai) (2015) 47:522–9. doi: 10.1093/abbs/gmv044
57. Lee L, Seager R, Nakamura Y, Wilkinson KA, Henley JM. Parkin-mediated ubiquitination contributes to the constitutive turnover of mitochondrial fission factor (Mff). PLos One (2019) 14:e0213116. doi: 10.1371/journal.pone.0213116
58. Cherok E, Xu S, Li S, Das S, Meltzer WA, Zalzman M, et al. Novel regulatory roles of mff and Drp1 in E3 ubiquitin ligase MARCH5-dependent degradation of MiD49 and Mcl1 and control of mitochondrial dynamics. Mol Biol Cell (2017) 28:396–410. doi: 10.1091/mbc.E16-04-0208
59. He L, Grammer AC, Wu X, Lipsky PE. TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF-{kappa}B activation. J Biol Chem (2004) 279:55855–65. doi: 10.1074/jbc.M407284200
60. Paz S, Vilasco M, Werden SJ, Arguello M, Joseph-Pillai D, Zhao T, et al. A functional c-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res (2011) 21:895–910. doi: 10.1038/cr.2011.2
61. Jacobs JL, Coyne CB. Mechanisms of MAVS regulation at the mitochondrial membrane. J Mol Biol (2013) 425:5009–19. doi: 10.1016/j.jmb.2013.10.007
62. Refolo G, Vescovo T, Piacentini M, Fimia GM, Ciccosanti F. Mitochondrial interactome: A focus on antiviral signaling pathways. Front Cell Dev Biol (2020) 8:8. doi: 10.3389/fcell.2020.00008
63. Thoresen D, Wang W, Galls D, Guo R, Xu L, Pyle AM. The molecular mechanism of RIG-I activation and signaling. Immunol Rev (2021) 304:154–68. doi: 10.1111/imr.13022
64. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science (2000) 287:664–6. doi: 10.1126/science.287.5453.664
65. Siegmund D, Wagner J, Wajant H. TNF receptor associated factor 2 (TRAF2) signaling in cancer. Cancers (2022) 14:4055. doi: 10.3390/cancers14164055
66. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell (2000) 102:33–42. doi: 10.1016/s0092-8674(00)00008-8
67. Ferri KF, Kroemer G. Mitochondria–the suicide organelles. Bioessays (2001) 23:111–5. doi: 10.1002/1521-1878(200102)23:2<111::AID-BIES1016>3.0.CO;2-Y
68. Gokhale S, Xie P. ChoK-full of potential: Choline kinase in b cell and T cell malignancies. Pharmaceutics (2021) 13:911. doi: 10.3390/pharmaceutics13060911
69. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer (2016) 16:635–49. doi: 10.1038/nrc.2016.77
70. Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab (2012) 16:751–64. doi: 10.1016/j.cmet.2012.10.017
71. Garcia Sanchez R, Hahn-Hagerdal B, Gorwa-Grauslund MF. PGM2 overexpression improves anaerobic galactose fermentation in saccharomyces cerevisiae. Microb Cell Fact (2010) 9:40. doi: 10.1186/1475-2859-9-40
72. Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed b cells. J Immunol (2014) 192:3626–36. doi: 10.4049/jimmunol.1302062
73. Matsumoto T, Jimi S, Migita K, Takamatsu Y, Hara S. Inhibition of glucose transporter 1 induces apoptosis and sensitizes multiple myeloma cells to conventional chemotherapeutic agents. Leuk Res (2016) 41:103–10. doi: 10.1016/j.leukres.2015.12.008
74. Liu T, Kishton RJ, Macintyre AN, Gerriets VA, Xiang H, Liu X, et al. Glucose transporter 1-mediated glucose uptake is limiting for b-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis (2014) 5:e1470. doi: 10.1038/cddis.2014.431
75. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc (2015) 90:927–63. doi: 10.1111/brv.12140
76. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi: 10.1038/nrc2981
77. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell (2014) 5:592–602. doi: 10.1007/s13238-014-0082-8
78. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large b-cell lymphoma identified by gene expression profiling. Nature (2000) 403:503–11. doi: 10.1038/35000501
79. Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in b cell chronic lymphocytic leukemia. J Exp Med (2001) 194:1639–47. doi: 10.1084/jem.194.11.1639
80. Andersson A, Ritz C, Lindgren D, Eden P, Lassen C, Heldrup J, et al. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia (2007) 21:1198–203. doi: 10.1038/sj.leu.2404688
81. Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood (2007) 109:1692–700. doi: 10.1182/blood-2006-07-037077
