 Dimitrios Troumpoukis1†
Dimitrios Troumpoukis1† Adriana Papadimitropoulou1†
Adriana Papadimitropoulou1† Chrysanthi Charalampous1
Chrysanthi Charalampous1 Paraskevi Kogionou1
Paraskevi Kogionou1 Kostas Palamaris2
Kostas Palamaris2 Panagiotis Sarantis3
Panagiotis Sarantis3 Ioannis Serafimidis1*
Ioannis Serafimidis1*- 1Center of Basic Research, Biomedical Research Foundation of the Academy of Athens, Athens, Greece
- 2First Department of Pathology, Medical School, National and Kapodistrian University of Athens, Athens, Greece
- 3Molecular Oncology Unit, Department of Biological Chemistry, Medical School, National and Kapodistrian University of Athens, Athens, Greece
Pancreatic cancer is currently the seventh leading cause of cancer-related deaths worldwide, with the estimated death toll approaching half a million annually. Pancreatic ductal adenocarcinoma (PDAC) is the most common (>90% of cases) and most aggressive form of pancreatic cancer, with extremely poor prognosis and very low survival rates. PDAC is initiated by genetic alterations, usually in the oncogene KRAS and tumor suppressors CDKN2A, TP53 and SMAD4, which in turn affect a number of downstream signaling pathways that regulate important cellular processes. One of the processes critically altered is autophagy, the mechanism by which cells clear away and recycle impaired or dysfunctional organelles, protein aggregates and other unwanted components, in order to achieve homeostasis. Autophagy plays conflicting roles in PDAC and has been shown to act both as a positive effector, promoting the survival of pancreatic tumor-initiating cells, and as a negative effector, increasing cytotoxicity in uncontrollably expanding cells. Recent findings have highlighted the importance of cancer stem cells in PDAC initiation, progression and metastasis. Pancreatic cancer stem cells (PaCSCs) comprise a small subpopulation of the pancreatic tumor, characterized by cellular plasticity and the ability to self-renew, and autophagy has been recognised as a key process in PaCSC maintenance and function, simultaneously suggesting new strategies to achieve their selective elimination. In this review we evaluate recent literature that links autophagy with PaCSCs and PDAC, focusing our discussion on the therapeutic implications of pharmacologically targeting autophagy in PaCSCs, as a means to treat PDAC.
Introduction
Pancreatic Ductal Adenocarcinoma (PDAC) is the most common form of pancreatic cancer and is expected to become the second-leading cause of cancer-related deaths worldwide by 2030 (1, 2). PDAC origin remains a controversial issue but most studies support the notion that it arises from the uncontrollable proliferation of the ductal cells of the exocrine compartment, resulting in the development of a highly aggressive neoplasm (3). The molecular profiling of PDAC includes multiple gene expression alterations and copy number aberrations with KRAS (Kirsten rat sarcoma viral oncogene homolog) mutations accounting for more than 90% of the cases. However, further cancer progression requires additional mutations including tumor suppressor protein p53 (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A) and SMAD family member 4 (SMAD4) (4, 5). Treatment strategies for PDAC include tumor resection in combination with chemo/radio therapies but the efficacy is very limited as most patients relapse and eventually die from metastasis within 5 years (2).
In PDAC, approximately 1% of the total tumor mass consists of Pancreatic Cancer Stem Cells (PaCSCs) that feature auto-renewal and differentiation characteristics that allow them to generate multiple and genetically diverse cancer cell lineages (6). PaCSCs were initially described by Li et al. as cells that uniquely express a combination of CD24/CD44/EpCAM surface markers, and display enriched tumor initiating capacity when transplanted in immunocompromised mice (7). Over the last few years, investigation of the PaCSC population has attracted significant attention and is now being recognised as the main source of tumor heterogeneity and plasticity, and a niche of major importance for pancreatic tumor initiation and progression (8). In addition to CD44, CD24 and EpCAM(ESA), other markers that have been subsequently employed to characterise PaCSCs include CD133, ALDH1, CXCR4 and DCLK1, however, expression of these proteins is not restricted to PaCSCs exclusively (9–12). Many diverse signaling pathways have been found to operate in PaCSCs, including Notch, WNT, Hippo, Sonic-Hedgehog, mTOR and PI3K/Akt. These pathways actively maintain stemness and ensure an increased metastatic potential, but at the same time promote chemoprevention and resistance to conventional therapies (13).
A major cellular process that was found to be critically altered during PDAC initiation and progression is autophagy (14). Autophagy is a highly conserved “self-digestion” process that involves the catabolism of dysfunctional compounds, damaged organelles and engulfed pathogens to maintain cellular integrity and survival. Autophagy is achieved by the enclosure of this cytoplasmic “waste” cargo inside double-membrane vesicles and their concomitant translocation to lysosomes for degradation. Normally, autophagy acts as a major regulator of homeostasis, however under stressful conditions (hypoxia, starvation etc) mainly induced by disease (including cancer), the degradation of subcellular elements is accelerated in order to recycle the macromolecules and render them available to fulfill energy requirements (15–17). Major autophagic signals and regulators include the mammalian target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), WNT and TGFβ (18–20).
In this concise review, we evaluate the literature, outline current evidence on the role of autophagy in PaCSCs, and discuss how this knowledge can lead to more effective therapies against PDAC.
