 Benjamin Gesundheit1*
Benjamin Gesundheit1* Eliel Ben-David2Yehudit Posen1Ronald Ellis1
Eliel Ben-David2Yehudit Posen1Ronald Ellis1 Guido Wollmann3,4
Guido Wollmann3,4 E. Marion Schneider5
E. Marion Schneider5 Karl Aigner6Lars Brauns7Thomas Nesselhut8
Karl Aigner6Lars Brauns7Thomas Nesselhut8 Ingrid Ackva9Christine Weisslein9
Ingrid Ackva9Christine Weisslein9 Arno Thaller9
Arno Thaller9- 1Rapo Yerape Ltd., Jerusalem, Israel
- 2Department of Radiology, Shaare Zedek Medical Center, Jerusalem, Israel
- 3Institute of Virology, Medical University of Innsbruck, Innsbruck, Austria
- 4Christian Doppler Laboratory for Viral Immunotherapy of Cancer, Innsbruck, Austria
- 5Division of Experimental Anesthesiology, University Hospital Ulm, Ulm, Germany
- 6Medias Klinik, Burghausen, Germany
- 7Arztpraxis am Regerplatz, Munich, Germany
- 8Institut für Tumor-Therapie, Duderstadt, Germany
- 9Praxisklinik fuer Allgemeinmedizin, Markt Berolzheim, Germany
Glioblastoma multiforme (GBM) remains an incurable condition, associated with a median survival time of 15 months with best standard of care and 5-year survival rate of <10%. We report on four GBM patients on combination treatment regimens that included oncolytic virus (OV) immunotherapy, who achieved clinical and radiological responses with long-term survival, thus far, of up to 14 years, and good quality of life. We discuss the radiological findings that provide new insights into this treatment, the scientific rationale of this innovative and promising therapy, and considerations for future research.
Introduction
Glioblastoma multiforme (GBM) represents ~50% of adult primary malignant brain tumors, which occur at an annual incidence of 2–3 per 100,000 adults (1), and is the most common cause of death among patients with central nervous system tumors. The standard treatment regimen includes resection followed by radiation and chemotherapy with temozolomide (TMZ) (2). Notwithstanding this aggressive approach, the median life expectancy for GBM patients is only 15 months (3), with limited treatment response after recurrence (4); only 5–10% of patients live for more than 5 years (5). With such a dismal prognosis, the need for new therapeutic approaches for GBM is significant.
For over a century, there have been anecdotal reports describing the coincidence of various viral or bacterial infections with tumor remission among cancer patients (6). Oncolytic viruses (OVs) have been characterized and defined as preferentially replicating in tumor cells and inducing their death while sparing normal cells (7). In addition to the direct lytic effect of OVs on tumor cells, a strong virus-activated innate and adaptive immune response contributes to the overall therapeutic outcome. These responses can overcome immunosuppressive forces in the tumor microenvironment, ultimately shifting “cold” tumors to “hot” tumors (8). The release of tumor-associated antigens and induction of immunogenic cell death subsequently stimulate anti-tumor immune responses with potential for long-lasting tumor control (9). Some OVs also infect tumor-associated endothelial cells, resulting in breakdown of the tumor vasculature and subsequent necrosis of uninfected tumor cells (10). Tumor cell preference for OV propagation is based on oncogenic signaling pathways or defects in innate antivirus responses frequently seen in malignant cells (11, 12). Recent years have seen significant breakthroughs in OV engineering, which has generated OVs encoding proteins that enhance their tropism for tumor cells (13–15).
While the first OV-based immunotherapy (virotherapy) has gained US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval for treatment of melanoma (16), oncolytic virotherapy for other tumor types is at various stages of clinical testing (17). Over the past three decades, OVs from >15 families have been preclinically assessed as potential treatment modalities for glioblastoma (18). Among these, nine have been included in numerous clinical trials (19). Importantly, these studies confirmed the general safety of OV application for brain tumors, with serious adverse effects rarely occurring. Durable complete responses were shown in up to 20% of patients, and regulatory fast-track designation by the FDA has been awarded to DNX-2401, Toca511, and PVS-RIPO (19).
