 Marc Cucurull1,2
Marc Cucurull1,2 Lucia Notario1,2Montse Sanchez-Cespedes3Cinta Hierro1,2Anna Estival1,2
Lucia Notario1,2Montse Sanchez-Cespedes3Cinta Hierro1,2Anna Estival1,2 Enric Carcereny1,2
Enric Carcereny1,2 Maria Saigí1,2*
Maria Saigí1,2*- 1Department of Medical Oncology, Catalan Institute of Oncology (ICO), Barcelona, Spain
- 2Badalona·Applied Research Group in Oncology (B·ARGO), Institut d’Investigació en Ciències de la Salut Germans Trias i Pujol (IGTP), Barcelona, Spain
- 3Cancer Genetics Group, Josep Carreras Leukemia Research Institute (IJC), Barcelona, Spain
Approximately 20% of lung adenocarcinomas harbor KRAS mutations, an oncogene that drives tumorigenesis and has the ability to alter the immune system and the tumor immune microenvironment. While KRAS was considered “undruggable” for decades, specific KRAS G12C covalent inhibitors have recently emerged, although their promising results are limited to a subset of patients. Several other drugs targeting KRAS activation and downstream signaling pathways are currently under investigation in early-phase clinical trials. In addition, KRAS mutations can co-exist with other mutations in significant genes in cancer (e.g., STK11 and KEAP1) which induces tumor heterogeneity and promotes different responses to therapies. This review describes the molecular characterization of KRAS mutant lung cancers from a biologic perspective to its clinical implications. We aim to summarize the tumor heterogeneity of KRAS mutant lung cancers and its immune-regulatory role, to report the efficacy achieved with current immunotherapies, and to overview the therapeutic approaches targeting KRAS mutations besides KRAS G12C inhibitors.
Introduction
After decades of research, the treatment efficacy of advanced lung cancer has remarkably improved, by incorporating novel therapeutic strategies including targeted therapies inhibiting specific genetically activated proteins, or immunotherapies such as immune-checkpoint inhibitors (ICI) (1). Mutations affecting members of the RAS family genes (KRAS, HRAS, NRAS) are the most frequent genetic alterations in human cancer, affecting about 27% of all tumors, including lung, colorectal and pancreatic ductal adenocarcinoma, among others (2, 3).
Lung adenocarcinoma (LuAD) is a type of cancer with the largest number of oncogenic alterations that are therapeutically approachable (4). Approximately 20-25% of the LuADs harbor KRAS mutations, most of them affecting codons 12 (~85%), 13 (~10%) or 61 (~5%). In LuADs from smokers, the vast majority of KRAS mutations consist on guanine to thymine transversions, an effect that is associated with the tobacco carcinogens (5). At the aminoacid level, most of these mutations replace the glycine (G) in codon 12 by a cysteine (C) (G12C) and occurs in almost 50% of KRAS mutant tumors. On the other hand, KRAS mutations in never-smokers are less frequent, and the most prevalent changes involve nucleotide transitions that replace the glycine in codon 12 by an aspartic acid (G12D) or a valine (G12V) (6, 7) (Figure 1).
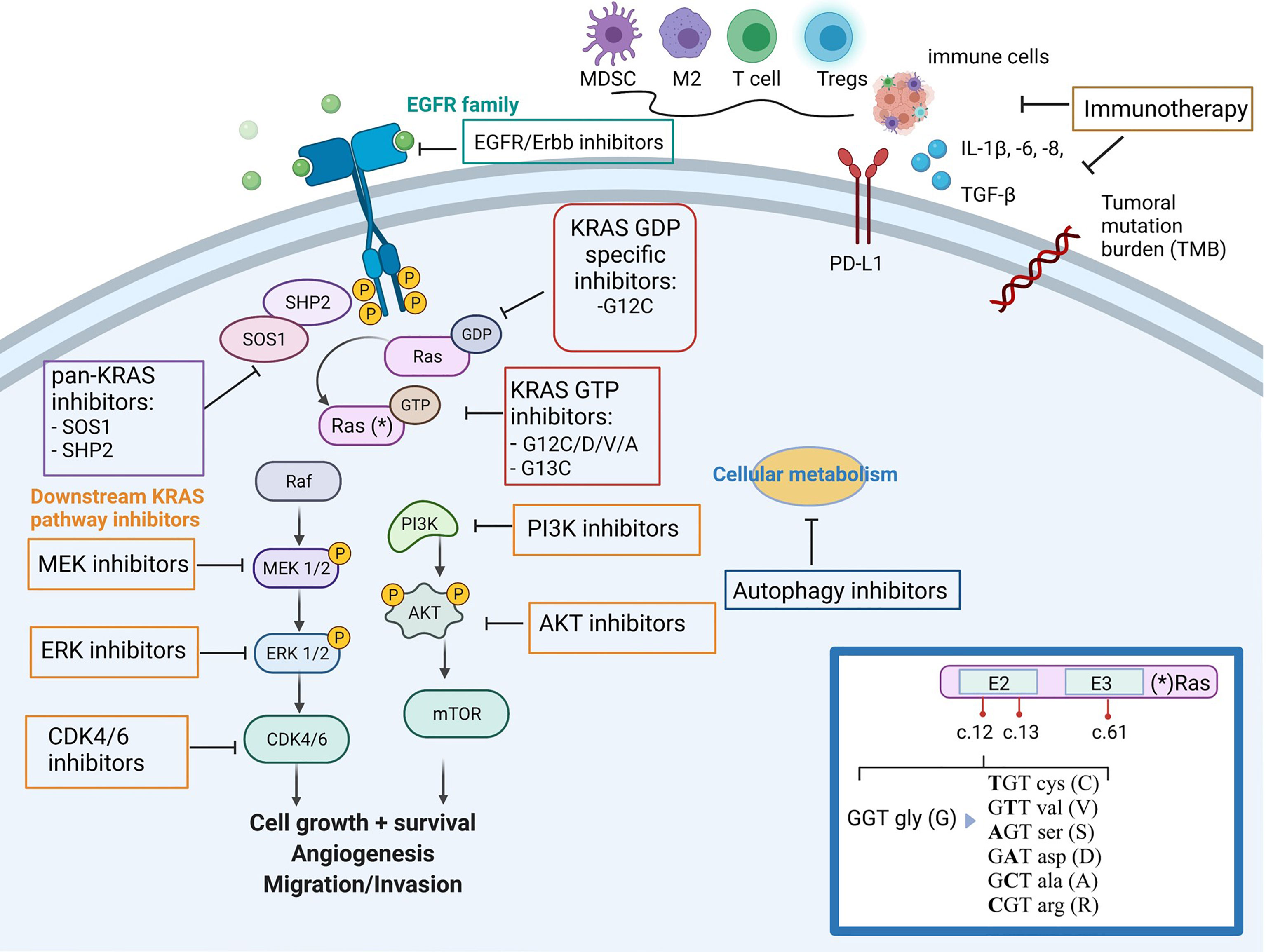
Figure 1 Simplified overview of KRAS mutant tumor, downstream/upstream KRAS pathways, its impact on the immune-microenvironment and family of drugs targeting KRAS mutant tumors. (*) In the inset box, the oncogenic mutations in codon 12 of KRAS and aminonacid changes. Figure has been created with BioRender.com.
