 Ruizhi Zhou
Ruizhi Zhou Tianqi Liang
Tianqi Liang Tianwen Li
Tianwen Li Junbin Huang*
Junbin Huang* Chun Chen
Chun Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 01 February 2023
Sec. Cancer Metabolism
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1070069
This article is part of the Research Topic Linking Cellular Metabolism to Hematological Malignancies, volume II View all 11 articles
L-asparaginase, which hydrolyzes asparagine into aspartic acid and ammonia, is frequently used to treat acute lymphoblastic leukaemia in children. When combined with other chemotherapy drugs, the event-free survival rate is 90%. Due to immunogenicity and drug resistance, however, not all patients benefit from it, restricting the use of L-asparaginase therapy in other haematological cancers. To solve the problem of immunogenicity, several L-ASNase variants have emerged, such as Erwinia-ASNase and PEG-ASNase. However, even when Erwinia-ASNase is used as a substitute for E. coli-ASNase or PEG-ASNase, allergic reactions occur in 3%-33% of patients. All of these factors contributed to the development of novel L-ASNases. Additionally, L-ASNase resistance mechanisms, such as the methylation status of ASNS promoters and activation of autophagy, have further emphasized the importance of personalized treatment for paediatric haematological neoplasms. In this review, we discussed the metabolic effects of L-ASNase, mechanisms of drug resistance, applications in non-ALL leukaemia, and the development of novel L-ASNase.
L-asparaginase (L-ASNase), an enzyme that hydrolyzes asparagine, is one of the most successful drugs for metabolic targeting to date and one of the most important chemotherapeutic drugs in standardized regimens for childhood ALL. L-ASNase is essential for improving the complete remission rate and long-term survival in children with ALL. In the moderate/low-risk group mainly according to the treatment response of 15-19 days and the level of minimal residual disease in 29-45days, event-free survival and overall survival rates can reach 90% when combined with other chemotherapeutic drugs (1–3). L-ASNase (L-ASNase) has been shown to have anticancer activity that depends on its ability to hydrolyse asparagine since it was discovered in guinea pig serum in 1953 (4–7). In 1966, Dolowy et al. first reported complete remission in a case of refractory childhood acute lymphoblastic leukaemia (ALL) treated with guinea pig-derived L-ASNase (8). In 1970, Clarkson et al. first reported the treatment of ALL with purified E. coli-derived L-ASNase (E. coli-ASNase) and the induction of remission (9). In the following decades, L-ASNase was widely used in the treatment of ALL. Currently, the clinically used L-ASNases include E. coli-ASNase, Erwinia-ASNase, and PEG-ASNase, among which PEG-ASNase has the longest half-life and lowest immunogenicity (5, 10). Nevertheless, allergic reactions to L-ASNase still occur in 30%-70% of patients, which limits its efficacy (11). To provide new insights into using L-ASNase in treating paediatric leukaemia, we discussed the metabolic effects of L-ASNase, mechanisms of drug resistance, applications in non-ALL leukaemia, and the development of novel L-ASNase in this review.
Tumour cells have different metabolic patterns compared to normal cells. This metabolic pattern is manifested by increased glycolysis, glucose uptake, and uptake and catabolism of amino acids (12–14). Metabolic reprogramming allows tumour cells to show resilience in hypoxic and nutrient-deficient environments. At the same time, however, such metabolic alterations also make tumour cells exhibit specific vulnerabilities, such as an increase in certain specific metabolic demands (15). The increased metabolic demands determine the importance of glucose and amino acids in tumour metabolism. Unlike normal cells, amino acids that are not essential to normal cells may be essential to tumour cells because tumour cells usually lose the ability to synthesize these amino acids de novo, enabling the amino acid deprivation therapy.
Asparagine is a nonessential amino acid involved in protein synthesis for normal cells, which can be obtained from food or produced by the combination of aspartate acid with ammonia catalysed by asparagine synthase (ASNS) (16, 17).Different from normal cells, due to the lack of ASNS, leukaemia cells frequently fail to synthesize asparagine and therefore must rely on the host to supply asparagine for their protein synthesis requirements. By catabolizing asparagine in serum, L-ASNase can expose leukaemia cells to an asparagine-deficient environment, and thus affecting protein synthesis in leukaemic cells and leading to their growth inhibition or death (5, 16–20). In addition, Hermanova et al. further demonstrated the molecular mechanism by which L-ASNase inhibits protein synthesis in leukaemic cells (21). The mammalian target of rapamycin protein complex 1 (mTORC1) plays a central role in the amino acid response. RagA/RagB switches from a GDP-bound state to a GTP-bound state as amino acid levels rise, which activates mTORC1, and in turn stimulates a series of downstream reactions, including protein synthesis (22). However, it was recently discovered that RagB-expressing cells can still activate mTORC1 even in an amino acid-deficient environment (23). By treating wild-type RagB cells and RagB-mutant cells (in a permanent GTP-bound state) separately with L-ASNase and assaying the levels of the mTORC1 downstream molecule p-S6 protein, Hermanova et al. found that wild-type RagB cells had significantly lower p-S6 protein levels, while RagB-mutant cells did not show significant changes in p-S6 protein levels. That is, L-ASNase can inhibit protein synthesis by inhibiting RagB-mTORC1 (21) (Figure 1).
