 Dominga Racanelli1†
Dominga Racanelli1† Monica Brenca1†
Monica Brenca1† Davide Baldazzi1†
Davide Baldazzi1† Frauke Goeman2†
Frauke Goeman2† Beatrice Casini2†
Beatrice Casini2† Biagio De Angelis3Marika Guercio3
Biagio De Angelis3Marika Guercio3 Giuseppe Maria Milano3Elena Tamborini4Adele Busico4Gianpaolo Dagrada4
Giuseppe Maria Milano3Elena Tamborini4Adele Busico4Gianpaolo Dagrada4 Cecilia Garofalo5
Cecilia Garofalo5 Chiara Caruso5
Chiara Caruso5 Antonella Brunello6
Antonella Brunello6 Ymera Pignochino7
Ymera Pignochino7 Enrico Berrino8Giovanni Grignani7
Enrico Berrino8Giovanni Grignani7 Katia Scotlandi9Alessandro Parra9
Katia Scotlandi9Alessandro Parra9 Claudia Maria Hattinger9
Claudia Maria Hattinger9 Toni Ibrahim10Laura Mercatali10
Toni Ibrahim10Laura Mercatali10 Alessandro De Vita10
Alessandro De Vita10 Maria Vincenza Carriero11Matteo Pallocca2Rossella Loria2Renato Covello2Marta Sbaraglia12Angelo Paolo Dei Tos12,13
Maria Vincenza Carriero11Matteo Pallocca2Rossella Loria2Renato Covello2Marta Sbaraglia12Angelo Paolo Dei Tos12,13 Rita Falcioni2,*,‡
Rita Falcioni2,*,‡ Roberta Maestro1,*,‡
Roberta Maestro1,*,‡- 1Unit of Oncogenetics and Functional Oncogenomics, Centro di Riferimento Oncologico di Aviano (CRO Aviano) IRCCS, National Cancer Institute, Aviano, Italy
- 2Department of Research, Diagnosis and Innovative Technology, IRCCS Regina Elena National Cancer Institute, Rome, Italy
- 3Department of Onco-Haematology and Cell and Gene Therapy Unit, Bambino Gesù Children's Hospital, IRCCS, Rome, Italy
- 4Department of Pathology, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy
- 5Advanced Translational Research Laboratory, Veneto Institute of Oncology IOV – IRCCS, Padua, Italy
- 6Medical Oncology 1, Department of Oncology, Veneto Institute of Oncology IOV – IRCCS, Padua, Italy
- 7Division of Medical Oncology, Candiolo Cancer Institute, FPO-IRCCS, Candiolo, Italy
- 8Unit of Pathology, Candiolo Cancer Institute FPO-IRCCS, Candiolo, Italy
- 9Laboratory of Experimental Oncology, IRCCS Istituto Ortopedico Rizzoli, Bologna, Italy
- 10Osteoncology and Rare Tumors Center, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, Meldola, Italy
- 11Tumor Progression Unit, Department of Experimental Oncology, Istituto Nazionale Tumori Fondazione “G. Pascale” IRCCS, Naples, Italy
- 12Department of Pathology, Azienda Ospedaliera Universitaria di Padova, Padua, Italy
- 13Department of Medicine, University of Padua School of Medicine, Padua, Italy
This work describes the set-up of a shared platform among the laboratories of the Alleanza Contro il Cancro (ACC) Italian Research Network for the identification of fusion transcripts in sarcomas by using Next Generation Sequencing (NGS). Different NGS approaches, including anchored multiplex PCR and hybrid capture-based panels, were employed to profile a large set of sarcomas of different histotypes. The analysis confirmed the reliability of NGS RNA-based approaches in detecting sarcoma-specific rearrangements. Overall, the anchored multiplex PCR assay proved to be a fast and easy-to-analyze approach for routine diagnostics laboratories.
Introduction
The term “sarcoma” identifies a heterogeneous group of rare tumors comprising over 60 different histologic variants (1). Due to their rarity and heterogeneity, the accuracy of sarcoma diagnosis remains challenging. In the diagnosis of sarcomas, tumor cell morphology (shape, pattern of growth, microenvironment contexture) and the expression of differentiation markers represent the most important factors, but molecular investigations are increasingly employed to complement these pathological assessments. Indeed, the identification of histotype-specific (pathognomonic) gene alterations is of paramount importance in the differential diagnosis among sarcoma variants, between malignant and benign mimics, as well as between sarcoma and other tumor types (1–3). In particular, about one third of all sarcomas presents pathognomonic chromosome rearrangements (translocations, deletions, insertions) that result in fusion genes and corresponding expression of fusion transcripts (4). Beside diagnostic relevance, the expression of fusion transcripts may have prognostic and/or predictive implications. For example, certain rearrangements, such as those involving ALK in inflammatory myofibroblastic tumors or COL1A1-PDGFB in dermatofibrosarcoma protuberans, are predictive of the response to tyrosine kinase inhibitors (5, 6). Moreover, the detection of NTRK fusions in a broad range of malignancies, including sarcomas, has gaining much attention due to the recent demonstration of therapeutic efficacy of a new class of tyrosine kinase inhibitors in NTRK rearranged tumors (7–9).
Commonly, FISH or RT-PCR are used to detect fusion events at the genomic or transcriptional level, respectively. However, both methods present limitations. In particular, since they are suited to investigate a specific pre-defined abnormality, they inevitably rely on a prior diagnostic hypothesis (reflex testing). The advent of technologies such as next generation sequencing (NGS), aka massive parallel sequencing, has laid down the bases to overcome this limitation. By allowing the simultaneous analysis of a large set of targets (from few genes to the whole transcriptome/genome) NGS has disclosed the possibility not only to reveal diagnostic/prognostic/predictive genetic abnormalities in the absence of a prior hypothesis but also to identify new aberrations (10–12).