In contrast to other oncogenic proteins activated in cancer and despite multiple attempts to harness it, KRAS was considered undruggable for a long time. The KRAS mutated proteins has a reduced capability to hydrolyze GTP or to interact with the GTPase-activating proteins, maintaining the oncogene and the downstream pathways constitutively activated. The lack of specific inhibitors targeting the KRAS hydrophobic pocket and the complexity of downstream pathways have contributed to the challenge of developing effective therapeutic strategies (8). After years of study, novel KRAS selective inhibitors became available for the KRAS G12C mutant protein, enabling a covalent binding that hinders downstream signaling, which led to promising results in the clinical setting. Two specific KRAS G12C inhibitors, first sotorasib (AMG510) and later adagrasib (MRTX849), earned the breakthrough designation by the US Food and Drugs Administration (FDA), to treat metastatic lung cancer patients harboring that particular KRAS mutation who have progressed to at least one prior systemic therapy. In the case of sotorasib, this was based on the efficacy results of the phase I/II CodeBreak-100 trial (NCT03600883) that reported an objective response rate (ORR) of 32% and disease control rate (DCR) of 88% among lung cancer patients. Thus, sotorasib has the FDA-approval for this clinical indication (9). On the other hand, data from the phase I/II KRYSTAL-1 trial (NCT03785249) for adagrasib showed significant benefit with an ORR of 45%, although the study is still ongoing and definitive conclusions cannot be inferred (10). Many other drugs targeting KRAS activation and other parallel and downstream pathways, as well as immunotherapeutic strategies, are currently under investigation in early-phase clinical trials (11).
Here, we aim to describe the tumor heterogeneity of KRAS mutant lung cancers and its immune-regulatory role, to report the efficacy with current immunotherapies, and to overview the therapeutic approaches targeting KRAS mutant tumors, other than KRAS G12C inhibitors.
KRAS-Mutant Lung Cancer Is a Heterogeneous Disease and Has a Characteristic Immune-Microenvironment
Mutations on KRAS and on other actionable oncogenic drivers, such as EGFR or ALK, are often mutually exclusive. However, they co-occur with mutations in important tumor suppressor genes, such as STK11 (also known as LKB1), KEAP1, TP53, or CDKN2A, whose inactivation cooperates with KRAS in the oncogenic process and, thus, characterize the heterogeneous nature of KRAS mutant tumors (12, 13). In addition, the different KRAS mutated proteins differ on their biologic properties to hydrolyze GTP and to activate downstream signaling pathways, which determines the differences in their therapeutic vulnerabilities (14, 15).
Skoulidis et al. described a molecular classification of KRAS mutant LuAD according to the presence of co-mutations in tumor suppressor genes TP53 (~30%) and/or LKB1 (~30% each) (16). These co-mutation partners lead to variances in gene expression and distinct patterns of inflammatory and immune-checkpoint molecules release, which models the tumor microenvironment and promotes different responses to therapies (12, 17). Because of this tumor heterogeneity, the prognostic role of KRAS mutant cancers remains uncertain, although most studies report a major aggressive behavior of this type of cancer (18–20).
KRAS mutant tumors are characterized by enabling tumor cells to escape immunosurveillance as one of the hallmarks of cancer (21). NF-κB, STAT3, and certain suppressive inflammatory cytokines such as IL-6, IL-1β and GM-CSF, are key transducers of the immunosuppressive properties of KRAS driven tumors (22–24). Other mechanisms in this tumor type consist in increasing the expression of immune-checkpoints (e.g., PD-L1) to prevent T-cell effector functions, eliciting the release of myeloid-derived suppressor cells, regulatory T cells, and M2-differentiated tumor-associated macrophages, all of which impair antitumor immunity and facilitates tumor growth (25, 26) (Figure 1).
In preclinical mice models, Stk11/Kras mutant tumors produced abundant IL-6 and were associated with neutrophil accumulation and inflammatory cytokines with immunosuppressive properties in the tumor microenvironment, along with increased levels of T-cell exhaustion markers, compared with normal lungs (27). Accordingly, KRAS/STK11-mutant tumors are associated to a “cold immunophenotype” with lower T-cell infiltration and lower rates of PD-L1 immunostaining, among other immunosuppressive features. Lung cancer patients with this tumor genetic profile showed a worse response to ICI than did patients with KRAS-mutant tumors without LKB1 co-mutations (27–30).
More recently, STK11 inactivation in KRAS-mutant tumors, has been shown to enhance the silencing of the STimulator of INterferon Genes (STING) protein, in part through epigenetic mechanisms (31). STING is a key component of the innate immunity and acts as a sensor for double-strand DNA (dsDNA) in the cytosol from virus and pathogens, which mediates the type I interferon production (32). By silencing STING expression, KRAS/STK11-mutant tumors become insensitive to cytoplasmatic dsDNA sensing, avoiding T-cell inflammation and promoting the recruitment of exhausted T-cell lymphocytes. Thus, restoring STING expression or activation of the cyclic GMP–AMP synthase (cGAS)-STING pathway with STING agonists, could induce T cell infiltration and turn this subset of tumors into more immunogenic, increasing the synergistic effects in combination with ICI (33).
