 Deshenyue Kong1,2,3†
Deshenyue Kong1,2,3† Jia-xue Sun2,3†Ji-qun Yang4†
Jia-xue Sun2,3†Ji-qun Yang4† Yuan-sen Li2,3
Yuan-sen Li2,3 Ke Bi2,3
Ke Bi2,3 Zun-yue Zhang2Kun-hua Wang2Hua-you Luo3*Mei Zhu3*
Zun-yue Zhang2Kun-hua Wang2Hua-you Luo3*Mei Zhu3* Yu Xu1,2,3*
Yu Xu1,2,3*- 1General Hospital of Eastern Theater Command, Nanjing, China
- 2Yunnan Technological Innovation Centre of Drug Addiction Medicine, Yunnan University, Kunming, China
- 3First Affiliated Hospital of Kunming Medical University, Kunming, China
- 4Third People’s Hospital of Kunming City/Drug Rehabilitation Hospital of Kunming City, Kunming, China
Substance use disorders (SUD) can lead to serious health problems, and there is a great interest in developing new treatment methods to alleviate the impact of substance abuse. In recent years, the ketogenic diet (KD) has shown therapeutic benefits as a dietary therapy in a variety of neurological disorders. Recent studies suggest that KD can compensate for the glucose metabolism disorders caused by alcohol use disorder by increasing ketone metabolism, thereby reducing withdrawal symptoms and indicating the therapeutic potential of KD in SUD. Additionally, SUD often accompanies increased sugar intake, involving neural circuits and altered neuroplasticity similar to substance addiction, which may induce cross-sensitization and increased use of other abused substances. Reducing carbohydrate intake through KD may have a positive effect on this. Finally, SUD is often associated with mitochondrial damage, oxidative stress, inflammation, glia dysfunction, and gut microbial disorders, while KD may potentially reverse these abnormalities and serve a therapeutic role. Although there is much indirect evidence that KD has a positive effect on SUD, the small number of relevant studies and the fact that KD leads to side effects such as metabolic abnormalities, increased risk of malnutrition and gastrointestinal symptoms have led to the limitation of KD in the treatment of SUD. Here, we described the organismal disorders caused by SUD and the possible positive effects of KD, aiming to provide potential therapeutic directions for SUD.
1. Introduction
Substance use disorders (SUD) are chronically relapsing conditions characterized by compulsive substance-seeking, lack of control in restricting intake, and the development of negative emotions during withdrawal (1). According to 2021 National Survey on Drug Use and Health in the United States, 46.3 million people aged 12 or older (16.5%) had SUD in the past year, including 29.5 million people with alcohol use disorders (AUD) and 24 million people with drug use disorders (DUD), and 7.3 million people have both AUD and DUD (2). A study based on the Global Burden of Disease showed that the global prevalence of substance use disorders was approximately 2.2%, with the prevalence of AUD (1.5%) being higher than other DUD (0.8%, including cannabis 0.32%, opioids 0.29%, amphetamines 0.10%, and cocaine 0.06%) (3). SUD is a significant contributor to the global burden of disease (4, 5). Deaths from SUD are increasing, rising from approximately 284,000 deaths in 2007 to 352,000 in 2017, creating a global health threat (6). Commonly abused substances include alcohol, opioids, stimulants, etc., which cause damage to various systems of the body, mainly the nervous system (7) and the immune system (8), etc. In addition, they often lead to metabolic disorders (9, 10). Many treatments for SUD are available, including non-pharmacological treatments, such as cognitive-behavioral therapy, and pharmacological treatments, such as drug replacement therapy including methadone, buprenorphine, or naltrexone maintenance (11). However, a radical cure for SUD usually means abstaining from substance abuse and dependence, but relapse is common and a radical cure for such disorders has not yet been discovered. Similar to other chronic conditions like heart disease or asthma, the treatment goal for SUD is typically not a radical cure but rather managing it as a chronic condition through a combination of complementary methods (12). However, the efficacy and sufficiency of SUD treatment strategies are affected by a multitude of factors. Firstly, SUD entails the interaction of multiple factors encompassing physical, psychological, and social domains; thus, a comprehensive treatment approach must account for these multifaceted dimensions. Second, there is individual variability in SUD, requiring individualized treatment plans. Third, different substance use disorders, such as alcohol and drugs, have specific treatment strategies and require targeted programs. Fourth, some individuals may encounter barriers to accessing appropriate treatment, such as limited medical resources, thereby impeding their ability to receive effective care. Furthermore, there exist challenges pertaining to individuals with SUD themselves; for example, an estimated 94% of individuals with SUD aged 12 or older in the United States do not receive any form of treatment, and almost all those who do not receive treatment at a specialty facility believe they do not need treatment (2). SUD remains severely undertreated, and it is estimated that only 11% of substance use individuals in need of treatment received appropriate care in the United States (13). Treatment of SUD is a long-term process, and it may be more effective to consider SUD as a chronic disease and to treat them in combination with multiple supporting approaches.
