 Kristina Aaltonen
Kristina Aaltonen Katarzyna Radke
Katarzyna Radke Aleksandra Adamska
Aleksandra Adamska Daniel Bexell
Daniel Bexell- Division of Translational Cancer Research, Department of Laboratory Medicine, Lund University, Lund, Sweden
Neuroblastoma is a childhood cancer derived from the sympathetic nervous system. High-risk neuroblastoma patients have a poor overall survival and account for ~15% of childhood cancer deaths. There is thus a need for clinically relevant and authentic models of neuroblastoma that closely resemble the human disease to further interrogate underlying mechanisms and to develop novel therapeutic strategies. Here we review recent developments in patient-derived neuroblastoma xenograft models and in vitro cultures. These models can be used to decipher mechanisms of metastasis and treatment resistance, for drug screening, and preclinical drug testing. Patient-derived neuroblastoma models may also provide useful information about clonal evolution, phenotypic plasticity, and cell states in relation to neuroblastoma progression. We summarize current opportunities for, but also barriers to, future model development and application. Integration of patient-derived models with patient data holds promise for the development of precision medicine treatment strategies for children with high-risk neuroblastoma.
1. Introduction
Despite significant academic, industrial, and clinical efforts, successfully translating preclinical findings to clinical trials and practice remains challenging (1–4) and less than 10% of drugs entering oncology clinical trials are eventually approved for clinical use (1, 5). Furthermore, these efforts and failures come at high financial and ethical costs. Pediatric malignancies have special considerations, since clinical drug testing is even more restricted by the relatively small number of patients and the ethics related to long-term side-effects in children. Involvement of multiple stakeholders is important to address the lack of childhood-specific drug development in the pharmaceutical sector (6, 7). Thus, there is an urgent need for clinically relevant and biologically accurate preclinical models to minimize these current bottlenecks to drug development and implementation (8).
Neuroblastoma (NB) is the most common solid extracranial pediatric tumor, accounting for ~15% of pediatric oncology deaths (9, 10). NB can be regarded as an aberration of neural crest development, and although it can arise anywhere along the sympathetic nervous system, most primary tumors are found in the adrenal gland (11). NB is biologically and clinically heterogenous, and patients are stratified into different risk groups based on tumor characteristics and disease presentation (12, 13). Clinical responses vary from spontaneous regression to metastatic and drug-resistant disease despite intensive treatment (13). Furthermore, patients often suffer from severe therapy-related long-term adverse effects (14).
NB is a copy number-driven disease with few targetable somatic mutations found at diagnosis, especially when compared with adult malignancies (15). The MYCN oncogene is amplified in ~20% of cases and is strongly correlated with aggressive phenotypes and unfavorable clinical outcomes (16). Other common chromosomal copy number changes, including 11q loss and 17q gain, are also poor prognostic features (17). Recurrent mutations are rare in NB but include ALK (9%), ATRX (7%), and PTEN (3%) mutations (15). In relapsed tumors, genome-wide sequencing has revealed a higher prevalence of recurrent mutations in targetable pathways, such as RAS-MAPK (18, 19), but at relatively low frequencies. Recent transcriptional and epigenetic analyses suggest that NB cells can adopt at least two phenotypic cell states, known as adrenergic (ADR)/differentiated and mesenchymal (MES)/immature (20–24). These findings highlight the heterogeneous and dynamic nature of NB.
Reliable, predictive, and authentic NB models are important because: (i) NB is uncommon, so sufficiently powered clinical trials are challenging and patient material is scarce for molecular studies (25), placing extra weight on the translatability of preclinical results; (ii) preclinical models that accurately recapitulate known clinical, genetic, and transcriptional intra- and inter-tumor heterogeneity are important for the identification of effective therapeutic targets; and (iii) patient-derived (PD) models resemble the clinical scenario better than conventional models and can therefore be used to screen for and test the most promising and safe novel therapies.
Here we discuss recent progress in patient-derived xenografts (PDXs) and PD in vitro cultures as preclinical NB models. We summarize the development of different PD models, their utility in studying biological mechanisms and treatment responses, and how they can be utilized for preclinical drug testing to improve treatment strategies against NB (Figure 1).
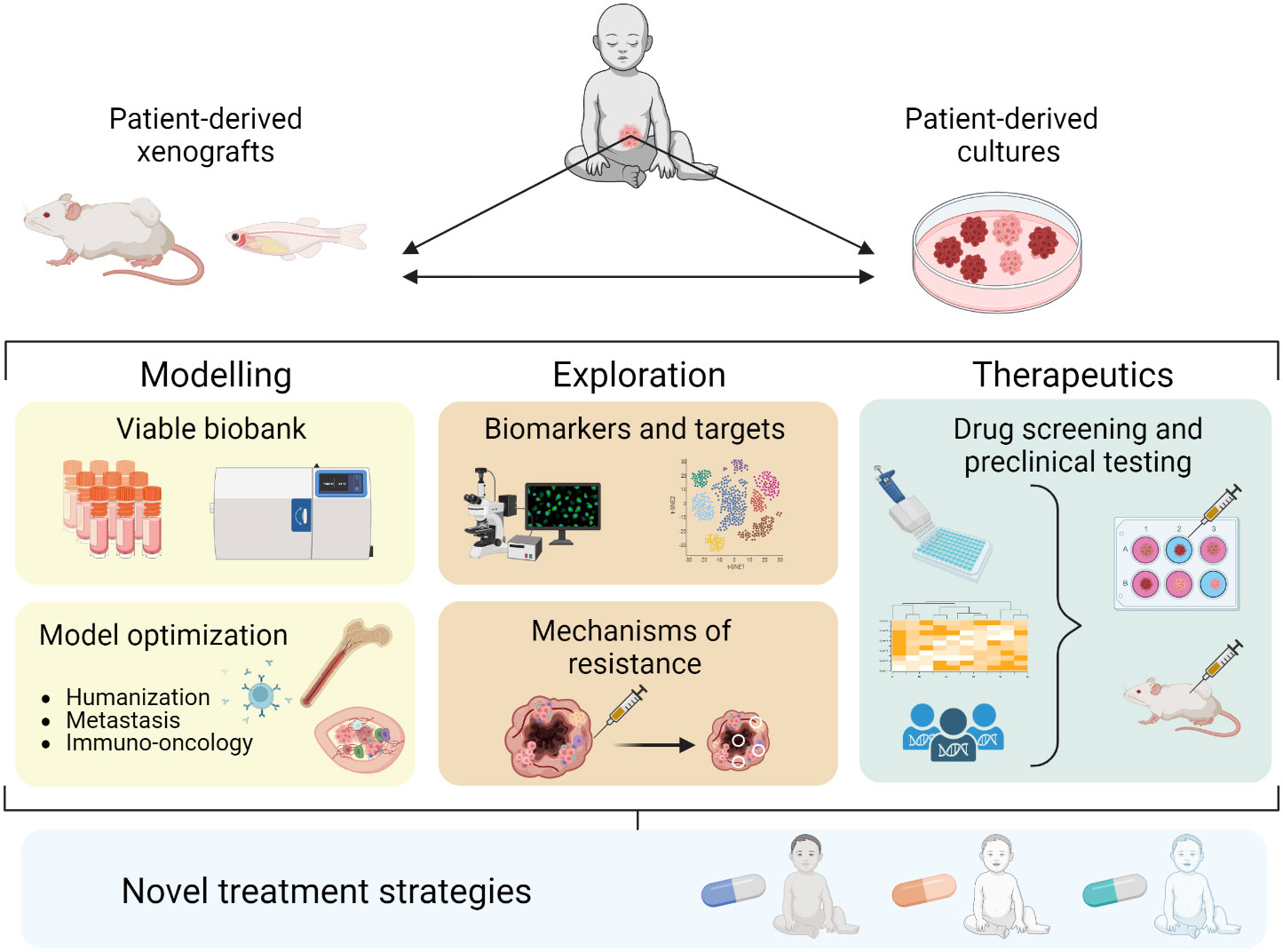
Figure 1 Establishment and application of PD models in NB contributing to novel treatment strategies.
