 Anand Khadse1,2Vilde D. Haakensen1,3*Laxmi Silwal-Pandit1Julian Hamfjord1,3Patrick Micke4Johan Botling4Odd Terje Brustugun1,5Ole Christian Lingjærde1,6Åslaug Helland1,3,7†Elin H. Kure1,2†
Anand Khadse1,2Vilde D. Haakensen1,3*Laxmi Silwal-Pandit1Julian Hamfjord1,3Patrick Micke4Johan Botling4Odd Terje Brustugun1,5Ole Christian Lingjærde1,6Åslaug Helland1,3,7†Elin H. Kure1,2†- 1Department of Cancer Genetics, Institute for Cancer Research, Oslo University Hospital, Oslo, Norway
- 2Faculty of Technology, Natural Sciences and Maritime Sciences, Department of Natural Sciences and Environmental Health, University of South-Eastern Norway, Bø i Telemark, Norway
- 3Department of Oncology, Oslo University Hospital, Oslo, Norway
- 4Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden
- 5Section of Oncology, Drammen Hospital, Vestre Viken Hospital Trust, Drammen, Norway
- 6Centre for Bioinformatics, Department of Informatics, University of Oslo, Oslo, Norway
- 7Department of Clinical Medicine, University of Oslo, Oslo, Norway
Lung cancer is a common disease with a poor prognosis. Genomic alterations involving the KRAS gene are common in lung carcinomas, although much is unknown about how different mutations, deletions, and expressions influence the disease course. The first approval of a KRAS-directed inhibitor was recently approved by the FDA. Mutations in the KRAS gene have been associated with poor prognosis for lung adenocarcinomas, but implications of the loss of heterozygosity (LOH) of KRAS have not been investigated. In this study, we have assessed the LOH of KRAS in early-stage lung adenocarcinoma by analyzing DNA copy number profiles and have investigated the effect on patient outcome in association with mRNA expression and somatic hotspot mutations. KRAS mutation was present in 36% of cases and was associated with elevated mRNA expression. LOH in KRAS was associated with a favorable prognosis, more prominently in KRAS mutated than in wild-type patients. The presence of both LOH and mutation in KRAS conferred a better prognosis than KRAS mutation alone. For wild-type tumors, no difference in prognosis was observed between patients with and without LOH in KRAS. Our study indicates that LOH in KRAS is an independent prognostic factor that may refine the existing prognostic groups of lung adenocarcinomas.
Introduction
Lung cancer is the leading cause of cancer-related deaths, causing an estimated 1.8 million deaths worldwide in 2018 (1). The overall 5-year survival rate for lung cancer patients during the period 2015–2019 in Norway was 22.7% in men and 29.2% in women, respectively (2). Lung adenocarcinoma is the most common type of non-small cell lung cancer (NSCLC) and accounts for 46% of all lung cancers in men and 52% in women (3). Adenocarcinoma is the most common type of lung cancer in never-smokers regardless of their age (4). Chromosomal abnormalities are frequent events in lung cancer, and both mutations and copy number aberrations can be the main drivers of the disease (5–7). Specific patterns of copy number gains and losses have been associated with histological subtypes of lung carcinomas, with lung adenocarcinoma displaying relatively fewer copy number alterations than lung squamous cell carcinoma and indicated to be mutation-driven (8, 9).
Deregulation of the Ras pathway by an activating Kirsten rat sarcoma viral oncogene (KRAS) mutation, occurring in about 30% of lung adenocarcinoma patients, is a hallmark of NSCLC (10). Previous studies have identified KRAS and TP53 mutations (46%) as early events in carcinogenesis in patients with early-stage lung adenocarcinoma (11, 12). EGFR mutations (14%) are more common in lung adenocarcinomas of patients who never smoked, and those who exhibit such mutations benefit from EGFR inhibitors. They are found mutually exclusive with KRAS mutations which are associated with significant tobacco exposure (13, 14). KRAS mutations were described as a negative prognostic marker in metastatic lung adenocarcinoma (12, 15); however, the results have been inconsistent for early-stage disease and are still debated (16–18). An independent prognostic impact of KRAS mutations has been difficult to establish in relation to confounding concurrent tobacco-associated mutations such as TP53, STK11, and KEAP1 (19). In recent years, tyrosine kinase inhibitors targeting ROS1 mutations and ALK translocations have been introduced as the standard of care. Multiple therapies targeting the alterations in the RTK/RAS/RAF and AKT/PI3K pathways have been in development. Amplifications in MET, PI3KCA, and ERBB2 were also in focus (7, 20). KRAS mutations have recently emerged as a useful negative predictive biomarker, predicting when therapy is unlikely to work. Despite decades of research, mutations in KRAS have been difficult to target due to the lack of surface targets for binding and its high affinity for GTP (21). Yet, recent results indicate the clinical effect of a selective KRASG12C inhibitor in a subgroup of patients with locally advanced or metastatic NSCLC (22, 23). The discovery provides an opportunity to selectively target KRASG12C in patients.
Loss of heterozygosity (LOH) is frequently observed in NSCLC, more frequent in squamous cell carcinomas than adenocarcinomas (24, 25). Several studies have reported allelic loss of chromosome 12p where KRAS resides, and that a loss correlates with the presence of KRAS mutation in human lung tumors (26). However, these studies did not investigate the prognostic impact of LOH in KRAS. Here, we have analyzed copy number profiles along with transcriptomic and mutation data of early-stage lung adenocarcinomas and assessed the prognostic significance of LOH in KRAS.
Materials and Methods
Patient Cohort
Participants included in the study were patients with operable early-stage lung adenocarcinomas surgically resected at Oslo University Hospital (OUH) from 2006 to 2011 (n = 133) and Uppsala University Hospital from 1995 to 2005 (n = 100). The study was approved by the Regional Committees for Medical and Health Research Ethics (REC) - South East Norway (reference no. S-06402b). We confirmed that all methods were performed in accordance with the relevant guidelines and regulations. All patients received oral and written information about the project and signed a written consent before entering the study. Clinical data from medical journals including follow-up were available for all patients. The main characteristics of the patients included are listed in Table 1. Some patients from stage II and stage III received adjuvant chemotherapy following standard guidelines. Tumor tissue was dissected and snap-frozen in liquid nitrogen and stored at −80°C until DNA and RNA isolation as previously described (27, 28). Tumor cellularity was estimated using the Allele-Specific Copy number Analysis of Tumors v2.3 (ASCAT) algorithm, and the samples with estimated tumor cell fraction greater than 20% were retained in the analysis.