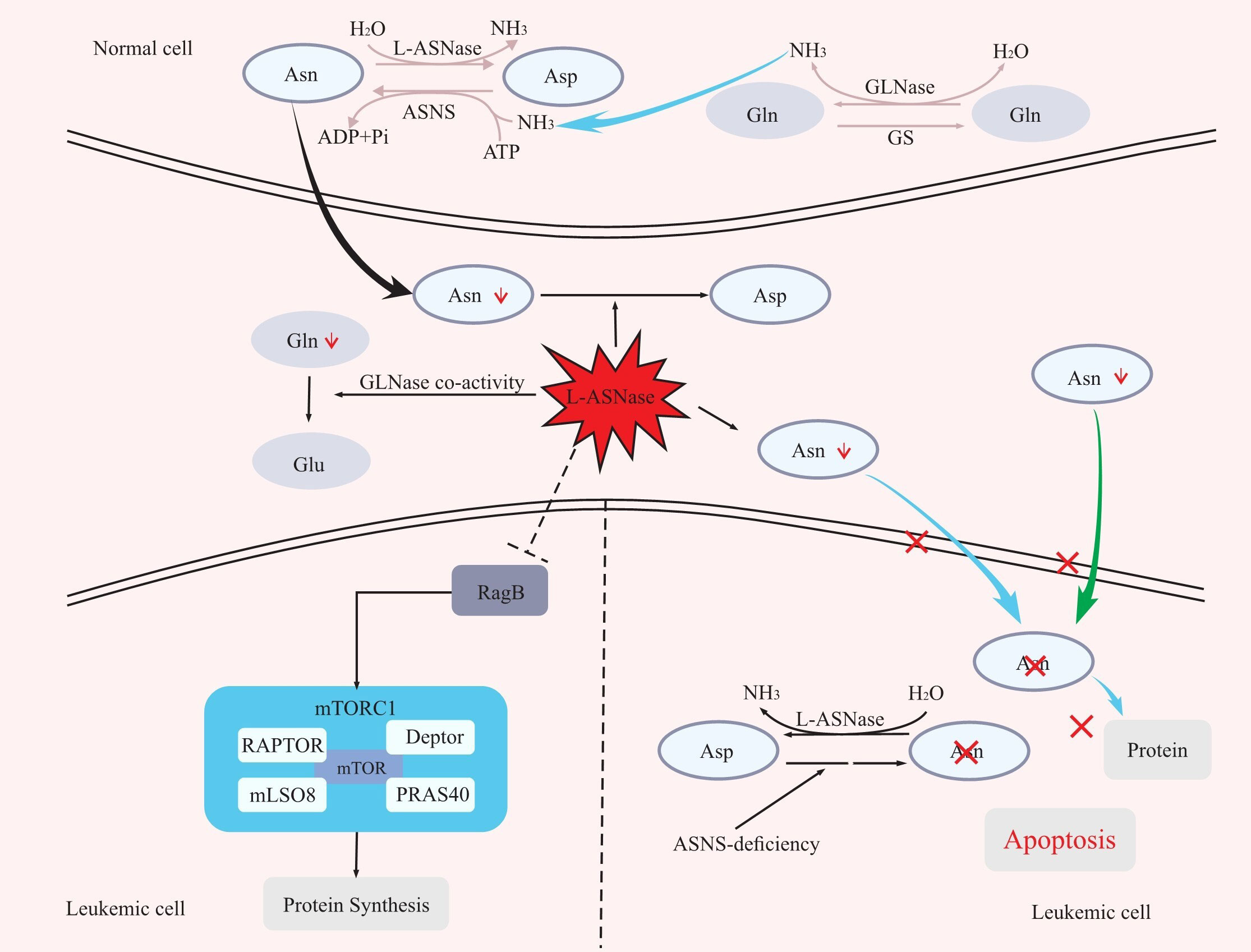
Figure 1 The mechanism of L-ASNase. L-ASNase depletes Asn, and GLNase coactivity hydrolyzes Gln, which further reduces Asn, leading to apoptosis of leukaemia cells. L-ASNase can inhibit RagB-mTORC1 and thus inhibit protein synthesis. Asp, aspartic acid; Asn, asparagine; Gln, glutamine; Glu, glutamic acid; ASNS, asparagine synthetase; L-ASNase, L-asparaginase; GS, glutamine synthetase; GLNase, glutaminase.
L-ASNase also has glutaminase (GLNase) activity that can hydrolyze glutamine. In the presence of ASNS, glutamine can act as an amino donor to facilitate the production of aspartic acid into asparagine. Therefore, the hydrolysis of glutamine by L-ASNase also contributes to the reduction of asparagine levels, improving the efficacy (16, 19, 24) (Figure 1). However, whether the anti-leukaemic effect of L-ASNase depends on GLNase activity is controversial (25–32). First, Offman and Parmentier et al. demonstrated that the killing effect of L-ASNase on leukaemic cells was reliant on GLNase activity and that the cytotoxicity of L-ASNase on leukaemic cells increased with increasing GLNase activity (25, 29). Chan et al. also demonstrated in a recent study that L-ASNase with GLNase activity was more cytotoxic to leukaemic cells and could better prolong the survival of mice in an ASNS-negative SUP- B15 xenograft model (32). In contrast, a previous study showed that L-ASNase without GLNase activity could achieve the same level of antitumour effects as wild-type L-ASNase in ASNS-negative leukaemia cell lines (30). Nguyen et al. also demonstrated that L-ASNase mutants with low GLNase activity had the same level of antitumour activity as L-ASNase mutants with high GLNase activity in an ASNS-negative SUP-B15 leukaemia cell xenograft model (31). However, the L-ASNase in their research is not completely devoid of GLNase activity. Therefore, we believe that GLNase activity of L-ASNase is required for the killing effect of L-ASNase in ASNS-negative tumour cells, but at what level of GLNase activity needs to be maintained is a question that require confirmation through more experiments.
In addition to protein synthesis and amino acid metabolism, Hermanova et al. found that L-ASNase can also affect the energy metabolism of leukaemic cells, including increased fatty acid oxidation and inhibition of glycolysis. They suggested that the inhibition of mTORC1 by L-ASNase was responsible for inducing fatty acid oxidation. Moreover, they found that fatty acid oxidation inhibitors and L-ASNase can act synergistically to kill cells (21). Therefore, the combination regimen of fatty acid oxidation inhibitors with L-ASNase may provide a brand-new option for the treatment of ALL. Furthermore, Takahashi et al. demonstrated in their study that L-ASNase can inhibit glycolysis in leukaemic cells (18), but the precise molecular mechanism of this is unknown. In general, L-ASNase can affect the energy metabolism of leukaemia cells, which in turn may hurt the efficacy of L-ASNase. Clarifying the specific mechanism will provide new selections to ALL treatment, and more research on combined treatment regimens targeting energy metabolism such as fatty acid oxidation will bring new hope to ALL treatment.
Studies on the mechanism of L-ASNase resistance have been widely conducted. Numerous studies have found elevated expression of ASNS in L-ASNase-resistant tumour cells. It has been confirmed that L-ASNase-resistant cells express higher levels of ASNS than L-ASNase-sensitive cells (33–36). Scherf and Holleman et al. reported that L-ASNase-sensitive cells express lower levels of ASNS mRNA in vitro (37, 38). In contrast, however, the results of Fine et al. did not find a correlation between the expression level of ASNS mRNA and sensitivity to L-ASNase (39). In B-lineage lymphocytic leukaemia cells carrying the TEL-AML 1 translocation, Stams et al. also obtained the same results as Fine et al. (40). Even in other studies, higher expression levels of ASNS mRNA were found in L-ASNase-sensitive TEL-AML1-positive cells compared with TEL-AML1-negative cells that were resistant to L-ASNase (41), but they did not further elucidate the relationship between TEL-AML1 fusion genes and ASNS gene expression. In addition, Su et al. stated that high ASNS expression did associate with resistance to L-ASNase. But they suggested that it should be the ASNS protein, rather than the mRNA, to be tested as indicators of L-ASNase resistance as there was no significant correlation between the levels of ASNS mRNA and ASNS protein (42). In any case, these studies illustrate the point that ASNS expression confers L-ASNase resistance in leukaemic cells.
