
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 13 November 2018
Sec. Molecular and Cellular Oncology
Volume 8 - 2018 | https://doi.org/10.3389/fonc.2018.00512
 Eva Eliassen1
Eva Eliassen1 Emily Lum1Joshua Pritchett2
Emily Lum1Joshua Pritchett2 Joseph Ongradi3
Joseph Ongradi3 Gerhard Krueger4
Gerhard Krueger4 John R. Crawford5
John R. Crawford5 Tuan L. Phan1,6
Tuan L. Phan1,6 Dharam Ablashi1*
Dharam Ablashi1* Stanley David Hudnall7
Stanley David Hudnall7In order to determine the role of human herpesvirus 6 (HHV-6) in human disease, several confounding factors, including methods of detection, types of controls, and the ubiquitous nature of the virus, must be considered. This is particularly problematic in the case of cancer, in which rates of detection vary greatly among studies. To determine what part, if any, HHV-6 plays in oncogenesis, a review of the literature was performed. There is evidence that HHV-6 is present in certain types of cancer; however, detection of the virus within tumor cells is insufficient for assigning a direct role of HHV-6 in tumorigenesis. Findings supportive of a causal role for a virus in cancer include presence of the virus in a large proportion of cases, presence of the virus in most tumor cells, and virus-induced in-vitro cell transformation. HHV-6, if not directly oncogenic, may act as a contributory factor that indirectly enhances tumor cell growth, in some cases by cooperation with other viruses. Another possibility is that HHV-6 may merely be an opportunistic virus that thrives in the immunodeficient tumor microenvironment. Although many studies have been carried out, it is still premature to definitively implicate HHV-6 in several human cancers. In some instances, evidence suggests that HHV-6 may cooperate with other viruses, including EBV, HPV, and HHV-8, in the development of cancer, and HHV-6 may have a role in such conditions as nodular sclerosis Hodgkin lymphoma, gastrointestinal cancer, glial tumors, and oral cancers. However, further studies will be required to determine the exact contributions of HHV-6 to tumorigenesis.
Human herpesvirus-6A and−6B (HHV-6A and HHV-6B) are linear, double-stranded DNA viruses and members of betaherpesvirinae, along with CMV and HHV-7. HHV-6A and HHV-6B were identified as two distinct herpesviruses as early as 1992 (1), and in 2014, they were formally classified as two separate species (2). While less is known about the epidemiology of HHV-6A, HHV-6B is a ubiquitous virus, with over 90% of the human population infected within the first 3 years of life. In 1986, HHV-6 was isolated from the peripheral blood mononuclear cells of AIDS-associated non-Hodgkin lymphomas by Robert Gallo and associates at the National Cancer Institute in their search for undiscovered herpesviruses that might be causing cancer in HIV-infected patients (3). Soon afterward, it was found that both HHV-6-transfected NIH3T3 fibroblasts (4) and human primary foreskin epidermal keratinocytes transfected with HHV-6 subgenomic clones PZV714 (5) and PZVB70 (6) produced tumors when injected into nude mice (7). The PZV714 and PZVB70 tumors were cytogenetically abnormal, with loss of chromosomes 12 and 13 and acquisition of extra marker chromosomes. These early results suggested that HHV-6 may be oncogenic in some settings. Decades later, several studies identified HHV-6A and HHV-6B (2) within a wide variety of tumors, including glioma, oral cancer, cervical cancer, adrenocortical tumors, gastrointestinal cancer, classical Hodgkin lymphoma, and non-Hodgkin lymphoma, as summarized in Tables 1–8 (99).
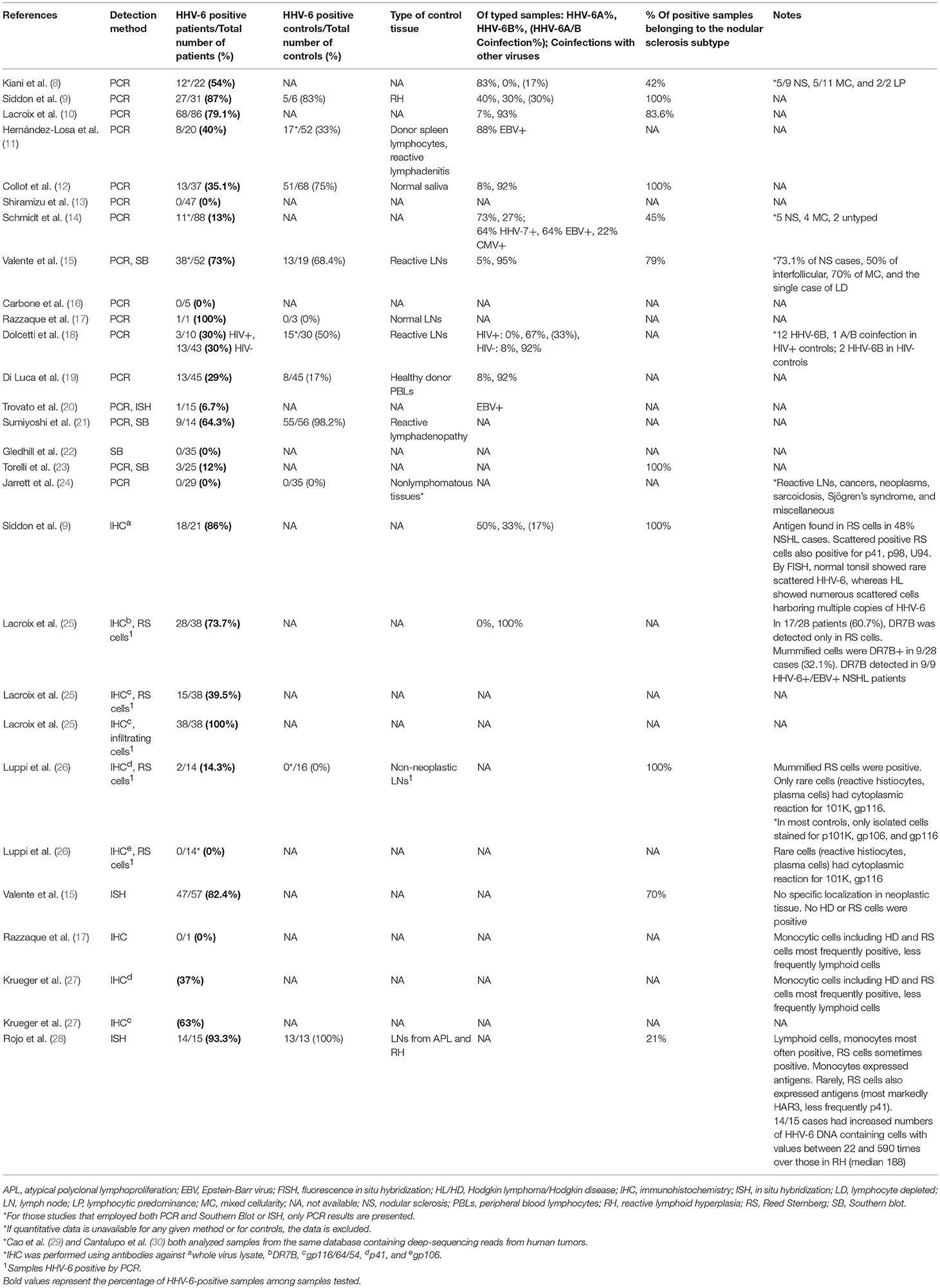
Table 1. Detection of HHV-6 in tumor tissue from patients with Hodgkin Lymphoma.
Both HHV-6A and HHV-6B share similar replication cycles: immediate-early (IE) proteins are synthesized within a few hours post-infection, which regulate the expression of the early and late genes. It takes ~72 h to complete a replication cycle (i.e., from infection to new virion release). It is now known, however, that these two species utilize distinct receptors for cellular entry: HHV-6A uses CD46, a ubiquitous complement regulatory protein, whereas HHV-6B primarily uses CD134, a molecule expressed only on activated T cells (100). Like other herpesviruses, HHV-6 displays broad cellular tropism, although it replicates most efficiently in CD4+ T cells in vitro. As is the case for other oncogenic human herpesviruses, including Epstein-Barr virus (EBV) and HHV-8, also known as Kaposi's sarcoma-associated herpesvirus (KSHV), HHV-6 establishes latency in lymphocytes and possesses a strong immunomodulatory capacity that can trigger both immunosuppressive and chronic inflammatory pathways (101).
HHV-6A/B are unique among human herpesviruses in their ability to integrate into the telomeres of chromosomes as a form of latency. They share significant homology with a neoplastic avian alpha herpesvirus, Marek's disease virus (MDV), which also integrates into the subtelomeric region of the chromosome and causes an aggressive T-cell lymphoma and immunosuppression in domestic chickens (102, 103). Approximately 1% of the world's population carries inherited chromosomally integrated HHV-6 (iciHHV-6), with the full genome integrated into the subtelomeric region of the chromosome in every nucleated cell (104). These integrated genomes can be activated by drugs (105), and immunocompromised patients with iciHHV-6 can develop symptomatic infections from the integrated strain (106). Inherited ciHHV-6 can affect telomeric stability (107), and telomeric disruption has been associated with hematologic diseases, such as aplastic anemia (108). Recent studies suggest that iciHHV-6 can be reactivated by HDAC inhibitors and can be horizontally transmitted through liver transplantation (109, 110).
Even if not directly oncogenic, HHV-6 may contribute to oncogenesis in cooperation with other viruses, such as EBV and human papillomavirus (HPV) (99). The molecular basis of this phenomenon is only partially understood. Herein, we review the wide range of neoplastic conditions associated with HHV-6 infection and the possible mechanisms by which HHV-6 might contribute to tumorigenesis.
The early use of such methods as serological testing and qualitative PCR represented pioneering steps in elucidating the roles of HHV-6 in a range of diseases, including several types of cancer. Data resulting from these techniques was largely inconclusive, and unfortunately, subsequent studies employing more targeted approaches, such as IHC and ISH, have not been carried out as frequently as would be needed to gain a better understanding of HHV-6 in cancer. Consequently, the progress of research in this field has been slow. As a ubiquitous virus, HHV-6 can often be found at a low viral load in the blood, latent or slightly reactivated without deleterious effects. It also tends to reactivate during periods of immunosuppression and stress. Thus, it is difficult to distinguish background reactivation from pathological reactivation using serological and qualitative PCR approaches that do not provide information on viral load, viral transcription, viral species, and localization of the virus. In tissues, HHV-6 may be found in infiltrating lymphocytes; because of this, staining techniques are preferred over whole-tissue PCR, as infection of tumor cells can thereby be distinguished from infection of infiltrating cells, and active infection can be differentiated from latent virus.
The role of HHV-6 in other illnesses is still under investigation, which can be problematic when choosing controls. For example, HHV-6 has been investigated as a contributing/causative factor in cases of Alzheimer's disease, benign lymphoproliferative disorders, and certain autoimmune conditions. Until the relationships between HHV-6 and these conditions (as well as others) is better understood, it is unclear whether using samples from patients with these disorders as controls is appropriate. Likewise, using samples from immunocompromised individuals may inaccurately represent the HHV-6-status of healthy controls, as patients with compromised immunity are at a much greater risk of HHV-6 reactivation, which can lead to clinical manifestations in some instances. Whenever possible, matched normal tissues from healthy individuals should be used as controls in order to avoid overestimation of the prevalence and activity of HHV-6 in healthy persons.
Since early research into the ability of HHV-6 to induce transformation, little work has been done on this topic. The use of animal models could be especially useful in this area, but to date, few such models exist for the study of HHV-6. In going forward, it will be necessary to standardize reference materials (111), emphasize collaboration among laboratories, and perform in vitro experiments to characterize the transforming potential of HHV-6, including further analysis of the functions of the viral DR-7 gene, as well as any effects the pair of viruses may have on other oncogenic agents.
In light of these limitations, this review will focus on studies and case reports that have employed robust techniques that contribute to our understanding of the correlates of HHV-6 infection. In each section, we will not focus on studies relying on standard serological techniques, instead focusing on studies using PCR, immunohistochemistry (IHC), in situ hybridization (ISH), sequencing techniques, and other targeted approaches, and the greatest attention will be paid to those studies that differentiate between active and latent infection, allow localization of viral DNA/mRNA/proteins, and distinguish between HHV-6A and HHV-6B. Likewise, we will include studies of HHV-6 detection in hematological, neurological, gastrointestinal, gynecological, and head and neck cancers in a series of tables, while studies detailing the presence of HHV-6 in other cancers that are, at present, more weakly associated with the virus, are not included. Those cancers that do not fall under these five systems, including skin, lung, and urological cancers, have fewer than five studies published on their association with HHV-6 and are not included in the tables. The tables illustrate the great variation in HHV-6 detection across studies, highlighting the challenges involved in drawing conclusions from many cases, particularly from detection rates alone.
Using PCR, HHV-6 was detected in the lymph nodes of 13/31 (12 HHV-6B, 1 HHV-6A) nodular sclerosis cases but 0/6 tissues from mixed-cellularity and lymphocyte-depleted HL (12), and the mean copy number was high at 6,711.4 per μg of DNA. Other PCR-based studies also found HHV-6 in a proportion of cases, but those based on ISH/IHC have proven to be more fruitful. In 1994, Krueger et al. reported HHV-6 p41 and gp116/64/54 antigen in Hodgkin Reed-Sternberg (RS) cells (27). Four years later, Luppi et al. detected HHV-6 p41 early antigen in the so-called “mummified” RS cells in two Hodgkin disease cases (26). The presence of viral antigen in these HL-associated cells indicated possible viral contribution to HL.
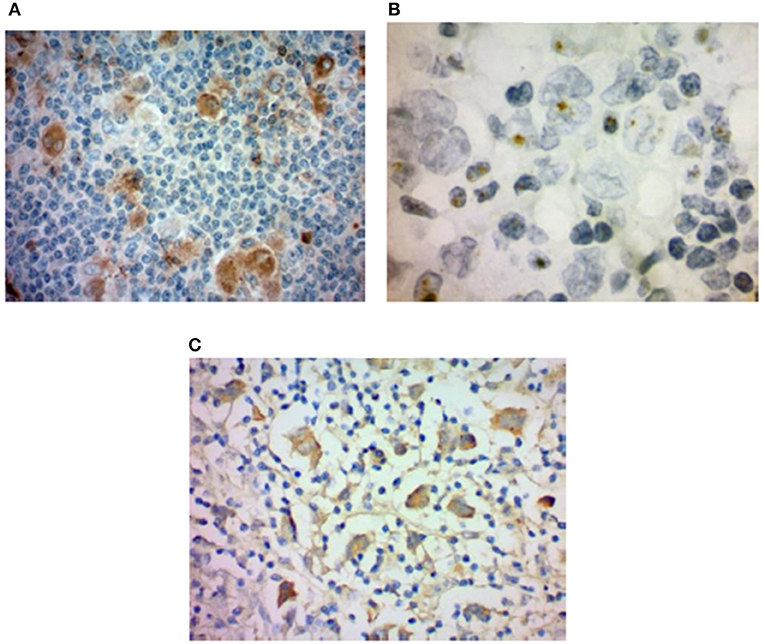
Figure 1. HHV-6 in nodular sclerosis Hodgkin lymphoma. (A) Presence of HHV-6 p41 by immunohistochemistry. Cytoplasmic staining of numerous large cells using a monoclonal antibody to HHV-6 p41 (3E3 clone). (B) HHV-6 U94 detection by immunohistochemistry. Staining with monoclonal antibody to HHV-6 U94 reveals positivity in the cytoplasm of numerous large cells. (C) Presence of HHV-6 by colorimetric in-situ hybridization with multiplex probe. HHV-6 DNA present in the nuclei of both small and large cells. From Eliassen et al. (112).
Later, Lacroix et al. analyzed lymph node biopsies from 48 patients, 38 of which were EBV-negative, and all but three of nodular sclerosis (NS) subtype (25). HHV-6 protein DR7B was detected by IHC in RS cells of 74% of EBV-negative cases, and in most of these cases (61%), DR7B was detected only in RS cells. Among the nine patients with HHV-6/EBV coinfections, the DR7B-positive RS cells were also positive for the EBV oncoprotein LMP-1, a key player in EBV-induced cell transformation. Notably, in 6 of the 9 patients with co-infections, DR7B was detected only in RS cells; all 6 patients had the NS subtype. In the single EBV/HHV-6+ patient with mixed cellularity subtype HL, only LMP-1 was found in RS cells, while DR7B was exclusively detected in infiltrating inflammatory cells. Both DR7A and DR7B have been shown to bind and inactivate the tumor suppressor p53 (113), suggesting that DR7 might play a role in lymphomagenesis (2).
The most recent study, conducted by Siddon et al., found HHV-6 by PCR in 27 of 31 (87%) NS subtype HL lymph node specimens. In nearly half of the HHV-6-positive cases (48%), U94 latent antigen, p41 early antigen, and/or p98 (HHV-6B) late antigen was expressed in scattered RS cells and leukocytes, suggesting viral replication within granulocytes and a few RS cells (9). Collectively, the use of immunohistochemical techniques has provided evidence suggestive of variable HHV-6 activity depending on HL subtype, with involvement of HHV-6 appearing more likely among NS cases. Additionally, greater viral load among NS cases than those of other subtypes supports the notion that there are subtype-specific differences in the role of HHV-6 in HL (10). Siddon et al. also reported that patients with HHV-6+ RS cells trended toward a younger age than those with EBV+ RS cells (p = 0.073).
The reported preferential localization of DR7 in RS cells, and not primarily among infiltrating inflammatory cells as would be expected as a normal distribution of latent virus or in the case of opportunistic reactivation, suggests that HHV-6 may be involved in HL pathogenesis (25). However, in some cases, HHV-6 was primarily located in infiltrating granulocytes, suggesting that HHV-6 may simply be reactivated within the immunodeficient microenvironment of HL. While greater sample sizes and further investigation is needed, the evidence suggestive of a role for HHV-6 in HL is presently strongest for NS cases.
In non-Hodgkin lymphoma (NHL), associations are weaker. The heterogeneity of methods, types of NHL, and types of controls included in past studies has compounded the difficulty in assigning a role to HHV-6 in this setting. PCR-based reports have documented an extreme range of HHV-6 positivity with 0–100% of blood/bone marrow/lymph nodes from cases testing positive. For instance, Usui et al. detected HHV-6 in up to 6.7% of ocular MALT tissues, while an earlier team documented a positive rate of 28.6% (31, 38). Among some studies, the ability to draw conclusions is limited by small sample sizes, even when the HHV-6 species is typed, the viral load is quantified, and types of lymphoma are differentiated (8, 12, 36). While the percentages of positive samples can be intriguing when viewed alone, the small sample sizes do not allow for adequate comparison. Lack of differentiation among NHL types in other studies also limits analysis.