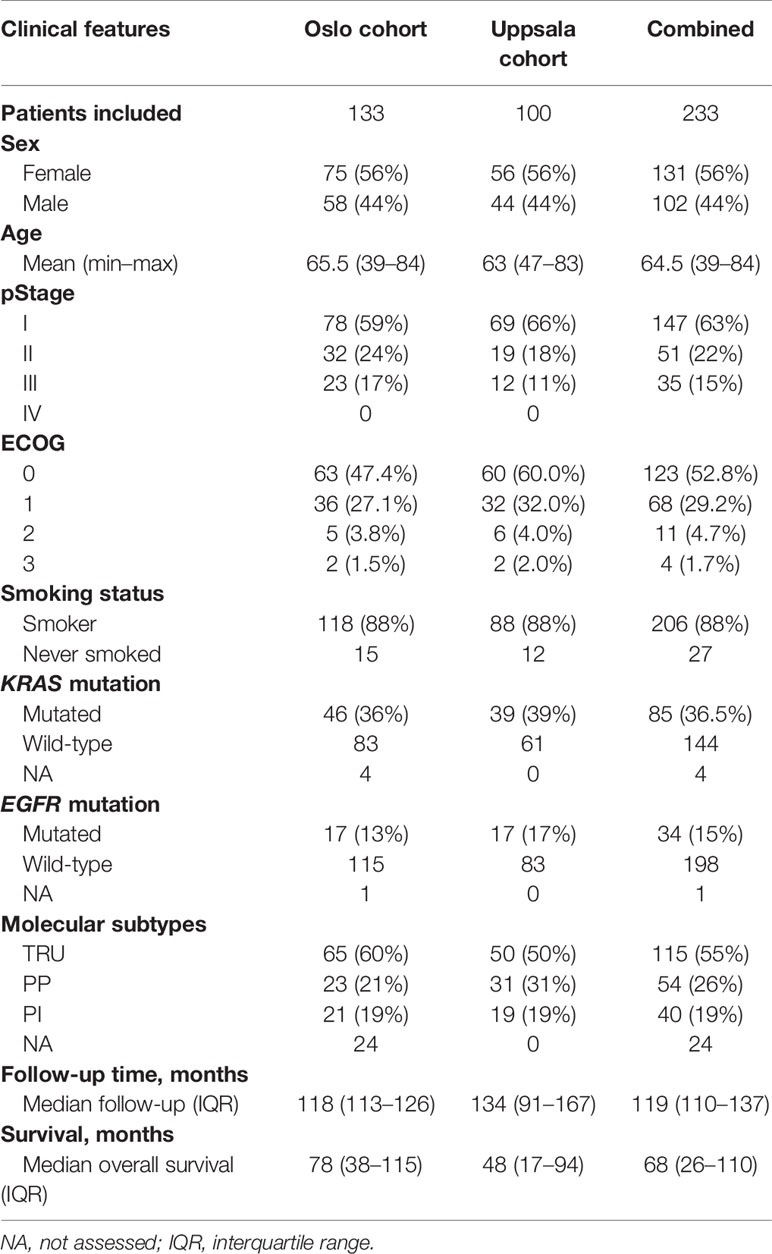
Table 1 Patient characteristics.
Mutation Data Acquisition
Genomic DNA was extracted from frozen tumor tissue using the Maxwell® 16 DNA purification kit (Promega, Madison, WI, USA) for the Oslo cohort and the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) for the Uppsala cohort, following the standard manufacturer’s protocol. EGFR mutation analyses of exons 18–21 were performed by real-time PCR using the therascreen EGFR mutation kit (Qiagen, Hilden, Germany) for the Oslo cohort. For the Uppsala cohort, PCR amplification of EGFR exons 18–21 was performed using the GeneAmp 2700 PCR cycler (Applied Biosystems, Waltham, MA, USA) and the ready-to-use ABgene PCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) to determine EGFR mutation status. The Oslo cohort was analyzed for KRAS mutations using the Wobble-enhanced ARMS (WE-ARMS) method (29), while pyrosequencing and the PyroMark Q24 KRAS Kit (Qiagen, Hilden, Germany) were used to detect mutations in KRAS codons 12/13 (exon 2) and 61 (exon 3) in the Uppsala cohort. Separate PCR reactions for codons 12/13 and 61 were performed on the GeneAmp 2700 PCR cycler (Applied Biosystems, Waltham, MA, USA) as described in the original article (9).
Gene Expression Profiling
The gene expression microarray SurePrint G3 Human GE, 8 × 60K (Agilent Technologies, Santa Clara, CA, USA) data for the subset of the Oslo cohort (n = 110) with GEO accession number GSE66863 was published previously (28). The data were log2-transformed and normalized between arrays by using the 75th percentile method in GeneSpring GX v.12.1 analysis software (Agilent Technologies, Santa Clara, CA, USA). The mRNA expression array includes 42,066 unique probes, and 30,370 probes remained after filtering out probes with no gene annotation or available gene names. The mRNA expression data for the Uppsala cohort (n = 100) were available on GEO (accession GSE37745). The samples were analyzed using Affymetrix Human Genome U133 Plus 2.0 Array and normalized using the Robust Multiarray Average (RMA) method. The average gene expression value was calculated when a gene mapped to more than one probe at the array.
Estimation of Allele-Specific Copy Numbers
In the Oslo cohort, genomic DNA was extracted from the frozen tumor tissue using the Maxwell® 16 DNA purification kit following standard protocol. The DNA was hybridized to Affymetrix genome-wide human SNP 6.0 arrays following the manufacturer’s instructions (Affymetrix, Santa Clara, CA, USA) at AROS Applied Biotechnology A/S (Aarhus, Denmark). Raw signal intensities were extracted and quantile-normalized using the Affymetrix Power Tools (APT) software and the PennCNV software to obtain log-transformed total signal intensities (LogR) for all probes and B allele frequencies (BAF) for SNP probes. After adjusting LogR for GC-binding artifacts, the LogR and BAF values were used as input for the allele-specific segmentation of normalized raw data by ASPCF with penalty parameter gamma = 70 and the subsequent analysis with ASCAT (30). The result was an allele-specific copy number profile of each tumor as well as estimates of tumor ploidy and tumor cell fraction (cellularity). ASCAT profiles were successfully obtained for 133 samples in the Oslo cohort, and these were used in the subsequent downstream analyses. Tumor samples of 104 lung adenocarcinomas from the Uppsala cohort were analyzed to obtain copy number profiles (31). Affymetrix Gene Chip Human Mapping 250K Nsp I arrays were used according to the manufacturer’s directions for the genomic DNA extracted from fresh frozen lung cancer tissue. Copy number analysis was performed using the ASCAT pipeline described above with changing platform parameter to “Affy250k_nsp” and obtained the ASCAT profiles for 100 samples by excluding four metastatic tumor profiles.
An ASCAT profile provides a segmentation of the genome into regions of constant allele-specific copy numbers. The total copy number of a segment is the sum of the major and minor allele copy numbers. Genomic regions with a total copy number greater or smaller than the tumor ploidy were considered as gains (amplifications) and losses (deletions), respectively. The ploidy-adjusted total copy number of a genomic region is determined by subtracting tumor ploidy from the total copy number of the region and rounded to the nearest integer. The genomic region with ploidy-adjusted total copy number 1 or above (i.e., 1, 2, 3…) was assigned as gain, whereas the region with a negative ploidy-adjusted total copy number (i.e., −1, −2, −3…) was assigned as loss. The hg19 genomic coordinates of SNP probes given in the array annotation file (release 35) were used to map aberrant genomic regions to the gene coordinates obtained from refFlat annotations. The chromosomal regions where only one allele (major or minor) was present were identified as regions with LOH. For each tumor, the genome instability index (GII) is defined as the fraction of the genome with loss or gain; in practice, this is calculated as the fraction of probes within segments with loss or gains.
Statistical Analyses
All statistical analyses were performed using R version 3.6.2. The Pearson correlation coefficient was used to estimate correlations, and the false discovery rate (FDR) was used to correct for multiple testing. Pearson’s Chi-squared test, Fisher’s exact test, t-test, or logistic regression was used when appropriate to test associations between different variables. Regions with a significant difference in gains or losses at a given position in two groups were determined using the two-proportion z-test implemented in the prop.test function.
Frequency Plot
Samples were divided into two groups based on LOH status (present or absent) in the KRAS region. The frequency of gain (or loss) at a given genomic position in a group was calculated as the proportion of samples in that group with the aberration. Frequencies of gains were plotted on the y-axis in a positive scale, while the frequencies of losses are plotted in a negative scale. Chromosome-wise genomic positions are plotted on the x-axis.