Here we wanted to assess feasibility, reliability, and applicability of NGS-based methods for the detection of sarcoma-associated fusion transcripts in a routine diagnostic setting. Our multicentric analysis confirms the sensitivity of anchored-based NGS profiling approaches and corroborates the suitability of these investigations in the diagnostic setting of sarcomas.
Materials and Methods
Case Selection
The study was conducted on a series of 150 sarcoma samples, representative of different sarcoma histotypes, retrieved from the pathological files of the participating institutions (Alleanza Contro il Cancro, ACC, Italian Research Network). Either Formalin-Fixed Paraffin-Embedded (FFPE) or frozen samples were analyzed. All sarcomas included in the study were histopathologically re-evaluated on hematoxylin-eosin stained slides, and representative areas were selected for molecular analyses.
NGS-based Fusion Transcript Identification
RNA was extracted from 5 to 10 μm-FFPE tissue sections using the Qiagen miRNeasy FFPE kit (Qiagen, Valencia, CA, USA) or the Invitrogen RecoverAll Total Nucleic Acid Isolation kit (Thermo Fisher Scientific, Waltham, MA, USA). For frozen samples the TRIzol reagent (Life Technologies Italia, Monza, Italy) followed by the RNeasy MinElute cleanup (Qiagen, Valencia, CA, USA) was used. Total RNA was quantified by using a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Quality was checked with the RNA 6000 Nano Kit on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), or by using the Archer PreSeqTM RNA QC qPCR Assay (ArcherDX, Boulder, CO, USA) and a threshold of DV200 >30 or PreSeq Cq <31 was used to identify high quality RNA, respectively.
FISH, RT-PCR, RT-qPCR, and IHC, used as primary detection approaches for the detection of possible fusion events, were performed during routine diagnostic procedures according to laboratory standard guidelines and validated reagents.
Three different commercially available NGS-based fusion panels were selected based on their capacity to cover most genes known to be involved in sarcoma-relevant fusions: an anchored multiplex PCR-based assay, namely the Archer FusionPlex Sarcoma kit (AMP-FPS)(ArcherDX, Boulder, CO, USA), covering 26 genes involved in sarcoma-associated fusions; two hybrid capture-based (HC) assays, namely the TruSight RNA Fusion Panel (TS-Fusion) (Illumina Inc., San Diego, CA, USA) and the TruSight RNA PanCancer Panel (TS-PanCancer) (Illumina Inc., San Diego, CA, USA) covering 507 and 1,385 genes commonly involved in cancer, respectively. Both HC assays included the 26 genes covered by the AMP-FPS kit. In a subset of samples, a customized version of the AMP-FPS panel was used to detect PAX3 fusion transcripts. Specifically, the assay was integrated with PAX3-specific primers (exons 6, 7 and 8) designed by using the Archer Assay Designer tool (ArcherDX, Boulder, CO, USA).
Libraries for all three panels were prepared and checked for quality according to the manufacturer's instructions, starting from 100 to 250 ng of RNA as input.
AMP-FPS libraries were run on either Illumina (MiSeq or NextSeq 500 Illumina Inc., San Diego, CA, USA) or Thermo (Ion S5 Thermo Fisher Scientific, Waltham, MA, USA) sequencing platforms, according to the manufacturer's instructions. HC-based libraries were sequenced on Illumina MiSeq instruments. Illumina TS-Fusion and TS-PanCancer sequencing data were analyzed by using the dedicated Illumina BaseSpace RNA-Seq Alignment tool (v.s.2.0.2), which relies on STAR and Manta algorithms (13, 14). PAR-masked/(RefSeq)hg19 was used as reference genome. A minimum of 3 million reads was obtained per sample (range 3007307–6284475). The mean percentage of reads aligned to the human genome was 98.9% (range 96.4–99.7%); the mean proportion of reads aligned to ribosomal RNA was below 2% (range 0.2–6.1%) and mean insert size was 134 bp (range 107–155 bp), in line with literature data (15). Only high-confidence fusions that passed default thresholds of the RNA-Seq Alignment tool (PASS) were recorded.
The Archer Analysis suite (v 5.1 or v 6.0) was exploited for the analysis of AMP-FPS panel results, using default settings. Default parameters (QC PASS) that, according to the Archer user manual, allow to achieve up to 95% of sensitivity in fusion detection, were employed to assess data quality. Samples included in the study met the quality cutoffs set by the Archer Analysis platform but in a few cases that, although not fulfilling all default criteria, nevertheless yielded high confidence fusion calls (cases #9, 31, 37, 47, 57, 60, 80, 126). Fusions were recorded as “high confidence calls”(strong = true in output table) if they passed all “strong evidence” default filters as described in the Archer analysis user manual (briefly: breakpoint spanning reads that support the candidate ≥ 5; “fusion_percent_of_GSP2_reads”, i.e., proportion of breakpoint spanning reads that support the candidate relative to the total number of reads spanning the breakpoint ≥10%; “min_unique_start_sites_for_strong_fusion” ≥3; fusion recorded in the Quiver database or not fulfilling the “negative evidence criteria”).