Other studies have revealed that the methylation status of the ASNS promoter region can affect the transcription of ASNS and thus affect the sensitivity of L-ASNase. ASNS is part of the amino acid response pathway that is activated by amino acid deficiency (43, 44). When asparagine is deprived, tumour cells can respond via the GCN2-ATF4 pathway. ATF4 binds to the ASNS promoter in a hypomethylated state and induces its expression (45). Jiang et al. found that the hypermethylated state of the ASNS promoter restricted the binding of the transcription factor ATF4 upon amino acid depletion, and thereby inhibiting ASNS expression (46). Overall, amino acid deficiency-induced ASNS expression requires both GCN2 activation and hypomethylation of the ASNS promoter region, which enable ATF4 binding to drive ASNS expression. A cohort study by Akahane et al. further confirmed that the hypomethylation status of the ASNS promoter region is associated with L-ASNase resistance. Their analysis of 75 Japanese children with T-ALL revealed an intermediate (33.3% < methylation <66.7%) or low (<33.3%) methylation status of the ASNS promoter region in 92% of refractory/relapsed cases (47). In addition, Touzart et al. found ASNS to be expressed at low levels in TLX1+ T-ALL cells (high ASNS methylation levels). TLX1+ T-ALL was more sensitive to L-ASNase than the TLX-CCRF-CEM cell line (low ASNS methylation level) (48). Recently, the important role of amino acid stress response genes in L-ASNase sensitivity was further confirmed by Ferguson et al., who identified a novel L-ASNase resistance gene, SLC7A11, whose high expression leads to L-ASNase sensitivity in cancer cells (49). In conclusion, these studies all suggested that the hypomethylation status of the ASNS promoter region contributes to the expression of ASNS induced by L-ASNase treatment, and thus conferring L-ASNase resistance to leukaemic cells. However, there are limited cohort studies to refer to at present, therefore, additional larger cohort studies are needed to further confirm the possibility of ASNS promoter region methylation as a predictor of treatment response.
Meanwhile, the factors affecting the methylation status of the ASNS promoter region have been reported. Worton et al. reported that L-ASNase induces ASNS promoter demethylation, which confers drug resistance to leukaemic cells (50). However, the mechanism by which L-ASNase induces demethylation has not been further confirmed. The study by Akahane et al. focused on the significance of SPI1 fusion in the methylation status of ASNS. In their cohort study, all seven SPI1 fusion cases had an ASNS promoter hypomethylation status, and the ASNS gene expression levels were significantly higher than those of SPI1 fusion-negative cases (47). This suggests that genetic modifications may play an important role in the methylation status of the ASNS promoter region. Yet, it is critical to confirm the relationship between poor prognosis-associated fusion genes and ASNS gene methylation status and the molecular mechanism of L-ASNase-induced demethylation, providing information for treatment option and improving the prognosis for ALL patients.
Several recent studies showed that L-ASNase resistance is related to phosphatase and tensin homologue (PTEN) deficiency and phosphatidylinositol-3 kinase (PI3K)/Akt/mTOR signalling pathway. PTEN is a major negative regulator of the PI3K/Akt/mTOR signalling pathway. Deletion of PTEN can occur in 20% of children with T-ALL and plays an important role in the development and prognosis of T-ALL in children (51–53). Hlozkova et al. proposed that the metabolic pattern of leukaemic cells is associated with L-ASNase resistance after investigating the effect of L-ASNase treatment on the extensive metabolic reprogramming of leukaemic cells. They found that cells with a high glycolytic response are resistant to L-ASNase (54). They subsequently confirmed the relationship between glycolytic levels and L-ASNase sensitivity by investigating the effects of PTEN deficiency on the metabolism of leukaemic cells and changes in L-ASNase sensitivity. Furthermore, a recent study by Hlozkova et al. found that, compared to PTEN wild-type cells, PTEN-deficient T-ALL cells have a higher glycolytic function and overactivated Akt, and these changes made T-ALL cells resistant to L-ASNase. Meanwhile, the resistance of PTEN-deficient cells to L-ASNase could be improved by inhibiting Akt signalling (53). These results suggest that Akt inhibitors may contribute to the treatment of T-ALL patients with PTEN mutations, but further experiments are still needed for verification.
Amino acid deprivation has been shown to induce the activation of autophagy, which is considered a self-protective mechanism in tumour cells (55–57). Hermanova et al. showed that L-ASNase can induce the activation of protective autophagy in leukaemia cells by inhibiting mTORC1 (21). As in previous studies (58), they suggested that autophagy could counteract nutrient imbalance by recycling amino acids, thus resisting the cytotoxicity of L-ASNase (21). Takahashi et al. also reported that L-ASNase treatment reduced glycolysis in leukaemia cells while causing mitochondrial damage and activating autophagy. However, they concluded that the function of L-ASNase-induced autophagy was to eliminate mitochondrial damage and thus reducing ROS production rather than amino acid recycling. Notably, in this study, they demonstrated that by inhibiting autophagy, the cytotoxicity of L-ASNase could be enhanced and such synergistic effect works through the ROS-p53 positive feedback loop (18). In addition, Takahashi et al. and Polak et al. further confirmed that autophagy inhibitors and L-ASNase have synergistic anti-leukaemic effects (59, 60). The activation of autophagy is one of the mechanisms leading to L-ASNase resistance, but the role of autophagy in this mechanism still needs to be further refined. The abovementioned studies suggest that timely detection of autophagy activation during L-ASNase treatment would be more helpful in the selection of treatment regimens, and the combination of L-ASNase with autophagy inhibitors may provide better clinical outcomes.
The tumour microenvironment affects the cytotoxicity of L-ASNase (61). A study by Iwamoto et al. revealed the interaction between leukaemic cells and their surrounding microenvironment. The expression of ASNS is much higher in normal bone marrow mesenchymal stem cells (MSCs) than in leukaemic lymphoblastoid cells. In vitro, leukaemic cells can acquire resistance to L-ASNase by receiving asparagine from MSCs (62). Glutamine synthetase expression is increased in bone marrow adipocytes after induction of chemotherapy with L-ASNase, producing more glutamine and thus protecting leukaemic cells from L-ASNase (63). Future studies focusing revealing the molecular mechanism of the interaction between leukaemia cells and the haematopoietic microenvironment in the bone marrow will further elucidate the anti-leukaemic effect of L-ASNase and hence improving the L-ASNase therapy.
Due to its immunogenicity, an L-ASNase treatment can cause an immune response, which is associated with the production of neutralizing antibodies. Neutralizing antibodies can inactivate L-ASNase, and thereby reducing efficacy. The production of neutralizing antibodies in patients without clinical symptoms is known as silent inactivation, which is usually not clinically evident and thus difficult for early detection (11, 64, 65). Although it has been suggested that patients with allergic reactions to E. coli-ASNase and PEG-ASNase should be switched to Erwinia-ASNase (66), 3-33% of patients can develop an immune response against Erwinia-ASNase, resulting in neutralizing antibodies against L-ASNase and thus resistance to L-ASNase (67, 68).
It has been shown that leukaemic cells can acquire L-ASNase resistance through the OPRM1-cAMP-caspase pathway. Kang et al. identified the opioid receptor μ1(OPRM1) as a key factor for L-ASNase resistance in paediatric ALL using unbiased genome-wide RNAi. They analysed OPRM1 expression levels in primary leukaemic cells from five children with ALL in relation to L-ASNase sensitivity and found that cells with low levels of OPRM1 were more resistant to L-ASNase (69). In addition, Lee et al. identified the Huntington-associated protein 1 gene (HAP1) as an L-ASNase resistance gene, and by examining the relationship between HAP1 levels and L-ASNase sensitivity in the cells of six ALL patients, they found that the lower HAP1 level, the more resistant they were. Furthermore, they found that HAP1 deletion prevented Ca2+ release from the endoplasmic reticulum and downregulated the Calpain-1-Bid-caspase-3/12 pathway, conferring L-ASNase resistance in leukaemic cells (70). Additionally, a recent study demonstrated that if the Wnt pathway is blocked, cells may degrade proteins via GSK3-dependent protein ubiquitination and proteasome degradation pathways to synthesize asparagine to counteract the cytotoxicity of L-ASNase (71).