Autophagy in PDAC – A double-edged sword
Increased autophagic activity in PDAC has long been recognized as a major contributing factor in tumor survival, progression and metastasis. Through autophagy, PDAC tumors gain vital resources for the maintenance of their integrity, and this is achieved via mechanisms that have been expertly reviewed in great detail elsewhere (more recently by Gillson et al, 2022) (14). Our intention here is to focus on the seemingly contradictory and often debated dual (positive and negative) role of autophagy in PDAC. In this context, autophagy has been shown to attenuate the development of preneoplastic lesions in early pancreatic carcinogenesis, whereas, at more advanced stages, it was shown to promote tumor development. Disruption of the vital autophagy related genes atg7 or atg5 induces the development of benign pancreatic intraepithelial neoplasia (PanIN) in mice harboring the oncogenic KrasG12D mutation. However, despite the fact that spontaneous adenomas do occur, these are not able to progress to a more malignant state (21–23). Autophagy is up-regulated in various PDAC cell lines and is found to be elevated in PanINs as they progress towards PDAC. Consequently, inhibition of autophagy with the use of chroloquine or with shRNA against atg5 suppresses their growth in vitro and attenuates tumor development in pancreatic cancer mouse models (24). This effect was attributed to elevation of ROS (Reactive Oxygen Species) levels, increased DNA damage and impaired mitochondrial function. Resulting tumors retain their benign character and do not evolve to a more malignant state, indicating that autophagy assists tumor initiation at first but then acts as tumor suppressor, blocking progression to more advanced stages. Similar observations were made with the loss of atg5 or atg7 in mice harboring oncogenic KRAS mutations. Biallelic loss of atg5/7 reinforced tumor initiation but repressed the transition of PanINs to PDAC in mice carrying a wild-type p53 allele. On the contrary, when p53 is deleted, loss of atg5/7 results in accelerated tumor progression indicating that p53 status can determine whether autophagy will act as a tumor suppressor or accelerate tumor development (22, 23). In a later study, Görgülü et al, showed that biallelic deletion of atg5 in the presence of oncogenic KRAS stimulated acinar-to-ductal metaplasia (ADM), which progressed to PanIN stage 1 but did not eventually lead to PDAC. In contrast, mice carrying just one wild-type copy of the atg5 gene were able to develop PDAC and exhibited increased invasive capacity with a higher frequency than their wild-type (KRAS : ATG5+/+) counterparts (25).
Interesting results regarding the cumulative role of autophagy in PDAC were also extracted from two mouse models, in which hallmark KRASG12D mutation was combined with two distinct heterozygous Trp53 loss-of-function mutations, frequently encountered in human pancreatic carcinomas (Trp53R172, Trp53172H) (26, 27). In these models, one Trp53 allele is inactivated via a loss-of-function mutation, while the second one is subsequently lost via loss of heterozygosity (LOH), at a later stage of tumor progression (28). These models do not bear a complete deletion of Trp53, and therefore emulate human disease pathology more faithfully, thus enabling the investigation of the complex interplay between mutant Trp53 and autophagy regulation. In both models, pancreas-specific autophagy inhibition was achieved via a Cre-mediated genetic ablation of ATG7 with conflicting results. While in KRasG12D/+; Trp53R172H/+ animals, ATG7 ablation impeded tumorigenesis, by reducing incidence of both pre-invasive and terminal PDAC lesions (27), KRasG12D/+; Trp53R172/+ mice were characterized by an increased abundance of both acinar-to-ductal metaplasia (ADM) and pancreatic-intraepithelial neoplasia (PanIN) foci as well as more foci of invasive pancreatic tumors (26). The contradictory data derived from the above studies confirm the context-dependent role of autophagy in tumor evolutionary routes, which seems to be largely affected by the genetic background of the tumor-initiating cells.
The dual role of autophagy in PDAC biology has highlighted the importance of deciphering whether this process has a predominantly positive or negative effect in tumor development. Clarification of this important point will determine whether autophagy should be pharmacologically stimulated or inhibited in order to provide an effective treatment option for PDAC, with consensus so far favoring the later hypothesis.
