 Rui He1
Rui He1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 23 November 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.764618
Colorectal cancer (CRC), a common malignant disease, has the second highest mortality rate among all cancer types. Due to the diversity and heterogeneity of CRC, few effective treatment strategies have been developed in recent years, except for surgical resection. As immunotherapy has become a revolutionary treatment after surgery, along with chemoradiotherapy and targeted therapy, numerous basic research studies and clinical trials have been conducted on CRC. Therefore, immune checkpoint inhibitor (ICI) therapy has become the main anti-CRC immunotherapy method used at present. With the rapid development of biotechnology and cell research, an increasing number of monotherapy or combination therapy strategies using ICIs for CRC have been designed in recent years. Methods to classify and review ICI strategies for different types of CRC to better guide treatment are continuously investigated. However, the identification of why the ICIs would be more effective in targeting particular subtypes of CRC such as high microsatellite instability (MSI-H) is more important because of the different immune backgrounds in patients. This review intends to classify different subtypes of CRC and summarizes the basic and clinical studies on ICIs for each subtype of CRC currently available. In addition, we also attempt to briefly discuss the progress in immunotherapy methods other than ICI therapy, such as chemoimmunotherapy strategy, chimeric antigen receptor-modified T (CAR-T) cells, or immunotherapy based on oncolytic viruses. Finally, we provide a perspective on the development of immunotherapy in the treatment of CRC and attempt to propose a new systematic classification of CRC based on immunological strategies, which may improve guidance for the selection of immunotherapy strategies for different subtypes of CRC in the future.
According to the latest global statistics, non-communicable diseases are still the leading cause of death worldwide, among which cancer is an important disease endangering human life. In 2018, the number of patients with a new cancer diagnosis reached more than 18 million, and 9.6 million patients died because of cancer, with continuous growth (1). As a life-threatening disease, colorectal cancer (CRC) was estimated to have the third highest incidence (6.1%) and the second highest mortality (9.4%) of all cancers. In America, in 2020, CRC remained a leading cause of cancer-related death, accounting for 40%–50% of all new diagnoses of breast, lung, and colorectal cancers (2, 3). Among the common sites of CRC, approximately 41% occur in the proximal colon, 22% in the distal colon, and 28% in the rectum (4, 5). Notably, 50% of patients will develop distant metastases known as metastatic CRC (mCRC), which has a high mortality rate. Therefore, new and effective treatment strategies for patients with CRC now need to be developed to reduce the mortality rate of CRC.
Immunotherapy has become the fourth largest cancer treatment program after surgery, chemoradiotherapy, and targeted therapy. The first clinical example of tumor immunotherapy dates to 1891, when Coley et al. accidentally observed a reduction in tumor volume by injecting Streptococcus into patients with inoperable osteosarcoma (6). Bacterial infection enhances the local immune response of tumor tissue, which induces activated immune cells such as T cells to destroy tumor cells, leading to the opening of a new chapter in cancer immunotherapy (6–8). In the past decade, immunotherapy using immune checkpoint inhibitors (ICIs) has attracted increasing attention due to its success in producing long-lasting responses to solid tumors such as melanoma and lung cancer (9). Since Le et al. found that patients with CRC and a deficient DNA mismatch repair (MMR) (dMMR) or high microsatellite instability (MSI) (MSI-H) benefit from ICI treatment in 2015 (10), ICIs such as programmed cell death 1/programmed cell death 1 ligand (PD-1/PD-L1) inhibitors and cytotoxic T lymphocyte (CTL) antigen 4 (CTLA4) inhibitors have been utilized to treat patients with CRC. However, although research suggests that different malignancies, such as melanoma, kidney cancer, bladder lung cancer, or Hodgkin’s disease, respond well to ICI immunotherapy, only a minor proportion of patients with mCRC benefit from it (10–13). With the rapid development of molecular biology and clinical trials concerning immunotherapy, more effective ICI strategies have been developed. In order to improve the effect of ICIs on CRC, more combination therapy strategies of ICIs and other drugs have become new research direction. In addition, immunotherapy methods other than ICIs, such as chimeric antigen receptor-modified T (CAR-T) cells or immunotherapy based on oncolytic viruses, have emerged rapidly in recent years. Cellular immunotherapy based on CAR-T cells has been successfully developed for the treatment of relapsed and refractory B-line lymphoblastic leukemia in recent years (14–16), which has encouraged the development of immunotherapy for CRC (17). Immunotherapy, a new and powerful antitumor therapy, would become an alternative treatment strategy for CRC patients.
In most CRC patients, tumor metastasis is always present. However, only patients with advanced CRC presenting with dMMR and MSI-H subtypes respond to ICIs (18). In contrast, the effect of immunotherapy on subtypes that are proficient in MMR (pMMR) and have microsatellite stability (MSS) and low MSI (MSI-L) has not been clearly determined. In patients with CRC, a high tumor mutational burden (TMB) has become a marker of immunotherapeutic responsiveness, while a lack of immune cell infiltration has been identified as a cause of tumor immune resistance (9, 19–21). Recently, immunotherapy strategies have become more diversified for patients with CRC (22). For CRC, which has multiple molecular subtypes, a more important goal is to review the optimal effects of different immunotherapies on different subtypes of CRC, which might guide the selection of clinical treatment strategies for patients with CRC. Therefore, we summarized the methods for classifying CRC in this review, and then we discussed different types of currently available ICI therapy strategies for each subtype of CRC. In addition, we discussed the progress in other immunotherapies for CRC in addition to ICI immunotherapy. Finally, we also provided a perspective on the development of immunotherapy for the treatment of CRC, and we attempted to propose a new systematic classification of CRC based on immunological strategies, which may guide the selection of immunotherapy strategies for different subtypes of CRC in the future (Figure 1).
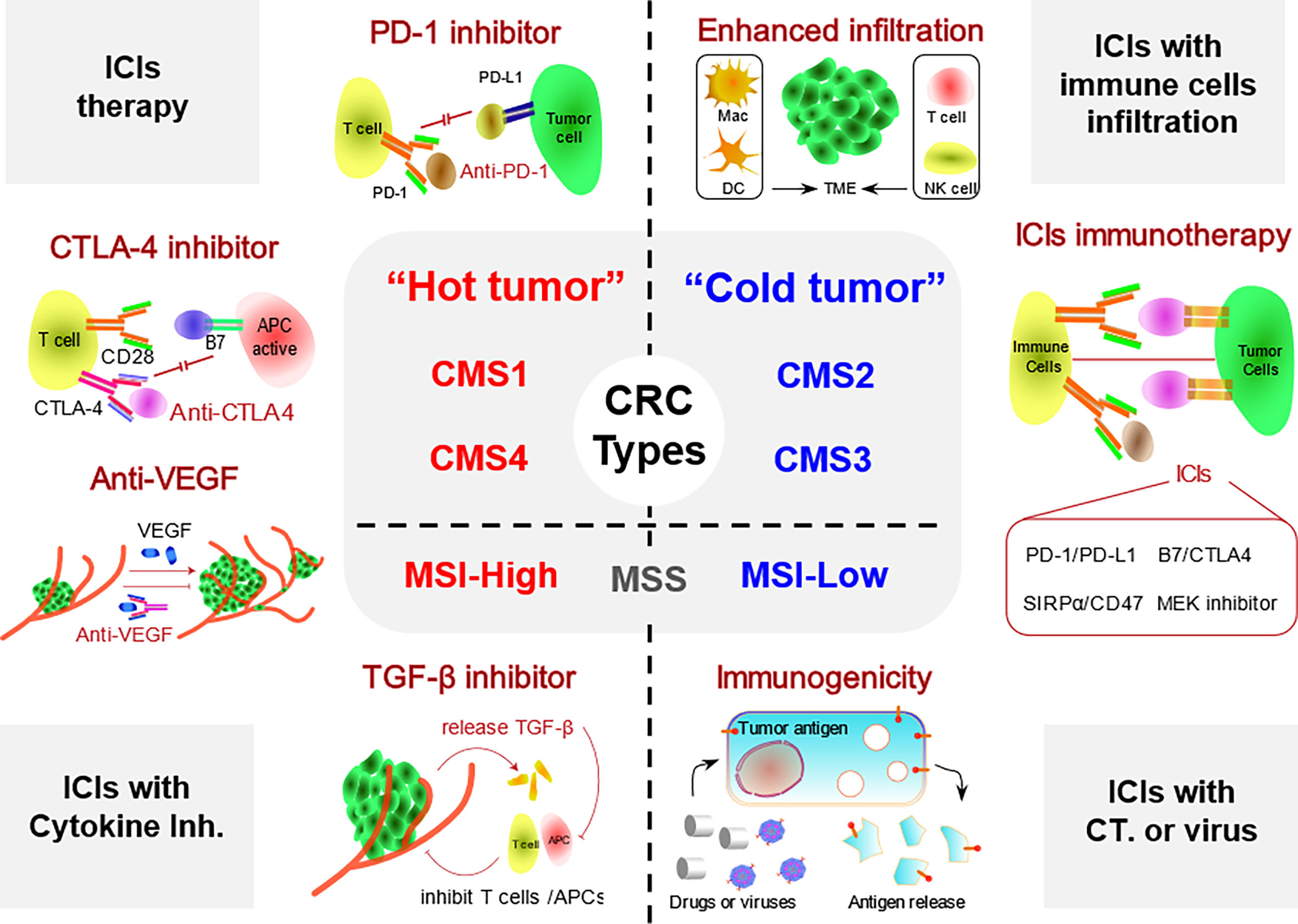
Figure 1 Different types of ICI-based immunotherapy strategies for patients with CRC. CRC is divided into hot and cold tumor subtypes. Hot CRC mainly includes the dMMR/MSI-H, CMS1, and CMS4 subtypes, while cold CRC includes the pMMR/MSI-L, CMS2, and CMS3 subtypes. TME, tumor microenvironment; Inh., inhibitor; CT, chemotherapy; ICI, immune checkpoint inhibitor; CRC, colorectal cancer; dMMR, deficient DNA mismatch repair; MSI-H, high microsatellite instability; pMMR, proficient mismatch repair.
The tumor microenvironment (TME), which is defined as tumor cells and the surrounding environment, includes tumor-related immune cells, blood vessels, cytokines, stroma, epidermal growth factor (EGF), transforming growth factor-beta (TGF-β), fibroblast growth factor (FGF), tumor necrosis factor-alpha (TNF-α), and other signaling molecules. The TME affects the occurrence and development of tumors (22–24). Among them, immune cells accurately monitor and remove tumor cells in vivo and play a crucial role in inhibiting tumor growth (25). However, the physical barrier, biochemical barrier, and physiological barrier produced by the TME induce a strong inhibitory effect on the immune system, resulting in the inability of immune cells to exert their antitumor activities and even promoting the occurrence of cancer (26). Therefore, the formation of an immunosuppressive TME is an important indicator of tumor deterioration (27, 28). Among the immune cells, T cells have a strong killing activity of tumor cells. However, abnormally high expression of immune checkpoint receptors such as PD-L1 in tumor cells inhibited the recognition and destruction of T cells. For reactivating T cells, the most mature method is to use ICIs to remove the immunosuppression effect of tumor cells and enable T cells to replay cytotoxicity. The strategy of immune regulation is expected to achieve the functional activation of the immune system in the TME without any external tools, only using the natural antitumor system of the organism, which is also the main advantage of immunotherapy (29).