In conclusion, ASNS expression remains a pivotal factor in the resistance of L-ASNase in leukaemic cells, and ASNS expression is closely related to the methylation status of its promoter region. In addition, activation of autophagy, high glycolysis levels, or inhibition of apoptotic signalling pathways can all promote L-ASNase resistance. The gradual uncovering of L-ASNase resistance mechanisms further emphasizes the significance of individualized therapy and continues to provide new ideas for the further development of individualized combination therapy regimens.
Although L-ASNase is currently used primarily for the treatment of ALL and some NK/T-cell lymphomas, there is growing evidence that L-ASNase can play a critical role in the treatment of other childhood leukaemias (26).
Dübbers et al. found that leukaemic cells from M1, M4, and M5 subtypes had negative ASNS staining among all FAB subtypes of AML and that AML-M5 had the lowest ASNS activity (72). This is in agreement with the results of Okada et al., who found L-ASNase to be effective against specific subtypes of AML (M1, M4, M5) in vitro (73). Additionally, according to Buaboonnam et al., patients with refractory/relapsed AML who received treatment with L-ASNase in combination with MTX had 1- and 2-year survival rates of 35.6% and 17.8%, respectively (74). Whether this regimen can be used as a treatment for patients with refractory/relapsed AML after intensive therapy still needs further study. More recently, Chen et al. reported that the combination of L-ASNase with MIT and Ara-C for AML could enhance the inhibition of tumour cell proliferation (75). It is the current belief that the toxic effect of L-ASNase on AML may be related to GLNase activity. Glutamine is a nutrient that AML cells require. L-ASNase can remove glutamine and thus inhibit the growth of AML cells. However, L-ASNase simultaneously promotes the production of glutamine synthetase, leading to L-ASNase resistance (76, 77). Thus, further research is still needed to clarify the role of the GLNase activity of L-ASNase. Furthermore, as in ALL, it has been shown that the bone marrow haematopoietic microenvironment protects AML cells. Cells in the bone marrow microenvironment can either release ASNS to counteract L-ASNase action or release lysosomal cysteine protease B (CTSB) to inactivate L-ASNase, which confers L-ASNase resistance (78)
The potential of L-ASNase in CML treatment has been uncovered. Song et al. found that L-ASNase inhibited growth and induced apoptosis in the human CML cell Lines K562 and KU812, among which the apoptosis induction of K562 cells by L-ASNase was dependent on caspase3 (79). This discovery makes it possible to use L-ASNase in the treatment of CML. Trinh et al. also demonstrated that L-ASNase could inhibit the growth of CML cells, and the combination of L-ASNase and imatinib can significantly induce CML cell death by downregulating antiapoptotic factors such as Bcl-2 and upregulating proapoptotic factors such as Bim, and thereby eradicating CML stem cells (80). A recent study by Konhauser et al. also demonstrated the synergistic effect of L-ASNase in combination with etoposide on killing K562 cells (81).
With the continuous development of studies on the metabolic and nonmetabolic effects of L-ASNase on paediatric leukaemia, studies on the therapeutic effects of L-ASNase on other non-ALL leukaemia are proliferating. These studies suggest that L-ASNase may provide a new option for the treatment of other paediatric leukaemias. These results are based on the enzymatic activity of L-ASNase, which depletes asparagine and glutamine in the blood and inhibits mTOR, which in turn affects protein synthesis and induces apoptosis. Meanwhile, these studies found that L-ASNase caused the activation of protective autophagy in tumour cells, so the combination of L-ASNase and autophagy inhibitors will benefit both ALL patients and non-ALL patients.
As mentioned above, L-ASNase is a xenogeneic protein agent that is highly immunogenic. Efficacy is compromised during L-ASNase treatment because of immunological or nonimmunological side effects. Erwinia-ASNase is often chosen as an alternative treatment for patients with E.coli-ASNase allergy (82), and PEG-ASNase has been introduced into the clinic for its longer half-life and lower immunogenicity. However, none of these variants can completely solve the problem. Neutralizing antibodies can still be produced and therefore inactivating L-ASNases (83, 84). To address these issues, several approaches have been used to develop novel L-ASNase preparations, such as reduced GLNase coactivity of L-ASNase, enzyme engineering modifications, and vector packaging.
Since most of the nonimmunological side effects of L-ASNase are attributed to GLNase activity, reducing the GLNase coactivity of L-ASNase may effectively ameliorate the side effects of L-ASNase. Consequently, L-ASNase variants with or without negligible GLNase activity were generated. Wolinella succinogenes-derived L-ASNase (WOA) was the first reported L-ASNase variant with low GLNase activity that did not suppress immune responses in mice (85–87). Reinert et al. showed no significant changes in glutamine in the liver and spleen of mice treated with the WOA variant compared to L-ASNase (88). Recent studies have also identified a guinea pig-derived humanized variant of L-ASNase that is completely devoid of GLNase activity. This variant has reduced immunogenicity while maintaining anti-leukaemic activity (89, 90).
Enzyme engineering has been widely employed to change the characteristics of L-ASNase in search of L-ASNase with low immunogenicity, a longer half-life, and lower GLNase activity. Since L-ASNase can be cleaved by CTSB and aspartate endopeptidase (78, 91), Offman et al. used site-directed mutagenesis to design an L-ASNase variant that is resistant to CTSB cleavage and has lower immunogenicity. They also designed a variant with low GLNase activity, N24A/R159S, which reduced the toxicity of L-ASNase (25). Furthermore, in a recent study, Maggi et al. designed an N24S mutant with improved protease resistance and thermostability in response to the instability and brief half-life of E. coli-ASNase (92).
In addition to the above methods, carrier packaging can also be used to reduce the immunogenicity of L-ASNase and make it more stable in vivo. Common carriers include erythrocytes, liposomes, nanocapsules, and microcapsules. The performance of these L-ASNases has also been demonstrated in vivo and in vitro (93–95). For instance, because it is encapsulated within cells, Eryaspase, a product that encapsulates E. coli-ASNase into erythrocytes, has a long half-life similar to that of erythrocytes and has low immunogenicity (96–98). Last year, Eryaspase was approved by the FDA for the treatment of ALL patients who are allergic to PEG-ASNase (99).
In summary, L-ASNase is still the cornerstone drug for the treatment of paediatric ALL. In addition to affecting the protein synthesis and amino acid metabolism of ALL cells, L-ASNase can affect energy metabolism. Also, changes in energy metabolism and autophagy in ALL cells may affect the efficacy of L-ASNase. The focus of current research on the mechanism of L-ASNase resistance is gradually shifting from the protein level to the gene expression regulation level. Meanwhile, there are studies that elucidate the relationship between leukaemia metabolic profiles and autophagy and L-ASNase resistance. Although the mechanism of L-ASNase resistance has not been fully elucidated to date, these studies suggested that the combination of fatty acid oxidation inhibitors or autophagy inhibitors and L-ASNase can provide better anti-leukaemic effects, which provide brand-new options for the future treatment of childhood leukaemia.