Although the initial response is geared toward antiviral defense, the OV-elicited immune activation plays a major role in the therapeutic outcome (20). Consequently, virotherapy has gained significant attention as a partner for other immunotherapeutic approaches, such as dendritic cell (DC) therapy, cancer vaccines, T-cell therapies and immune checkpoint inhibitors (CPI) (21–23). CPIs selectively target immune inhibitory signals that contribute to the immune suppressive tumor environment, and thereby reinvigorate anti-tumor T-cell responses. CPIs have been shown to be particularly effective in combating tumors that are hypermutated or with specific neoantigen signatures (24), including recurrent, multifocal biallelic mismatch repair deficiency (bMMRD)-associated GBM (25). Tumoral OV infection precipitates endogenous DC migration and activation, which elicit a shift toward antitumor immunity. DC-based immunotherapies have been proposed to synergize with OVs (21, 26).
This case series presents the clinical and radiological outcomes of four patients with histologically-confirmed GBM treated with experimental combination virotherapy regimens as compassionate treatment. Given the nature of this early clinical experience and significant socio-economic factors, different exploratory treatment regimens involving a range of generically available OV strains were used. These cases are instructive for documenting clinical and radiological responses to virotherapy as an important basis for developing standardized and improved protocols for future clinical research.
Case Presentation
Informed consent for publication was obtained from all patients in this case series. Regulatory approval for compassionate use was within the framework of the German Individueller Heilversuch. Patients were treated with individualized regimens comprised of three OVs: wild-type Newcastle disease virus (NDV) (Wageningen University, Netherlands), wild-type parvovirus (PV) (University Marburg, Germany), and wild-type vaccinia virus (VV) (Paul Ehrlich Institut Berlin, Germany). Each virus was used at a clinical dosage level of 109 TCID50 (tissue culture infectious doses), as quantified by virus-specific cell-culture assays. Viruses were prepared for clinical use in conditioned cell culture medium that had been clarified by centrifugation, diluted to final TCID50 level as needed, sterile-filtered, stored frozen, and thawed on the day of injection. Viruses were injected to patients at intervals of ~2–3 weeks, administered by sequential 10 mL injections via the same catheter. A summary of the treatments and clinical and radiological presentations of the four patients is shown in Table 1 and Figure 1.
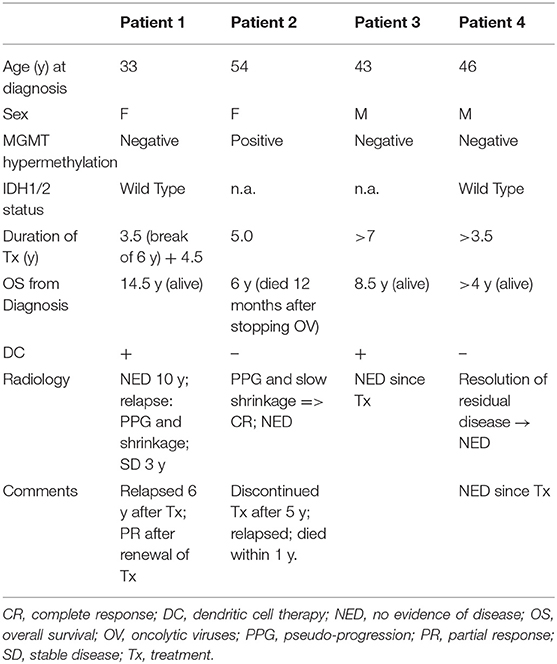
Table 1. Clinical summary of GBM patients undergoing OV therapy.