Molecular Subtype Assignment
Adenocarcinomas were classified as terminal respiratory unit (TRU, formerly bronchioid), proximal-proliferative (PP, formerly magnoid), or proximal-inflammatory (PI, formerly squamoid) using the previously published centroid classifiers for adenocarcinomas (32). The subtype predictor centroids of 506 genes were used and samples were assigned to the closest centroid subtype.
Genome-Wide Correlation Analysis
The Pearson correlation coefficient was calculated to estimate the correlation between the copy number alteration and mRNA expression in 197 samples for which both copy number and expression data were available. The allele-specific copy number values of the genes were compared against normalized mRNA expression data. The correlation coefficients and p-values were reported for the regions where significant association (adjusted p < 0.05) was found between the gene expression and copy number state. The functional enrichment analysis of correlating genes was carried out using the Database for Annotation, Visualization and Integrated Discovery (DAVID) Functional Annotation Tool v.6.8.
Survival Analysis
The Kaplan–Meier estimator was used for the visualization of survival curves, the Log-rank test was used for testing differences between survival curves, and Cox proportional hazards (PH) regression was used to model and investigate survival as a function of covariates. All analyses were performed using the R package survival version 3.1.8. Overall survival (OS) time was calculated from the date of surgery to the time of death or censoring. Relapse-free survival (RFS) time was calculated from the date of surgery to the date of recurrence of disease or censoring. Patients were censored as of March 2020. Factors predicting the outcomes were assessed using the Cox proportional hazards model. Multivariate Cox proportional hazards regression analyses (adjusted for age and tumor stage) were used to analyze the correlation between KRAS LOH and survival in lung adenocarcinoma patients.
Results
Association of KRAS LOH to Clinicopathological and Molecular Features
In order to investigate the possible genetic changes associated with LOH in KRAS, the tumors were further divided into two groups based on LOH at the KRAS locus (12p12.1). No significant association was observed between KRAS LOH and clinicopathological characteristics in this patient cohort (Table 2). Mutations and GII in the samples are illustrated in Figure 1A. There was no clear association between KRAS LOH and mutation (odds ratio = 0.51, Fisher’s exact p = 0.057). Tumors with LOH at the KRAS locus had an overall higher genomic instability than tumors without LOH (average GII of 0.57 compared to 0.46; p < 0.001, t-test), although high GII was not found to be associated with the overall survival [HR = 0.913; 95% confidence interval (CI) 0.427–1.951; p = 0.814].

Table 2 Association between KRAS LOH status and clinicopathological characteristics.
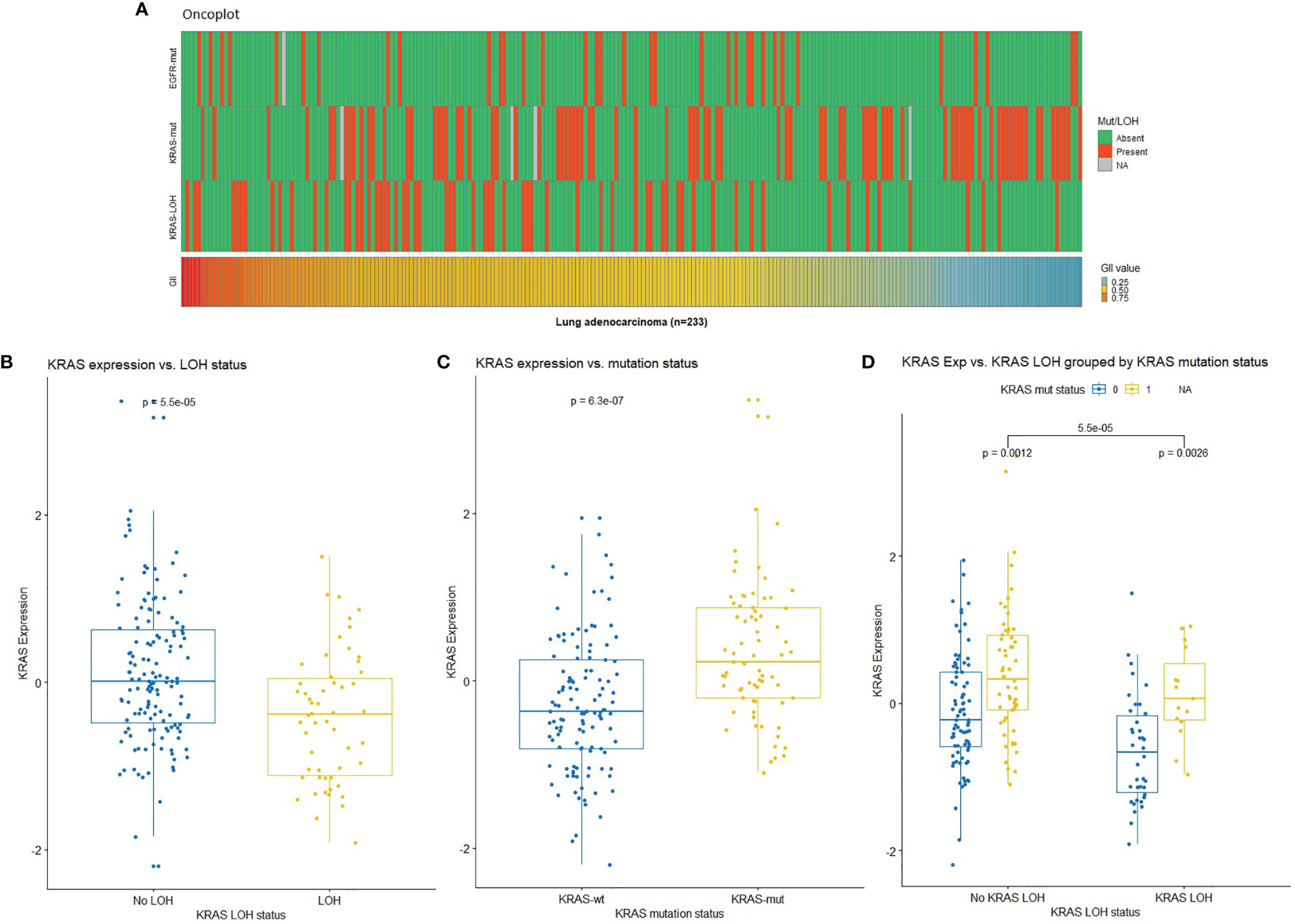
Figure 1 (A) Oncoplot showing mutation status in EGFR and KRAS (red: mutated, green: wild-type, gray: missing), loss of heterozygosity (LOH) at the KRAS locus (red: LOH, green: no LOH), and genomic instability index (GII) in the Oslo cohort (red: high, blue: low). (B) KRAS expression by KRAS LOH status indicating higher expression in samples with no LOH in KRAS. (C) KRAS expression by KRAS mutation status indicating high expression in samples with KRAS mutation (wt: wild-type; mut: mutation). (D) KRAS expression by KRAS LOH status grouped by KRAS mutation status in lung adenocarcinomas with respect to KRAS LOH and mutation status [0: wild-type (blue); 1: mutation (yellow)]. NA, Not available.
Genomic Aberrations in Early-Stage Lung Adenocarcinomas
The lung adenocarcinomas displayed overall complex DNA copy number profiles with recurrent aberrations in almost all chromosomes. Recurrent gains were observed on 1q, 5p, 6p, 7p, 8q, 14p, 17q, and 20p in more than 25% of cases, and similarly, recurrent losses on 3p, 5q, 6q, 8p, 9p, 9q, 10q, 13q, 15q, 17p, 18q, 19p, 21q, and 22q were observed in more than 25% of cases. LOH was observed in the KRAS gene in 26.6% of cases. Frequencies of genomic alterations, i.e., the percentage of patients with gains or losses across the genome, are shown for the complete lung adenocarcinoma cohort (n = 233) (Figure S1A) and in individual cohorts (Figure S1B) in Supplementary Figure S1. Hotspot mutation frequency of the total number of samples was 36.5% for KRAS and 14.6% for EGFR. EGFR and KRAS mutations were mutually exclusive. The most frequent KRAS mutations observed in the samples were G12C (37%), G12V (21%), and G12D (19%) (Supplementary Figure S2).