As a type of cancer with high morbidity and mortality rates, more effective treatment strategies have not been developed for CRC, except for surgical resection or classical chemotherapy strategies such as FOLFOX. Therefore, the effective formulation of antitumor strategies for CRC is important to reduce mortality. The MMR/MSI system is the most important indicator of CRC classification and is used to develop treatment strategies. Microsatellites are tandem repeats of dozens of nucleotides, comprising one to six nucleotides as repeat units (30). MSI is a frameshift mutation of a microsatellite in tumor cells due to the insertion or deletion of repeated units (31, 32). The MMR system works to combat these errors by identifying and repairing DNA damage and correcting insertions, deletions, or mismatched bases that result from the error cycle that occurs during DNA replication (33–35). Regarding the MMR/MSI system, MMR is divided into dMMR and pMMR. Notably, dMMR is manifested as the absence of MMR proteins. When the MMR system is dysfunctional or mutated, these genetic errors are not corrected, thus allowing them to be permanently integrated into the tumor DNA, which is called MSI-H (31). However, MMR protein expression is normal in pMMR, which is mainly divided into MSI-L or MSS (36, 37). The dMMR/MSI-H subtype of CRC accounts for approximately 15% of all cases and 5% of mCRC cases (38–40). Due to the high mutation rate in dMMR/MSI-H, the tumor has high immunogenicity, enabling it to activate the antitumor effect of the immune system. Patients with dMMR/MSI-H are more responsive to ICI-based immunotherapy, which has sparked strong interest in immunotherapy for CRC. Therefore, the search for new and more effective immunotherapy strategies for the treatment of different CRC subtypes has become mainstream (Table 1).
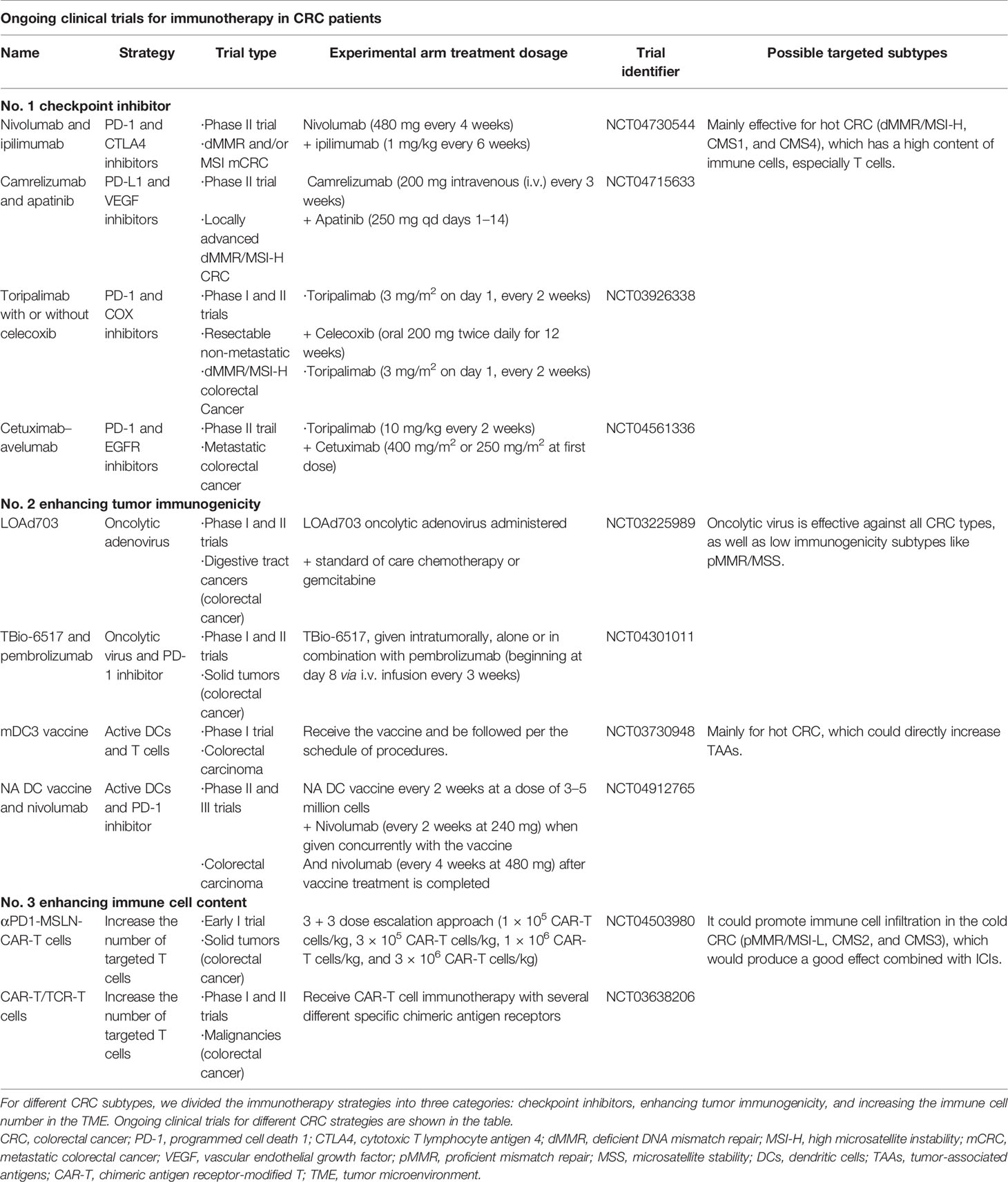
Table 1 Ongoing clinical trials for immunotherapy in patients with CRC.
Genes involved in MMR regulate DNA MMR, and a loss of expression causes the accumulation of mismatches during DNA replication, resulting in MSI. Approximately 15% of CRC cases are caused by the MSI pathway (41). According to the different states of the MMR/MSI system, CRC is divided into two subsets. The DNA MMR system relies on some key genes, such as MLH1, MSH2, MSH6, PMS2, or MSH3, which correct mismatched, misinserted, or deleted bases in DNA (42, 43). If the gene repair function is inactivated due to MMR protein defects, these errors in DNA synthesis may freely and permanently integrate into the cellular DNA, resulting in MSI of CRC. As an important marker of MMR protein deficiency, MSI stability has become another major indicator to guide treatment strategies for patients with CRC (44–46). According to the degree of instability in detection markers, MSI is divided into MSI-H, MSI-L, and MSS. dMMR is lacking MMR proteins, which is mainly manifested as MSI-H. pMMR shows normal expression of MMR proteins and includes MSI-L and MSS.
In recent years, as in-depth studies on immune markers have been conducted, a large number of studies have shown that a high TMB predicts the efficacy of ICIs in the treatment of CRC (7, 39, 47). In the MMR/MSI classification system of CRC, patients with the dMMR/MSI-H subtype have a higher TMB, which might be associated with higher expression of neoantigens on MHC-I molecules (48). From The Cancer Genome Atlas (TCGA)-CRC cohort of 276 patients with CRC published in 2012, 44 (16%) CRC specimens showed a hypermutant phenotype (defined as greater than 12 TMB mutations per 106 bases). Meanwhile, 37 patients presented the MSI-H subtype of CRC (49). In addition, POLE mutant CRC usually presents with the pMMR-MSS phenotype, which also belongs to the hypermutant phenotype. The TMB in MSI-H and MSS is higher than that in MSI-L. All these subtypes of CRC are highly sensitive to ICI treatment strategies (50), indicating that the MMR/MSI classification system is also helpful in guiding decisions on immunotherapy strategies for patients with CRC. In this section, we distinguished patients with three subsets of CRC based on the MMR/MSI system and summarized the different ICI strategies for three of the CRC subtypes.
As mentioned above, based on the MMR/MSI classification system, dMMR is mainly characterized by MSI-H, which is defined as instability at two or more sites that results in a large number of DNA replication errors, highlighted by genetic and accidental changes in the MMR gene (51). Through whole exome sequencing of the same number of CRC tumor cells, an average of 1,782 individual cell mutations were found in dMMR/MSI-H tumors, while only 73 individual cell mutations were detected in pMMR/MSS tumors (10). Since most of the dMMR/MSI-H CRC subtypes have a high TMB, ICIs and immunotherapy exert excellent therapeutic effects on patients with a high TMB (52). Therefore, ICI immunotherapy strategies have become the primary clinical treatment for patients with the dMMR/MSI-H subtype, including PD-1 inhibitor (pembrolizumab or nivolumab) monotherapy (19, 53), combination therapy with a PD-1 inhibitor (nivolumab), and CTLA4 inhibitor (ipilimumab) (54) and combination therapy with a PD-L1 inhibitor (atezolizumab) and anti-angiogenic vascular endothelial growth factor (VEGF) antibodies (bevacizumab) (55). For example, Overman enrolled 74 patients with the MSI-H subtype in a clinical study (NCT02060188) and treated them with nivolumab, a PD-1 inhibitor (56, 57). After treatment with nivolumab, approximately 68.9% of patients required longer than 12 weeks for disease control. In addition, eight patients (34.8%) experienced an immune response lasting longer than 12 months, indicating that nivolumab provides long-lasting response and disease control in patients with dMMR/MSI-H mCRC. More interestingly, in patients with the MSI-H subtype, the combination strategy incorporating ICIs appears to exert a greater antitumor effect. In 2018, Overman and colleagues further explored the therapeutic effect of the combination of the PD-1 inhibitor nivolumab and the CTLA4 inhibitor ipilimumab on MSI-H tumors (54). Among 119 patients, the combination inhibitor treatment achieved 80% effective tumor control at 12 weeks, and more than 94% of the immune response was sustained. All these data showed that the combination strategy of a PD-1 inhibitor and CTLA4 inhibitor results in a higher immune response rate and longer overall survival (OS) and progression-free survival (PFS) durations for patients with the MSI-H subtype, prompting the Food and Drug Administration (FDA) to approve nivolumab and ipilimumab as treatments for patients with the dMMR/MSI-H subtype (22). In conclusion, significant therapeutic efficacy has been documented for ICIs for the MSI-H subtype, providing a promising new treatment option for patients.
Recently, a randomized trial was conducted with the PD-L1 and CTLA4 inhibitors durvalumab and tremelimumab, respectively, for supportive pMMR/MSS CRC therapy (58). One hundred eighty patients with CRC were divided into the D+T group (durvalumab (D) and tremelimumab (T)) and BSC group (best supportive care). Although the objective response rate (ORR) and PFS were similar between the two groups, the OS was improved in the D+T group, indicating that the survival time was extended using durvalumab and tremelimumab immunotherapy. This study is the first to combine a PD-L1 inhibitor and CTLA4 inhibitor to prolong the survival of patients with pMMR/MSS advanced refractory CRC, raising new hopes for immunotherapy for pMMR/MSS mCRC. However, the clinical benefit is limited to a small subset of patients with the pMMR/MSS CRC subtype, accounting for approximately 4% of all patients with CRC (59, 60). For pMMR/MSS CRC subtype, ICI treatment cannot achieve the best therapeutic effect. When MSI is present, tumor cells release many tumor-associated antigens (TAAs) that are normally located inside tumor membranes, which are then taken up and presented by antigen-presenting cells (APCs) located in the tumor immune microenvironment, enhancing the antitumor ability of T cells (61, 62). Nevertheless, for the pMMR/MSS subtype of CRC, the DNA structures are too stable to release TAAs, thus blocking immune system activation or inducing a failure of activated immune cells to recognize tumor cells. Therefore, some studies have indicated that PD-1 inhibitors are less effective in patients with pMMR/MSS CRC subtypes (10, 63–66). New therapeutic strategies are urgently needed to enhance tumor immunity in patients with pMMR/MSS CRC. To date, many studies have indicated that chemotherapy, molecular targeted therapy, and radiotherapy cause immunogenic cell death (ICD) in cancer cells (67). After ICD, tumor cells are exposed to a large number of TAAs, and damage-related molecular patterns and proinflammatory cytokines are released and effectively promote immune cell infiltration and activate APCs (such as dendritic cells (DCs) and macrophages). Then, DCs and macrophages gradually mature and cross-present tumor antigens after their uptake, resulting in antigen-specific immune responses to tumors (68). The immunogenic-based treatment strategy covers all associated antigens of tumor cells and minimizes the incidence of immune escape, thereby mediating a systemic immune response through site-specific in situ cell death (69–72). These theories might provide new immunotherapy strategies for patients with pMMR/MSS CRC presenting a low immune response.
