 Raymond S. Traweek1†Brandon M. Cope1†Christina L. Roland1Emily Z. Keung1Elise F. Nassif1Derek J. Erstad2*
Raymond S. Traweek1†Brandon M. Cope1†Christina L. Roland1Emily Z. Keung1Elise F. Nassif1Derek J. Erstad2*- 1Department of Surgical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Division of Surgical Oncology, Baylor College of Medicine, Houston, TX, United States
Dedifferentiated liposarcoma (DDLPS) is an aggressive adipogenic cancer with poor prognosis. DDLPS tumors are only modestly sensitive to chemotherapy and radiation, and there is a need for more effective therapies. Genetically, DDLPS is characterized by a low tumor mutational burden and frequent chromosomal structural abnormalities including amplification of the 12q13-15 chromosomal region and the MDM2 gene, which are defining features of DDLPS. The MDM2 protein is an E3 ubiquitin ligase that targets the tumor suppressor, p53, for proteasomal degradation. MDM2 amplification or overexpression in human malignancies is associated with cell-cycle progression and worse prognosis. The MDM2–p53 interaction has thus garnered interest as a therapeutic target for DDLPS and other malignancies. MDM2 binds p53 via a hydrophobic protein interaction that is easily accessible with synthetic analogues. Multiple agents have been developed, including Nutlins such as RG7112 and small molecular inhibitors including SAR405838 and HDM201. Preclinical in vitro and animal models have shown promising results with MDM2 inhibition, resulting in robust p53 reactivation and cancer cell death. However, multiple early-phase clinical trials have failed to show a benefit with MDM2 pathway inhibition for DDLPS. Mechanisms of resistance are being elucidated, and novel inhibitors and combination therapies are currently under investigation. This review provides an overview of these strategies for targeting MDM2 in DDLPS.
Introduction
Liposarcomas are rare, adipogenic cancers that typically arise in the extremities and retroperitoneum. They are composed of several histologic subtypes, of which well-differentiated and dedifferentiated tumors are the most common. Well-differentiated liposarcomas (WDLPS) exhibit a more indolent behavior with frequent local recurrence. Dedifferentiated liposarcomas (DDLPS) are of a higher grade with potential for distant metastasis (1). DDLPS can arise de novo, in mixed states concomitantly with WDLPS, or upon recurrence after resection of WDLPS (2).
Surgical resection remains the definitive management for DDLPS in appropriate candidates. However, due to the frequent invasion of surrounding structures and the large size of these tumors, achieving a microscopically negative resection margin remains difficult. Local recurrence is common, and perioperative therapies are frequently utilized (3, 4). First-line chemotherapy is anthracycline based, usually doxorubicin in combination with ifosfamide. Unfortunately, DDLPS are relatively chemoresistant, with radiographic responses observed in less than one-third of patients (5). Consequently, there is no consensus regarding systemic therapy in the neoadjuvant or adjuvant setting. Preoperative radiation therapy is frequently utilized in academic treatment centers for lower-grade tumors, as it may reduce local recurrence (6, 7). Overall, outcomes remain poor with high rates of recurrence and a limited overall survival of approximately 50% following the first local recurrence (8–10). For these reasons, there has been increasing interest in targeted therapies in the treatment of DDLPS.
The tumor biology of DDLPS is characterized by a low tumor mutational burden (TMB) with high somatic copy-number alterations (SCNAs) (11). Amplification of chromosome 12q13-15 results in amplification of MDM2, a defining feature of DDLPS (12). The MDM2 protein has E3 ubiquitin ligase activity that targets the tumor suppressor p53, for degradation. Overexpression of the MDM2 protein is associated with cell-cycle progression and malignant proliferation, thus offering a potential treatment target. While MDM2 inhibitors have been associated with promising preclinical findings, results from early-phase clinical trials have thus far been disappointing (13, 14). Nonetheless, MDM2 inhibition remains an area of ongoing investigation, including the development of novel inhibitors and combination therapies (15, 16).
In this review, we discuss the unique genetic and epigenetic characteristics of DDLPS and the rationale for targeting MDM2. We build on this information by describing the MDM2–p53 pathway and provide a summary of approaches to MDM2 drug design. Finally, we review preclinical and clinical trials focused on MDM2 inhibition, examining mechanisms of failure and future directions for improved pathway inhibition and therapeutic efficacy.
The genomic landscape of DDLPS
DDLPS are characterized by a mutational signature defined by 12q13-15 amplification, as well as dysregulated epigenetic patterns associated with downregulation of tumor suppressors and increased expression of proliferative genes. While most DDLPS occur de novo, they can also present with WDLPS in mixed states for both primary and recurrent tumors. Dedifferentiated tumors are histologically characterized by a transition from mature adipocytes to cells with marked atypia (17). The molecular pathogenesis of dedifferentiation remains poorly understood, although it is of great interest to researchers given its association with adverse tumor biology and worse prognosis.
The genomic landscape of DDLPS is characterized by low TMB and frequent SCNAS. TMB has been identified as a predictive biomarker for response to immunotherapy, supported by the premise that highly mutated tumors are more likely to have detectable neoantigens, although this relationship is not reliable for all cancers (18, 19). In a recent analysis, Chalmers et al. performed whole exome sequencing across multiple cancer types to characterize TMB (20). In this study, undifferentiated soft-tissue sarcomas demonstrated low TMB with a median of 2.5 mutations/Mb, placing them in the bottom 40th percentile among 167 tumor types. These findings were recapitulated by Liu et al., in which a median TMB of 1.97 mutations/Mb was demonstrated for a cohort of DDLPS (21). Accordingly, immune checkpoint blockade with anti-CTLA-4 monotherapy has yielded minimal tumor response in both preclinical and clinical models for DDLPS, which may be partly related to low mutational burden (22, 23).
Compared with other liposarcoma histologic subtypes, DDLPS demonstrates the highest frequency of SCNAs (11). SCNAs have been shown to support tumorigenesis and mutagenic processes that drive genomic instability during tumor growth and evolution (24). Certain patterns of SCNAs appear conserved across different cancers. In a recent pooled analysis of focal SCNAs among 17 tumor types, Beroukhim et al. demonstrated a median overlap of 79% across epithelial, gastrointestinal, and genitourinary malignancies (25). For WDLPS and DDLPS, the highly recurrent focal amplification of chromosome 12q13-15 is a defining SCNA that appears unique to these histologic subtypes (Figure 1). Amplification of the 12q arm may be related to incurred chromosomal structural abnormalities, and initial work to characterize the cytogenetics of retroperitoneal liposarcomas by Dal Cin et al. identified supernumerary rings and long marker chromosomes (“rods”) as the primary karyotypic abnormalities within their cohort of WDLPS and DDLPS (11, 26, 27). A subsequent work by Pedeutour et al. demonstrated that these ring and rod structures contained amplified segments of the 12q chromosome (28). Taken together, the finding of abnormal ring/rod chromosomal structures containing amplification of the 12q13-15 arm is a defining feature for WDLPS and DDLPS.
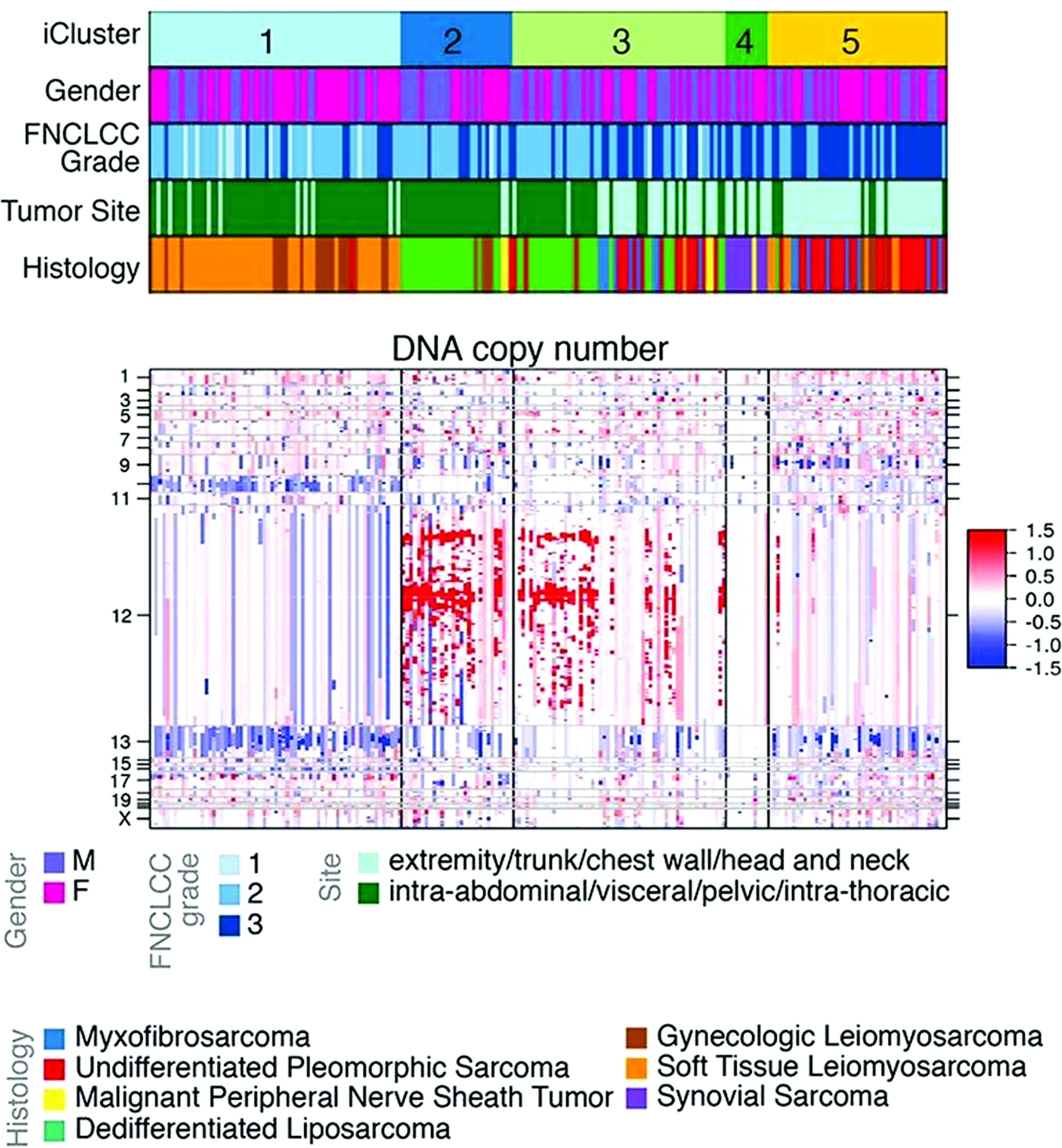
Figure 1 Clustered analysis of DNA copy number among soft-tissue sarcomas. Cluster C1 is primarily composed of leiomyosarcoma. Clusters C2 and C3 are primarily composed of dedifferentiated liposarcoma. Cluster C4 is composed of synovial sarcomas and malignant peripheral nerve sheath tumors. Cluster C5 is composed of high-grade undifferentiated pleomorphic sarcomas. Amplification in red, deletion in blue. Adapted from Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas (11).
Subsequent investigations have focused on individual genes located within the 12q13-15 arm, including MDM2, CDK4, HMGA2, and YEATS4 (Figure 2) (29). Of these, MDM2 is a known oncogene amplified across multiple cancer types (30, 31). Oliner et al. identified a 5–50-fold MDM2 amplification in both WD and DD LPS with 12q amplification, which has been recapitulated in other studies (12, 31, 32). MDM2 encodes an E3 ubiquitin ligase that negatively regulates the tumor suppressor p53 by marking it for proteasomal degradation, inhibiting cell-cycle arrest and apoptosis in response to DNA damage (33). Loss of p53 function is a crucial step in malignant transformation for multiple cancer types and has been shown to adversely impact tumor biology (34, 35). Therefore, the MDM2–p53 interaction is a target of interest in cancer therapeutics.
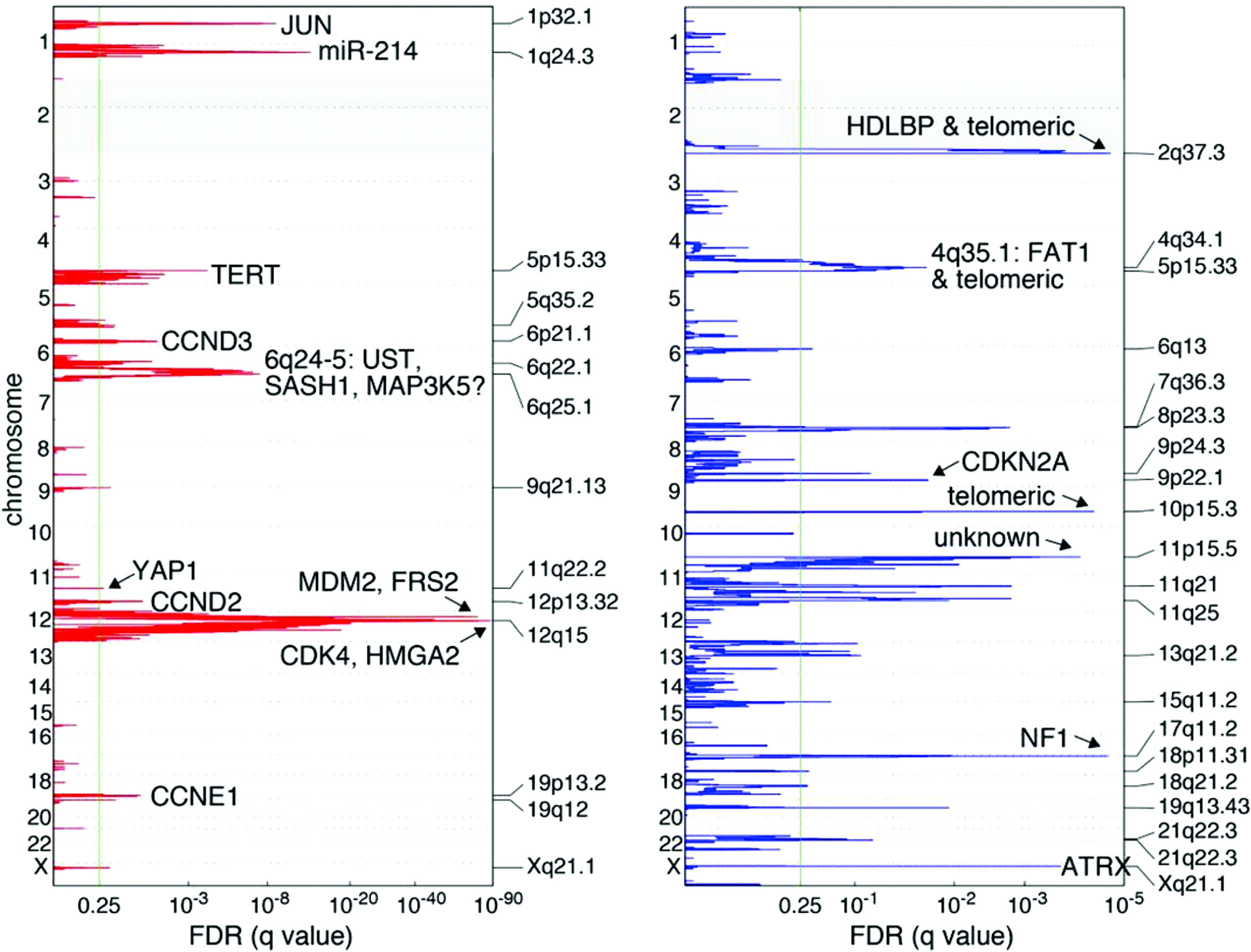
Figure 2 Recurrent genetic alterations among 50 DDLPS samples. Amplification in red, deletions in blue. Green line indicates the significance threshold for focally amplified or deleted genes. Adapted from Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas (11).