Significant differences in gains or losses across 15-kb genomic intervals were identified using prop.test (p < 0.05) by comparing the proportion of amplification or deletion at a given interval in the two groups. Chromosomes 1p, 7p/q, 8p, 12p, 13q, and 16q showed significant differences in gains between the two groups. Similarly, significant differences in losses were identified at 1p, 2q, 5p/q, 9p/q, 10q, 12 p/q, 13q, 15q, 17p/q, 19q, and Xp region (Figure S1B).
None of the samples had a complete loss of KRAS. Twelve samples had copy-neutral LOH in KRAS. These samples had loss of one KRAS allele, but the total copy number at the locus was equal to tumor ploidy. The expression of KRAS was lower in the samples with LOH at KRAS compared with samples without LOH (Figure 1B). We observed higher KRAS expression in KRAS mutated compared with wild-type samples (Figure 1C). When the samples were stratified by KRAS mutation status, mutant KRAS samples showed higher expression than wild-type KRAS samples in KRAS LOH-positive samples (Figure 1D).
Prognostic Significance of Loss of Heterozygosity in KRAS
Lung adenocarcinoma patients with LOH in KRAS had significantly better OS (HR = 0.65; 95% CI 0.45–0.95; p = 0.025) (Figure 2A) compared with patients with no KRAS LOH. A similar trend was also seen in RFS although statistical significance was not reached (HR = 0.64; 95% CI 0.39–1.06; p = 0.08) (Figure 2B). Multivariate analyses also suggest a tendency of improved OS for patients with LOH in KRAS compared with no LOH (HR = 0.69; 95% CI 0.46–1.06; p = 0.09) (Table 3).
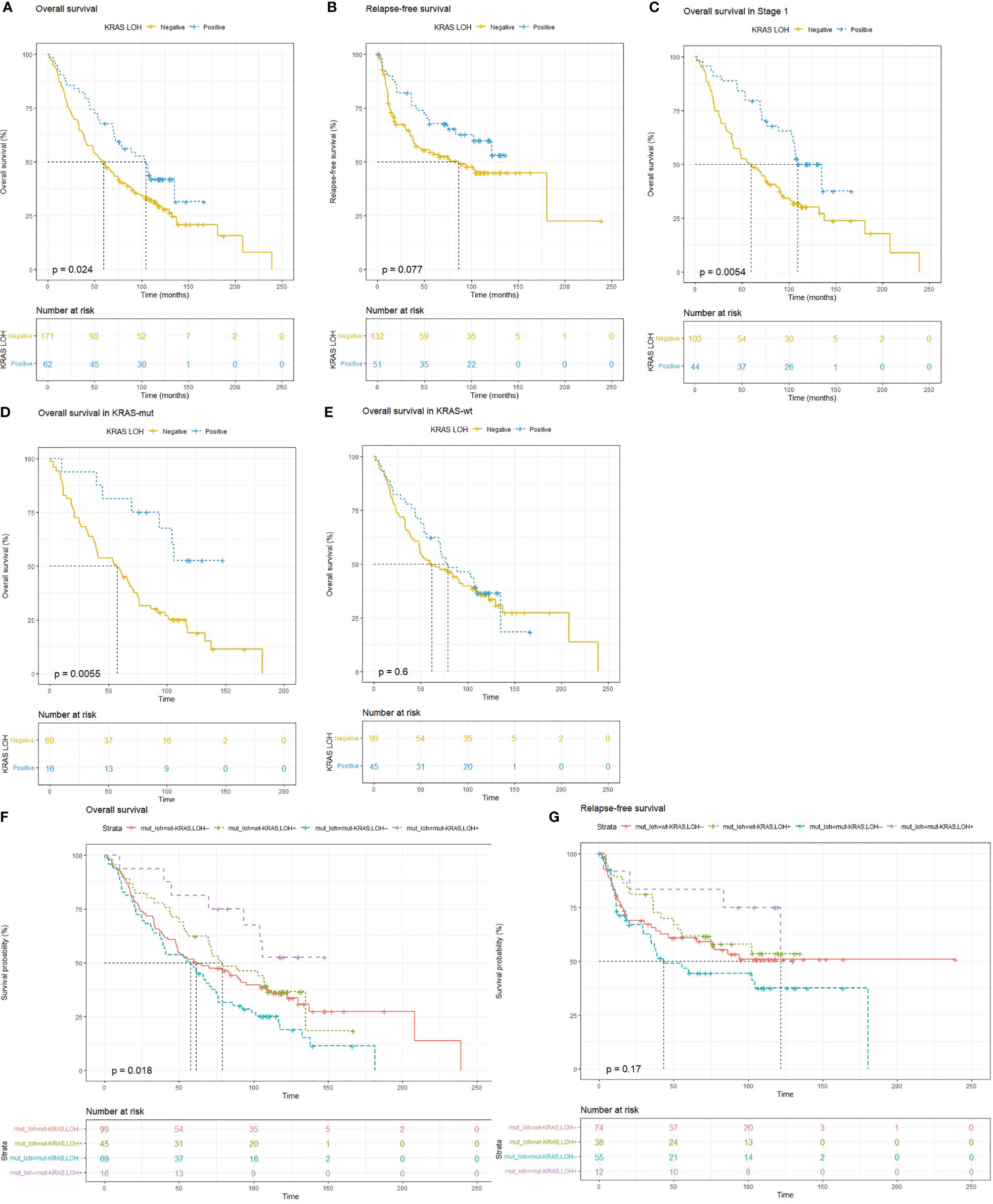
Figure 2 Kaplan–Meier survival plots for (A) overall survival (OS) and (B) relapse-free survival (RFS) in patients with lung adenocarcinomas with and without LOH in KRAS indicating improved survival with LOH in KRAS. (C) OS in patients with stage I disease. (D) OS in wild-type KRAS tumors and (E) KRAS mutated tumors. (F, G) OS and RFS in patients based on combined KRAS mutation and LOH status, where patients with both KRAS mutation and KRAS LOH (purple) have better OS and RFS, whereas patients with only KRAS mutation and no KRAS LOH (blue) have the worst OS as well as RFS.