2. PD NB models
Conventional cell lines have been used as laboratory models for decades, and they have provided valuable knowledge about tumor biology and drug efficacy in many cancer types. However, cancer cell lines are usually passaged under serum-containing conditions for years and thus their molecular profiles often differ from the original patient tumor (26, 27). This matters in terms of model fidelity, especially when considering clinical applicability; for example, clinically important features such as drug resistance might be lost after long-term in vitro passaging (27). PD NB models are established directly from tumor material obtained from children after parental informed consent. PD models have been shown to better reflect the features (e.g., treatment response) of their original tumors, compared with conventional models (28–31). PDXs have now been established from many diverse tumor types of adult and pediatric cancers including NB (32–35). Over the last few decades, the cancer research community has gradually turned towards PD model systems (8), and the US National Cancer Institute recently decided to replace its panel of human cancer cell lines (NCI-60) with well-characterized PDX models (36) for drug screening.
2.1. Establishment of NB PDXs in vivo
NB PDXs have been established in immunocompromised mice, mainly by implanting tumor samples or cells obtained from patients next to the adrenal gland (orthotopic implantation) or subcutaneously (ectopic/heterologous implantation). Established orthotopic PDX tumors can be monitored by clinical imaging techniques such as FDG-PET or MRI (37), and they have been shown to retain important patient tumor characteristics such as invasive growth patterns into surrounding tissues and spontaneous metastatic capacity to the bone marrow, lungs, and liver (37–39). PDX models retain NB-specific molecular features, including cellular differentiation status, protein marker expression (synaptophysin, chromogranin A, NCAM/CD56), chromosomal copy number changes (including 1p loss, MYCN amplification, 17q gain), mutational profiles, and DNA methylation status (32, 34, 37–41). Transcriptional analysis of orthotopic NB PDXs has shown that they also retain a certain degree of patient-specific gene expression, indicating transcriptional stability, from the corresponding NBs (32, 39, 40). Thus, although a PDX is established from only a fragment of the original patient tumor, data from multiple laboratories have shown that NB PDXs represent the main and clinically relevant features of NB patient tumors. There are now several sources of NB PDX tumors (detailed in (42)), including the US Pediatric Preclinical In Vivo Testing Consortium (PIVOT) and the European ITCC-P4 - Pediatric Preclinical Proof of Concept Platform.
The site of implantation affects the PDX model: orthotopic implantation has a higher engraftment rate and tumors grow faster than those implanted subcutaneously (32, 41). The human tumor microenvironment (TME) is gradually lost in vivo. Instead, orthotopic NB PDXs have been shown to contain a murine TME including for example vascularization, pericytes, macrophages, and extracellular matrix resembling the architecture in the parental NB (38). Potential functional differences between human and mouse TMEs are not fully elucidated and this uncertainty is important to consider (43). It has been debated whether the use of mice as hosts leads to murine-specific tumor evolution during PDX engraftment and propagation (44, 45). However, serial in vivo passaging of orthotopic NB PDXs for up to two years has shown that PDXs retain key genetic aberrations (e.g., 1p loss, MYCN amplification, and 17q gain) and acquire only minor genetic changes over time, as would be expected from their natural evolution (39). Clonal dynamics studies during tumor progression in PDXs have shown the presence of branched evolution, clonal sweeps, and convergent evolution of specific small deletions in potentially tumor-associated genes (46), a pattern similar to tumor evolution in NB patients (47).
There have been cases where human lymphomas have developed at the site of NB-cell injection (41, 48), or when murine-derived tumors have replaced the human PDX (49, 50), so thorough and frequent characterization of PDXs is necessary. Setting up robust biobanks for storage of well-characterized, early passage PDX-tumors will be of great benefit to the research community (8).
While most PDX models have been established in mice, zebrafish are increasingly used as hosts for implantation of PD tumor cells, including NB. Zebrafish allow for rapid and low-cost preclinical drug screening in an intact organism that may inform about precision medicine strategies in NB (51, 52). Furthermore, genetically-modified strains are available and tumors can be visualized from an early stage and followed dynamically. However, challenges in translating drug testing findings to patients include limited toxicity and pharmacodynamic data (51), temperature differences, and the non-mammalian TME.
PD cells and tumor biopsies have also been implanted into chick embryos with a high engraftment rate, forming metastases only from tumor cells from patients with metastatic NB and not from localized disease (53). NB cells migrated along the embryonic aorta and along peripheral nerves, demonstrating these as major routes for metastatic dissemination. This model allows for investigation of tumor progression and metastasis in an embryonic environment in vivo (53). However, the clinical relevance of the models remains uncertain.
2.2. PD cultures in vitro
PD tumor cultures are established in vitro directly from patients and can be grown as tumor organoids (PDOs), spheroids, or as semi-attached or attached cultures. PD cultures provide an opportunity to test potential therapeutics in a faster, high-throughput manner compared to PDXs.
PD NB cultures are isolated directly from primary or metastatic tumors from patients and are cultured in serum-free medium with defined growth factors to avoid neurospecific differentiation. This is best achieved on low-attachment plastics, with or without Matrigel or other scaffolding materials. Several groups have shown that PD NB cultures retain the copy-number profiles, mutation patterns, and other genetic and phenotypic characteristics of the tumor of origin (54–57) in both Matrigel and as free-floating spheres, but PDOs in Matrigel have better self-organization (56). Establishing PD cultures from different stages and subgroups of NB has been challenging. In general, more aggressive, MYCN-amplified, and metastatic tumors are easier to propagate in vitro. Recent advances in 3D scaffolding with hydrogels and porous scaffolding (reviewed in (58)) together with further optimization of culture conditions might increase the probability of successful establishment. Characterization of culture conditions is also important to understand how different transcriptional cell states might be maintained in vitro. Notably, it is important to verify NB identity and lack of contamination with other cells, which can otherwise overtake PD NB cultures (59, 60).
Since the limited number of NB patients restricts the number of models, a complementary approach is to use PDX-derived in vitro cultures after expansion of patient material in vivo (37, 40, 57, 61–63). Similar to PD cultures, PDX-derived NB cells can be grown adherent or as free-floating 3D cultures, and they retain patient-specific genomic aberrations as well as tumorigenic and metastatic capacity in vivo (62). Drug responses between NB PD- and PDX-derived cultures are highly correlated, suggesting that these models can be used interchangeably for drug testing (41). Biobanking of PD- and PDX-derived NB cultures will be a very important tool for future drug screening and larger preclinical drug testing (8).
3. Applications of PD NB models
Conventional cell lines, cell-line derived xenografts, and genetically engineered mouse models have been the main preclinical tools used to study resistance mechanisms and for drug testing. By using PD models that retain the main characteristics and heterogeneity of the original tumors, patient tumors and their treatment response can be better represented in the laboratory, thereby bridging the gap between preclinical models and the clinic.