The ketogenic diet (KD) is a diet with a reduced proportion of carbohydrates and a relatively increased proportion of fat (14). KD causes a fasting-like effect and puts the body into a state of ketosis. Normally, carbohydrates are converted to glucose as the main source of energy for the brain. However, in the absence of carbohydrates, the body begins to look for alternative sources of energy (15), namely from acetyl coenzyme A producing excess ketone bodies, including acetoacetate, β-hydroxybutyric acid (BHBA), and acetone. This process is called ketogenesis (16). Unlike pathological keto acidosis, ketosis is a physiological mechanism, as these ketone bodies can be efficiently used without excessive concentrations (17). In the past, KD has been widely and successfully used in the treatment of epileptic disorders and obesity (18, 19). In addition, there has been increasing evidence in recent years of the therapeutic potential of KD in other diseases, such as diabetes (20), Alzheimer’s disease (21), Parkinson’s disease (22), cancer (23), autism (24), multiple sclerosis (25), and headache (26). Most of them were neurological disorders, suggesting a potential role for the ketogenic diet in these disorders (27). Recent research has found that KD may also have a positive effect on SUD, mainly in the treatment of AUD (28). In addition, cocaine-related animal studies have shown that KD decreased cocaine-induced stereotyped responses in rats, and also disrupted the sensitization of ambulatory responses (29). Here, we summarize the current studies on the treatment of SUD by KD (Table 1). These have led to a further understanding of the therapeutic potential of KD.
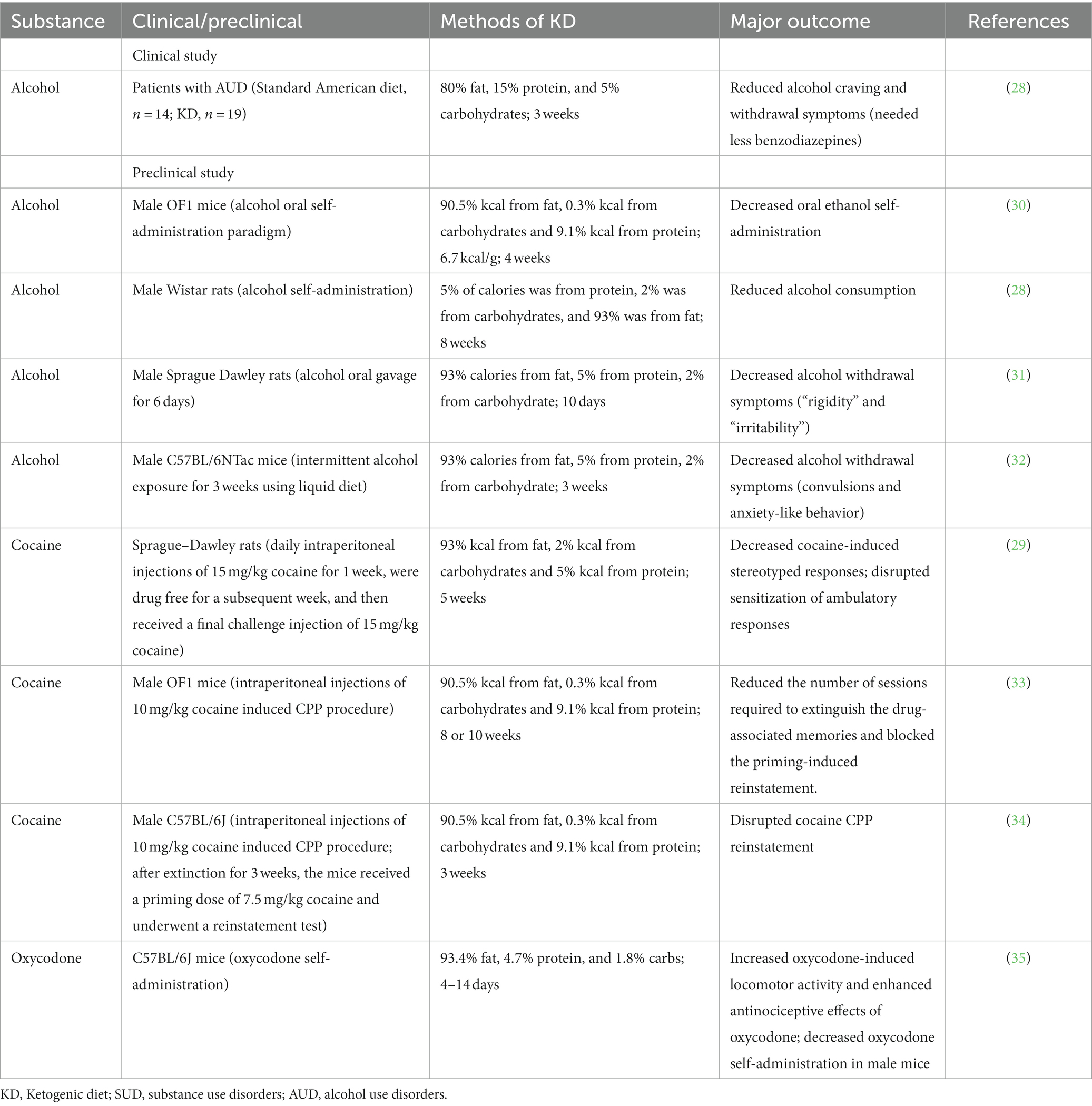