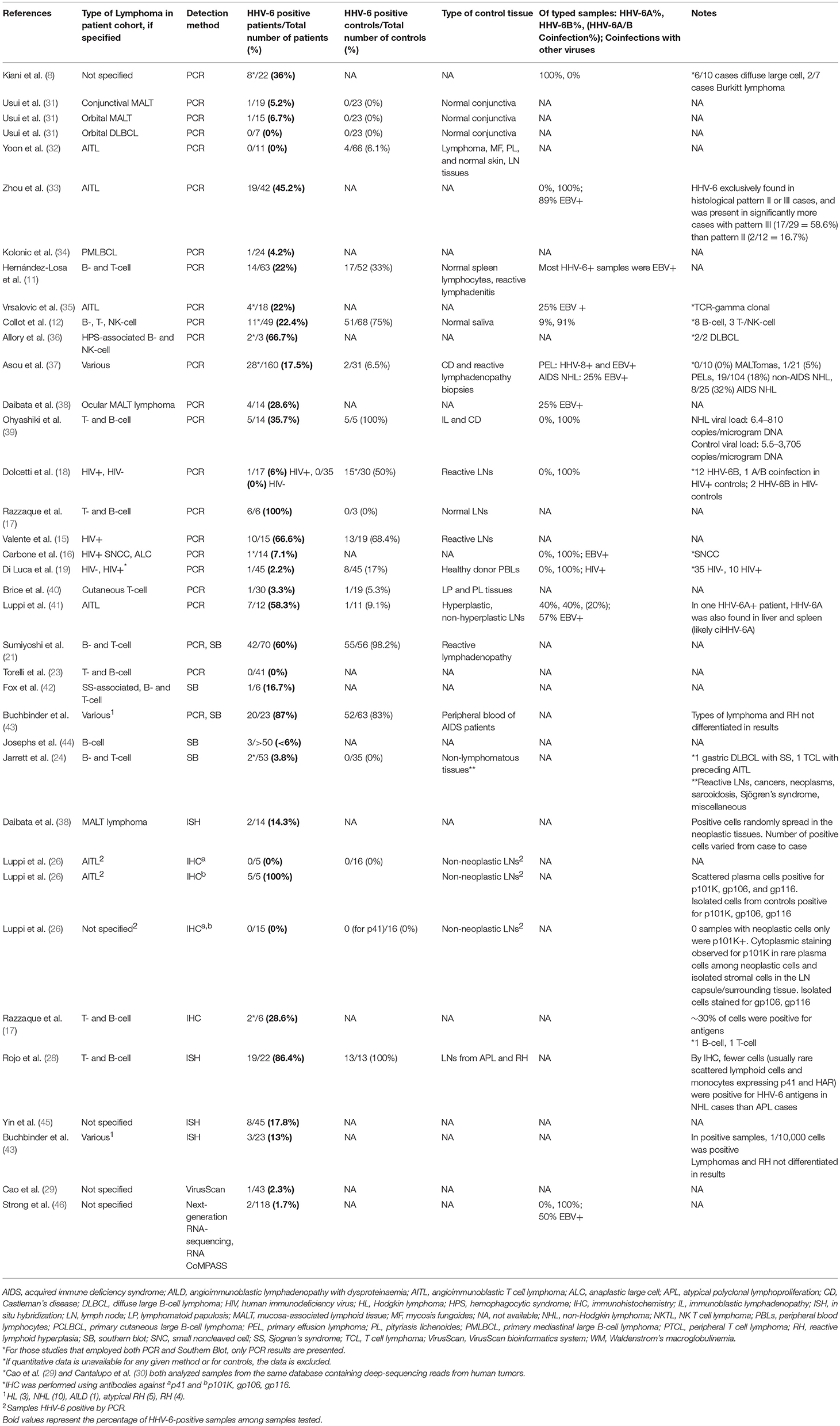
Table 2. Detection of HHV-6 in tumor tissue from patients with Non-Hodgkin Lymphoma.
By IHC/ISH, HHV-6 DNA and antigens have been detected in AITL, MALT lymphoma, unspecified NHL, and unspecified T- and B-cell lymphomas, but the signal was localized in scattered cells, often plasma cells, rather than neoplastic cells (17, 26, 28, 38, 43). RNA and DNA sequencing has identified HHV-6 in only up to 2.3% of NHL cases. On the whole, neither HHV-6A nor HHV-6B appear to be causative agents in NHL development, but investigation into specific subtypes of NHL, such as DLBCL, may be more constructive than viewing all types together. Moreover, the absence of viral DNA and antigens in neoplastic cells suggests that HHV-6 likely does not cause the development of most NHL, but it remains to be seen whether the virus might contribute to local immune dysfunction, or exacerbation of oncogenic effects of other agents, that predisposes to lymphomagenesis.
To date, the preponderance of data suggests no association between HHV-6 and leukemia. Contrasting findings, and differences in HHV-6 species predominating in bone marrow of leukemia patients, may stem from the use of different probes for HHV-6A and/or divergence in HHV-6A across geographical areas (50, 54). If this is the case, false negatives may have resulted. Likewise, PCR testing of blood samples has failed to demonstrate a strong association between HHV-6 viral load and leukemia (47, 52, 114, 115), and while serological testing has shown increased titers of HHV-6 antibody in leukemia (116–124), this is expected since HHV-6 reactivates in response to immunosuppression and chemotherapy (50). The inclusion of different subtypes of leukemia may result in skewed conclusions because HHV-6 may not have the same role across subtypes, as in the case of HL.
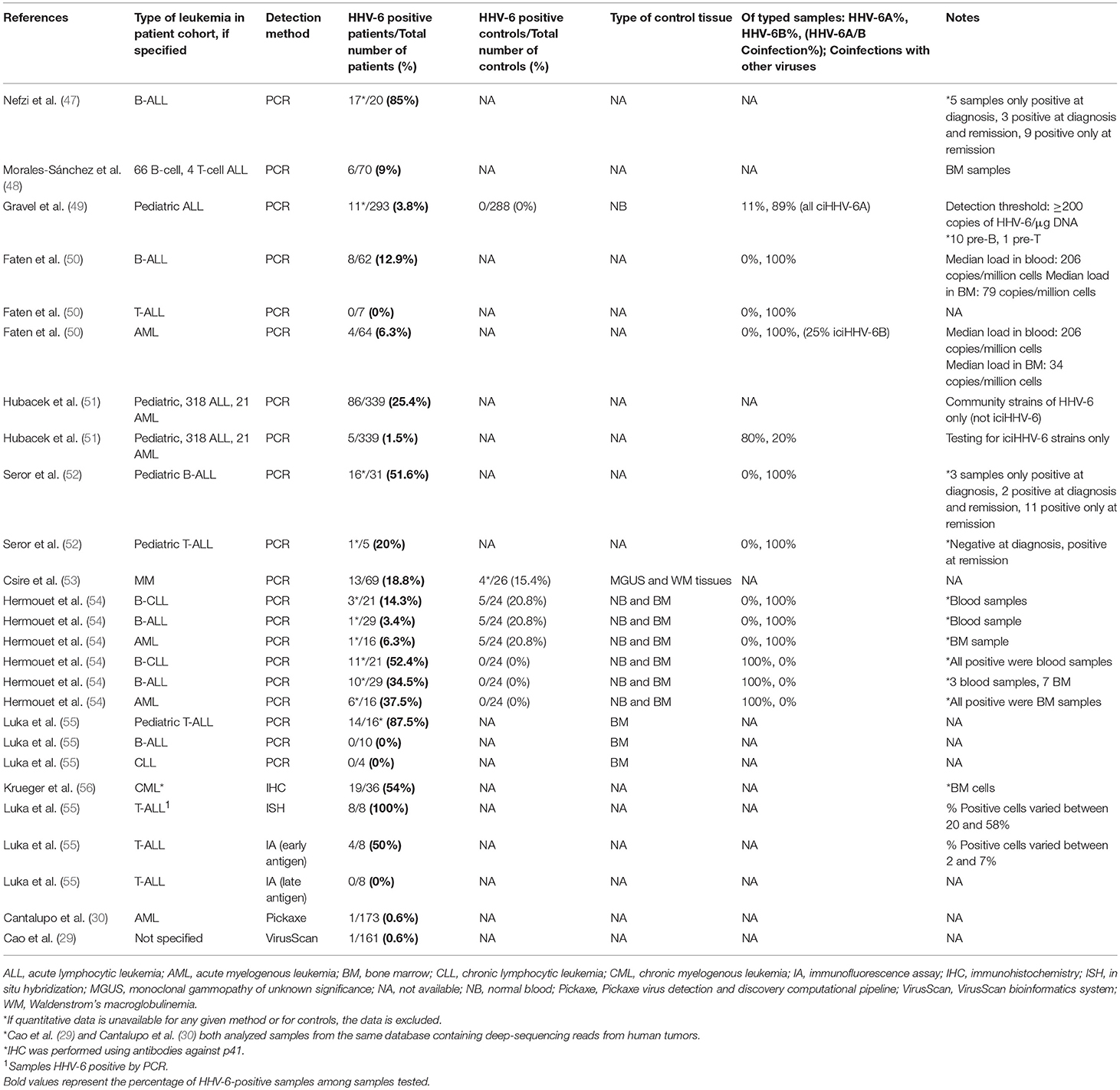
Table 3. Detection of HHV-6 in blood/bone marrow from patients with Leukemia.
Only one group has investigated HHV-6 antigen expression in leukemia, with intriguing results: HHV-6 early antigen p41 was detected in bone marrow cells—blasts and megakaryocytes—of 54% of 36 patients with chronic myelogenous leukemia (56). Since bone marrow biopsies from healthy individuals were not readily available, comparison between these two populations were not carried out. While this has been one of the strongest studies on HHV-6 and leukemia as it revealed active infection in approximately half of patients, standing alone, it is not enough to draw any clinically significant conclusions per se. Unfortunately, serological testing and PCR analysis of blood are inadequate in determining the role of HHV-6 in leukemia, and these methods fall short in differentiating between pathological and background HHV-6 infections. As blood is a relevant sample used in evaluating leukemia, this presents another hurdle; latent HHV-6 or HHV-6 undergoing low-level reactivation is occasionally present in healthy individuals. For these reasons, although many studies have made preliminary investigations into HHV-6 in several types of leukemia, forward progress of our understanding of the virus in these cancers has been stunted by the reliance upon blood PCR and serological analysis.
Both HHV-6A and HHV-6B are neurotropic viruses, with glial cells being the most common viral reservoir in the nervous system. HHV-6B infection of the brain is associated with encephalitis and febrile seizures, while the presence of HHV-6A has been studied in the context of multiple sclerosis. Several studies have shown that HHV-6 infects human astrocytes and oligodendrocytes (125–128), and both species impair glutamate uptake (129) and induce chemokine/cytokine dysregulation in persistently infected glial cells (130). Long-term latent infection is also implicated in demyelination, especially in white matter (131).
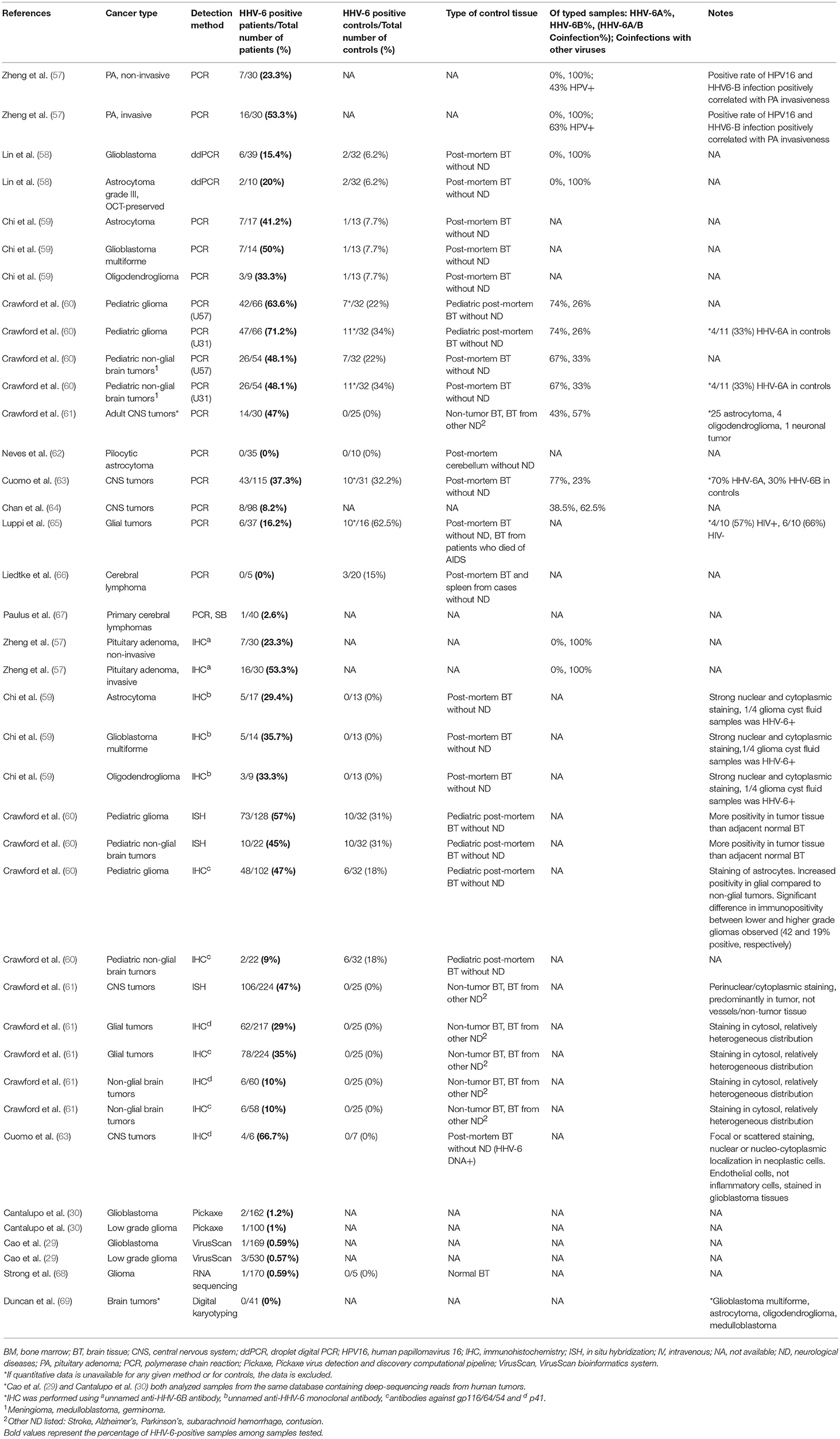
Table 4. Detection of HHV-6 in tumor tissue from patients with cancers of the nervous system.
One small study using droplet digital PCR (ddPCR) reported HHV-6B in 3 of 19 (15.8%) formalin-fixed paraffin embedded (FFPE) glioblastoma samples, 3 of 20 (15%) frozen glioblastoma samples, and 2 of 10 (20%) frozen astrocytoma grade III specimens. All were negative for cytomegalovirus (CMV) and HHV-6A (58). The viral loads of HHV-6B in tumors ranged from <100 to >20,000 copies/million cells. While the low viral loads likely indicate latency, the high loads suggest active infection, or potentially iciHHV-6. Of note, nested PCR is more sensitive, but also more prone to false positives, than ddPCR or sequencing analysis (132).
Analysis of 120 pediatric gliomas from 88 untreated patients using ISH, IHC, and nested PCR revealed higher rates of expression of the proteins U57 (major capsid protein) and U31 (large tegument protein) in tumors than in non-tumor controls (60). HHV-6 antigens were identified in 58% of low-grade gliomas compared to 19% of high-grade gliomas and 25% of non-glial tumors. The same group investigated the presence of HHV-6 in adult glial tumors and found that 47% of tumors tested positive for U57 compared to 0 of 25 controls (61). Moreover, HHV-6A early and HHV-6B late antigens were detected three times more frequently in glial tumors than in non-glial tumors, signifying more frequent active infection among those with glial tumors. Whether this correlation represents opportunistic reactivation due to the tumor will be an important avenue to explore.
These findings are supported by a more recent study, which analyzed 40 glioma tissue specimens and 13 normal brain tissue specimens (59). Using nested PCR, HHV-6 DNA was identified in 17 of 40 (42.5%) glioma tissue samples, compared to only 1 of 13 (7.7%) control samples. Along the same lines, 13 of 40 (32.5%) other glioma specimens stained positively for HHV-6, while no HHV-6 immunoreactivity was detected in normal brain tissues. Of note, HHV-6A was isolated from the glioma cyst fluids, and the HHV-6-infected specimens displayed higher levels of interleukin-6 (IL-6), IL-8, and transforming growth factor-β (TGF-β).
Taken together, the in vitro and in vivo data suggests that both species of HHV-6 are present in glioma. The greater prevalence of HHV-6 DNA and proteins among glial tumors compared to controls and the altered cytokine profile in HHV-6-positive specimens is suggestive of a potential role in gliomagenesis. To confirm this hypothesis, further studies are necessary to determine the localization of HHV-6 antigens, longitudinally investigating the effects of HHV-6 infection in animal models of glioma. Ultimately, a clinical trial is warranted to determine what role, if any HHV-6 plays in adult and pediatric glioma.
HHV-6B was recently implicated in the progression of invasive pituitary adenomas (PAs) through the toll-like receptor 3 (TLR3) signaling pathway. TLR3 is known to recognize double stranded RNA; however, studies have shown that the TLR3 cascade can also be activated by herpesviruses (133). Among 30 patients with invasive PAs and 30 patients with non-invasive PAs, HHV-6B DNA was detected in biopsy samples from 53.55% of invasive cases and 30% of non-invasive cases (57). Similarly, in invasive PA, TLR3 mRNA and protein were significantly higher than in noninvasive PA. TLR3 activity contributes to the innate immune response during viral infection via recognition of virus-associated double stranded RNA, but it has also been noted for its involvement in generating a pro-tumorigenic local environment and in promoting tumor cell invasion and proliferaation.
Rat pituitary adenoma G3 cells challenged with polyinosinic:polycytidylic acid (Poly(I:C)), a TLR3 agonist serving as a viral mimic, increased TLR3 expression and cell proliferation through the TLR3 signaling pathway (57). In addition, these cells expressed higher levels of Bcl-2, which can inhibit apoptosis, and lower levels of cleaved caspase 3. NF-κB, which is involved in cell proliferation, promotion of tumor invasion, and activation of inflammatory cytokines, was also activated, as were NF-κB-regulated genes for the inflammatory cytokines MMP9, IL-6, IL-1β, and TNF-α. Further studies on whether HHV-6 plays a direct role in TLR3 activation are necessary to confirm the significance of this association. This is the only study of its kind, but it suggests that perhaps the immunomodulatory capacity of HHV-6, and particularly its effects on innate immune effectors and mediators, leads to neurological dysfunction over time, as has recently been suggested in Alzheimer's disease (Readhead 2018), that may promote oncogenesis.