3.1. Identification of treatment resistance mechanisms and biomarkers
Treatment resistance, relapse after therapy, and metastasis are urgent clinical problems in NB. A few studies have used PD models to identify diverse mechanisms implicated in NB invasion, migration, metastasis (64, 65), and resistance to specific chemotherapies (66). Using a clinically relevant treatment protocol (COJEC-induction therapy), NB PDXs show similar chemotherapy responses to their corresponding patients, suggesting that NB PDXs are useful for modelling chemoresistance and relapse (46). The models showed that chemoresistant NBs have a lower ADR signature and enrichment for an immature MES-like phenotype, suggesting an association between the MES cell state and relapse (46). These results are consistent with recent findings in the clinical setting (24, 67). The ability to accurately model treatment responses and their association with phenotypic cell states make in vivo PDXs a very promising tool to explore NB phenotypes in a reproducible manner, as well as characterizing the role of phenotypic plasticity in acquired and intrinsic resistance.
Reliable biomarkers for monitoring tumor responses are important for longer-term studies of relapse and resistance, and in clinical diagnostics. NB PDXs reproduce the patient’s relative levels of circulating metanephrines (68). Given that metanephrines are tumor progression biomarkers [plasma levels correlate with tumor volume (69)], this could pave the way for a minimally invasive method of monitoring tumor response/resistance in orthotopic PDX models. Another approach for monitoring responses is with gene signatures as recently optimized and used in a therapeutic study for high-risk relapsed NB in PD models (70).
3.2. Drug testing
3.2.1. Application in the preclinical setting
Preclinical PD model testing is now highly recommended for proof-of-concept studies of new drugs and drug combinations aiming for clinical trials in the pediatric population (8). Many NB targets identified in patients have been tested in PD models in vitro and in vivo, allowing the evaluation of specific responses in tumors harboring different underlying, molecular alterations. Some known genetic vulnerabilities in NB are still under investigation, while others, for example ALK, have been clinically tested (71, 72). Table 1 presents an overview of recent preclinical drug investigations of established and novel NB targets that were identified and tested in PD models.
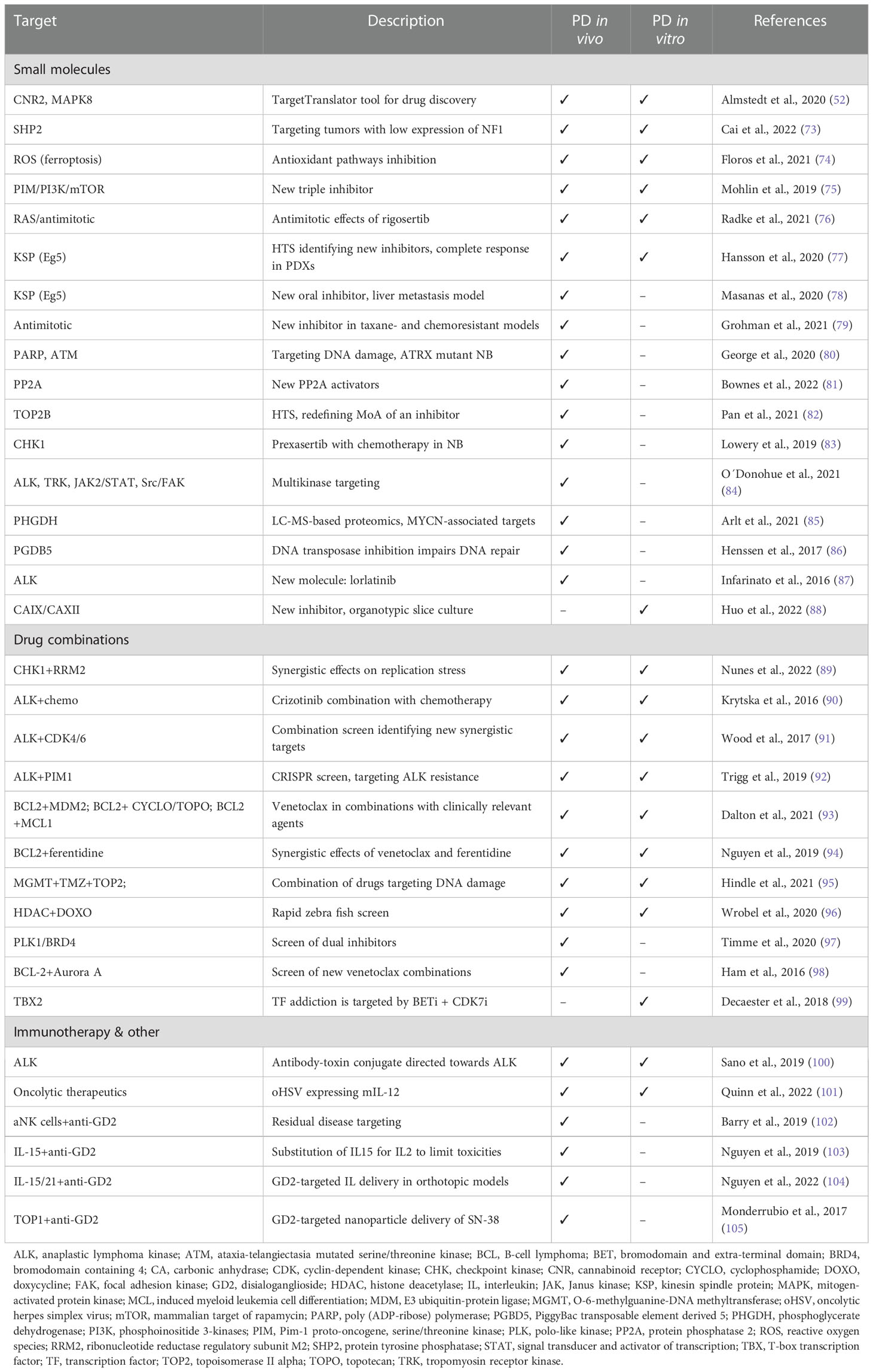
Table 1 Selected preclinical drug testing studies using NB patient-derived models.
High-throughput screens (HTS) in vitro can facilitate the discovery of specific targets and/or drugs using CRISPR/siRNA or phenotypic response. Most screening approaches still use conventional cell lines, but more recently PDX-derived cultures of high-risk NB have been used for the initial identification, for example for a KSP inhibitor (77). Compounds identified in drug screens can be further verified in vivo in PDXs.
In our experience, PD models show high intra-model variability in drug response (46, 75, 77) and are often less responsive to different treatments than conventional cell lines and xenografts [discussed also in (29)]. The lower sensitivity of PD models could indicate an even smaller effect in patients, thus providing important information with respect to optimal clinical implementation.
3.2.2. Application for precision medicine in the clinic
The possibility of identifying actionable genetic alterations in pediatric cancers has contributed to optimism that the approach is useful for clinical trial design and target identification for high-risk and relapsed pediatric tumors, including high-risk NB (71, 72). Langenberg et al. thoroughly summarized current pediatric precision medicine programs around the world (106). Many of the programs/consortia [Pediatric MATCH (US) or INFORM (Europe)] have enabled patients to receive treatments tailored to the individual tumor’s molecular profile (107–109). However, relatively few identified mutations (<30%) have led to targeted therapies (106, 107, 110). This highlights the need for molecular profiling of patients to be backed up by real-time functional testing of drug sensitivities in PD models.
Both the PIVOT (US, earlier PPTC) and ITCC-P4 (Europe) repositories hold PDXs. Considering that PDXs take time to establish, co-clinical avatar studies are generally very difficult. Nevertheless, the rarity of pediatric cancers and scarcity of models representing specific subtypes within pediatric tumors makes those repositories a valuable resource for the accelerated development and translation of novel therapeutics into early phase trials (34, 111, 112). Lau et al. developed a pediatric precision medicine platform (including a few high-risk NBs) of PDX models and HTS in PDOs, observing a correlation between PDX results, HTS-PDOs, and the clinical responses in patients (113). Importantly, the addition of functional drug testing to a genome-only analysis increased the number of patients with drug options by also identifying drug sensitivities not associated with molecular hallmarks (113).