Table 1. Clinical and preclinical studies reporting the effect of the KD on SUD.
KD may be effective as a potential adjunctive therapy for SUD treatment. Importantly, it may have targeted protective effects. Here, we summarized the possible positive effects of KD in reducing carbohydrate intake, ketosis status, neuroprotection, protecting glial cells, reducing inflammation, and regulating gut microbiota, against the negative effects caused by SUD (Figure 1). However, there are few relevant studies besides AUD, and the role of KD in the treatment of SUD has not been clarified. Although SUD treatments exist, current treatment strategies are still not effective and sufficient, and there is a clear need for more effective treatments (13). The purpose of this review is to evaluate the viability of KD as a potential therapeutic option, to provide potential directions for future treatment of SUD, and to alleviate the suffering caused by SUDs to individuals, as well as the huge toll they take on a social level.
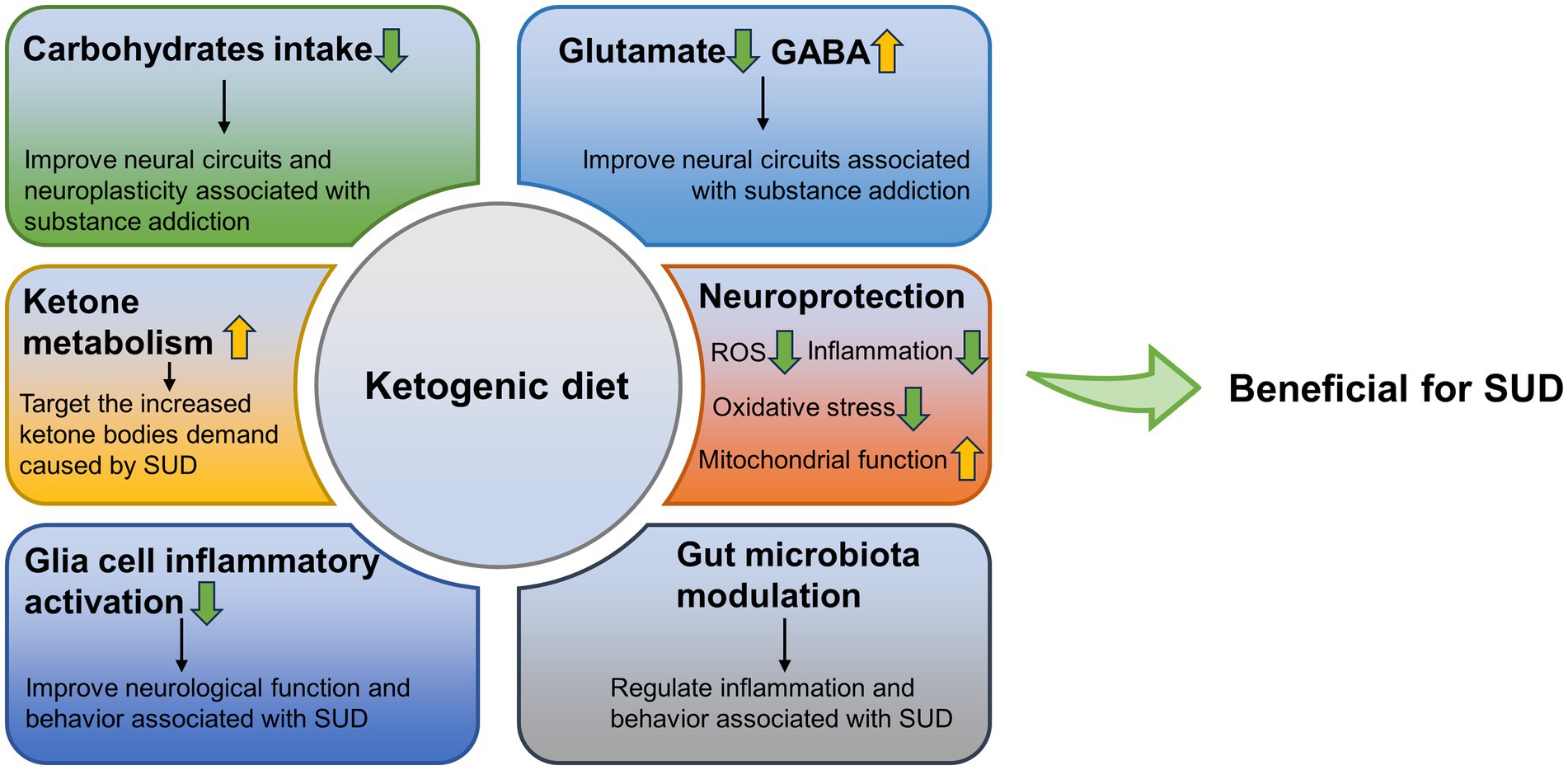
Figure 1. Possible positive effects of ketogenic diet against the negative effects caused by SUD. GABA, γ-aminobutyric acid; SUD, substance use disorders; ROS, reactive oxygen species.
2. Ketogenic diet decreases the intake of carbohydrates
Since the nutritional status of the SUD population usually appears abnormal (36), some studies have evaluated the dietary patterns of this population. The results show that SUD, particularly opioid use disorder, such as heroin use, and treatment with methadone, are strongly associated with increased consumption of sugar or sweets (37–40). These people preferred sucrose-rich foods to protein-and fat-rich foods (37). Similarly, cocaine and alcohol abusers also preferred higher