Gastrointestinal cancer (GIC) typically develops from a benign polyp, becomes an adenoma, and finally transforms into a carcinoma (134). One study detected HHV-6 in the GI tract of 63% of healthy individuals (135), whereas another study detected HHV-6 in 23% of liver transplant patients and 19% of immunocompetent patients (136). HHV-6 causes a range of GI symptoms, including diarrhea, colitis, and bacterial infection of the digestive tract (137), and an association between biliary complications and HHV-6 has been reported in many studies (136, 138).
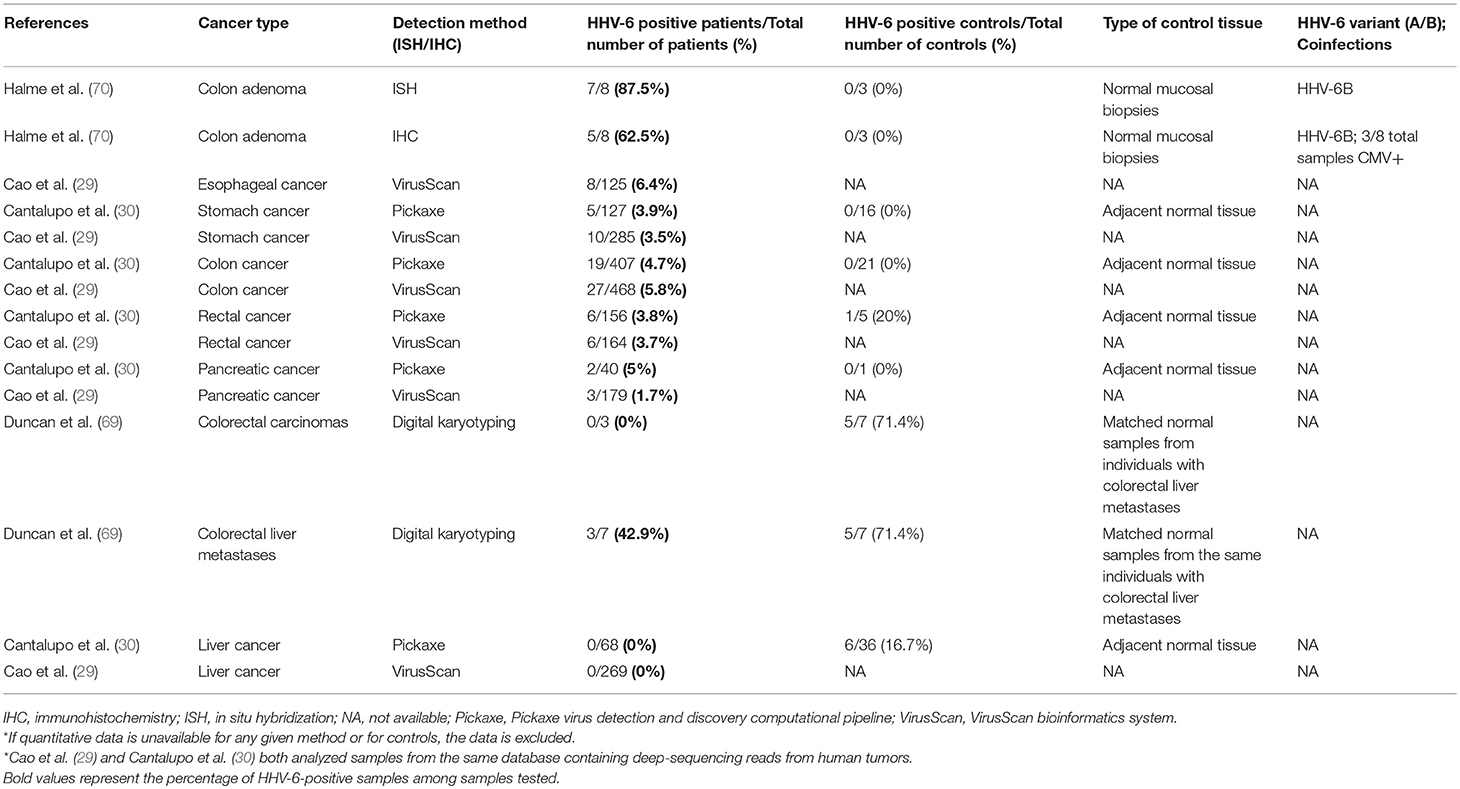
Table 5. Detection of HHV-6 in tumor tissue from patients with gastrointestinal cancers.
Using a bioinformatics and sequencing approach to investigate the viral basis of 6,813 tumors and 559 adjacent normal samples, Cao et al. found HHV-6 at a relatively high prevalence in GICs (29). Specifically, HHV-6 was detected in 26 cases of colon adenocarcinomas (5.8% of total cases), 9 cases of stomach adenocarcinomas (3.5%), 7 cases of esophageal cancer (6.4%), and 6 cases of rectal adenocarcinomas (3.7%). The virus was not detected in paired adjacent normal samples. Cantalupo et al. analyzed tumor sample sequences from The Cancer Genome Atlas (TCGA) as well and identified HHV-6 sequences in 3.9% of stomach cancer samples and 4.7% of colon cancers. In contrast, the virus was neither found in 16 paired normal stomach samples nor in 21 normal colon samples (30). Rectal cancer samples were positive at a comparable frequency (3.8%), but 1 of 5 normal controls was also positive. EBV and CMV were also frequently detected in GIC, and together with HHV-6, they were the most commonly identified viruses in stomach and colorectal tumors. In some cases, HHV-6 was detected by both DNA-sequencing and RNA-sequencing. This sequencing approach has a low risk of false-positives.
In addition, HHV-6B was frequently detected in adenomatous polyps of the colon, while mucosal biopsies from patients without adenomas revealed neither IHC nor ISH positivity (70). Gastrointestinal cancers, in addition to oral cancer, brain tumors (particularly glial tumors), and NSHL represent malignancies that deserve priority for future studies on the oncogenic capacity of HHV-6A and HHV-6B.
HHV-6A also emerged as a pathogen of interest in ovarian cancer in view of a broad-scale investigation of the ovarian cancer oncobiome using the microarray system PathoChip, which identified species of bacteria, fungi, parasites, and viruses that predominated in cancer tissue when compared to control tissue (71). Conserved and specific probes for both HHV-6A and−6B were found in the cancer biopsies but were absent in the 20 matched ovarian tissue samples and 20 non-matched ovarian biopsies. Ten instances of HHV-6A integration in various chromosomes were detected, with U47, encoding envelope glycoprotein O, identified as the most commonly inserted viral sequence. Most integrations were found in intronic or intergenic regions, but some were present at exonic and sub-telomeric loci. Of the genes that HHV-6A sequences were found to be integrated into or located near, most were significantly associated with cancers, and six, including CPLX1, IGFBP3, and the oncogene SH3RF2, were associated with malignant tumor formation (p = 8.45 × 10−7). Although HHV-6A was one of many pathogens detected more frequently in cancer tissues than in matched controls, the association between the genes that the virus integrated into or near to and the development of cancer, as well as the lack of HHV-6 in non-cancerous samples, makes a case for closer examination of integrated HHV-6 in ovarian cancer. A single study is insufficient to draw strong conclusions from, so reproduction of these initial findings is a logical next step.
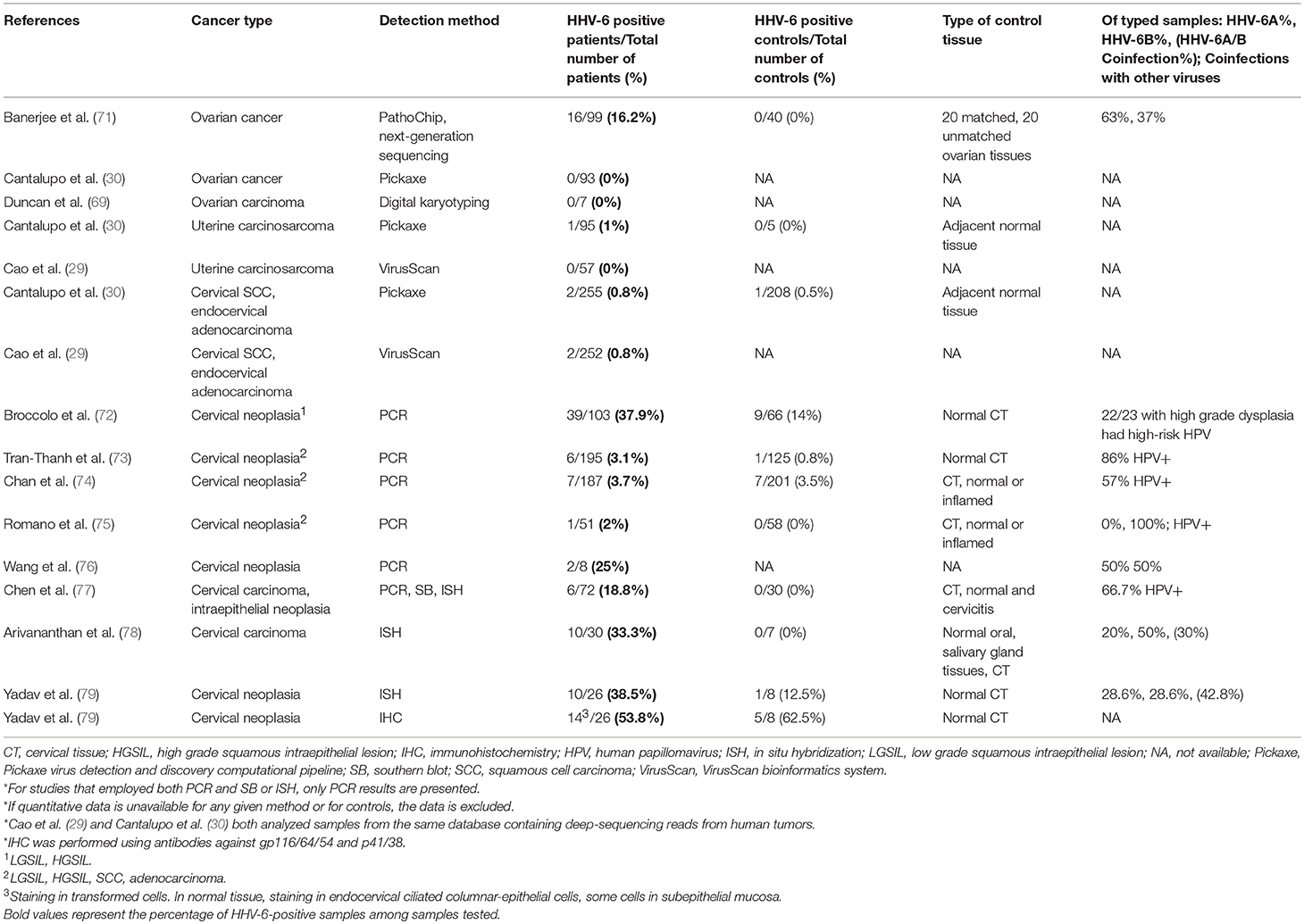
Table 6. Detection of HHV-6 in tumor tissue from patients with gynecological cancers.
Oral cancer is often linked to tobacco and/or alcohol use, as well as to HPV infections. The salivary glands serve as a reservoir for HHV-6, which is persistently shed in the saliva. While HHV-6 has not been shown to cause oral cancer, by facilitating the transformation process, HHV-6 may serve as a co-factor with HPV and chemical carcinogens found in tobacco and alcohol.
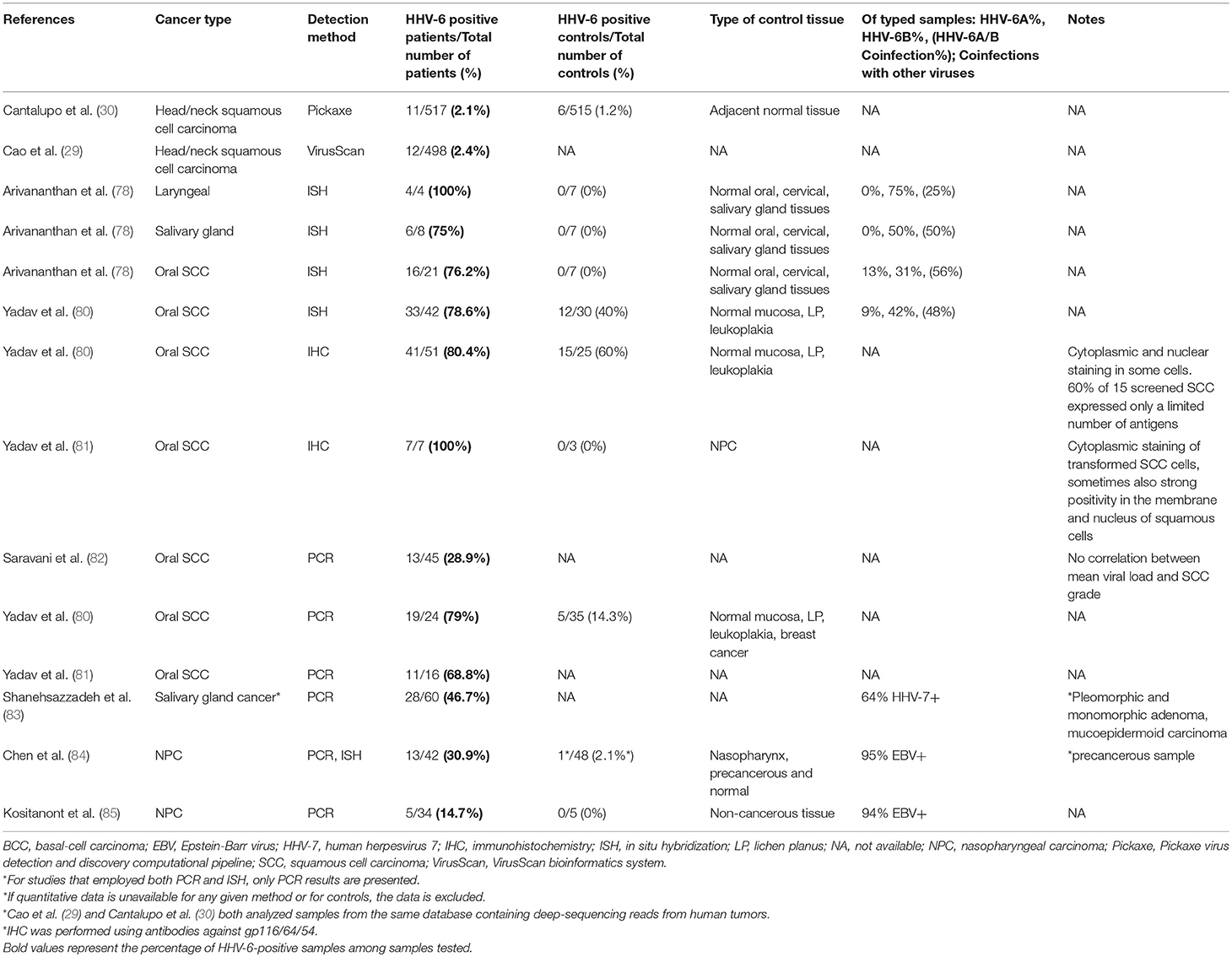
Table 7. Detection of HHV-6 in tumor tissue from patients with head and neck cancers.
Both HHV-6 DNA and antigens were detected in early studies using tissue samples from oral mucosal tumors (80, 81), but they were undetectable in controls. Antigen (HHV-6 glycoprotein) was detected by immunoperoxidase staining in transformed squamous cells of all samples, where it was localized in the cytoplasm, cell membrane, and nucleus; it was not found in cells surrounding the carcinomas. Of seven HHV-6 antigen-positive tumor samples, only five were positive for HHV-6 DNA, suggesting relatively low levels of HHV-6 DNA within these cells (81). The failure to detect DNA in the antigen-expressing cells could also mean that the virus acts using a “hit-and-run” method, in which it participates in the early phases of tumor development by spurring oncogenic changes within the cell, but then effectively disappears (139). Additionally, elevated levels of IgA antibodies to HHV-6 were detected in five patients with advanced oral carcinoma, but in none of the controls.
Follow up investigations revealed HHV-6 DNA and antigens by PCR, ISH, and IHC in oral squamous cell, laryngeal, and salivary gland carcinomas but not in normal oral and salivary gland biopsies (78, 80). Many HHV-6-positive carcinoma samples revealed HHV-6A/HHV-6B co-infection. Most recently, HHV-6 DNA was found in 27% of FFPE tissue samples from oral squamous cell carcinomas (82). No association with mean viral load or histopathologic grade was identified. The level of contrast in HHV-6 DNA and proteins between oral cancer and control tissues is intriguing and merits further study. The presence of the virus in transformed cells- and not in cells outside of the tumor- is of particular consequence, and efforts should be made to replicate this finding with larger sample sizes. As only a single report on HHV-6 in oral cancer has been published since the turn of the century, renewed efforts are due to detect the presence of the virus- active or latent- in tumor cells.
Qualitative PCR performed on non-melanoma skin cancer specimens revealed that 32% of tumors were HHV-6 positive as compared to 15% of healthy skin specimens (140), and Cao et al. found HHV-6 in only 0.4% of melanomas via sequencing (29). The current data, though very limited in scope, does not favor the involvement of HHV-6 in skin cancers and points instead to opportunistic reactivation. A similar situation is seen in such heterogeneous conditions as multiple myeloma, sarcoma, and prostate, uterine, liver, kidney, thyroid, and pancreatic cancers, for which few studies have been carried out, and associations are weak and not supportive of an etiological role for HHV-6.
Several studies indicate that HHV-6 does not contribute to breast cancer (78, 80, 141). Interestingly, the HHV-6B U54 tegument protein inhibits MCF-7 breast cancer cell proliferation in vitro by inhibiting NFAT activity (142).
HHV-6 has been detected in conjunction with several other pathogens in a range of conditions. Describing each such study is beyond the scope of this review, so only those studies providing the most meaningful data, in terms of their contributions to an understanding of cooperative activity between HHV-6 and other viruses, will be detailed.
HHV-6 DNA has been detected in many cases of B- and T-cell NHL (24, 44), sometimes in addition to other viruses. Nakayama-Ichiyama et al. (88, 89) reported dual HHV-6/HHV-8 infection in 2 cases, a primary cutaneous large B cell lymphoma and a diffuse large B-cell lymphoma (DLBCL) with dual HHV-6/HHV-8 positive, EBV-negative tumor cells, and negative serology for HIV and human T lymphotropic virus 1 (HTLV-1). The co-localization of these viruses in tumor cells of B-cell lymphomas is quite compelling and deserves follow-up. In a study of 191 primary effusion lymphoma (PEL) cases, HHV-6 was detected in 18% of HHV-8-negative tumors and in 5% of HHV-8-positive tumors (37). Since all PEL tumors are currently defined as HHV-8-positive, the diagnosis of PEL is questionable in some of these cases. HHV-6 infection of the HHV-8 positive B-cell lymphoma line BCBL-1 induces HHV-8 lytic activation (143), and it has been proposed that the simultaneous activity of an HHV-6 gene homologous to the adeno-associated virus (AAV) type 2 rep gene (144) and HHV-8 regulatory genes could play a role in lymphomagenesis (145, 146).