In addition, a recent study identified a key mechanism of T-cell failure and resistance to checkpoint blockade in patients with MSS CRC. The depletion of tumor-infiltrating T cells and a simultaneous increase in VEGF-α levels were observed in MSS colorectal tumors, which might explain why human T cells deplete the associated transcriptional programming in a virulence-dependent manner (73–75). After VEGF-α was inhibited, T cells restored antitumor viability. The combination of a PD-1 inhibitor and VEGF-α inhibitor effectively restores the antitumor function of T cells in MSS CRC, achieving a better therapeutic effect (76).
For patients with the pMMR/MSI-L CRC subtype, the microsatellite stability of the cancer cells ranges between MSI-H and MSS subtypes. Therefore, this subtype has few specific tumor characteristics. To date, commonly used immunotherapy strategies for the MSI-L subtype are divided into three categories: CTLA4 inhibitors (ipilimumab), PD-1 inhibitors (pembrolizumab or nivolumab), and PD-L1 inhibitors (atezolizumab or durvalumab), as well as their combinations. Therefore, the combination of pembrolizumab and nivolumab and the combination of nivolumab and ipilimumab have been approved for the treatment of CRC by the FDA (77). ICIs are not absolutely effective against the MSI-L subtype. In a study published in 2015, Le et al. observed that pembrolizumab treatment did not exert favorable immune-mediated antitumor effects on patients with PMMR MSI-L tumors (10). In addition, in the study by Overman, 142 patients with pMMR/MSI-L had a limited response to immunotherapy. Only one of 20 patients exhibited an immune-mediated antitumor response to the combination of PD-1 and CTLA4 antibodies (54). In recent years, with the discovery of new ICI combination strategies, substantial progress has been achieved in improving the efficacy of combination immunotherapy for patients with this type of tumor. According to the study by Liu published in 2015, the combination of a PD-L1 inhibitor and MEK inhibitor showed significant efficacy in patients with pMMR/MSI-L CRC (78). The RAS–MAPK pathway is the intersection or the last common pathway of transmembrane transduction of growth signals by multiple membrane receptors. Activation is related to the reduction of tumor infiltration by T cells and directly promotes the proliferation of tumor cells. MEK is the downstream effector of the RAS–MAPK pathway. Mekinist, a MEK inhibitor, increases the tumor infiltration of CD4+ T lymphocytes but does not affect CD8+ T cells. More importantly, in murine models of KRAS-mutated CT26 colorectal tumors, when the two drugs were used together, more potent and long-lasting antitumor activity was observed than in mice treated with either of the two drugs alone (18, 78, 79).
Endocellular peptides are processed and expressed on major histocompatibility complex class I (MHC I) molecules on the surface of almost all human cells, including cancer cells. This type of peptide is specifically recognized by T-cell receptors (TCRs) (18). The response of T cells is modulated by a series of coinhibitory or costimulatory signals. Among them, the membrane-binding ligands CD80 and CD86 of the B7 family bind to the costimulatory protein CD28, especially in activated T cells, resulting in an interaction with CTLA4 and the inhibition of T-cell activity (80). Similarly, membrane-bound PD-L1 and programmed cell death 2 ligand (PD-L2) bind to PD-1 to further incapacitate T cells and induce apoptosis (81–83). When these inhibitor signals are antagonized by the corresponding antibodies, T cells are activated and produce an antitumor effect. However, the T-cell content appears to be deficient in the MSI-L subtype, which may be the root cause of the poor response of this type to immunotherapy (56). In addition, the combination of a PD-1 inhibitor and other modulators of immune checkpoints, such as CTLA4, may benefit a small subset of patients with pMMR/MSI-L tumors. This benefit might be associated with the low microsatellite instability of the tumor cells themselves. Due to its poor specificity, this subtype displays low sensitivity to ICIs. Currently, few studies have been conducted on immunotherapy for this type of CRC, and more effective strategies should be designed that mainly focus on approach to increase the infiltration of immune cells into pMMR/MSI-L tumors.
As a highly heterogeneous malignant tumor disease, the biological behaviors of any two different CRC lesions vary widely in terms of the genetics and epigenetics of different lesions (84). Therefore, a method that can classify CRC according to tumor heterogeneity at the molecular biological level in the current era of precision treatment development is urgently needed. Consensus molecular subgroups (CMS) are a consensus classification based on cancer gene expression proposed by Guinney (85). Due to the extensive chromosomal alterations and dMMRs in CRC, genetic heterogeneity exists among different CRC cells. Based on pathology and molecular biological data from 3,000 patients with CRC, Guinney classified patients with CRC into four subtypes: CMS1, microsatellite unstable immunotype (14%), which is characterized by high MSI-H mutations and exhibits both BRAF mutations and strong immune cell infiltration; CMS2, the most common type (37%), which is characterized by activation of the WNT and MYC pathways and chromosomal instability; CMS3, the metabolic type (13%), which is mainly characterized by mutations in KRAS, mixed MSI status, and abnormal metabolic pathways; and CMS4, a mesenchymal subtype (3%), is characterized by activation of transforming factor TGF-β and enhanced angiogenesis, interstitial infiltration, and inflammatory infiltration (20, 85, 86). Among the four subtypes, the CMS1 and CMS4 subtypes are characterized by more extensive lymphocyte infiltration and a higher distribution of inflammatory cytokines around the tumor, while the CMS2 and CMS3 subtypes exhibit almost no lymphocyte or inflammatory cell infiltration. However, in the TME, the number of infiltrated immune cells directly modulates the effect of ICIs. As a result, the four subtypes of CRC based on the CMS system were artificially divided into two types, hot CRC and cold CRC, according to the presence of lymphocyte infiltration and the inflammatory environment around the tumor (39).
The TME of hot CRC contains many lymphocytes and inflammatory infiltrates, which mainly include the CMS1 and CMS4 subtypes. As the main antitumor cells, T cells are abundant in hot tumors and are more easily activated, and patients with these subtypes are more likely to benefit from immunotherapy (86). Although a large number of immune cells are present around both the CMS1 and CMS4 subtypes, the CMS4 mesenchymal subtype seems to be more prone to an adverse inflammatory immunophenotype characterized by the activation of transforming factor TGF-β and enhanced tumor angiogenesis, tumor growth, and metastasis. The malignant inflammatory environment will block the antitumor effect of immune cells, resulting in immunosuppression. Therefore, different immunotherapy strategies for the two subtypes of hot tumors are expected to obtain the maximum therapeutic benefits from personalized immunotherapy.
CMS1, also known as the MSI-like subtype, is the main potential beneficiary of immunotherapy for CRC. In the TME of the CMS1 subtype, the infiltration of a large number of immune cells, such as T cells, and a high BRAF gene mutation burden increase the effectiveness of immunotherapy. In 2016, Becht et al. showed that when a large number of invasive T cells, especially cytotoxic CD8+ T cells, accumulate in the TME, patients experience longer PFS and OS (21, 87). The authors suggested that the effective release of TAAs by CRC tumor cells induces a locally adaptive immune response (88). Meanwhile, they also detected large numbers of infiltrating T and B cells in tumors of the CMS1 subtype, which produced a strong guarantee for treatment strategies of ICIs such as PD-1/PD-L1 inhibitors. Tumor cells of the CMS1 subtype overexpress CXCL9, CXCL10 (specific chemokines that recruit T cells), and CXCL13 (protein that recruits B cells) and release IFNG and IL-15 (89). These characteristics promote the recruitment and activation of immune cells such as APCs or T cells, which are closely associated with a good prognosis in patients with CMS1 (87, 89, 90). In addition, some studies found that PD-1 is also expressed at high levels on the surface of CMS1 tumors. Therefore, for CMS1 tumors with a high level of T-cell infiltration, the use of ICIs such as PD-1 inhibitors might effectively assist infiltrating T cells in escaping the immune suppression of tumor cells, thus activating an effective antitumor immune response (13, 91). In general, the CMS1 subtype of CRC with a high level of immune cell infiltration has a better immune response and immunotherapy with ICI exerting a better antitumor effect (92).
CMS4 is the second largest subtype, accounting for approximately 23% of the total number of CRC cases (85). The main manifestations are the activation of transforming factor TGF-β, enhanced angiogenesis, interstitial infiltration, and inflammatory infiltration. However, unlike CMS1, CMS4 has an adverse inflammatory immunophenotype, which leads to a poor immune microenvironment, and active T cells are unable to kill tumor cells. The antitumor response of potential immune cells in the TME of CMS4 is potentially blocked by adverse inflammatory infiltration in the stroma, thus inhibiting the immune response to cancer cells. Therefore, little therapeutic strategies by using ICIs were studied in CMS4 subtype. According to the underlying mechanism of immunosuppression, more newly approaches were explored to reactivate antitumor immunity with the aim of determining the main mechanism of immunosuppression in patients with CMS4, described as follows: 1) by changing the immunosuppressive environment of the TME, immune cells are remodeled, and the antitumor functions of macrophages, DCs, and T cells are reactivated to transform the TME of CMS4 into a hot tumor-like subtype (93). 2) Significant infiltration of fibroblasts and innate immune cells is observed in CMS4, and the increase in TGF-β signaling exerts a significant inhibitory effect on immune cells. Therefore, the use of selective TGF-β inhibitors in combination with ICIs might be useful for immunotherapy in patients with CMS4 (94, 95). 3) CMS4 promotes angiogenesis and increased expression of VEGF-related factors, such as FGF, in the microenvironment, which might promote tumor growth. Among them, the proangiogenic molecule VEGF has been shown to play an important role in promoting tumor development in the immunosuppressive microenvironment. Methods to eliminate VEGF or reduce its expression and release have become another research direction. Currently, many clinical studies are investigating the efficacy of VEGF inhibitors for CRC. For example, many clinical trials have attempted to use bevacizumab (VEGF inhibitor) in combination with classic chemotherapy regimens, such as FOLFOX and FOLFIRI (NCT03635021 and NCT02339116), which exert a better antitumor effect. In a phase III clinical trial published in 2020, the authors reported a significantly longer PFS in the combination treatment group than in the control group (96). The results indicated that initial treatment with FOLFOXIRI plus bevacizumab followed by the reintroduction of the same regimen after disease progression seems to be a preferable therapeutic strategy and has an acceptable safety profile. However, the real function of the combination strategy also must be confirmed in appropriate, larger randomized clinical trials.