concentrations of sucrose and highly sweet foods (41–43). And after withdrawal, the dietary patterns improved (38). It has been shown that excessive intake of carbohydrates, whether monosaccharides (glucose and fructose), disaccharides (sucrose), or polysaccharides (starch and glycogen), can injure human health (44). Sugar intake is also associated with increased vulnerability to SUD. By analyzing data from 17 countries worldwide, sugar and sweetener supply quantity were significantly and positively associated with anxiety disorders, mood disorders, impulse control disorders, and SUD (45). Patients undergoing bariatric or weight loss surgery have a higher incidence of substance use disorders, which may be attributed to the intake of high-sugar/low-fat foods or foods with a high glycemic index (46). In addition, sweet liking status predicts an increased risk of having alcohol-related problems in young adults (47). Likewise, there is similar evidence in animal experiments. For example, high saccharin intake rats are a well-recognized model of vulnerability to drug abuse. High saccharin rats are more likely to consume sweets and cocaine and are more vulnerable to the negative emotional effects of morphine withdrawal (48, 49). Excessive sucrose intake disrupts reward processing and reduces the reward value of sucrose in rats (50). Mice fed a 10% sucrose solution for 4 weeks showed greater locomotor responses to acute cocaine use compared to mice consuming standard food and water (51). Therefore, this particular dietary preference should receive attention, and exploring the reasons for increased sugar consumption and the mechanisms underlying the association between sugar intake and SUD may provide potential information to improve health.