B-cell (38, 46) and T-cell (35, 41) lymphomas with dual HHV-6 and EBV positivity have also been described, often using PCR. Standing alone, these PCR-based studies have not provided overly compelling evidence for or against a cooperative relationship in lymphomagenesis. In a recent case-control study of 214 NHL cases and 214 matched controls from three population-based prospective cohorts, seropositivity for HHV-6A immediate-early 1 protein (IE1A), but not HHV-6B IE1, was significantly associated with a greater risk of developing NHL of any type (OR 1.85, 95% CI 1.04–3.29) (147). However, levels of antibodies against EBV proteins ZEBRA and EA-D were slightly correlated with IE1A seropositivity, and the correlation between HHV-6A IE1A and NHL development was somewhat weaker after adjusting for these EBV-specific antibodies, suggesting that HHV-6A activity may interact with EBV and increase the risk of NHL. Alternatively, an immunosuppressive or otherwise altered environment in individuals at risk for NHL may set the stage for HHV-6A reactivation, although the finding that HHV-6B IE1 was not correlated with NHL argues against this. However, if this is the case, HHV-6A activity may serve as a biomarker that can be a predictive factor for NHL. The longitudinal nature of this study, the differentiation between HHV-6A and HHV-6B, and the use of a newly developed serological assay that can identify active HHV-6 were definite strengths. Subtype-specific analysis would be a worthwhile avenue to pursue going forward.
Among lymphomas demonstrating coinfection, the potential of a cooperative role has been most thoroughly described in the setting of angioimmunoblastic T-cell lymphoma (AITL). EBV and HHV-6 have been detected in B cells and plasma cells, respectively, of patients with AITL; neither virus was detected in these patients' neoplastic CD4+ T cells (35, 41). Among a cohort of EBV-positive AITL cases with advanced pattern III histology, some were HHV-6B-positive, whereas none of the dual-infected cases were typed as early pattern I histology (33). While the presence of both HHV-6 and EBV may indicate a cooperative pathogenic role, it is also possible that it merely indicates a level of immune dysfunction that allows for viral reactivation from latency. Interestingly, however, HHV-6 and EBV viral loads in the dual-positive tumors displayed an inverse relationship (i.e., samples with the highest EBV or HHV-6 load had relatively low levels of the other virus) (33). Early studies also reported evidence of HHV-6 infection in HIV-associated non-Hodgkin lymphoma (NHL). In this immunocompromised population, HHV-6 was found in up to 32% of B-cell lymphomas (16, 18, 19).
HHV-6 has been isolated from NHL biopsies at highly variable rates, and the wide range of detection methods used on neoplastic tissues, as well as the difficulty in obtaining healthy lymph nodes for use as controls, limits the ability to unequivocally confirm the involvement of HHV-6 in triggering malignant transformation in NHL. However, in the face of such ambiguity, comparison of other relevant factors can be helpful in determining how HHV-6 affects the development of NHL. For example, Zhou et al. found that HHV-6B viral load was dramatically higher in AITL tissue with pattern III histology (median 40 copies/103 cells) than in samples with pattern II histology (median 0.7 copies/103 cells) (33). While a high rate of AITL tissue samples were positive for HHV-6, the virus was not detected within malignant T cells; instead, infection of plasma cells was apparent in these samples (26). These results suggest that HHV-6-infected plasma cells may thrive in the immunosuppressive tumor microenvironment. Similarly, EBV-infected large B cells are typically present within AITL tumor tissue. The presence of these two herpesviruses (HHV-6 and EBV) in non-malignant cells within the tumor lends credence to the notion that these viruses are opportunistic pathogens that thrive in the immunosuppressive tumor microenvironment. Nevertheless, a secondary supportive or contributory role for these viruses in tumor growth cannot be ruled out.
A trend of HHV-6/EBV coinfection has been observed in EBV-associated nasopharyngeal carcinoma (NPC). Among 34 NPC tumor samples tested by PCR, all of which were either World Health Organization type 2 or 3, 14.7 and 94.1% were positive for HHV-6 and EBV DNA, respectively, while none of the nasopharyngeal tissue samples from five controls were positive (85). HHV-6 infection of NPC specimens has also been correlated with heightened expression of EBV LMP-1 (84). Whether this phenomenon is a result of HHV-6 activity or whether HHV-6 reactivation results from the effects of NPC remains to be determined. The difference in prevalence between HHV-6 and EBV suggests that, if HHV-6 is involved in some cases of NPC, it is involved in a more limited subset, and likely is not sufficient for transformation.
Neoplastic cells isolated from pulmonary adenocarcinomas that contained HHV-6 by PCR (25%) were all co-infected with EBV (148). Cantalupo et al. identified HHV-6 DNA and/or RNA sequences in a small number (3.5%) of lung tumors and paired normal lung tissue (30). HHV-6 is likely not a primary carcinogenic agent in lung cancer, but its detection in some tumor cells supports a possibly contributory role.
There are over 100 types of HPV, of which at least 13 are important in the etiology of cervical cancer. HHV-6 has been shown to productively infect HPV-immortalized cells, transactivate HPV gene expression of oncoproteins, and enhance the expression of HPV RNA (75, 76, 149). Notably, two HHV-6 clones that were previously found to upregulate the expression of HPV-transforming genes were also found to transactivate the long terminal repeat of HIV-1 when combined with the HIV-1 transactivator TAT-1 in cervical carcinoma cell lines (150).
In an early analysis of squamous cell carcinoma (SCC) and cervical intraepithelial neoplasia, HHV-6A and/or B were detected by PCR in 8% of neoplastic cases compared to 0% of negative controls (77). In another early study, HHV-6 was detected by IHC in 54% of cervical carcinoma samples vs. 63% of normal tissues. However, in HHV-6+ carcinomas, virus was present within tumor cells, raising the possibility of differential viral activity across individuals through which HHV-6 may be relatively common in healthy cervical tissues, but may act as a contributory factor in cervical carcinogenesis in predisposed individuals. Of note, while HHV-6B was found in both cancerous and control samples, HHV-6A was only found in the cancerous tissues (79). Recent literature has implicated HHV-6A in cases of unexplained female infertility (151) and tissue taken from infertile women with endometrial HHV-6A infections has revealed an altered immune profile that points to local immune dysregulation in response to infection. HHV-6B has been found in the semen of healthy sperm donors as well as in the vaginal canal of healthy women (152, 153). It is possible that HHV-6A may be more pathogenic than HHV-6B in the female reproductive tract.
The presence of HHV-6 in HPV-infected women has also been correlated with a higher grade of squamous intraepithelial lesions; while 41% of high-grade lesions were HHV-6+, none of the normal HPV+ cervixes were HHV-6+ (72). A previous cohort, however, did not express this pattern, and although HHV-6 was found in 8.3% (2/24 samples) of cervical carcinomas, no carcinoma was coinfected with HPV (74).
Conflicting PCR results have been presented on the presence of HHV-6 in bladder cancer specimens. In one cohort, 7% of cases were HHV-6B+, compared to 0% of non-malignant bladder tissue. Of the HHV-6B+ positive cases, 40% were HPV co-infected (154). In another study, HHV-6 was found in both tumor and normal bladder tissues in equal proportion, and HPV was not detected in any sample (155). At present, these results are too limited in scope to draw firm conclusions but point to a bystander role for HHV-6.
It has been suggested that iciHHV-6 might predispose the formation of marker chromosomes. In the case of a patient with DLBCL and iciHHV-6 integration in chromosome 17p, 10% of the metaphases analyzed contained a clone with a second HHV-6 signal in an extra marker chromosome (86) (Table 8). The authors pointed out that structurally abnormal chromosomes are commonly found in DLBCL, and that the high expression of HHV-6 U94, a protein with DNA-binding, exonuclease, and helicase-ATP activities, may have been involved in tumorigenesis.
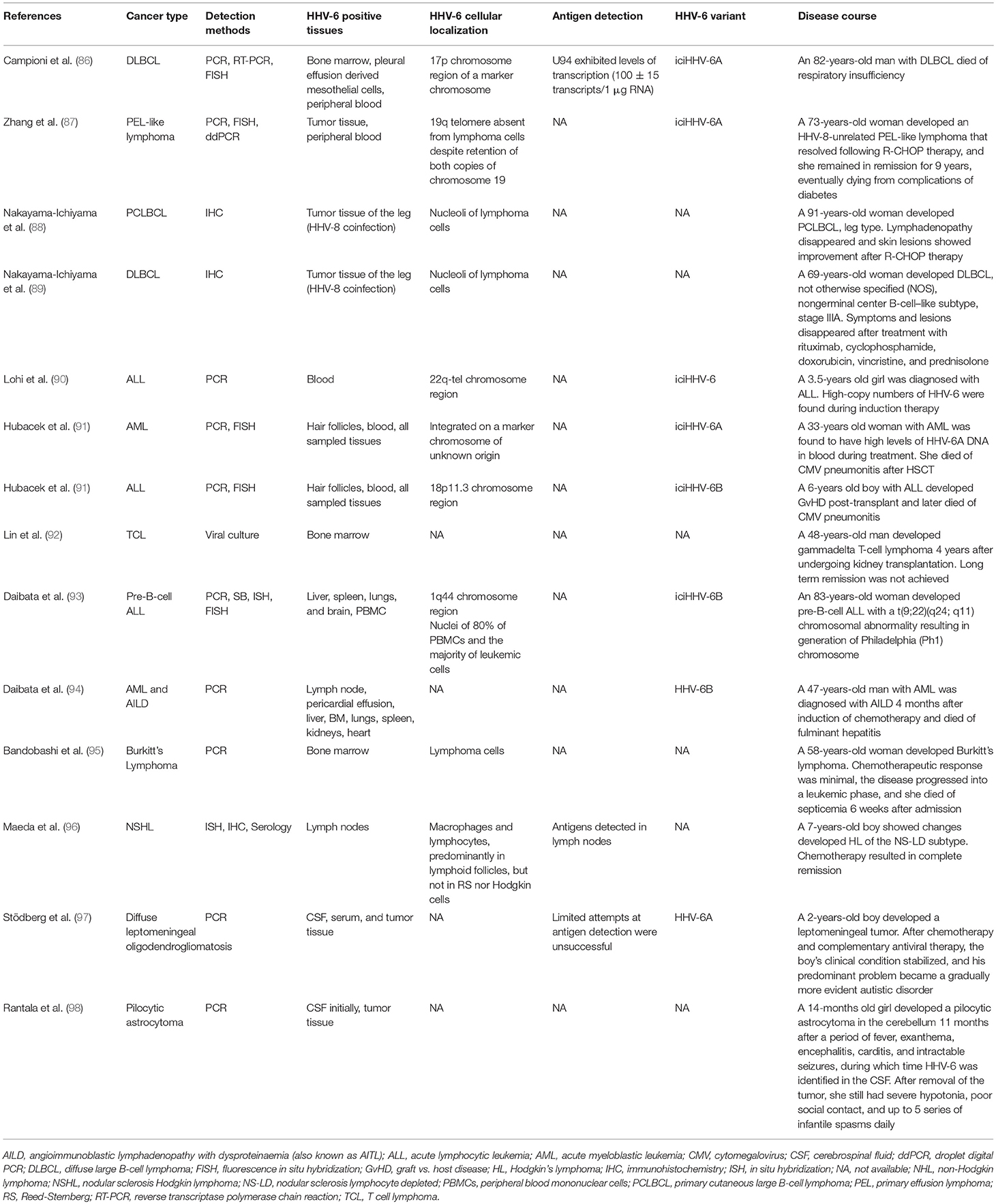
Table 8. HHV-6 detection in individual cancer patients.
Recently, iciHHV-6 was found in 2 of 35 pediatric adrenocortical tumors, and fluorescence in situ hybridization (FISH) revealed that the HHV-6 sequences were integrated at the telomeric region of 11p15, a site that is commonly implicated in this condition (156). In both cases, loss of heterozygosity (LOH) of chromosome 11 and heightened expression of IGF2 were demonstrated, as was paternal transmission of the integrated virus. LOH of chromosome 11p, via duplication of the paternal chromosome and loss of the maternal chromosome, as well as LOH at chromosome 17 appear to be early markers of adrenocortical carcinoma progression (157, 158). Integration of HHV-6 can also occur at chromosome 17p13 (159, 160), a locus that contains the TP53 tumor suppressor gene.
On the other end of the spectrum, a single report has documented the loss of iciHHV-6A from the telomere of chromosome 19q in an HHV-8-negative PEL-like lymphoma (87). Notably, loss of the integrated HHV-6A appeared to have occurred in the early stages of lymphoma development, raising the possibility of HHV-6A involvement in early chromosomal dysfunction. The unique nature of iciHHV-6 among human herpesviruses is an area deserving of greater investigation, and the effects of integration on chromosomal stability may be among the most intriguing subtopics in this field.
Transfection of normal mouse and human cells with HHV-6 DNA fragments, and with the entire genome, led to the induction of tumors in nude mice, as initially shown by Puri, Razzaque et al. (4, 7, 161). Although the mechanism of transformation was not determined, loss of normal chromosomes and the presence of extra marker chromosomes were detected. In examining viral genes (Figure 2), Kashanchi et al. demonstrated that HHV-6 ORF-1 (DR-7) gene expression led to tumor production (113). The ORF-1-associated oncoprotein has been detected in non-Hodgkin lymphoma, glioblastoma, and in RS cells of Hodgkin lymphoma (9, 25, 99), and it is thought that it may act by binding to and inhibiting expression of the tumor suppressor p53, which can also be bound by the HHV-6 U14 protein (162). Specifically, HHV-6 infection is thought to inhibit p53 nuclear localization, thereby limiting its ability to inhibit cell growth and promote apoptosis (163). These findings support the possibility that HHV-6 is oncogenic (113).
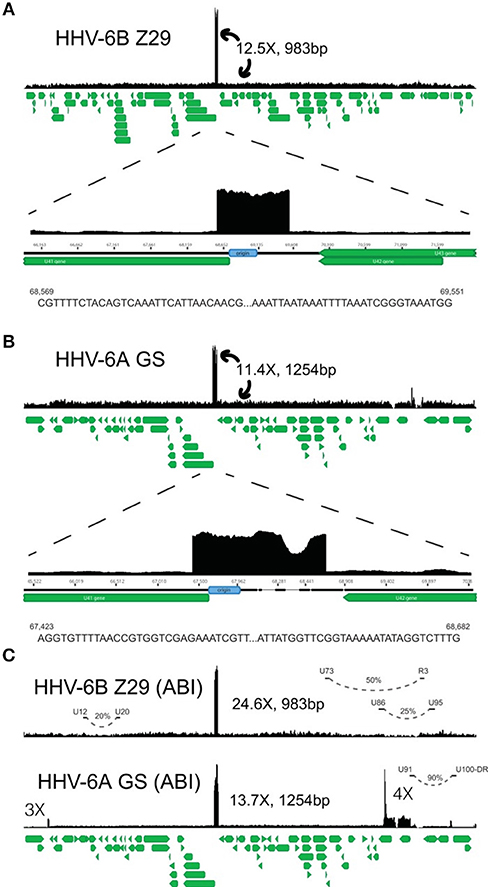
Figure 2. HHV-6A and HHV-6B genomic map. Representative coverage maps of HHV-6B Z29 and HHV-6A GS reference strains. Shotgun DNAsequencing reads from cultured virus were mapped to the NCBI HHV-6B and HHV-6A reference genomes, NC_000898 and NC_001664, respectively. The green stacked lines indicate the gene models for the respective viral species. (A) HHV-6B strain Z29 yielded a homogeneous 983-bp tandem repeat that was present at ~12.5 times higher coverage than the rest of the genome. Sequences at the 5 = and 3 = ends of the tandem repeat in strain Z29 are depicted and are different than those indicated previously (31). (B) HHV-6A strain GS yielded a heterogeneous 1,254-bp tandem repeat that was present at ~11.4 times higher coverage than the rest of the genome. Sequences at the 5 = and 3 = ends of the heterogeneous tandem repeat in strain GS are depicted. (C) ABI quantitative DNA material for HHV-6A GS and HHV-6B Z29 also demonstrated similar origin tandem repeats with additional loci with copy number differences in the GS strain. Long-distance rearrangements between U12 to U20, U73 to R3, U86 to U95, and the U91-to-U100/DR intergenic region are represented by curved dashed lines, and the estimated viral subpopulation containing the respective deletion is indicated by the percentage. From Greninger et al. (111).
Another HHV-6 protein, U24, has weaker associations with oncogenicity, but recent developments merit its mention. HNedd4L-WW3* domain (human neural precursor cell expressed developmentally down-regulated protein 4-like) has newly been identified as a cognate ligand of the HHV-6A U24 protein, which is expressed in the early stages of infection (164). Notably, Nedd4 dysregulation has been observed in glioma (165, 166) and glioblastoma multiforme (167).
The HHV-6B immediate-early gene U95 protein interacts with GRIM-19 and is associated with loss of mitochondrial membrane potential (168). GRIM-19 is involved in the oxidative phosphorylation system and in regulation of cell death, and it is also bound by KSHV, HPV type 16, and simian virus 40 LT proteins, as well as CMV RNA (169). The HHV-6B U95 promoter is regulated by R3, a repetitive region of HHV-6B with NF-κB-binding sites (170). NF-κB is important in the control of cellular proliferation and survival. As noted previously, initial data pointed to possible HHV-6B-induced TLR3, Bcl-2, and NF-κB up-regulation as well as down-regulation of cleaved caspase 3 in rat pituitary adenoma cells. However, in order to distinguish causation from correlation, this requires follow up and use of HHV-6B rather than a viral mimic (57).
IHC performed on some cancer samples has revealed an absence of HHV-6 antigens in neoplastic cells, suggesting that if HHV-6 plays a role in neoplasia, it may participate indirectly (26), perhaps by modulating the tumor microenvironment or enhancing the potential for a primary tumorigenic virus to induce neoplasia (99). In one study, adult T cell leukemia cells that were persistently infected with HHV-6B triggered the growth of uninfected, neighboring cells, indicating that HHV-6 may affect inter-cellular signaling pathways to stimulate the growth of transformed cells that are not themselves infected (171). Both species have robust immunomodulatory abilities and are able to impact cytokine and chemokine expression and networks (172). The ways by which each virus may produce both immunosuppressive and inflammatory responses, both of which can impact carcinogenesis, are myriad and too extensive to comprehensively describe in this review.