CMS2 and CMS3 subtypes are the main representative types of cold CRC tumors. The CMS2 subtype is the most common subtype, accounting for approximately 37% of all CRC cases (85). The subtype is mainly gene-specific, showing activation of WNT and MYC pathways and chromosomal instability. CMS3 mainly presents as an epithelial neoplasm. This subtype is characterized by KRAS gene mutations, a mixed MSI status and abnormal metabolic pathways. Both of these types exhibit gene-specific expression and methylation abnormalities. It has been explored that ICIs drugs alone have poor efficacy, which is related to the immunosuppressive nature of cold CRC. Currently, a variety of immunotherapeutic strategies have been developed, such as oncolytic virus, cytokine therapy, CAR-T therapy, and passive immunotherapy against TAAs (97), which have contributed to the immune-activated antitumor therapy of cold tumors.
The main characteristic of the CMS2 and CMS3 subtypes is the lack of tumor immunogenicity in the TME (98). The immunosuppression mechanism is different from that of hot tumors. Therefore, methods to activate the tumor cells of CMS2 and CMS3 to release TAAs are the starting point of immunotherapy for cold tumors. ICD is mediated by the release of TAAs when tumor cells undergo apoptosis, which stimulates long-lasting antitumor immune effects in the body (99). ICD of tumor cells substantially improves the low immunogenic microenvironment of cold tumors and promotes immune activation. Chemotherapy is one of the main methods to induce ICD in tumor cells. Some anthracyclines and oxaliplatin not only induce tumor cell apoptosis but also induce ICD (67). When TAAs are released, APCs such as DCs and macrophages are activated, along with the activation of T cells (100, 101). In addition, the efficacy of chemotherapy combined with ICIs becomes an important therapeutic strategy for cold CRC and has been well established. In 2017, chemotherapy combined with pembrolizumab (Keytruda) was approved by the FDA as a first-line treatment for metastatic non-small cell lung cancer (NSCLC) (102, 103). The effective rate of Keytruda combined with chemotherapy was 55%, while the effective rate of chemotherapy alone was only 29%. Combination therapy reduced the risk of disease progression. Since then, the FDA approved Keytruda in combination with pemetrexed and platinum-based chemotherapy as a first-line treatment for metastatic non-squamous NSCLC in patients without EGFR and ALK cancer genetic variants in August 2018 and approved Keytruda in combination with standard chemotherapy (carboplatin and paclitaxel/albumin paclitaxel) as a first-line treatment for squamous NSCLC in October 2018.
In addition, lower immune cell infiltration is another major factor contributing to a poor immune response (104), which is also a reason for ineffective ICIs. The low immune cell infiltration of cold tumors results in the insufficient effect of the activated immune system on eradicating the tumor tissue. Therefore, approaches that promote the enrichment of immune cells and activate the strong and lasting immune antitumor response have become the main challenge to overcome in immunotherapy for cold CRC. In a study presented at the ASCO-SITC Symposium on Clinical Immuno-Oncology in 2020, Shen and colleagues found that the methylation and expression levels of ARHGAP9, TBX21 (T-bet), and LAG3 genes were associated with the infiltration of CTLs (105). In CMS2 tumor cells with low T-cell infiltration, the expression of ARHGAP9 and T-bet genes was downregulated, and the methylation level was low. The expression of ARHGAP9, T-bet, FML1, HLA-DPB1, and STX11 genes in the CMS3 subtype was also downregulated, and the methylation level was low (105). However, in the CMS1 subtype with high infiltration of immune cells, the expression level and methylation level of T-bet gene were higher. Therefore, the expression levels of these genes differ between CMS subtypes, and the methylation level is related to the extent of CTL infiltration. T-bet gene was the only gene that was strongly correlated with CTL infiltration, which might directly reflect the degree of T-cell infiltration in patients with CRC. Therefore, the T-bet protein is likely to be a key regulator of T-cell infiltration in CRC (106). An increase in the methylation level of T-bet gene in the CMS2 and CMS3 subtypes and upregulation of the expression level of T-bet gene have become effective strategies for cold tumor immunotherapy.
ICIs has always been the most mature and effective immunotherapy for CRC. However, it appears to be limited for CRC subtypes with low immunogenicity or low immune cell infiltration. Therefore, immunotherapy by using ICIs alone is not effective in some specific CRC subtypes. Chemoimmunotherapy has become an important therapeutic strategy in the treatment of CRC. CAVE-Colon study has been proposed in recent years (NCT04561336). The researcher found that the combination strategy of cetuximab, an EGFR-targeted drug, and avelumab, an ICI drug, would significantly improve survival of CRC patients. Cetuximab improved the T-cell activity by increasing immune cell invasion and inducing APC maturation, while ICIs eliminated the tumor immunosuppressive effect on T cells.
In addition, in order to further improve the survival of CRC patients treated by FOLFOX or FOLFIRI strategies, a new GOLFIG was developed (107, 108). This kind of strategy combined low-dose recombinant interleukin-2 (rIL-2) and granulocyte macrophage colony-stimulating factor (GM-CSF) based on FOLFOX and gemcitabine, a chemotherapy drug. In the GOLFLG strategy, multiple chemotherapy drugs can successfully destroy tumor cells in large quantities, while rIL-2 and GM-CSF would promote the activation of APCs, then activate CTLs, and then accurately destroy the remaining tumor cells (109, 110). Correale et al. firstly explored the effect of FOLFIG trials. They found the remission rate (RR) and disease control rate (DCR) of patients with mCRC were higher than those of FOLFOX strategy, indicating significant immune response and antitumor activities (111). In addition, PFS was significantly lower in patients treated with GOLFIG than with FOLFOX, indicating GOLFIG could effectively reduce the recurrence rate (112, 113). Chemoimmunotherapy has broadened the scope of application of ICIs and maximized the antitumor effect.
Oncolytic virus immunotherapy is a promising approach for the treatment of solid tumors and has the ability to selectively replicate the tumor, achieve ideal immunogenicity, and deliver foreign genes to the tumor in a targeted manner (114, 115). In 1991, Martuza et al. showed that transgenic HSV was an effective treatment for glioblastoma (116). Since then, oncolytic virus therapy based on HSV has been developed. The antitumor strategy of oncolytic viruses is mainly mediated by the genetic modification of some of the less virulent viruses existing in nature. These approaches use the inactivation or defect of tumor suppressor genes in target cells to produce therapeutic oncolytic viruses with a specific recognition ability, which selectively infect tumor cells, then replicate in large numbers, and eventually destroy tumor cells. Meanwhile, damaged tumor cells release a large number of TAAs, and thus the immunogenic death of tumor cells is induced to attract more immune cells and clear the tumor tissue (116). In recent decades, oncolytic virus therapy has attracted increasing attention and achieved great progress in related research. The oncolytic vaccinia virus (VV) treatment strategy has been shown to transform cold tumors of the CRC type into hot tumors by promoting the infiltration of immune cells, resulting in effective antitumor immunity (117, 118). However, the therapeutic potential of oncolytic viruses is also affected by the immunosuppression of immune cells by tumors, such as immune checkpoints. According to the study published by Wei, compared with oncolytic VV therapy alone, the strategy of VV combined with monoclonal antibody against T-cell immunoglobulin and ITIM domain (TIGIT) exerts better effects on reducing the tumor burden and prolonging survival (119). They found that the number of CD8+ T cells increased and the microvascular density decreased in the TME of CRC using the VV-ATIGIT strategy for immunohistochemistry, which proved that the combination strategy significantly inhibited tumor growth in mice with colon cancer. In conclusion, oncolytic viruses combined with ICIs may be a new direction for the treatment of CRC (Figure 2) (119).
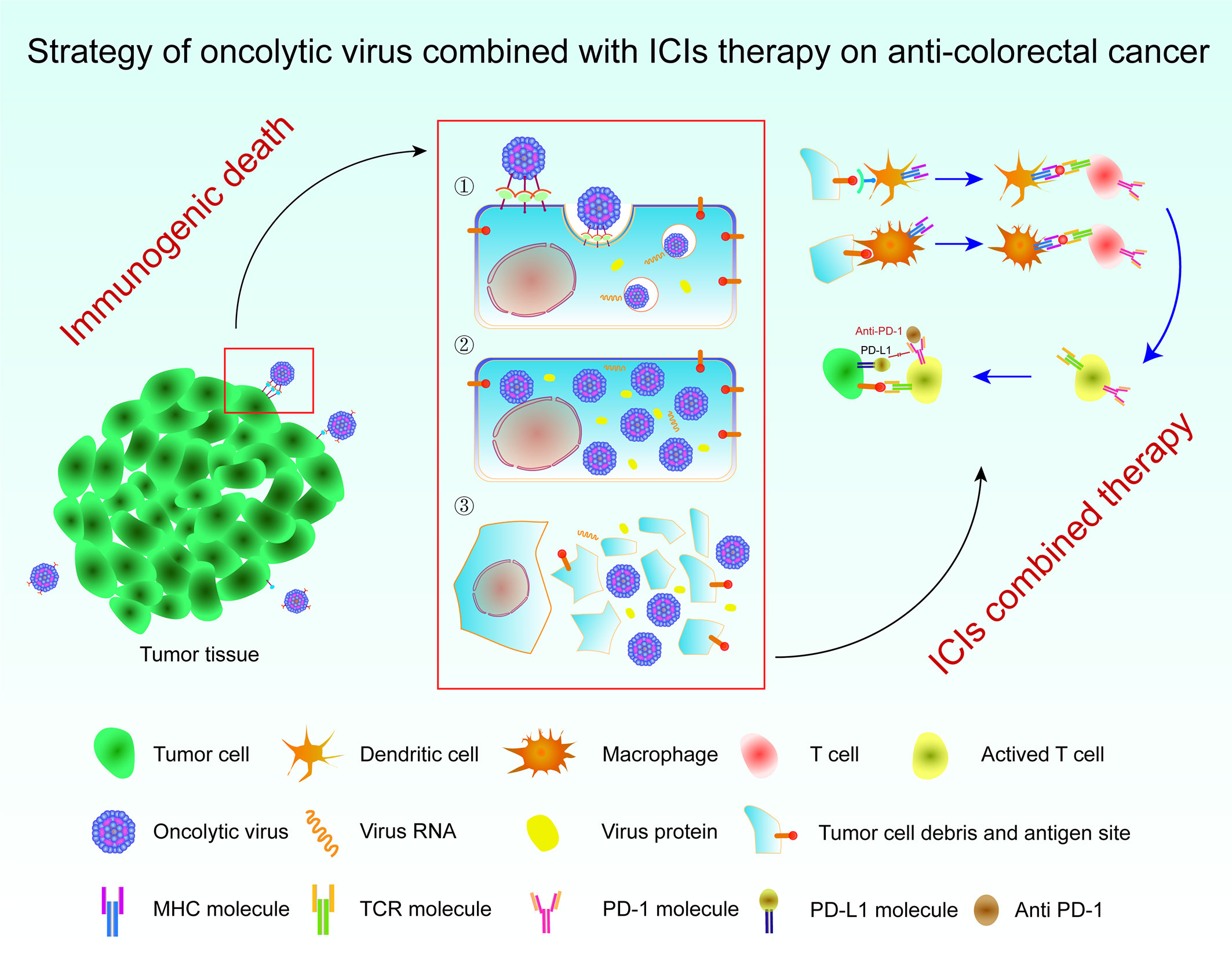
Figure 2 Strategy of an oncolytic virus combined with ICI therapy for colorectal cancer. Immunogenic cell death occurs in tumor cells upon the action of oncolytic viruses, which release TAAs to activate APCs and then promote T cells to attack tumor cells. The combination of an oncolytic virus with ICIs removes the inhibition of T cells by tumor cells through the immune checkpoint. The tumor tissue is then removed to the maximum extent possible. ICI, immune checkpoint inhibitor; TAAs, tumor-associated antigens; APCs, antigen-presenting cells.