Of 48 cases (12 of the first set and 36 of the second set) where a fusion was detected by NGS but the partner genes had not been previously determined by the primary detection method, material was available for orthogonal validations (RT-PCR) in 39 cases, confirming NGS results. The involvement of SSX4 (SS18-SSX4), called sometime by the AMP-FPS assay in synovial sarcoma samples, was checked by nested RT-PCR (primers: Fw-SS18 GGACCACCACAGCCACCCCA, Rev-SSX ATGTTTCCCCCTTTTGGGTC; Rev-SSX4 GTCTTGTTAATCTTCTCCAAGG) and Sanger sequencing on a single index case.
For second level bioinformatic analyses of HC library raw data, Arriba, STAR-Fusion and Pizzly (16–18), administered through a command line interface, were employed for fusion calling using default settings.
Results
NGS-based Identification of Fusion Transcripts: Panel Comparison
As a first step toward the assessment of suitability of NGS-based approaches for the detection of pathognomonic fusions in sarcomas, performance and ease-of-use (library preparation complexity, hands-on time, user-friendly dedicated bioinformatic analysis tool) of three different NGS fusion panels were evaluated on a set of sarcoma samples previously characterized by either FISH or RT-qPCR for gene fusions (Table 1). Twenty-six samples were analyzed with a hybrid capture-based panel (HC) (Illumina TS-Fusion). Twenty samples were analyzed with an anchored multiplex PCR panel (Archer AMP-FPS), 19 of which investigated also with the Illumina TS-Fusion. In addition, 9 samples were profiled with a more comprehensive HC panel (Illumina TS-PanCancer).
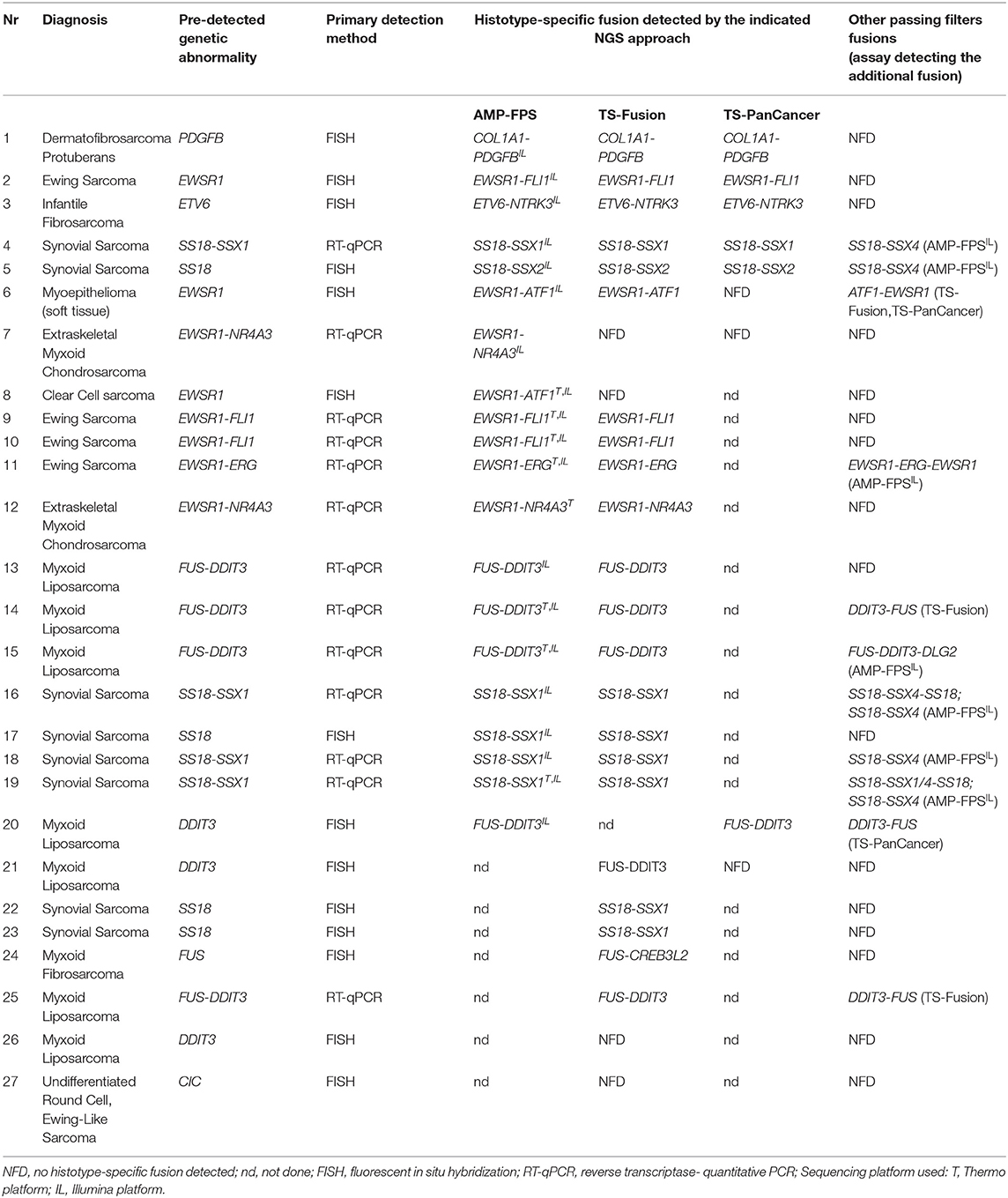
Table 1. NGS fusion profiling: panel comparison.