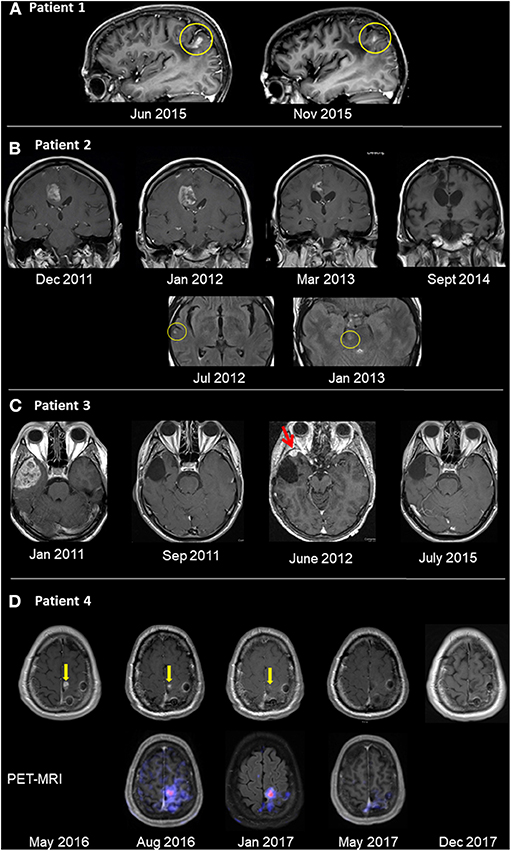
Figure 1. Radiological follow-up of GBM tumors treated with oncolytic virotherapy. (A) Patient 1 was diagnosed with GBM in 7/05 and underwent total surgical resection in 8/05. OV therapy was initiated in 5/06 and given for 3.5 years, after which patient remained with NED for 6.0 years. Recurrence in the left parietal lobe appeared on sagittal T1 contrast-enhanced images in 6/15 (left). The patient improved clinically after renewing OV alone. Five months thereafter, lesion shrinkage was observed (right). (B) Patient 2 was diagnosed with GBM in 10/10, underwent surgical resection and chemoradiotherapy. Relapse occurred (12/11) in the right frontal lobe (left image top row, T1 contrast enhanced images), with clinical deterioration. OV therapy was initiated. Follow-up imaging showed shrinkage until disappearance of the pathological enhancement. Concurrent to OV therapy and shrinkage, multiple scattered FLAIR hyperintense foci (with and without enhancement) were seen (bottom row, FLAIR images, yellow circle), possibly an immune-mediated response. (C) Patient 3 was diagnosed with a right temporal lesion (1/11; left image, T1 post contrast), underwent surgical resection and chemoradiotherapy, and no residual tumor mass was seen (9/11). Relapse occurred (6/12; red arrow), and following a second resection, OV therapy was initiated. The patient has no residual tumor (7/15; right image) and has remained with no radiological or clinical evidence of disease. (D) Patient 4 was diagnosed with GBM (10/15) and underwent resection and chemoradiotherapy. Following relapse in the surgical bed (5/16; yellow arrow, left image of T1 contrast enhanced images), OV was initiated. The focus of pathological enhancement decreased in size until disappearance (1/17). Concurrent PET-MRI images (bottom row) showed hypermetabolic activity even while Gd enhancement was decreasing.