For CMS2 and CMS3 CRC subtypes, the low level of immune cell infiltration is the main factor contributing to a poor immune response. The emergence of CAR-T cell therapy has become the main and precise treatment strategy to solve this difficulty. The CAR-T strategy is a classic adoptive cellular immunotherapy that kills tumor cells by injecting genetically modified and amplified immune effector T cells (120). CAR-T immunotherapy has many advantages, such as strong specificity, strong targeting, a high fatality rate, few side effects, and no drug resistance (119). For some CRC subtypes with low immune cell infiltration, CAR-T therapy with high efficiency and specific targeting of tumors is undoubtedly an appropriate immunotherapy strategy. Carcinoembryonic antigen (CEA) is a sensitive biomarker for gastrointestinal tumors that is widely expressed in CRC tissues and serum and not expressed in many other normal tissues or organisms (121), making it an effective target for CAR-T therapy. In 2017, Zhang et al. conducted a phase I clinical trial of systemic intravenous CAR-T therapy for CRC with high positive CEA expression (NCT02349724). In the trial, five stepped-up CAR-T cell doses were administered to 10 patients with CRC, and no serious adverse events associated with CAR-T therapy were observed during follow-up. Seven of the 10 patients had progressed at the time of previous treatment but were stable after CAR-T treatment. Imaging analysis showed that tumors shrank after treatment in two patients. After long-term observation, the serum CEA level decreased significantly in most patients. In addition, CAR-T persisted in the peripheral blood of patients who received high doses of CAR-T. More importantly, proliferation of CAR-T was observed in the trial, especially in patients following the second CAR-T treatment. CEA-targeted CAR-T therapy was well tolerated by patients with CEA-positive CRC, even at high doses. Some efficacy was observed in the majority of patients who received the therapy (17).
In 2019, Ying et al. loaded the CD19 truncated (CD19T) gene in an oncolytic virus to increase the expression of the CD19 antigen on the tumor cell membrane surface by taking advantage of the transgenic transmission potential of the oncolytic virus (122). Meanwhile, they combined CAR-T immunotherapy with the chimeric CD19 gene to enhance the specific targeting effect of CAR-T. The combination of the oncolytic virus and truncated CD19 (OV-CD19T) specifically replicated and expressed CD19T antigen in different solid tumor cells. The combination of OV-CD19T and CD19 CAR-T therapy is a safe and effective treatment for multiple solid tumors both in vitro and in vivo, without causing neurotoxicity or severe cytokine release syndrome (CRS), suggesting long-lasting tumor killing effects (122). In the next year, Priceman further explored a novel and effective combination immunotherapy based on the combination method (123). They used a genetically engineered oncolytic virus to overexpress the CD19 antigen on the surface of tumor cell membranes, thereby enhancing the antitumor response of CD19 CAR-T therapy to solid tumors. CD19 has long been an ideal target for CAR-T against hematological malignancies due to its highly restricted expression on B cells (124–126). CD19-mediated CAR-T therapy has been approved by the FDA for B-cell malignancies. However, in solid tumors, the coexpression of most tumor antigens in normal tissues limits the potential of CAR-T in solid tumors. The strategy of introducing specific targets into solid tumors to promote CAR-T immune activation might advance the suitability of CAR-T therapy for solid tumors.
The biological process of ideal immune-activated antitumor therapy is that ICD tumor cells release TAAs, damage-associated molecular patterns, and proinflammatory cytokines, which function as an alarm to effectively promote immune cell infiltration and activation. APCs promote cross-presentation of tumor antigens, triggering a wide range of tumor antigen-specific immune responses (127). T cells, the terminal killer cells of immune antitumor cells, are of decisive significance for immune efficacy. However, cold CRC tumors have low immunogenicity or less T-cell infiltration in the TME, which may be due to the presence of initiation defects or the absence of high-affinity T cells. Therapeutic cancer vaccines with TAAs or tumor-specific antigens (TSAs) might directly stimulate the immune system and induce an immune response by delivering these antigens directly to professional APCs, thus activating CD4+ and CD8+ T cells to kill tumors (128, 129). Many cancer vaccines have been developed, including cell vaccines, nucleic acid vaccines, protein peptide vaccines, and genetically engineered vaccines (130–134). Adoptive cell immunotherapy (ATT) is a major cellular vaccine strategy and a novel approach to TSA presentation. When the immune cell antigen load is low or the relevant tumor antigen load is not available, ATT might achieve better tumor targeting and killing effects by directly overexpressing tumor cell antigen antibodies in immune cells (135). For example, CD8+ T cells targeting k-ras or TP53 mutations have been identified (136, 137). In addition, recent studies have shown that circulating PD-1-positive lymphocytes recognize neoantigens in human digestive tract tumors (32, 138).
Thymidylate synthase poly-epitope-peptide (TSPP) is an anticancer poly-epitope peptide vaccine, which can promote the promote cross-presentation of tumor antigens through APCs and trigger a highly specific immune response with multi-antigen specificity (139). TSPP has shown good antitumor activity in preclinical studies. However, the antitumor effect of TSPP is significantly influenced by the TME such as inflammation and immune response. K-ras and IL17/A have been found to be important factors to predict the resistance of TSPP. Patients with k-ras mutation have no statistically significant inflammatory markers and also have poorer immune response (139). IL17/A, as an inflammatory cytokine independent of k-ras status, could amplify and enhance the cytotoxic effects of CTLs at CRC tumor sites (140). By predicting k-ras mutation and IL-17/A level, the immunoreactivity and antitumor efficacy of immunotherapy like TSPP vaccine against CRC could be evaluated.
In addition, the immunotherapy response is usually associated with the TMB, and patients with dMMR/MSI-H have higher genetic mutation characteristics (52). Therefore, vaccination against the associated antigens of specific genetic mutations has proven effective against this type. In 2017, Maletzki et al. constructed a mouse model of dMMR induced by knockout of MLH1. After the treatment dMMR cancer vaccine was administered to the mice, the OS time was prolonged, and the tumor mutation load was reduced, indicating that the specific vaccine is a viable option for the treatment of the dMMR subtype (141, 142). Similarly, human clinical trials of therapeutic cancer vaccines have shown good results based on the MSI status (143). The main question to be answered in order to promote increased immune efficacy is whether the combined ICI immunotherapy and vaccine strategy is more effective than ICIs alone in dMMR/MSI-H CRC or whether it is capable of causing a response in pMMR/MSI-L CRC that does not respond to ICIs.
The TME is complex and changeable, and the immune response is affected by many factors. Different types of CRC are heterogeneous. Even for the same solid tumor, a large difference in gene or protein expression may be observed. Therefore, extensive clearance of tumor cells is difficult to achieve with a single immunotherapy (144). Immune adjuvant, a non-specific immunoproliferative agent, is an adjuvant that effectively activates the immune response to antigens or changes the type of immune response, which is important for enhancing the efficacy of immunotherapy for CRC. At present, immune adjuvants are mainly divided into biological and abiotic adjuvants, and the latter exerts a better effect on immune sensitization. In a phase I clinical trial, Hou used Mn2+ combined with an anti-PD-1 antibody to safely enhance the antitumor efficacy of treatment for advanced mCRC (145). A lack of Mn2+ promotes tumor growth. When B16F10 melanoma cells were implanted in Mn2+-deficient mice, at least 2.5 * 103 cells were needed. However, seven times more tumor cells were needed in normal mice to achieve 100% tumor incidence. Moreover, the tumor volume and weight of Mn2+-deficient mice were significantly increased. Tumor infiltration of CD4+ and CD8+ T cells was significantly decreased (146, 147). After the Mn2+ injection, tumor growth was inhibited. At the same time, Mn2+ injection on one side of the tumor also inhibited tumor growth on the other side, suggesting a systemic antitumor response. Moreover, Mn2+ promoted DC maturation and enhanced antitumor effects through the cGAS–STING pathway, which also exerts an antitumor effect on patients with immune-tolerant tumors. From 2018 to 2019, Lv et al. initiated a phase I clinical trial on the effect of Mn2+ on advanced mCRC. In this study, a MnCl2 solution was administered intranasally or inhaled. The authors documented a clinical RR of 45.5% and a tumor control rate of 90.9% after treatment with the MnCl2 solution (148). More importantly, five patients who had previously failed to respond to anti-PD-1 antibodies or chemotherapy in combination with radiation showed good DCRs during the treatment, suggesting that Mn2+ restored immunotherapeutic efficacy in immune treatment-resistant patients. As an inorganic adjuvant, Mn2+ has undoubtedly become the most important immune sensitizer for immunotherapy of CRC. It potentially activates the immune response of patients with immune treatment-resistant CRC, providing a new and more effective therapeutic strategy (149).
As an effective treatment strategy following surgery, radiotherapy, chemotherapy, and targeted therapy, ICI immunotherapy strategies displays powerful antitumor efficacy and strong therapeutic potential in the treatment of CRC tumors. However, as an emerging treatment, many challenges remain in terms of safety and efficacy. For ICI treatment strategies, although good therapeutic effect has been shown in part of CRC subtypes, it is not ideal for some other CRC subtypes such as pMMR/MSS and dMMR/MSI-L. More new ICI combination strategies need to be developed by exploring their specific mechanisms. On the other hand, due to the heterogeneity of solid tumors and the external microenvironment, the efficacy of immunotherapy for solid tumors is not as expected (150). Low clinical target response rates and the risk of autoimmune diseases remain major limitations. An overactive immune system during treatment might cause severe adverse reactions in patients with CRC (151). In recent years, more combination therapies for ICIs have been developed, which solved some limitations of ICIs only for CRC therapy. For example, ICIs combined with chemotherapy could improve the immunogenicity of CRC subtypes. ICIs combined with CAR-T cell therapy could improve T-cell enrichment at the tumor site, exerting synergistic effects. However, although more CRC immunotherapy strategies are being developed, there is still no detailed classification targeted at the characteristics of CRC immune environment. A more meaningful approach would be to formulate the classification based on the different immune response characteristics of CRC. First, for the subtypes with high immune cell infiltration (dMMR/MSI-H, CMS1, and CMS4), treatment strategies for CRC could be modified according to the main molecular mechanisms of immunosuppression between tumor cells and immune cells, such as immune checkpoints. For example, patients with dMMR/MSI-H CRC have benefited the most from ICI immunotherapy. In addition, for patients with CMS4, TGF-β is a major signal that enhances tumor metastasis (152). Preclinical models suggest that TGF-β suppression resets immune rejection signals and may restore susceptibility to checkpoint suppression, suggesting that it represents a good target for future CRC immunotherapy research. Second, for low immune cell-infiltrating tumor subtypes (pMMR/MSS, dMMR/MSI-L, CMS2, and CMS3), improving the immunogenicity of the tumor while increasing the targeting of the immune system and improving immune cell invasion have become the most important immunotherapy strategies instead of using ICIs. Promising treatment options for patients with pMMR/MSS CRC include chemotherapy, targeted therapy, oncolytic virus, local ablation, and the combination of TLR agonists with checkpoint inhibitors. Although treatment strategies have shown good efficacy in early clinical or preclinical studies, they have not yet been used in clinical trials. In conclusion, as the most promising antitumor therapy currently available, immunotherapy has great potential to be explored as a therapeutic strategy for CRC, which may bring new hope to patients with CRC in the future.