The quantity of 12q13-15 amplification and MDM2 upregulation appears to be associated with the degree of tumor dedifferentiation (36). Horvai et al. analyzed 29 WD and DD liposarcomas, the majority of which were retroperitoneal with concomitant WD and DD components (37). They observed amplification of 12q13-15 in all tumors, with dedifferentiated components having significantly more total amplifications by comparative genomic hybridization. Similarly, in studies using FISH analysis, MDM2 amplification was identified in abnormal ring/rod structures, and the quantity of protein expression was associated with the degree of cellular atypia (27). The association between 12q13-15/MDM2 amplification and dedifferentiation raises the question of whether this mutation supports the dedifferentiation process. Beird et al. analyzed 17 liposarcomas with both WD and DD components and observed an overlap in 8.3% of somatic mutations, suggesting shared clonal ancestry (38). Furthermore, it has been shown that WDLPS frequently precedes dedifferentiation, which may be in part due to the subsequent amplification of c-Jun, a proto-oncogene involved in adipocyte differentiation that is primarily observed in ring and rod structures associated with DDLPS (39, 40). Thus, it is reasonable to hypothesize that the phenotypic sequelae from 12q13-15 amplification impact dedifferentiation programs.
Given the low TMB observed in DDLPS, it is thought that epigenetic changes such as aberrant methylation might have an important role in disease pathogenesis. Demicco et al. performed an unsupervised clustering analysis of 50 DDLPS tumor samples and identified hyper- and hypomethylated phenotypes (11). Disease-specific survival (DSS) was significantly worse in association with hypermethylated tumors, suggesting a potential role as an adverse prognostic biomarker. Hypermethylation of genes and proteins affecting adipocyte differentiation has been identified within DDLPS. Taylor et al. demonstrated methylation of CEBPA, a core promoter of adipocyte differentiation, within 24% of 80 dedifferentiated tumors, and this was not observed in the analyzed WDLPS tumors or non-neoplastic adipocyte tissue controls (41). The authors confirmed that CEBPA hypermethylation was associated with a statistically significant reduction in identifiable CEBPA mRNA. Additional work by Keung et al. identified trimethylation of histone H3 at the lysine 9 residue (H3K9me3) among DDLPS cells, which was not observed in WDLPS (42). The authors demonstrated an association between H3K9me3 and downregulation of KLF6, a known tumor suppressor, suggesting that histone modification may play a role in driving oncogenesis.
Based on the observation that dedifferentiation is associated with both higher rates of DNA methylation and greater quantity of MDM2 amplification, it is conceivable that MDM2 might influence epigenetic modification processes. In this regard, Cao et al., using non-sarcomatous cell lines in vitro, observed an inverse association between MDM2 and HBP1 protein expression by IHC (43). HBP1 is a transcription factor that acts as an epigenetic regulator via both activator and repressor roles of several key cell-cycle regulator genes. High-affinity binding elements within HBP1 repress cell-cycle regulator genes N-MYC, C-MYC, DNMT1, and EZH2 at the transcriptional level, whereas downstream effector targets of HBP1 activate genes encoding p21, p16, and histone H.1 (44–48) In this study, the authors identified MDM2-mediated ubiquitination and proteasomal degradation of HBP1, which was associated with increased expressions of DNMT1 and EZH2, resulting in global DNA and histone hypermethylation with subsequent genomic instability. These findings provide an example of a potential mechanism for MDM2-driven epigenetic modification. However, there remains a paucity of information regarding MDM2 activity on epigenetic modifications in DDLPS. In a separate study, Stocker et al. characterized genomic signatures, gene mutations, and methylation profiles for a small sample of DDLPS patients. They identified MDM2 amplifications in all cases but observed hypermethylated genomes in only 30% of tumors, and there was no significant association between methylation status and mutational signatures (49). Given these observations, further investigation to better characterize potential interactions between MDM2 hyperfunction and epigenetic modifications in DDLPS is warranted.
In addition, it remains difficult to associate epigenetic chromatin modifications with downstream transcriptional changes in vivo, as the two often appear unrelated. Moreover, medications that alter DNA methylation status, such as histone deacetylase inhibitors (HDACs), have provided only modest clinical benefit in soft-tissue sarcomas in early-phase clinical trials despite efficacy in hematologic malignancies (50–52). Ultimately, the epigenomics driving the dedifferentiation of liposarcomas are poorly understood and remain an area of ongoing research (53).
Finally, it is worth noting that DDLPS are genetically distinct from pleomorphic liposarcomas (PLPS), although both exhibit dedifferentiated histology and aggressive behavior. PLPS are mutationally characterized by high aneuploidy and complex karyotypic rearrangements (54–56). Unlike WDLPS/DDLPS, amplification of the 12q13-15 region is not a defining feature, although somatic mutations in TP53 have been observed in up to 17% of cases (17, 56). Barretina et al. evaluated genomic alterations in 24 PLPS compared with 50 DDLPS tumors (26). In this study, pleomorphic tumors demonstrated significant chromosomal alterations and somatic mutations in NF1, RB1, and PIK3CA, but they lacked 12q13-15 amplification (57). In another study, Schmidt et al. evaluated 36 patients with multiple liposarcoma histologic subtypes including WDLPS, DDLPS, myxoid, and PLPS (58). Using comparative genomic hybridization, the authors demonstrated that PLPS tumors contained frequent genomic imbalances occurring in almost all chromosomal regions. In particular, PLPS has been shown to frequently contain copy number gains of chromosomes 1, 5q, 20q, and 22q (59). Clinically, PLPS are more chemosensitive than DDLPS but appear to have less tumor immune infiltration (60–63). Both tumor types have a similar propensity for local recurrence, but pleomorphic LPS are at a higher risk for distant dissemination (64). Importantly, due to the lack of 12q13-15 amplification, pleomorphic sarcomas are not good candidates for targeted MDM2 inhibition.
Mechanism of the MDM2–p53 protein interaction
The tumor-suppressor protein p53 is a critical regulator of cellular processes including division, DNA repair, apoptosis, and senescence. Under physiologic conditions, p53 is maintained at low levels to facilitate appropriate cellular turnover (65). Activity of p53 is primarily regulated by MDM2, an E3 ubiquitin ligase that promotes p53 proteasomal degradation (Figure 3) (66). Loss-of-function mutations in MDM2 potentiate p53 acetylation and protein stability, resulting in upregulated downstream effectors (67). Conversely, MDM2 amplification is associated with reduced p53 activity and has been implicated in multiple cancers including DDLPS (68, 69).
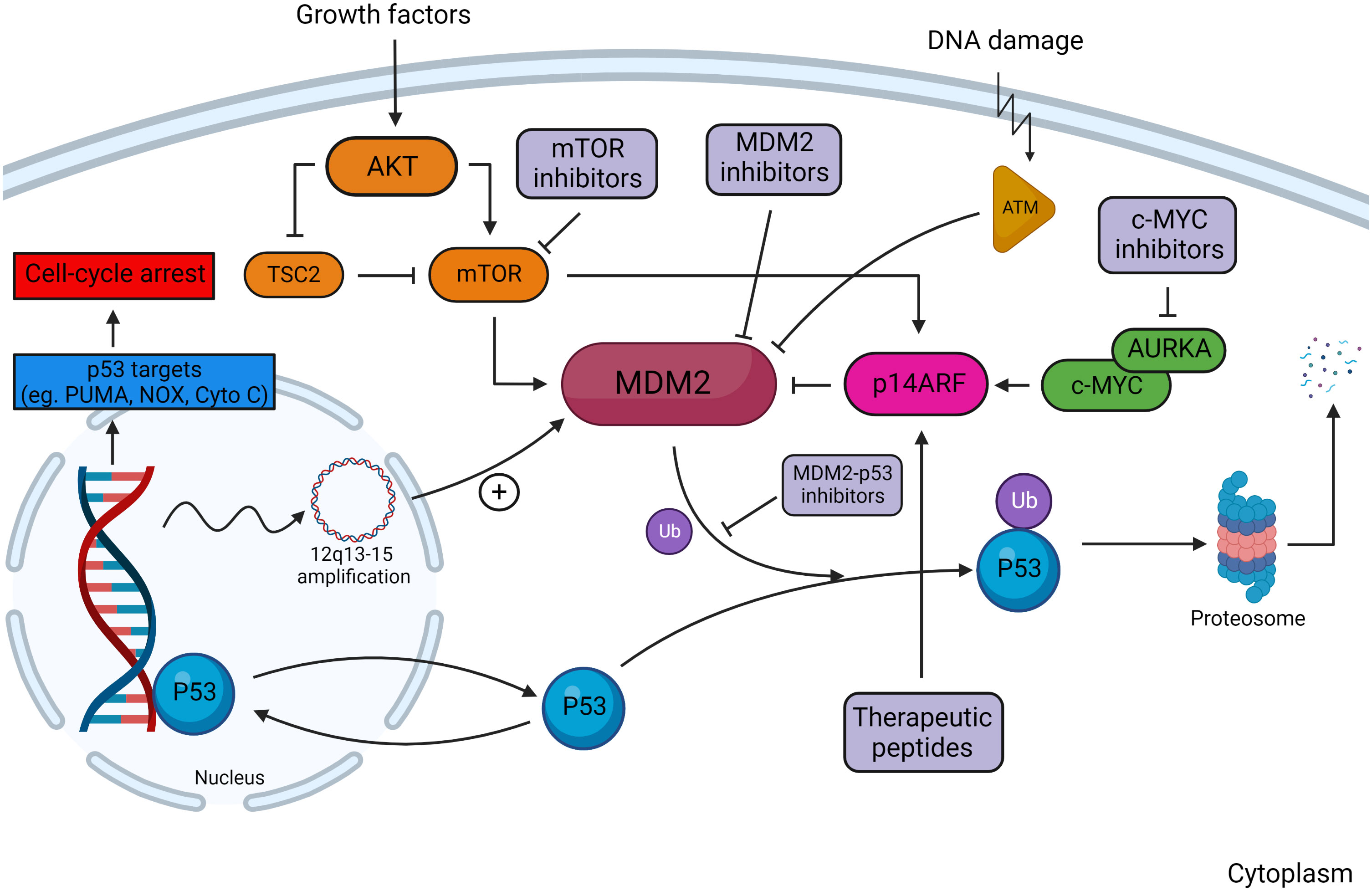
Figure 3 Overview of the MDM2–p53 pathway in response to DNA damage. In the absence of DNA damage, MDM2 facilitates ubiquitination of p53, tagging it for proteosomal degradation. Damage to DNA activates kinases, including ataxia telangiectasia-mutated (ATM) kinase, which are responsible for phosphorylating MDM2 and reducing its affinity to bind cytosolic p53. Regulators of the MDM2–p53 interaction include the AKT/mTOR pathway, ARF, and c-MYC. Downstream transcriptional targets of p53 serve to arrest cell-cycle progression and induce cellular apoptosis. Multiple sites of action are targeted by MDM2 and MDM2–p53 inhibitors, which decrease p53 degradation in tumors with wild-type p53. Created with BioRender.com.
The MDM2 protein contains several structural elements that are necessary to inhibit p53. MDM2 physically interacts with p53 through its NH2 terminal domain, which forms a hydrophobic cleft that binds a helical region of the p53 transactivation domain (70). This conceals the transcriptionally active domain of p53 from co-regulatory proteins through a key-and-lock configuration. Additionally, MDM2 contains a C-terminus RING (zinc-finger) domain that ubiquitinates p53 at six separate lysine residues along its C-terminus, marking it for proteasomal degradation (71, 72). Nuclear localization signal (NLS) and nuclear export signal (NES) domains allow for the shuttling of MDM2 into the nucleus as well as into the cytoplasm for protein turnover, respectively (73). Because of this import/export function, MDM2 has been observed in both the nucleus and cytoplasm. In addition, the function of MDM2 is partly dependent upon the activity of its homolog, MDMX, which shares structural similarity within the N-terminus p53-binding domain (74). MDMX attenuates p53 activity through heterodimerization with MDM2 resulting in protein complex stabilization (75, 76).
Under physiologic conditions, DNA damage attenuates the MDM2–p53 interaction by two primary mechanisms. First, MDM2 can undergo inactivating kinase-dependent phosphorylation. The phosphorylation status of the central acidic and C-terminal RING domains of MDM2 are associated with the efficacy of MDM2-mediated p53 degradation (77). It has been shown that the ataxia telangiectasia-mutated (ATM) kinase, itself activated primarily by DNA double-stranded breaks, phosphorylates both the serine 395 residue within the central acidic domain and five sites near the C-terminal RING domain of MDM2, impairing MDM2-mediated nuclear exportation and the proteosomal degradation of p53 (78, 79). Similar inactivating kinase functions have been described for glycogen synthase kinase 3β, casein kinase 1, and casein kinase 2 (80–82). Second, MDM2 can undergo proteasomal degradation via self-ubiquitination. Oxidative by-products of DNA damage stimulate ubiquitin-activating and conjugating enzymes that drive MDM2 self-ubiquitination, which is dependent on the C-terminal RING finger domain (83, 84). DNA damage, particularly dsDNA breaks, also stimulate the activation of other E3 ligases that target MDM2 such as P300-CBP-associated factor (PCAF), a histone acetyltransferase and transcriptional co-activator of p53 (85, 86). Similarly, it has also been shown that phosphorylation of MDM2 by casein kinase 1 promotes SCFβ-TrCP-dependent MDM2 ubiquitination and turnover (87).
Other posttranslational modifications that influence MDM2 regulation have been identified that may represent actionable targets for directed therapy. The addition of a small ubiquitin-like modifier (SUMO) protein to lysine residues, termed SUMOylation, occurs in response to DNA damage and abrogates the ability for MDM2 to bind p53, resulting in increased cellular p53 protein levels (88, 89). More recent work has characterized lysine modification by the small ubiquitin-like molecule, NEDD8. Watson et al. observed that NEDD8 stabilizes MDM2, termed NEDDylation, via binding to the MDM2 RING domain and preventing ubiquitination (90). The authors additionally identified that the NEDD8-specific isopeptide, NEDP1, which serves to de-NEDDylate MDM2, was induced by doxorubicin in vitro. NEDP1 activity was associated with an increased auto-ubiquitination of MDM2, a reduction in detectable MDM2 protein, and an increase in p53 protein expression by Western blot. As these findings indicate, there are multiple MDM2–p53 regulators, many of which require further investigation, and some of these regulators may potentially serve as useful targets for combination therapies.