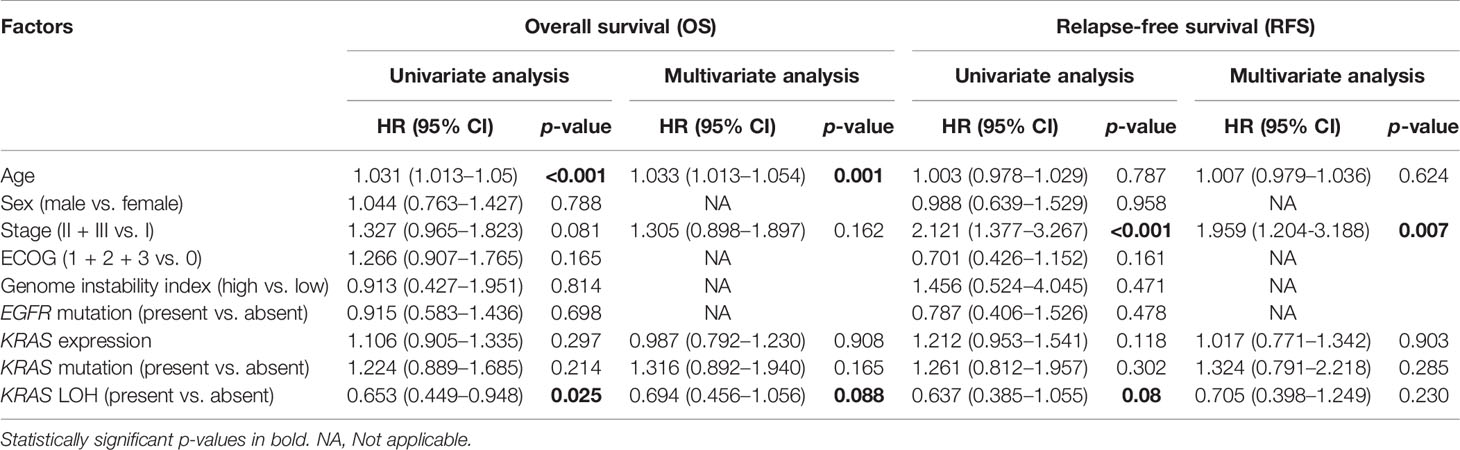
Table 3 Univariate and multivariate Cox proportional hazards regression analyses of different prognostic variables for overall survival (OS) and relapse-free survival (RFS) in early-stage lung adenocarcinoma.
In order to adjust the effect of any potential confounder on survival associated with LOH in KRAS, we performed bivariate Cox proportional hazards regression analyses with LOH in KRAS as a fixed independent variable including potential covariate in the bivariate model (Supplementary Table S1). The analysis showed that the effect of LOH in KRAS on OS as well as on RFS was unaffected even after adjusting the effect of confounders such as genomic instability and KRAS mutations, suggesting LOH in KRAS as an independent prognostic factor.
KRAS LOH in Stage I Lung Adenocarcinomas
The Cox proportional hazards regression model for patients with LOH in KRAS adjusted for progression stage suggested reduced hazard for OS [HR = 0.682 (0.46–0.99), p = 0.04]. The difference in the fraction of samples with KRAS LOH in different stages can be observed in Table 2. In stage I, a greater number of samples had LOH in KRAS. To remove the stage bias, we assessed the effect of KRAS LOH in stage I patients separately. The Kaplan–Meier estimator showed that patients with stage I disease and LOH at KRAS had better OS (p = 0.005) than patients with no KRAS LOH. The median OS in the patients with LOH in KRAS was 9 years, compared with 5 years in the patients with both alleles intact (Figure 2C).
KRAS LOH According to KRAS Mutation Status
To test whether LOH in the KRAS locus influenced prognosis in KRAS mutated patients, we performed Kaplan–Meier analysis on the patients with KRAS mutation and patients with wild-type KRAS separately. In the subgroup of patients with KRAS mutation, we found that LOH in KRAS conferred better OS (p < 0.01) compared with no LOH. No such difference in survival was observed in patients with wild-type KRAS (Figures 2D, E). We further evaluated the combined effect of KRAS LOH and KRAS mutation on overall survival and recurrence-free survival of the patients (Figures 2F, G). Kaplan–Meier curves suggest a significantly shorter survival time in patients with mutated KRAS without KRAS LOH, whereas patients with mutated KRAS with LOH have the most favorable OS as well as RFS.
Alterations in Hotspots
We assessed common hotspot mutation regions for copy number alterations in lung adenocarcinomas to determine their concurrences and associations with LOH in KRAS. The copy number changes and the LOH statistics for EGFR, TP53, ALK, ERBB2, BRAF, MET, RET, ROS1, HER2, NTRK, STK11, and PIK3CA are given in Table 4. The copy number changes and the LOH in common hotspot gene regions relative to KRAS in lung adenocarcinoma samples are shown in Figure 3. The chi-square test shows the LOH in TP53, ERBB2, BRAF, RET, NTRK3, and PIK3CA associated with LOH in KRAS. Sixty percent of the samples had TP53 copy number loss and 85% of the KRAS LOH samples also had LOH in TP53 with statistically significant association. Similarly, STK11 had a high percentage (64%) of LOH in the samples, but they are not associated with LOH in KRAS.
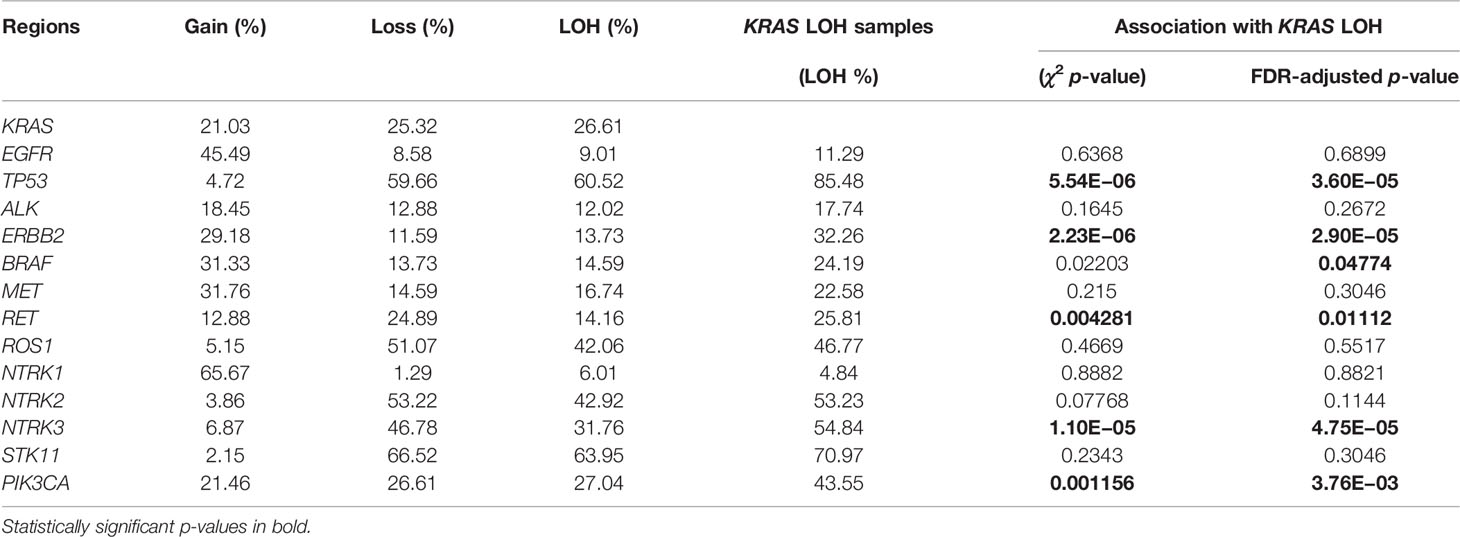
Table 4 Copy number changes in common hotspots in lung adenocarcinomas and associations between LOH in genetic hotspot regions and KRAS LOH.