RH conceived of the presented idea and researched the background of the study. RH prepared the figures and tables. RH, MJ, YL, WY, and XZ wrote the manuscript. RH, MJ, and CZ checked the manuscript. All authors contributed to the article and approved the submitted version.
The authors thank the financial support by the transverse project “Studies on the effect and mechanism of GSG2 in the occurrence and development of colon cancer” (No. Z0S0001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
3. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal Cancer Statistics, 2020. CA Cancer J Clin (2020) 70:145–64. doi: 10.3322/caac.21601
4. Cheng L, Eng C, Nieman LZ, Kapadia AS, Du XL. Trends in Colorectal Cancer Incidence by Anatomic Site and Disease Stage in the United States From 1976 to 2005. Am J Clin Oncol (2011) 34:573–80. doi: 10.1097/COC.0b013e3181fe41ed
5. Thanikachalam K, Khan G. Colorectal Cancer and Nutrition. Nutrients (2019) 11:164. doi: 10.3390/nu11010164
6. Coley WB. IiContribution to the Knowledge of Sarcoma. Ann Surg (1891) 14:199–220. doi: 10.1097/00000658-189112000-00015
7. Tintelnot J, Stein A. Immunotherapy in Colorectal Cancer: Available Clinical Evidence, Challenges and Novel Approaches. World J Gastroenterol (2019) 25:3920–8. doi: 10.3748/wjg.v25.i29.3920
8. McCarthy EF. The Toxins of William B. Coley and the Treatment of Bone and Soft-Tissue Sarcomas. Iowa Orthop J (2006) 26:154–8.
9. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of Tumor Mutation Burden as an Immunotherapy Biomarker: Utility for the Oncology Clinic. Ann Oncol (2019) 30:44–56. doi: 10.1093/annonc/mdy495
10. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
11. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and Activity of Anti-PD-L1 Antibody in Patients With Advanced Cancer. N Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
12. Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep (2016) 17:1206. doi: 10.1016/j.celrep.2016.10.009
13. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer Is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discov (2015) 5:43–51. doi: 10.1158/2159-8290.CD-14-0863
14. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma. Clin Cancer Res (2019) 25:1702–8. doi: 10.1158/1078-0432.CCR-18-2743
15. O’Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients With Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Clin Cancer Res (2019) 25:1142–6. doi: 10.1158/1078-0432.CCR-18-2035
16. Pellegrino M, Del Bufalo F, De Angelis B, Quintarelli C, Caruana I, de Billy E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells (2020) 10:14. doi: 10.3390/cells10010014
17. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol Ther (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
18. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in Colorectal Cancer: Rationale, Challenges and Potential. Nat Rev Gastroenterol Hepatol (2019) 16:361–75. doi: 10.1038/s41575-019-0126-x
19. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
21. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science (2006) 313:1960–4. doi: 10.1126/science.1129139
22. Franke AJ, Skelton WP, Starr JS, Parekh H, Lee JJ, Overman MJ, et al. Immunotherapy for Colorectal Cancer: A Review of Current and Novel Therapeutic Approaches. J Natl Cancer Inst (2019) 111:1131–41. doi: 10.1093/jnci/djz093
23. Korneev KV, Atretkhany KN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. TLR-Signaling and Proinflammatory Cytokines as Drivers of Tumorigenesis. Cytokine (2017) 89:127–35. doi: 10.1016/j.cyto.2016.01.021
24. Junttila MR, de Sauvage FJ. Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response. Nature (2013) 501:346–54. doi: 10.1038/nature12626
25. Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J, et al. Immune Cells Within the Tumor Microenvironment: Biological Functions and Roles in Cancer Immunotherapy. Cancer Lett (2020) 470:126–33. doi: 10.1016/j.canlet.2019.11.009
26. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell (2019) 35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009
27. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
28. Quail DF, Joyce JA. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat Med (2013) 19:1423–37. doi: 10.1038/nm.3394
29. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-Based Prognostic Factors of Colorectal Cancers are Associated With the State of the Local Immune Reaction. J Clin Oncol (2011) 29:610–8. doi: 10.1200/JCO.2010.30.5425
30. Avvaru AK, Sharma D, Verma A, Mishra RK, Sowpati DT. MSDB: A Comprehensive, Annotated Database of Microsatellites. Nucleic Acids Res (2020) 48:D155–9. doi: 10.1093/nar/gkz886
31. Wheeler JM, Bodmer WF, Mortensen NJ. DNA Mismatch Repair Genes and Colorectal Cancer. Gut (2000) 47:148–53. doi: 10.1136/gut.47.1.148
32. Lichtenstern CR, Ngu RK, Shalapour S, Karin M. Immunotherapy, Inflammation and Colorectal Cancer. Cells (2020) 9:618. doi: 10.3390/cells9030618
33. Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and Characterization of Microsatellite Instability Across 18 Cancer Types. Nat Med (2016) 22:1342–50. doi: 10.1038/nm.4191
34. de la Chapelle A, Hampel H. Clinical Relevance of Microsatellite Instability in Colorectal Cancer. J Clin Oncol (2010) 28:3380–7. doi: 10.1200/JCO.2009.27.0652
35. Chen W, Swanson BJ, Frankel WL. Molecular Genetics of Microsatellite-Unstable Colorectal Cancer for Pathologists. Diagn Pathol (2017) 12:24. doi: 10.1186/s13000-017-0613-8
36. Temraz S, Nassar F, Nasr R, Charafeddine M, Mukherji D, Shamseddine A. Gut Microbiome: A Promising Biomarker for Immunotherapy in Colorectal Cancer. Int J Mol Sci (2019) 20:4155. doi: 10.3390/ijms20174155
37. Gologan A, Sepulveda AR. Microsatellite Instability and DNA Mismatch Repair Deficiency Testing in Hereditary and Sporadic Gastrointestinal Cancers. Clin Lab Med (2005) 25:179–96. doi: 10.1016/j.cll.2004.12.001
38. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, et al. Screening for the Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). N Engl J Med (2005) 352:1851–60. doi: 10.1056/NEJMoa043146
39. Ciardiello D, Vitiello PP, Cardone C, Martini G, Troiani T, Martinelli E, et al. Immunotherapy of Colorectal Cancer: Challenges for Therapeutic Efficacy. Cancer Treat Rev (2019) 76:22–32. doi: 10.1016/j.ctrv.2019.04.003
40. Fleisher AS, Esteller M, Harpaz N, Leytin A, Rashid A, Xu Y, et al. Microsatellite Instability in Inflammatory Bowel Disease-Associated Neoplastic Lesions is Associated With Hypermethylation and Diminished Expression of the DNA Mismatch Repair Gene, Hmlh1. Cancer Res (2000) 60:4864–8.
41. Pino MS, Chung DC. Microsatellite Instability in the Management of Colorectal Cancer. Expert Rev Gastroenterol Hepatol (2011) 5:385–99. doi: 10.1586/egh.11.25
42. Vilar E, Tabernero J. Molecular Dissection of Microsatellite Instable Colorectal Cancer. Cancer Discov (2013) 3:502–11. doi: 10.1158/2159-8290.CD-12-0471
43. Goel A, Arnold CN, Boland CR. Multistep Progression of Colorectal Cancer in the Setting of Microsatellite Instability: New Details and Novel Insights. Gastroenterology (2001) 121:1497–502. doi: 10.1053/gast.2001.29978
44. Greenson JK, Bonner JD, Ben-Yzhak O, Cohen HI, Miselevich I, Resnick MB, et al. Phenotype of Microsatellite Unstable Colorectal Carcinomas: Well-Differentiated and Focally Mucinous Tumors and the Absence of Dirty Necrosis Correlate With Microsatellite Instability. Am J Surg Pathol (2003) 27:563–70. doi: 10.1097/00000478-200305000-00001
45. Klingbiel D, Saridaki Z, Roth AD, Bosman FT, Delorenzi M, Tejpar S. Prognosis of Stage II and III Colon Cancer Treated With Adjuvant 5-Fluorouracil or FOLFIRI in Relation to Microsatellite Status: Results of the PETACC-3 Trial. Ann Oncol (2015) 26:126–32. doi: 10.1093/annonc/mdu499
46. Pecina-Slaus N, Kafka A, Salamon I, Bukovac A. Mismatch Repair Pathway, Genome Stability and Cancer. Front Mol Biosci (2020) 7:122. doi: 10.3389/fmolb.2020.00122
47. Innocenti F, Ou FS, Qu X, Zemla TJ, Niedzwiecki D, Tam R, et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J Clin Oncol (2019) 37:1217–27. doi: 10.1200/JCO.18.01798
48. Goodman AM, Castro A, Pyke RM, Okamura R, Kato S, Riviere P, et al. MHC-I Genotype and Tumor Mutational Burden Predict Response to Immunotherapy. Genome Med (2020) 12:45. doi: 10.1186/s13073-020-00743-4
49. Cancer Genome Atlas N. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
50. Cohen R, Shi Q, Andre T. Immunotherapy for Early Stage Colorectal Cancer: A Glance Into the Future. Cancers (Basel) (2020) 12:1990. doi: 10.3390/cancers12071990
51. De’ Angelis GL, Bottarelli L, Azzoni C, De’ Angelis N, Leandro G, Di Mario F, et al. Microsatellite Instability in Colorectal Cancer. Acta Biomed (2018) 89:97–101. doi: 10.23750/abm.v89i9-S.7960
52. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor Mutational Load Predicts Survival After Immunotherapy Across Multiple Cancer Types. Nat Genet (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
53. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in Patients With Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (Checkmate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol (2017) 18:1182–91. doi: 10.1016/S1470-2045(17)30422-9
54. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol (2018) 36:773–9. doi: 10.1200/JCO.2017.76.9901
55. Hochster HS, Bendell JC, Cleary JM, Foster P, Eckhardt SG. Efficacy and Safety of Atezolizumab (Atezo) and Bevacizumab (Bev) in a Phase Ib Study of Microsatellite Instability (MSI)-High Metastatic Colorectal Cancer (Mcrc). J Clin Oncol (2017) 35:673–. doi: 10.1200/JCO.2017.35.4_suppl.673
56. Lin A, Zhang J, Luo P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front Immunol (2020) 11:2039. doi: 10.3389/fimmu.2020.02039
57. Zhang L, Zhao Y, Dai Y, Cheng JN, Gong Z, Feng Y, et al. Immune Landscape of Colorectal Cancer Tumor Microenvironment From Different Primary Tumor Location. Front Immunol (2018) 9:1578. doi: 10.3389/fimmu.2018.01578