The Efficacy of Current Immunotherapies in Lung Cancers With KRAS Mutations
Besides the immune-related nature of KRAS driven tumors, the smoking habit of patients with KRAS mutations has been associated with higher tumor mutational burden (TMB), which might predict better responses to ICI (34, 35).
Mazieres et al. retrospectively evaluated a cohort of non-small cell lung cancer (NSCLC) harboring different oncogenic driver mutations. Among them, the subset of KRAS mutant tumors expressed higher rates of PD-L1 and responded better to ICI than tumors with other oncogenic driver alterations (36). On the other hand, most phase 3 clinical trials evaluating all-comers with NSCLC treated with ICI did not stratify by KRAS status, and only post-hoc analyses have been performed on that subset. The following section compiles the evidence derived from exploratory analyses of the most relevant phase 3 clinical trials evaluating ICI and real-world data from retrospective cohorts of NSCLC patients treated with ICI.
The efficacy of nivolumab has been reported in two international studies. In the Italian Expanded Access Program out of the 530 patients who received 2nd or 3rd line nivolumab, 206 (39%) were positive for KRAS mutation. KRAS status did not influence nivolumab efficacy in terms of ORR (20% vs 17%, p = 0.39) nor DCR (47% vs 41%, p = 0.23) in patients with KRAS-mutant tumors when compared to KRAS-wild type (wt). No statistically significant differences were found in the median progression-free survival (mPFS) nor in the median overall survival (mOS) between both groups, although the 3-months PFS was significantly higher in KRAS-mutant patients (53% vs 42%, p = 0.01) (37). On the other hand, in the phase 3 study CA209-057, patients who had progressed to previous platinum-based chemotherapy (ChT), were randomized to receive either nivolumab or docetaxel. Among the 582 patients studied, KRAS was tested in 185 patients and 33% showed a KRAS mutation. When comparing the latter with KRAS-wt patients in the groups who received ICI, the hazard ratio (HR) for OS and PFS were 0.52 (95% confidence interval [CI]: 0.29-0.95) and 0.82 (95% CI: 0.47-1.43), respectively, in favor to KRAS mutant patients (38).
Regarding pembrolizumab, the clinical outcomes of two phase 3 clinical trials including KRAS mutant population have been reported. The KEYNOTE-042 trial (NCT0222089) evaluated pembrolizumab compared to platinum-based ChT as first-line treatment for PD-L1 positive tumors. The exploratory analysis presented by Herbst et al. showed that, out of 301 patients, 22.9% harbored KRAS mutations (9.6% were KRAS G12C) and presented higher levels of PD-L1 and tissue TMB. Both mOS and mPFS favored KRAS mutant tumors when treated with pembrolizumab compared to KRAS-wt, with a HR of 0.42 (95% CI: 0.22-0.81) and 0.51 (95% CI: 0.29-0.87), respectively (39). On the other hand, the KEYNOTE-189 trial evaluated platinum-based ChT either alone or in combination with pembrolizumab as first-line setting in advanced disease. Among 289 patients, 30.8% harbored a KRAS mutation and 12.8% a KRAS G12C. These tumors also presented higher levels of PD-L1 and higher tissue TMB. Pembrolizumab-based therapy was associated with improved clinical outcomes in terms of OS, PFS, and ORR regardless of KRAS status (40). Finally, results from real-world data published by Frost et al. from a multicenter, retrospective study evaluated the efficacy of first-line pembrolizumab in 119 patients with KRAS mutant LuAD with high PD-L1 expression (≥50%). Co-mutations in TP53 were also evaluated, and patients with KRAS G12C/TP53 had significantly higher ORR (100% vs 27.3%; p = 0.003) and longer mPFS (33.3 vs 2.8 months; HR, 0.18; 95% CI: 0.06-0.53; p = 0.002) than tumors with KRAS nonG12C/TP53 mutations (41), suggesting that KRAS G12C present better outcomes to immune-based therapies. Furthermore, Noordhof et al. reported another retrospective study that evaluated the outcomes of first-line pembrolizumab in 595 patients with metastatic LuAD and high PD-L1 expression. KRAS mutations were found in 57% of the cases. Although not statistically significant, mOS with ICI was higher in KRAS mutant patients than in those with KRAS-wt (19.2 vs 16.8 months; p = 0.86) (42).
In relation to atezolizumab, efficacy results are derived from a single trial, IMpower 150 (NCT02366143), a phase 3 study with first-line ChT and bevacizumab in combination or not with atezolizumab in patients with metastatic NSCLC. An exploratory analysis evaluated atezolizumab efficacy in KRAS mutant population according to STK11/KEAP1 mutation status. Among 920 evaluable patients, 24.5% harbored a KRAS mutation, which in 45% of the cases were co-mutated with STK11 and/or KEAP1. Greater benefits in terms of OS and PFS were observed in patients harboring KRAS mutations in the immunotherapy-based arm, regardless of STK11 and KEAP1 status (43).
Finally, results on durvalumab in stage III NSCLC patients after ChT-radiotherapy are available from a retrospective study that was carried out in 134 patients from MD Anderson Hospital. Patients with driver oncogenic mutations, including KRAS mutations (n=26) and targetable driver mutations (n=24) in EGFR, ALK translocations, ROS1 fusions, MET exon 14 skipping, RET fusion, and/or BRAF, had significantly worse mPFS compared to those without driver mutations (n=84) (8.9 months vs 26.6 months; HR 2.62 p < 0.001), particularly in cases with KRAS mutant tumors (mPFS 7.9 months, HR 3.34, p < 0.001), with no impact on OS based on driver mutation status (44).
Targeting KRAS Beyond KRAS G12C Mutations
KRAS is a GTPase that, when mutated, loses the ability to turn back to the GDP-bound state and leads to a constitutively active GTP-bound state. This, in turn, activates downstream signaling pathways, including MAPK, PI3K/AKT/mTOR, and Ras-like GEF, among others, all of them responsible for cell proliferation and survival (45, 46) (Figure 1). In addition, the heterogeneity of KRAS mutations results in a variety of different diseases, which hinders the finding of a unique common therapy to address all of them. Thus, while KRAS G12C specific inhibitors have proven efficacy against their target, many other therapeutic strategies are currently under development for those KRAS mutant tumors with no druggable genetic alteration (47). Clinical trials addressed to KRAS mutant NSCLC non specific for KRAS G12C are listed in Table 1.