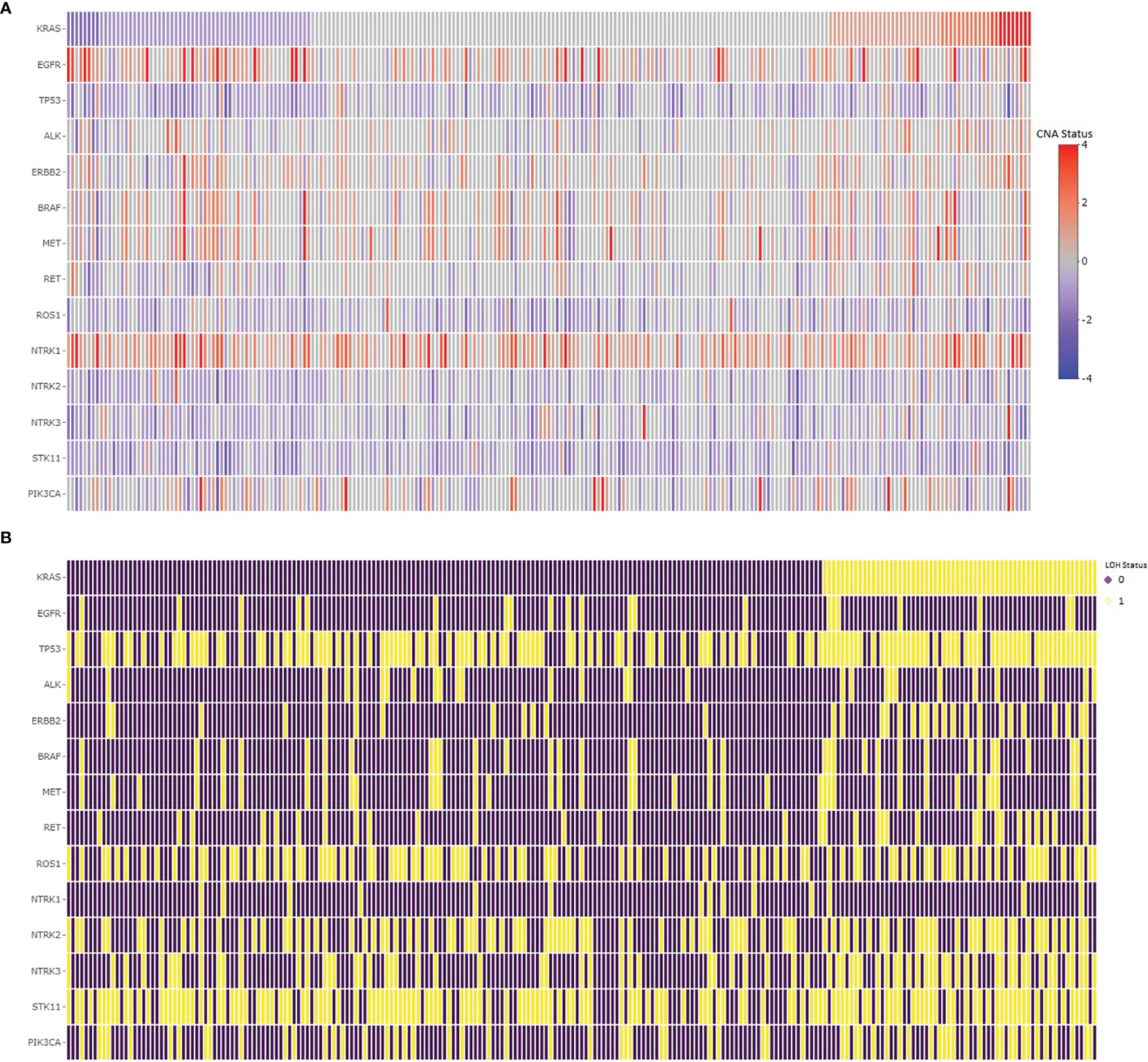
Figure 3 Copy number changes (A) and LOH (B) in common hotspots in lung adenocarcinomas. (A) The copy number changes in the hotspot gene regions sorted by copy number changes in the KRAS gene, where the red color represents copy number gain, whereas the blue color represents copy number loss in the regions. (B) The loss of heterozygosity in the hotspot gene regions, where the purple color shows no loss, whereas the yellow color represents loss of heterozygosity in the region.
Correlating Genes and Association With Survival
Somatic copy number changes may contribute to the development and progression of cancer by affecting gene expression level (33). Improved knowledge about genomic alterations and expression profiles in the subgroups defined by KRAS LOH may identify genes involved in disease development or outcome. We performed a genome-wide correlation analysis to identify genomic aberrations associated with KRAS LOH with an effect on gene expression. We found the mRNA expressions of 9,663 genes to be significantly correlated with KRAS LOH (adjusted p < 0.05), of which 2,474 genes had a moderately high correlation (r > 0.4) to their ploidy-adjusted copy number state. Correlation analysis showed a moderate correlation (r = 0.4) between KRAS expression and its copy number in the genome. The detailed list of the significantly correlated genes is given in Supplementary Table S2. From the 2,474 significantly correlated genes, we found 1,371 deleted genes, while 852 genes showed significant gains in copy number in the samples with LOH in KRAS. Functional analysis using DAVID showed that genes with copy number losses were involved in biological processes such as alternate splicing, protein transport, ubiquitin-mediated proteolysis, cell cycle, and DNA repair. Genes with gains in copy number were enriched for genes involved in transcription regulation, ribosomes, and mRNA processing. This overview of the genomic and transcriptional landscape in lung adenocarcinoma stratified by LOH in KRAS suggests that the overall genomic and transcriptional landscape of lung adenocarcinoma is affected to some extent by the KRAS LOH status.
We further conducted differential expression analysis between samples with and without KRAS LOH and identified 197 genes that were significantly differentially expressed in both cohorts. From the differentially expressed genes, 38 genes were overexpressed, of which 15 genes were significantly amplified, and 82 genes were underexpressed, of which 70 were significantly deleted in the samples with KRAS LOH. Significantly amplified and deleted gene regions with significant correlation for their mRNA–copy number in samples with LOH in KRAS with respect to no LOH samples are listed in Supplementary Table S3. Gene set enrichment analysis of these genes shows their role in RNA transport pathways and protein-binding function (Supplementary Table S4).
Eight of the amplified genes and 66 of the deleted genes showed a significant correlation between their copy number and expression. We investigated the association of these genes to patient outcome and found that overexpression of CDC14A and downregulation of GABARAPL1 and RFK in the KRAS LOH-positive samples were significantly associated with OS. The survival analysis showed that high CDC14A expression was significantly associated with improved OS. Similarly, the lower expression of GABARAPL1 and RFK was associated with improved OS in lung adenocarcinoma patients (Supplementary Figure S3). The genes functionally separate the two subgroups and may be involved in the outcome.
Discussion
In this study, we identified the number of recurrent genomic aberrations in a lung adenocarcinoma subgroup defined by KRAS LOH status. The study demonstrates that for patients with early-stage lung adenocarcinomas, loss of heterozygosity in the KRAS region is associated with improved OS as well as RFS. The genome-wide copy number analysis showed gains and losses consistent with the results reported in previously published studies (34, 35). In order to identify the genes whose mRNA expression had changed due to change in copy number, we performed a correlation analysis between copy number and mRNA expression data and found 2,474 genes with statistically significant correlation (r > 0.4, p.adjust < 0.05). We found relatively few genes with correlation between mRNA expression and copy number compared to a previously published study (31). KRAS gene expression showed a moderately high correlation to its copy number, which signifies the importance of copy number analysis in relation to KRAS.
We identified significant differences in aberration pattern between patients with and without KRAS LOH. To our knowledge, no previous studies have investigated the aberration pattern of tumor DNA based on LOH in the KRAS region. Survival analysis showed that LOH in the KRAS region correlates with survival and is associated with improved prognosis more specifically in patients with KRAS mutated tumors. We tested other clinicopathological parameters for their association with KRAS LOH and found no significant association, suggesting KRAS LOH as an independent prognostic factor. Previous studies found KRASG12C mutations associated with negative clinical outcomes in advanced cancers. The recent finding of the selective KRASG12C inhibitor (23) shows that KRAS is no longer undruggable. This shows the importance of molecular characterization of the tumor of each patient diagnosed with lung cancer to identify druggable targets. Our study identified an improved OS in KRAS mutated tumors with LOH in KRAS, irrespective of which hotspot KRAS mutation was present. We also found that a relatively small number of KRAS mutated tumors had LOH in the gene, with a slightly negative correlation for their co-occurrence.