Autophagy in pancreatic cancer stem cells
Cancer stem cells have been shown to rely heavily on autophagic processes for the maintenance of their stemness, their survival under hypoxic and other stress conditions and the development of resistance to therapies (29). The study of autophagy in PaCSCs has been attracting increasing attention over the last decade due to the realization of its crucial involvement in PDAC development and its potential role in PDAC therapies (Figure 1). Rausch et al. was the first to demonstrate an interaction between PaCSCs, hypoxia and autophagy, initially by studying fixed patient-derived PDAC samples where he revealed strong co-expression of markers for CSCs (CD44, CD24), hypoxia (CA-IX) and autophagy (BECLIN1, LC3) (30). During hypoxia, cells from the established cell line MIA-PaCa2 that features high CSC properties are able to survive and migrate, whereas BxPc-3 cells with limited CSC properties cannot respond efficiently and eventually undergo apoptosis (30). The higher stemness of MIA-PaCa2 cells is due to several key mutations that they carry. These include amino-acid substitutions in KRAS and TP53 (G12D and R248W, respectively) and a complete loss of CDKN2A (31, 32). On the contrary, BxPc-3 cells appear to have physiological KRAS while they carry a different amino-acid substitution in TP53 (Y220C) and display complete loss of both CDKN2A and SMAD4 proteins. These genotypic differences are considered to be responsible for the higher stemness of MIA-PaCa2 cells, indirectly leading to the suppression of non-canonical Wnt signaling (33). In this context, autophagosomes and expression of autophagy-related genes (beclin1, atg3, atg4b, and atg12) were found to be significantly elevated in CSCs of the MIA-PaCa2 cell-line but not in BxPc-3 cells. Similarly, Zhu et al. has shown that in the stressful environment of hypoxia, pancreatic cancer cells with stem cell-like properties (isolated by means of CD133 expression) displayed a significant degree of metastatic potential, self-renewal ability and elevated expression of the autophagy-related proteins LC3-II and BECLIN1 (34). Moreover, HIF-1a has been positively correlated with autophagy (35) as migrating cells with stem cell features exhibited concurrent up-regulation of HIF-1a and autophagy-related genes (ATGs). This correlation was further confirmed by evidence that HIF-1a down-regulation by RNA silencing results in reduced autophagy (34). Co-expression of the autophagy marker LC3 with stem cell markers CD133, CD44 and ALDH1 in pancreatic cancer tissue samples is another example in support of the positive correlation between autophagy and cancer cell stemness (36). Interestingly, the same study showed that pancreatic cancer patients with increased co-expression of LC3 and ALDH1 correlated with poor Progression Free Survival (PFS) and worse Overall Survival (OS). PANC-1 cells transfected with lentivirus carrying shRNA against atg5, atg7 and becn1 exhibited a significant decrease in CD44, CD133 and ALDH1 expression. Reduced levels of expression of these CSC markers concomitantly affected the stem cell properties of the transfected cells. Their self-renewal capacity and their proliferation potential were reduced, as these cells formed less spheres and exhibited impaired growth in culture compared to their control counterparts (36). Furthermore, when the transfected cells were transplanted into NOD/SCID mice, tumor volume was consistently lower compared to control mice and displayed resistance to gemcitabine. These results were further confirmed by pharmacological regulation of autophagy with the addition of the autophagy inhibitor chloroquine (CQ) and the inducer rapamycin. Levels of LC3/ALDH1 and CD44/CD133 expression were reduced by CQ treatment but showed significant increase in the presence of rapamycin (36). Additionally, osteopontin (OPN), which was previously reported to positively regulate sphere forming ability of cancer cells, was found to increase the levels of CD44, CD133, ALDH1 and LC3-II in various pancreatic cell lines, an effect that can be reversed by attenuating osteopontin activity or blocking autophagy altogether. After suppression of the OPN-mediated signaling pathways, it was revealed that NF-kB is a major contributor of OPN-stimulated autophagy and CSC activity. This observation is in agreement