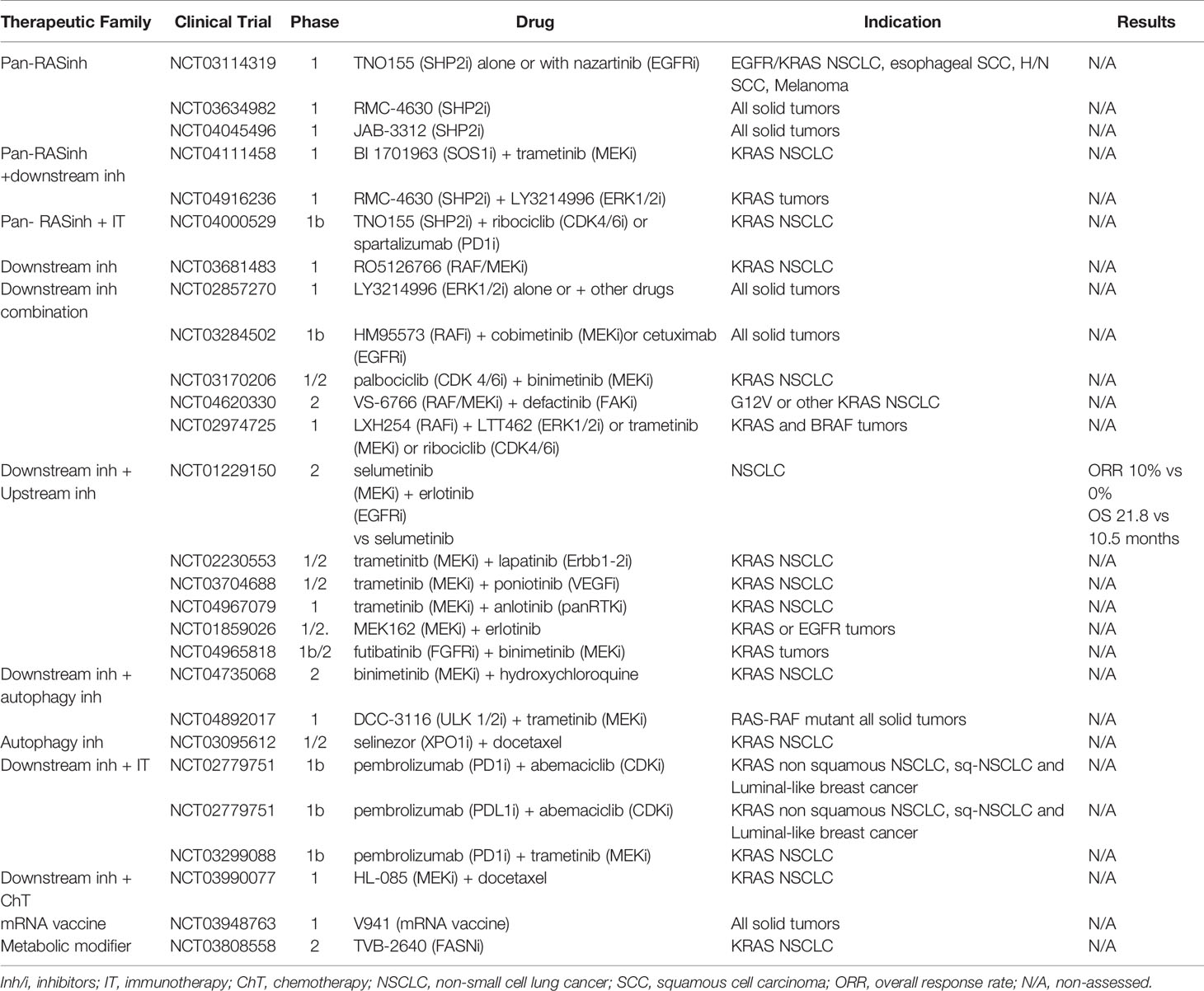
Table 1 Clinical trials of drugs targeting KRAS.
Before the advent of KRAS G12C inhibitors, several strategies were tested to target the broad spectrum of KRAS mutant tumors, but most of them failed. Historically, the majority of these strategies focused on downstream effectors. For instance, the MEK inhibitor selumetinib combined with docetaxel showed good responses in early trials but failed to improve survival in a randomized phase 3 trial (48, 49). The cyclin-dependent kinase (CDK4/6) inhibitor abemaciclib was also tested in a randomized phase 3 trial against erlotinib but could not achieve its OS primary endpoint (50). Combining downstream effector inhibitors targeting MEK/PI3K demonstrated moderate responses but unacceptable toxicity profiles (51). Another therapeutic strategy focusing on the RAS family was the interruption of its anchoring to the cell membrane by inhibiting the post-translational farnesylation (e.g. tipifarnib). Unfortunately, despite a seemingly effective strategy in early phase trials, phase 2 and 3 trials assessing the effectivity in KRAS mutant tumors could not meet the expected outcomes (52). Hence, ChT remained the main treatment for these tumors for a long time, albeit with limited success.
Since KRAS mutant tumors form a heterogenous disease, nowadays, investigational efforts are focused on a subset of targeted therapies that can be further classified in different families according to their mechanism of action. Furthermore, to achieve better outcomes, the different families can be combined with themselves, with KRAS G12C inhibitors, or with immunotherapy. We will subsequently develop further each one of these potential options:
i) KRAS Mutation-Specific Inhibitors
Besides KRAS G12C inhibitors, no other point mutations of KRAS have been successfully targeted in human trials yet. However, the artificial cyclic peptide KS-58 enhanced anti-cancer activity in vitro and in vivo in KRAS G12D mutant tumors by blocking intracellular Ras-effector protein interactions (53). Also, the new molecule MRTX1133 has shown promising results, binding to G12D in lung and pancreatic tumor models (54).