Of interest, the HHV-6A/B gene U94 exhibits anti-tumorigenic effects in some in-vitro carcinoma models. Expression of HHV-6B U94 in breast cancer and cervical cancer cell lines decreased the migration and invasiveness of the cells in vitro, and the injection of U94-expressing cancer cells into mice resulted in reduced tumor growth and more limited vasculature in the resulting tumors (173). Earlier, HHV-6A was found to reduce lymphangiogenesis and angiogenesis in vitro (174) and limit migration of endothelial cells (174) and oligodendrocyte progenitor cells (175). As noted previously, U54 also inhibited breast cancer cell proliferation in vitro (142). While the express use of the U94 gene in vector form, in the absence of the rest of the genome, may hold potential in arresting the development of certain cancers, it is unclear what these anti-oncogenic properties mean when viewed holistically, and how this particular viral gene interacts with others that are expressed during the infectious course.
In spite of high molecular homology between the two HHV-6 species, it is obvious that their pathomechanisms, including carcinogenic and co-carcinogenic effects, are different. The two species utilize different receptors for cellular entry; consequently, their intracellular signal transduction pathways differ on a basic level. HHV-6A binds to CD46, which is a complement regulatory protein (176). CD46 signal transduction exerts positive regulation on T cell proliferation, regulatory T cell differentiation, and IL-10 production, which is regarded as immunosuppressive. Expression of CD46 is upregulated in many tumors (177).
HHV-6B, on the other hand, typically binds to CD134, also known as OX40 or tumor necrosis factor receptor superfamily member 4 (TNFRSF4) (178, 179). Binding of a ligand to CD134 blocks apoptosis, preventing the death of proliferating T cells and positively regulating B cell proliferation by modulating Th1 and Th2 mediated function (180, 181). These effects on the immune system might relate to immune dysregulation and lymphomagenesis.
Lately, there has been increased recognition of the importance of regulation of gene expression at the posttranscriptional level. Among small non-coding RNA (sncRNA) products, microRNA (miRNA) produced by both infected cells and by viruses are able to mutually regulate cellular and viral mRNA translation through base-pair complementarity. Abnormal activity of cellular miRNA has been associated with cancer and viral immune evasion. Viruses express functional miRNA that target both viral and cellular transcripts to suppress or destabilize mRNA. The net result of either process is reduced synthesis of proteins encoded by target mRNA.
Roseoloviruses, which are phylogenetically ancient viruses, have not developed miRNA-associated regulatory mechanisms as sophisticated as those of the gamma herpesviruses. Both EBV and KSHV encode viral miRNAs (44 and 25, respectively) that have been directly linked with malignant transformation. Of interest, some herpesvirus miRNA (EBV-miR-BART2-5p and KSHV-miR-K12-7), despite having evolved independently, suppress identical cellular mRNA targets, thus developing similar strategies to modulate host cell transformation (182).
Little attention has been paid to roseolovirus miRNA. Only recently, five novel HHV-6A encoded sncRNAs have been identified in Jjhan cells infected by strain U1102, one of which is a miRNA. This HHV-6 miRNA targets the HHV-6A immediate early (IE) gene U86 and is consequently named miR-U86. Overexpression of miR-U86 in transduced Jjhan cells results in significant growth retardation and reduced cell viability (183). Current data suggest that miR-U86 has no role in tumorigenesis. HHV-6A alters cellular miRNA expression in infected T cells, but too little information is currently available to draw conclusions about the relationship between these changes and oncogenesis (184).
Infection of Sup-T1 cells with HHV-6B, on the other hand, results in an abundant production of sncRNAs derived from either the direct repeat regions or the lytic origin of replication that give rise to smaller RNA species. Four miRNAs were identified and named hhv6b-miR-Ro6-1,−2,−3, and−6. Ro6-2 is a seed ortholog of the human miR-582-5p, which is upregulated in certain pituitary adenomas and downregulates expression of TGF-β (185). Similar to the HHV-8 miR-155 ortholog miR-K12, HHV-6B Ro6-2 may share a broad range of cellular mRNA targets with human miR-582-5p and may modulate the host-pathogen relationship. In cells (e.g., CD4+ T cells) that do not normally express cellular miRNA-582-5p, HHV-6B infection may lead to Ro6-2 miRNA-mediated suppression of TGF-β. HHV-6B encoded miRNAs are expressed in low abundance, similar to the oncogenic Marek's disease virus, but in contrast to other herpesviruses (186). All in all, the similarities between HHV-6B encoded miRNAs and oncogenic KSHV and human miRNAs suggest that they may play roles in tumorigenesis, though more work is needed to clarify their effects.
Roseoloviruses can also regulate cell-virus interactions through the release of microvesicles. Viruses have evolved to insert viral components into uninfected cells via microvesicles (MV) that are shed from the plasma membrane, as well as exosomes, which originate within microvesicular bodies (MVBs) and are released into the extracellular environment. Both can be isolated from any bodily fluid or excretion, and they can cross the blood-brain-barrier. They can include cellular and viral proteins, mRNAs, miRNAs, lipids, and carbohydrates. Once released, they can be taken up through phagocytosis or pinocytosis by neighboring or distant cells, where the contents can have direct consequences by manipulating gene expression of the recipient cells. HHV-6 infection dramatically increases MVB formation. HHV-6B redirects MHC class I molecules into MVBs, and along with gB viral proteins, they are released in exosomes. It is possible that the MHC molecules can assist viral entry in other cells (187). A reduction in MHC class I molecules may also contribute to ineffective immune targeting and clearance of transformed cells (188). In addition, HHV-6A infected HSB-2 cells tend to form MVBs, which can contain mature virions, and the small vesicles inside the MVBs carry the envelope glycoproteins gM and gB. It is presumed that viral glycoproteins expressed in exosomes may interact to form a virological synapse and promote the efficient spreading of HHV-6A from infected to uninfected cells. Furthermore, several reports on other viruses have shown T cell activation in response to exosomes secreted by antigen presenting cells (189).
Lately, studies on extracellular vesicles have indicated that they play a significant role in tumor progression (190). Although data on HHV-6-associated microvesicle pathomechanisms is limited, comparisons with other carcinogenic herpesviruses might help to elucidate their possible tumorigenic effects. EBV, for example, can introduce viral antigens, RNA, and growth factors into other cells. Unfortunately, the absence of active viral infection in these targeted cells may hamper the verification of a causal link between the virus and diseases in which viral infection has been implicated. KSHV was also found to modulate the microenvironment through packaging of viral factors in exosomes. The viral factors increased cell migration and IL-6 production, and promoted a switch to glycolytic metabolism in recipient endothelial cells (191), all of which have been implicated in carcinogenesis (192, 193). Further studies on HHV-6A and HHV-6B associated MVs and exosomes are required to verify their direct or indirect carcinogenic effects.
Finally, both iciHHV-6 and community strains (71) of HHV-6 may affect chromosomal stability or transcription of genes associated with malignancy through integration into the cellular genome. Depending on the site of integration, telomeric disruption, loss of chromosomal integrity, and/or disruption of genes associated with cancers may result, in turn promoting oncogenesis. Proposed mechanisms of HHV-6-associated oncogenesis and oncomodulation are summarized in Figures 3, 4.
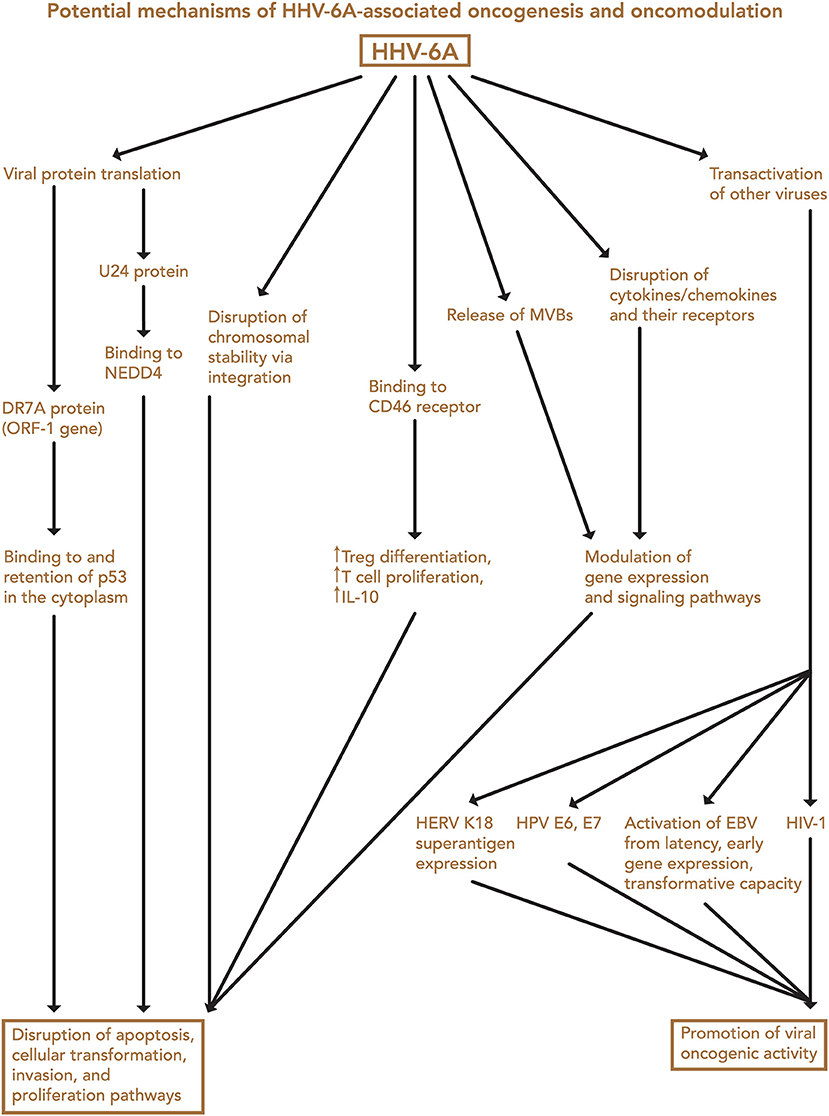
Figure 3. Potential mechanisms of HHV-6A-associated oncogenesis and oncomodulation. Several HHV-6A-mediated changes may contribute to disruption of apoptosis, cellular transformation, invasion, and proliferation pathways, and transactivation of other viruses may contribute to their oncogenic activity. EBV, Epstein-Barr virus; HERV, human endogenous retrovirus; HPV, human papillomavirus; HIV-1, human immunodeficiency virus-1; MVB, microvesicular bodies; Treg, regulatory T cell.
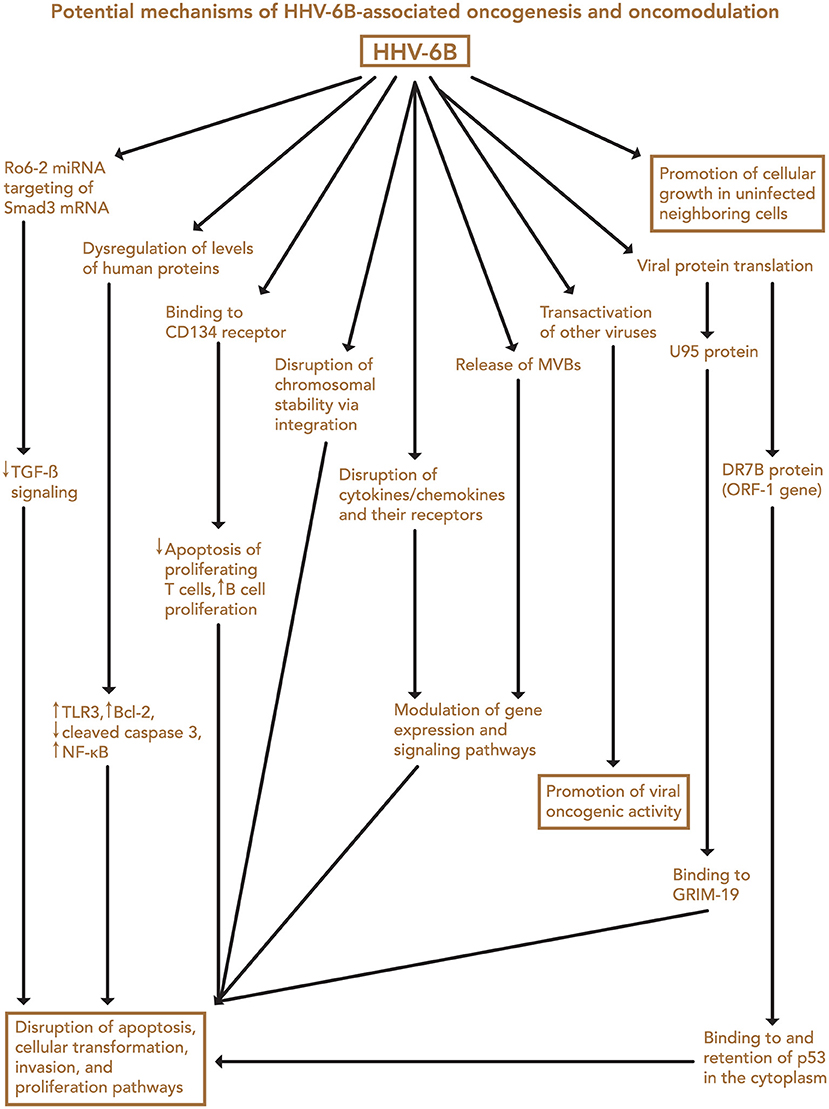
Figure 4. Potential mechanisms of HHV-6B-associated oncogenesis and oncomodulation. Several HHV-6B-mediated changes may contribute to disruption of apoptosis, cellular transformation, invasion, and proliferation pathways, and transactivation of other viruses may contribute to their oncogenic activity. In addition, HHV-6B-infected cells may promote cellular growth in neighboring uninfected cells through a presently unknown mechanism. MVB, microvesicular bodies.
HHV-6 tends to reactivate during periods of stress and immunosuppression. Like HHV-8, immunosuppressive conditions- present through inherited, environmental, or age-associated means may be key to pathogenic reactivation of HHV-6 resulting in oncogenic changes. While immunosuppression triggers viral reactivation from latency, both HHV-6A and HHV-6B are also immunomodulatory viruses that affect the functions of natural killer cells and T cells, as well as other immune cells, cytokines, and chemokines (172). In establishing persistence, the viruses employ strategies to evade detection and elimination by immune cells, which can result in down-modulation of immune cell activities. The effects of infection are myriad, time-dependent, and vary across individuals. Genetic factors are likely to affect the extent of HHV-6-asscoiated immunomodulation and outcomes during and after primary HHV-6 infection, although the contribution of genetics in HHV-6-associated illness is largely unknown. Likewise, environmental influences may impact the types of changes that the viral infection induces in individuals. In terms of HHV-6A and HHV-6B themselves, it is possible that certain variants are more prone to contributing to oncogenesis or in dampening the immune response.
HHV-6B reactivates frequently in patients after receiving hematopoietic stem cell transplantation for hematological malignancies (194) and has been shown to impair late reconstitution of CD4+ T cells, likely by affecting thymopoiesis (195–197). A weaker immune response as a consequence of HHV-6 infection may allow for circumstances beneficial for the continuation of neoplasia, as immune cells may be less effective in detecting and destroying abnormal cells. It has been suggested that HHV-6 infection contributes to the progression of GIC through the promotion of lymphopenia and immunosuppression (134), and in some instances, atypical lymphoproliferative disorders progress to overt malignant lymphoma, particularly in the setting of immunodeficiency (198, 199). Virus-mediated dysregulation of cell proliferation, differentiation, and cell death may play a role (200, 201).
While both viruses have shown oncogenic potential themselves, HHV-6A and –B-mediated transactivation of other oncogenic agents may also play an indirect role in tumorigenesis. In general, herpesviruses, but especially beta herpesviruses, are known for transactivating each other and several other heterologous viruses. HHV-6A U16 and U30 gene products transactivate E6 and E7 of HPV-16 in cervical epithelial cells (149). Additionally, HHV-6A activates EBV from latency (202), increases EBV early gene expression (203), and bolsters its transformative capacity. Meanwhile, EBV renders B cells susceptible to HHV-6 infection (203, 204). HHV-6A has also been shown to transactivate the long terminal repeat of HIV-1 in double infected cells (205), and several gene products of HHV-6A have been identified as possessing HIV-1 activating potential independently of each other (206). Soluble mediators (e.g., TNF-α) released from HHV-6A–infected lymphoid cells also upregulated HIV-1 infection in other cells carrying the virus (207). All of these effects could consequently facilitate AIDS and AIDS–associated tumor progression. HHV-6A (208) and HHV-6B (209) activate human endogenous retrovirus (HERV) K18 superantigen expression, and some HERV species have been implicated in teratocarcinoma and other germline tumors.
Exploring biomarkers and performing functional studies to investigate interactions between HHV-6 activity and genetic variants related to molecular/cellular pathways involved in immune surveillance and clearance of transformed cells, as well as to susceptibility to other infections, will be valuable going forward.
Detection of viral RNA/DNA in tumor tissue by PCR and sequencing is the first step in elucidating the role of a virus in tumors. Given the presence of numerous reactive inflammatory cells and stromal cells within tumor tissue, PCR positivity may arise from non-tumor cells. As such, the next step would be direct localization of virus to tumor cells by immunohistochemistry, in-situ hybridization, and/or electron microscopy. These techniques require high quality tumor tissue, high sensitivity and specificity of the detection reagents, and simultaneous running of proper positive and negative controls. Even if the virus is localized to tumor cells, the percent positivity of tumor cells is likely important since one would expect that if the virus is involved in tumor growth, then it should be present in at least a significant fraction of tumor cells.
Cancer development follows a multistep selection course with acquisition of genetic defects leading from a benign polyclonal process to a malignant monoclonal process (210). In some settings, HHV-6A/B may play a supportive role in this process. Even if not overtly oncogenic in terms of inducing or activating oncogenes in susceptible cells, HHV-6 may indirectly stimulate growth and/or block apoptosis in infected cells, interfere with epigenetic regulation and post-translational events, or potentiate the role of other viruses, in susceptible individuals (211). Although no definitive evidence of a direct role for HHV-6 as a tumorigenic virus has been produced, further investigation is warranted, especially for nodular sclerosis Hodgkin Lymphoma, glial tumors, gastrointestinal tumors, and oral cancers.
EE, EL, JP, JO, GK, JC, TP, DA, and SH: all contributed to the content of the manuscript and edited as needed.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Paul Levine, Dr. Mario Luppi, and Kristin Loomis for their valuable input in the preparation of this manuscript. We also thank Elaine O'Leary for her work in obtaining permission for our figures, and Sheryl Lunsford for her work on the mechanism figures.
1. Ablashi D, Agut A, Berneman Z, Campadelli-Fiume G, Carrigan D, Ceccerini-Nelli L, et al. Human herpesvirus-6 strain groups: a nomenclature. Arch Virol. (1993) 129:363–6.
2. Ablashi D, Agut H, Alvarez-Lafuente R, Clark DA, Dewhurst S, DiLuca D, et al. Classification of HHV-6A and HHV-6B as distinct viruses. Arch Virol. (2014) 159:863–70. doi: 10.1007/s00705-013-1902-5
3. Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, et al. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science (1986) 234:596–601.