MDM2 cross talk with oncogenic pathways
The MDM2–p53 axis is also regulated by multiple, parallel oncogenic signaling pathways implicated in sarcomagenesis, and these pathways might represent valuable targets for combination therapeutic strategies in DDLPS. Among these, ARF-mediated MDM2 inhibition is well described. ARF, also referred to as p14ARF in humans, is encoded from the partially open reading frame p14ARF located on the CDKN2A locus and acts as a direct inhibitor of MDM2 (91, 92). Specifically, the N-terminal domain of p14ARF binds to MDM2, sequestering MDM2 to the nucleus (93, 94). Epigenetic alterations in p14ARF have been implicated in oncogenesis, particularly myxoid and pleomorphic sarcomas. Davidović et al. identified epigenetic silencing via methylation of the promoter region of p14ARF in 83% and 50% of myxoid and pleomorphic sarcoma samples, respectively (95). Similarly, Oda et al. identified hypermethylation of the p14ARF promoter with a subsequent reduced expression of the p14ARF protein and an overexpression of p53 in their cohort of round-cell liposarcoma samples (96). Variable levels of p14ARF expression have been observed in DDLPS (97). Taken together, p14ARF epigenetic silencing might represent a valuable target for affecting MDM2 function in sarcoma.
The proto-oncogene MYC—and its product c-MYC—is another critical regulator of MDM2 function via ARF signaling (98–100). c-MYC is a transcription factor that regulates numerous downstream targets supporting cellular survival and proliferation (101, 102). Under physiologic conditions, c-MYC activation has been observed to induce the expression of p14ARF, which in turn inhibits MDM2 (103, 104). However, the aberrant overexpression of c-MYC has been associated with a reduced expression of p53 by IHC and has been identified in numerous human cancers (105). Furthermore, high levels of c-MYC have been closely associated with hypermethylation of CDKN2A and loss of p14ARF, suggesting that overexpression of c-MYC may contribute to enhanced MDM2 activity (106, 107). Despite these data, there is a paucity of studies corroborating these findings in DDLPS. In this regard, Kim et al. used in vitro mesenchymal stem cells to co-express MDM2 and CDK4 in cell lines containing several known oncogenic mutational signatures, including stabilized c-MYC resistant to proteasomal degradation. The authors noted that c-MYC stabilized cell lines overexpressing MDM2 and CDK4 developed DDLPS like-morphology in vitro (108). These findings were recapitulated in vivo, wherein nude mice that were inoculated with c-MYC-stabilized cell lines overexpressing MDM2 and CDK4 exhibited tumor growth with dedifferentiated lipoblasts on histologic analysis. This tumor growth was significantly larger and more accelerated when compared with nude mice harboring cell lines that lacked c-MYC stabilization. Taken together, these data suggest that c-MYC dysregulation in the context of MDM2 overexpression might influence lipoblast dedifferentiation and warrants further investigation in human DDLPS.
Finally, the aberrant activation of the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway has gained increasing interest for its potential role in DDLPS. AKT is a serine/threonine kinase which, under physiologic conditions in response to both growth factor signaling and cell stress signaling, regulates a number of downstream cellular functions including cell-cycle progression, apoptosis, and DNA repair (109). AKT interacts with the MDM2–p53 pathway in two primary ways. First, in response to cellular growth factors, AKT phosphorylates MDM2 at one of two serine residues, causing nuclear localization and intranuclear binding of MDM2 to p53, and thereby facilitating p53 degradation (109, 110). Second, AKT has been observed to regulate the downstream transcription factor mTOR. The direct activation of mTOR by AKT via phosphorylation at the Ser2448 residue activates signaling pathways propagating cellular proliferation (111). AKT has additionally been observed to phosphorylate and subsequently repress tuberous sclerosis complex 2 (TSC2), which in turn suppresses mTOR signaling (112–114). Phosphorylation of mTOR results in an increased translation of MDM2, in turn promoting the degradation of p53 (115). Overexpression of this pathway is associated with unchecked progression through the cell cycle and has become increasingly identified as a driver of oncogenesis in DDLPS. Ishii et al. identified a significantly increased level of mTOR activation in DDLPS when compared with WDLPS by PCR, which was correlated with an increased mTOR expression by IHC (116). Using an in vitro model, the authors then cultured DDLPS cell lines with the mTOR inhibitor RAD001 and observed a decrease in cellular proliferation by 20%. Similar findings were observed by Gutierrez et al. wherein the authors documented evidence of aberrant AKT pathway activation via detection of phosphorylated Ser473-AKT in 47% of DDLPS samples (117). Tumor growth in vitro was abrogated by BEZ235, an inhibitor of the AKT pathway, suggesting that aberrant AKT activation influences DDLPS proliferation.
Taken together, these data illustrate the complex interplay of parallel oncogenic pathways that impact MDM2–p53 signaling in DDLPS. Co-inhibition of these pathways might be an effective strategy for addressing signaling redundancy and improving response rates to MDM2-targeted therapy.
Targeting the MDM2 pathway: drug design and preclinical studies
Microscopic and genetic investigations of DDLPS over the last three decades have provided strong supporting evidence to target the MDM2 pathway (118, 119). MDM2 amplification is associated with worse clinical outcomes, including a significantly shorter time to disease recurrence and reduced overall survival following oncologic resection (120). MDM2 amplification is also associated with reduced sensitivity to doxorubicin chemotherapy (121). Accordingly, this oncogenic pathway has garnered significant interest as a therapeutic target.
Regarding drug design, MDM2–p53 binding is characterized by hydrophobic amino acid interactions for an energetically relaxed state (122). The p53 binding interface is located within the NH2 terminal transactivational domain, which contains an amphipathic alpha-helix with multiple hydrophobic residues (aa 18-26) (123). Using X-ray crystallography, Phe19, Trp23, and Leu26 within this helix have been identified as critical to stable binding of MDM2 (70). Conversely, the MDM2 binding interface is located within the NH2 terminal domain (aa 25–109), which creates a structural cleft that allows for the intercalation of hydrophobic residues on the p53 amphipathic helix (124). Within the crystal structure of MDM2, residues Gly58, Glu68, Val75, and Cys77 appear essential to p53 binding (125). The characterization of this binding interaction has led to the development of multiple synthetic compounds, most of which are defined by aromatic structures with amphipathic features that mimic the MDM2–p53 protein–protein interaction.
One group of such compounds includes Nutlins, which are cis-imidazoline analogs that rely on two bromophenyl groups and an ethyl ether side chain to mimic the p53 residues Trp23, Leu26, and Phe19, respectively. Nutlins have been extensively studied in preclinical models. Vassilev et al. utilized wild-type p53 and mutant p53 cell lines to demonstrate that specific Nutlin enantiomers, primarily Nutlin-3a, initiated apoptotic caspase activation in 45% of MDM2-amplified osteosarcoma cell lines at 48 h (125). Subsequent in vivo mouse xenograft models bearing the same wild-type p53 osteosarcoma cell lines were treated with Nutlin-3, which was associated with a 90% inhibition of tumor growth relative to vehicle controls. In a separate analysis, Tovar et al., using both in vitro cell lines and xenograft-bearing mice, demonstrated that RG7112, a second-generation Nutlin, induced a dose-dependent blockade of cell-cycle progression that was associated with increased p53 protein expression (126). Additional preclinical testing of Nutlin-3 has been examined in hematologic malignancies with MDM2 amplification including multiple myeloma, acute myeloid leukemia, and chronic lymphocytic leukemia (127–129). It has also been reported that Nutlin-3a is able to induce p53-independent mechanisms of cell death by enhancing the stability of the tumor suppressor, p73, a member of the p53 family with pro-apoptotic activities (130). Additional p53-independent regulatory targets of MDM2 have been reported such as pRb and E2F/DP (131).
SAR405838, a non-Nutlin small molecular inhibitor of MDM2, has also been investigated in preclinical experiments and clinical trials. Wang et al. administered SAR405838 to mice bearing xenografted leukemia and solid tumor implants (132). The authors identified intracellular accumulation of p53 within tumor tissue and increased transcriptional activity of wild-type p53, which was associated with an increase in pro-apoptotic proteins and cell-cycle arrest. Bill et al. recapitulated these findings in vitro, showing that MDM2 inhibition with SAR405838 induced cell-cycle arrest and apoptosis in wild-type p53 human DDLPS tumor cells, which was associated with enrichment of p53-mediated gene expression patterns (133). The authors also showed that SAR405838 was associated with significant reductions in tumor volume in DDLPS xenograft-bearing mice.
HDM201, a more recent MDM2 inhibitor, has shown improved potency and selectivity. Preclinical data from Jeay et al. demonstrated cell growth inhibition in p53 wild-type, Mdm2-amplified osteosarcoma cells utilizing both continuous and pulsed HDM201 treatment methods (15). Compared with controls, HDM201 treatment in vitro was associated with induction of p53 downstream markers. This included a 16-fold induction of P21 mRNA, which encodes for a protein that is a potent cyclin-dependent kinase inhibitor that plays a crucial role in DNA-damage-related cell-cycle arrest (134, 135). Mdm2-amplified osteosarcoma cells were then xenografted into rats, and the animals were treated with HDM201. The investigators observed induction of multiple cell-cycle arrest-related genes in a dose-dependent manner. These included Puma, p21, and Gdf, which are well-described downstream proapoptotic targets of p53 that contribute to cell-cycle arrest and apoptosis (136, 137). High-dose HDM201 was associated with tumor cellular apoptosis and complete tumor regression, which was sustained in all rats for 30 days after stopping treatment. The authors performed an additional study using Mdm2-amplified WDLPS cells that were xenografted into mice, and they observed similar rates of tumor regression in response to HDM201 treatment.
Finally, the efficacy of HDM201 in preclinical studies may be partly due to its effect on the tumor immune microenvironment. Wang et al. investigated HDM201-mediated immunologic changes within the tumor microenvironment using an immunocompetent, syngeneic mouse tumor model (16). HDM201 treatment was associated with increased tumoral levels of CD103+ antigen-presenting dendritic cells, which are thought to play a critical role in tumor antigen presentation and priming of cytotoxic T lymphocytes (138). The authors additionally observed an increase in Tbet+Eomes+CD8+ cytotoxic T cells, a subset of cytotoxic T cells which have demonstrated potent cytotoxic activity with an exhausted phenotype (139, 140). T-cell exhaustion has been closely associated with response to anti-PD-1 therapy, particularly in combination with chemotherapy or other immunotherapeutics (141, 142). The authors therefore combined HDM201 with an anti-PDL-1 antibody, which was associated with significantly longer survival when compared with HDM201 monotherapy. Antitumor activity with combination therapy was abrogated with Tp53 knockout, indicating that HDM201 treatment was required for induction of an immunologic response. These data suggest that modulation of the tumor microenvironment may synergize with MDM2-targeted therapies; as such, this remains an area of ongoing investigation.
Clinical trial results for MDM2 pathway inhibitors in liposarcoma and solid tumors
Despite the promising preclinical data, MDM2 inhibition has yielded mixed results in clinical trials (Table 1). Ray-Coquard et al. reported findings from a phase I clinical trial evaluating RG7112, in which patients with chemotherapy-naïve primary or relapsed WDLPS and DDLPS received neoadjuvant treatment prior to surgical resection (14). Twenty patients were enrolled, of which 18 were p53 wild-type and 14 had MDM2 amplification identified via silver in-situ hybridization. Nineteen of 20 patients completed at least one preoperative treatment cycle. Four patients received only one cycle of treatment; two were discontinued for neutropenia and thrombocytopenia, one for progressive disease and one for phlebitis. Five patients received two cycles, and 10 patients received three cycles. All patients had at least one adverse event, and there were 12 serious adverse events in eight patients. The authors raised concerns about gastrointestinal and hematologic toxicity with long-term usage. Measured p53 protein concentrations were significantly increased 4.86-fold from pretreatment levels, and p21 protein concentrations were significantly increased 3.48-fold. However, treatment response as measured by Response Evaluation Criteria in Solid Tumors (RECIST) criteria included only one confirmed partial response, stable disease in 14 patients, and progressive disease in five patients (all DDLPS).
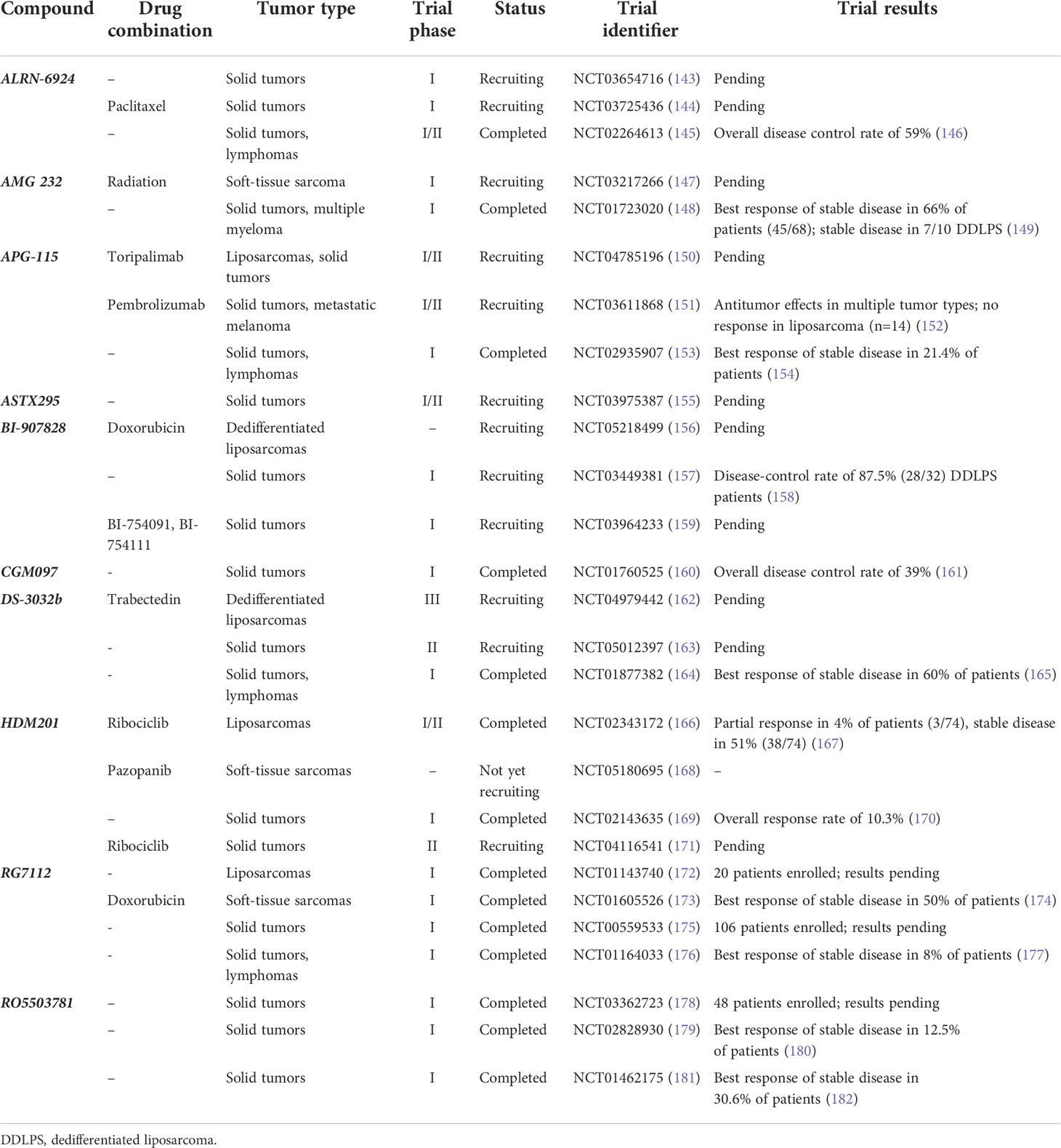
Table 1 List of clinical trials investigating MDM2 inhibition in liposarcomas or other solid tumors.