ii) Pan-KRAS Inhibitors
SOS1 is a guanine exchange factor for KRAS promoting the phosphorylation of GDP to GTP by binding to its catalytic site. Moreover, SOS1 can bind to the allosteric site of KRAS that potentiates its GEF function, increasing its positive feedback regulation (54). SOS1 inhibition has demonstrated a depletion effect on tumors that depend on KRAS activation. Recently, a new potent and selective SOS1 inhibitor, BI-3406, has shown in vitro and in vivo antitumor activity (55). The drug decreased GTP-loaded KRAS and attenuated feedback reactivation by MEK inhibitors, suggesting that this combination may be a promising treatment option. In fact, that specific combination was tested in vitro in cell lines resistant to KRAS G12C inhibitors with satisfactory results and is currently ongoing phase 1 trials (56). SHP2 is a protein tyrosine phosphatase existing either bound to the cytoplasmatic portion of an activated RTK or as a component of the RAS activating complex (57). SHP2 allosteric inhibitors, such as TNO155 and RMC-4630, have shown activity on ERK inhibition; ongoing trials combining both treatments are currently assessing their potential to overcome drug resistance to RTK/RAS/MAPK inhibitors (58, 59).
iii) Downstream KRAS Inhibitors
The immediate effect of activated RAS is the interaction between the RAS switched region with the RAS binding domain (RBD) conserved in multiple proteins of multiple signaling pathways. A RAS mimetic small molecule, rigosertib, targets these RBD by interrupting RAF, RALGDS, and PI3Ks signaling cascades (60). So far, RAF and MEK inhibitors alone or in combination with RTK inhibitors have failed to prove effective in KRAS mutant tumors. However, a novel RAF/MEK potent inhibitor showed a 60% tumor reduction in RAS-RAF mutated tumors and is being tested in combination with a FAK inhibitor in KRAS mutant LuAD (61). ERK 1/2 inhibitors are yet to be approved but are expected to directly suppress the MAPK pathway’s effector node. When administered alone, only achieved disease stabilization and the phase I trial testing LT462 (NCT02711345) was terminated earlier. ERK inhibitors could further be combined with drugs targeting upstream nodes in the MAPK pathway to reduce the incidence of acquired resistance. Besides, the PI3K/mTOR/AKT pathway is a downstream pathway activated by the RAS family. In this early phase clinical trial (NCT00933777), the combination of sorafenib (a multi-TKI) with everolimus (mTOR inhibitor) did not achieve any partial response in NSCLC patients harboring KRAS mutation, assessed by CT-scans (62). However, combinations of MEK and PI3K/mTOR/AKT inhibitors have demonstrated better efficacy in KRAS mutant LuADthan either one alone, but their potential toxicity has to be addressed by different treatment schemes (e.g. intermittent dosage) (63, 64).
iv) Upstream KRAS Inhibitors
KRAS-mutant tumors are still sensitive to extracellular growth factors and not completely independent of growth factor receptors (ERBB proteins), since both have a role in tumorigenesis (65). Their activity could be tackled by blocking upstream effectors and the combination of pan-Erbb family inhibitors with KRAS pathway inhibitors could increase the efficacy as well as contribute to overcome the drug resistance enhancing the outcomes observed with inhibiting each target alone, although at cost of increased toxicities. Two preclinical studies further support the idea that mutant KRAS demands activation of ERBB receptors to facilitate lung tumorigenesis (65, 66). One approach was focused on combining KRAS inhibition with epidermal growth factor receptor (EGFR) TKIs, as the EGFR signaling pathway is often activated in tumor cells to bypass KRAS inhibition. However, past clinical trials combining MEK inhibitor, selumetinib with EGFR inhibitor, Erlotinib were largely unsuccessful with an ORR of 10% (95% CI 2.1 to 26.3%) in KRAS mutant patients (67).
v) Cellular Metabolism and Autophagy
KRAS mutant tumors present a high glucose metabolism, so that, multiple glycolytic genes are upregulated and its suppression could prevent tumor growth (68). Autophagy is a strategy to overcome starvation in healthy cells and it has been observed to be increased in many cancer types. Autophagy prevents cells from undergoing programmed cell death. Moreover, it has been shown that the RAF/MEK/ERK cascade leads to autophagy via STK11/AMPkinase-activated (AMPK) protein that activates the autophagy kinase 1 signaling axis (69). Combined inhibition of autophagy and MAPK signaling is nowadays being studied in phase 1 and 2 trials (NCT04892017) (70).
Conclusions
The high frequency of KRAS mutations in cancer justifies the multiple efforts invested in developing novel therapeutic strategies targeting KRAS. A deeper understanding of the cancer biology and immune system interactions that fuel carcinogenesis in KRAS mutant tumors is essential for developing new drugs and improving disease prognosis. Besides KRAS G12C specific inhibitors, several other drugs targeting KRAS directly or indirectly are being investigated. In addition, the list of actionable KRAS mutations in lung cancer will likely increase in the upcoming years.
Current immunotherapies seem to be effective for subset of KRAS mutant tumors, due in part, by the influence of smoking related nature of KRAS G12C mutations. The presence of co-mutations such as STK11 or KEAP1 shape the tumor immune microenvironment and might has an impact on treatment efficacy. Incorporating these genetic alterations in diagnostic panels as predictive markers represent a useful strategy for therapeutic decisions, including immunotherapy-based regimens.
Finally, the genomic complexity of KRAS mutant tumors will ultimately require tailored application of therapeutic approaches and upcoming data from clinical trials will contribute to provide the most promising strategies.
Author Contributions
Conceptualization, EC, MC, and MS. Writing—original draft preparation, MC, EC, and MS. Writing—review and editing, MS-C, CH, LN, and AE. Supervision, MS, MC, EC, and MS-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. MS is supported by a Juan-Rodés contract from the Instituto de Salud Carlos III (JR20/00015).