Patient 1
A previously healthy 33-year old woman presented in July 2005 (7/05) with slurred speech and left-arm weakness. CT studies showed a space-occupying lesion (SOL) in the left parietal lobe. Following total surgical resection (8/05), full neurological recovery was observed and corresponded with no radiological evidence of disease (NED) in the post-surgical MRI. Retrospective pathological evaluations revealed wild-type sequences for both isocitrate dehydrogenase 1 and 2 (IDH1/2) and O6-methylguanine-DNA methyltransferase (MGMT) promoter. Routine postoperative radiation (60 Gy) combined with oral TMZ (75 mg/m2/d for 42d) (12/05-1/06) was discontinued after 20 (instead of the usual 30) doses due to severe thrombocytopenia (6,000*109/L) and extreme weakness (3/06). OV therapy was initiated (5/06) and administered via an intraarterial (IA) port system implanted end-to-side into the carotid artery. The patient remained stable with normal quality of life (QoL) and NED for 3.5 years after initiation treatment, which was discontinued due to pregnancy (10/09). After an uneventful pregnancy, she gave birth to a healthy baby. After a normal QoL for 6 years, she presented (6/15) with blurred vision, headache, stuttering, right hemiplegia, paranesthesia, hemianopsia, Jacksonian seizure, confusion, and disorientation in space and time. MRI (6/15; Figure 1A) confirmed relapse in the left parietal lesion measuring 9 × 7 × 15 mm. OV therapy was initiated (7/15) and resulted in prompt clinical improvement, including resolution of seizure activities and EEG findings of Jacksonian seizures. MRI (11/15) showed shrinkage of the tumor to 6 × 7 × 8 mm (Figure 1A). Virotherapy was continued with combinations of various viruses and switched (3/16) to IV administration due to complications with the IA port system. The patient has remained stable for 4.5 years after initiating the second phase of OV and 14.5 years after initiating OV treatment.
Patient 2
A previously healthy 54-year-old woman presented with grand-mal seizures and left hemiplegia. She had gross surgical resection, and the tumor was found to be MGMT promoter methylation-positive. She received routine treatment (2/11) with radiation (60 Gy) and TMZ (75 mg/m2/d for 42 d, followed by 200 mg/m2 for 5 days/month). Full neurological recovery was observed. She relapsed radiologically, with an MRI (11/11) showing an irregular right frontal enhancing lesion (25 × 11 × 5 mm). The lesion did not respond to a second round of radio-chemotherapy, and clinically she progressed to left hemiplegia. There was no response to second-line chemotherapy with lomustine (110 mg/m2, Day 1/42) and procarbazine (60 mg/m2, Days 8–21/42), which was discontinued due to thrombocytopenia. The lesion grew rapidly (12/11; Figure 1B) to 29 × 18 × 25 mm and was accompanied by clinical deterioration. The patient then started IA OV treatment. Hemiplegia improved clinically 3 weeks after initiating OV therapy, although surveillance MRI 1 month after OV therapy (1/12; Figure 1B) showed an initial increase in tumor size to 30 × 32 × 30 mm, compatible with either true progression or pseudo-progression. However, follow-up MRI scans were compatible with central necrosis, followed by a slow decrease in the size of the enhancing lesion, until no radiological evidence of disease was seen (9/14; Figure 1B). Fluid-attenuated inversion, recovery (FLAIR) images captured during OV treatment showed multiple diffuse foci of FLAIR abnormalities (without clinical embolic causes, such as atrial fibrillation). For socio-economic reasons, the treatment protocol was modified to longer treatment intervals; the patient remained stable with NED. Five years from diagnosis, she decided to discontinue treatment for economic and psychological reasons. She then deteriorated clinically and radiologically and died 12 months later.
Patient 3
A previously healthy 43-year-old man complained for 2 months of increasing headaches and weakness of his left leg. MRI (1/11) showed a SOL in the right temporal lobe (48 × 42 × 36 mm), which was surgically resected. Histology revealed MGMT-negative GBM. After standard chemo-radiation with TMZ (75 mg/m2/d for 42 d), he received five additional cycles of TMZ (200 mg/m2/d × 5 d/month) and recovered clinically. Follow-up MRI showed radiological relapse (13 × 10 × 10 mm) in the initial tumor bed (6/12; Figure 1C), and subsequently he underwent another surgery. Postoperatively, he refused further chemo-radiation and started OV treatment instead (8/12), which he continues to receive. The patient works, enjoys a normal QoL, and remains radiologically stable with NED (7/15; Figure 1C) for 8.5 years from initial diagnosis.