82. Lautner-Csorba O, Gezsi A, Erdelyi DJ, Hullam G, Antal P, Semsei AF, et al. Roles of genetic polymorphisms in the folate pathway in childhood acute lymphoblastic leukemia evaluated by Bayesian relevance and effect size analysis. PLos One (2013) 8:e69843. doi: 10.1371/journal.pone.0069843
83. Weiner AS, Beresina OV, Voronina EN, Voropaeva EN, Boyarskih UA, Pospelova TI, et al. Polymorphisms in folate-metabolizing genes and risk of non-hodgkin's lymphoma. Leuk Res (2011) 35:508–15. doi: 10.1016/j.leukres.2010.10.004
84. De Silva NS, Silva K, Anderson MM, Bhagat G, Klein U. Impairment of mature b cell maintenance upon combined deletion of the alternative NF-kappaB transcription factors RELB and NF-kappaB2 in b cells. J Immunol (2016) 196:2591–601. doi: 10.4049/jimmunol.1501120
85. Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, et al. Germinal center b cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J Exp Med (2014) 211:2103–18. doi: 10.1084/jem.20132613
86. Almaden JV, Liu YC, Yang E, Otero DC, Birnbaum H, Davis-Turak J, et al. B-cell survival and development controlled by the coordination of NF-kappaB family members RelB and cRel. Blood (2016) 127:1276–86. doi: 10.1182/blood-2014-10-606988
87. Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol (2011) 192:7–16. doi: 10.1083/jcb.201006159
88. Basu Ball W, Neff JK, Gohil VM. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett (2018) 592:1273–90. doi: 10.1002/1873-3468.12887
89. van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr (2017) 1859:1558–72. doi: 10.1016/j.bbamem.2017.04.006
90. Leibowitz B, Yu J. Mitochondrial signaling in cell death via the bcl-2 family. Cancer Biol Ther (2010) 9:417–22. doi: 10.4161/cbt.9.6.11392
91. Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell (2011) 21:92–101. doi: 10.1016/j.devcel.2011.06.017
92. Sen R. Control of b lymphocyte apoptosis by the transcription factor NF-kappaB. Immunity (2006) 25:871–83. doi: 10.1016/j.immuni.2006.12.003
93. Mambetsariev N, Lin WW, Stunz LL, Hanson BM, Hildebrand JM, Bishop GA. Nuclear TRAF3 is a negative regulator of CREB in b cells. Proc Natl Acad Sci U.S.A. (2016) 113:1032–7. doi: 10.1073/pnas.1514586113
94. Seo JH, Chae YC, Kossenkov AV, Lee YG, Tang HY, Agarwal E, et al. MFF regulation of mitochondrial cell death is a therapeutic target in cancer. Cancer Res (2019) 79:6215–26. doi: 10.1158/0008-5472.CAN-19-1982
95. Rovira-Llopis S, Banuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol (2017) 11:637–45. doi: 10.1016/j.redox.2017.01.013
96. Abdelzaher E, Mostafa MF. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) upregulation in breast carcinoma contributes to tumor progression and predicts early tumor recurrence. Tumour Biol (2015) 36:5473–83. doi: 10.1007/s13277-015-3214-8
97. Grupp K, Sanader S, Sirma H, Simon R, Koop C, Prien K, et al. High lysophosphatidylcholine acyltransferase 1 expression independently predicts high risk for biochemical recurrence in prostate cancers. Mol Oncol (2013) 7:1001–11. doi: 10.1016/j.molonc.2013.07.009
98. Uehara T, Kikuchi H, Miyazaki S, Iino I, Setoguchi T, Hiramatsu Y, et al. Overexpression of lysophosphatidylcholine acyltransferase 1 and concomitant lipid alterations in gastric cancer. Ann Surg Oncol (2016) 23 Suppl 2):S206–13. doi: 10.1245/s10434-015-4459-6
99. Ding J, Ding X, Leng Z. LPCAT1 promotes gefitinib resistance via upregulation of the EGFR/PI3K/AKT signaling pathway in lung adenocarcinoma. J Cancer (2022) 13:1837–47. doi: 10.7150/jca.66126
100. Lee WN, Guo P, Lim S, Bassilian S, Lee ST, Boren J, et al. Metabolic sensitivity of pancreatic tumour cell apoptosis to glycogen phosphorylase inhibitor treatment. Br J Cancer (2004) 91:2094–100. doi: 10.1038/sj.bjc.6602243
101. Figlak K, Williams G, Bertolini M, Paus R, Philpott MP. Human hair follicles operate an internal cori cycle and modulate their growth via glycogen phosphorylase. Sci Rep (2021) 11:20761. doi: 10.1038/s41598-021-99652-8