Similar findings were observed in a phase I trial for SAR405838, in which patients with locally advanced or metastatic, medically refractory solid tumors with a p53 wild-type or a marginal p53 mutation (<40%) received SAR405838 monotherapy (13). Of the 74 patients enrolled, the most common primary tumor type was liposarcoma (n = 35, 47%). The study contained a second expansion cohort composed of 21 patients with DDLPS to determine the maximum tolerated dose (MTD). Of this expansion cohort, 89% of tumors exhibited MDM2 amplification and no TP53 mutations were observed. Regarding the total cohort, the most frequent treatment-related adverse effects were nausea (59%), fatigue (58%), and vomiting (42%). Serious adverse effects occurred in 30% of patients, including thrombocytopenia (8%), neutropenia (4%), and lymphopenia (16%). Eleven patients required drug discontinuation, and 12 required drug dose modification. In this study, there were no objective responses, although 58% of subjects had a stable disease per RECIST criteria. On subgroup analysis of the primary cohort, DDLPS patients had a higher percentage of stable disease (22/31, 71%). The MTD expansion cohort of DDLPS patients similarly had a 56% rate of stable disease, and progression-free survival at 3 months was 32%. The authors concluded that despite the lack of objective response, evidence of disease stabilization and p53 activation in the majority of patients warranted further evaluation, particularly for combination regimens.
HDM201 was recently investigated in a phase I, multicenter, open-label trial (170). In total, 115 patients with treatment-refractory locally advanced or metastatic solid tumors and 93 patients with relapsed or refractory hematologic malignancies were included. Treatment-related adverse events, most commonly gastrointestinal disorders and cytopenias, were observed in 90% patients with solid tumors and 88% of patients with hematologic malignancies. Approximately 40% of patients experienced at least one adverse event that required dose adjustment or interruption. Baseline biomarker status was evaluated in 48 patients using the FoundationOne panel, which demonstrated MDM2 amplification in 16 (33%). MDM2 amplification was more prevalent in patients who experienced either a partial response or stable disease (53%) compared with patients with progressive disease (23%). Among patients with progressive disease, mutations in CDKN2A, ATRX, KRAS, AKT1, and CDK6 were also observed. For solid tumors, the objective response rate was 3.5% and the stable disease rate was 36.5% by RECIST criteria. Twelve patients with liposarcoma were treated in a dose-expansion phase of the study. Of these, one patient experienced a partial response (8%) and nine (75%) patients had a stable disease. The median progression-free survival for these 12 patients was 5.6 months, and one-third of patients had a stable disease for greater than 6 months. Taken together, despite the limited efficacy of HDM201 among the total cohort of patients with solid tumors, the authors noted that patients with liposarcoma in the expansion group experienced noticeable disease control. Favorable responses among those treated for hematologic malignancies were also observed. In conclusion, the authors recommended further investigation of HDM201 therapy, alone or in combination; there are currently two ongoing HDM201 clinical trials for patients with liposarcoma.
Finally, phase I trials investigating the MDM2 inhibitors CGM097 and DS3032b have been studied in solid tumors and lymphoma. CGM097 was evaluated in 51 patients with locally advanced or metastatic, TP53 wild-type solid tumors (161). While no dose-limiting toxicities were reported, delayed-onset thrombocytopenia, nausea, leukopenia, vomiting, and fatigue were all recorded at incidences of greater than 20%. Stable disease was observed in 19/51 (37%), and one melanoma patient experienced a partial response as measured by RECIST criteria. The trial investigators observed evidence of p53 reactivation via induction of downstream molecular targets but without an associated clinically significant tumor response. DS3032b was evaluated in a phase I study of patients with WDLPS/DDLPS, solid tumors, and lymphomas (165). In this study, 63% of patients had been treated with three or more prior therapies and 87% of tumors were documented TP53 wild types. Of the 94 patients enrolled in the study, 60% of patients achieved stable disease for a median duration of 6.7 months with three partial responses observed among patients with DDLPS, synovial sarcoma, and squamous cell lung cancer by RECIST criteria. Adverse events included thrombocytopenia (61%) and neutropenia (28%), with 8% of patients experiencing dose-limiting toxicities.
Inhibition of co-activators of the MDM2–p53 pathway has also been investigated in clinical trials. Of these, mTOR inhibition is well studied, and initial trials examining mTOR inhibition monotherapy using either temsirolimus or ridaforolimus yielded limited clinical efficacy. Okuno et al. conducted a phase II single-institution clinical trial examining the utilization of temsirolimus monotherapy in patients with advanced STS. Of the 40 patients that were enrolled, 95% experienced progression while on treatment and median overall survival was 7.6 months (183). Analysis of posttreatment peripheral blood samples demonstrated suppressed mTOR signaling in approximately 66% of patients, indicating adequate therapeutic levels of temsirolimus. Similar findings were observed in subsequent clinical trials. Chawla et al. conducted a phase II multicenter clinical trial examining the use of ridaforolimus monotherapy in patients with advanced STS. The median overall survival in their cohort was 10 months, and the overall response rate was 1.9% as determined by RECIST 1.1 criteria (184). Additionally, 98% of patients reported some form of adverse event, with 11% of patients discontinuing therapy due to poor tolerance of side effects.
Due to the disappointing results of mTOR inhibitor monotherapy and guided by preclinical observations that tumor cell growth of breast and prostate cancer cell lines in vitro was abrogated when treated with combination mTOR/MAP-kinase inhibition, several early-phase clinical trials examined combination therapy with mTOR inhibition (107, 117, 185, 186). Eroglu et al. examined MAP-kinase inhibition monotherapy versus combination mTOR/MAP-kinase inhibition among a cohort of STS patients in a randomized, phase II clinical trial (185–187). There was no significant difference in progression-free survival between the two treatment arms; however, the authors noted that combination therapy was associated with significantly improved median PFS in the leiomyosarcoma subgroup. Based on these data, there was increased interest in the combination of mTOR inhibitors as an adjunct to first-line chemotherapy for the treatment of STS. Trucco et al. conducted a phase II clinical trial examining mTOR inhibition with temsirolimus in combination with doxorubicin in patients with advanced, non-resectable STS. The median PFS for the study population was 10.3 months, and the best response was partial response in 13% of patients as determined by RECIST 1.1 criteria (188). The authors did not report histology-specific outcomes but did note that median PFS was slightly better with combination therapy when compared with mTOR inhibitor monotherapy. Taken together, these data suggest that mTOR inhibition may provide some clinical benefit in select histologic subtypes when combined with other systemic therapies. However, the utility of mTOR inhibition in DDLPS remains unclear and the combination of mTOR and MDM2 inhibition has not been evaluated.
c-MYC represents another co-activator of MDM2, and direct c-MYC inhibitors have demonstrated low cellular affinity and minimal in vivo potency (189–191). This is thought to be a result of difficulties with targeting the transactivation domain. Nuclear magnetic resonance studies have been used to demonstrate that the transactivation domain of c-MYC exhibits a heterogenous conformational space depending on the phosphorylation status of its N-terminus (192). However, transient structural elements of the MYC protein stabilize when bound to other cofactors, notably Aurora kinase A (AURKA) (193). Heterodimeric modeling has allowed for the development of indirect MYC inhibitors through targeted AURKA inhibition (194). In this regard, Dickson et al. conducted a multicenter phase II clinical trial investigating the use of AURKA inhibitor MLN8237 in patients with advanced or metastatic STS. The overall response rate was 2.8%, and the median PFS was 11.7 weeks (195). Among liposarcoma patients specifically, median PFS was 13 weeks and median OS was 68 weeks. The authors identified that 73% of liposarcoma patients were progression-free at 12 weeks, which they suggest is promising based on previous reports designating favorable drug response in STS as 40% progression-free at 12 weeks (196). Of note, p53 activity was not significantly changed in the posttreatment correlative specimen analysis of DDLPS as determined by Western blot. Given these findings, there is ongoing work to elucidate the clinical benefit of various c-MYC cofactor inhibitors.
There have also been studies investigating cyclin-dependent kinase (CDK) inhibitors as modulators of the p53 pathway. Specifically, CDK4/6, which share overlapping function and structural homology, have been robustly examined as a targetable modulators of the cell cycle (197). CDK4/6 phosphorylates the retinoblastoma tumor-suppressor protein (Rb1), which in turn dissociates Rb1 from the transcription factor E2F and allows progression of the cell cycle from the G1 to S phases (198). CDK4/6 has been identified as a downstream target of the AKT/mTOR pathway, suggesting that CDK4/6 may have synergistic effects with signaling molecules co-expressed in the AKT-MDM2 axis (199). Additionally, CDK4/6 is often upregulated alongside MDM2 due to 12q13-15 amplification, although not as frequently; Binh et al. demonstrated CDK4 amplification in 90% of DDLPS samples when compared with the near 100% prevalence of MDM2 amplification (200). Taken together, CDK4/6 inhibition represents an alternative mechanism for p53 reactivation in addition to MDM2 inhibition.
Clinical trials investigating the use of CDK4/6 inhibitor palbociclib has yielded modest clinical benefit. Dickson et al. conducted a phase II, non-randomized clinical trial in which patients with advanced WDLPS and DDLPS received palbociclib. Of the 28 patients, 57% achieved progression-free survival at 12 weeks and one patient achieved durable complete response 2 years after treatment (201). However, the authors did not include histology-specific outcomes and close to 80% of patients experienced tumor growth while on treatment. Due to the disappointing results of CDK4/6 inhibitor monotherapy, there has been increasing interest in combination CDK4/6 and MDM2 inhibition. Razak et al. conducted a phase Ib proof-of-concept trial in which patients with advanced WDLPS and DDLPS received concomitant siremadlin, a selective MDM2 inhibitor, with ribociclib, a selective CDK4/6 inhibitor. In this study, no patients achieved complete response and three patients achieved partial response (202). The overall disease control rate remained poor at 27% among the cohort, and one patient died of treatment related hematotoxicity.
In summary, the majority of targeted therapy studies to date in DDLPS demonstrate inferior clinical performance when compared with the estimated objective response rate of 26% for first-line anthracycline-based chemotherapy for DDLPS (203). Therefore, the majority of studied targeted inhibitors are currently not recommended as an adjunct to systemic therapy. Per NCCN guidelines, palbociclib may have utility under certain circumstances (204). Despite these discouraging results, it should be noted that most of these studies were performed in advanced-stage disease refractory to multiple lines of therapy, and the therapeutic potential of combination therapy requires further investigation.
Mechanisms of resistance to MDM2 inhibition
Given the poor performance of MDM2 inhibitors in clinical trials for solid tumors, certain research efforts have shifted focus toward elucidating mechanisms of resistance. One commonly identified mechanism of resistance involves mutation in the TP53 gene that either renders the protein inactive or mitigates binding affinity for MDM2. Jung et al. evaluated liposarcoma tumor biopsy samples from patients treated in the SAR405838 trial over multiple time points, including a pretreatment baseline and every 6 weeks thereafter for 36 weeks (205). Upon examination of circulating cell-free DNA, the authors identified 26 acquired TP53 missense mutations during the study interval. Variants appeared within 6 weeks after treatment initiation and occurred with increasing frequency with cumulative therapy. Most mutations were in the p53-DNA-binding domain, rendering the tumor suppressor ineffective. The authors concluded that MDM2 inhibition exerted selective pressure on malignant cells, driving clonal expansion of lineages with inactivating TP53 mutations. This observation corresponded clinically with an early, modest clinical benefit that diminished over time.
In a separate study, Chapeau et al. performed a genetic screen to identify additional mechanisms of resistance to MDM2 inhibition (206). Using a piggyBac insertional mutagenesis system, the authors performed mutagenesis on genetically modified Arf -/- mice (207). CDKN2A deletion with subsequent p14ARF inactivation results in MDM2 activation in certain human malignancies. Tumors derived from these mice were allografted, and mice were then treated with the MDM2 inhibitor, HDM201. In total, 87 genes were significantly altered by mutagenesis. Sixteen of 21 tumors initially responded, but all relapsed over time. Refractory tumors displayed outgrowth of unique subclones resistant to MDM2 inhibition. Among these, multiple genetic mechanisms of acquired treatment resistance were identified, including both somatic and insertional changes. Loss-of-function mutation in Tp53 was observed in 54% of resistant tumors, and upregulation of antiapoptotic B-cell lymphoma-extra-large (Bcl-xL) and Mdmx gain-of-function mutations were also frequently identified. On subsequent combination therapy testing, the authors observed an additive effect using a Bcl-xL- selective inhibitor (ABT-263) in combination with MDM2 inhibition in 35/138 Tp53 wild-type cell lines, providing further support for investigation of combination therapies.
MDMX upregulation and gain of function represent another mechanism of resistance to MDM2 inhibition. Although MDMX does not have E3 ubiquitin ligase activity, it shares structural homology with MDM2 in the NH2 terminus p53-binding cleft, and it is able to independently sequester the p53 transactivational domain (208–210). MDMX also contains a C-terminus RING finger domain that allows for binding and stabilization of the MDM2 protein, and both functions contribute toward p53 inactivation (211). These effects have been demonstrated in vitro, in which human fibroblast cells were transformed to overexpress MDMX, resulting in acquired resistance to Nutlin-3a-mediated MDM2 inhibition. Conversely, Nutlin-3a treatment was rescued by siRNA knockdown of MDMX. Deletion of the MDMX C-terminus RING finger domain similarly rescued Nutlin-3a treatment, suggesting its structural role in MDM2 binding and stabilization. Based on these findings, combination approaches to simultaneously address MDM2 and MDMX function may improve treatment response rates and mitigate acquired resistance.