Conflict of Interest
MC reports advisory/consultancy: Roche, Bristol-Myers Squibb, AstraZeneca; Travel/expenses: Pfizer, Roche. MSC reports a sponsored research agreement with Merck Serono Pharmaceuticals. CH reports research grants from Merck, and speaker’s bureau from MSD, Lilly and Ipsen. AE reports honoraris from Roche, MSD, AstraZeneca and Pharmamar. Travel/expenses: Roche, MSD, AstraZeneca, Lilly, Pfizer, Pharmamar. EC reports advisory/consultancy: AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, MSD, Novartis, Roche, Takeda. Speaker bureau: AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb. MSD, Novartis, Pfizer, Roche, Takeda, Amgen. Travel/accommodation/expenses: Bristol Myers Squibb, Pfizer, Roche, Takeda. MS reports a sponsored research agreement with Merck Serono Pharmaceuticals. Advisory/consultancy: Roche, Bristol-Myers Squibb, AstraZeneca, Takeda; Travel/expenses: Pfizer, Roche.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Herbst RS, Morgensztern D, Boshoff C. The Biology and Management of Non-Small Cell Lung Cancer. Nature (2018) 553(7689):446–54. doi: 10.1038/nature25183
2. Pakkala S, Ramalingam S. Personalized Therapy for Lung Cancer: Striking a Moving Target. JCI Insight (2018) 3(15):e12085. doi: 10.1172/jci.insight.120858
3. Drosten M, Barbacid M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell (2020) 37(4):543–50. doi: 10.1016/j.ccell.2020.03.013
4. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in Developmental Disorders and Cancer. Nat Rev Cancer (2007) 7(4):295–308. doi: 10.1038/nrc2109
5. Ahrendt SA, Decker PA, Alawi EA, Zhu Yr YR, Sanchez-Cespedes M, Yang SC, et al. Cigarette Smoking Is Strongly Associated With Mutation of the K-Ras Gene in Patients With Primary Adenocarcinoma of the Lung. Cancer (2001) 92(6):1525–30. doi: 10.1002/cncr1478
6. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, et al. Genomic Landscape of Non-Small Cell Lung Cancer in Smokers and Never-Smokers. Cell (2012) 150(6):1121–34. doi: 10.1016/j.cell.2012.08.024
7. Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and Distinctive Spectrum of KRAS Mutations in Never Smokers With Lung Adenocarcinoma. Clin Cancer Res (2008) 14(18):5731–4. doi: 10.1158/1078-0432.ccr-08-0646
8. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C Inhibition With Sotorasib in Advanced Solid Tumors. N Engl J Med (2020) 383(13):1207–17. doi: 10.1056/NEJMoa1917239
9. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers With KRAS P.G12C Mutation. N Engl J Med (2021) 384(25):2371–81. doi: 10.1056/nejmoa2103695
10. Riely G, Ou SI, Rybkin I, Spira A, Papadopoulos K, Sabari JK, et al. 99o_PR - KRYSTAL-1: Activity and Preliminary Pharmacodynamic (PD) Analysis of Adagrasib (MRTX849) in Patients (Pts) With Advanced Non-Small-Cell Lung Cancer (NSCLC) Harboring KRASG12C Mutation. J Thorac Oncol (2021) 16(suppl_4):S748–802. doi: 10.1016/S1556-0864(21)01941-9
11. Kidger AM, Sipthorp J, Cook SJ. ERK1/2 Inhibitors: New Weapons to Inhibit the RAS-Regulated RAF-MEK1/2-ERK1/2 Pathway. Pharmacol Ther (2018) 187:45–60. doi: 10.1016/j.pharmthera.2018.02.007
12. Skoulidis F, Heymach JV. Co-Occurring Genomic Alterations in Non-Small-Cell Lung Cancer Biology and Therapy. Nat Rev Cancer (2019) 19(9):495–509. doi: 10.1038/s41568-019-0179-8
13. Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, et al. Inactivation of LKB1/STK11 Is a Common Event in Adenocarcinomas of the Lung. Cancer Res (2002) 62(13):3659–62.
14. Haigis K. KRAS Alleles: The Devil Is in the Detail. Trends Cancer (2017) 3(10):686–97. doi: 10.1016/j.trecan.2017.08.006
15. Gibbs JB, Sigal IS, Poe M, Scolnick EM. Intrinsic GTPase Activity Distinguishes Normal and Oncogenic Ras P21 Molecules. Proc Natl Acad Sci (1984) 81(18):5704–8. doi: 10.1073/pnas.81.18.5704
16. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-Occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma With Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov (2015) 5(8):860–77. doi: 10.1158/2159-8290.cd-14-1236
17. Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, et al. KRAS Mutation-Induced Upregulation of PD-L1 Mediates Immune Escape in Human Lung Adenocarcinoma. Cancer Immunol Immunother (2017) 66(9):1175–87. doi: 10.1007/s00262-017-2005-z
18. Finn SP, Addeo A, Dafni U, Thunnissen E, Bubendorf L, Madsen LB, et al. Prognostic Impact of KRAS G12C Mutation in Patients With NSCLC: Results From the European Thoracic Oncology Platform Lungscape Project. J Thorac Oncol (2021) 16(6):990–1002. doi: 10.1016/j.jtho.2021.02.016
19. El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol (2019) 14(5):876–89. doi: 10.1016/j.jtho.2019.01.020
20. Nadal E, Chen G, Prensner JR, Shiratsuchi H, Sam C, Zhao L, et al. KRAS-G12C Mutation Is Associated With Poor Outcome in Surgically Resected Lung Adenocarcinoma. J Thorac Oncol (2014) 9(10):1513–22. doi: 10.1097/JTO.0000000000000305
21. Hanahan D, Weinberg R. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
22. Caetano MS, Zhang H, Cumpian AM, Gong L, Unver N, Ostrin EJ, et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-Ras-Mutant Lung Cancer. Cancer Res (2016) 76(11):3189–99. doi: 10.1158/0008-5472.CAN-15-2840
23. Dias Carvalho P, Guimarães CF, Cardoso AP, Mendonça S, Costa ÂM, Oliveira MJ, et al. KRAS Oncogenic Signaling Extends Beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res (2018) 78(1):7–14. doi: 10.1158/0008-5472.CAN-17-2084