Univariate analysis using Cox proportional hazards regression showed that only age, stage, and KRAS LOH were the significant factors for OS. Multivariate Cox proportional hazards regression analysis showed that KRAS LOH has a weak significance as a prognostic factor for OS (p-value = 0.08), but not for RFS. LOH in KRAS was a statistically significant prognostic factor in stage I disease with mutant KRAS. The mRNA expression and copy number state of KRAS have a moderately high correlation, and the loss of an allele in KRAS resulted in a lower overall expression of the gene. While LOH in KRAS is associated with reduced expression of the KRAS gene in KRAS wild-type samples, this is not the case in the KRAS mutated samples. This could indicate that mutant KRAS with LOH leads to an always-ON state of the gene, and the interaction with wild-type KRAS in no LOH samples may be regulating the expression of mutant KRAS. This phenomenon may be attributed to the repressive effect of the wild-type allele in the KRAS mutated sample when both alleles are present (36, 37). This indicates that the association between LOH in KRAS and survival is not explained by the expression of the KRAS gene.
Hotspot mutations in lung cancer are well characterized for their concurrences (38). We analyzed copy number changes in these hotspot genes to determine their concurrences and associations with LOH in KRAS. TP53 mutations are known to be associated with smoking and KRAS mutation. We found LOH in TP53 to be associated with LOH in KRAS. We also found that mutation in the hotspot genes such as RET, NTRK3, and PIK3CA regions is associated with LOH in KRAS. We did integrative analyses to investigate the differential gene expression and copy number changes in the samples with KRAS LOH compared with those without KRAS LOH. We found 66 genes deleted with lowered mRNA expression, while 10 genes were amplified along with an increase in their mRNA expression levels in the samples with KRAS LOH. We found that the CDC14A gene that plays a role in cell cycle regulation was amplified along with increased mRNA expression level in KRAS LOH-positive samples, and its expression was positively associated with OS. Previous studies suggested that the gene was differentially expressed in cancers and it can interact with the tumor suppressor p53 (39, 40). An autophagy-related gene GABARAPL1 was found deleted with lowered expression in the samples with KRAS LOH. Studies have shown that its expression is associated with better outcome in breast cancers (41). In contrast, we found the lower expression of GABARAPL1 to be associated with better outcome in our study. We believe that these genetic differences can provide new approaches to refine prognostication of lung adenocarcinomas and deserve to be explored further.
In conclusion, our study shows that LOH in KRAS is associated with a favorable prognosis in patients with early-stage lung adenocarcinomas, particularly in patients with KRAS mutated tumors. Our study indicates that LOH in KRAS is a prognostic factor that can refine the existing prognostic groups of lung adenocarcinomas.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Regional Committees for Medical and Health Research Ethics Norway (ref: S-06402b). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AK, VDH, OCL, JH, and EHK designed the study. AK performed the bioinformatics analyses with the help of LS-P and OCL. OTB, PM, JB, and ÅH provided the clinical data. AK and VDH wrote the first draft of the manuscript. EHK and ÅH supervised the study and were responsible for the funding. All authors critically reviewed the manuscript.
Funding
This research was supported by the University of South-Eastern Norway and Institute for Cancer Research, Oslo University Hospital, Oslo, Norway, Grant Number: 88503-2013, the Norwegian Cancer Society.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would also like to thank Åsa Öjlert for the estimation of the previously published lung adenocarcinoma subtypes. We thank all the patients who participated in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.873532/full#supplementary-material
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Cancer Registry of Norway. Cancer in Norway 2019 - Cancer Incidence, Mortality, Survival and Prevalence in Norway. Cancer Regist Norw (2020) 98–100.
3. Brustugun OT, Grønberg BH, Fjellbirkeland L, Helbekkmo N, Aanerud M, Grimsrud TK, et al. Substantial Nation-Wide Improvement in Lung Cancer Relative Survival in Norway From 2000 to 2016. Lung Cancer (2018) 122:138–45. doi: 10.1016/j.lungcan.2018.06.003
4. Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung Cancer in Never Smokers - A Review. Eur J Cancer (2012) 48(9):1299–311. doi: 10.1016/j.ejca.2012.03.007
5. Varella-Garcia M. Chromosomal and Genomic Changes in Lung Cancer. Cell Adhesion Migration (2010) 4:100–6. doi: 10.4161/cam.4.1.10884
6. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature (2013) 499(7457):214–8. doi: 10.1038/nature12213
7. Cancer Genome Atlas Research N. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature (2014) 511(7511):543–50. doi: 10.1038/nature13385
8. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging Landscape of Oncogenic Signatures Across Human Cancers. Nat Genet (2013) 45(10):1127–33. doi: 10.1038/ng.2762
9. Planck M, Edlund K, Botling J, Micke P, Isaksson S, Staaf J. Genomic and Transcriptional Alterations in Lung Adenocarcinoma in Relation to EGFR and KRAS Mutation Status. PloS One (2013) 8(10):e78614. doi: 10.1371/journal.pone.0078614
10. Meuwissen R, Berns A. Mouse Models for Human Lung Cancer. Genes Dev (2005) 19:643–64. doi: 10.1101/gad.1284505
11. Yokota J, Kohno T. Molecular Footprints of Human Lung Cancer Progression. Cancer Sci (2004) 95(3):197–204. doi: 10.1111/j.1349-7006.2004.tb02203.x
12. Scoccianti C, Vesin A, Martel G, Olivier M, Brambilla E, Timsit JF, et al. Prognostic Value of TP53, KRAS and EGFR Mutations in Nonsmall Cell Lung Cancer: The EUELC Cohort. Eur Respir J (2012) 40(1):177–84. doi: 10.1183/09031936.00097311
13. Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and Distinctive Spectrum of KRAS Mutations in Never Smokers With Lung Adenocarcinoma. Clin Cancer Res (2008) 14(18):5731–4. doi: 10.1158/1078-0432.CCR-08-0646
14. Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence That Synthetic Lethality Underlies the Mutual Exclusivity of Oncogenic KRAS and EGFR Mutations in Lung Adenocarcinoma. Elife (2015) 4:1–23. doi: 10.7554/eLife.06907.015
15. Ziv E, Erinjeri JP, Yarmohammadi H, Boas FE, Petre EN, Gao S, et al. Lung Adenocarcinoma: Predictive Value of KRAS Mutation Status in Assessing Local Recurrence in Patients Undergoing Image-Guided Ablation. Radiology (2017) 282(1):251–8. doi: 10.1148/radiol.2016160003