The immunogenicity of L-ASNase is a reason for its drug resistance. Using carrier packaging L-ASNase such as erythrocytes and nanocapsules can effectively reduce its immunogenicity and therefore L-ASNase can work better. The performance of these L-ASNases has also been demonstrated in vivo. Moreover, the necessity of GLNase activity for the anticancer effect of L-ASNase is still highly controversial. Although the development of L-ASNase variants with low GLNase activity continues, the necessity of GLNase activity and the level of GLNase activity that should be maintained for L-ASNase still needs to be further investigated. Moreover, some glutamine-dependent haematological tumours may not benefit from L-ASNase variants without GLNase activity.
Finally, addressing the above issues will not only help to solve the problem of ALL resistance to L-ASNase but also help to explain the potential application of L-ASNase in other tumours.
TLia and TLi performed the collection and interpretations of all relevant literature. RZ wrote the manuscript. CC and JH critically read and revised the manuscript. All authors contributed to the article and approved the submitted version.
We thank Sanming Project of Medicine in Shenzhen (No.SZSM202011004), Shenzhen Science and Technology Innovation Commission (JCYJ20210324123004011, JCYJ20180307150419435) and Shenzhen Healthcare Research Project (Grant No. SZLY2018001) for supporting the manuscript preparation and publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pui CH, Bowman WP, Ribeiro RC, Onciu M, Jeha S, Simmons V, et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. n Engl J Med (2009) 12:2730–41. doi: 10.1056/NEJMoa0900386
2. Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L, Baruchel A, et al. L-asparaginase treatment in acute lymphoblastic leukemia: A focus on erwinia asparaginase. Cancer (2011) 117(2):238–49. doi: 10.1002/cncr.25489
3. Egler RA, Ahuja SP, Matloub Y. L-asparaginase in the treatment of patients with acute lymphoblastic leukemia. J Pharmacol Pharmacother. (2016) 7(2):62–71. doi: 10.4103/0976-500X.184769
4. Kidd JG. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. i. course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J Exp Med (1953) 98(6):565–82. doi: 10.1084/jem.98.6.565
5. Rizzari C, Putti MC, Colombini A, Casagranda S, Ferrari GM, Papayannidis C, et al. Rationale for a pediatric-inspired approach in the adolescent and young adult population with acute lymphoblastic leukemia, with a focus on asparaginase treatment. Hematol Rep (2014) 6(3):5554. doi: 10.4081/hr.2014.5554
6. Broome JD. Evidence that the l-asparaginase of guinea pig serum is responsible for its antilymphoma effects. II. lymphoma 6C3HED cells cultured in a medium devoid of l-asparagine lose their susceptibility to the effects of guinea pig serum in vivo. J Exp Med (1963) 118:121–48. doi: 10.1084/jem.118.1.121
7. Yellin TO, Wriston JC. Purification and properties of guinea pig serum asparaginase. Biochemistry (1966) 5(5):1605–12. doi: 10.1021/bi00869a022
8. Dolowy WC, Henson D, Cornet J, Sellin H. Toxic and antineoplastic effects of l-asparaginase. study of mice with lymphoma and normal monkeys and report on a child with leukemia. Cancer (1966) 19(12):1813–9. doi: 10.1002/1097-0142(196612)19:12<1813::aid-cncr2820191208>3.0.co;2-e
9. Clarkson B, Krakoff I, Burchenal J, Karnofsky D, Golbey R, Dowling M, et al. Clinical results of treatment with e. coli l-asparaginase in adults with leukemia, lymphoma, and solid tumors. Cancer (1970) 25(2):279–305. doi: 10.1002/1097-0142(197002)25:2<279::AID-CNCR2820250205>3.0.CO;2-7
10. Tong WH, Pieters R, Kaspers GJL, te Loo DMWM, Bierings MB, van den Bos C, et al. A prospective study on drug monitoring of PEGasparaginase and erwinia asparaginase and asparaginase antibodies in pediatric acute lymphoblastic leukemia. Blood (2014) 123(13):2026–33. doi: 10.1182/blood-2013-10-534347
11. Cecconello DK, de Magalhães MR, Werlang ICR, Lee ML de M, Michalowski MB, Daudt LE. Asparaginase: An old drug with new questions. Hematol Transfus Cell Ther (2020) 42(3):275–82. doi: 10.1016/j.htct.2019.07.010
12. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. (2021) 20(1):28. doi: 10.1186/s12943-021-01316-8
13. Fadaka A, Ajiboye B, Ojo O, Adewale O, Olayide I, Emuowhochere R. Biology of glucose metabolization in cancer cells. J Oncological Sci (2017) 3(2):45–51. doi: 10.1016/j.jons.2017.06.002
14. Lunt SY, Vander Heiden MG. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27(1):441–64. doi: 10.1146/annurev-cellbio-092910-154237
15. Vettore L, Westbrook RL, Tennant DA. New aspects of amino acid metabolism in cancer. Br J Cancer. (2020) 122(2):150–6. doi: 10.1038/s41416-019-0620-5
16. Choi BH, Coloff JL. The diverse functions of non-essential amino acids in cancer. Cancers (Basel). (2019) 11(5):675. doi: 10.3390/cancers11050675
17. Lomelino CL, Andring JT, McKenna R, Kilberg MS. Asparagine synthetase: Function, structure, and role in disease. J Biol Chem (2017) 292(49):19952–8. doi: 10.1074/jbc.R117.819060
18. Takahashi H, Inoue J, Sakaguchi K, Takagi M, Mizutani S, Inazawa J. Autophagy is required for cell survival under l-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene (2017) 36(30):4267–76. doi: 10.1038/onc.2017.59
19. Jiang J, Batra S, Zhang J. Asparagine: A metabolite to be targeted in cancers. Metabolites (2021) 11(6):402. doi: 10.3390/metabo11060402
20. Batool T, Makky EA, Jalal M, Yusoff MM. A comprehensive review on l-asparaginase and its applications. Appl Biochem Biotechnol (2016) 178(5):900–23. doi: 10.1007/s12010-015-1917-3
21. Hermanova I, Arruabarrena-Aristorena A, Valis K, Nuskova H, Alberich-Jorda M, Fiser K, et al. Pharmacological inhibition of fatty-acid oxidation synergistically enhances the effect of l-asparaginase in childhood ALL cells. Leukemia (2016) 30(1):209–18. doi: 10.1038/leu.2015.213
22. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
23. Gollwitzer P, Grützmacher N, Wilhelm S, Kümmel D, Demetriades C. A rag GTPase dimer code defines the regulation of mTORC1 by amino acids. Nat Cell Biol (2022) 24(9):1394–406. doi: 10.1038/s41556-022-00976-y
24. Miller HK, Balis ME. Glutaminase activity of l-asparagine amidohydrolase. Biochem Pharmacol (1969) 18(9):2225–32. doi: 10.1016/0006-2952(69)90329-3
25. Offman MN, Krol M, Patel N, Krishnan S, Liu J, Saha V, et al. Rational engineering of l-asparaginase reveals importance of dual activity for cancer cell toxicity. Blood (2011) 117(5):1614–21. doi: 10.1182/blood-2010-07-298422
26. Emadi A, Zokaee H, Sausville EA. Asparaginase in the treatment of non-ALL hematologic malignancies. Cancer Chemother Pharmacol (2014) 73(5):875–83. doi: 10.1007/s00280-014-2402-3
27. Ollenschläger G, Roth E, Linkesch W, Jansen S, Simmel A, Mödder B. Asparaginase-induced derangements of glutamine metabolism: the pathogenetic basis for some drug-related side-effects. Eur J Clin Invest (1988) 18(5):512–6. doi: 10.1111/j.1365-2362.1988.tb01049.x
28. Panosyan EH, Grigoryan RS, Avramis IA, Seibel NL, Gaynon PS, Siegel SE, et al. Deamination of glutamine is a prerequisite for optimal asparagine deamination by asparaginases In vivo (CCG-1961). Anticancer Res (2004) 24(2C):1121–6.