All three targeted RNA-sequencing panels permit the identification of common and known fusions involved in sarcomas, but also the discovery of novel fusions. The AMP-FPS panel targets a limited set of genes (26 target genes) that are commonly involved in sarcoma-associated fusions. This AMP-FPS panel employs unidirectional gene-specific primers to detect fusion transcripts involving target genes. In addition, molecular barcodes are included to enable single molecule counting, de-duplication and error correction, thus allowing quantitative analysis and confident mutation calling.
In HC-based panels the transcripts of interest are enriched by hybridization and capture with biotinylated probes (507 genes in TS-Fusion, 1385 genes in TS-PanCancer, in both cases including the 26 genes targeted by the AMP-FPS panel).
Raw data obtained with the different panels were then analyzed using the dedicated bioinformatic suite (BaseSpace RNA-Seq Alignment for Illumina HC panels, Archer Analysis platform for the AMP-FPS panel). The AMP-FPS assay correctly identified the pathognomonic fusion in all samples analyzed (20/20), irrespective of the sequencing platform used (Thermo and/or Illumina), demonstrating an excellent sensitivity. The pathognomonic fusion was correctly called in 22/26 samples analyzed with the TS-Fusion HC assay. Of the 9 cases analyzed with the TS-PanCancer HC panel, the dedicated bioinformatic tool identified the diagnostic fusion in 7 cases, in one of these as a reciprocal fusion. To further explore the performance of HC panels, data generated with TS-Fusion and TS-PanCancer panels were re-evaluated with additional algorithms, namely Arriba, STAR-Fusion and Pizzly (16–18). Although impractical in a routine diagnostic setting, as they rely on a command line interface, these tools are reported to have high fusion detection rates (16–18). With the exception of case #27, for which no algorithm detected, as high confidence calls, fusions involving the CIC gene, apparently rearranged according to FISH, at least one fusion caller was capable of detecting, among others, a fusion transcript involving the target gene in cases previously scored negative with the BaseSpace RNA-Seq Alignment tool, emphasizing the importance of software sensitivity in data analysis (Supplemental Tables 1–3).
Additional passing filters fusions (in frame and out of frame) were occasionally called beside the pathognomonic one, but the actual biological significance of these alterations is unclear. For instance, beside the canonical fusion involving SS18 and SSX1 or SSX2, additional fusions involving SSX4 were called in 5/6 synovial sarcomas analyzed with the AMP-FPS panel. It should be pointed out that the AMP-FPS approach relies on relatively small amplicons. Thus, in the presence of highly homologous genes (e.g., SSX1, SSX2, SSX4), this technique may fail to properly distinguish the target (19). Indeed, a deeper analysis of an index case confirmed the expression of SS18-SSX1, suggesting that the alleged SS18-SSX4 fusion was likely an alignment artifact.
Overall, both AMP-FPS and HC assays demonstrated a good detection capability. The HC assays were definitively more comprehensive and suitable for a research environment. In contrast, the AMP-FPS panel was limited in breath (only 26 target genes), and hence with reduced capacity of discovering new fusions, but definitively provided for a better ease-of-use. In particular, the hands-on-time for library preparation was reduced. Moreover, compared to the BaseSpace RNA-Seq Alignment, the AMP-FPS dedicated bioinformatic analysis tool (Archer Analysis platform) featured a more user-friendly graphical interface with detailed and straightforward information about the fusion (exons involved, in frame/out of frame, confidence of the call) (Figure 1).
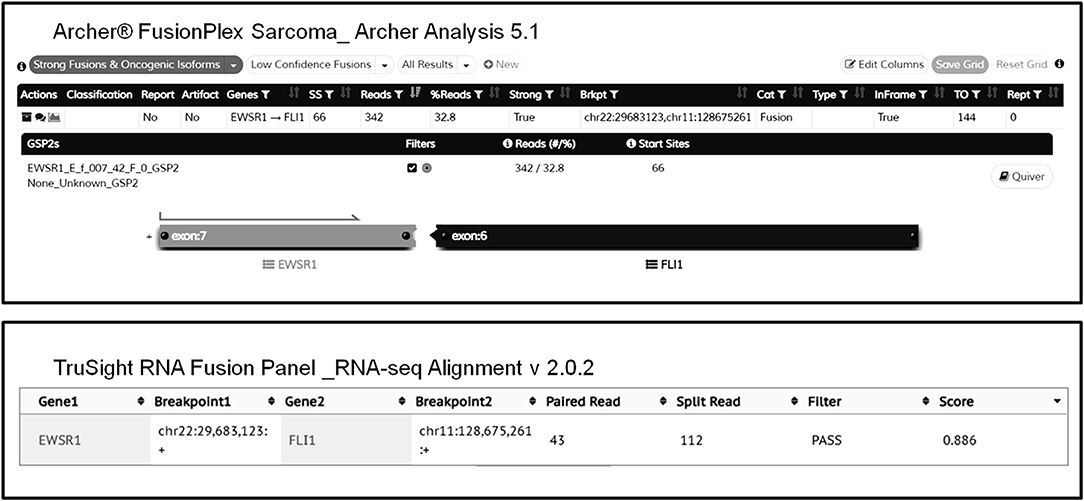
Figure 1. Representative graphical output of Archer Analysis (top) and Illumina BaseSpace RNA-Seq Alignment (bottom) tools. The EWSR1-FLI1 fusion detected in sample #2 by both AMP-FPS and HC panels is shown.
On the whole, we considered the AMP-FPS assay more suitable for routine diagnostics.
Validation on a Larger Set of Cases of the AMP-FPS Fusion Transcript Assay
Based on these results, with a view to translating NGS-based fusion identification in a routine diagnostic setting, we sought to extend the evaluation of the AMP-FPS panel (on either a Thermo or an Illumina sequencing platform) to 123 additional cases (Table 2).