16. Tsao MS, Aviel-Ronen S, Ding K, Lau D, Liu N, Sakurada A, et al. Prognostic and Predictive Importance of P53 and RAS for Adjuvant Chemotherapy in Non-Small-Cell Lung Cancer. J Clin Oncol (2007) 25(33):5240–7. doi: 10.1200/JCO.2007.12.6953
17. Capelletti M, Wang XF, Gu L, Graziano SL, Kratzke RA, Strauss GM, et al. Impact of KRAS Mutations on Adjuvant Carboplatin/Paclitaxel in Surgically Resected Stage IB NSCLC: CALGB 9633. J Clin Oncol (2010) 28(15_suppl):7008–8. doi: 10.1200/jco.2010.28.15_suppl.7008
18. Shepherd FA, Domerg C, Hainaut P, Jänne PA, Pignon JP, Graziano S, et al. Pooled Analysis of the Prognostic and Predictive Effects of KRAS Mutation Status and KRAS Mutation Subtype in Early-Stage Resected Non–Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J Clin Oncol (2013) 31(17):2173–81. doi: 10.1200/JCO.2012.48.1390
19. La Fleur L, Falk-Sörqvist E, Smeds P, Berglund A, Sundström M, Mattsson JS, et al. Mutation Patterns in a Population-Based Non-Small Cell Lung Cancer Cohort and Prognostic Impact of Concomitant Mutations in KRAS and TP53 or STK11. Lung Cancer (2019) 130:50–8. doi: 10.1016/j.lungcan.2019.01.003
20. West L, Vidwans SJ, Campbell NP, Shrager J, Simon GR, Bueno R, et al. A Novel Classification of Lung Cancer Into Molecular Subtypes. PloS One (2012) 7(2):e31906. doi: 10.1371/journal.pone.0031906
21. Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an Undruggable Pocket on KRAS. Proc Natl Acad Sci USA (2019) 116(32):15823–9. doi: 10.1073/pnas.1904529116
22. Fakih M, O’Neil B, Price TJ, Falchook GS, Desai J, Kuo J, et al. Phase 1 Study Evaluating the Safety, Tolerability, Pharmacokinetics (PK), and Efficacy of AMG 510, a Novel Small Molecule KRAS G12C Inhibitor, in Advanced Solid Tumors. J Clin Oncol (2019) 37(15_suppl):3003–3. doi: 10.1200/JCO.2019.37.15_suppl.3003
23. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS G12C Inhibition With Sotorasib in Advanced Solid Tumors. N Engl J Med (2020) 383(13):1207–17. doi: 10.1056/NEJMoa1917239
24. Yoshino I, Osoegawa A, Yohena T, Kameyama T, Oki E, Oda S, et al. Loss of Heterozygosity (LOH) in Non-Small Cell Lung Cancer: Difference Between Adenocarcinoma and Squamous Cell Carcinoma. Respir Med (2005) 99(3):308–12. doi: 10.1016/j.rmed.2004.08.008
25. Staaf J, Isaksson S, Karlsson A, Jönsson M, Johansson L, Jönsson P, et al. Landscape of Somatic Allelic Imbalances and Copy Number Alterations in Human Lung Carcinoma. Int J Cancer (2013) 132(9):2020–31. doi: 10.1002/ijc.27879
26. Yu CC, Qiu W, Juang CS, Mansukhani MM, Halmos B, Su GH. Mutant Allele Specific Imbalance in Oncogenes With Copy Number Alterations: Occurrence, Mechanisms, and Potential Clinical Implications. Cancer Lett (2017) 384:86–93. doi: 10.1016/j.canlet.2016.10.013
27. Bjaanæs MM, Nilsen G, Halvorsen AR, Russnes HG, Solberg S, Jørgensen L, et al. Whole Genome Copy Number Analyses Reveal a Highly Aberrant Genome in TP53 Mutant Lung Adenocarcinoma Tumors. BMC Cancer (2021) 21(1):1089. doi: 10.1186/s12885-021-08811-7
28. Bjaanaes MM, Fleischer T, Halvorsen AR, Daunay A, Busato F, Solberg S, et al. Genome-Wide DNA Methylation Analyses in Lung Adenocarcinomas: Association With EGFR, KRAS and TP53 Mutation Status, Gene Expression and Prognosis. Mol Oncol (2016) 10(2):330–43. doi: 10.1016/j.molonc.2015.10.021
29. Hamfjord J, Stangeland AM, Skrede ML, Tveit KM, Ikdahl T, Kure EH. Wobble-Enhanced ARMS Method for Detection of KRAS and BRAF Mutations. Diagn Mol Pathol (2011) 20(3):158–65. doi: 10.1097/PDM.0b013e31820b49e2
30. Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, et al. Allele-Specific Copy Number Analysis of Tumors. Proc Natl Acad Sci (2010) 107(39):16910–5. doi: 10.1073/pnas.1009843107
31. Jabs V, Edlund K, König H, Grinberg M, Madjar K, Rahnenführer J, et al. Integrative Analysis of Genome-Wide Gene Copy Number Changes and Gene Expression in Non-Small Cell Lung Cancer. PloS One (2017) 12(11):e0187246. doi: 10.1371/journal.pone.0187246
32. Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, et al. Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PloS One (2012) 7(5):e36530. doi: 10.1371/journal.pone.0036530
33. Bhattacharya A, Bense RD, Urzúa-Traslaviña CG, de Vries EGE, van Vugt MATM, Fehrmann RSN. Transcriptional Effects of Copy Number Alterations in a Large Set of Human Cancers. Nat Commun (2020) 11111(1):1–12:2020. doi: 10.1038/s41467-020-14605-5
34. Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the Cancer Genome in Lung Adenocarcinoma. Nature (2007) 450(7171):893–8. doi: 10.1038/nature06358
35. Micke P, Edlund K, Holmberg L, Kultima HG, Mansouri L, Ekman S, et al. Gene Copy Number Aberrations Are Associated With Survival in Histologic Subgroups of Non-Small Cell Lung Cancer. J Thorac Oncol (2011) 6(11):1833–40. doi: 10.1097/JTO.0b013e3182295917
36. To MD, Rosario RD, Westcott PMK, Banta KL, Balmain A. Interactions Between Wild-Type and Mutant Ras Genes in Lung and Skin Carcinogenesis. Oncogene (2013) 32(34):4028–33. doi: 10.1038/onc.2012.404
37. Zhou B, Der CJ, Cox AD. The Role of Wild Type RAS Isoforms in Cancer. Semin Cell Dev Biol (2016) 58:60–9. doi: 10.1016/j.semcdb.2016.07.012
38. Mäki-Nevala S, Sarhadi VK, Rönty M, Kettunen E, Husgafvel-Pursiainen K, Wolff H, et al. Hot Spot Mutations in Finnish Non-Small Cell Lung Cancers. Lung Cancer (2016) 99:102–10. doi: 10.1016/j.lungcan.2016.06.024
39. Paulsen MT, Starks AM, Derheimer FA, Hanasoge S, Li L, Dixon JE, et al. The P53-Targeting Human Phosphatase Hcdc14a Interacts With the CdkI/cyclin B Complex and Is Differentially Expressed in Human Cancers. Mol Cancer (2006) 5(1):1–11. doi: 10.1186/1476-4598-5-25
40. Chen N-P, Uddin B, Voit R, Schiebel E. Human Phosphatase CDC14A Is Recruited to the Cell Leading Edge to Regulate Cell Migration and Adhesion. Proc Natl Acad Sci (2016) 113(4):990–5. doi: 10.1073/pnas.1515605113
Keywords: KRAS, LOH, prognostic marker, copy number aberration, NSCLC
Citation: Khadse A, Haakensen VD, Silwal-Pandit L, Hamfjord J, Micke P, Botling J, Brustugun OT, Lingjærde OC, Helland Å and Kure EH (2022) Prognostic Significance of the Loss of Heterozygosity of KRAS in Early-Stage Lung Adenocarcinoma. Front. Oncol. 12:873532. doi: 10.3389/fonc.2022.873532
Received: 10 February 2022; Accepted: 31 March 2022;
Published: 29 April 2022.
Edited by:
Rajiv Kumar, German Cancer Research Center (DKFZ), GermanyReviewed by:
Gang Zheng, Mayo Clinic, United StatesFrancesco Pepe, University of Naples Federico II, Italy
Copyright © 2022 Khadse, Haakensen, Silwal-Pandit, Hamfjord, Micke, Botling, Brustugun, Lingjærde, Helland and Kure. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vilde D. Haakensen, vilde.haakensen@gmail.com
†These authors share senior authorship