Stapled peptides, which are composed of stabilized alpha-helical peptides, are one potential strategy to target shared protein–protein interactions between MDM2–p53 and MDMX–p53, acting as a dual MDM2/MDMX inhibitor (212). ALRN-6924 was the first clinical drug of this type to be studied in a phase I, open-label, multicenter clinical trial (213). Seventy-one patients were enrolled, of which 17 had sarcomas, 60 had other solid tumors, and four had lymphoma. Ninety percent of tumors were p53 wild types. In total, 41 patients were evaluable for treatment efficacy, and of these, 24 patients (59%) demonstrated disease control by RECIST criteria. Four patients achieved a response: two complete responses and two partial responses; one of the partial responses was liposarcoma of unspecified subtype. The median duration of clinical benefit was 7.5 months. There were 10 patients with p53 wild-type and MDM2 tumor amplification, of which five patients (71%) achieved disease control. A trend was observed in which patients with lower expression levels of MDMX were more likely to achieve disease control than were patients with higher MDMX levels, although this association did not reach statistical significance. ALRN-6924 showed a good safety profile with the most common toxicities consisting of mild to moderate nausea, fatigue, vomiting, and myelosuppression. The limited side effect profile combined with improved rates of disease control provides rationale for further study (213).
Genomic and transcriptomic analyses of MDM2-resistant cells have revealed additional potential resistance mechanisms (214–216). N-RAS, MAPK/ERK, IGFBP1, and NF-kB, which are known drivers of cell proliferation and survival, are frequently upregulated in response to MDM2 inhibition. Increased drug export is an additional form of treatment resistance observed with MDM2 inhibitors. Grigoreva et al. compared the expression levels of the ABC gene encoding P-glycoprotein export protein in colon cancer cell lines treated with Nutlin-3a compared with controls and demonstrated that resistant lines exhibited 3–19-fold increased mRNA expression, which correlated with increased protein expression by Western blot (217). The authors utilized a fluorescent dye that binds to the intracellular H-site of P-glycoprotein and is actively released from cells. In so doing, the authors observed a two-fold increase in fluorescence intensity within wild-type cells when compared with resistant cell lines, suggesting greater intracellular accumulation of dye within non-resistant cell lines and increased dye efflux within chemoresistant cell lines. Taken together, these data suggest that increased P-glycoprotein-mediated efflux may contribute to drug resistance. While the molecular pathogenesis of these mechanisms requires further characterization and validation, these findings indicate that there are myriad ways in which cancers can evade targeted therapy. In this context, there has been increasing interest in combination therapy to overcome common mechanisms of resistance.
To date, there has been some success with combination techniques, mostly in preclinical studies. Saiki et al. conducted a cell-based screen to identify compounds that synergize with MDM2 inhibition in TP53 wild-type cells (218). In total, 13/1,169 library compounds were potential candidates, including PI3K/MAPK inhibitors, BH3 mimetics, BCR-ABL kinase antagonists, and HDAC inhibitors. Triple-combination studies using MDM2, PI3K, and BRAF/MEK inhibitors demonstrated even greater suppression of cell growth than all permutations of two-way combinations. In a separate in vitro analysis, it was shown that combination MDM2 inhibition plus doxorubicin was associated with significantly reduced tumor growth compared with doxorubicin monotherapy, suggesting that the addition of MDM2 inhibition to conventional cytotoxic therapy might also be beneficial (120). Finally, 12q13-15 co-amplification of CDK4 and MDM2 may represent therapeutic targets for combination therapy. While mixed results have been observed in preclinical application of CDK4/MDM2 combined targeting, there is an ongoing multicenter phase II clinical trial investigating co-administration of HDM201 with LEE011, a known CDK4 inhibitor (NCT02343172) (219, 220). In all instances of proposed combination strategies, the main barrier to implementation remains a concern for significant clinical side effects, which can be observed with even single-agent use of targeted treatments.
Key implications and future directions
When thinking to the future of targeted therapy for DDLPS, the findings from this review imply several key considerations. First, while MDM2 amplification is convincingly implicated in DDLPS oncogenesis, further investigation is necessary to better assess its importance in established tumors. In clinically detectable disease, it is possible that MDM2 hyperfunction is non-essential given the relative ease for tumors to develop resistance mechanisms in response to targeted therapy. Further studies in this regard would help to better appraise the utility of continuing to pursue the MDM2 pathway as a pharmacologic target. Second, regarding mechanisms of resistance, based on the seminal studies by Jung and Chapeu et al., selection for p53 inactivating mutations is a common phenomenon to bypass targeted MDM2 inhibition in sarcoma. As a key regulator of cell-cycle arrest, inactivating p53 mutations further limit the potential for activation of tumor-suppressive programs. Thus, co-targeting of parallel, interacting oncogenic pathways including PI3K/AKT/mTOR, MYC, and Bcl-x would seem a reasonable approach to mitigating MDM2 bypass mechanisms. Third, regarding the use of targeted therapies, whereas there have been preliminary studies investigating combination approaches for sarcomas, these have often been based on drug availability in the setting of advanced disease and not necessarily designed for synergistic or cooperative function, likely limiting the potential for this approach. Furthermore, whereas increasing the combination of inhibited targets would potentially lead to greater efficacy, toxicity remains an ongoing and major challenge for targeted therapies. Most combinatorial studies to date involve two agents, but it is conceivable going forward to have studies with multiple drugs designed in a rational cocktail, toxicities permitting. In this regard, development of more accurate real-time biomarkers to measure treatment response in DDLPS—such examples might include cell-free DNA (cfDNA) and advanced radiomics—is essential helping to monitor for resistant clonal outgrowth with targeted agents. Finally, in the current era, targeted therapy inevitably fails for most malignancies, but the phenotypic impacts of targeted therapy remain poorly understood and could potentially be used to leverage other treatment modalities. For example, regarding MDM2 inhibition in DDLPS, by virtue of selecting for specific clonal populations with phenotypic features such as inactivating p53 mutations, important questions arise regarding the impact on tumor heterogeneity, sensitivity to cytotoxic therapy, or changes in immunogenicity. While still entirely hypothetical, characterizing patterns of iatrogenic tumor evolution are minimally described to date in DDLPS, and this would be potentially useful when thinking about the implications of MDM2 inhibition.
Discussion
MDM2 amplification in DDLPS represents an excellent therapeutic target in theory, although clinical applications have thus far been disappointing. For reasons that are not fully understood, MDM2 inhibition has been more successful in other malignancies such as acute myelogenous leukemia, chronic lymphocytic leukemia, and multiple myeloma, in which early-phase clinical trials have demonstrated efficacy (127–129). In both preclinical experiments and clinical trials for solid tumors, MDM2 inhibition has been associated with an increase in p53 protein expression, yet this observation has not been associated with improved outcomes such as tumor growth inhibition or longer survival. Findings from recent studies indicate that this observed discrepancy is a result of multiple factors, including clonal expansion of cell lines with inactivating p53 mutations, augmentation of co-activators such as MDMX, and other unforeseen mechanisms of resistance (205). The tumor suppressor p53 is one of the key regulators of cell-cycle progression and is thus influenced by numerous signaling pathways beyond MDM2 (221, 222). As might be expected, MDM2 inhibition alone appears insufficient in the face of oncogenic redundancy.
Despite these challenges, MDM2-targeted therapies remain of interest in the treatment of DDLPS. Attempts to optimize the pharmacokinetics and dynamics of novel MDM2 inhibitors are underway. Novel drugs designed to simultaneously inhibit both MDM2 and MDMX represent an area of active research. In addition, alternative targets amplified on the 12q arm, including CDK4, HMGA2, and YEATS4, are also being investigated for combination therapies (223, 224). Advancements in peptide synthesis and cell-free DNA represent promising future directions in the targeted treatment and surveillance of these tumors. Taken together, these promising avenues of research will hopefully lead to less toxic and more efficacious treatments that mitigate the severity of this deadly disease.
Author contributions
RT, BC CR, EK, EN, and DE performed data acquisition, data interpretation, and manuscript construction. All authors contributed to the article and approved the submitted version.
Funding
Supported in part by grant from the National Institute of Health (T32CA009599 to RT). There are no other financial relationships related to the design or execution of this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Thway K. Well-differentiated liposarcoma and dedifferentiated liposarcoma: An updated review. Semin Diagn Pathol (2019) 36(2):112–21. doi: 10.1053/j.semdp.2019.02.006
2. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol (1997) 21(3):271–81. doi: 10.1097/00000478-199703000-00002
3. Singer S, Corson JM, Demetri GD, Healey EA, Marcus K, Eberlein TJ, et al. Prognostic factors predictive of survival for truncal and retroperitoneal soft-tissue sarcoma. Ann Surg (1995) 221(2):185–95. doi: 10.1097/00000658-199502000-00009
4. Alvarenga JC, Ball AB, Fisher C, Fryatt I, Jones L, Thomas JM, et al. Limitations of surgery in the treatment of retroperitoneal sarcoma. Br J Surg (1991) 78(8):912–6. doi: 10.1002/bjs.1800780806
5. Livingston JA, Bugano D, Barbo A, Lin H, Madewell J.E, Wang W.L, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep (2017) 7(1):11836. doi: 10.1038/s41598-017-12132-w
6. Haas RLM, Bonvalot S, Miceli R, Strauss D.C, Swallow C.J, Hohenberger P, et al. Radiotherapy for retroperitoneal liposarcoma: A report from the transatlantic retroperitoneal sarcoma working group. Cancer (2019) 125(8):1290–300. doi: 10.1002/cncr.31927
7. Nussbaum DP, Rushing CN, Lane W.O, Cardona D.M, Kirsch D.G, Peterson B.L, et al. Preoperative or postoperative radiotherapy versus surgery alone for retroperitoneal sarcoma: a case-control, propensity score-matched analysis of a nationwide clinical oncology database. Lancet Oncol (2016) 17(7):966–75. doi: 10.1016/S1470-2045(16)30050-X
8. Bonvalot S, Gronchi A, Péchoux Le C, Swallow C.J, Strauss D, Meeus P, et al. Preoperative radiotherapy plus surgery versus surgery alone for patients with primary retroperitoneal sarcoma (EORTC-62092: STRASS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol (2020) 21(10):1366–77. doi: 10.1016/S1470-2045(20)30446-0
9. Keung EZ, Hornick J.L, Bertagnolli M.M, Baldini E.H, Raut CP, et al. Predictors of outcomes in patients with primary retroperitoneal dedifferentiated liposarcoma undergoing surgery. J Am Coll Surg (2014) 218(2):206–17. doi: 10.1016/j.jamcollsurg.2013.10.009
10. MacNeill AJ, Miceli R, Strauss D.C, Bonvalot S, Hohenberger P, Coevorden Van F. Post-relapse outcomes after primary extended resection of retroperitoneal sarcoma: A report from the trans-Atlantic RPS working group. Cancer (2017) 123(11):1971–8. doi: 10.1002/cncr.30572
11. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell (2017) 171(4):950–965.e28. doi: 10.1016/j.cell.2017.10.014
12. Oliner JD, Pietenpol J.A, Thiagalingam S, Gyuris J, Kinzler K.W, Vogelstein B, et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature (1993) 362(6423):857–60. doi: 10.1038/362857a0
13. de Jonge M, Weger V.A, Dickson M.A, Langenberg M, Cesne Le A, Wagner A.J, et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer (2017) 76:144–51. doi: 10.1016/j.ejca.2017.02.005
14. Ray-Coquard I, Blay J.Y, Italiano A, Cesne Le A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol (2012) 13(11):1133–40. doi: 10.1016/S1470-2045(12)70474-6
15. Jeay S, Ferretti S, Holzer P, Fuchs J, Chapeau EA, Wartmann M, et al. Dose and schedule determine distinct molecular mechanisms underlying the efficacy of the p53-MDM2 inhibitor HDM201. Cancer Res (2018) 78(21):6257–67. doi: 10.1158/0008-5472.CAN-18-0338
16. Wang HQ, Mulford I.J, Sharp F, Liang J, Kurtulus S, Trabucco G, et al. Inhibition of MDM2 promotes antitumor responses in p53 wild-type cancer cells through their interaction with the immune and stromal microenvironment. Cancer Res (2021) 81(11):3079–91. doi: 10.1158/0008-5472.CAN-20-0189
17. Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology (2014) 46(2):95–104. doi: 10.1097/PAT.0000000000000050
18. Rizvi NA, Hellmann M.D, Snyder A, Kvistborg P, Makarov V, Havel J.J, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
19. Snyder A, Makarov Merghoub Yuan Zaretsky Desrichard T. J.J.M.A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi: 10.1056/NEJMoa1406498
20. Chalmers ZR, Connelly C.F, Fabrizio D, Gay L, Ali S.M, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med (2017) 9(1):34. doi: 10.1186/s13073-017-0424-2
21. Liu W, Tong H, Zhang C, Zhuang R, Guo H, Lv C, et al. Integrated genomic and transcriptomic analysis revealed mutation patterns of de-differentiated liposarcoma and leiomyosarcoma. BMC Cancer (2020) 20(1):1035. doi: 10.1186/s12885-020-07456-2
22. Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun (2013) 13:5.
23. Maki RG, Jungbluth A.A, Gnjatic S, Schwartz G.K, D R, Keohan M.L, et al. A pilot study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma 2013 (2013) p:168145. doi: 10.1155/2013/168145
24. Harbers L, Agostini F, Nicos M, Poddighe D, Bienko M, Crosetto N. Somatic copy number alterations in human cancers: An analysis of publicly available data from the cancer genome atlas. Front Oncol (2021) 11:700568. doi: 10.3389/fonc.2021.700568
25. Beroukhim R, Mermel C.H, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463(7283):899–905. doi: 10.1038/nature08822
26. Barretina J, Taylor B.S, Banerji S, Ramos A.H, Lagos-Quintana M, Decarolis P.L, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet (2010) 42(8):715–21. doi: 10.1038/ng.619
27. Dal Cin P, Kools P, Sciot R, De Wever I, Van Damme B, Van de Ven W, et al. Cytogenetic and fluorescence in situ hybridization investigation of ring chromosomes characterizing a specific pathologic subgroup of adipose tissue tumors. Cancer Genet Cytogenet (1993) 68(2):85–90. doi: 10.1016/0165-4608(93)90001-3
28. Pedeutour F, Forus A, Coindre J.M, Berner J.M, Nicolo G, Michiels J.F, et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer (1999) 24(1):30–41. doi: 10.1002/(SICI)1098-2264(199901)24:1<30::AID-GCC5>3.0.CO;2-P
29. Creytens D, Gorp Van J, Speel E.J, Ferdinande L. Characterization of the 12q amplicons in lipomatous soft tissue tumors by multiplex ligation-dependent probe amplification-based copy number analysis. Anticancer Res (2015) 35(4):1835–42.