with other studies that have identified NF-kB as an inducer of autophagy under stressful environmental conditions such as heat shock (37). Finally, recent studies by Qin et al. showed that the stem cell-like properties of pancreatic cancer cells were reduced after the inhibition of the Nutrient-deprivation Autophagy Factor-1 (NAF-1), an autophagy-related protein localized in the outer mitochondrial membrane that is mainly involved in the maintenance of mitochondrial integrity (38–41). Pancreatic CSCs display an increased sensitivity to defects in mitochondrial homeostasis due to their predominant dependence on oxidative phosphorylation (OXPHOS) for their survival (42). NAF-1 inhibition therefore significantly affects stem cell properties of PaCSCs and mitigates their invasive capacity (41).
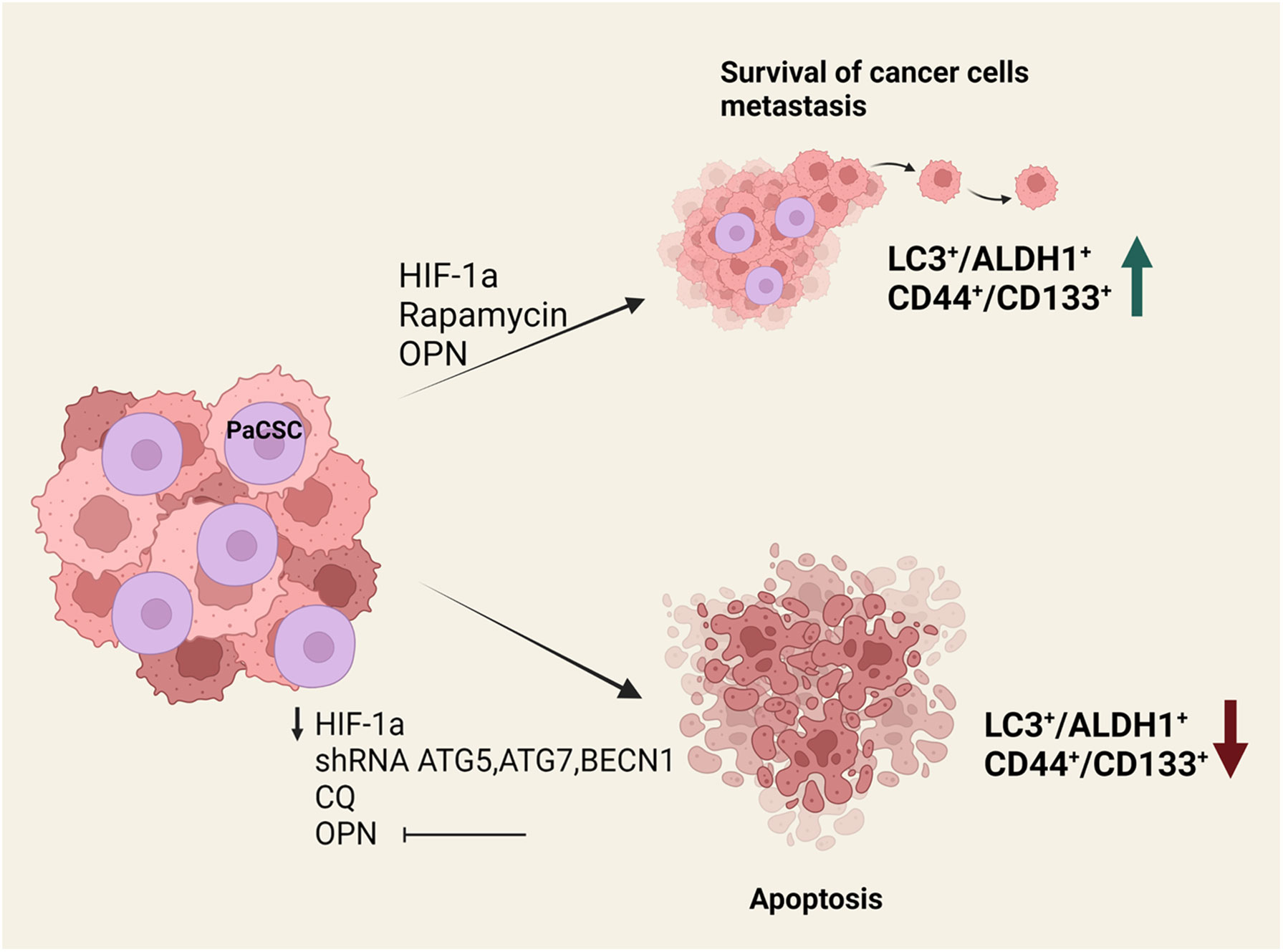
Figure 1 Effects of autophagy perturbation in PaCSCs maintenance and function. In vitro and in vivo experiments have shown that the presence of HIF-1a, rapamycin or osteopontin increase the survival of cancer stem cells and help metastasis. This is associated with an increase in LC3+/ALDH1+ and CD44+/CD133+ surface marker expression. On the contrary, when there is a reduction of HIF-1a or osteopontin, when atg5, atg7 and becn1 are silenced, or in the presence of chloroquine, cancer stem cells undergo apoptosis.
Targeting autophagy in PDAC
The majority of patients with PDAC are diagnosed in advanced stages of the disease, when the tumor is inoperable and surgery is no longer a viable treatment option. Chemotherapy and radiotherapy are first-line palliative treatments for PDAC patients and the standard chemotherapeutic drug commonly used was Gemcitabine until Folfirinox (a combination of 5-fluorouracil, folinic acid, irinotecan and oxaliplatin) proved to be more efficient in increasing OS (43).
Targeting autophagy in combination with chemotherapy has emerged as an attractive and very promising strategy in confronting PDAC. A variety of autophagy inhibitors, with distinct substrate specificities, have been developed and evaluated in vitro on cancer cell-lines, as well as in vivo on mouse models of PDAC. Based on their target molecules, the existing inhibitors can be segregated into two main classes: those that aim at constraining the initial step of autophagosome formation (44, 45) and those that block the merging of autophagosomes with lysosomes by altering the acidic environment of lysosomes (46, 47). The first class encompasses agents that impede the generation of the two core autophagy initiation multi-protein complexes, ULK1 and PI3KC3-C1 (48, 49). The main representatives of this class are MRT68921 (50, 51), SBI-0206965 (52–54) and SBI-7455 (55), which selectively target ULK1 kinase, and Spautin-1 (56, 57) and SAR405 (58, 59), which selectively suppress the PI3KC3-C1 complex. To this day, none of these compounds have progressed further into clinical studies on human patients.
On the contrary, the compound Hydroxychloroquine (HCQ) -a derivative of chloroquine and the major representative of the second class of autophagy inhibitors (60) - has been assessed in a number of clinical trials for PDAC, either as a monotherapy, or in combination with standard chemotherapy regimens or novel classes of anti-proliferative drugs (Table 1). Initially, a clinical trial investigated the effect of monotherapy using daily administrations of Hydroxychloroquine, however the drug yielded no significant responses (61). On the contrary, two phase 1/phase 1b/2 clinical trials that used a combination of gemcitabine and HCQ, provided encouraging data as there was reduction of the CA19.9 tumor marker, an increase in LC3-II staining and an improved PFS. Despite these very promising observations however, no safe conclusions could be drawn by these two studies due to the small number of patients and the lack of randomization (62, 63). More recently, a randomized phase II clinical trial was conducted using Gemcitabine together with nab-paclitaxel, with or without HCQ, in patients with advanced pancreatic cancer. Although the OS or PFS were not significantly improved, the study showed that the response rate was improved in HCQ-treated patients, therefore it was proposed that HCQ be included at the preoperative stage (61, 64).

Table 1 Clinical trials for PDAC that include targeting of autophagy.