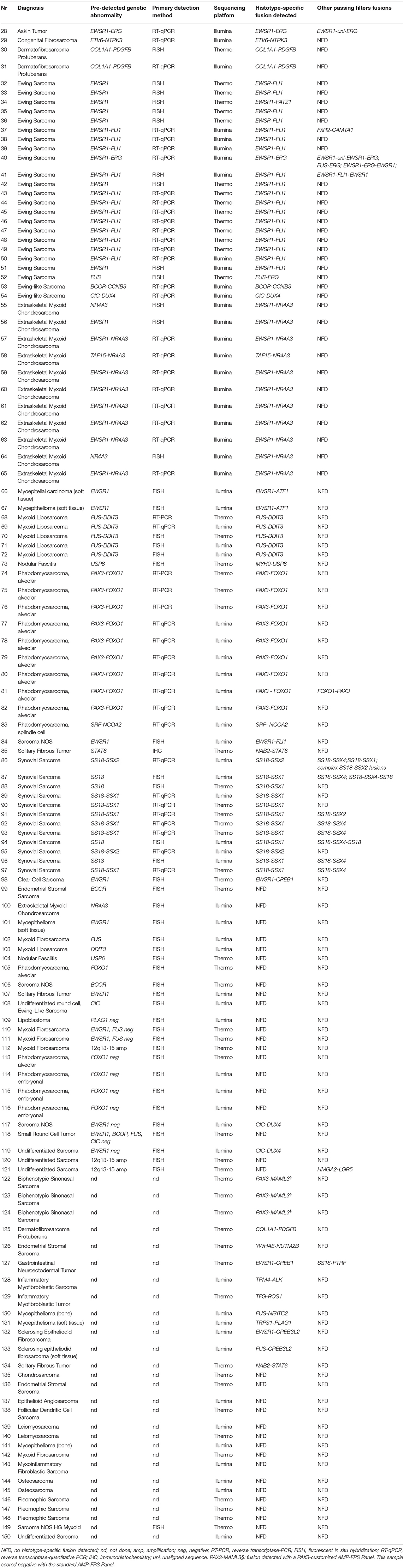
Table 2. Validation of the AMP-FPS fusion transcript assay.
Overall, the AMP-FPS panel confirmed the good performance. Of 81 cases with a pre-detected genetic abnormality suggestive of a fusion event, this NGS assay proved effective in 71, with orthogonal validations (RT-PCR) confirming the NGS result where appropriate (see Material and Methods). In the remaining 10 cases, a gene rearrangement was suggested by FISH. Nevertheless, although samples passed quality filters, the AMP-FPS assay failed to detect a fusion transcript. There are several possible explanations for this discrepancy including inadequate tumor cell fraction or low expression levels of the fusion transcript, chromosome rearrangements not yielding a fusion transcript, unusual breakpoints not covered by the assay or lack of primers covering the target gene. For instance, in two tumors (one endometrial stromal sarcoma and one sarcoma NOS) FISH indicated a rearrangement of the BCOR gene with an unknown partner. It is worth noting that the commercial AMP-FPS panel used in this study does not include primers for BCOR. Moreover, beside the common CCNB3 partner (covered by the panel), BCOR has been reported to fuse with other genes which are also not targeted by the AMP-FPS assay (e.g., ZC3H7B, MAML3, CIITA) (20–23). Thus, in the absence of probes for BCOR and potential partner genes, the failure of the assay in the 2 BCOR rearranged tumors of our series is not surprising. The same holds true for rearrangements involving NR4A3 in extraskeletal myxoid chondrosarcomas: while the AMP-FPS assay covers the most NR4A3 common partners (EWSR1, TAF15, TCF12, TFG) it lacks probes for both NR4A3 and uncommon partners (24), thus scoring negative in the presence of alternative fusions.
The AMP-FPS assay failed to detect any fusion also in 3 cases of biphenotypic sinonasal sarcoma. Although in these cases no prior investigation (FISH or RT-PCR) was performed, this tumor is known to be typified by gene fusions involving the PAX3 gene (25). Since the PAX3 gene is not covered by the commercial AMP-FPS panel, we commissioned a customization of the assay by spiking-in primers to cover PAX3 fusions. By using this customized AMP-FPS assay we were able to demonstrate and validate that all 3 cases expressed a PAX3-MAML3 chimeric transcript (Figure 2).