30. Dei Tos AP, Doglioni C, Piccinin S, Sciot R, Furlanetto A, Boiocchi M, et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-c genes in atypical lipomatous tumours. J Pathol (2000) 190(5):531–6. doi: 10.1002/(SICI)1096-9896(200004)190:5<531::AID-PATH579>3.0.CO;2-W
31. Italiano A, Bianchini L, Keslair F, Bonnafous S, Cardot-Leccia N, Coindre J.M, et al. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. Int J Cancer (2008) 122(10):2233–41. doi: 10.1002/ijc.23380
32. Ware PL, Snow AN, Gvalani M, Pettenati MJ, Qasem SA. MDM2 copy numbers in well-differentiated and dedifferentiated liposarcoma: characterizing progression to high-grade tumors. Am J Clin Pathol (2014) 141(3):334–41. doi: 10.1309/AJCPLYU89XHSNHQO
33. Mendrysa SM, McElwee MK, Michalowski J, O'Leary KA, Young KM, Perry ME. mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol (2003) 23(2):462–72. doi: 10.1128/MCB.23.2.462-473.2003
34. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer (2006) 6(12):909–23. doi: 10.1038/nrc2012
35. Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol Metab (2020) 33:2–22. doi: 10.1016/j.molmet.2019.10.002
36. Fletcher CD, Akerman M, Cin Dal P, Wever I, Mandahl N, Mertens F, et al. Correlation between clinicopathological features and karyotype in lipomatous tumors. a report of 178 cases from the chromosomes and morphology (CHAMP) collaborative study group. Am J Pathol (1996) 148(2):623–30.
37. Horvai AE, DeVries S, Roy R, O'Donnell RJ, Waldman F. Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma. Mod Pathol (2009) 22(11):1477–88. doi: 10.1038/modpathol.2009.119
38. Beird HC, Wu C.C, Ingram D.R, Wang W.L, Alimohamed A, Gumbs C, et al. Genomic profiling of dedifferentiated liposarcoma compared to matched well-differentiated liposarcoma reveals higher genomic complexity and a common origin. Cold Spring Harb Mol Case Stud (2018) 4(2): 1477–88. doi: 10.1101/mcs.a002386
39. Snyder EL, Sandstrom D.J, Law K, Fiore C, Sicinska E, Brito J, et al. C-jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme. J Pathol (2009) 218(3):292–300. doi: 10.1002/path.2564
40. Saâda-Bouzid E, Burel-Vandenbos F, Ranchère-Vince D, Birtwisle-Peyrottes I, Chetaille B, Bouvier C, et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod Pathol (2015) 28(11):1404–14. doi: 10.1038/modpathol.2015.96
41. Taylor BS, DeCarolis PL, Angeles CV, Brenet F, Schultz N, Antonescu CR, et al. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discovery (2011) 1(7):587–97. doi: 10.1158/2159-8290.CD-11-0181
42. Keung EZ, Akdemir KC, Sannaa Al GA, Garnett J, Lev D, Torres KE, et al. Increased H3K9me3 drives dedifferentiated phenotype via KLF6 repression in liposarcoma. J Clin Invest (2015) 125(8):2965–78. doi: 10.1172/JCI77976
43. Cao Z, Xue J, Cheng Y, Wang J, Liu Y, Li H, et al. MDM2 promotes genome instability by ubiquitinating the transcription factor HBP1. Oncogene (2019) 38(24):4835–55. doi: 10.1038/s41388-019-0761-2
44. Pan K, Chen Y, Roth M, Wang W, Wang S, Yee AS, et al. HBP1-mediated transcriptional regulation of DNA methyltransferase 1 and its impact on cell senescence. Mol Cell Biol (2013) 33(5):887–903. doi: 10.1128/MCB.00637-12
45. Zhang X, Kim J, Ruthazer R, McDevitt MA, Wazer DE, Paulson KE, et al. The HBP1 transcriptional repressor participates in RAS-induced premature senescence. Mol Cell Biol (2006) 26(22):8252–66. doi: 10.1128/MCB.00604-06
46. Chen Y, Pan K, Wang P, Cao Z, Wang W, Wang S, et al. HBP1-mediated regulation of p21 protein through the Mdm2/p53 and TCF4/EZH2 pathways and its impact on cell senescence and tumorigenesis. J Biol Chem (2016) 291(24):12688–705. doi: 10.1074/jbc.M116.714147
47. Escamilla-Powers JR, Daniel CJ, Farrell A, Taylor K, Zhang X, Byers S, et al. The tumor suppressor protein HBP1 is a novel c-myc-binding protein that negatively regulates c-myc transcriptional activity 2. J Biol Chem (2010) 285(7):4847–58. doi: 10.1074/jbc.M109.074856
48. Li H, Wang W, Liu X, Paulson K, Yee A, Zhang X. Transcriptional factor HBP1 targets p16INK4A, upregulating its expression and consequently is involved in ras-induced premature senescence. Oncogene (2010) 29(36):5083–94. doi: 10.1038/onc.2010.252
49. Stocker M, Nail Le LR, Belenet De H, Wunder JS, Andrulis IL, Gokgoz N, et al. Inhibition of P53-mediated cell cycle control as the determinant in dedifferentiated liposarcomas development. Am J Cancer Res (2021) 11(6):3271–84.
50. Chu QS, Nielsen TO, Alcindor T, Gupta A, Endo M, Goytain A, et al. A phase II study of SB939, a novel pan-histone deacetylase inhibitor, in patients with translocation-associated recurrent/metastatic sarcomas-NCIC-CTG IND 200†. Ann Oncol (2015) 26(5):973–81. doi: 10.1093/annonc/mdv033
51. Schmitt T, Mayer-Steinacker R, Mayer F, Grünwald V, Schütte J, Hartmann JT, et al. Vorinostat in refractory soft tissue sarcomas - results of a multi-centre phase II trial of the German soft tissue sarcoma and bone tumour working group (AIO). Eur J Cancer (2016) 64:74–82. doi: 10.1016/j.ejca.2016.05.018
52. Yan B, Chen Q, Shimada K, Tang M, Li H, Gurumurthy A, et al. Histone deacetylase inhibitor targets CD123/CD47-positive cells and reverse chemoresistance phenotype in acute myeloid leukemia. Leukemia (2019) 33(4):931–44. doi: 10.1038/s41375-018-0279-6
53. Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F, Reuss DE, et al. Sarcoma classification by DNA methylation profiling. Nat Commun (2021) 12(1):498. doi: 10.1038/s41467-020-20603-4
54. Mertens F, Fletcher CD, Cin Dal P, Wever De I, Mandahl N, Mitelman F, et al. Cytogenetic analysis of 46 pleomorphic soft tissue sarcomas and correlation with morphologic and clinical features: a report of the CHAMP study group. chromosomes and MorPhology. Genes Chromosomes Cancer (1998) 22(1):16–25. doi: 10.1002/(SICI)1098-2264(199805)22:1<16::AID-GCC3>3.0.CO;2-A
55. Gebhard S, Coindre JM, Michels JJ, Terrier P, Bertrand G, Trassard M, et al. Pleomorphic liposarcoma: clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases: a study from the French federation of cancer centers sarcoma group. Am J Surg Pathol (2002) 26(5):601–16. doi: 10.1097/00000478-200205000-00006
56. Szymanska J, Tarkkanen M, Wiklund T, Virolainen M, Blomqvist C, Asko-Seljavaara S, et al. Gains and losses of DNA sequences in liposarcomas evaluated by comparative genomic hybridization. Genes Chromosomes Cancer (1996) 15(2):89–94. doi: 10.1002/(SICI)1098-2264(199602)15:2<89::AID-GCC2>3.0.CO;2-#
57. May CD, Landers SM, Bolshakov S, Ma X, Ingram DR, Kivlin CM, et al. Co-Targeting PI3K, mTOR, and IGF1R with small molecule inhibitors for treating undifferentiated pleomorphic sarcoma. Cancer Biol Ther (2017) 18(10):816–26. doi: 10.1080/15384047.2017.1373230
58. Schmidt H, Bartel F, Kappler M, Würl P, Lange H, Bache M, et al. Gains of 13q are correlated with a poor prognosis in liposarcoma. Mod Pathol (2005) 18(5):638–44. doi: 10.1038/modpathol.3800326
59. Rieker RJ, Joos S, Bartsch C, Willeke F, Schwarzbach M, Otaño-Joos M, et al. Distinct chromosomal imbalances in pleomorphic and in high-grade dedifferentiated liposarcomas. Int J Cancer (2002) 99(1):68–73. doi: 10.1002/ijc.10287
60. Jeon HM, Lee JS, Kim SH, Yun KH, Park KH, Jeon MK, et al. Comprehensive immuno-molecular profiles for liposarcoma: Roles of programmed death ligand 1, microsatellite instability, and PIK3CA. Oncology (2020) 98(11):817–26. doi: 10.1159/000509004
61. Vargas AC, Maclean FM, Sioson L, Tran D, Bonar F, Mahar A, et al. Prevalence of PD-L1 expression in matched recurrent and/or metastatic sarcoma samples and in a range of selected sarcomas subtypes. PloS One (2020) 15(4):e0222551. doi: 10.1371/journal.pone.0222551
62. Yan L, Wang Z, Cui C, Guan X, Dong B, Zhao M, et al. Comprehensive immune characterization and T-cell receptor repertoire heterogeneity of retroperitoneal liposarcoma. Cancer Sci (2019) 110(10):3038–48. doi: 10.1111/cas.14161
63. Dancsok AR, Gao D, Lee AF, Steigen SE, Blay JY, Thomas DM, et al. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology (2020) 9(1):1747340. doi: 10.1080/2162402X.2020.1747340
64. Keung EZ, Somaiah N. Overview of liposarcomas and their genomic landscape. J Trans Genet Genomics (2019) 3:8. doi: 10.20517/jtgg.2019.03
65. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer (2009) 9(10):691–700. doi: 10.1038/nrc2715
66. Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell (2006) 21(3):307–15. doi: 10.1016/j.molcel.2006.01.020
67. Tian H, Tackmann NR, Jin A, Zheng J, Zhang Y. Inactivation of the MDM2 RING domain enhances p53 transcriptional activity in mice. J Biol Chem (2017) 292(52):21614–22. doi: 10.1074/jbc.RA117.000122
68. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer (2013) 13(2):83–96. doi: 10.1038/nrc3430
69. Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res (1998) 26(15):3453–9. doi: 10.1093/nar/26.15.3453
70. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science (1996) 274(5289):948–53. doi: 10.1126/science.274.5289.948
71. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature (1997) 387(6630):296–9. doi: 10.1038/387296a0
72. Chao CC. Mechanisms of p53 degradation. Clin Chim Acta (2015) 438:139–47. doi: 10.1016/j.cca.2014.08.015
73. Tao W, Levine AJ. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci U.S.A. (1999) 96(12):6937–41. doi: 10.1073/pnas.96.12.6937
74. Popowicz GM, Czarna A, Holak TA. Structure of the human mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle (2008) 7(15):2441–3. doi: 10.4161/cc.6365
75. Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem (1999) 274(53):38189–96. doi: 10.1074/jbc.274.53.38189
76. Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. RING domain-mediated interaction is a requirement for MDM2's E3 ligase activity. Cancer Res (2007) 67(13):6026–30. doi: 10.1158/0008-5472.CAN-07-1313
77. Blattner C, Hay T, Meek DW, Lane DP. Hypophosphorylation of Mdm2 augments p53 stability. Mol Cell Biol (2002) 22(17):6170–82. doi: 10.1128/MCB.22.17.6170-6182.2002
78. Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, et al. ATM-Dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev (2001) 15(9):1067–77. doi: 10.1101/gad.886901
79. Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM Activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J (2009) 28(24):3857–67. doi: 10.1038/emboj.2009.294
80. Allende-Vega N, Dias S, Milne D, Meek D. Phosphorylation of the acidic domain of Mdm2 by protein kinase CK2. Mol Cell Biochem (2005) 274(1-2):85–90. doi: 10.1007/s11010-005-3074-4
81. Kulikov R, Boehme KA, Blattner C. Glycogen synthase kinase 3-dependent phosphorylation of Mdm2 regulates p53 abundance. Mol Cell Biol (2005) 25(16):7170–80. doi: 10.1128/MCB.25.16.7170-7180.2005
82. Riley MF, Lozano G. The many faces of MDM2 binding partners. Genes Cancer (2012) 3(3-4):226–39. doi: 10.1177/1947601912455322
83. Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM, et al. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem (2000) 275(12):8945–51. doi: 10.1074/jbc.275.12.8945
84. Honda R, Yasuda H. Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene (2000) 19(11):1473–6. doi: 10.1038/sj.onc.1203464
85. Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J (2004) 23(7):1547–56. doi: 10.1038/sj.emboj.7600145
86. Linares LK, Kiernan R, Triboulet R, Chable-Bessia C, Latreille D, Cuvier O, et al. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat Cell Biol (2007) 9(3):331–8. doi: 10.1038/ncb1545
87. Inuzuka H, Tseng A, Gao D, Zhai B, Zhang Q, Shaik S, et al. Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCF(beta-TRCP) ubiquitin ligase. Cancer Cell (2010) 18(2):147–59. doi: 10.1016/j.ccr.2010.06.015
88. Miyauchi Y, Yogosawa S, Honda R, Nishida T, Yasuda H. Sumoylation of Mdm2 by protein inhibitor of activated STAT (PIAS) and RanBP2 enzymes. J Biol Chem (2002) 277(51):50131–6. doi: 10.1074/jbc.M208319200
89. Jiang M, Chiu SY, Hsu W. SUMO-specific protease 2 in Mdm2-mediated regulation of p53. Cell Death Differ (2011) 18(6):1005–15. doi: 10.1038/cdd.2010.168
90. Watson IR, Li BK, Roche O, Blanch A, Ohh M, Irwin MS. Chemotherapy induces NEDP1-mediated destabilization of MDM2. Oncogene (2010) 29(2):297–304. doi: 10.1038/onc.2009.314
91. Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U.S.A. (1998) 95(14):8292–7. doi: 10.1073/pnas.95.14.8292
92. Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell (1997) 91(5):649–59. doi: 10.1016/S0092-8674(00)80452-3
93. Rizos H, Darmanian AP, Mann GJ, Kefford RF. Two arginine rich domains in the p14ARF tumour suppressor mediate nucleolar localization. Oncogene (2000) 19(26):2978–85. doi: 10.1038/sj.onc.1203629
94. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar arf sequesters Mdm2 and activates p53. Nat Cell Biol (1999) 1(1):20–6. doi: 10.1038/8991
95. Davidović R, Sopta J, Mandušić V, Krajnović M, Stanojević M, Tulić G, et al. p14(ARF) methylation is a common event in the pathogenesis and progression of myxoid and pleomorphic liposarcoma. Med Oncol (2013) 30(3):682. doi: 10.1007/s12032-013-0682-9
96. Oda Y, Yamamoto H, Takahira T, Kobayashi C, Kawaguchi K, Tateishi N, et al. Frequent alteration of p16(INK4a)/p14(ARF) and p53 pathways in the round cell component of myxoid/round cell liposarcoma: p53 gene alterations and reduced p14(ARF) expression both correlate with poor prognosis. J Pathol (2005) 207(4):410–21. doi: 10.1002/path.1848
97. Italiano A, Bianchini L, Gjernes E, Keslair F, Ranchere-Vince D, Dumollard JM, et al. Clinical and biological significance of CDK4 amplification in well-differentiated and dedifferentiated liposarcomas. Clin Cancer Res (2009) 15(18):5696–703. doi: 10.1158/1078-0432.CCR-08-3185