Many attempts have been made in order to combine autophagy inhibition with novel cancer immunotherapy protocols that are currently under development. Yamamoto et al. showed that autophagy inhibition increases expression of MHC-I molecules on the surface of PDAC cells, leading to increased infiltration of immune cells, thus demonstrating that autophagy inhibition combined with immunotherapy could be a promising therapeutic option against PDAC (65). Nevertheless, the sole study to this day performed on pancreatic cancer patients treated with a combination of HCQ, gemcitabine, nab-paclitaxel and avelumab (an anti-PD-L1 antibody) was terminated due to undesirable side effects. Further clinical trials that combine HCQ with MAPK inhibitors for advanced and metastatic pancreatic cancer are currently under way (66).
In addition to the synthetic agents mentioned above, a number of natural derivatives have demonstrated a capacity to interact in multiple ways with the autophagic pathways, and can potentially be used in PDAC treatment. Even though their exact mechanism of action is unclear, they seem to converge to a common path of apoptosis inhibition. Fudan-Yueyang-Ganoderma lucidum (FYGL; a proteoglycan extracted from Ganoderma lucidum) and Alantolactone demonstrated inhibitory effect in late-stage of autophagy along with increased apoptotic potential, mediated by elevated ROS production and suppression of STAT3 and Bcl-2 respectively (67–70). Curcumin is also capable of inducing pro-apoptotic effects, by increasing the BAX/Bcl-2 ratio (71), while ursolic acid (UA) downregulated autophagy, predominantly via inducing cell cycle arrest (72).
Discussion – prospects of targeting autophagy in PaCSCs
The destruction of CSC niches is essential in order to achieve PDAC therapy and avoid cancer recurrence, however CSC niches in PDAC tumors are protected by the low-oxygen tumor environment and this is a major contributing factor to anti-cancer therapy resistance (29). Possible treatments could arise from the targeting of autophagy in PaCSCs and several preclinical studies are exploring the possibility of eliminating PaCSC niches by perturbing autophagy. Signaling pathways that are active in PaCSCs, such as NOTCH, WNT and SHH, are being explored as possible targets for impairing pluripotency in combination with autophagy targeting (9). On the other hand, the fact that PaCSCs reside in the anatomically distinct regions of a niche, which is often pharmacologically inaccessible, necessitates the exploration of alterative properties of PaCSCs as targets for therapy. Cancer dormancy is characterized by attenuation of cell proliferation and the transition to a quiescence-like state. It has been shown that dormancy contributes significantly to cancer relapse and induction of metastasis. Conventional anticancer therapies usually target proliferating cells hence the acquisition of a dormant phenotype results to the evasion of treatments and worse overall survival. The biological mechanisms and signaling pathways regulating the dormant phenotype of tumor cells also apply to CSC behavior, and recent studies indicate that autophagy plays a vital role in the entrance of CSCs to a dormant state, the maintenance of their survival and their reactivation under specific conditions (73).
In addition to the pre-clinical studies, efforts to effectively tackle PDAC currently explore the clinical use of small molecules that target autophagy, in combination with chemotherapy and/or immunotherapy (14). Targeting cancer stemness is currently the basis of ongoing clinical trials on Phase III with paclitaxel/nab paclitaxel and gemcitabine with the addition of napabucasin, a small molecule that inhibits STAT3-mediated gene transcription. The results from phase 1b/2 achieved a Partial Response (PR) of almost 42% and approximate 3% of Complete Response (CR) (74). This enhanced chemotherapy scheme could be complemented with selected compounds that target key components of the autophagic process, in order not only to eliminate tumor-cells but also to eradicate hidden and evading PaCSCs.
Despite the undoubted benefits of autophagy inhibition in cancer treatment, the complex role of this “self-digestion” process in tumorigenesis, coupled with the capacity of tumor stem cells to utilize alternative nutrient sources under starvation conditions, could impose some restrictions to the clinical efficacy of this therapeutic approach. While in the majority of cases autophagy is considered a cytoprotective mechanism that serves as an adaptation system of neoplastic cells in conditions of nutrient scarcity, there is still controversy regarding its overall effects in cancer initiation and progression. Recent experimental evidence, from both in vitro and in vivo studies, suggests a context-dependent anti-tumor effect of autophagy, mediated by a detrimental effect on cancer stem cell survival and metastasis. Even though autophagy is a process antagonistic to apoptosis, overt autophagic influx can in fact trigger apoptosis under certain conditions by activation of caspase-8 and the diminution of endogenous apoptosis inhibitors (75). Moreover, genetic ablation of ATG5 in a KRAS-driven mouse model of PDAC enhances the metastatic potential of tumor stem cells, implying an anti-metastatic effect of autophagy under certain circumstances, especially during the initial stages of tumor development (25). It has also been shown that neoplastic cells are equipped with an intrinsic ability to circumvent dependency on autophagy and exploit compensatory signal transduction systems, such as Nrf2 signalling and deployment of complementary nutritional sources through the process of micropinocytosis (76). These strategies enable cancer stem cells to survive nutrient stress conditions arising from starvation, thus ensuring their survival and growth. It is therefore implied that potential anti-PDAC pharmacotherapy schemes relying on autophagy targeting in PaCSCs should carefully consider both the tumor-suppressive and the tumor-promoting effects of this action.
Author contributions
DT, AP and IS wrote the first draft of the manuscript. All authors contributed to the article, reviewed and edited the final manuscript and approved the submitted version.