4. Puri RK, Leland P, Razzaque A. Antigen(s)-specific tumour-infiltrating lymphocytes from tumour induced by human herpes virus-6 (HHV-6) DNA transfected NIH 3T3 transformants. Clin Exp Immunol. (1991) 83:96–101.
5. Josephs SF, Salahuddin SZ, Ablashi DV, Schachter F, Wong-Staal F, Gallo RC. Genomic analysis of the human B-lymphotropic virus (HBLV). Science (1986) 234:601–3.
6. Kishi M, Harada H, Takahashi M, Tanaka A, Hayashi M, Nonoyama M, et al. A repeat sequence, GGGTTA, is shared by DNA of human herpesvirus 6 and Marek's disease virus. J Virol. (1988) 62:4824–7.
7. Razzaque A, Williams O, Wang J, Rhim JS. Neoplastic transformation of immortalized human epidermal keratinocytes by two HHV-6 DNA clones. Virology (1993) 195:113–20. doi: 10.1006/viro.1993.1351
8. Kiani H, Makvandi M, Samarbafzadeh A, Teimoori A, Nisi N, Mehravaran H, et al. Association of HHV-6 with Hodgkin and non Hodgkin lymphoma. Iran J Microbiol. (2016) 8:153–8.
9. Siddon A, Lozovatsky L, Mohamed A, Hudnall SD. Human herpesvirus 6 positive Reed-Sternberg cells in nodular sclerosis Hodgkin lymphoma. Br J Haematol. (2012) 158:635–43. doi: 10.1111/j.1365-2141.2012.09206.x
10. Lacroix A, Jaccard A, Rouzioux C, Piguet C, Petit B, Bordessoule D, et al. HHV-6 and EBV DNA quantitation in lymph nodes of 86 patients with Hodgkin's lymphoma. J Med Virol. (2007) 79:1349–56. doi: 10.1002/jmv.20868
11. Hernandez-Losa J, Fedele CG, Pozo F, Tenorio A, Fernandez V, Castellvi J, et al. Lack of association of polyomavirus and herpesvirus types 6 and 7 in human lymphomas. Cancer (2005) 103:293–8. doi: 10.1002/cncr.20801
12. Collot S, Petit B, Bordessoule D, Alain S, Touati M, Denis F, et al. Real-time PCR for quantification of human herpesvirus 6 DNA from lymph nodes and saliva. J Clin Microbiol. (2002) 40:2445–51. doi: 10.1128/JCM.40.7.2445-2451.2002
13. Shiramizu B, Chang CW, Cairo MS. Absence of human herpesvirus-6 genome by polymerase chain reaction in children with Hodgkin disease: a Children's Cancer Group Lymphoma Biology Study. J Pediatr Hematol Oncol. (2001) 23:282–5.
14. Schmidt CA, Oettle H, Peng R, Binder T, Wilborn F, Huhn D, et al. Presence of human beta- and gamma-herpes virus DNA in Hodgkin's disease. Leuk Res. (2000) 24:865–70. doi: 10.1016/S0145-2126(00)00042-4
15. Valente G, Secchiero P, Lusso P, Abete MC, Jemma C, Reato G, et al. Human herpesvirus 6 and Epstein-Barr virus in Hodgkin's disease: a controlled study by polymerase chain reaction and in situ hybridization. Am J Pathol. (1996) 149:1501–10.
16. Carbone A, Dolcetti R, Gloghini A, Maestro R, Vaccher E, di Luca D, et al. Immunophenotypic and molecular analyses of acquired immune deficiency syndrome-related and Epstein-Barr virus-associated lymphomas: a comparative study. Hum Pathol. (1996) 27:133–46.
17. Razzaque A, Francillon Y, Jilly PN, Varricchio F. Detection of human herpesvirus 6 sequences in lymphoma tissues by immunohistochemistry and polymerase chain reactions. Cancer Lett. (1996) 106:221–6.
18. Dolcetti R, Di Luca D, Carbone A, Mirandola P, De Vita S, Vaccher E, et al. Human herpesvirus 6 in human immunodeficiency virus-infected individuals: association with early histologic phases of lymphadenopathy syndrome but not with malignant lymphoproliferative disorders. J Med Virol. (1996) 48:344–53. doi: 10.1002/(SICI)1096-9071(199604)48:4<344::AID-JMV8>3.0.CO;2-7
19. Di Luca D, Dolcetti R, Mirandola P, De Re V, Secchiero P, Carbone A, et al. Human herpesvirus 6: a survey of presence and variant distribution in normal peripheral lymphocytes and lymphoproliferative disorders. J Infect Dis. (1994) 170:211–5.
20. Trovato R, Di Lollo S, Calzolari A, Torelli G, Ceccherini-Nelli L. Detection of human herpesvirus-6 and Epstein-Barr virus genome in childhood Hodgkin's disease. Pathologica (1994) 86:500–3.
21. Sumiyoshi Y, Kikuchi M, Ohshima K, Takeshita M, Eizuru Y, Minamishima Y. Analysis of human herpes virus-6 genomes in lymphoid malignancy in Japan. J Clin Pathol. (1993) 46:1137–8.
22. Gledhill S, Gallagher A, Jones DB, Krajewski AS, Alexander FE, Klee E, et al. Viral involvement in Hodgkin's disease: detection of clonal type A Epstein-Barr virus genomes in tumour samples. Br J Cancer (1991) 64:227–32.
23. Torelli G, Marasca R, Luppi M, Selleri L, Ferrari S, Narni F, et al. Human herpesvirus-6 in human lymphomas: identification of specific sequences in Hodgkin's lymphomas by polymerase chain reaction. Blood (1991) 77:2251–8.
24. Jarrett RF, Gledhill S, Qureshi F, Crae SH, Madhok R, Brown I, et al. Identification of human herpesvirus 6-specific DNA sequences in two patients with non-Hodgkin's lymphoma. Leukemia (1988) 2:496–502.
25. Lacroix A, Collot-Teixeira S, Mardivirin L, Jaccard A, Petit B, Piguet C, et al. Involvement of human herpesvirus-6 variant B in classic Hodgkin's lymphoma via DR7 oncoprotein. Clin Cancer Res. (2010) 16:4711–21. doi: 10.1158/1078-0432.CCR-10-0470
26. Luppi M, Barozzi P, Garber R, Maiorana A, Bonacorsi G, Artusi T, et al. Expression of human herpesvirus-6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. (1998) 153:815–23. doi: 10.1016/S0002-9440(10)65623-4
27. Krueger GR, Guenther A, Knuefermann R, Kluppelberg U, Luka J, Pearson GR, et al. Human herpesvirus-6 (HHV-6) in Hodgkin's disease: cellular expression of viral antigens as compared to oncogenes met and fes, tumor suppressor gene product p53, and interleukins 2 and 6. in vivo (1994) 8:501–16.
28. Rojo J, Ferrer Argote VE, Klueppelberg U, Krueger GR, Eidt E, Ablashi DV, et al. Semi-quantitative in situ hybridization and immunohistology for antigen expression of human herpesvirus-6 in various lymphoproliferative diseases. in vivo (1994) 8:517–26.
29. Cao S, Wendl MC, Wyczalkowski MA, Wylie K, Ye K, Jayasinghe R, et al. Divergent viral presentation among human tumors and adjacent normal tissues. Sci Rep. (2016) 6:28294. doi: 10.1038/srep28294
30. Cantalupo PG, Katz JP, Pipas JM. Viral sequences in human cancer. Virology (2018) 513:208–16. doi: 10.1016/j.virol.2017.10.017
31. Usui Y, Rao NA, Takase H, Tsubota K, Umazume K, Diaz-Aguilar D, et al. Comprehensive polymerase chain reaction assay for detection of pathogenic DNA in lymphoproliferative disorders of the ocular adnexa. Sci Rep. (2016) 6:36621. doi: 10.1038/srep36621
32. Yoon GS, Choi YK, Bak H, Kim BJ, Kim MN, Choi J, et al. Angioimmunoblastic T cell lymphomas: frequent cutaneous skin lesions and absence of human herpes viruses. Ann Dermatol. (2009) 21:1–5. doi: 10.5021/ad.2009.21.1.1
33. Zhou Y, Attygalle AD, Chuang SS, Diss T, Ye H, Liu H, et al. Angioimmunoblastic T-cell lymphoma: histological progression associates with EBV and HHV6B viral load. Br J Haematol. (2007) 138:44–53. doi: 10.1111/j.1365-2141.2007.06620.x
34. Kolonic SO, Dzebro S, Kusec R, Planinc-Peraica A, Dominis M, Jaksic B. Primary mediastinal large B-cell lymphoma: a single-center study of clinicopathologic characteristics. Int J Hematol. (2006) 83:331–6. doi: 10.1532/IJH97.E0529
35. Vrsalovic MM, Korac P, Dominis M, Ostojic S, Mannhalter C, Kusec R. T- and B-cell clonality and frequency of human herpes viruses-6,−8 and Epstein Barr virus in angioimmunoblastic T-cell lymphoma. Hematol Oncol. (2004) 22:169–77. doi: 10.1002/hon.740
36. Allory Y, Challine D, Haioun C, Copie-Bergman C, Delfau-Larue MH, Boucher E, et al. Bone marrow involvement in lymphomas with hemophagocytic syndrome at presentation: a clinicopathologic study of 11 patients in a Western institution. Am J Surg Pathol. (2001) 25:865–74.
37. Asou H, Tasaka T, Said JW, Daibata M, Kamada N, Koeffler HP. Co-infection of HHV-6 and HHV-8 is rare in primary effusion lymphoma. Leuk Res. (2000) 24:59–61. doi: 10.1016/S0145-2126(99)00144-7
38. Daibata M, Komatsu T, Taguchi H. Human herpesviruses in primary ocular lymphoma. Leuk Lymphoma (2000) 37:361–5. doi: 10.3109/10428190009089436
39. Ohyashiki JH, Abe K, Ojima T, Wang P, Zhou CF, Suzuki A, et al. Quantification of human herpesvirus 6 in healthy volunteers and patients with lymphoproliferative disorders by PCR-ELISA. Leuk Res. (1999) 23:625–30.
40. Brice SL, Jester JD, Friednash M, Golitz LE, Leahy MA, Stockert SS, et al. Examination of cutaneous T-cell lymphoma for human herpesviruses by using the polymerase chain reaction. J Cutan Pathol. (1993) 20:304–7.
41. Luppi M, Marasca R, Barozzi P, Artusi T, Torelli G. Frequent detection of human herpesvirus-6 sequences by polymerase chain reaction in paraffin-embedded lymph nodes from patients with angioimmunoblastic lymphadenopathy and angioimmunoblastic lymphadenopathy-like lymphoma. Leuk Res. (1993) 17:1003–11.
42. Fox RI, Saito I, Chan EK, Josephs S, Salahuddin SZ, Ahlashi DV, et al. Viral genomes in lymphomas of patients with Sjogren's syndrome. J Autoimmun. (1989) 2:449–55.
43. Buchbinder A, Josephs SF, Ablashi D, Salahuddin SZ, Klotman ME, Manak M, et al. Polymerase chain reaction amplification and in situ hybridization for the detection of human B-lymphotropic virus. J Virol Methods (1988) 21:191–7.
44. Josephs SF, Buchbinder A, Streicher HZ, Ablashi DV, Salahuddin SZ, Guo HG, et al. Detection of human B-lymphotropic virus (human herpesvirus 6) sequences in B cell lymphoma tissues of three patients. Leukemia (1988) 2:132–5.
45. Yin SY, Ming HA, Jahan N, Manak M, Jaffe ES, Levine PH. In situ hybridization detection of human herpesvirus 6 in biopsy specimens from Chinese patients with non-Hodgkin's lymphoma. Arch Pathol Lab Med. (1993) 117:502–6.
46. Strong MJ, O'Grady T, Lin Z, Xu G, Baddoo M, Parsons C, et al. Epstein-Barr virus and human herpesvirus 6 detection in a non-Hodgkin's diffuse large B-cell lymphoma cohort by using RNA sequencing. J Virol. (2013) 87:13059–62. doi: 10.1128/JVI.02380-13
47. Nefzi F, Lambert C, Gautheret-Dejean A, Fisson S, Khebizi Q, Khelif A, et al. Cytokine and cellular responses to human herpesvirus-6B in patients with B-acute lymphoblastic leukemia. Microbiol Immunol. (2016) 60:770–7. doi: 10.1111/1348-0421.12452
48. Morales-Sanchez A, Pompa-Mera EN, Fajardo-Gutierrez A, Alvarez-Rodriguez FJ, Bekker-Mendez VC, Flores-Chapa Jde D, et al. EBV, HCMV, HHV6, and HHV7 screening in bone marrow samples from children with acute lymphoblastic leukemia. Biomed Res Int. (2014) 2014:548097. doi: 10.1155/2014/548097
49. Gravel A, Sinnett D, Flamand L. Frequency of chromosomally-integrated human herpesvirus 6 in children with acute lymphoblastic leukemia. PLoS ONE (2013) 8:e84322. doi: 10.1371/journal.pone.0084322
50. Faten N, Agnes GD, Nadia BF, Nabil AB, Monia Z, Abderrahim K, et al. Quantitative analysis of human herpesvirus-6 genome in blood and bone marrow samples from Tunisian patients with acute leukemia: a follow-up study. Infect Agent Cancer (2012) 7:31. doi: 10.1186/1750-9378-7-31
51. Hubacek P, Muzikova K, Hrdlickova A, Cinek O, Hyncicova K, Hrstkova H, et al. Prevalence of HHV-6 integrated chromosomally among children treated for acute lymphoblastic or myeloid leukemia in the Czech Republic. J Med Virol. (2009) 81:258–63. doi: 10.1002/jmv.21371
52. Seror E, Coquerel B, Gautheret-Dejean A, Ballerini P, Landman-Parker J, Leverger G, et al. Quantitation of Human herpes virus 6 genome in children with acute lymphoblastic leukemia. J Med Virol. (2008) 80:689–93. doi: 10.1002/jmv.21118
53. Csire M, Mikala G, Peto M, Janosi J, Juhasz A, Tordai A, et al. Detection of four lymphotropic herpesviruses in Hungarian patients with multiple myeloma and lymphoma. FEMS Immunol Med Microbiol. (2007) 49:62–7. doi: 10.1111/j.1574-695X.2006.00171.x
54. Hermouet S, Sutton CA, Rose TM, Greenblatt RJ, Corre I, Garand R, et al. Qualitative and quantitative analysis of human herpesviruses in chronic and acute B cell lymphocytic leukemia and in multiple myeloma. Leukemia (2003) 17:185–95. doi: 10.1038/sj.leu.2402748
55. Luka J, Pirruccello SJ, Kersey JH. HHV-6 genome in T-cell acute lymphoblastic leukaemia. Lancet (1991) 338:1277–8.
56. Krueger GR, Kudlimay D, Ramon A, Klueppelberg U, Schumacher K. Demonstration of active and latent Epstein-Barr virus and human herpevirus-6 infections in bone marrow cells of patients with myelodysplasia and chronic myeloproliferative diseases. in vivo (1994) 8:533–42.
57. Zheng X, Li S, Zang Z, Hu J, An J, Pei X, et al. Evidence for possible role of toll-like receptor 3 mediating virus-induced progression of pituitary adenomas. Mol Cell Endocrinol. (2016) 426:22–32. doi: 10.1016/j.mce.2016.02.009
58. Lin CT, Leibovitch EC, Almira-Suarez MI, Jacobson S. Human herpesvirus multiplex ddPCR detection in brain tissue from low- and high-grade astrocytoma cases and controls. Infect Agent Cancer (2016) 11:32. doi: 10.1186/s13027-016-0081-x
59. Chi J, Gu B, Zhang C, Peng G, Zhou F, Chen Y, et al. Human herpesvirus 6 latent infection in patients with glioma. J Infect Dis. (2012) 206:1394–8. doi: 10.1093/infdis/jis513
60. Crawford JR, Santi MR, Thorarinsdottir HK, Cornelison R, Rushing EJ, Zhang H, et al. Detection of human herpesvirus-6 variants in pediatric brain tumors: association of viral antigen in low grade gliomas. J Clin Virol. (2009) 46:37–42. doi: 10.1016/j.jcv.2009.05.011
61. Crawford JR, Santi MR, Cornelison R, Sallinen SL, Haapasalo H, MacDonald TJ. Detection of human herpesvirus-6 in adult central nervous system tumors: predominance of early and late viral antigens in glial tumors. J Neurooncol. (2009) 95:49–60. doi: 10.1007/s11060-009-9908-2
62. Neves AM, Thompson G, Carvalheira J, Trindade JC, Rueff J, Caetano JM, et al. Detection and quantitative analysis of human herpesvirus in pilocytic astrocytoma. Brain Res. (2008) 1221:108–14. doi: 10.1016/j.brainres.2008.05.009
63. Cuomo L, Trivedi P, Cardillo MR, Gagliardi FM, Vecchione A, Caruso R, et al. Human herpesvirus 6 infection in neoplastic and normal brain tissue. J Med Virol. (2001) 63:45–51 doi: 10.1002/1096-9071(200101)63:1<45::AID-JMV1006>3.0.CO;2-K
64. Chan PK, Ng HK, Cheng AF. Detection of human herpesviruses 6 and 7 genomic sequences in brain tumours. J Clin Pathol. (1999) 52:620–3.
65. Luppi M, Barozzi P, Maiorana A, Marasca R, Trovato R, Fano R, et al. Human herpesvirus-6: a survey of presence and distribution of genomic sequences in normal brain and neuroglial tumors. J Med Virol. (1995) 47:105–11.
66. Liedtke W, Trubner K, Schwechheimer K. On the role of human herpesvirus 6 in viral latency in nervous tissue and in cerebral lymphoma. J Neurol Sci. (1995) 134:184–8.
67. Paulus W, Jellinger K, Hallas C, Ott G, Muller-Hermelink HK. Human herpesvirus-6 and Epstein-Barr virus genome in primary cerebral lymphomas. Neurology (1993) 43:1591–3.
68. Strong MJ, Blanchard ET, Lin Z, Morris CA, Baddoo M, Taylor CM, et al. A comprehensive next generation sequencing-based virome assessment in brain tissue suggests no major virus–tumor association. Acta Neuropathol Commun. (2016) 4:71. doi: 10.1186/s40478-016-0338-z
69. Duncan CG, Leary RJ, Lin JC, Cummins J, Di C, Schaefer CF, et al. Identification of microbial DNA in human cancer. BMC Med Genomics (2009) 2:22. doi: 10.1186/1755-8794-2-22.