Patient 4
A previously healthy 46-year-old male presented with right hemiparesis and unstable gait. MRI detected a left superior fronto-parietal SOL, which was surgically resected (10/15), resulting in clinical recovery. Pathology confirmed GBM with wild-type IDH. Despite negative MGMT, standard chemo-radiation with TMZ was given. Follow-up MRI showed suspicious findings of radiological relapse, with nodular enhancement, superior and inferior to the surgical bed (5/16; Figure 1D). Thus, the patient decided to proceed with monthly OV therapy. While remaining clinically asymptomatic, conventional MRI studies showed continued resolution of pathological enhancement over the next 12 months (8/16–12/17; Figure 1D). Interestingly, PET-MRI scans (8/16; 1/17; Figure 1D, bottom row) revealed diminishing gadolinium (Gd) enhancement and relatively stable hypermetabolic activity until disappearing during virotherapy (5/17). The patient remains clinically stable, conducts full physical work, and enjoys normal QoL >4 years after diagnosis.
Discussion
The four presented GBM cases, all of which relapsed after standard treatments, benefited from treatment with various OV strains. The patients achieved complete response or stable disease, with long-term overall survival ranging between 4 and 14 years. Oncolytic virotherapy was well-tolerated and enabled improved QoL for years after its initiation. While the nature of this case presentation precludes a direct comparison to empiric median survival rates (5), the prolonged survival shows the potential of OV therapy for the management of GBM. These promising observations, corroborated by well-documented radiological responses, justify further development of standardized protocols for clinical trials in GBM patients. Combination therapy, integration of various types of OVs, and extended duration of treatment after full radiological response must be carefully considered, given that late relapse can occur (patients #1 and #2) despite NED in radiological surveillance. The choice of OV types, dosage and treatment frequency for each of the four patients was personalized, based on clinical responses to virotherapy (mainly body temperature, rash or clinical improvement) as well as socioeconomic factors. Despite these significant limitations of non-standardized treatment regimens and the very small number of patients, the radiological evidence and promising clinical outcomes illustrate the contribution of OV to GBM management, even at an advanced stage. As is the case of many protocols in oncology, different regimens can be clinically effective. Due to the heterogenicity of the four individual regimens administered, it is not yet possible to define the critical component(s) for future clinical study protocols. For example, regarding DC administration, patients #2 and #4 showed a good response without DCs, suggesting that DCs could be excluded from future protocols.
Monitoring tools remain to be established and optimized to quantify the “virogram,” tumor tissue OV sensitivity, viremia levels, circulating tumor cells and tumor biomarkers in the cerebrospinal fluid, immune responses to the OVs, and radiological observations monitoring responses to OV. Most importantly, monitoring specific immune profiles before and after OV immunotherapy might detect a modulation of the immune system indicative of activation of anti-tumor immune responses. Indeed, long-term survival among GBM patients has been correlated with improved immune status (27–29), which might explain the apparent success of OV immunotherapy in managing GBM.
At the time of initial diagnosis, brain micro-tumors are present. Thus, even advanced surgical techniques can provide only limited disease control, which might account for short-term extension of patient lifespan but with little impact on long-term survival. Current standard therapy leads ideally to minimal residual disease (MRD), which inevitably results in relapse. Consequently, future clinical trial designs should administer OV immunotherapies immediately following initial standard treatment with surgery and chemo-radiation, at which point patients are closest to MRD (30).
Future radiological protocols should place more focus on documenting unique immune responses such as those identified in the presented cases. Punctate foci with abnormal enhancement and FLAIR signals were seen in patient 2, both adjacent to as well as distant from the relapse site, a few months after initiation of OV therapy. MRI of patient 4 showed decreased pathological Gd enhancement following initiation of OV therapy, until it disappeared 12 months later. On the other hand, concurrent PET-MRI images showed hypermetabolic activity. Conventionally, these findings are interpreted as possible disease progression or alternative diagnoses (e.g., embolic stroke). However, since these foci disappeared over the subsequent few months, this phenomenon might be a pseudo-progression reflecting an inflammatory response to OV infection, as described for some immune therapies (29). Thus, optimal radiological surveillance is crucial for identification of the desired and unique immune responses to OV (31).