102. Garcia SN, Guedes RC, Marques MM. Unlocking the potential of HK2 in cancer metabolism and therapeutics. Curr Med Chem (2019) 26:7285–322. doi: 10.2174/0929867326666181213092652
103. Shan W, Zhou Y, Tam KY. The development of small-molecule inhibitors targeting hexokinase 2. Drug Discovery Today (2022) 27:2574–85. doi: 10.1016/j.drudis.2022.05.017
104. Sdelci S, Rendeiro AF, Rathert P, You W, Lin JG, Ringler A, et al. MTHFD1 interaction with BRD4 links folate metabolism to transcriptional regulation. Nat Genet (2019) 51:990–8. doi: 10.1038/s41588-019-0413-z
105. Rehei AL, Zhang L, Fu YX, Mu WB, Yang DS, Liu Y, et al. MicroRNA-214 functions as an oncogene in human osteosarcoma by targeting TRAF3. Eur Rev Med Pharmacol Sci (2018) 22:5156–64. doi: 10.26355/eurrev_201808_15711
106. Network TCGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature (2015) 517:576–82. doi: 10.1038/nature14129
107. Zhang J, Chen T, Yang X, Cheng H, Spath SS, Clavijo PE, et al. Attenuated TRAF3 fosters activation of alternative NF-kappaB and reduced expression of antiviral interferon, TP53, and RB to promote HPV-positive head and neck cancers. Cancer Res (2018) 78:4613–26. doi: 10.1158/0008-5472.CAN-17-0642
108. Georgopoulos NT, Steele LP, Thomson MJ, Selby PJ, Southgate J, Trejdosiewicz LK. A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ (2006) 13:1789–801. doi: 10.1038/sj.cdd.4401859
109. Dunnill CJ, Ibraheem K, Mohamed A, Southgate J, Georgopoulos NT. A redox state-dictated signalling pathway deciphers the malignant cell specificity of CD40-mediated apoptosis. Oncogene (2017) 36:2515–28. doi: 10.1038/onc.2016.401
110. Sirinian C, Papanastasiou AD, Karayel O, Degn SE, Peroukidis S, Chaniotis D, et al. Analysis of RANK-c interaction partners identifies TRAF3 as a critical regulator of breast cancer aggressiveness. Neoplasia (2022) 33:100836. doi: 10.1016/j.neo.2022.100836
111. Shiode Y, Kodama T, Shigeno S, Murai K, Tanaka S, Newberg JY, et al. TNF receptor-related factor 3 inactivation promotes the development of intrahepatic cholangiocarcinoma through NF-kappaB-inducing kinase-mediated hepatocyte transdifferentiation. Hepatology (2022). doi: 10.1002/hep.32317
112. Zhang B, Yang C, Wang R, Wu J, Zhang Y, Liu D, et al. OTUD7B suppresses smac mimetic-induced lung cancer cell invasion and migration via deubiquitinating TRAF3. J Exp Clin Cancer Res (2020) 39:244. doi: 10.1186/s13046-020-01751-3
113. Li J, Ayoub A, Xiu Y, Yin X, Sanders JO, Mesfin A, et al. TGFbeta-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat Commun (2019) 10:2795. doi: 10.1038/s41467-019-10677-0
114. Boyce BF, Li J, Xing L, Yao Z. Bone remodeling and the role of TRAF3 in osteoclastic bone resorption. Front Immunol (2018) 9:2263. doi: 10.3389/fimmu.2018.02263
115. Shi JH, Ling C, Wang TT, Zhang LN, Liu WW, Qin Y, et al. TRK-fused gene (TFG) regulates ULK1 stability via TRAF3-mediated ubiquitination and protects macrophages from LPS-induced pyroptosis. Cell Death Dis (2022) 13:93. doi: 10.1038/s41419-022-04539-9
116. Liu HJ, Qin Y, Zhao ZH, Zhang Y, Yang JH, Zhai DH, et al. Lentinan-functionalized selenium nanoparticles target tumor cell mitochondria via TLR4/TRAF3/MFN1 pathway. Theranostics (2020) 10:9083–99. doi: 10.7150/thno.46467
Keywords: TRAF3, mitochondria, metabolism, B lymphocytes, lymphomas
Citation: Jung J, Gokhale S and Xie P (2023) TRAF3: A novel regulator of mitochondrial physiology and metabolic pathways in B lymphocytes. Front. Oncol. 13:1081253. doi: 10.3389/fonc.2023.1081253
Received: 27 October 2022; Accepted: 13 January 2023;
Published: 27 January 2023.
Edited by:
Enrico Milan, San Raffaele Scientific Institute (IRCCS), ItalyReviewed by:
Jian-Hong Shi, Affiliated Hospital of Hebei University, ChinaMala Shanmugam, Emory University, United States
Copyright © 2023 Jung, Gokhale and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Xie, eGllQGRscy5ydXRnZXJzLmVkdQ==
†These authors have contributed equally to this work