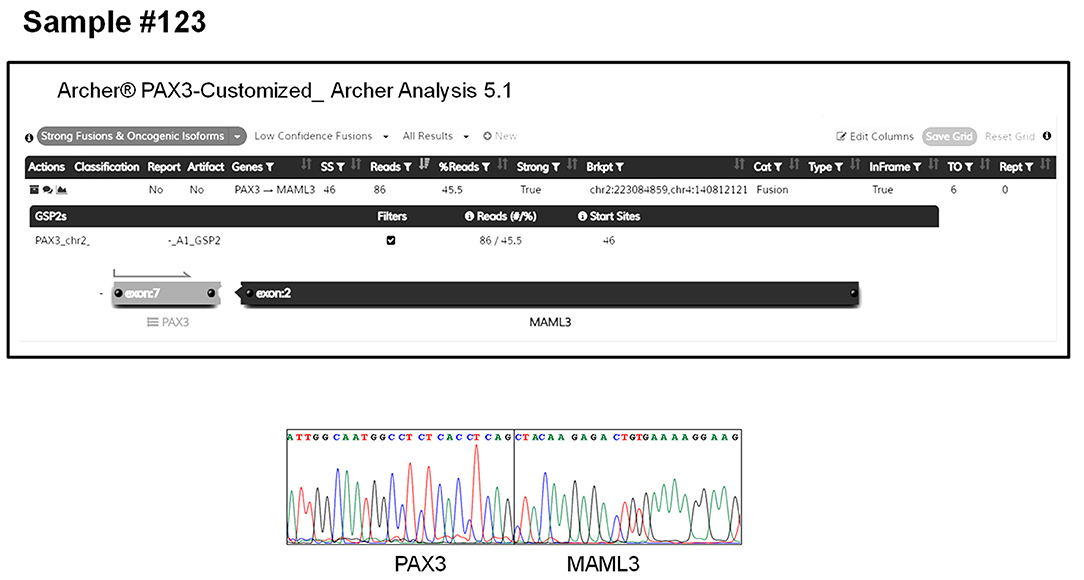
Figure 2. PAX3-MAML3 fusion detected by the customized AMP-FPS panel in a representative case of biphenotypic sinonasal sarcoma (sample #123). The top panel shows the output of the Archer Analysis tool. The bottom panel shows the validation of the fusion by RT-PCR sequencing.
Interestingly, a rare EWSR1-PATZ1 fusion was detected by AMP-FPS in one EWSR1 FISH-positive Ewing sarcoma (case #34). This fusion had been previously described in rare cases of spindled or small round cell sarcomas and it is considered to identify a distinct, Ewing-like entity (26). Moreover, the NGS profiling allowed the detection of disease-associated fusion transcripts also in a set of cases for which no prior molecular data was available or scored negative for FISH. These included one dermatofibrosarcoma protuberans (COL1A1-PDGFB), one endometrial stromal sarcoma (YWHAE-NUTM2B, aka YWHAE-FAM22B), one gastrointestinal neuroectodermal tumor (EWSR1-CREB1), one inflammatory myofibroblastic sarcoma (TPM4-ALK), one inflammatory myofibroblastic tumor (TFG-ROS1), 2 myoepitheliomas (one FUS-NFATC2 and one TRPS1-PLAG1), 2 sclerosing epithelioid fibrosarcomas (one EWSR1-CREB3L2 and one FUS-CREB3L2) and one solitary fibrous tumor (NAB2-STAT6). In addition, 2/5 tumors negative for EWSR1 rearrangements according to FISH, turned out to express a CIC-DUX4 fusion, leading to the diagnosis of CIC-DUX4 fusion-positive undifferentiated round cell sarcoma (27). In all these cases the identified fusions were confirmed by RT-PCR.
Finally, the series analyzed included also sarcoma variants typically devoid of pathognomonic fusions (e.g., leiomyosarcoma, osteosarcoma). Thus, the negative result of the NGS profiling in these cases may be considered compatible with the pathological diagnosis.
Discussion
The expression of fusion transcripts characterizes over a third of sarcomas where it may provide diagnostic, prognostic and predictive information. The cooperative effort described in this work was aimed at assessing feasibility, reliability, and applicability of NGS-based approaches for the detection of pathognomonic fusion transcripts in a routine diagnostic setting.
In line with recent reports (12, 19), our study corroborates the robustness of NGS, and in particular of AMP-FPS profiling, for the detection of clinically relevant fusions in sarcomas. On one hand, our analysis emphasizes the worth of implementing this type of approach in routine diagnostics. On the other hand, it underlines the importance of being aware of the actual detection capability of the panel used (genes covered by the assay) in relation to the specific tumor variant under investigation.
Our study demonstrates also the versatility of certain NGS fusion commercial panels to respond to specific diagnostic needs. In fact, the possibility of further implementing commercially available panels by spiking-in probes for genetic targets not included in the standard version of the assay allows to expand its detection capability. Indeed, beside PAX3, due to the recent therapeutic successes of NTRK fusions targeting drugs in solid tumors (7, 8), we are in the process of customizing the AMP-FPS panel by including primers for NTRK1 and NTRK2 (currently only NTRK3 is covered by the AMP-FPS assay).
Importantly, in the presence of a negative result, a re-evaluation of RNA and library quality is mandatory as highly degraded RNA and poor quality libraries may affect the sensitivity of the assay. Nonetheless, we found that apparently low quality samples may still be effective for fusion detection. Indeed, a few cases included in this study (cases #9, 31, 37, 47, 57, 60, 80, 126), although not fulfilling all quality criteria, nevertheless yielded a correct fusion call. This indicates that this type of assay may work even in suboptimal conditions.
Finally, when reporting the result of this type of NGS analysis, especially if negative, a statement specifying the characteristics and the limits of the assay employed (type of NGS panel, number of target genes, website of the provider for the list of targeted fusions) and the actual performance of the test according to the manufacturer's standards (fulfillment of quality parameters) should always be included in the pathology report. It is worth reaffirming that the AMP-FPS assay is designed to target the most common breakpoint regions of the genes covered by the assay. Thus, unusual breakpoints may be source of “false negative” results. Moreover, when dealing with sarcoma variants expressing uncommon fusions, the presence of primers for the target genes should be verified prior to setting up the profiling because the lack of appropriate primers will yield a false negative result. The negativity in the AMP-FPS assay of the two BCOR rearranged tumors, included in this series, is instructive in this regard.
In the case of a positive result, beside the genes involved in the fusion, the inclusion in the pathology report of details about the fusion variant detected, including reading frame of the chimeric transcript (in frame/out of frame) and exons involved might be useful. This is of particular importance if the fusion protein is potentially actionable and the retention of specific domains in the chimeric protein is crucial for drug sensitivity, as in the case of NTRK fusions (7–9).