70. Halme L, Loginov R, Arola J, Turunen U, Lautenschlager I. HHV-6 antigen and HHV-6 DNA expression in sporadic adenomatous polyps of the colon. Scand J Gastroenterol. (2013) 48:1423–7. doi: 10.3109/00365521.2013.843200
71. Banerjee S, Tian T, Wei Z, Shih N, Feldman MD, Alwine JC, et al. The ovarian cancer oncobiome. Oncotarget (2017) 8:36225–45. doi: 10.18632/oncotarget.16717
72. Broccolo F, Cassina G, Chiari S, Garcia-Parra R, Villa A, Leone BE, et al. Frequency and clinical significance of human beta-herpesviruses in cervical samples from Italian women. J Med Virol. (2008) 80:147–53. doi: 10.1002/jmv.21054
73. Tran-Thanh D, Koushik A, Provencher D, Drouin P, Dubuc-Lissoir J, Gauthier P, et al. Detection of human herpes virus type 6 DNA in precancerous lesions of the uterine cervix. J Med Virol. (2002) 68:606–10. doi: 10.1002/jmv.10229
74. Chan PK, Chan MY, Li WW, Chan DP, Cheung JL, Cheng AF. Association of human beta-herpesviruses with the development of cervical cancer: bystanders or cofactors. J Clin Pathol. (2001) 54:48–53. doi: 10.1136/jcp.54.1.48
75. Romano N, Romano FM, Viviano E, Vitale F, Villafrate MR, Perna AM, et al. Rare association of human herpesvirus 6 DNA with human papillomavirus DNA in cervical smears of women with normal and abnormal cytologies. J Clin Microbiol. (1996) 34:1589–91.
76. Wang H, Chen M, Berneman ZN, Delgado G, DiPaolo JA. Detection of human herpesvirus-6 in paraffin-embedded tissue of cervical cancer by polymerase chain reaction. J Virol Methods (1994) 47:297–305.
77. Chen M, Wang H, Woodworth CD, Lusso P, Berneman Z, Kingma D, et al. Detection of human herpesvirus 6 and human papillomavirus 16 in cervical carcinoma. Am J Pathol. (1994) 145:1509–16.
78. Arivananthan M, Yadav M, Kumar S. Detection of HHV-6 genotypes by in situ hybridization with variant-specific oligonucleotide probes. J Virol Methods (1997) 66:5–14.
79. Yadav M, Arivananthan M, Kumar S. HHV-6 antigen and viral DNA detected in cervical cells from archived tissue using histochemical staining and hybridization. Clin Diagn Virol. (1996) 7:23–33.
80. Yadav M, Arivananthan M, Chandrashekran A, Tan BS, Hashim BY. Human herpesvirus-6 (HHV-6) DNA and virus-encoded antigen in oral lesions. J Oral Pathol Med. (1997) 26:393–401.
81. Yadav M, Chandrashekran A, Vasudevan DM, Ablashi DV. Frequent detection of human herpesvirus 6 in oral carcinoma. J Natl Cancer Inst. (1994) 86:1792–4.
82. Saravani S, Miri-Moghaddam E, Sanadgol N, Kadeh H, Nazeri MR. Human herpesvirus-6 and epstein-barr virus infections at different histopathological grades of oral squamous cell carcinomas. Int J Prev Med. (2014) 5:1231–8.
83. Shanehsazzadeh M, Rad JS, Pourazar A, Behbahani M. Epidemiology of herpes human virus 6 and 7 infections in salivary gland neoplasms in Isfahan, Iran. Med Arch. (2014) 68:276–8. doi: 10.5455/medarh.2014.68.276-278
84. Chen X, Jian S, Wang H. The study on clinical significance of human herpesvirus 6 infection of NPC tissues. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi (1999) 13:262–5.
85. Kositanont U, Kondo K, Chongkolwatana C, Metheetrairut C, Puthavathana P, Chantarakul N, et al. Detection of Epstein-Barr virus DNA and HHV-6 DNA in tissue biopsies from patients with nasopharyngeal carcinoma by polymerase chain reaction. Southeast Asian J Trop Med Public Health (1993) 24:455–60.
86. Campioni D, Gentili V, Cavazzini F, Bortolotti D, Nacheva EP, Cuneo A, et al. Detection of inherited chromosomally integrated HHV-6 (ciHHV-6) in a marker chromosome. Eur J Haematol. (2017) 98:635–7. doi: 10.1111/ejh.12872
87. Zhang E, Cotton VE, Hidalgo-Bravo A, Huang Y, Bell AJ, Jarrett RF, et al. HHV-8-unrelated primary effusion-like lymphoma associated with clonal loss of inherited chromosomally-integrated human herpesvirus-6A from the telomere of chromosome 19q. Sci Rep. (2016) 6:22730. doi: 10.1038/srep22730
88. Nakayama-Ichiyama S, Yokote T, Iwaki K, Miyoshi T, Takubo T, Tsuji M, et al. Co-infection of human herpesvirus-6 and human herpesvirus-8 in primary cutaneous diffuse large B-cell lymphoma, leg type. Br J Haematol. (2011) 155:514–6. doi: 10.1111/j.1365-2141.2011.08702.x
89. Nakayama-Ichiyama S, Yokote T, Oka S, Iwaki K, Kobayashi K, Hirata Y, et al. Diffuse large B-cell lymphoma, not otherwise specified, associated with coinfection of human herpesvirus 6 and 8. J Clin Oncol. (2011) 29:e636–637. doi: 10.1200/JCO.2011.35.1254
90. Lohi O, Arola M, Lautenschlager I, Nacheva EP, Vettenranta K. A high circulating copy number of HHV-6 due to chromosomal integration in a child with acute lymphoblastic leukemia. Pediatr Blood Cancer (2010) 55:1236–8. doi: 10.1002/pbc.22671
91. Hubacek P, Virgili A, Ward KN, Pohlreich D, Keslova P, Goldova B, et al. HHV-6 DNA throughout the tissues of two stem cell transplant patients with chromosomally integrated HHV-6 and fatal CMV pneumonitis. Br J Haematol. (2009) 145:394–8. doi: 10.1111/j.1365-2141.2009.07622.x
92. Lin WC, Moore JO, Mann KP, Traweek ST, Smith C. Post transplant CD8+ gammadelta T-cell lymphoma associated with human herpes virus-6 infection. Leuk Lymphoma (1999) 33:377–84.
93. Daibata M, Taguchi T, Kamioka M, Kubonishi I, Taguchi H, Miyoshi I. Identification of integrated human herpesvirus 6 DNA in early pre-B cell acute lymphoblastic leukemia. Leukemia (1998) 12:1002–4.
94. Daibata M, Ido E, Murakami K, Kuzume T, Kubonishi I, Taguchi H, et al. Angioimmunoblastic lymphadenopathy with disseminated human herpesvirus 6 infection in a patient with acute myeloblastic leukemia. Leukemia (1997) 11:882–5.
95. Bandobashi K, Daibata M, Kamioka M, Tanaka Y, Kubonishi I, Taguchi H, et al. Human herpesvirus 6 (HHV-6)-positive Burkitt's lymphoma: establishment of a novel cell line infected with HHV-6. Blood (1997) 90:1200–7.
96. Maeda A, Sata T, Enzan H, Tanaka K, Wakiguchi H, Kurashige T, et al. The evidence of human herpesvirus 6 infection in the lymph nodes of Hodgkin's disease. Virchows Arch A Pathol Anat Histopathol. (1993) 423:71–5.
97. Stödberg T, Deniz Y, Esteitie N, et al. A case of diffuse leptomeningeal oligodendrogliomatosis associated with HHV-6 variant A. Neuropediatrics (2002) 33:266–70. doi: 10.1055/s-2002-36739
98. Rantala H, Mannonen L, Ahtiluoto S, et al. Human herpesvirus-6 associated encephalitis with subsequent infantile spasms and cerebellar astrocytoma. Dev Med Child Neurol. (2000) 42:418–21. doi: 10.1111/j.1469-8749.2000.tb00123.x
99. Crawford JR. HHV-6A and HHV-6B in cancer. In: Flamand LI, Krueger GRF, Ablashi DV, editors. Human Herpesviruses HHV-6A, HHV-6B & HHV-7, 3rd ed. Boston, MA: Elsevier (2014). p. 297–310.
100. De Bolle L, Naesens L, De Clercq E. Update on human herpesvirus 6 biology, clinical features, and therapy. Clin Microbiol Rev. (2005) 18:217–45. doi: 10.1128/CMR.18.1.217-245.2005
101. Alibek K, Baiken Y, Kakpenova A, Mussabekova A, Zhussupbekova S, Akan M, et al. Implication of human herpesviruses in oncogenesis through immune evasion and supression. Infect Agent Cancer (2014) 9:3. doi: 10.1186/1750-9378-9-3
102. Biggs PM. Oncogenic herpesvirus infections of animals. J Clin Pathol Suppl (R Coll Pathol). (1972) 6:65–72.
103. Kaufer BB, Flamand L. Chromosomally integrated HHV-6: impact on virus, cell and organismal biology. Curr Opin Virol. (2014) 9:111–8. doi: 10.1016/j.coviro.2014.09.010
104. Pellett PE, Ablashi DV, Ambros PF, Agut H, Caserta MT, Descamps V, et al. Chromosomally integrated human herpesvirus 6: questions and answers. Rev Med Virol. (2012) 22:144–55. doi: 10.1002/rmv.715
105. Arbuckle JH, Medveczky MM, Luka J, Hadley SH, Luegmayr A, Ablashi D, et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc Natl Acad Sci USA. (2010) 197:5563–8. doi: 10.1073/pnas.0913586107
106. Endo A, Watanabe K, Ohye T, Suzuki K, Matsubara T, Shimizu N, et al. Molecular and virological evidence of viral activation from chromosomally integrated human herpesvirus 6A in a patient with X-linked severe combined immunodeficiency. Clin Infect Dis. (2014) 59:545–8. doi: 10.1093/cid/ciu323
107. Huang Y, Hidalgo-Bravo A, Zhang E, Cotton VE, Mendez-Bermudez A, Wig G, et al. Human telomeres that carry an integrated copy of human herpesvirus 6 are often short and unstable, facilitating release of the viral genome from the chromosome. Nucleic Acids Res. (2014) 42:315–27. doi: 10.1093/nar/gkt840
108. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. (2005) 352:1413–24. doi: 10.1056/NEJMoa042980
109. Politikos I, McMasters M, Bryke C, Avigan D, Boussiotis VA. Possible reactivation of chromosomally integrated human herpesvirus 6 after treatment with histone deacetylase inhibitor. Blood Adv. (2018) 2:1367–70. doi: 10.1182/bloodadvances.2018015982
110. Bonnafous P, Marlet J, Bouvet D, Salamé E, Tellier AC, Guyetant S, et al. Fatal outcome after reactivation of inherited chromosomally integrated HHV-6A (iciHHV-6A) transmitted through liver transplantation. Am J Transplant. (2018) 18:1548–51. doi: 10.1111/ajt.14657
111. Greninger AL, Roychoudhury P, Makhsous N, Hanson D, Chase J, Krueger G, et al. Copy number heterogeneity, large origin tandem repeats, and interspecies recombination in human herpesvirus 6A (HHV-6A) and HHV-6B reference strains. J Virol. (2018) 92:e00135-18. doi: 10.1128/JVI.00135-18
112. Eliassen E, Krueger G, Luppi M, Ablashi D. Lymphoproliferative syndromes associated with human herpesvirus-6A and human herpesvirus-6B. Mediterr J Hematol Infect Dis. (2018) 10:e2018035. doi: 10.4084/MJHID.2018.035
113. Kashanchi F, Araujo J, Doniger J, Muralidhar S, Hoch R, Khleif S, et al. Human herpesvirus 6 (HHV-6) ORF-1 transactivating gene exhibits malignant transforming activity and its protein binds to p53. Oncogene (1997) 14:359–67. doi: 10.1038/sj.onc.1200840.
114. Mori S, Sugahara K, Uemura A, Akamatsu N, Hirakata Y, Murata K, et al. Usefulness of a comprehensive PCR-based assay for human herpes viral DNA in blood mononuclear cell samples. Lab Hematol. (2005) 11:163–70. doi: 10.1532/LH96.05027
115. Bogdanovic G, Pou C, Barrientos-Somarribas M, Bjerkner A, Honkaniemi E, Allander T, et al. Virome characterisation from Guthrie cards in children who later developed acute lymphoblastic leukaemia. Br J Cancer (2016) 115:1008–14. doi: 10.1038/bjc.2016.261
116. Clark DA, Alexander FE, McKinney PA, Roberts BE, O'Brien C, Jarrett RF, et al. The seroepidemiology of human herpesvirus-6 (HHV-6) from a case-control study of leukaemia and lymphoma. Int J Cancer (1990) 45:829–33.
117. Levine PH, Ablashi DV, Saxinger WC, Connelly RR. Antibodies to human herpes virus-6 in patients with acute lymphocytic leukemia. Leukemia (1992) 6:1229–31.
118. Lyall EG. Serological response of paediatric oncology patients to human herpesvirus-6. J Med Virol. (1994) 43:373–9.
119. Schlehofer B, Blettner M, Geletneky K, Haaf HG, Kaatsch P, Michaelis J, et al. Sero-epidemiological analysis of the risk of virus infections for childhood leukaemia. Int J Cancer (1996) 65:584–90. doi: 10.1002/(sici)1097-0215(19960301)65:5<584::aid-ijc5>3.0.co;2-z
120. Gentile G, Mele A, Ragona G, Faggioni A, Zompetta C, Tosti ME, et al. Human herpes virus-6 seroprevalence and leukaemias: a case-control study. GIMEMA (Gruppo Italiano Malattie Ematologiche dell' Adulto). Br J Cancer (1999) 80:1103–6. doi: 10.1038/sj.bjc.6690471
121. Salonen MJ, Siimes MA, Salonen EM, Vaheri A, Koskiniemi M. Antibody status to HHV-6 in children with leukaemia. Leukemia (2002) 16:716–9. doi: 10.1038/sj.leu.2402437.
122. Assem MM, Gad WH, El-Sharkawy NM, El-Rouby MN, Ghaleb FM, Tarek H, et al. Prevalence of anti human herpes virus-6 IgG and its receptor in acute leukemia (membrane cofactor protein: MCP, CD46). J Egypt Natl Canc Inst. (2005) 17:29–34.
123. Steininger C, Rassenti LZ, Vanura K, Eigenberger K, Jager U, Kipps TJ, et al. Relative seroprevalence of human herpes viruses in patients with chronic lymphocytic leukaemia. Eur J Clin Invest. (2009) 39:497–506. doi: 10.1111/j.1365-2362.2009.02131.x
124. Tacyildiz N, Dincaslan HU, Ozdemir H, Yavuz G, Unal E, Ikinciogullari A, et al. The seroprevalence of Kaposi's sarcoma associated herpes virus and human herpes virus-6 in pediatric patients with cancer and healthy children in a Turkish pediatric oncology center. Indian J Med Paediatr Oncol. (2014) 35:221–5. doi: 10.4103/0971-5851.142039
125. Albright AV, Lavi E, Black JB, Goldberg S, O'Connor MJ, Gonzalez-Scarano F. The effect of human herpesvirus-6 (HHV-6) on cultured human neural cells: oligodendrocytes and microglia. J Neurovirol. (1998) 4:486–94.
126. Ahlqvist J, Fotheringham J, Akhyani N, Yao K, Fogdell-Hahn A, Jacobson S. Differential tropism of human herpesvirus 6 (HHV-6) variants and induction of latency by HHV-6A in oligodendrocytes. J Neurovirol. (2005) 11:384–94. doi: 10.1080/13550280591002379
127. De Bolle L, Van Loon J, De Clercq E, Naesens L. Quantitative analysis of human herpesvirus 6 cell tropism. J Med Virol. (2005) 75:76–85. doi: 10.1002/jmv.20240
128. Donati D, Martinelli E, Cassiani-Ingoni R, Ahlqvist J, Hou J, Major EO, et al. Variant-specific tropism of human herpesvirus 6 in human astrocytes. J Virol. (2005) 79:9439–48. doi: 10.1128/JVI.79.15.9439-9448.2005
129. Fotheringham J, Williams EL, Akhyani N, Jacobson S. Human herpesvirus 6 (HHV-6) induces dysregulation of glutamate uptake and transporter expression in astrocytes. J Neuroimmune Pharmacol. (2008) 3:105–16. doi: 10.1007/s11481-007-9084-0
130. Reynaud JM, Jegou JF, Welsch JC, Horvat B. Human herpesvirus 6A infection in CD46 transgenic mice: viral persistence in the brain and increased production of proinflammatory chemokines via Toll-like receptor 9. J Virol. (2014) 88:5421–36. doi: 10.1128/JVI.03763-13
131. Skuja S, Zieda A, Ravina K, Chapenko S, Roga S, Teteris O, et al. Structural and Ultrastructural Alterations in Human Olfactory Pathways and Possible Associations with Herpesvirus 6 Infection. PLoS ONE (2017) 12:e0170071. doi: 10.1371/journal.pone.0170071
132. Flamand L, Gravel A, Boutolleau D, Alvarez-Lafuente R, Jacobson S, Malnati MS, et al. Multicenter Comparison of PCR Assays for Detection of Human Herpesvirus 6 DNA in Serum. J Clin Microbiol. (2008) 46:2700–6. doi: 10.1128/JCM.00370-08
133. Peri P, Mattila RK, Kantola H, Broberg E, Karttunen HS, Waris M, et al. Herpes simplex virus type 1 Us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol J. (2008) 5:140. doi: 10.1186/1743-422X-5-140
134. Sultanova A, Chistjakovs M, Chapenko S, Donina S, Murovska M. Possible interference of human beta-herpesviruses-6 and−7 in gastrointestinal cancer development. Exp Oncol. (2013) 35:93–6.