24. Ancrile B, Lim KH, Counter CM. Oncogenic Ras-Induced Secretion of IL6 Is Required for Tumorigenesis. Genes Dev (2007) 21(14):1714–9. doi: 10.1101/gad.1549407
25. Brooks GD, McLeod L, Alhayyani S, Miller A, Russell PA, Ferlin W, et al. IL6 Trans-Signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res (2016) 76(4):866–76. doi: 10.1158/0008-5472.CAN-15-2388
26. Tani T, Kitajima S, Conway EB, Knelson EH, Barbie DA. KRAS G12C Inhibition and Innate Immune Targeting. Expert Opin Ther Targets (2021) 25(3):167–74. doi: 10.1080/14728222.2021.1902991
27. Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-Cell Activity in the Lung Tumor Microenvironment. Cancer Res (2016) 76(5):999–1008. doi: 10.1158/0008-5472.CAN-15-1439
28. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov (2018) 8(7):822–35. doi: 10.1158/2159-8290.cd-18-0099
29. Skoulidis F, Arbour KC, Hellmann MD, Patil PD, Marmarelis ME, Awad MM, et al. Association of STK11/LKB1 Genomic Alterations With Lack of Benefit From the Addition of Pembrolizumab to Platinum Doublet Chemotherapy in Non-Squamous Non-Small Cell Lung Cancer. J Clin Oncol (2019) 37(15_suppl):102–2. doi: 10.1200/jco.2019.37.15_suppl.102
30. Saigi M, Alburquerque-Bejar JJ, Sanchez-Cespedes M. Determinants of Immunological Evasion and Immunecheckpoint Inhibition Response in Non-Small Cell Lung Cancer: The Genetic Front. Oncogene (2019) 38(31):5921–32. doi: 10.1038/s41388-019-0855-x
31. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated With LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov (2019) 9(1):34–45. doi: 10.1158/2159-8290.CD-18-0689
32. Della Corte C, Sen T, Gay CM, Ramkumar K, Diao L, Cardnell RJ, et al. STING Pathway Expression Identifies NSCLC With an Immune-Responsive Phenotype. J Thorac Oncol (2020) 15(5):777–91. doi: 10.1016/j.jtho.2020.01.009
33. Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. Preliminary Results of the First-In-Human (FIH) Study of MK-1454, an Agonist of Stimulator of Interferon Genes (STING), as Monotherapy or in Combination With Pembrolizumab in Patients With Advanced Solid Tumors or Lymphomas. Ann Oncol (2018) 29(1):viii712. doi: 10.1093/annonc/mdy424.015
34. Rodenhuis S, van de Wetering ML, Mooi WJ, Evers SG, van Zandwijk N, Bos JL. Mutational Activation of the K-Ras Oncogene. A Possible Pathogenetic Factor in Adenocarcinoma of the Lung. N Engl J Med (1987) 317(15):929–35. doi: 10.1056/NEJM198710083171504
35. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther (2017) 16(11):2598–608. doi: 10.1158/1535-7163.mct-17-0386
36. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune Checkpoint Inhibitors for Patients With Advanced Lung Cancer and Oncogenic Driver Alterations: Results From the IMMUNOTARGET Registry. Ann Oncol (2019) 30(8):1321–8. doi: 10.1093/annonc/mdz167
37. Passiglia F, Cappuzzo F, Alabiso O, Bettini AC, Bidoli P, Chiari R, et al. Efficacy of Nivolumab in Pre-Treated Non-Small-Cell Lung Cancer Patients Harbouring KRAS Mutations. Br J Cancer (2019) 120(1):57–62. doi: 10.1038/s41416-018-0234-3
38. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab Versus Docetaxel in Advanced Non-Squamous Non–Small-Cell Lung Cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/nejmoa1507643
39. Herbst RS, Lopes G, Kowalski DM, Kasahara K, Wu Y, De Castro G Jr, et al. Association of KRAS Mutation Status With Response to Pembrolizumab Monotherapy Given as First-Line Therapy for PD-L1 Positive Advanced Non-Squamous NSCLC in KEYNOTE-042. (2019), Oncologypro.esmo.org.
40. Gadgeel S, Rodriguez-Abreu D, Felip E, Esteban E, Speranza G, Reck M, et al. KRAS Mutational Status and Efficacy in KEYNOTE-189: Pembrolizumab Plus Chemotherapy (vs Placebo Plus Chemo as First-Line Therapy for Metastatic Non-Squamous NSCLC. (2019), Oncologypro.esmo.org.
41. Frost N, Kollmeier J, Vollbrecht C, Grah C, Matthes B, Pultermann D, et al. KRASG12C/TP53 Co-Mutations Identify Long-Term Responders to First Line Palliative Treatment With Pembrolizumab Monotherapy in PD-L1 High (≥50%) Lung Adenocarcinoma. Trans Lung Cancer Res (2021) 10(2):737–52. doi: 10.21037/tlcr-20-958
42. Noordhof AL, Damhuis RAM, Hendriks LEL, de Langen AJ, Timens W, Venmans BJW, et al. Prognostic Impact of KRAS Mutation Status for Patients With Stage IV Adenocarcinoma of the Lung Treated With First-Line Pembrolizumab Monotherapy. Lung Cancer (2021) 155(2):163–9. doi: 10.1016/j.lungcan.2021.04.001
43. West H, Cappuzzo F, Reck M, Mok T, Jotte RM, Nishio M, et al. 1265p IMpower150: A Post Hoc Analysis of Efficacy Outcomes in Patients With KRAS, STK11 and KEAP1 Mutations. Ann Oncol (2020) 31(2):S817–8. doi: 10.1016/j.annonc.2020.08.1579
44. Liu Y, Zhang Z, Rinsurongkawong W, Le X, Gay CM, Ning MS, et al. Driver Mutations to Predict for Poorer Outcomes in Non-Small Cell Lung Cancer Patients Treated With Concurrent Chemoradiation and Consolidation Durvalumab. J Clin Oncol (2021) 39(15_suppl):8528–8. doi: 10.1200/jco.2021.39.15_suppl.8528
45. Pantsar T. The Current Understanding of KRAS Protein Structure and Dynamics. Comput Struct Biotechnol J (2020) 18(1):189–98. doi: 10.1016/j.csbj.2019.12.004
46. Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output With Small Molecules Targeting the Inactive State. Cancer Discov (2016) 6(3):316–29. doi: 10.1158/2159-8290.CD-15-1105
47. Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, Roszik J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers (2021) 13(6):1204. doi: 10.3390/cancers13061204