While mixtures of OV strains may convey additive clinical effects, systematic assessment in controlled clinical trials with multiple OVs remains challenging. To date, combinations of OVs in clinical trials have been limited to one oncolytic Maraba vesiculovirus component combined with an adenoviral vaccine vector (32). Preclinically, a few studies have addressed potential synergy of OV combinations, such as adenovirus and vaccinia (33), mumps and measles virus (34), and Newcastle disease virus (NDV) with reovirus and parvovirus (35). Furthermore, combination of multiple therapeutic modalities including IM with DCs and/or CPI, might provide superior results, and the combined or sequential use of various OVs might boost the therapeutic efficacy by avoiding neutralizing antibody responses elicited against a specific virus strain. Thus, it is important to monitor immune activation of patients undergoing OV therapy by testing for antibodies against original tumor tissue as well as OV-specific neutralizing antibodies. Moreover, activation of NK and T cells can be monitored and may be relevant in understanding states of clinical remission. Immune phenotypes may vary in individual patients when tested shortly after diagnosis and before surgery (36). Accordingly, we found individual variations, which may imply that sustained remission in GBM patients may be based on different effector cells. Ideally, each patient should be tested before and at different time points during OV therapy.
Engineered OV strains might further enhance direct tumor killing and tumor-specific immune activation (37). The feasibility of these options needs to be explored first in xenograft mouse models and syngeneic tumor models to validate the mode of therapeutic activity in controlled settings (38, 39). All of these considerations need to be investigated in phase 1 trials followed by randomized large multi-center, controlled clinical trials, with the ultimate aim of developing standardized clinical protocols to further improve the outcomes of GBM patients.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
Written informed consent for publication was obtained from all patients in this case series.
Author Contributions
BG reviewed all data and prepared the manuscript. EB-D reviewed all radiological studies and prepared them for the manuscript. YP, RE GW, ES, LB, and TN contributed to the preparation of the manuscript and review of the literature. KA performed the surgeries. AT treated the patients with the help of IA and CW.
Conflict of Interest
BG is the founder and CEO, RE is CTO and YP is researcher and medical writer of Rapo Yerape Ltd., KA is the director of the Medias Klinik for surgical oncology, and AT is the founder and director of the Praxisklinik für Allgemeinmedizin.
The authors declare that this study received funding from Rapo Yerape Ltd., which had the following involvement in the study: analysis of data and preparation of the manuscript.
Acknowledgments
The authors thank Prof. John M. Gomori for his insightful input and discussions regarding the radiology data.
References
1. American Association of Neurological Surgeons (2020). Glioblastoma Multiforme. Available online at: https://www.aans.org/en/Patients/Neurosurgical-Conditions-and-Treatments/Glioblastoma-Multiforme (accessed May 27, 2019).
2. Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. (2015) 314:2535–43. doi: 10.1001/jama.2015.16669
3. Thakkar JP, Dolecek TA, Horbinski C, Ostrom QT, Lightner DD, Barnholtz-Sloan JS, et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. (2014) 23:1985–96. doi: 10.1158/1055-9965.EPI-14-0275
4. Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. (2014) 15:943–53. doi: 10.1016/S1470-2045(14)70314-6
5. Gallego O. Nonsurgical treatment of recurrent glioblastoma. Curr Oncol. (2015) 22:e273–81. doi: 10.3747/co.22.2436
6. Zamarin D, Palese P. Oncolytic Newcastle disease virus for cancer therapy: old challenges and new directions. Fut Microbiol. (2012) 7:347–67. doi: 10.2217/fmb.12.4
7. Guo ZS, Liu Z, Kowalsky S, Feist M, Kalinski P, Lu B, et al. Oncolytic immunotherapy: conceptual evolution, current strategies, future perspectives. Front Immunol. (2017) 8:555. doi: 10.3389/fimmu.2017.00555
8. Gujar S, Pol JG, Kroemer G. Heating it up: oncolytic viruses make tumors 'hot' and suitable for checkpoint blockade immunotherapies. Oncoimmunology. (2018) 7:e1442169. doi: 10.1080/2162402X.2018.1442169
9. Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. (2019) 18:689–706. doi: 10.1038/s41573-019-0029-0
10. Angarita FA, Acuna SA, Ottolino-Perry K, Zerhouni S, McCart JA. Mounting a strategic offense: fighting tumor vasculature with oncolytic viruses. Trends Mol Med. (2013) 19:378–92. doi: 10.1016/j.molmed.2013.02.008
11. Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. (2000) 6:821–5. doi: 10.1038/77558
12. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. (2015) 14:642–62. doi: 10.1038/nrd4663
13. Schirrmacher V. Fifty years of clinical application of newcastle disease virus: time to celebrate! Biomedicines. (2016) 4:16. doi: 10.3390/biomedicines4030016
14. Maroun J, Munoz-Alia M, Ammayappan A, Schulze A, Peng KW, Russell S. Designing and building oncolytic viruses. Future Virol. (2017) 12:193–213. doi: 10.2217/fvl-2016-0129
15. Gesundheit B, Rosenzweig JP. Editorial: oncolytic viruses-genetically engineering the future of cancer therapy. Front Oncol. (2017) 7:271. doi: 10.3389/fonc.2017.00271
16. Rehman H, Silk AW, Kane MP, Kaufman HL. Into the clinic: talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J Immunother Cancer. (2016) 4:53. doi: 10.1186/s40425-016-0158-5
17. Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer. (2018) 6:140. doi: 10.1186/s40425-018-0458-z
18. Wollmann G, Ozduman K, van den Pol NA. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. (2012) 18:69–81. doi: 10.1097/PPO.0b013e31824671c9
19. Martikainen M, Essand M. Virus-based immunotherapy of glioblastoma. Cancers. (2019) 11:186. doi: 10.3390/cancers11020186
20. Filley AC, Dey M. Immune system, friend or foe of oncolytic virotherapy? Front Oncol. (2017) 7:106. doi: 10.3389/fonc.2017.00106
21. Kim Y, Clements DR, Sterea AM, Jang HW, Gujar SA, Lee PW. Dendritic cells in oncolytic virus-based anti-cancer therapy. Viruses. (2015) 7:6506–25. doi: 10.3390/v7122953
22. Marchini A, Scott EM, Rommelaere J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses. (2016) 8:9. doi: 10.3390/v8010009
23. Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. (2011) 19:1008–16. doi: 10.1038/mt.2011.65
24. Gubin MM, Schreiber RD. Cancer. The odds of immunotherapy success. Science. (2015) 350:158–9. doi: 10.1126/science.aad4140
25. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. (2016) 34:2206–11. doi: 10.1200/JCO.2016.66.6552
26. Panek WK, Kane JR, Young JS, Rashidi A, Kim JW, Kanojia D, et al. Hitting the nail on the head: combining oncolytic adenovirus-mediated virotherapy and immunomodulation for the treatment of glioma. Oncotarget. (2017) 8:89391–405. doi: 10.18632/oncotarget.20810