Funding
Work in the senior author’s laboratory is supported by funding provided by Boehringer Ingelheim GmbH (CRA: 493341) and the Greek General Secretariat of Research and Innovation (GSRI) (Code: T1EDK-03532). The funders had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
Acknowledgments
The authors thank all members of the Stem Cell and Cancer Biology laboratory at BRFAA for critical discussions over the manuscript and wish to apologise to all investigators whose work has not been cited here due to space restrictions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Quante AS, Ming C, Rottmann M, Engel J, Boeck S, Heinemann V, et al. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med (2016) 5(9):2649–56. doi: 10.1002/cam4.767
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
3. Adamska A, Domenichini A, Falasca M. Pancreatic ductal adenocarcinoma: Current and evolving therapies. Int J Mol Sci (2017) 18(7):1338. doi: 10.3390/ijms18071338
4. Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet (2020) 395(10242):2008–20. doi: 10.1016/S0140-6736(20)30974-0
5. Qian Y, Gong Y, Fan Z, Luo G, Huang Q, Deng S, et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol Oncol (2020) 13(1):130. doi: 10.1186/s13045-020-00958-3
6. Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. J Clin Oncol (2008) 26(17):2806–12. doi: 10.1200/JCO.2008.16.6702
7. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res (2007) 67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030
8. Subramaniam D, Kaushik G, Dandawate P, Anant S. Targeting cancer stem cells for chemoprevention of pancreatic cancer. Curr Med Chem (2018) 25(22):2585–94. doi: 10.2174/0929867324666170127095832
9. Barman S, Fatima I, Singh AB, Dhawan P. Pancreatic cancer and therapy: Role and regulation of cancer stem cells. Int J Mol Sci (2021) 22(9):4765. doi: 10.3390/ijms22094765
10. Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer (2009) 125(10):2323–31. doi: 10.1002/ijc.24573
11. Maréchal R, Demetter P, Nagy N, Berton A, Decaestecker C, Polus M, et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br J Cancer (2009) 100(9):1444–51. doi: 10.1038/sj.bjc.6605020
12. Ito H, Tanaka S, Akiyama Y, Shimada S, Adikrisna R, Matsumura S, et al. Dominant expression of DCLK1 in human pancreatic cancer stem cells accelerates tumor invasion and metastasis. PloS One (2016) 11(1):e0146564. doi: 10.1371/journal.pone.0146564
13. Hermann PC, Sainz B. Pancreatic cancer stem cells: A state or an entity? Semin Cancer Biol (2018) 53:223–31. doi: 10.1016/j.semcancer.2018.08.007
14. Gillson J, Abd El-Aziz YS, Leck LYW, Jansson PJ, Pavlakis N, Samra JS, et al. Autophagy: A key player in pancreatic cancer progression and a potential drug target. Cancers (Basel) (2022) 14(14):3528. doi: 10.3390/cancers14143528
15. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell (2007) 1(3):313–23. doi: 10.1016/j.stem.2007.06.002
16. Ikenaga N, Ohuchida K, Mizumoto K, Yu J, Kayashima T, Hayashi A, et al. Characterization of CD24 expression in intraductal papillary mucinous neoplasms and ductal carcinoma of the pancreas. Hum Pathol (2010) 41(10):1466–74. doi: 10.1016/j.humpath.2010.04.004
17. Hou YC, Chao YJ, Tung HL, Wang HC, Shan YS. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer (2014) 120(17):2766–77. doi: 10.1002/cncr.28774
18. Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U.S.A. (2011) 108(12):4788–93. doi: 10.1073/pnas.1100844108
19. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell (2010) 40(2):280–93. doi: 10.1016/j.molcel.2010.09.023
20. Tan VP, Miyamoto S. Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J Mol Cell Cardiol (2016) 95:31–41. doi: 10.1016/j.yjmcc.2016.01.005
21. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev (2011) 25(8):795–800. doi: 10.1101/gad.2016211
22. Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature (2013) 504(7479):296–300. doi: 10.1038/nature12865
23. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discovery (2014) 4(8):905–13. doi: 10.1158/2159-8290.CD-14-0362
24. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev (2011) 25(7):717–29. doi: 10.1101/gad.2016111
25. Görgülü K, Diakopoulos KN, Ai J, Schoeps B, Kabacaoglu D, Karpathaki AF, et al. Levels of the autophagy-related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology (2019) 156(1):203–217.e20. doi: 10.1053/j.gastro.2018.09.053