48. Blumenschein GR Jr, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A Randomized Phase II Study of the MEK1/MEK2 Inhibitor Trametinib (GSK1120212) Compared With Docetaxel in KRAS-Mutant Advanced Non-Small-Cell Lung Cancer (NSCLC)†. Ann Oncol (2015) 26(5):894–901. doi: 10.1093/annonc/mdv072
49. Jänne P, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib Plus Docetaxel for KRAS-Mutant Advanced Non-Small-Cell Lung Cancer: A Randomised, Multicentre, Placebo-Controlled, Phase 2 Study. Lancet Oncol (2013) 14(1):38–47. doi: 10.1016/s1470-2045(12)70489-8
50. Goldman JW, Mazieres J, Barlesi F, Dragnev KH, Koczywas M, Göskel T, et al. A Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients With Stage IV Non-Small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: Juniper. Front Oncol (2020) 10:578756(1). doi: 10.3389/fonc.2020.578756
51. Ramanathan R, Von Hoff DD, Eskens F, Blumenschein G Jr, Richards D, Genvresse I, et al. Phase Ib Trial of the PI3K Inhibitor Copanlisib Combined With the Allosteric MEK Inhibitor Refametinib in Patients With Advanced Cancer. Target Oncol (2020) 15(2):163–74. doi: 10.1007/s11523-020-00714-0
52. Mazieres J, Pradines A, Favre G. Perspectives on Farnesyl Transferase Inhibitors in Cancer Therapy. Cancer Lett (2004) 206(2):159–67. doi: 10.1016/j.canlet.2003.08.033
53. Sakamoto K, Masutani T, Hirokawa T. Generation of KS-58 as the First K-Ras(G12D)-Inhibitory Peptide Presenting Anti-Cancer Activity In Vivo. Sci Rep (2020) 10(1):21671. doi: 10.1038/s41598-020-78712-5
54. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J Med Chem (2021). doi: 10.1021/acs.jmedchem.1c01688
55. Jeng HH, Taylor LJ, Bar-Sagi D. Sos-Mediated Cross-Activation of Wild-Type Ras by Oncogenic Ras Is Essential for Tumorigenesis. Nat Commun (2012) 3(1):1168. doi: 10.1038/ncomms2173
56. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers Through Combined MEK Inhibition. Cancer Discov (2021) 11(1):142–57. doi: 10.1158/2159-8290.CD-20-0142
57. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From in Vitro Experiments. J Thorac Oncol (2021) 16(8):1321–32. doi: 10.1016/j.jtho.2021.04.015
58. Kerr DL, Haderk F, Bivona TG. Allosteric SHP2 Inhibitors in Cancer: Targeting the Intersection of RAS, Resistance, and the Immune Microenvironment. Curr Opin Chem Biol (2021) 62(1):1–12. doi: 10.1016/j.cbpa.2020.11.007
59. LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J Med Chem (2020) 63(22):13578–94. doi: 10.1021/acs.jmedchem.0c01170
60. Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, et al. A Small Molecule RAS-Mimetic Disrupts RAS Association With Effector Proteins to Block Signaling. Cell (2016) 165(3):643–55. doi: 10.1016/j.cell.2016.03.045
61. Chenard-Poirier M, Kaiser M, Boyd K, Sriskandarajah P, Constantinidou A, Harris SJ, et al. Results From the Biomarker-Driven Basket Trial of RO5126766 (CH5127566), a Potent RAF/MEK Inhibitor, in RAS- or RAF-Mutated Malignancies Including Multiple Myeloma. J Clin Oncol (2017) 35(15_suppl):2506–6. doi: 10.1200/jco.2017.35.15_suppl.2506
62. Nogova L, Mattonet C, Scheffler M, Taubert M, Gardizi M, Sos ML, et al. Sorafenib and Everolimus in Patients With Advanced Solid Tumors and KRAS-Mutated NSCLC: A Phase I Trial With Early Pharmacodynamic FDG-PET Assessment. Cancer Med (2020) 14(9):4991–5007. doi: 10.1002/cam4.3131
63. Broutin S, Stewart A, Thavasu P, Paci A, Bidart JM, Banerji U. Insights Into Significance of Combined Inhibition of MEK and M-TOR Signalling Output in KRAS Mutant Non-Small-Cell Lung Cancer. Br J Cancer (2016) 115(5):549–52. doi: 10.1038/bjc.2016.220
64. Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, et al. Antitumor Activity in RAS-Driven Tumors by Blocking AKT and MEK. Clin Cancer Res (2015) 21(4):739–48. doi: 10.1158/1078-0432.CCR-14-1901
65. Kruspig B, Monteverde T, Neidler S, Hock A, Kerr E, Nixon C, et al. The ERBB Network Facilitates KRAS-Driven Lung Tumorigenesis. Sci Trans Med (2018) 10(446):eaao2565. doi: 10.1126/scitranslmed.aao2565
66. Moll HP, Pranz K, Musteanu M, Grabner B, Hruschka N, Mohrherr J, et al. Afatinib Restrains K-RAS–driven Lung Tumorigenesis. Sci Trans Med (2018) 10(446):eaao2301. doi: 10.1126/scitranslmed.aao2301
67. Carter CA, Rajan A, Keen C, Szabo E, Khozin S, Thomas A, et al. Selumetinib With and Without Erlotinib in KRAS Mutant and KRAS Wild-Type Advanced Nonsmall-Cell Lung Cancer. Ann Oncol (2016) 27(4):693–9. doi: 10.1093/annonc/mdw008
68. Kerk S, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic Networks in Mutant KRAS-Driven Tumours: Tissue Specificities and the Microenvironment’. Nat Rev Cancer (2021) 21(8):510–25. doi: 10.1038/s41568-021-00375-9
69. Shackelford D, Shaw RJ. The LKB1–AMPK Pathway: Metabolism and Growth Control in Tumour Suppression. Nat Rev Cancer (2009) 9(8):563–75. doi: 10.1038/nrc2676
Keywords: KRAS, NSCLC, ICI, PD-L1, STK11
Citation: Cucurull M, Notario L, Sanchez-Cespedes M, Hierro C, Estival A, Carcereny E and Saigí M (2022) Targeting KRAS in Lung Cancer Beyond KRAS G12C Inhibitors: The Immune Regulatory Role of KRAS and Novel Therapeutic Strategies. Front. Oncol. 11:793121. doi: 10.3389/fonc.2021.793121
Received: 11 October 2021; Accepted: 21 December 2021;
Published: 13 January 2022.
Edited by:
Laura Mezquita, Hospital Clínic de Barcelona, SpainReviewed by:
Matthias Scheffler, University Hospital of Cologne, GermanyMohamed I. Saad, Hudson Institute of Medical Research, Australia
Copyright © 2022 Cucurull, Notario, Sanchez-Cespedes, Hierro, Estival, Carcereny and Saigí. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Saigí, bXNhaWdpQGljb25jb2xvZ2lhLm5ldA==