58. Chen EX, Jonker DJ, Kennecke HF, Berry SR, O’Callaghan CJ. CCTG CO.26 Trial: A Phase II Randomized Study of Durvalumab (D) Plus Tremelimumab (T) and Best Supportive Care (BSC) Versus BSC Alone in Patients (Pts) With Advanced Refractory Colorectal Carcinoma (Rcrc). J Clin Oncol (2019) 37:481–. doi: 10.1200/JCO.2019.37.4_suppl.481
59. Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, et al. Tumor Microsatellite Instability and Clinical Outcome in Young Patients With Colorectal Cancer. N Engl J Med (2000) 342:69–77. doi: 10.1056/NEJM200001133420201
60. Kawakami H, Zaanan A, Sinicrope FA. Microsatellite Instability Testing and its Role in the Management of Colorectal Cancer. Curr Treat Options Oncol (2015) 16:30. doi: 10.1007/s11864-015-0348-2
61. Steinman RM. The Dendritic Cell System and its Role in Immunogenicity. Annu Rev Immunol (1991) 9:271–96. doi: 10.1146/annurev.iy.09.040191.001415
62. Steinman RM, Banchereau J. Taking Dendritic Cells Into Medicine. Nature (2007) 449:419–26. doi: 10.1038/nature06175
63. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
64. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J Clin Oncol (2010) 28:3167–75. doi: 10.1200/JCO.2009.26.7609
65. Yamamoto N, Nokihara H, Yamada Y, Shibata T, Tamura Y, Seki Y, et al. Phase I Study of Nivolumab, an Anti-PD-1 Antibody, in Patients With Malignant Solid Tumors. Invest New Drugs (2017) 35:207–16. doi: 10.1007/s10637-016-0411-2
66. O’Neil BH, Wallmark JM, Lorente D, Elez E, Raimbourg J, Gomez-Roca C, et al. Safety and Antitumor Activity of the Anti-PD-1 Antibody Pembrolizumab in Patients With Advanced Colorectal Carcinoma. PLoS One (2017) 12:e0189848. doi: 10.1371/journal.pone.0189848
67. Vanmeerbeek I, Sprooten J, De Ruysscher D, Tejpar S, Vandenberghe P, Fucikova J, et al. Trial Watch: Chemotherapy-Induced Immunogenic Cell Death in Immuno-Oncology. Oncoimmunology (2020) 9:1703449. doi: 10.1080/2162402X.2019.1703449
68. Kelly B, O’Neill LA. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res (2015) 25:771–84. doi: 10.1038/cr.2015.68
69. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-Based Mechanisms of Cytotoxic Chemotherapy: Implications for the Design of Novel and Rationale-Based Combined Treatments Against Cancer. Cell Death Differ (2014) 21:15–25. doi: 10.1038/cdd.2013.67
70. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic Cell Death and Damps in Cancer Therapy. Nat Rev Cancer (2012) 12:860–75. doi: 10.1038/nrc3380
71. Yong SB, Chung JY, Song Y, Kim J, Ra S, Kim YH. Non-Viral Nano-Immunotherapeutics Targeting Tumor Microenvironmental Immune Cells. Biomaterials (2019) 219:119401. doi: 10.1016/j.biomaterials.2019.119401
72. Rong L, Zhang Y, Li WS, Su Z, Fadhil JI, Zhang C. Iron Chelated Melanin-Like Nanoparticles for Tumor-Associated Macrophage Repolarization and Cancer Therapy. Biomaterials (2019) 225:119515. doi: 10.1016/j.biomaterials.2019.119515
73. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A Transcriptionally and Functionally Distinct PD-1(+) CD8(+) T Cell Pool With Predictive Potential in Non-Small-Cell Lung Cancer Treated With PD-1 Blockade. Nat Med (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
74. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-a Modulates Expression of Inhibitory Checkpoints on CD8+ T Cells in Tumors. J Exp Med (2015) 212:139–48. doi: 10.1084/jem.20140559
75. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 With Ramucirumab Strongly Impacts Effector/Activated Regulatory T Cells and CD8(+) T Cells in the Tumor Microenvironment. J Immunother Cancer (2018) 6:106. doi: 10.1186/s40425-018-0403-1
76. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-a Drives TOX-Dependent T Cell Exhaustion in Anti-PD-1-Resistant Microsatellite Stable Colorectal Cancers. Sci Immunol (2019) 4:eaay0555. doi: 10.1126/sciimmunol.aay0555
77. Morse MA, Hochster H, Benson A. Perspectives on Treatment of Metastatic Colorectal Cancer With Immune Checkpoint Inhibitor Therapy. Oncologist (2020) 25:33–45. doi: 10.1634/theoncologist.2019-0176
78. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination With Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res (2015) 21:1639–51. doi: 10.1158/1078-0432.CCR-14-2339
79. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP Kinase Inhibition Promotes T Cell and Anti-Tumor Activity in Combination With PD-L1 Checkpoint Blockade. Immunity (2016) 44:609–21. doi: 10.1016/j.immuni.2016.01.024
80. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity (2016) 44:955–72. doi: 10.1016/j.immuni.2016.05.002
81. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 Is a Second Ligand for PD-1 and Inhibits T Cell Activation. Nat Immunol (2001) 2:261–8. doi: 10.1038/85330
82. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova TG, et al. Endothelial Expression of PD-L1 and PD-L2 Down-Regulates CD8+ T Cell Activation and Cytolysis. Eur J Immunol (2003) 33:3117–26. doi: 10.1002/eji.200324270
83. Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J, et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin Cancer Res (2017) 23:3158–67. doi: 10.1158/1078-0432.CCR-16-1761
84. Sagaert X, Vanstapel A, Verbeek S. Tumor Heterogeneity in Colorectal Cancer: What do We Know So Far? Pathobiology (2018) 85:72–84. doi: 10.1159/000486721
85. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat Med (2015) 21:1350–6. doi: 10.1038/nm.3967
86. Roelands J, Kuppen PJK, Vermeulen L, Maccalli C, Decock J, Wang E, et al. Immunogenomic Classification of Colorectal Cancer and Therapeutic Implications. Int J Mol Sci (2017) 18:2229. doi: 10.3390/ijms18102229
87. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity (2013) 39:782–95. doi: 10.1016/j.immuni.2013.10.003
88. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary Lymphoid Structures in Cancer and Beyond. Trends Immunol (2014) 35:571–80. doi: 10.1016/j.it.2014.09.006
89. Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular Network Reconstruction Identifies T-Cell Homing Factors Associated With Survival in Colorectal Cancer. Gastroenterology (2010) 138:1429–40. doi: 10.1053/j.gastro.2009.10.057
90. Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, et al. Functional Network Pipeline Reveals Genetic Determinants Associated With In Situ Lymphocyte Proliferation and Survival of Cancer Patients. Sci Transl Med (2014) 6:228ra37. doi: 10.1126/scitranslmed.3007240
91. Asaoka Y, Ijichi H, Koike K. PD-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 373:1979. doi: 10.1056/NEJMc1510353
92. Becht E, de Reynies A, Giraldo NA, Pilati C, Buttard B, Lacroix L, et al. Immune and Stromal Classification of Colorectal Cancer is Associated With Molecular Subtypes and Relevant for Precision Immunotherapy. Clin Cancer Res (2016) 22:4057–66. doi: 10.1158/1078-0432.CCR-15-2879
93. Janji B, Hasmim M, Parpal S, Berchem G, Noman MZ. Firing Up the Cold Tumors by Targeting Vps34. Oncoimmunology (2020) 9:1809936. doi: 10.1080/2162402X.2020.1809936
94. Jackstadt R, van Hooff SR, Leach JD, Cortes-Lavaud X, Lohuis JO, Ridgway RA, et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell (2019) 36:319–36.e7. doi: 10.1016/j.ccell.2019.08.003
95. Mishra S, Bernal C, Silvano M, Anand S, Ruiz IAA. The Protein Secretion Modulator TMED9 Drives CNIH4/Tgfalpha/GLI Signaling Opposing TMED3-WNT-TCF to Promote Colon Cancer Metastases. Oncogene (2019) 38:5817–37. doi: 10.1038/s41388-019-0845-z
96. Cremolini C, Antoniotti C, Rossini D, Lonardi S, Loupakis F, Pietrantonio F, et al. Upfront FOLFOXIRI Plus Bevacizumab and Reintroduction After Progression Versus Mfolfox6 Plus Bevacizumab Followed by FOLFIRI Plus Bevacizumab in the Treatment of Patients With Metastatic Colorectal Cancer (TRIBE2): A Multicentre, Open-Label, Phase 3, Randomised, Controlled Trial. Lancet Oncol (2020) 21:497–507. doi: 10.1016/S1470-2045(19)30862-9
97. Shih YC, Elting LS, Pavluck AL, Stewart A, Halpern MT. Immunotherapy in the Initial Treatment of Newly Diagnosed Cancer Patients: Utilization Trend and Cost Projections for non-Hodgkin’s Lymphoma, Metastatic Breast Cancer, and Metastatic Colorectal Cancer. Cancer Invest (2010) 28:46–53. doi: 10.3109/07357900902783187
98. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer. Nat Rev Cancer (2017) 17:268. doi: 10.1038/nrc.2017.24
99. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic Cell Death in Cancer Therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
100. Willemen Y, Van den Bergh JM, Lion E, Anguille S, Roelandts VA, Van Acker HH, et al. Engineering Monocyte-Derived Dendritic Cells to Secrete Interferon-Alpha Enhances Their Ability to Promote Adaptive and Innate Anti-Tumor Immune Effector Functions. Cancer Immunol Immunother (2015) 64:831–42. doi: 10.1007/s00262-015-1688-2
101. Idorn M, Olsen M, Halldorsdottir HR, Skadborg SK, Pedersen M, Hogdall C, et al. Improved Migration of Tumor Ascites Lymphocytes to Ovarian Cancer Microenvironment by CXCR2 Transduction. Oncoimmunology (2018) 7:e1412029. doi: 10.1080/2162402X.2017.1412029
102. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab Plus Chemotherapy in Metastatic non-Small-Cell Lung Cancer. N Engl J Med (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
103. Doroshow DB, Sanmamed MF, Hastings K, Politi K, Rimm DL, Chen L, et al. Immunotherapy in non-Small Cell Lung Cancer: Facts and Hopes. Clin Cancer Res (2019) 25:4592–602. doi: 10.1158/1078-0432.CCR-18-1538
104. Zhang X, Shi M, Chen T, Zhang B. Characterization of the Immune Cell Infiltration Landscape in Head and Neck Squamous Cell Carcinoma to Aid Immunotherapy. Mol Ther Nucleic Acids (2020) 22:298–309. doi: 10.1016/j.omtn.2020.08.030
105. Shen YY, Hummel J, Trindade IC, Papageorgiou C, Shyu CR, Mitchem JB. Regulation of Gene Expression by DNA Methylation With Cytotoxic T Lymphocytes Evaluation in Consensus Molecular Subtypes of Colorectal Cancer. J Clin Oncol (2020) 38. doi: 10.1200/JCO.2020.38.5_suppl.25
106. Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and Function of Regulatory T Cells Expressing the Transcription Factor T-Bet. Nature (2017) 546:421–5. doi: 10.1038/nature22360