Real-time drug testing for the immediate benefit of the patient is likely to be more feasible in PDO models where the time for establishment is much shorter and the readout can be performed with a higher throughput. The COMPASS consortium (Clinical implementation Of Multidimensional PhenotypicAl drug SenSitivities in paediatric precision oncology) is a large-scale effort to implement HTS in PD models. This European collaborative platform aims to implement PDO screening for individualized drug sensitivity assessment and therapy (114). Recently, the network also standardized drug scoring tools and developed machine learning approaches (115, 116).
4. Current and future model optimization
Although the successful establishment of PD NB models is encouraging, certain aspects can still be improved. For example, the distribution and function of the extracellular matrix (ECM) has been shown to influence NB progression in patient samples (117, 118). Consistently, modulation of ECM components induces specific cell behaviors of PD NB cultures (63). Optimization of ECM conditions could thus contribute to improved NB modelling.
The lack of a complete immune system is a limitation of most PDX models, since PD tumors are generally implanted in immunocompromised mouse strains (e.g., NSG) to permit tumor engraftment. Reconstitution of a humanized immune system, for example by injection of human hematopoietic stem cells into sub-lethally irradiated mice, could improve the immune status of the models (119). A technically advanced humanized mouse strain (MISTRG) supports the intrinsic development of human natural killer (NK) cells after bone marrow transplantation (120). When combined with orthotopic NB PDXs, these mice have allowed the identification of immune modulating functions in common between PDXs and patient tumors and suggest that the model is useful for immuno-oncology studies in general and in NB in particular (121). The use of PDO and stromal/immune cell co-cultures can be applied in vitro, where it has been very challenging to optimize culture conditions for multiple cell types over longer periods (122, 123). Co-cultures of NB organoids and peripheral blood mononuclear cells (from a healthy donor) were recently used to test a novel immunotherapy (124).
Innovative technological advances have suggested that microfluidics (lab-on-a-chip) and bioprinting may provide future systems for studying tumor cell and stromal/immune cell interactions. Functional short-time cultures of both tumor cells and immune cells in a microfluidic system have been reported for adult cancers (125, 126) and might be applicable also to pediatric cancers. Very recently, the first bioprinted, vascularized NB microenvironment on a fluidic chip was reported (127). Implantation of cell line-derived NB spheroids led to NB cell survival for two weeks and successful micro-vessel infiltration of the spheroids (127). A different study managed to establish PD NB organotypic slice cultures that could potentially preserve an intact NB tumor microenvironment (88). This study used a perfusion-based bioreactor to force medium through the tissue, thereby providing continuous nutrient delivery to the whole tumor.
Orthotopic NB PDXs retain spontaneous metastatic capacity in vivo to the lungs, liver, and bone marrow, mimicking the entire process from primary tumor growth to invasion and metastasis (37, 39). However, the TME is generally murine (38) and there are uncertainties about cross reactivity between human NB cells and the mouse TME. The presence of human mesenchymal stem cells can increase growth and metastasis of NB cells in vivo (128), suggesting species preference. Recent advances in tissue engineering have produced in vivo models of humanized bone (so-called ossicles) in mice. Implanted PDX-derived NB cells form osteolytic tumor lesions in the ossicles and display higher and faster engraftment rates than in mouse bone (129). This model could thus be valuable for the investigation of human NB growth and treatment responses in a humanized metastatic niche.
Further optimization of PD models to account for more patient-like microenvironmental factors both in vivo and in vitro is ongoing and will likely contribute to improved translatability.
5. Conclusions
The use of clinically relevant preclinical models is of immense importance in childhood cancers, such as high-risk NB, where the access to patient material is limited. PD models reflect the characteristics of the tumor of origin better than conventional in vivo and in vitro models. Nevertheless, ongoing efforts may further optimize their translational relevance. Existing NB PDXs and PD cultures have been – and continue to be – used to decipher therapy resistance and for target identification and drug testing. Future studies will need to investigate how PD models can be used to exploit phenotypic plasticity and NB cell states in preclinical studies to better benefit NB patients.
Author contributions
KA, KR, AA, AS, AM and DB wrote and reviewed the manuscript. KA and DB supervised the project. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the following grants and fellowships: Swedish Cancer Society grant 20 0897 PjF (DB). Swedish Cancer Society postdoctoral fellowship 21 0346 PT (AM). The Swedish Childhood Cancer Foundation grant PR2020-0018 (DB). The Swedish Research Council grant 2021-02597 (DB).
Acknowledgments
Figure 1 was created using Biorender.com.
Conflict of interest
DB has received research funding from Healx, aPODD foundation, and Captor Therapeutics for unrelated work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics (2019) 20(2):273–86. doi: 10.1093/biostatistics/kxx069
2. Sharpless NE, Depinho RA. The mighty mouse: Genetically engineered mouse models in cancer drug development. Nat Rev Drug discovery (2006) 5(9):741–54. doi: 10.1038/nrd2110
3. Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J cancer (2001) 84(10):1424–31. doi: 10.1054/bjoc.2001.1796
4. Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML, et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med (2019) 11(509):eaaw8412. doi: 10.1126/scitranslmed.aaw8412
5. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotechnol (2014) 32(1):40–51. doi: 10.1038/nbt.2786
6. Moreno L, Barone G, DuBois SG, Molenaar J, Fischer M, Schulte J, et al. Accelerating drug development for neuroblastoma: Summary of the second neuroblastoma drug development strategy forum from innovative therapies for children with cancer and international society of paediatric oncology Europe neuroblastoma. Eur J Cancer (2020) 136:52–68. doi: 10.1016/j.ejca.2020.05.010
7. Pearson AD, Heenen D, Kearns PR, Goeres A, Marshall LV, Blanc P, et al. 10-year report on the European paediatric regulation and its impact on new drugs for children's cancers. Lancet Oncol (2018) 19(3):285–7. doi: 10.1016/S1470-2045(18)30105-0