99. Fallah Y, Brundage J, Allegakoen P, Shajahan-Haq AN. MYC-driven pathways in breast cancer subtypes. Biomolecules (2017) 7(3). doi: 10.3390/biom7030053
100. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. C-myc and cancer metabolism. Clin Cancer Res (2012) 18(20):5546–53. doi: 10.1158/1078-0432.CCR-12-0977
101. Dang CV, K A, Zeller KI, Nguyen T, Osthus RC, Li F. The c-myc target gene network. Semin Cancer Biol (2006) 16(4):253–64. doi: 10.1016/j.semcancer.2006.07.014
102. Dang CV. C-myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol (1999) 19(1):1–11. doi: 10.1128/MCB.19.1.1
103. Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in myc-induced lymphomagenesis. Genes Dev (1999) 13(20):2658–69. doi: 10.1101/gad.13.20.2658
104. Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev (1998) 12(15):2424–33. doi: 10.1101/gad.12.15.2424
105. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature (2001) 411(6835):342–8. doi: 10.1038/35077213
106. Saporita AJ, Jr Maggi LB, Apicelli AJ, Weber JD. Therapeutic targets in the ARF tumor suppressor pathway. Curr Med Chem (2007) 14(17):1815–27. doi: 10.2174/092986707781058869
107. Lowe SW, Sherr CJ. Tumor suppression by Ink4a-arf: progress and puzzles. Curr Opin Genet Dev (2003) 13(1):77–83. doi: 10.1016/S0959-437X(02)00013-8
108. Kim YJ, Kim M, Park HK, Yu DB, Jung K, Song K, et al. Co-Expression of MDM2 and CDK4 in transformed human mesenchymal stem cells causes high-grade sarcoma with a dedifferentiated liposarcoma-like morphology. Lab Invest (2019) 99(9):1309–20. doi: 10.1038/s41374-019-0263-4
109. Manning BD, Toker A. AKT/PKB signaling: Navigating the network. Cell (2017) 169(3):381–405. doi: 10.1016/j.cell.2017.04.001
110. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via akt-mediated MDM2 phosphorylation. Nat Cell Biol (2001) 3(11):973–82. doi: 10.1038/ncb1101-973
111. Wendel H-G, Stanchina Ed, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by akt and eIF4E in oncogenesis and cancer therapy. Nature (2004) 428(6980):332–7. doi: 10.1038/nature02369
112. Osaki M, Oshimura M, Ito H. PI3K-akt pathway: its functions and alterations in human cancer. Apoptosis (2004) 9(6):667–76. doi: 10.1023/B:APPT.0000045801.15585.dd
113. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discovery (2005) 4(12):988–1004. doi: 10.1038/nrd1902
114. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by akt and suppresses mTOR signalling. Nat Cell Biol (2002) 4(9):648–57. doi: 10.1038/ncb839
115. Moumen A, Patané S, Porras A, Dono R, Maina F. Met acts on Mdm2 via mTOR to signal cell survival during development. Development (2007) 134(7):1443–51. doi: 10.1242/dev.02820
116. Ishii T, Kohashi K, Iura K, Maekawa A, Bekki H, Yamada Y, et al. Activation of the akt-mTOR and MAPK pathways in dedifferentiated liposarcomas. Tumour Biol (2016) 37(4):4767–76. doi: 10.1007/s13277-015-4232-2
117. Gutierrez A, Snyder EL, Marino-Enriquez A, Zhang YX, Sioletic S, Kozakewich E, et al. Aberrant AKT activation drives well-differentiated liposarcoma. Proc Natl Acad Sci U.S.A. (2011) 108(39):16386–91. doi: 10.1073/pnas.1106127108
118. Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer (2003) 3(9):685–94. doi: 10.1038/nrc1168
119. Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer (2011) 11(8):541–57. doi: 10.1038/nrc3087
120. Bill KLJ, Seligson ND, Hays JL, Awasthi A, Demoret B, Stets CW, et al. Degree of MDM2 amplification affects clinical outcomes in dedifferentiated liposarcoma. Oncologist (2019) 24(7):989–96. doi: 10.1634/theoncologist.2019-0047
121. Ricciotti RW, Baraff AJ, Jour G, Kyriss M, Wu Y, Liu Y, et al. High amplification levels of MDM2 and CDK4 correlate with poor outcome in patients with dedifferentiated liposarcoma: A cytogenomic microarray analysis of 47 cases. Cancer Genet (2017) 218-219:69–80. doi: 10.1016/j.cancergen.2017.09.005
123. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science (1994) 265(5170):346–55. doi: 10.1126/science.8023157
124. Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of the interaction of p53 with MDM2;–fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene (1994) 9(9):2523–9.
125. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science (2004) 303(5659):844–8. doi: 10.1126/science.1092472
126. Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res (2013) 73(8):2587–97. doi: 10.1158/0008-5472.CAN-12-2807
127. Stühmer T, Chatterjee M, Hildebrandt M, Herrmann P, Gollasch H, Gerecke C, et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood (2005) 106(10):3609–17. doi: 10.1182/blood-2005-04-1489
128. Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood (2005) 106(9):3150–9. doi: 10.1182/blood-2005-02-0553
129. Saddler C, Ouillette P, Kujawski L, Shangary S, Talpaz M, Kaminski M, et al. Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood (2008) 111(3):1584–93. doi: 10.1182/blood-2007-09-112698
130. Lau LM, Nugent JK, Zhao X, Irwin MS. HDM2 antagonist nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene (2008) 27(7):997–1003. doi: 10.1038/sj.onc.1210707
131. Zhang Z, Wang H, Li M, Rayburn ER, Agrawal S, Zhang R. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene (2005) 24(48):7238–47. doi: 10.1038/sj.onc.1208814
132. Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barrière C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res (2014) 74(20):5855–65. doi: 10.1158/0008-5472.CAN-14-0799
133. Bill KL, Garnett J, Meaux I, Ma X, Creighton CJ, Bolshakov S, et al. SAR405838: A novel and potent inhibitor of the MDM2:p53 axis for the treatment of dedifferentiated liposarcoma. Clin Cancer Res (2016) 22(5):1150–60. doi: 10.1158/1078-0432.CCR-15-1522
134. Reisman D, Takahashi P, Polson A, Boggs K. Transcriptional regulation of the p53 tumor suppressor gene in s-phase of the cell-cycle and the cellular response to DNA damage. Biochem Res Int 2012 (2012) 2012: 808934. doi: 10.1155/2012/808934
135. Fischer M, Quaas M, Steiner L, Engeland K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res (2016) 44(1):164–74. doi: 10.1093/nar/gkv927
136. Kuribayashi K, Finnberg N, Jeffers JR, Zambetti GP, El-Deiry WS. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle (2011) 10(14):2380–9. doi: 10.4161/cc.10.14.16588
137. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol (2008) 9(5):402–12. doi: 10.1038/nrm2395
138. Sharma MD, Rodriguez PC, Koehn BH, Baban B, Cui Y, Guo G, et al. Activation of p53 in immature myeloid precursor cells controls differentiation into Ly6c(+)CD103(+) monocytic antigen-presenting cells in tumors. Immunity (2018) 48(1):91–106.e6. doi: 10.1016/j.immuni.2017.12.014
139. Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U.S.A. (2008) 105(39):15016–21. doi: 10.1073/pnas.0801497105
140. Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science (2012) 338(6111):1220–5. doi: 10.1126/science.1229620
141. Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, et al. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol (2011) 41(10):2977–86. doi: 10.1002/eji.201141639
142. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother (2011) 34(5):409–18. doi: 10.1097/CJI.0b013e31821ca6ce
143. Shulman DSPhase 1 study of the dual MDM2/MDMX inhibitor ALRN-6924 in pediatric cancer. ClinicalTrials.gov Identifier: NCT03654716. First posted August 31, 2018. https://clinicaltrials.gov/ct2/show/NCT03654716
144. Dumbrava EEALRN-6924 and paclitaxel in treating patients with advanced, metastatic, or unresectable solid tumors. ClinicalTrials.gov Identifier: NCT03725436. First posted October 31, 2018. https://clinicaltrials.gov/ct2/show/NCT03725436
145. Selivanova GALRN-6924 in patients with advanced solid tumors or lymphomas. ClinicalTrials.gov Identifier: NCT02264613. First posted October 15, 2014. https://clinicaltrials.gov/ct2/show/NCT02264613
146. Saleh MN, Patel MR, Bauer TM, Goel S, Falchook GS, Shapiro GI, et al. Phase 1 trial of ALRN-6924, a dual inhibitor of MDMX and MDM2, in patients with solid tumors and lymphomas bearing wild-type TP53. Clin Cancer Res (2021) 27(19):5236–47. doi: 10.1158/1078-0432.CCR-21-0715
147. Welliver MX. Navtemadlin and radiation therapy in treating patients with soft tissue sarcoma. ClinicalTrials.gov Identifier: NCT03217266. First posted July 14, 2017. https://clinicaltrials.gov/ct2/show/NCT03217266
148. Amgen MDA phase 1 study evaluating AMG 232 in advanced solid tumors or multiple myeloma. ClinicalTrials.gov Identifier: NCT01723020. First posted November 7, 2012. https://clinicaltrials.gov/ct2/show/NCT01723020
149. Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA, et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drugs (2020) 38(3):831–43. doi: 10.1007/s10637-019-00840-1
150. Guo Y. APG-115 in combination with PD-1 inhibitor in patients with advanced liposarcoma or advanced solid tumors. ClinicalTrials.gov Identifier: NCT04785196. First posted March 5, 2021. https://clinicaltrials.gov/ct2/show/NCT04785196
151. Zhai Y. A study of APG-115 in combination with pembrolizumab in patients with metastatic melanomas or advanced solid tumors. ClinicalTrials.gov Identifier: NCT03611868. First posted August 2, 2018. https://clinicaltrials.gov/ct2/show/NCT03611868
152. Tolcher AW, Reeves JA, McKean M, Chmielowski B, Beck JT, Shaheen MF, et al. Preliminary results of a phase II study of alrizomadlin (APG-115), a novel, small-molecule MDM2 inhibitor, in combination with pembrolizumab in patients (pts) with unresectable or metastatic melanoma or advanced solid tumors that have failed immuno-oncologic (I-O) drugs. J Clin Oncol (2021) 39(15_suppl):2506–6. doi: 10.1200/JCO.2021.39.15_suppl.2506
153. Zhai YAPG-115 in patients with advanced solid tumors or lymphomas. ClinicalTrials.gov Identifier: NCT02935907. First posted October 18, 2016. https://clinicaltrials.gov/ct2/show/NCT02935907
154. Rasco DW, Lakhani NJ, Li Y, Men L, Wang H, Ji J, et al. A phase I study of a novel MDM2 antagonist APG-115 in patients with advanced solid tumors. J Clin Oncol (2019) 37(15_suppl):3126–6. doi: 10.1200/JCO.2019.37.15_suppl.3126
155. Astex Pharmaceuticals, Inc. Study of ASTX295 in patients with solid tumors with wild-type p53. ClinicalTrials.gov Identifier: NCT03975387. FIrst posted June 5, 2019. https://clinicaltrials.gov/ct2/show/NCT03975387.
156. Boehringer IngelheimBrightline-1: A study to compare BI 907828 with doxorubicin in people with a type of cancer called dedifferentiated liposarcoma. ClinicalTrials.gov Identifier: NCT05218499. First posted February 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05218499
157. Boehringer Ingelheim. This study aims to find the best dose of BI 907828 in patients with different types of advanced cancer (Solid tumors). ClinicalTrials.gov Identifier: NCT03449381. First posted February 28, 2018. https://clinicaltrials.gov/ct2/show/NCT03449381
158. Gounder MM, Yamamoto N, Patel MR, Bauer TM, Schöffski P, Grempler R, et al. A phase Ia/Ib, dose-escalation/expansion study of the MDM2–p53 antagonist BI 907828 in patients with solid tumors, including advanced/metastatic liposarcoma (LPS). J Clin Oncol (2022) 40(16_suppl):3004–4. doi: 10.1200/JCO.2022.40.16_suppl.3004
159. Boehringer Ingelheim. A study in patients with different types of advanced cancer (Solid tumors) to test different doses of BI 907828 in combination with BI 754091 (Ezabenlimab) and BI 754111 or BI 907828 in combination with BI 754091 (Ezabenlimab). ClinicalTrials.gov Identifier: NCT03964233. First posted May 28, 2019. https://clinicaltrials.gov/ct2/show/NCT03964233
160. Novartis Pharmaceuticals. A phase I dose escalation study of CGM097 in adult patients with selected advanced solid tumors. ClinicalTrials.gov Identifier: NCT01760525. First posted January 4, 2013. https://clinicaltrials.gov/ct2/show/NCT01760525
161. Bauer S, Demetri GD, Halilovic E, Dummer R, Meille C, Tan DSW, et al. Pharmacokinetic-pharmacodynamic guided optimisation of dose and schedule of CGM097, an HDM2 inhibitor, in preclinical and clinical studies. Br J Cancer (2021) 125(5):687–98. doi: 10.1038/s41416-021-01444-4
162. Rain Therapeutics Inc. Treatment of milademetan versus trabectedin in patient with dedifferentiated liposarcoma (MANTRA). ClinicalTrials.gov Identifier: NCT04979442. First posted July 28, 2021. https://clinicaltrials.gov/ct2/show/NCT04979442
163. Rain Therapeutics Inc. Milademetan in Advanced/Metastatic solid tumors. ClinicalTrials.gov Identifier: NCT05012397. First posted August 19, 2021. https://clinicaltrials.gov/ct2/show/NCT05012397
164. Daiichi Sankyo, Inc. A phase 1 multiple ascending dose study of milademetan in subjects with advanced solid tumors or lymphomas. ClinicalTrials.gov Identifier: NCT01877382. First posted June 13, 2013. https://clinicaltrials.gov/ct2/show/NCT01877382
165. Bauer TM, Gounder MM, Weise AM, Schwartz GK, Carvajal RD, Kumar P, et al. A phase 1 study of MDM2 inhibitor DS-3032b in patients with well/de-differentiated liposarcoma (WD/DD LPS), solid tumors (ST) and lymphomas (L). J Clin Oncol (2018) 36(15_suppl):11514–4. doi: 10.1200/JCO.2018.36.15_suppl.11514
166. Novartis Pharmaceuticals. Study of safety and efficacy of HDM201 in combination with LEE011 in patients with liposarcoma. ClinicalTrials.gov Identifier: NCT02343172. First posted January 21, 2015. https://clinicaltrials.gov/ct2/show/NCT02343172
167. Pharmeceuticals, N. Clinical trials results CHDM201X2103C (2020). Available at: https://www.novctrd.com/ctrdweb/trialresult/trialresults/pdf?trialResultId=17708.