26. Long JS, Kania E, McEwan DG, Barthet VJA, Brucoli M, Ladds MJGW, et al. ATG7 is a haploinsufficient repressor of tumor progression and promoter of metastasis. Proc Natl Acad Sci U.S.A. (2022) 119(28):e2113465119. doi: 10.1073/pnas.2113465119
27. Mainz L, Sarhan MAFE, Roth S, Sauer U, Maurus K, Hartmann EM, et al. Autophagy blockage reduces the incidence of pancreatic ductal adenocarcinoma in the context of mutant. Front Cell Dev Biol (2022) 10:785252. doi: 10.3389/fcell.2022.785252
28. Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol (2018) 15(1):13–30. doi: 10.1038/nrclinonc.2017.151
29. Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ (2019) 26(4):690–702. doi: 10.1038/s41418-019-0292-y
30. Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S, et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol (2012) 227(3):325–35. doi: 10.1002/path.3994
31. Sun C, Yamato T, Furukawa T, Ohnishi Y, Kijima H, Horii A, et al. Characterization of the mutations of the K-ras, p53, p16, and SMAD4 genes in 15 human pancreatic cancer cell lines. Oncol Rep (2001) 8(1):89–92. doi: 10.3892/or.8.1.89
32. Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas (2010) 39(4):425–35. doi: 10.1097/MPA.0b013e3181c15963
33. Wang MT, Holderfield M, Galeas J, Delrosario R, To MD, Balmain A, et al. K-Ras promotes tumorigenicity through suppression of non-canonical wnt signaling. Cell (2015) 163(5):1237–51. doi: 10.1016/j.cell.2015.10.041
34. Zhu H, Wang D, Zhang L, Xie X, Wu Y, Liu Y, et al. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol Rep (2014) 32(3):935–42. doi: 10.3892/or.2014.3298
35. Wang P, Long M, Zhang S, Cheng Z, Zhao X, He F, et al. Hypoxia inducible factor-1α regulates autophagy via the p27-E2F1 signaling pathway. Mol Med Rep (2017) 16(2):2107–12. doi: 10.3892/mmr.2017.6794
36. Yang MC, Wang HC, Hou YC, Tung HL, Chiu TJ, Shan YS. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer (2015) 14:179. doi: 10.1186/s12943-015-0449-3
37. Nivon M, Richet E, Codogno P, Arrigo AP, Kretz-Remy C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy (2009) 5(6):766–83. doi: 10.4161/auto.8788
38. Lin CC, Lin MS. New insight into curcumin-based therapy in spinal cord injuries: CISD2 regulation. Neural Regener Res (2016) 11(2):222–3. doi: 10.4103/1673-5374.177718
39. Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, et al. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci U.S.A. (2007) 104(36):14342–7. doi: 10.1073/pnas.0707189104
40. Chen YF, Kao CH, Chen YT, Wang CH, Wu CY, Tsai CY, et al. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev (2009) 23(10):1183–94. doi: 10.1101/gad.1779509
41. Qin T, Cheng L, Xiao Y, Qian W, Li J, Wu Z, et al. NAF-1 inhibition by resveratrol suppresses cancer stem cell-like properties and the invasion of pancreatic cancer. Front Oncol (2020) 10:1038. doi: 10.3389/fonc.2020.01038
42. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab (2015) 22(4):590–605. doi: 10.1016/j.cmet.2015.08.015
43. Chandana S, Babiker HM, Mahadevan D. Therapeutic trends in pancreatic ductal adenocarcinoma (PDAC). Expert Opin Investig Drugs (2019) 28(2):161–77. doi: 10.1080/13543784.2019.1557145
44. Hamacher-Brady A. Autophagy regulation and integration with cell signaling. Antioxid Redox Signal (2012) 17(5):756–65. doi: 10.1089/ars.2011.4410
45. Shang L, Wang X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy (2011) 7(8):924–6. doi: 10.4161/auto.7.8.15860
46. Gonzalez-Noriega A, Grubb JH, Talkad V, Sly WS. Chloroquine inhibits lysosomal enzyme pinocytosis and enhances lysosomal enzyme secretion by impairing receptor recycling. J Cell Biol (1980) 85(3):839–52. doi: 10.1083/jcb.85.3.839
47. Yayon A, Cabantchik ZI, Ginsburg H. Susceptibility of human malaria parasites to chloroquine is pH dependent. Proc Natl Acad Sci U.S.A. (1985) 82(9):2784–8. doi: 10.1073/pnas.82.9.2784
48. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell (2008) 19(12):5360–72. doi: 10.1091/mbc.e08-01-0080
49. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol (2013) 15(7):741–50. doi: 10.1038/ncb2757
50. Petherick KJ, Conway OJ, Mpamhanga C, Osborne SA, Kamal A, Saxty B, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem (2015) 290(48):28726. doi: 10.1074/jbc.A114.627778
51. Singha B, Laski J, Ramos Valdés Y, Liu E, DiMattia GE, Shepherd TG. Inhibiting ULK1 kinase decreases autophagy and cell viability in high-grade serous ovarian cancer spheroids. Am J Cancer Res (2020) 10(5):1384–99. Available at: https://e-century.us/files/ajcr/10/5/ajcr0108592.pdf.
52. Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell (2015) 59(2):285–97. doi: 10.1016/j.molcel.2015.05.031
53. Lu J, Zhu L, Zheng LP, Cui Q, Zhu HH, Zhao H, et al. Overexpression of ULK1 represents a potential diagnostic marker for clear cell renal carcinoma and the antitumor effects of SBI-0206965. EBioMedicine (2018) 34:85–93. doi: 10.1016/j.ebiom.2018.07.034
54. Tang F, Hu P, Yang Z, Xue C, Gong J, Sun S, et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol Rep (2017) 37(6):3449–58. doi: 10.3892/or.2017.5635
55. Ren H, Bakas NA, Vamos M, Chaikuad A, Limpert AS, Wimer CD, et al. Design, synthesis, and characterization of an orally active dual-specific ULK1/2 autophagy inhibitor that synergizes with the PARP inhibitor olaparib for the treatment of triple-negative breast cancer. J Med Chem (2020) 63(23):14609–25. doi: 10.1021/acs.jmedchem.0c00873
56. Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell (2011) 147(1):223–34. doi: 10.1016/j.cell.2011.08.037
57. Xiao J, Feng X, Huang XY, Huang Z, Huang Y, Li C, et al. Spautin-1 ameliorates acute pancreatitis via inhibiting impaired autophagy and alleviating calcium overload. Mol Med (2016) 22:643–52. doi: 10.2119/molmed.2016.00034
58. Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol (2014) 10(12):1013–9. doi: 10.1038/nchembio.1681
59. Zhou P, Li Y, Li B, Zhang M, Xu C, Liu F, et al. Autophagy inhibition enhances celecoxib-induced apoptosis in osteosarcoma. Cell Cycle (2018) 17(8):997–1006. doi: 10.1080/15384101.2018.1467677
60. Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang Y, et al. Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer Res (2011) 17(10):3248–58. doi: 10.1158/1078-0432.CCR-10-0890
61. Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist (2014) 19(6):637–8. doi: 10.1634/theoncologist.2014-0086
62. Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol (2015) 22(13):4402–10. doi: 10.1245/s10434-015-4566-4
63. Samaras P, Tusup M, Nguyen-Kim TDL, Seifert B, Bachmann H, von Moos R, et al. Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother Pharmacol (2017) 80(5):1005–12. doi: 10.1007/s00280-017-3446-y
64. Karasic TB, O'Hara MH, Loaiza-Bonilla A, Reiss KA, Teitelbaum UR, Borazanci E, et al. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: A phase 2 randomized clinical trial. JAMA Oncol (2019) 5(7):993–8. doi: 10.1001/jamaoncol.2019.0684
65. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 581(7806):100–5. doi: 10.1038/s41586-020-2229-5
66. Reyes-Castellanos G, Abdel Hadi N, Carrier A. Autophagy contributes to metabolic reprogramming and therapeutic resistance in pancreatic tumors. Cells (2022) 11(3):426. doi: 10.3390/cells11030426
67. Bao S, Zheng H, Ye J, Huang H, Zhou B, Yao Q, et al. Dual targeting EGFR and STAT3 with erlotinib and alantolactone Co-loaded PLGA nanoparticles for pancreatic cancer treatment. Front Pharmacol (2021) 12:625084. doi: 10.3389/fphar.2021.625084
68. He R, Shi X, Zhou M, Zhao Y, Pan S, Zhao C, et al. Alantolactone induces apoptosis and improves chemosensitivity of pancreatic cancer cells by impairment of autophagy-lysosome pathway via targeting TFEB. Toxicol Appl Pharmacol (2018) 356:159–71. doi: 10.1016/j.taap.2018.08.003
69. Wu X, Jiang L, Zhang Z, He Y, Teng Y, Li J, et al. Pancreatic cancer cell apoptosis is induced by a proteoglycan extracted from. Oncol Lett (2021) 21(1):34. doi: 10.3892/ol.2020.12295
70. Zheng H, Yang L, Kang Y, Chen M, Lin S, Xiang Y, et al. Alantolactone sensitizes human pancreatic cancer cells to EGFR inhibitors through the inhibition of STAT3 signaling. Mol Carcinog (2019) 58(4):565–76. doi: 10.1002/mc.22951
71. Zhu Y, Bu S. Curcumin induces autophagy, apoptosis, and cell cycle arrest in human pancreatic cancer cells. Evid Based Complement Alternat Med (2017) 2017:5787218. doi: 10.1155/2017/5787218
72. Lin JH, Chen SY, Lu CC, Lin JA, Yen GC. Ursolic acid promotes apoptosis, autophagy, and chemosensitivity in gemcitabine-resistant human pancreatic cancer cells. Phytother Res (2020) 34(8):2053–66. doi: 10.1002/ptr.6669
73. Akkoc Y, Peker N, Akcay A, Gozuacik D. Autophagy and cancer dormancy. Front Oncol (2021) 11:627023. doi: 10.3389/fonc.2021.627023
74. Wang S, Zheng Y, Yang F, Zhu L, Zhu XQ, Wang ZF, et al. The molecular biology of pancreatic adenocarcinoma: translational challenges and clinical perspectives. Signal Transduct Target Ther (2021) 6(1):249. doi: 10.1038/s41392-020-00422-1
75. Lim J, Murthy A. Targeting autophagy to treat cancer: Challenges and opportunities. Front Pharmacol (2020) 11:590344. doi: 10.3389/fphar.2020.590344
Keywords: autophagy, pancreas, pancreatic cancer, PDAC - pancreatic ductal adenocarcinoma, Cancer Stem Cell (CSC), pancreatic cancer stem cells, hydroxychloroquine
Citation: Troumpoukis D, Papadimitropoulou A, Charalampous C, Kogionou P, Palamaris K, Sarantis P and Serafimidis I (2022) Targeting autophagy in pancreatic cancer: The cancer stem cell perspective. Front. Oncol. 12:1049436. doi: 10.3389/fonc.2022.1049436
Received: 20 September 2022; Accepted: 09 November 2022;
Published: 24 November 2022.
Edited by:
Guan Wang, Sichuan University, ChinaReviewed by:
Wanli Yang, Fourth Military Medical University, ChinaSiddharth Mehra, University of Miami Health System, United States
Copyright © 2022 Troumpoukis, Papadimitropoulou, Charalampous, Kogionou, Palamaris, Sarantis and Serafimidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ioannis Serafimidis, aXNlcmFmQGJpb2FjYWRlbXkuZ3I=
†These authors have contributed equally to this work and share first authorship