8. Vassal G, Houghton PJ, Pfister SM, Smith MA, Caron HN, Li XN, et al. International consensus on minimum preclinical testing requirements for the development of innovative therapies for children and adolescents with cancer. Mol Cancer Ther (2021) 20(8):1462–8. doi: 10.1158/1535-7163.MCT-20-0394
9. Gatta G, Botta L, Rossi S, Aareleid T, Bielska-Lasota M, Clavel J, et al. Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5–a population-based study. Lancet Oncol (2014) 15(1):35–47. doi: 10.1016/S1470-2045(13)70548-5
10. Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am (2010) 24(1):65–86. doi: 10.1016/j.hoc.2009.11.011
11. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers (2016) 2:16078. doi: 10.1038/nrdp.2016.78
12. Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33(27):3008–17. doi: 10.1200/JCO.2014.59.4648
13. Tolbert VP, Matthay KK. Neuroblastoma: Clinical and biological approach to risk stratification and treatment. Cell Tissue Res (2018) 372(2):195–209. doi: 10.1007/s00441-018-2821-2
14. Cohen LE, Gordon JH, Popovsky EY, Gunawardene S, Duffey-Lind E, Lehmann LE, et al. Late effects in children treated with intensive multimodal therapy for high-risk neuroblastoma: High incidence of endocrine and growth problems. Bone Marrow Transplant (2014) 49(4):502–8. doi: 10.1038/bmt.2013.218
15. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45(3):279–84. doi: 10.1038/ng.2529
16. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of n-myc in untreated human neuroblastomas correlates with advanced disease stage. Sci (New York NY) (1984) 224(4653):1121–4. doi: 10.1126/science.6719137
17. Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The international neuroblastoma risk group (INRG) classification system: an INRG task force report. J Clin Oncol Off J Am Soc Clin Oncol (2009) 27(2):289–97. doi: 10.1200/JCO.2008.16.6785
18. Schramm A, Koster J, Assenov Y, Althoff K, Peifer M, Mahlow E, et al. Mutational dynamics between primary and relapse neuroblastomas. Nat Genet (2015) 47(8):872–7. doi: 10.1038/ng.3349
19. Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet (2015) 47(8):864–71. doi: 10.1038/ng.3333
20. van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet (2017) 49(8):1261–6. doi: 10.1038/ng.3899
21. Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugène C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet (2017) 49(9):1408–13. doi: 10.1038/ng.3921
22. Jansky S, Sharma AK, Körber V, Quintero A, Toprak UH, Wecht EM, et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat Genet (2021) 53(5):683–93. doi: 10.1038/s41588-021-00806-1
23. Bedoya-Reina OC, Li W, Arceo M, Plescher M, Bullova P, Pui H, et al. Single-nuclei transcriptomes from human adrenal gland reveal distinct cellular identities of low and high-risk neuroblastoma tumors. Nat Commun (2021) 12(1):5309. doi: 10.1038/s41467-021-24870-7
24. Gartlgruber M, Sharma AK, Quintero A, Dreidax D, Jansky S, Park Y-G, et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat Cancer (2021) 2(1):114–28. doi: 10.1038/s43018-020-00145-w
25. Fletcher JI, Ziegler DS, Trahair TN, Marshall GM, Haber M, Norris MD. Too many targets, not enough patients: Rethinking neuroblastoma clinical trials. Nat Rev Cancer (2018) 18(6):389–400. doi: 10.1038/s41568-018-0003-x
26. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell (2006) 9(5):391–403. doi: 10.1016/j.ccr.2006.03.030
27. Gillet JP, Calcagno AM, Varma S, Marino M, Green LJ, Vora MI, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci United States America (2011) 108(46):18708–13. doi: 10.1073/pnas.1111840108
28. Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer discovery (2014) 4(9):998–1013. doi: 10.1158/2159-8290.CD-14-0001
29. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med (2015) 21(11):1318–25. doi: 10.1038/nm.3954
30. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell (2015) 161(4):933–45. doi: 10.1016/j.cell.2015.03.053
31. Calandrini C, Schutgens F, Oka R, Margaritis T, Candelli T, Mathijsen L, et al. An organoid biobank for childhood kidney cancers that captures disease and tissue heterogeneity. Nat Commun (2020) 11(1):1310. doi: 10.1038/s41467-020-15155-6
32. Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature (2017) 549(7670):96–100. doi: 10.1038/nature23647
33. Brabetz S, Leary SES, Gröbner SN, Nakamoto MW, Şeker-Cin H, Girard EJ, et al. A biobank of patient-derived pediatric brain tumor models. Nat Med (2018) 24(11):1752–61. doi: 10.1038/s41591-018-0207-3
34. Rokita JL, Rathi KS, Cardenas MF, Upton KA, Jayaseelan J, Cross KL, et al. Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep (2019) 29(6):1675–89.e9. doi: 10.1016/j.celrep.2019.09.071
35. Tucker ER, George S, Angelini P, Bruna A, Chesler L. The promise of patient-derived preclinical models to accelerate the implementation of personalised medicine for children with neuroblastoma. J Pers Med (2021) 11(4):248. doi: 10.3390/jpm11040248
36. Ledford H. US Cancer institute to overhaul tumour cell lines. Nature (2016) 530(7591):391. doi: 10.1038/nature.2016.19364
37. Braekeveldt N, Wigerup C, Gisselsson D, Mohlin S, Merselius M, Beckman S, et al. Neuroblastoma patient-derived orthotopic xenografts retain metastatic patterns and geno- and phenotypes of patient tumours. Int J cancer (2015) 136(5):E252–61. doi: 10.1002/ijc.29217
38. Braekeveldt N, Wigerup C, Tadeo I, Beckman S, Sanden C, Jonsson J, et al. Neuroblastoma patient-derived orthotopic xenografts reflect the microenvironmental hallmarks of aggressive patient tumours. Cancer letters (2016) 375(2):384–9. doi: 10.1016/j.canlet.2016.02.046
39. Braekeveldt N, von Stedingk K, Fransson S, Martinez-Monleon A, Lindgren D, Axelson H, et al. Patient-derived xenograft models reveal intratumor heterogeneity and temporal stability in neuroblastoma. Cancer Res (2018) 78(20):5958–69. doi: 10.1158/0008-5472.CAN-18-0527
40. Stewart E, Shelat A, Bradley C, Chen X, Federico S, Thiagarajan S, et al. Development and characterization of a human orthotopic neuroblastoma xenograft. Dev Biol (2015) 407(2):344–55. doi: 10.1016/j.ydbio.2015.02.002
41. Kamili A, Gifford AJ, Li N, Mayoh C, Chow SO, Failes TW, et al. Accelerating development of high-risk neuroblastoma patient-derived xenograft models for preclinical testing and personalised therapy. Br J cancer (2020) 122(5):680–91. doi: 10.1038/s41416-019-0682-4
42. Ornell KJ, Coburn JM. Developing preclinical models of neuroblastoma: Driving therapeutic testing. BMC BioMed Eng (2019) 1:33. doi: 10.1186/s42490-019-0034-8
43. Bleijs M, van de Wetering M, Clevers H, Drost J. Xenograft and organoid model systems in cancer research. EMBO J (2019) 38(15):e101654. doi: 10.15252/embj.2019101654
44. Ben-David U, Ha G, Tseng Y-Y, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet (2017) 49:1567. doi: 10.1038/ng.3967
45. Woo XY, Giordano J, Srivastava A, Zhao Z-M, Lloyd MW, de Bruijn R, et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat Genet (2021) 53(1):86–99. doi: 10.1038/s41588-020-00750-6