Data Availability Statement
Sequencing data files are available in the NCBI-SRA (http://www.ncbi.nlm.nih.gov/sra) database under the accession number PRJNA608250.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethic committee Istituto Ortopedico Rizzoli IRCCS, Regina Elena National Cancer Institute IRCCS, Bambino Gesù Children's Hospital IRCCS and by the proper institutional review boards of the CRO Aviano IRCCS National Cancer Institute, Veneto Institute of Oncology (IOV) IRCCS, University of Padua, Candiolo Cancer Institute FPO-IRCCS, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) Meldola IRCCS, Istituto Nazionale dei Tumori di Milano Fondazione IRCCS. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
RM conceived the work on the behalf of the ACC sarcoma working group. All authors contributed to the generation of molecular profiling data. Each center involved in panel sequencing was responsible for generation, analyses and sharing of data. RF and RM coordinated the collection and integration of data. DR, MB, DB, FG, and BC were in charge of panel comparison. DR, MB, and DB were in charge of second-level bioinformatic analyses. RM and RF wrote the first draft of the manuscript with the support of DR and MB. All authors revised and approved the final version of the manuscript.
Funding
This work was supported by the Ministry of Health and Alleanza Contro il Cancro (ACC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with two of the authors ET and ABu.
Acknowledgments
For their suggestions and support, the authors are grateful to: Valentina Laquintana (Regina Elena National Cancer Institute, Rome); Sara Piccinin, Daniela Gasparotto, Kelly Fassetta, Beatrice Valenti (Centro di Riferimento Oncologico, CRO Aviano); Franco Locatelli, Simona Caruso, Ida Russo, Rita Alaggio, Rita De Vito, Emanuele Agolini, Martina Rinelli (Bambino Gesù Children's Hospital, IRCCS, Rome); Carolina Zamuner (Veneto Institute of Oncology, Padua, Italy); Massimo Serra, Laura Pazzaglia, Marco Gambarotti, Stefania Benini, Alberto Righi (Istituto Ortopedico Rizzoli, Bologna); Federica Pieri, Michela Tebaldi, Elisa Chiadini (Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, Meldola). Special thanks for their work go to secretaries, preclinical, and clinical coordinators of the ACC sarcoma working group, the Italian Sarcoma Group (ISG), the Rizzoli and the CRO Aviano Institutes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00489/full#supplementary-material
Supplemental Table 1. Fusion transcripts called by the Arriba algorithm.
Supplemental Table 2. Fusion transcripts called by the Pizzly algorithm.
Supplemental Table 3. Fusion transcripts called by the STAR-Fusion algorithm.
Abbreviations
NGS, next generation sequencing; FFPE, Formalin-Fixed Paraffin-Embedded; FISH, fluorescence in situ hybridization; RT-PCR, reverse transcriptase-PCR; RT-qPCR, reverse transcriptase-quantitative PCR; IHC, immunohistochemistry; HC, hybrid capture-based panel; AMP-FPS, Anchored Multiplex PCR FusionPlex Sarcoma panel; TS-Fusion, TruSight RNA Fusion panel; TS-PanCancer, TruSight RNA PanCancer panel
References
1. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. WHO Classification of Tumors of Soft Tissue and Bone. International Agency for Research on Cancer (2013).
2. Schaefer I-M, Cote GM, Hornick JL. Contemporary sarcoma diagnosis, genetics, and genomics. J Clin Oncol. (2018) 36:101–10. doi: 10.1200/JCO.2017.74.9374
3. Sbaraglia M, Dei Tos AP. The pathology of soft tissue sarcomas. Radiol Med. (2019) 124:266–81. doi: 10.1007/s11547-018-0882-7
4. Mertens F, Antonescu CR, Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer. (2016) 55:291–310. doi: 10.1002/gcc.22335
5. Ugurel S, Mentzel T, Utikal J, Helmbold P, Mohr P, Pföhler C, et al. Neoadjuvant imatinib in advanced primary or locally recurrent dermatofibrosarcoma protuberans: a multicenter phase II DeCOG trial with long-term follow-up. Clin Cancer Res. (2014) 20:499–510. doi: 10.1158/1078-0432.CCR-13-1411
6. Schöffski P, Sufliarsky J, Gelderblom H, Blay J-Y, Strauss SJ, Stacchiotti S, et al. Crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumours with and without anaplastic lymphoma kinase gene alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): a multicentre, single-drug, prospective, non-randomised phase 2 trial. Lancet Respir Med. (2018) 6:431–41. doi: 10.1016/S2213-2600(18)30116-4
7. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. (2018) 19:705–14. doi: 10.1016/S1470-2045(18)30119-0
8. Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. (2020) 21:271–82. doi: 10.1016/S1470-2045(19)30691-6
9. Solomon JP, Linkov I, Rosado A, Mullaney K, Rosen EY, Frosina D, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. (2020) 33:38–46. doi: 10.1038/s41379-019-0324-7