168. Centre Leon Berard. HDM201 and pazopanib in patients with P53 wild-type Advanced/Metastatic soft tissue sarcomas. ClinicalTrials.gov Identifier: NCT05180695. First posted January 6, 2022. https://clinicaltrials.gov/ct2/show/NCT05180695
169. Novartis Pharmaceuticals. Study to determine and evaluate a safe and tolerated dose of HDM201 in patients with selected advanced tumors that are TP53wt. ClinicalTrials.gov Identifier: NCT02143635. First posted May 21, 2014. https://clinicaltrials.gov/ct2/show/NCT02143635
170. Stein EM, DeAngelo DJ, Chromik J, Chatterjee M, Bauer S, Lin CC, et al. Results from a first-in-Human phase I study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin Cancer Res (2022) 28(5):870–81. doi: 10.1158/1078-0432.CCR-21-1295
171. Blay JY. A study evaluating the activity of anti-cancer treatments targeting tumor molecular Alterations/Characteristics in advanced metastatic tumors. ClinicalTrials.gov Identifier: NCT04116541. First posted October 4, 2019. https://clinicaltrials.gov/ct2/show/NCT04116541
172. Hoffmann-La Roche. A study of RO5045337 in patients with liposarcomas prior to debulking surgery. ClinicalTrials.gov Identifier: NCT01143740. First posted June 14, 2010. https://clinicaltrials.gov/ct2/show/NCT01143740
173. Hoffmann-La Roche. A study of RO5045337 in combination with doxorubicin in patients with soft tissue sarcoma. ClinicalTrials.gov Identifier: NCT01605526. FIrst posted May 25, 2012. https://clinicaltrials.gov/ct2/show/NCT01605526
174. Chawla SP, Blay J-Y, Italiano A, Gutierrez M, Cesne AL, Gomez-Roca CA, et al. Phase ib study of RG7112 with doxorubicin (D) in advanced soft tissue sarcoma (ASTS). J Clin Oncol (2013) 31(15_suppl):10514–4. doi: 10.1200/jco.2013.31.15_suppl.10514
175. Hoffmann-La Roche. A study of RO5045337 RG7112 in patients with advanced solid tumors. ClinicalTrials.gov Identifier: NCT00559533. First posted November 16, 2007. https://clinicaltrials.gov/ct2/show/NCT00559533
176. Hoffmann-La Roche. A study of RO5045337 in patients with solid tumors. ClinicalTrials.gov Identifier: NCT01164033. First posted July 16, 2010. https://clinicaltrials.gov/ct2/show/NCT01164033
177. Patnaik A, Tolcher A, Beeram M, Nemunaitis J, Weiss GJ, Bhalla K, et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother Pharmacol (2015) 76(3):587–95. doi: 10.1007/s00280-015-2830-8
178. Hoffmann-La Roche. A study to investigate the bioequivalence or relative bioavailability of three new idasanutlin tablet variants following oral administration in participants with solid tumors. ClinicalTrials.gov Identifier: NCT03362723. First posted December 5, 2017. https://clinicaltrials.gov/ct2/show/NCT03362723
179. Hoffmann-La Roche. A study to determine the excretion balance, pharmacokinetics, metabolism and absolute oral bioavailability of a single oral dose of 14C -labeled idasanutlin and an intravenous tracer dose of 13C -labeled idasanutlin in a single cohort of participants with solid tumors (Malignancies). ClinicalTrials.gov Identifier: NCT02828930. First posted July 12, 2016. https://clinicaltrials.gov/ct2/show/NCT02828930
180. Pápai Z, Chen LC, Costa Da D, Blotner S, Vazvaei F, Gleave M, et al. A single-center, open-label study investigating the excretion balance, pharmacokinetics, metabolism, and absolute bioavailability of a single oral dose of [(14)C]-labeled idasanutlin and an intravenous tracer dose of [(13)C]-labeled idasanutlin in a single cohort of patients with solid tumors. Cancer Chemother Pharmacol (2019) 84(1):93–103. doi: 10.1007/s00280-019-03851-0
181. Hoffmann-La Roche. A first-In-Human study of RO5503781 in participants with advanced malignancies except leukemia. ClinicalTrials.gov Identifier: NCT01462175. First posted October 31, 2011. https://clinicaltrials.gov/ct2/show/NCT01462175
182. Italiano A, Jr Miller WH, Blay JY, Gietema JA, Bang YJ, Mileshkin LR, et al. Phase I study of daily and weekly regimens of the orally administered MDM2 antagonist idasanutlin in patients with advanced tumors. Invest New Drugs (2021) 39(6):1587–97. doi: 10.1007/s10637-021-01141-2
183. Okuno S, Bailey H, Mahoney MR, Adkins D, Maples W, Fitch T, et al. A phase 2 study of temsirolimus (CCI-779) in patients with soft tissue sarcomas: a study of the Mayo phase 2 consortium (P2C). Cancer (2011) 117(15):3468–75. doi: 10.1002/cncr.25928
184. Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, G Z, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol (2012) 30(1):78–84. doi: 10.1200/JCO.2011.35.6329
185. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest (2008) 118(9):3065–74. doi: 10.1172/JCI34739
186. Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest (2008) 118(9):3051–64. doi: 10.1172/JCI34764
187. Eroglu Z, Tawbi HA, Hu J, Guan M, Frankel PH, Ruel NH, et al. A randomised phase II trial of selumetinib vs selumetinib plus temsirolimus for soft-tissue sarcomas. Br J Cancer (2015) 112(10):1644–51. doi: 10.1038/bjc.2015.126
188. Trucco MM, Meyer CF, Thornton KA, Shah P, Chen AR, Wilky BA, et al. A phase II study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcomas. Clin Sarcoma Res (2018) 8:21. doi: 10.1186/s13569-018-0107-9
189. Carabet LA, Rennie PS, Cherkasov A. Therapeutic inhibition of myc in cancer. structural bases and computer-aided drug discovery approaches. Int J Mol Sci (2018) 20(1). doi: 10.3390/ijms20010120
190. Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell (2019) 36(5):483–497.e15. doi: 10.1016/j.ccell.2019.10.001
191. Kato GJ, Barrett J, Villa-Garcia M, Dang CV. An amino-terminal c-myc domain required for neoplastic transformation activates transcription. Mol Cell Biol (1990) 10(11):5914–20. doi: 10.1128/mcb.10.11.5914-5920.1990
192. Andresen C, Helander S, Lemak A, Farès C, Csizmok V, Carlsson J, et al. Transient structure and dynamics in the disordered c-myc transactivation domain affect Bin1 binding. Nucleic Acids Res (2012) 40(13):6353–66. doi: 10.1093/nar/gks263
193. Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, et al. Structural basis of n-myc binding by aurora-a and its destabilization by kinase inhibitors. Proc Natl Acad Sci U.S.A. (2016) 113(48):13726–31. doi: 10.1073/pnas.1610626113
194. Bayliss R, Burgess SG, Leen E, Richards MW. A moving target: structure and disorder in pursuit of myc inhibitors. Biochem Soc Trans (2017) 45(3):709–17. doi: 10.1042/BST20160328
195. Dickson MA, Mahoney MR, Tap WD, D'Angelo SP, Keohan ML, Van Tine BA, et al. Phase II study of MLN8237 (Alisertib) in advanced/metastatic sarcoma. Ann Oncol (2016) 27(10):1855–60. doi: 10.1093/annonc/mdw281
196. Van Glabbeke M, Verweij J, Judson I, Nielsen OS. Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer (2002) 38(4):543–9. doi: 10.1016/S0959-8049(01)00398-7
197. Liao Y, Feng Y, Shen J, Hornicek FJ, Duan Z. The roles and therapeutic potential of cyclin-dependent kinases (CDKs) in sarcoma. Cancer Metastasis Rev (2016) 35(2):151–63. doi: 10.1007/s10555-015-9601-1
198. Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer (2001) 1(3):222–31. doi: 10.1038/35106065
199. Du Q, Guo X, Wang M, Li Y, Sun X, Li Q. The application and prospect of CDK4/6 inhibitors in malignant solid tumors. J Hematol Oncol (2020) 13(1):41. doi: 10.1186/s13045-020-00880-8
200. Binh MB, Sastre-Garau X, Guillou L, Pinieux G, Terrier P, Lagacé R, et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol (2005) 29(10):1340–7. doi: 10.1097/01.pas.0000170343.09562.39
201. Dickson MA, Schwartz GK, Keohan ML, D'Angelo SP, Gounder MM, Chi P, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: A phase 2 clinical trial. JAMA Oncol (2016) 2(7):937–40. doi: 10.1001/jamaoncol.2016.0264
202. Abdul Razak AR, Bauer S, Suarez C, Lin CC, Quek R, Hütter-Krönke ML, et al. Co-Targeting of MDM2 and CDK4/6 with siremadlin and ribociclib for the treatment of patients with well-differentiated or dedifferentiated liposarcoma: Results from a proof-of-Concept, phase ib study. Clin Cancer Res (2022) 28(6):1087–97. doi: 10.1158/1078-0432.CCR-21-1291
203. Judson I, Verweij J, Gelderblom H, Hartmann JT, Schöffski P, Blay JY, et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol (2014) 15(4):415–23. doi: 10.1016/S1470-2045(14)70063-4
204. von Mehren M, Kane JM, Bui MM, Choy E, Connelly M, Dry S, et al. NCCN guidelines insights: Soft tissue sarcoma, version 1.2021. J Natl Compr Canc Netw (2020) 18(12):1604–12. doi: 10.6004/jnccn.2020.0058
205. Jung J, Lee JS, Dickson MA, Schwartz GK, Cesne Le A, Varga A, et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun (2016) 7:12609. doi: 10.1038/ncomms12609
206. Chapeau EA, Gembarska A, Durand EY, Mandon E, Estadieu C, Romanet V, et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an arf(-/-) mouse model. Proc Natl Acad Sci U.S.A. (2017) 114(12):3151–6. doi: 10.1073/pnas.1620262114
207. Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature (2005) 436(7048):272–6. doi: 10.1038/nature03681
208. Graves B, Thompson T, Xia M, Janson C, Lukacs C, Deo D, et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc Natl Acad Sci U.S.A. (2012) 109(29):11788–93. doi: 10.1073/pnas.1203789109
209. ElSawy KM, Verma CS, Joseph TL, Lane DP, Twarock R, Caves LS. On the interaction mechanisms of a p53 peptide and nutlin with the MDM2 and MDMX proteins: a Brownian dynamics study. Cell Cycle (2013) 12(3):394–404. doi: 10.4161/cc.23511
210. Yu DH, Xu ZY, Mo S, Yuan L, Cheng XD, Qin JJ. Targeting MDMX for cancer therapy: Rationale, strategies, and challenges. Front Oncol (2020) 10:1389. doi: 10.3389/fonc.2020.01389
211. Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to nutlin-3. Cancer Res (2006) 66(6):3169–76. doi: 10.1158/0008-5472.CAN-05-3832
212. Iyer VV. A review of stapled peptides and small molecules to inhibit protein-protein interactions in cancer. Curr Med Chem (2016) 23(27):3025–43. doi: 10.2174/0929867323666160627103134
213. Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med (2018) 10(436). doi: 10.1126/scitranslmed.aao3003
214. Berberich A, Kessler T, Thomé CM, Pusch S, Hielscher T, Sahm F, et al. Targeting resistance against the MDM2 inhibitor RG7388 in glioblastoma cells by the MEK inhibitor trametinib. Clin Cancer Res (2019) 25(1):253–65. doi: 10.1158/1078-0432.CCR-18-1580
215. Deben C, Boullosa LF, Domen A, Wouters A, Cuypers B, Laukens K, et al. Characterization of acquired nutlin-3 resistant non-small cell lung cancer cells. Cancer Drug Resistance (2021) 4(1):233–43. doi: 10.20517/cdr.2020.91
216. Haronikova L, Bonczek O, Zatloukalova P, Kokas-Zavadil F, Kucerikova M, Coates PJ, et al. Resistance mechanisms to inhibitors of p53-MDM2 interactions in cancer therapy: can we overcome them? Cell Mol Biol Lett (2021) 26(1):53. doi: 10.1186/s11658-021-00293-6
217. Grigoreva T, Sagaidak A, Romanova A, Novikova D, Garabadzhiu A, Tribulovich V, et al. Establishment of drug-resistant cell lines under the treatment with chemicals acting through different mechanisms. Chem Biol Interact (2021) 344:109510. doi: 10.1016/j.cbi.2021.109510
218. Saiki AY, Caenepeel S, Yu D, Lofgren JA, Osgood T, Robertson R, et al. MDM2 antagonists synergize broadly and robustly with compounds targeting fundamental oncogenic signaling pathways. Oncotarget (2014) 5(8):2030–43. doi: 10.18632/oncotarget.1918
219. Laroche-Clary A, Chaire V, Algeo MP, Derieppe MA, Loarer FL, Italiano A, et al. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol (2017) 10(1):123. doi: 10.1186/s13045-017-0482-3
220. Sriraman A, Dickmanns A, Najafova Z, Johnsen SA, Dobbelstein M. CDK4 inhibition diminishes p53 activation by MDM2 antagonists. Cell Death Dis (2018) 9(9):918. doi: 10.1038/s41419-018-0968-0
221. Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol (2019) 20(4):199–210. doi: 10.1038/s41580-019-0110-x
222. Jung JH, Lee H, Zeng SX, Lu H. RBM10, a new regulator of p53. Cells (2020) 9(9). doi: 10.3390/cells9092107
223. Vilgelm AE, Saleh N, Shattuck-Brandt R, Riemenschneider K, Slesur L, Chen SC, et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci Transl Med (2019) 11(505). doi: 10.1126/scitranslmed.aav7171
Keywords: dedifferentiated liposarcoma, MDM2, p53, targeted therapy, clinical trial, small molecular inhibitor
Citation: Traweek RS, Cope BM, Roland CL, Keung EZ, Nassif EF and Erstad DJ (2022) Targeting the MDM2-p53 pathway in dedifferentiated liposarcoma. Front. Oncol. 12:1006959. doi: 10.3389/fonc.2022.1006959
Received: 29 July 2022; Accepted: 19 October 2022;
Published: 10 November 2022.
Edited by:
Jon Kim, University of Florida, United StatesReviewed by:
Min Lu, Icahn School of Medicine at Mount Sinai, United StatesChuanxi Zheng, Shenzhen University, China
Copyright © 2022 Traweek, Cope, Roland, Keung, Nassif and Erstad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derek J. Erstad, Derek.erstad@bcm.edu
†These authors share first authorship