107. Fernando NH, Hurwitz HI. Targeted Therapy of Colorectal Cancer: Clinical Experience With Bevacizumab. Oncologist (2004) 9 Suppl 1:11–8. doi: 10.1634/theoncologist.9-suppl_1-11
108. Meyerhardt JA, Mayer RJ. Systemic Therapy for Colorectal Cancer. N Engl J Med (2005) 352:476–87. doi: 10.1056/NEJMra040958
109. Caraglia M, Correale P, Giannicola R, Staropoli N, Botta C, Pastina P, et al. GOLFIG Chemo-Immunotherapy in Metastatic Colorectal Cancer Patients. A Critical Review on a Long-Lasting Follow-Up. Front Oncol (2019) 9:1102. doi: 10.3389/fonc.2019.01102
110. Correale P, Botta C, Rotundo MS, Guglielmo A, Conca R, Licchetta A, et al. Gemcitabine, Oxaliplatin, Levofolinate, 5-Fluorouracil, Granulocyte-Macrophage Colony-Stimulating Factor, and Interleukin-2 (GOLFIG) Versus FOLFOX Chemotherapy in Metastatic Colorectal Cancer Patients: The GOLFIG-2 Multicentric Open-Label Randomized Phase III Trial. J Immunother (2014) 37:26–35. doi: 10.1097/CJI.0000000000000004
111. Correale P, Cusi MG, Del Vecchio MT, Aquino A, Prete SP, Tsang KY, et al. Dendritic Cell-Mediated Cross-Presentation of Antigens Derived From Colon Carcinoma Cells Exposed to a Highly Cytotoxic Multidrug Regimen With Gemcitabine, Oxaliplatin, 5-Fluorouracil, and Leucovorin, Elicits a Powerful Human Antigen-Specific CTL Response With Antitumor Activity In Vitro. J Immunol (2005) 175:820–8. doi: 10.4049/jimmunol.175.2.820
112. Correale P, Tagliaferri P, Fioravanti A, Del Vecchio MT, Remondo C, Montagnani F, et al. Immunity Feedback and Clinical Outcome in Colon Cancer Patients Undergoing Chemoimmunotherapy With Gemcitabine + FOLFOX Followed by Subcutaneous Granulocyte Macrophage Colony-Stimulating Factor and Aldesleukin (GOLFIG-1 Trial). Clin Cancer Res (2008) 14:4192–9. doi: 10.1158/1078-0432.CCR-07-5278
113. Tenti S, Correale P, Conca R, Pastina P, Fioravanti A. Occurrence of Sjogren Syndrome in a Long-Term Survivor Patient With Metastatic Colon Carcinoma Treated With GOLFIG Regimen. J Chemother (2012) 24:245–6. doi: 10.1179/1973947812Y.0000000008
114. Hermiston T. Gene Delivery From Replication-Selective Viruses: Arming Guided Missiles in the War Against Cancer. J Clin Invest (2000) 105:1169–72. doi: 10.1172/JCI9973
115. Hill C, Carlisle R. Achieving Systemic Delivery of Oncolytic Viruses. Expert Opin Drug Deliv (2019) 16:607–20. doi: 10.1080/17425247.2019.1617269
116. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science (1991) 252:854–6. doi: 10.1126/science.1851332
117. Bommareddy PK, Shettigar M, Kaufman HL. Integrating Oncolytic Viruses in Combination Cancer Immunotherapy. Nat Rev Immunol (2018) 18:498–513. doi: 10.1038/s41577-018-0014-6
118. Ge Y, Wang H, Ren J, Liu W, Chen L, Chen H, et al. Oncolytic Vaccinia Virus Delivering Tethered IL-12 Enhances Antitumor Effects With Improved Safety. J Immunother Cancer (2020) 8:e000710. doi: 10.1136/jitc-2020-000710
119. Zuo S, Wei M, He B, Chen A, Wang S, Kong L, et al. Enhanced Antitumor Efficacy of a Novel Oncolytic Vaccinia Virus Encoding a Fully Monoclonal Antibody Against T-Cell Immunoglobulin and ITIM Domain (TIGIT). EBioMedicine (2021) 64:103240. doi: 10.1016/j.ebiom.2021.103240
120. Marei HE, Althani A, Caceci T, Arriga R, Sconocchia T, Ottaviani A, et al. Recent Perspective on CAR and Fcgamma-CR T Cell Immunotherapy for Cancers: Preclinical Evidence Versus Clinical Outcomes. Biochem Pharmacol (2019) 166:335–46. doi: 10.1016/j.bcp.2019.06.002
121. Nap M, Mollgard K, Burtin P, Fleuren GJ. Immunohistochemistry of Carcino-Embryonic Antigen in the Embryo, Fetus and Adult. Tumour Biol (1988) 9:145–53. doi: 10.1159/000217555
122. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A Safe and Potent Anti-CD19 CAR T Cell Therapy. Nat Med (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
123. Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, et al. Effective Combination Immunotherapy Using Oncolytic Viruses to Deliver CAR Targets to Solid Tumors. Sci Transl Med (2020) 12:eaaz1863. doi: 10.1126/scitranslmed.aaz1863
124. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
125. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
126. Gopal AK, Chen R, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, et al. Durable Remissions in a Pivotal Phase 2 Study of Brentuximab Vedotin in Relapsed or Refractory Hodgkin Lymphoma. Blood (2015) 125:1236–43. doi: 10.1182/blood-2014-08-595801
127. Duan X, Chan C, Lin W. Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angew Chem Int Ed Engl (2019) 58:670–80. doi: 10.1002/anie.201804882
128. Sahin U, Tureci O. Personalized Vaccines for Cancer Immunotherapy. Science (2018) 359:1355–60. doi: 10.1126/science.aar7112
129. Hollingsworth RE, Jansen K. Turning the Corner on Therapeutic Cancer Vaccines. NPJ Vaccines (2019) 4:7. doi: 10.1038/s41541-019-0103-y
130. Shang N, Figini M, Shangguan J, Wang B, Sun C, Pan L, et al. Dendritic Cells Based Immunotherapy. Am J Cancer Res (2017) 7:2091–102.
131. Obara W, Kanehira M, Katagiri T, Kato R, Kato Y, Takata R. Present Status and Future Perspective of Peptide-Based Vaccine Therapy for Urological Cancer. Cancer Sci (2018) 109:550–9. doi: 10.1111/cas.13506
132. Toussaint B, Chauchet X, Wang Y, Polack B, Le Gouellec A. Live-Attenuated Bacteria as a Cancer Vaccine Vector. Expert Rev Vaccines (2013) 12:1139–54. doi: 10.1586/14760584.2013.836914
133. Gatti-Mays ME, Redman JM, Collins JM, Bilusic M. Cancer Vaccines: Enhanced Immunogenic Modulation Through Therapeutic Combinations. Hum Vaccin Immunother (2017) 13:2561–74. doi: 10.1080/21645515.2017.1364322
134. McNamara MA, Nair SK, Holl EK. RNA-Based Vaccines in Cancer Immunotherapy. J Immunol Res (2015) 2015:794528. doi: 10.1155/2015/794528
135. Blankenstein T, Leisegang M, Uckert W, Schreiber H. Targeting Cancer-Specific Mutations by T Cell Receptor Gene Therapy. Curr Opin Immunol (2015) 33:112–9. doi: 10.1016/j.coi.2015.02.005
136. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
137. Malekzadeh P, Pasetto A, Robbins PF, Parkhurst MR, Paria BC, Jia L, et al. Neoantigen Screening Identifies Broad TP53 Mutant Immunogenicity in Patients With Epithelial Cancers. J Clin Invest (2019) 129:1109–14. doi: 10.1172/JCI123791
138. Gros A, Tran E, Parkhurst MR, Ilyas S, Pasetto A, Groh EM, et al. Recognition of Human Gastrointestinal Cancer Neoantigens by Circulating PD-1+ Lymphocytes. J Clin Invest (2019) 129:4992–5004. doi: 10.1172/JCI127967
139. Correale P, Botta C, Staropoli N, Nardone V, Pastina P, Ulivieri C, et al. Systemic Inflammatory Status Predict the Outcome of K-RAS WT Metastatic Colorectal Cancer Patients Receiving the Thymidylate Synthase Poly-Epitope-Peptide Anticancer Vaccine. Oncotarget (2018) 9:20539–54. doi: 10.18632/oncotarget.24993
140. Kuwabara T, Ishikawa F, Kondo M, Kakiuchi T. The Role of IL-17 and Related Cytokines in Inflammatory Autoimmune Diseases. Mediators Inflamm (2017) 2017:3908061. doi: 10.1155/2017/3908061
141. Maletzki C, Gladbach YS, Hamed M, Fuellen G, Semmler ML, Stenzel J, et al. Cellular Vaccination of MLH1(-/-) Mice - an Immunotherapeutic Proof of Concept Study. Oncoimmunology (2018) 7:e1408748. doi: 10.1080/2162402X.2017.1408748
142. de Weger VA, Turksma AW, Voorham QJ, Euler Z, Bril H, van den Eertwegh AJ, et al. Clinical Effects of Adjuvant Active Specific Immunotherapy Differ Between Patients With Microsatellite-Stable and Microsatellite-Instable Colon Cancer. Clin Cancer Res (2012) 18:882–9. doi: 10.1158/1078-0432.CCR-11-1716
143. Kloor M, Reuschenbach M, Pauligk C, Karbach J, Rafiyan MR, Al-Batran SE, et al. A Frameshift Peptide Neoantigen-Based Vaccine for Mismatch Repair-Deficient Cancers: A Phase I/Iia Clinical Trial. Clin Cancer Res (2020) 26:4503–10. doi: 10.1158/1078-0432.CCR-19-3517
144. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfbeta Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature (2018) 554:538–43. doi: 10.1038/nature25492
145. Hou L, Tian C, Yan Y, Zhang L, Zhang H, Zhang Z. Manganese-Based Nanoactivator Optimizes Cancer Immunotherapy via Enhancing Innate Immunity. ACS Nano (2020) 14:3927–40. doi: 10.1021/acsnano.9b06111
146. Maltzman JS, Carman JA, Monroe JG. Role of EGR1 in Regulation of Stimulus-Dependent CD44 Transcription in B Lymphocytes. Mol Cell Biol (1996) 16:2283–94. doi: 10.1128/mcb.16.5.2283
147. Smialowicz RJ, Rogers RR, Riddle MM, Luebke RW, Rowe DG, Garner RJ. Manganese Chloride Enhances Murine Cell-Mediated Cytotoxicity: Effects on Natural Killer Cells. J Immunopharmacol (1984) 6:1–23. doi: 10.3109/08923978409026455
148. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese Is Critical for Antitumor Immune Responses via Cgas-STING and Improves the Efficacy of Clinical Immunotherapy. Cell Res (2020) 30:966–79. doi: 10.1038/s41422-020-00395-4
149. Yang Y. Cancer Immunotherapy: Harnessing the Immune System to Battle Cancer. J Clin Invest (2015) 125:3335–7. doi: 10.1172/JCI83871
150. Chen Q, Chen M, Liu Z. Local Biomaterials-Assisted Cancer Immunotherapy to Trigger Systemic Antitumor Responses. Chem Soc Rev (2019) 48:5506–26. doi: 10.1039/c9cs00271e
151. Brudno JN, Kochenderfer JN. Recent Advances in CAR T-Cell Toxicity: Mechanisms, Manifestations and Management. Blood Rev (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
Keywords: immunotherapy, colorectal cancer, immune checkpoint inhibitors, microsatellite, ICI combination therapy
Citation: He R, Lao Y, Yu W, Zhang X, Jiang M and Zhu C (2021) Progress in the Application of Immune Checkpoint Inhibitor-Based Immunotherapy for Targeting Different Types of Colorectal Cancer. Front. Oncol. 11:764618. doi: 10.3389/fonc.2021.764618
Received: 25 August 2021; Accepted: 01 November 2021;
Published: 23 November 2021.
Edited by:
Eyad Elkord, University of Salford, United KingdomReviewed by:
Farhad Jadidi-Niaragh, Tabriz University of Medical Sciences, IranCopyright © 2021 He, Lao, Yu, Zhang, Jiang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Jiang, jiangmin1023@suda.edu.cn; Chunrong Zhu, zcr050311@163.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.