10. Brenca M, Maestro R. Massive parallel sequencing in sarcoma pathobiology: state of the art and perspectives. Expert Rev Anticancer Ther. (2015) 15:1473–88. doi: 10.1586/14737140.2015.1108192
11. Xiao X, Garbutt CC, Hornicek F, Guo Z, Duan Z. Advances in chromosomal translocations and fusion genes in sarcomas and potential therapeutic applications. Cancer Treat Rev. (2018) 63:61–70. doi: 10.1016/j.ctrv.2017.12.001
12. Pei J, Zhao X, Patchefsky AS, Flieder DB, Talarchek JN, Testa JR, et al. Clinical application of RNA sequencing in sarcoma diagnosis: an institutional experience. Medicine. (2019) 98:e16031. doi: 10.1097/MD.0000000000016031
13. Dobin A, Gingeras TR. Optimizing RNA-Seq Mapping with STAR. Methods Mol Biol. (2016) 1415:245–62. doi: 10.1007/978-1-4939-3572-7_13
14. Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. (2016) 32:1220–2. doi: 10.1093/bioinformatics/btv710
15. Kim B, Lee H, Shin S, Lee S-T, Choi JR. Clinical evaluation of massively parallel RNA sequencing for detecting recurrent gene fusions in hematologic malignancies. J Mol Diagn. (2019) 21:163–70. doi: 10.1016/j.jmoldx.2018.09.002
16. Uhrig S. Arriba - Fast and Accurate Gene Fusion Detection from RNA-Seq Data. (2019). Available online at: https://github.com/suhrig/arriba
17. Haas BJ, Dobin A, Li B, Stransky N, Pochet N, Regev A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. (2019) 20:213. doi: 10.1186/s13059-019-1842-9
18. Melsted P, Hateley S, Joseph IC, Pimentel H, Bray N, Pachter L. Fusion detection and quantification by pseudoalignment. bioRxiv. 166322. (2017). doi: 10.1101/166322
19. Lam SW, Cleton-Jansen A-M, Cleven AHG, Ruano D, van Wezel T, Szuhai K, et al. Molecular analysis of gene fusions in bone and soft tissue tumors by anchored multiplex PCR-based targeted next-generation sequencing. J Mol Diagn. (2018) 20:653–63. doi: 10.1016/j.jmoldx.2018.05.007
20. Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen-Gogo S, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. (2012) 44:461–6. doi: 10.1038/ng.1107
21. Panagopoulos I, Thorsen J, Gorunova L, Haugom L, Bjerkehagen B, Davidson B, et al. Fusion of the ZC3H7B and BCOR genes in endometrial stromal sarcomas carrying an X;22-translocation. Genes Chromosomes Cancer. (2013) 52:610–8. doi: 10.1002/gcc.22057
22. Specht K, Zhang L, Sung Y-S, Nucci M, Dry S, Vaiyapuri S, et al. Novel BCOR-MAML3 and ZC3H7B-BCOR Gene Fusions in Undifferentiated Small Blue Round Cell Sarcomas. Am J Surg Pathol. (2016) 40:433–42. doi: 10.1097/PAS.0000000000000591
23. Yoshida A, Arai Y, Hama N, Chikuta H, Bando Y, Nakano S, et al. Expanding the clinicopathologic and molecular spectrum of BCOR-associated sarcomas in adults. Histopathology. (2020) 76:509–20. doi: 10.1111/his.14023
24. Urbini M, Astolfi A, Pantaleo MA, Serravalle S, Dei Tos AP, Picci P, et al. HSPA8 as a novel fusion partner of NR4A3 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. (2017) 56:582–6. doi: 10.1002/gcc.22462
25. Carter CS, East EG, McHugh JB. Biphenotypic sinonasal sarcoma: a review and update. Arch Pathol Laboratory Med. (2018) 142:1196–201. doi: 10.5858/arpa.2018-0207-RA
26. Bridge JA, Sumegi J, Druta M, Bui MM, Henderson-Jackson E, Linos K, et al. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol. (2019) 32:1593–604. doi: 10.1038/s41379-019-0301-1
Keywords: sarcoma, molecular diagnosis, fusion transcripts, NGS, anchored multiplex PCR, hybrid capture-based panel
Citation: Racanelli D, Brenca M, Baldazzi D, Goeman F, Casini B, De Angelis B, Guercio M, Milano GM, Tamborini E, Busico A, Dagrada G, Garofalo C, Caruso C, Brunello A, Pignochino Y, Berrino E, Grignani G, Scotlandi K, Parra A, Hattinger CM, Ibrahim T, Mercatali L, De Vita A, Carriero MV, Pallocca M, Loria R, Covello R, Sbaraglia M, Dei Tos AP, Falcioni R and Maestro R (2020) Next-Generation Sequencing Approaches for the Identification of Pathognomonic Fusion Transcripts in Sarcomas: The Experience of the Italian ACC Sarcoma Working Group. Front. Oncol. 10:489. doi: 10.3389/fonc.2020.00489
Received: 13 December 2019; Accepted: 18 March 2020;
Published: 15 April 2020.
Edited by:
Massimo Broggini, Istituto Di Ricerche Farmacologiche Mario Negri, ItalyReviewed by:
Don A. Baldwin, Fox Chase Cancer Center, United StatesTricarico Rossella, Fox Chase Cancer Center, United States
Copyright © 2020 Racanelli, Brenca, Baldazzi, Goeman, Casini, De Angelis, Guercio, Milano, Tamborini, Busico, Dagrada, Garofalo, Caruso, Brunello, Pignochino, Berrino, Grignani, Scotlandi, Parra, Hattinger, Ibrahim, Mercatali, De Vita, Carriero, Pallocca, Loria, Covello, Sbaraglia, Dei Tos, Falcioni and Maestro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rita Falcioni, cml0YS5mYWxjaW9uaUBpZm8uZ292Lml0; Roberta Maestro, bWFlc3Ryb0Bjcm8uaXQ=
†These authors have contributed equally to this work and share first authorship