46. Mañas A, Aaltonen K, Andersson N, Hansson K, Adamska A, Seger A, et al. Clinically relevant treatment of PDX models reveals patterns of neuroblastoma chemoresistance. Sci Adv (2022) 8(43):eabq4617. doi: 10.1016/j.celrep.2019.09.071
47. Karlsson J, Valind A, Holmquist Mengelbier L, Bredin S, Cornmark L, Jansson C, et al. Four evolutionary trajectories underlie genetic intratumoral variation in childhood cancer. Nat Genet (2018) 50(7):944–50. doi: 10.1038/s41588-018-0131-y
48. Williams AP, Stewart JE, Stafman LL, Aye JM, Mroczek-Musulman E, Ren C, et al. Corruption of neuroblastoma patient derived xenografts with human T cell lymphoma. J Pediatr Surg (2019) 54(10):2117–9. doi: 10.1016/j.jpedsurg.2018.10.051
49. Moyer AM, Yu J, Sinnwell JP, Dockter TJ, Suman VJ, Weinshilboum RM, et al. Spontaneous murine tumors in the development of patient-derived xenografts: a potential pitfall. Oncotarget (2019) 10(39):3924–30. doi: 10.18632/oncotarget.27001
50. Tillman H, Janke LJ, Funk A, Vogel P, Rehg JE. Morphologic and immunohistochemical characterization of spontaneous Lymphoma/Leukemia in NSG mice. Vet Pathol (2020) 57(1):160–71. doi: 10.1177/0300985819882631
51. Gatzweiler C, Ridinger J, Herter S, Gerloff XF, ElHarouni D, Berker Y, et al. Functional therapeutic target validation using pediatric zebrafish xenograft models. Cancers (2022) 14(3):849. doi: 10.3390/cancers14030849
52. Almstedt E, Elgendy R, Hekmati N, Rosen E, Warn C, Olsen TK, et al. Integrative discovery of treatments for high-risk neuroblastoma. Nat Commun (2020) 11(1):71. doi: 10.1038/s41467-019-13817-8
53. Delloye-Bourgeois C, Bertin L, Thoinet K, Jarrosson L, Kindbeiter K, Buffet T, et al. Microenvironment-driven shift of Cohesion/Detachment balance within tumors induces a switch toward metastasis in neuroblastoma. Cancer Cell (2017) 32(4):427–43.e8. doi: 10.1016/j.ccell.2017.09.006
54. Bate-Eya LT, Ebus ME, Koster J, den Hartog IJ, Zwijnenburg DA, Schild L, et al. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur J Cancer (2014) 50(3):628–37. doi: 10.1016/j.ejca.2013.11.015
55. Barton J, Pacey K, Jain N, Kasia T, Edwards D, Thevanesan C, et al. Establishment and phenotyping of neurosphere cultures from primary neuroblastoma samples. F1000Res (2019) 8:823. doi: 10.12688/f1000research.18209.1
56. Fusco P, Parisatto B, Rampazzo E, Persano L, Frasson C, Di Meglio A, et al. Patient-derived organoids (PDOs) as a novel in vitro model for neuroblastoma tumours. BMC cancer (2019) 19(1):970. doi: 10.1186/s12885-019-6149-4
57. Thole TM, Toedling J, Sprüssel A, Pfeil S, Savelyeva L, Capper D, et al. Reflection of neuroblastoma intratumor heterogeneity in the new OHC-NB1 disease model. Int J cancer (2020) 146(4):1031–41. doi: 10.1002/ijc.32572
58. Nolan JC, Frawley T, Tighe J, Soh H, Curtin C, Piskareva O. Preclinical models for neuroblastoma: Advances and challenges. Cancer letters (2020) 474:53–62. doi: 10.1016/j.canlet.2020.01.015
59. Hansford LM, McKee AE, Zhang L, George RE, Gerstle JT, Thorner PS, et al. Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Res (2007) 67(23):11234–43. doi: 10.1158/0008-5472.CAN-07-0718
60. Mohlin S, Pietras A, Wigerup C, Ora I, Andang M, Nilsson K, et al. Tumor-initiating cells in childhood neuroblastoma–letter. Cancer Res (2012) 72(3):821–2. author reply 3. doi: 10.1158/0008-5472.CAN-11-1761
61. Coulon A, Flahaut M, Mühlethaler-Mottet A, Meier R, Liberman J, Balmas-Bourloud K, et al. Functional sphere profiling reveals the complexity of neuroblastoma tumor-initiating cell model. Neoplasia (2011) 13(10):991–1004. doi: 10.1593/neo.11800
62. Persson CU, von Stedingk K, Bexell D, Merselius M, Braekeveldt N, Gisselsson D, et al. Neuroblastoma patient-derived xenograft cells cultured in stem-cell promoting medium retain tumorigenic and metastatic capacities but differentiate in serum. Sci Rep (2017) 7(1):10274. doi: 10.1038/s41598-017-09662-8
63. Gavin C, Geerts N, Cavanagh B, Haynes M, Reynolds CP, Loessner D, et al. Neuroblastoma invasion strategies are regulated by the extracellular matrix. Cancers (2021) 13(4):736. doi: 10.3390/cancers13040736
64. Ben Amar D, Thoinet K, Villalard B, Imbaud O, Costechareyre C, Jarrosson L, et al. Environmental cues from neural crest derivatives act as metastatic triggers in an embryonic neuroblastoma model. Nat Commun (2022) 13(1):2549. doi: 10.1038/s41467-022-30237-3
65. Bownes LV, Williams AP, Marayati R, Stafman LL, Markert H, Quinn CH, et al. EZH2 inhibition decreases neuroblastoma proliferation and in vivo tumor growth. PloS One (2021) 16(3):e0246244. doi: 10.1371/journal.pone.0246244
66. Koneru B, Farooqi A, Nguyen TH, Chen WH, Hindle A, Eslinger C, et al. ALT neuroblastoma chemoresistance due to telomere dysfunction-induced ATM activation is reversible with ATM inhibitor AZD0156. Sci Transl Med (2021) 13(607):eabd5750. doi: 10.1126/scitranslmed.abd5750
67. Sengupta S, Das S, Crespo AC, Cornel AM, Patel AG, Mahadevan NR, et al. Mesenchymal and adrenergic cell lineage states in neuroblastoma possess distinct immunogenic phenotypes. Nat Cancer (2022) 3(10):1228–46. doi: 10.1038/s43018-022-00427-5
68. Abid K, Popovic MB, Bourloud KB, Schoumans J, Grand-Guillaume J, Grouzmann E, et al. The noradrenergic profile of plasma metanephrine in neuroblastoma patients is reproduced in xenograft mice models and arise from PNMT downregulation. Oncotarget (2021) 12(1):49–60. doi: 10.18632/oncotarget.27858
69. Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, et al. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem (2005) 51(4):735–44. doi: 10.1373/clinchem.2004.045484
70. Müller M, Rösch L, Najafi S, Gatzweiler C, Ridinger J, Gerloff XF, et al. Combining APR-246 and HDAC-inhibitors: A novel targeted treatment option for neuroblastoma. Cancers (2021) 13(17):4476. doi: 10.3390/cancers13174476
71. Mosse YP, Fox E, Teachey DT, Reid JM, Safgren SL, Carol H, et al. A phase II study of alisertib in children with Recurrent/Refractory solid tumors or leukemia: Children's oncology group phase I and pilot consortium (ADVL0921). Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(11):3229–38. doi: 10.1158/1078-0432.CCR-18-2675
72. Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A children's oncology group phase 1 consortium study. Lancet Oncol (2013) 14(6):472–80. doi: 10.1016/S1470-2045(13)70095-0
73. Cai J, Jacob S, Kurupi R, Dalton KM, Coon C, Greninger P, et al. High-risk neuroblastoma with NF1 loss of function is targetable using SHP2 inhibition. Cell Rep (2022) 40(4):111095. doi: 10.1016/j.celrep.2022.111095
74. Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, et al. MYCN-amplified neuroblastoma is addicted to iron and vulnerable to inhibition of the system xc-/Glutathione axis. Cancer Res (2021) 81(7):1896–908. doi: 10.1158/0008-5472.CAN-20-1641