27. Antonopoulos M, Swang V, Dionysiou D, Graf N, Stamatakos G. Immune phenotype correlates with survival in patients with GBM treated with standard temozolomide-based therapy and immunotherapy. Anticancer Res. (2019) 39:2043–51. doi: 10.21873/anticanres.13315
28. Aide N, Hicks RJ, Le Tourneau C, Lheureux S, Fanti S, Lopci E. FDG PET/CT for assessing tumour response to immunotherapy: report on the EANM symposium on immune modulation and recent review of the literature. Eur J Nucl Med Mol Imaging. (2019) 46:238–50. doi: 10.1007/s00259-018-4171-4
29. Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. (2013) 19:462–8. doi: 10.1158/1078-0432.CCR-12-2625
30. Alban TJ, Alvarado AG, Sorensen MD, Bayik D, Volovetz J, Serbinowski E, et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight. (2018) 3:e122264. doi: 10.1172/jci.insight.122264
31. Strauss SB, Meng A, Ebani EJ, Chiang GC. Imaging glioblastoma posttreatment: progression, pseudoprogression, pseudoresponse, radiation necrosis. Radiol Clin North Am. (2019) 57:1199–216. doi: 10.1016/j.rcl.2019.07.003
32. Pol JG, Atherton MJ, Bridle BW, Stephenson KB, Le Boeuf F, Hummel JL, et al. Development and applications of oncolytic Maraba virus vaccines. Oncolytic Virother. (2018) 7:117–28. doi: 10.2147/OV.S154494
33. Vaha-Koskela M, Tahtinen S, Gronberg-Vaha-Koskela S, Taipale K, Saha D, Merisalo-Soikkeli M, et al. Overcoming tumor resistance by heterologous adeno-poxvirus combination therapy. Mol Ther Oncolytics. (2015) 1:14006. doi: 10.1038/mto.2014.6
34. Zhang LF, Tan DQ, Jeyasekharan AD, Hsieh WS, Ho AS, Ichiyama K, et al. Combination of vaccine-strain measles and mumps virus synergistically kills a wide range of human hematological cancer cells: Special focus on acute myeloid leukemia. Cancer Lett. (2014) 354:272–80. doi: 10.1016/j.canlet.2014.08.034
35. Alkassar M, Gartner B, Roemer K, Graesser F, Rommelaere J, Kaestner L, et al. The combined effects of oncolytic reovirus plus Newcastle disease virus and reovirus plus parvovirus on U87 and U373 cells in vitro and in vivo. J Neurooncol. (2011) 104:715–27. doi: 10.1007/s11060-011-0606-5
36. Mostafa H, Pala A, Hogel J, Hlavac M, Dietrich E, Westhoff MA, et al. Immune phenotypes predict survival in patients with glioblastoma multiforme. J Hematol Oncol. (2016) 9:77. doi: 10.1186/s13045-016-0272-3
37. de Graaf JF, de Vor L, Fouchier RAM, van den Hoogen GB. Armed oncolytic viruses: a kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. (2018) 41:28–39. doi: 10.1016/j.cytogfr.2018.03.006
38. Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. (2017) 5:29–41. doi: 10.1158/2326-6066.CIR-16-0114
Keywords: glioblastoma, immunotherapy, oncolytic virotherapy, biological therapy, Newcastle disease virus (NDV)
Citation: Gesundheit B, Ben-David E, Posen Y, Ellis R, Wollmann G, Schneider EM, Aigner K, Brauns L, Nesselhut T, Ackva I, Weisslein C and Thaller A (2020) Effective Treatment of Glioblastoma Multiforme With Oncolytic Virotherapy: A Case-Series. Front. Oncol. 10:702. doi: 10.3389/fonc.2020.00702
Received: 17 December 2019; Accepted: 15 April 2020;
Published: 14 May 2020.
Edited by:
Zhi Sheng, Virginia Tech, United StatesReviewed by:
Peter M. Chumakov, Engelhardt Institute of Molecular Biology (RAS), RussiaChristian Badr, Massachusetts General Hospital and Harvard Medical School, United States
Braden C. McFarland, University of Alabama at Birmingham, United States
Copyright © 2020 Gesundheit, Ben-David, Posen, Ellis, Wollmann, Schneider, Aigner, Brauns, Nesselhut, Ackva, Weisslein and Thaller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin Gesundheit, Z2VzdW5kaGVpdG1kQGF0Z2VzdW5kaGVpdGluc3RpdHV0ZS5jb20=