135. Falasca F, Maida P, Gaeta A, Verzaro S, Mezzaroma I, Fantauzzi A, et al. Detection and quantification of EBV, HHV-6 and CMV DNA in the gastrointestinal tract of HIV-positive patients. Infection (2014) 42:1033–7. doi: 10.1007/s15010-014-0690-5
136. Halme L, Arola J, Höckerstedt KH, Lautenschlager I. Human Herpesvirus 6 Infection of the Gastroduodenal Mucosa. Clin Infect Dis. (2008) 46:434–9. doi: 10.1086/525264
137. Revest M, Minjolle S, Veyer D, Lagathu GL, Michelet C, Colimon R. Detection of HHV-6 in over a thousand samples: New types of infection revealed by an analysis of positive results. J Clin Virol. (2011) 51:20–4. doi: 10.1016/j.jcv.2011.02.001
138. Yasui T, Suzuki T, Yoshikawa T, Yatsuya H, Kawamura Y, Miura H, et al. Clinical course of human herpesvirus 6 infection in pediatric living donor liver transplantation. Pediatr Transplant. (2018) 22:e13239. doi: 10.1111/petr.13239
139. Niller HH, Wolf H, Minarovits J. Viral hit and run-oncogenesis: genetic and epigenetic scenarios. Cancer Lett. (2011) 305:200–17. doi: 10.1016/j.canlet.2010.08.007
140. Leite JL, Stolf HO, Reis NA, Ward LS. Human herpesvirus type 6 and type 1 infection increases susceptibility to nonmelanoma skin tumors. Cancer Lett. (2005) 224:213–9. doi: 10.1016/j.canlet.2004.11.010
141. Gravel A, Dubuc I, Brooks-Wilson A, Aronson KJ, Simard J, Velasquez-Garcia HA, et al. Inherited chromosomally integrated human herpesvirus 6 and breast cancer. Cancer Epidemiol Biomarkers Prev. (2017) 26:425–7. doi: 10.1158/1055-9965.EPI-16-0735
142. Iampietro M, Gravel A, Flamand L. Inhibition of breast cancer cell proliferation through disturbance of the calcineurin/NFAT pathway by human herpesvirus 6B U54 tegument protein. J Virol. (2014) 88:12910–4. doi: 10.1128/JVI.02107-14
143. Lu C, Zeng Y, Huang Z, Huang L, Qian C, Tang G, et al. Human herpesvirus 6 activates lytic cycle replication of Kaposi's sarcoma-associated herpesvirus. Am J Pathol. (2005) 166:173–83. doi: 10.1016/S0002-9440(10)62242-0
144. Thomson BJ, Efstathiou S, Honess RW. Acquisition of the human adeno-associated virus type-2 rep gene by human herpesvirus type-6. Nature (1991) 351:78–80. doi: 10.1038/351078a0
145. Cesarman E, Knowles DM. The role of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Semin Cancer Biol. (1999) 9:165–74. doi: 10.1006/scbi.1998.0118
146. Sarid R, Olsen SJ, Moore PS. Kaposi's sarcoma-associated herpesvirus: epidemiology, virology, and molecular biology. Adv Virus Res. (1999) 52:139–232.
147. Bassig BA, Willhauck-Fleckenstein M, Shu XO, Koh WP, Gao YT, Purdue MP, et al. Serologic markers of viral infection and risk of non-Hodgkin lymphoma: A pooled study of three prospective cohorts in China and Singapore. Int J Cancer (2018). doi: 10.1002/ijc.31385
148. Gomez-Roman JJ, Martinez MN, Fernandez SL, Val-Bernal JF. Epstein-Barr virus-associated adenocarcinomas and squamous-cell lung carcinomas. Mod Pathol. (2009) 22:530–7. doi: 10.1038/modpathol.2009.7
149. Chen M, Popescu N, Woodworth C, Berneman Z, Corbellino M, Lusso P, et al. Human herpesvirus 6 infects cervical epithelial cells and transactivates human papillomavirus gene expression. J Virol. (1994) 68:1173–8.
150. Garzino-Demo A, Chen M, Lusso P, Berneman Z, DiPaolo JA. Enhancement of TAT-induced transactivation of the HIV-1 LTR by two genomic fragments of HHV-6. J Med Virol. (1996) 50:20–4. doi: 10.1002/(SICI)1096-9071(199609)50:1<20::AID-JMV5>3.0.CO;2-V
151. Marci R, Gentili V, Bortolotti D, Lo Monte G, Caselli E, Bolzani S, et al. Presence of HHV-6A in endometrial epithelial cells from women with primary unexplained infertility. PLoS ONE (2016) 11:e0158304. doi: 10.1371/journal.pone.0158304
152. Maeda T, Okuno T, Hayashi K, Nagata M, Ueda M, Terashima K, et al. Outcomes of infants whose mothers are positive for human herpesvirus-6 DNA within the genital tract in early gestation. Acta Paediatr Jpn. (1997) 39:653–7.
153. Neofytou E, Sourvinos G, Asmarianaki M, Spandidos DA, Makrigiannakis A. Prevalence of human herpes virus types 1-7 in the semen of men attending an infertility clinic and correlation with semen parameters. Fertil Steril. (2009) 91:2487–94. doi: 10.1016/j.fertnstert.2008.03.074
154. Escudero AL, Luque RJ, Quintero A, Alvarez-Kindelan J, Requena MJ, Montironi R, et al. Association of human herpesvirus type 6 DNA with human bladder cancer. Cancer Lett. (2005) 230:20–4. doi: 10.1016/j.canlet.2004.12.014
155. Panagiotakis GI, Papadogianni D, Chatziioannou MN, Lasithiotaki I, Delakas D, Spandidos DA. Association of human herpes, papilloma and polyoma virus families with bladder cancer. Tumour Biol. (2013) 34:71–9. doi: 10.1007/s13277-012-0512-2
156. Pinto EM, Chen X, Easton J, Finkelstein D, Liu Z, Pounds S, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun. (2015) 6:6302. doi: 10.1038/ncomms7302
157. Letouze E, Rosati R, Komechen H, Doghman M, Marisa L, Fluck C, et al. SNP array profiling of childhood adrenocortical tumors reveals distinct pathways of tumorigenesis and highlights candidate driver genes. J Clin Endocrinol Metab. (2012) 97:E1284–93. doi: 10.1210/jc.2012-1184
158. Giovannoni I, Boldrini R, Benedetti MC, Inserra A, De Pasquale MD, Francalanci P. Pediatric adrenocortical neoplasms: immunohistochemical expression of p57 identifies loss of heterozygosity and abnormal imprinting of the 11p15.5. Pediatr Res. (2017) 81:468–72. doi: 10.1038/pr.2016.239
159. Tweedy J, Spyrou MA, Pearson M, Lassner D, Kuhl U, Gompels UA. Complete genome sequence of germline chromosomally integrated human herpesvirus 6A and analyses integration sites define a new human endogenous virus with potential to reactivate as an emerging infection. Viruses (2016) 8:E19. doi: 10.3390/v8010019
160. Engdahl E, Dunn N, Niehusmann P, Wideman S, Wipfler P, Becker AJ, et al. Human herpesvirus 6B induces hypomethylation on chromosome 17p13.3, correlating with increased gene expression and virus integration. J Virol. (2017) 91:e02105-16. doi: 10.1128/JVI.02105-16.
162. Takemoto M, Koike M, Mori Y, Yonemoto S, Sasamoto Y, Kondo K, et al. Human herpesvirus 6 open reading frame U14 protein and cellular p53 interact with each other and are contained in the virion. J Virol (2005) 79:13037–46. doi: 10.1128/JVI.79.20.13037-13046.2005.
163. Takemoto M, Mori Y, Ueda K, Kondo K, Yamanishi K. Productive human herpesvirus 6 infection causes aberrant accumulation of p53 and prevents apoptosis. J Gen Virol. (2004) 85(Pt 4), 869–79. doi: 10.1099/vir.0.19626-0
164. Sang Y, Zhang R, Scott WR, Creagh AL, Haynes CA, Straus SK. U24 from Roseolovirus interacts strongly with Nedd4 WW domains. Sci Rep. (2017) 7:39776. doi: 10.1038/srep39776
165. He S, Deng J, Li G, Wang B, Cao Y, Tu Y. Down-regulation of Nedd4L is associated with the aggressive progression and worse prognosis of malignant glioma. Jpn J Clin Oncol. (2012) 42:196–201. doi: 10.1093/jjco/hyr195
166. Zhang H, Nie W, Zhang X, Zhang G, Li Z, Wu H, et al. NEDD4-1 regulates migration and invasion of glioma cells through CNrasGEF ubiquitination in vitro. PLoS ONE (2013) 8:e82789. doi: 10.1371/journal.pone.0082789
167. Dai B, Pieper RO, Li D, Wei P, Liu M, Woo SY, et al. FoxM1B regulates NEDD4-1 expression, leading to cellular transformation and full malignant phenotype in immortalized human astrocytes. Cancer Res. (2010) 70:2951–61. doi: 10.1158/0008-5472.CAN-09-3909
168. Yeo WM, Isegawa Y, Chow VT. The U95 protein of human herpesvirus 6B interacts with human GRIM-19: silencing of U95 expression reduces viral load and abrogates loss of mitochondrial membrane potential. J Virol. (2008) 82:1011–20. doi: 10.1128/JVI.01156-07
169. Kalvakolanu DV, Nallar SC, Kalakonda S. Cytokine-induced tumor suppressors: a GRIM story. Cytokine (2010) 52:128–42. doi: 10.1016/j.cyto.2010.03.009
170. Takemoto M, Shimamoto T, Isegawa Y, Yamanishi K. The R3 region, one of three major repetitive regions of human herpesvirus 6, is a strong enhancer of immediate-early gene U95. J Virol. (2001) 75:10149–60. doi: 10.1128/JVI.75.21.10149-10160.2001
171. Ojima T, Abe K, Ohyashiki JH, Shirakata M, Yamamoto K. IL-2-regulated persistent human herpesvirus-6B infection facilitates growth of adult T cell leukemia cells. J Med Dent Sci. (2005) 52:135–41. doi: 10.11480/jmds.520206
172. Lusso P. HHV-6 and the immune system: mechanisms of immunomodulation and viral escape. J Clin Virol. (2006) 37(Suppl. 1):S4–10. doi: 10.1016/S1386-6532(06)70004-X
173. Caccuri F, Ronca R, Laimbacher AS, Berenzi A, Steimberg N, Campilongo F, et al. U94 of human herpesvirus 6 down-modulates Src, promotes a partial mesenchymal-to-epithelial transition and inhibits tumor cell growth, invasion and metastasis. Oncotarget (2017) 8:44533–49. doi: 10.18632/oncotarget.17817
174. Caruso A, Caselli E, Fiorentini S, Rotola A, Prandini A, Garrafa E, et al. U94 of human herpesvirus 6 inhibits in vitro angiogenesis and lymphangiogenesis. Proc Natl Acad Sci USA. (2009) 106:20446–51. doi: 10.1073/pnas.0905535106
175. Campbell A, Hogestyn JM, Folts CJ, Lopez B, Proschel C, Mock D, et al. Expression of the human herpesvirus 6A latency-associated transcript U94A disrupts human oligodendrocyte progenitor migration. Sci Rep. (2017) 7:3978. doi: 10.1038/s41598-017-04432-y
176. Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P. CD46 is a cellular receptor for human herpesvirus 6. Cell (1999) 99:817–27.
177. Hutzen B, Bid HK, Houghton PJ, Pierson CR, Powell K, Bratasz A, et al. Treatment of medulloblastoma with oncolytic measles viruses expressing the angiogenesis inhibitors endostatin and angiostatin. BMC Cancer (2014) 14:206. doi: 10.1186/1471-2407-14-206
178. Tang H, Serada S, Kawabata A, Ota M, Hayash E, Naka T, et al. CD134 is a cellular receptor specific for human herpesvirus-6B entry. Proc Natl Acad Sci USA. (2013) 110:9096–9. doi: 10.1073/pnas.1305187110
179. Hansen AS, Bundgaard BB, Biltoft M, Rossen LS, Hollsberg P. Divergent tropism of HHV-6AGS and HHV-6BPL1 in T cells expressing different CD46 isoform patterns. Virology (2017) 502:160–70. doi: 10.1016/j.virol.2016.12.027
180. Flynn S, Toellner KM, Raykundalia C, Goodall M, Lane P. CD4 T cell cytokine differentiation: the B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, Blr-1. J Exp Med. (1998) 188:297–304.
181. So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci USA. (2006) 103:3740–5. doi: 10.1073/pnas.0600205103
182. Piedade D, Azevedo-Pereira JM. The role of microRNAs in the pathogenesis of herpesvirus infection. Viruses (2016) 8:E156. doi: 10.3390/v8060156
183. Nukui M, Mori Y, Murphy EA. A human herpesvirus 6A-encoded microRNA: role in viral lytic replication. J Virol. (2015) 89:2615–27. doi: 10.1128/JVI.02007-14
184. Caselli E, D'Accolti M, Soffritti I, Zatelli MC, Rossi R, Degli Uberti E, et al. HHV-6A in vitro infection of thyrocytes and T cells alters the expression of miRNA associated to autoimmune thyroiditis. Virol J. (2017) 14:3. doi: 10.1186/s12985-016-0672-6
185. Butz H, Liko I, Czirjak S, Igaz P, Korbonits M, Racz K, et al. MicroRNA profile indicates downregulation of the TGFbeta pathway in sporadic non-functioning pituitary adenomas. Pituitary (2011) 14:112–24. doi: 10.1007/s11102-010-0268-x
186. Tuddenham L, Jung JS, Chane-Woon-Ming B, Dolken L, Pfeffer S. Small RNA deep sequencing identifies microRNAs and other small noncoding RNAs from human herpesvirus 6B. J Virol. (2012) 86:1638–49. doi: 10.1128/JVI.05911-11
187. Ota M, Serada S, Naka T, Mori Y. MHC class I molecules are incorporated into human herpesvirus-6 viral particles and released into the extracellular environment. Microbiol Immunol. (2014) 58:119–25. doi: 10.1111/1348-0421.12121
188. Bubeník J. MHC class I down-regulation: tumour escape from immune surveillance?. Int J Oncol. (2004) 25:487–491. doi: 10.3892/ijo.25.2.487
189. Mori Y, Koike M, Moriishi E, Kawabata A, Tang H, Oyaizu H, et al. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic (2008) 9:1728–42. doi: 10.1111/j.1600-0854.2008.00796.x
190. Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. (2017) 27:172–88. doi: 10.1016/j.tcb.2016.11.003
191. Anderson MR, Kashanchi F, Jacobson S. Exosomes in viral disease. Neurotherapeutics (2016) 13:535–46. doi: 10.1007/s13311-016-0450-6
192. Mathupala SP, Rempel A, Pedersen PL. Aberrant glycolytic metabolism of cancer cells: a remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J Bioenerg Biomembr. (1997) 29:339–43.
193. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. (2016) 37:11553–72. doi: 10.1007/s13277-016-5098-7
194. Inazawa N, Hori T, Hatakeyama N, Yamamoto M, Yoto Y, Nojima M, et al. Large-scale multiplex polymerase chain reaction assay for diagnosis of viral reactivations after allogeneic hematopoietic stem cell transplantation. J Med Virol. (2015) 87:1427–35. doi: 10.1002/jmv.24161
195. de Koning C, Admiraal R, Nierkens S, Boelens JJ. Human herpesvirus 6 viremia affects T-cell reconstitution after allogeneic hematopoietic stem cell transplantation. Blood Adv. (2018) 2:428–32. doi: 10.1182/bloodadvances.2017012724
196. Phan TL, Carlin K, Ljungman P, Politikos I, Boussiotis V, Boeckh M, et al. Human herpesvirus-6B (HHV-6B) reactivation is a risk factor for grade 2-4 acute graft-versus-host disease (aGVHD) after hematopoietic stem cell transplantation (HCT): a systematic review and meta-analysis. Biol Blood Marrow Transplant. (2018). doi: 10.1016/j.bbmt.2018.04.021. [Epub ahead of print].
197. Phan TL, Pritchett JC, Leifer C, Zerr DM, Koelle DM, Di Luca D, et al. HHV-6B infection, T-cell reconstitution, and graft-vs-host disease after hematopoietic stem cell transplantation. Bone Marrow Transplant. (2018). doi: 10.1038/s41409-018-0225-2
198. Krueger GR, Ablashi DV, Salahuddin SZ, Josephs SF. Diagnosis and differential diagnosis of progressive lymphoproliferation and malignant lymphoma in persistent active herpesvirus infection. J Virol Methods (1988) 21:255–64.
199. Krueger GR, Manak M, Bourgeois N, Ablashi DV, Salahuddin SZ, Josephs SS, et al. Persistent active herpes virus infection associated with atypical polyclonal lymphoproliferation (APL) and malignant lymphoma. Anticancer Res. (1989) 9:1457–76.
200. Brandt ME, Krueger G, Wang G. (Eds.). A Biodynamical Model of Human T-Cell Development and Pathology: Design, Testing and Validation. Dordrecht: Springer Netherlands (2008).
201. Krueger GRF, Buja L. (Eds.). Abnormal Variation of the Immune Response as Related to Cancer. Dordrecht: Springer Netherlands (2008).
202. Flamand L, Menezes J. Cyclic AMP-responsive element-dependent activation of Epstein-Barr virus zebra promoter by human herpesvirus 6. J Virol. (1996) 70:1784–91.
203. Cuomo L, Angeloni A, Zompetta C, Cirone M, Calogero A, Frati L, et al. Human herpesvirus 6 variant A, but not variant B, infects EBV-positive B lymphoid cells, activating the latent EBV genome through a BZLF-1-dependent mechanism. AIDS Res Hum Retroviruses (1995) 11:1241–5. doi: 10.1089/aid.1995.11.1241
204. Cuomo L, Trivedi P, de Grazia U, Calogero A, D'Onofrio M, Yang W, et al. Upregulation of Epstein-Barr virus-encoded latent membrane protein by human herpesvirus 6 superinfection of EBV-carrying Burkitt lymphoma cells. J Med Virol. (1998) 55:219–26.
205. Lusso P, Ensoli B, Markham PD, Ablashi DV, Salahuddin SZ, Tschachler E, et al. Productive dual infection of human CD4+ T lymphocytes by HIV-1 and HHV-6. Nature (1989) 337:370–3. doi: 10.1038/337370a0
206. Ongradi J, Kovesdi V, Nagy K, Matteoli B, Ceccherini-Nelli L, Ablashi DV. In vitro and in vivo Transactivation of HIV by HHV-6. Vienna: InTech (2011).
207. Ongradi J, Ceccherini-Nelli L, Soldaini E, Bendinelli M. Endotoxin Suppresses Indirect Activation of HIV-1 by Human Herpesvirus 6. Amsterdam: Elsevier Science Publishers, B.V. (1990).
208. Tai AK, Luka J, Ablashi D, Huber BT. HHV-6A infection induces expression of HERV-K18-encoded superantigen. J Clin Virol. (2009) 46:47–8. doi: 10.1016/j.jcv.2009.05.019
209. Turcanova VL, Bundgaard B, Hollsberg P. Human herpesvirus-6B induces expression of the human endogenous retrovirus K18-encoded superantigen. J Clin Virol. (2009) 46:15–9. doi: 10.1016/j.jcv.2009.05.015
210. Singh RK, T J. The Evolution of Diversity Within Tumors and Metastases. Dordrecht: Springer Netherlands (2008).
Keywords: HHV-6, herpesvirus, human herpesvirus 6, HHV6, oncogenic, cancer, malignant, transformation
Citation: Eliassen E, Lum E, Pritchett J, Ongradi J, Krueger G, Crawford JR, Phan TL, Ablashi D and Hudnall SD (2018) Human Herpesvirus 6 and Malignancy: A Review. Front. Oncol. 8:512. doi: 10.3389/fonc.2018.00512
Received: 13 July 2018; Accepted: 19 October 2018;
Published: 13 November 2018.
Edited by:
Daniel Christian Hoessli, University of Karachi, PakistanReviewed by:
Saraswati Sukumar, Johns Hopkins University School of Medicine, United StatesCopyright © 2018 Eliassen, Lum, Pritchett, Ongradi, Krueger, Crawford, Phan, Ablashi and Hudnall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dharam Ablashi, ZGhhcmFtX2FibGFzaGlAaGh2LTZmb3VuZGF0aW9uLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.