75. Mohlin S, Hansson K, Radke K, Martinez S, Blanco-Apiricio C, Garcia-Ruiz C, et al. Anti-tumor effects of PIM/PI3K/mTOR triple kinase inhibitor IBL-302 in neuroblastoma. EMBO Mol Med (2019) 11(8):e10058. doi: 10.15252/emmm.201810058
76. Radke K, Hansson K, Sjölund J, Wolska M, Karlsson J, Esfandyari J, et al. Anti-tumor effects of rigosertib in high-risk neuroblastoma. Transl Oncol (2021) 14(8):101149. doi: 10.1016/j.tranon.2021.101149
77. Hansson K, Radke K, Aaltonen K, Saarela J, Mañas A, Sjölund J, et al. Therapeutic targeting of KSP in preclinical models of high-risk neuroblastoma. Sci Transl Med (2020) 12(562):eaba4434. doi: 10.1126/scitranslmed.aba4434
78. Masanas M, Masiá N, Suárez-Cabrera L, Olivan M, Soriano A, Majem B, et al. The oral KIF11 inhibitor 4SC-205 exhibits antitumor activity and potentiates standard and targeted therapies in primary and metastatic neuroblastoma models. Clin Transl Med (2021) 11(10):e533. doi: 10.1002/ctm2.533
79. Grohmann C, Walker F, Devlin M, Luo MX, Chüeh AC, Doherty J, et al. Preclinical small molecule WEHI-7326 overcomes drug resistance and elicits response in patient-derived xenograft models of human treatment-refractory tumors. Cell Death disease (2021) 12(3):268. doi: 10.1038/s41419-020-03269-0
80. George SL, Lorenzi F, King D, Hartlieb S, Campbell J, Pemberton H, et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine (2020) 59:102971. doi: 10.1016/j.ebiom.2020.102971
81. Bownes LV, Marayati R, Quinn CH, Beierle AM, Hutchins SC, Julson JR, et al. Pre-clinical study evaluating novel protein phosphatase 2A activators as therapeutics for neuroblastoma. Cancers (2022) 14(8):1952. doi: 10.3390/cancers14081952
82. Pan M, Wright WC, Chapple RH, Zubair A, Sandhu M, Batchelder JE, et al. The chemotherapeutic CX-5461 primarily targets TOP2B and exhibits selective activity in high-risk neuroblastoma. Nat Commun (2021) 12(1):6468. doi: 10.1038/s41467-021-26640-x
83. Lowery CD, Dowless M, Renschler M, Blosser W, VanWye AB, Stephens JR, et al. Broad spectrum activity of the checkpoint kinase 1 inhibitor prexasertib as a single agent or chemopotentiator across a range of preclinical pediatric tumor models. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(7):2278–89. doi: 10.1158/1078-0432.CCR-18-2728
84. O'Donohue TJ, Ibáñez G, Coutinho DF, Mauguen A, Siddiquee A, Rosales N, et al. Translational strategies for repotrectinib in neuroblastoma. Mol Cancer Ther (2021) 20(11):2189–97. doi: 10.1158/1535-7163.MCT-21-0126
85. Arlt B, Zasada C, Baum K, Wuenschel J, Mastrobuoni G, Lodrini M, et al. Inhibiting phosphoglycerate dehydrogenase counteracts chemotherapeutic efficacy against MYCN-amplified neuroblastoma. Int J cancer (2021) 148(5):1219–32. doi: 10.1002/ijc.33423
86. Henssen AG, Reed C, Jiang E, Garcia HD, von Stebut J, MacArthur IC, et al. Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Sci Transl Med (2017) 9(414):eaam9078. doi: 10.1126/scitranslmed.aam9078
87. Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer discovery (2016) 6(1):96–107. doi: 10.1158/2159-8290.CD-15-1056
88. Huo Z, Bilang R, Supuran CT, von der Weid N, Bruder E, Holland-Cunz S, et al. Perfusion-based bioreactor culture and isothermal microcalorimetry for preclinical drug testing with the carbonic anhydrase inhibitor SLC-0111 in patient-derived neuroblastoma. Int J Mol Sci (2022) 23(6):3128. doi: 10.3390/ijms23063128
89. Nunes C, Depestel L, Mus L, Keller KM, Delhaye L, Louwagie A, et al. RRM2 enhances MYCN-driven neuroblastoma formation and acts as a synergistic target with CHK1 inhibition. Sci Adv (2022) 8(28):eabn1382. doi: 10.1126/sciadv.abn1382
90. Krytska K, Ryles HT, Sano R, Raman P, Infarinato NR, Hansel TD, et al. Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2016) 22(4):948–60. doi: 10.1158/1078-0432.CCR-15-0379
91. Wood AC, Krytska K, Ryles HT, Infarinato NR, Sano R, Hansel TD, et al. Dual ALK and CDK4/6 inhibition demonstrates synergy against neuroblastoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23(11):2856–68. doi: 10.1158/1078-0432.CCR-16-1114
92. Trigg RM, Lee LC, Prokoph N, Jahangiri L, Reynolds CP, Amos Burke GA, et al. The targetable kinase PIM1 drives ALK inhibitor resistance in high-risk neuroblastoma independent of MYCN status. Nat Commun (2019) 10(1):5428. doi: 10.1038/s41467-019-13315-x
93. Dalton KM, Krytska K, Lochmann TL, Sano R, Casey C, D'Aulerio A, et al. Venetoclax-based rational combinations are effective in models of MYCN-amplified neuroblastoma. Mol Cancer Ther (2021) 20(8):1400–11. doi: 10.1158/1535-7163.MCT-20-0710
94. Nguyen TH, Koneru B, Wei SJ, Chen WH, Makena MR, Urias E, et al. Fenretinide via NOXA induction, enhanced activity of the BCL-2 inhibitor venetoclax in high BCL-2-Expressing neuroblastoma preclinical models. Mol Cancer Ther (2019) 18(12):2270–82. doi: 10.1158/1535-7163.MCT-19-0385
95. Hindle A, Koneru B, Makena MR, Lopez-Barcons L, Chen WH, Nguyen TH, et al. The O6-methyguanine-DNA methyltransferase inhibitor O6-benzylguanine enhanced activity of temozolomide + irinotecan against models of high-risk neuroblastoma. Anticancer Drugs (2021) 32(3):233–47. doi: 10.1097/CAD.0000000000001020
96. Wrobel JK, Najafi S, Ayhan S, Gatzweiler C, Krunic D, Ridinger J, et al. Rapid In vivo validation of HDAC inhibitor-based treatments in neuroblastoma zebrafish xenografts. Pharm (Basel) (2020) 13(11):345. doi: 10.3390/ph13110345
97. Timme N, Han Y, Liu S, Yosief HO, García HD, Bei Y, et al. Small-molecule dual PLK1 and BRD4 inhibitors are active against preclinical models of pediatric solid tumors. Transl Oncol (2020) 13(2):221–32. doi: 10.1016/j.tranon.2019.09.013
98. Ham J, Costa C, Sano R, Lochmann TL, Sennott EM, Patel NU, et al. Exploitation of the apoptosis-primed state of MYCN-amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell (2016) 29(2):159–72. doi: 10.1016/j.ccell.2016.01.002
99. Decaesteker B, Denecker G, Van Neste C, Dolman EM, Van Loocke W, Gartlgruber M, et al. TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat Commun (2018) 9(1):4866. doi: 10.1038/s41467-018-06699-9
100. Sano R, Krytska K, Larmour CE, Raman P, Martinez D, Ligon GF, et al. An antibody-drug conjugate directed to the ALK receptor demonstrates efficacy in preclinical models of neuroblastoma. Sci Transl Med (2019) 11(483):eaau9732. doi: 10.1126/scitranslmed.aau9732
101. Quinn CH, Beierle AM, Hutchins SC, Marayati R, Bownes LV, Stewart JE, et al. Targeting high-risk neuroblastoma patient-derived xenografts with oncolytic virotherapy. Cancers (2022) 14(3):762. doi: 10.3390/cancers14030762
102. Barry WE, Jackson JR, Asuelime GE, Wu HW, Sun J, Wan Z, et al. Activated natural killer cells in combination with anti-GD2 antibody dinutuximab improve survival of mice after surgical resection of primary neuroblastoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(1):325–33. doi: 10.1158/1078-0432.CCR-18-1317