29. Parmentier JH, Maggi M, Tarasco E, Scotti C, Avramis VI, Mittelman SD. Glutaminase activity determines cytotoxicity of l-asparaginases on most leukemia cell lines. Leuk Res (2015) 39(7):757–62. doi: 10.1016/j.leukres.2015.04.008
30. Chan WK, Lorenzi PL, Anishkin A, Purwaha P, Rogers DM, Sukharev S, et al. The glutaminase activity of l-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood (2014) 123(23):3596–606. doi: 10.1182/blood-2013-10-535112
31. Nguyen HA, Su Y, Zhang JY, Antanasijevic A, Caffrey M, Schalk AM, et al. A novel l-asparaginase with low l-glutaminase coactivity is highly efficacious against both T and b cell acute lymphoblastic leukemias in vivo. Cancer Res (2018) 78(6):1549–60. doi: 10.1158/0008-5472.CAN-17-2106
32. Chan WK, Horvath TD, Tan L, Link T, Harutyunyan KG, Pontikos MA, et al. Glutaminase activity of l-asparaginase contributes to durable preclinical activity against acute lymphoblastic leukemia. Mol Cancer Ther (2019) 18(9):1587–92. doi: 10.1158/1535-7163.MCT-18-1329
33. Aslanian AM, Kilberg MS. Multiple adaptive mechanisms affect asparagine synthetase substrate availability in asparaginase-resistant MOLT-4 human leukaemia cells. Biochem J (. 2001) 358(Pt 1):59–67. doi: 10.1042/bj3580059
34. Horowitz B, Madras BK, Meister A, Old LJ, Boyes EA, Stockert E. Asparagine synthetase activity of mouse leukemias. Science (1968) 160(3827):533–5. doi: 10.1126/science.160.3827.533
35. Haskell CM, Canellos GP. L-asparaginase resistance in human leukemia–asparagine synthetase. Biochem Pharmacol (1969) 18(10):2578–80. doi: 10.1016/0006-2952(69)90375-X
36. Kiriyama Y, Kubota M, Takimoto T, Kitoh T, Tanizawa A, Akiyama Y, et al. Biochemical characterization of U937 cells resistant to l-asparaginase: the role of asparagine synthetase. Leukemia (1989) 3(4):294–7.
37. Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet (2000) 24(3):236–44. doi: 10.1038/73439
38. Holleman A, Cheok MH, den Boer ML, Yang W, Veerman AJP, Kazemier KM, et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med (2004) 351(6):533–42. doi: 10.1056/NEJMoa033513
39. Fine BM, Kaspers GJL, Ho M, Loonen AH, Boxer LM. A genome-wide view of the in vitro response to l-asparaginase in acute lymphoblastic leukemia. Cancer Res (2005) 65(1):291–9. doi: 10.1158/0008-5472.291.65.1
40. Stams WAG, den Boer ML, Holleman A, Appel IM, Beverloo HB, van Wering ER, et al. Asparagine synthetase expression is linked with l-asparaginase resistance in TEL-AML1–negative but not TEL-AML1–positive pediatric acute lymphoblastic leukemia. Blood (2005) 105(11):4223–5. doi: 10.1182/blood-2004-10-3892
41. Stams WAG, den Boer ML, Beverloo HB, Meijerink JPP, Stigter RL, van Wering ER, et al. Sensitivity to l-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+pediatric ALL. Blood (2003) 101(7):2743–7. doi: 10.1182/blood-2002-08-2446
42. Su N, Pan YX, Zhou M, Harvey RC, Hunger SP, Kilberg MS. Correlation between asparaginase sensitivity and asparagine synthetase protein content, but not mRNA, in acute lymphoblastic leukemia cell lines. Pediatr Blood Cancer. (2008) 50(2):274–9. doi: 10.1002/pbc.21213
43. Kilberg MS, Balasubramanian M, Fu L, Shan J. The transcription factor network associated with the amino acid response in mammalian Cells12. Adv Nutr (2012) 3(3):295–306. doi: 10.3945/an.112.001891
44. Balasubramanian MN, Butterworth EA, Kilberg MS. Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am J Physiol Endocrinol Metab (2013) 304(8):E789–99. doi: 10.1152/ajpendo.00015.2013
45. Ren Y, Roy S, Ding Y, Iqbal J, Broome JD. Methylation of the asparagine synthetase promoter in human leukemic cell lines is associated with a specific methyl binding protein. Oncogene (2004) 23(22):3953–61. doi: 10.1038/sj.onc.1207498
46. Jiang J, Srivastava S, Seim G, Pavlova NN, King B, Zou L, et al. Promoter demethylation of the asparagine synthetase gene is required for ATF4-dependent adaptation to asparagine depletion. J Biol Chem (2019) 294(49):18674–84. doi: 10.1074/jbc.RA119.010447
47. Akahane K, Kimura S, Miyake K, Watanabe A, Kagami K, Yoshimura K, et al. Association of allele-specific methylation of the ASNS gene with asparaginase sensitivity and prognosis in T-ALL. Blood Adv (2022) 6(1):212–24. doi: 10.1182/bloodadvances.2021004271
48. Touzart A, Lengliné E, Latiri M, Belhocine M, Smith C, Thomas X, et al. Epigenetic silencing affects l -asparaginase sensitivity and predicts outcome in T-ALL. Clin Cancer Res (2019) 25(8):2483–93. doi: 10.1158/1078-0432.CCR-18-1844
49. Ferguson DC, McCorkle JR, Barnett KR, Bonten EJ, Bergeron BP, Bhattarai KR, et al. Amino acid stress response genes promote l-asparaginase resistance in pediatric acute lymphoblastic leukemia. Blood Adv (2022) 6(11):3386–97. doi: 10.1182/bloodadvances.2022006965
50. Worton KS, Kerbel RS, Andrulis IL. Hypomethylation and reactivation of the asparagine synthetase gene induced by l-asparaginase and ethyl methanesulfonate. Cancer Res (1991) 51(3):985–9.
51. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell (2017) 170(4):605–35. doi: 10.1016/j.cell.2017.07.029
52. Martelli AM, Paganelli F, Fazio A, Bazzichetto C, Conciatori F, McCubrey JA. The key roles of PTEN in T-cell acute lymphoblastic leukemia development, progression, and therapeutic response. Cancers (Basel). (2019) 11(5):629. doi: 10.3390/cancers11050629
53. Hlozkova K, Hermanova I, Safrhansova L, Alquezar-Artieda N, Kuzilkova D, Vavrova A, et al. PTEN/PI3K/Akt pathway alters sensitivity of T-cell acute lymphoblastic leukemia to l-asparaginase. Sci Rep (2022) 12:4043. doi: 10.1038/s41598-022-08049-8
54. Hlozkova K, Pecinova A, Alquezar-Artieda N, Pajuelo-Reguera D, Simcikova M, Hovorkova L, et al. Metabolic profile of leukemia cells influences treatment efficacy of l-asparaginase. BMC Cancer (2020) 20(1):526. doi: 10.21203/rs.2.17519/v1
55. García Ruiz O, Sánchez-Maldonado JM, López-Nevot MÁ, García P, Macauda A, Hernández-Mohedo F, et al. Autophagy in hematological malignancies. Cancers (2022) 14(20):5072. doi: 10.3390/cancers14205072
56. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol (2007) 8(9):741–52. doi: 10.1038/nrm2239
57. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell (2006) 10(1):51–64. doi: 10.1016/j.ccr.2006.06.001
58. Ajoolabady A, Aghanejad A, Bi Y, Zhang Y, Aslkhodapasandhukmabad H, Abhari A, et al. Enzyme-based autophagy in anti-neoplastic management: From molecular mechanisms to clinical therapeutics. Biochim Biophys Acta (BBA) - Rev Cancer. (2020) 1874(1):188366. doi: 10.1016/j.bbcan.2020.188366
59. Takahashi H, Inoue J, Sakaguchi K, Takagi M, Mizutani S, Inazawa J. Autophagy inhibition sensitizes acute lymphoblastic leukemia cells to l-asparaginase. Blood (2015) 126(23):3772. doi: 10.1182/blood.V126.23.3772.3772
60. Polak R, Bierings MB, van der Leije CS, Sanders MA, Roovers O, Marchante JRM, et al. Autophagy inhibition as a potential future targeted therapy for ETV6-RUNX1-driven b-cell precursor acute lymphoblastic leukemia. Haematologica (2019) 104(4):738–48. doi: 10.3324/haematol.2018.193631
61. Chiu M, Franchi-Gazzola R, Bussolati O, D’Amico G, Dell’Acqua F, Rizzari C. Asparagine levels in the bone marrow of patients with acute lymphoblastic leukemia during asparaginase therapy. Pediatr Blood Cancer. (2013) 60(11):1915–5. doi: 10.1002/pbc.24663
62. Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. (2007) 117(4):1049–57. doi: 10.1172/JCI30235
63. Steiner M, Hochreiter D, Kasper DC, Kornmüller R, Pichler H, Haas OA, et al. Asparagine and aspartic acid concentrations in bone marrow versus peripheral blood during Berlin–Frankfurt–Münster-based induction therapy for childhood acute lymphoblastic leukemia. Leukemia Lymphoma. (2012) 53(9):1682–7. doi: 10.3109/10428194.2012.668681
64. Zalewska-Szewczyk B, Gach A, Wyka K, Bodalski J, Młynarski W. The cross-reactivity of anti-asparaginase antibodies against different l-asparaginase preparations. Clin Exp Med (2009) 9(2):113–6. doi: 10.1007/s10238-008-0026-9
65. Bernard C, Hall MP, Doede T. Intravenous and intramuscular administration of asparaginase in pediatric patients with acute lymphoblastic leukemia: Treatment patterns and perceptions. Blood (2015) 126(23):4914. doi: 10.1182/blood.V126.23.4914.4914
66. van der Sluis IM, Vrooman LM, Pieters R, Baruchel A, Escherich G, Goulden N, et al. Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation. Haematologica (2016) 101(3):279–85. doi: 10.3324/haematol.2015.137380
67. Vrooman LM, Kirov II, Dreyer ZE, Kelly M, Hijiya N, Brown P, et al. Activity and toxicity of intravenous erwinia asparaginase following allergy to e. coli-derived asparaginase in children and adolescents with acute lymphoblastic leukemia. Pediatr Blood Cancer. (2016) 63(2):228–33. doi: 10.1002/pbc.25757
68. Van Trimpont M, Peeters E, De Visser Y, Schalk AM, Mondelaers V, De Moerloose B, et al. Novel insights on the use of l-asparaginase as an efficient and safe anti-cancer therapy. Cancers (2022) 14(4):902. doi: 10.3390/cancers14040902
69. Kang SM, Rosales JL, Meier-Stephenson V, Kim S, Lee KY, Narendran A. Genome-wide loss-of-function genetic screening identifies opioid receptor μ1 as a key regulator of l-asparaginase resistance in pediatric acute lymphoblastic leukemia. Oncogene (2017) 36(42):5910–3. doi: 10.1038/onc.2017.211
70. Lee JK, Kang S, Wang X, Rosales JL, Gao X, Byun HG, et al. HAP1 loss confers l-asparaginase resistance in ALL by downregulating the calpain-1-Bid-caspase-3/12 pathway. Blood (2019) 133(20):2222–32. doi: 10.1182/blood-2018-12-890236
71. Hinze L, Pfirrmann M, Karim S, Degar J, McGuckin C, Vinjamur D, et al. Synthetic lethality of wnt pathway activation and asparaginase in drug-resistant acute leukemias. Cancer Cell (2019) 35(4):664–676.e7. doi: 10.1016/j.ccell.2019.03.004
72. Dübbers A, Würthwein G, Müller HJ, Schulze-Westhoff P, Winkelhorst M, Kurzknabe E, et al. Asparagine synthetase activity in paediatric acute leukaemias: AML-M5 subtype shows lowest activity. Br J Haematol (2000) 109(2):427–9. doi: 10.1046/j.1365-2141.2000.02015.x
73. Okada S, Hongo T, Yamada S, Watanabe C, Fujii Y, Ohzeki T, et al. In vitro efficacy of l-asparaginase in childhood acute myeloid leukaemia. Br J Haematol (2003) 123(5):802–9. doi: 10.1046/j.1365-2141.2003.04703.x
74. Buaboonnam J, Cao X, Pauley JL, Pui CH, Ribeiro RC, Rubnitz JE, et al. Sequential administration of methotrexate and asparaginase in relapsed or refractory pediatric acute myeloid leukemia. Pediatr Blood Cancer. (2013) 60(7):1161–4. doi: 10.1002/pbc.24470
75. Chen T, Zhang J, Zeng H, Zhang Y, Zhang Y, Zhou X, et al. Antiproliferative effects of l-asparaginase in acute myeloid leukemia. Exp Ther Med (2020) 20(3):2070–8. doi: 10.3892/etm.2020.8904
76. Willems L, Jacque N, Jacquel A, Neveux N, Trovati Maciel T, Lambert M, et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood (2013) 122(20):3521–32. doi: 10.1182/blood-2013-03-493163