103. Nguyen R, Moustaki A, Norrie JL, Brown S, Akers WJ, Shirinifard A, et al. Interleukin-15 enhances anti-GD2 antibody-mediated cytotoxicity in an orthotopic PDX model of neuroblastoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(24):7554–64. doi: 10.1158/1078-0432.CCR-19-1045
104. Nguyen R, Zhang X, Sun M, Abbas S, Seibert C, Kelly MC, et al. Anti-GD2 antibodies conjugated to IL15 and IL21 mediate potent antitumor cytotoxicity against neuroblastoma. Clin Cancer Res an Off J Am Assoc Cancer Res (2022) 28(17):3785–96. doi: 10.1158/1078-0432.CCR-22-0717
105. Monterrubio C, Paco S, Olaciregui NG, Pascual-Pasto G, Vila-Ubach M, Cuadrado-Vilanova M, et al. Targeted drug distribution in tumor extracellular fluid of GD2-expressing neuroblastoma patient-derived xenografts using SN-38-loaded nanoparticles conjugated to the monoclonal antibody 3F8. J Control Release (2017) 255:108–19. doi: 10.1016/j.jconrel.2017.04.016
106. Langenberg KPS, Looze EJ, Molenaar JJ. The landscape of pediatric precision oncology: Program design, actionable alterations, and clinical trial development. Cancers (2021) 13(17):4324. doi: 10.3390/cancers13174324
107. Parsons DW, Janeway KA, Patton DR, Winter CL, Coffey B, Williams PM, et al. Actionable tumor alterations and treatment protocol enrollment of pediatric and young adult patients with refractory cancers in the national cancer institute-children's oncology group pediatric MATCH trial. J Clin Oncol Off J Am Soc Clin Oncol (2022) 40(20):2224–34. doi: 10.1200/JCO.21.02838
108. van Tilburg CM, Pfaff E, Pajtler KW, Langenberg KPS, Fiesel P, Jones BC, et al. The pediatric precision oncology INFORM registry: Clinical outcome and benefit for patients with very high-evidence targets. Cancer discovery (2021) 11(11):2764–79. doi: 10.1158/2159-8290.CD-21-0094
109. Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalised medicine for high-risk paediatric cancer patients - the INFORM pilot study. Eur J Cancer (2016) 65:91–101. doi: 10.1016/j.ejca.2016.06.009
110. Newman S, Nakitandwe J, Kesserwan CA, Azzato EM, Wheeler DA, Rusch M, et al. Genomes for kids: The scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer discovery (2021) 11(12):3008–27. doi: 10.1158/2159-8290.CD-20-1631
111. Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: Description of models and early testing results. Pediatr Blood cancer (2007) 49(7):928–40. doi: 10.1002/pbc.21078
112. Zwaan CM, Kearns P, Caron H, Verschuur A, Riccardi R, Boos J, et al. The role of the 'innovative therapies for children with cancer' (ITCC) European consortium. Cancer Treat Rev (2010) 36(4):328–34. doi: 10.1016/j.ctrv.2010.02.008
113. Lau LMS, Mayoh C, Xie J, Barahona P, MacKenzie KL, Wong M, et al. In vitro and in vivo drug screens of tumor cells identify novel therapies for high-risk child cancer. EMBO Mol Med (2022) 14(4):e14608. doi: 10.15252/emmm.202114608
114. Langenberg K, Dolman E, Molenaar J. Abstract A40: Integration of high-throughput drug screening on patient-derived organoids into pediatric precision medicine programs: The future is now! Cancer Res (2020) 80(s):A40. doi: 10.1158/1538-7445.PEDCA19-A40
115. ElHarouni D, Berker Y, Peterziel H, Gopisetty A, Turunen L, Kreth S, et al. iTReX: Interactive exploration of mono- and combination therapy dose response profiling data. Pharmacol Res (2022) 175:105996. doi: 10.1016/j.phrs.2021.105996
116. Berker Y, ElHarouni D, Peterziel H, Fiesel P, Witt O, Oehme I, et al. Patient-by-Patient deep transfer learning for drug-response profiling using confocal fluorescence microscopy of pediatric patient-derived tumor-cell spheroids. IEEE Trans Med Imaging (2022) 41(12):3981–99. doi: 10.1109/TMI.2022.3205554
117. Tadeo I, Berbegall AP, Castel V, García-Miguel P, Callaghan R, Påhlman S, et al. Extracellular matrix composition defines an ultra-high-risk group of neuroblastoma within the high-risk patient cohort. Br J cancer (2016) 115(4):480–9. doi: 10.1038/bjc.2016.210
118. Burgos-Panadero R, El Moukhtari SH, Noguera I, Rodríguez-Nogales C, Martín-Vañó S, Vicente-Munuera P, et al. Unraveling the extracellular matrix-tumor cell interactions to aid better targeted therapies for neuroblastoma. Int J Pharm (2021) 608:121058. doi: 10.1016/j.ijpharm.2021.121058
119. Martinov T, McKenna KM, Tan WH, Collins EJ, Kehret AR, Linton JD, et al. Building the next generation of humanized hemato-lymphoid system mice. Front Immunol (2021) 12:643852. doi: 10.3389/fimmu.2021.643852
120. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol (2014) 32(4):364–72. doi: 10.1038/nbt.2858
121. Nguyen R, Patel AG, Griffiths LM, Dapper J, Stewart EA, Houston J, et al. Next-generation humanized patient-derived xenograft mouse model for pre-clinical antibody studies in neuroblastoma. Cancer Immunol Immunother (2021) 70(3):721–32. doi: 10.1007/s00262-020-02713-6
122. Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer (2018) 18(7):407–18. doi: 10.1038/s41568-018-0007-6
123. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Sci (New York NY) (2019) 364(6444):952–5. doi: 10.1126/science.aaw6985
124. Kholosy WM, Derieppe M, van den Ham F, Ober K, Su Y, Custers L, et al. Neuroblastoma and DIPG organoid coculture system for personalized assessment of novel anticancer immunotherapies. J Pers Med (2021) 11(9):869. doi: 10.3390/jpm11090869
125. Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer discovery (2018) 8(2):196–215. doi: 10.1158/2159-8290.CD-17-0833
126. Cui X, Ma C, Vasudevaraja V, Serrano J, Tong J, Peng Y, et al. Dissecting the immunosuppressive tumor microenvironments in glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife (2020) 9:e52253. doi: 10.7554/eLife.52253
127. Nothdurfter D, Ploner C, Coraça-Huber DC, Wilflingseder D, Müller T, Hermann M, et al. 3D bioprinted, vascularized neuroblastoma tumor environment in fluidic chip devices for precision medicine drug testing. Biofabrication (2022) 14(3):035002. doi: 10.1088/1758-5090/ac5fb7
128. Yu JL, Chan S, Fung MK, Chan GC. Mesenchymal stem cells accelerated growth and metastasis of neuroblastoma and preferentially homed towards both primary and metastatic loci in orthotopic neuroblastoma model. BMC cancer (2021) 21(1):393. doi: 10.1186/s12885-021-08090-2
Keywords: drug screening, neuroblastoma, patient-derived models, patient-derived xenograft, pediatric cancer, precision medicine, tumor organoids
Citation: Aaltonen K, Radke K, Adamska A, Seger A, Mañas A and Bexell D (2023) Patient-derived models: Advanced tools for precision medicine in neuroblastoma. Front. Oncol. 12:1085270. doi: 10.3389/fonc.2022.1085270
Received: 31 October 2022; Accepted: 21 December 2022;
Published: 19 January 2023.
Edited by:
Shiv K. Gupta, Mayo Clinic, United StatesReviewed by:
John Morton, University of Colorado Anschutz Medical Campus, United StatesLizzie Tucker, Institute of Cancer Research (ICR), United Kingdom
Copyright © 2023 Aaltonen, Radke, Adamska, Seger, Mañas and Bexell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Bexell, ZGFuaWVsLmJleGVsbEBtZWQubHUuc2U=