77. Emadi A, Law JY, Strovel ET, Lapidus RG, Jeng LJB, Lee M, et al. Asparaginase erwinia chrysanthemi effectively depletes plasma glutamine in adult patients with relapsed/refractory acute myeloid leukemia. Cancer Chemother Pharmacol (2018) 81(1):217–22. doi: 10.1007/s00280-017-3459-6
78. Kaspers GJL. Acute myeloid leukaemia niche regulates response to l-asparaginase. Br J Haematol (2019) 186(3):397–9. doi: 10.1111/bjh.15924
79. Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z, et al. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget (2015) 6(6):3861–73. doi: 10.18632/oncotarget.2869
80. Trinh A, Khamari R, Fovez Q, Mahon FX, Turcq B, Bouscary D, et al. Antimetabolic cooperativity with the clinically approved l-asparaginase and tyrosine kinase inhibitors to eradicate CML stem cells. Mol Metab (2022) 55:101410. doi: 10.1016/j.molmet.2021.101410
81. Konhäuser M, Kannaujiya VK, Steiert E, Schwickert K, Schirmeister T, Wich PR. Co-Encapsulation of l-asparaginase and etoposide in dextran nanoparticles for synergistic effect in chronic myeloid leukemia cells. Int J Pharmaceutics. (2022) 622:121796. doi: 10.1016/j.ijpharm.2022.121796
82. Salzer W, Seibel N, Smith M. Erwinia asparaginase in pediatric acute lymphoblastic leukemia. Expert Opin Biol Ther (2012) 12(10):1407–14. doi: 10.1517/14712598.2012.718327
83. Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, et al. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer (2007) 110(1):103–11. doi: 10.1002/cncr.22739
84. Kloos R, van der Sluis IM, Mastrobattista E, Hennink W, Pieters R, Verhoef JJ. Acute lymphoblastic leukaemia patients treated with PEGasparaginase develop antibodies to PEG and the succinate linker. Br J Haematol (2020) 189(3):442–51. doi: 10.1111/bjh.16254
85. Durden DL, Distasio JA. Comparison of the immunosuppressive effects of asparaginases from escherichia coli and vibrio succinogenes1. Cancer Res (1980) 40(4):1125–9.
86. Distasio JA, Niederman RA, Kafkewitz D, Goodman D. Purification and characterization of l-asparaginase with anti-lymphoma activity from vibrio succinogenes. J Biol Chem (1976) 251(22):6929–33. doi: 10.1016/S0021-9258(17)32924-1
87. Distasio JA, Durden DL, Paul RD, Nadji M. Alteration in spleen lymphoid populations associated with specific amino acid depletion during l-asparaginase Treatment1. Cancer Res (1982) 42(1):252–8.
88. Reinert RB, Oberle LM, Wek SA, Bunpo P, Wang XP, Mileva I, et al. Role of glutamine depletion in directing tissue-specific nutrient stress responses to l-asparaginase *. J Biol Chem (2006) 281(42):31222–33. doi: 10.1016/S0021-9258(19)84035-8
89. Schalk AM, Lavie A. Structural and kinetic characterization of Guinea pig l-asparaginase type III. Biochemistry (2014) 53(14):2318–28. doi: 10.1021/bi401692v
90. Rigouin C, Nguyen HA, Schalk AM, Lavie A. Discovery of human-like l-asparaginases with potential clinical use by directed evolution. Sci Rep (2017) 7:10224. doi: 10.1038/s41598-017-10758-4
91. Patel N, Krishnan S, Offman MN, Krol M, Moss CX, Leighton C, et al. A dyad of lymphoblastic lysosomal cysteine proteases degrades the antileukemic drug l-asparaginase. J Clin Invest. (2009) 119(7):1964–73. doi: 10.1172/JCI37977
92. Maggi M, Mittelman SD, Parmentier JH, Colombo G, Meli M, Whitmire JM, et al. A protease-resistant escherichia coli asparaginase with outstanding stability and enhanced anti-leukaemic activity in vitro. Sci Rep (2017) 7:14479. doi: 10.1038/s41598-017-15075-4
93. Karamitros CS, Yashchenok AM, Möhwald H, Skirtach AG, Konrad M. Preserving catalytic activity and enhancing biochemical stability of the therapeutic enzyme asparaginase by biocompatible multilayered polyelectrolyte microcapsules. Biomacromolecules (2013) 14(12):4398–406. doi: 10.1021/bm401341k
94. da S, Pereira A, Souza CPL, Moraes L, Fontes-Sant’Ana GC, Amaral PFF. Polymers as encapsulating agents and delivery vehicles of enzymes. Polymers (2021) 13(23):4061. doi: 10.3390/polym13234061
95. Sindhu R, Manonmani HK. L-asparaginase-mediated therapy in l-asparagine auxotrophic cancers: A review. Anti-Cancer Agents Medicinal Chem (2022) 22(13):2393–410. doi: 10.2174/1871520622666220106103336
96. Kwon YM, Chung HS, Moon C, Yockman J, Park YJ, Gitlin SD, et al. L-asparaginase encapsulated intact erythrocytes for treatment of acute lymphoblastic leukemia (ALL). J Control Release. (2009) 139(3):182–9. doi: 10.1016/j.jconrel.2009.06.027
97. Rossi L, Pierigè F, Aliano MP, Magnani M. Ongoing developments and clinical progress in drug-loaded red blood cell technologies. BioDrugs (2020) 34(3):265–72. doi: 10.1007/s40259-020-00415-0
98. Koleva L, Bovt E, Ataullakhanov F, Sinauridze E. Erythrocytes as carriers: From drug delivery to biosensors. Pharmaceutics (2020) 12(3):276. doi: 10.3390/pharmaceutics12030276
99. ERYTECH granted U.S. FDA fast track designation for eryaspase in hypersensitive ALL. ERYTECH PHARMA SA. Available at: https://investors.erytech.com/news-releases/news-release-details/erytech-granted-us-fda-fast-track-designation-eryaspase.
Keywords: L-asparaginase, asparagine synthetase, metabolic, drug resistance, childhood leukaemia
Citation: Zhou R, Liang T, Li T, Huang J and Chen C (2023) Possible mechanism of metabolic and drug resistance with L-asparaginase therapy in childhood leukaemia. Front. Oncol. 13:1070069. doi: 10.3389/fonc.2023.1070069
Received: 14 October 2022; Accepted: 20 January 2023;
Published: 01 February 2023.
Edited by:
Jian Yu, Beihang University, ChinaReviewed by:
Angelo Brunelli Albertoni Laranjeira, Washington University in St. Louis, United StatesCopyright © 2023 Zhou, Liang, Li, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junbin Huang, aHVhbmdqYjM3QG1haWwuc3lzdS5lZHUuY24=; Chun Chen, Y2hlbmNodW5AbWFpbC5zeXN1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.