The sweet taste preference of the SUD population makes it tempting to wonder whether their increased preference and consumption of sugar are associated with changes in gustatory responses. However, several studies on taste perception in SUD populations have shown different results, making it unclear whether SUD causes changes in taste perception that give rise to a preference process for sugar (52–54). These differential results may be attributed to the small sample size of the experiment, as well as differences in gender, age, education level, and income of participants. In addition, there are methodological differences between studies in terms of sucrose pleasantness and intensity measurements.
Importantly, the neural circuits associated with sugar intake may be similar to SUD. The mu-opioid receptor (MOR) is a G protein-coupled receptor that is associated with pain perception. It is not only a major target for heroin or other opioids, but also for most non-opioid substances of abuse, such as alcohol, cocaine, and nicotine (55, 56). Interestingly, several studies suggest a link between sugar or sweet foods intake and MOR. In rodent models, MOR knockout or antagonism leads to reduced hedonic responses to sweet stimuli and consumption of sweet solutions or sucrose (57–59). Similarly, in humans, morphine stimulation of MOR increased the pleasure of the sucrose solutions (60). In contrast, a MOR inverse agonist, GSK1521498, selectively reduced sensory hedonic ratings and intake of high-sugar foods (61). MOR in the nucleus accumbens (NAc) may partially mediate the motivation and hedonic experience of sucrose consumption (62). Specifically, the rostrodorsal quadrant of the NAc contains an opioid hedonic hotspot that mediates the enhancements of sucrose “liking” (63). Also, this is important for the endocannabinoid-enhanced preference for sucrose (64). Suggesting the interdependence of opioid and cannabinoid signaling in enhancing taste hedonic.
Another important neural circuit is the dopaminergic system. The dopaminergic system is a key regulatory component of behavior associated with SUD. Almost all known addictive substances can cause a significant increase in dopamine (DA) release and activate reward areas in the brain (65, 66). The effects of substances of abuse include direct activation of DA neurons, increased DA release, blockade of DA reuptake, and DA neuron de-inhibition (67). Excessive DA signaling may regulate gene expression and modify the synaptic function and circuit activity (68). In addition, the substance of abuse act on the nucleus accumbens DA signaling, inducing glutamatergic-mediated neural adaptations in the DA striatal-thalamo-cortical and limbic pathways (69). These effects eventually lead to substance addiction. It has been shown that intermittent sucrose use and drug abuse are similar in that both can repeatedly increase extracellular DA in the NAc shell (70). And this sucrose-induced DA release in the NAc may be sucrose concentration-dependent (71).
Sucrose consumption may lead to pathophysiological consequences similar to those produced by substance abuse (72). High-fat and high-sugar diet alters glutamate, DA, and opioid signaling in the dorsal striatum of mice. Specifically, a high-fat and high-sugar diet increased the α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid – to – N-methyl-D-aspartic acid receptor current ratio in medium spiny neurons and prolonged spontaneous glutamate-mediated currents, increased DA release and slower DA reuptake in the striatum, and reduced MOR-mediated synaptic plasticity (73). In this study, both high fat and high sugar were added to the diet, and it is not clear which of these factors played a major role. However, Study shows that sucrose intake reduces the availability of MOR and DA D2/3 receptors in the porcine brain (74). In addition, in rats, casual exposure to 1% sucrose for 3 weeks resulted in altered MOR and D1/D2 receptor mRNA and protein expression in the NAc (75). And repeated sucrose intake decreased the density of DA D2 receptors in the striatum (76). Thus, sucrose may alter the neural circuits that encode reward and enhance the SUD drive.
The SUD and increased sugar intake appear to be a vicious cycle due to similar neural circuits and altered neuroplasticity. Sensitivity to one substance may lead to cross-sensitization to another substance. For instance, amphetamine treatment produced psychomotor sensitization and accelerated the subsequent escalation of cocaine intake (77), and nicotine-sensitive animals may consume more alcohol (78). Hence, increased sugar intake may also cause sensitivity to SUD. Reducing sugar intake may be an important complement to the treatment of SUD. And the very low carbohydrate intake of the ketogenic diet may offer help for this potential treatment (Figure 2A). However, the role of the KD in SUD treatment by lowering carbohydrate intake is largely unknown. Conversely, for example, fat consumption was also associated with the DA and opioid signaling pathways in the hypothalamus (79). Therefore, the improved effect of KD with low-carbohydrate and high-fat intake in the neural circuits of addiction remains to be investigated.
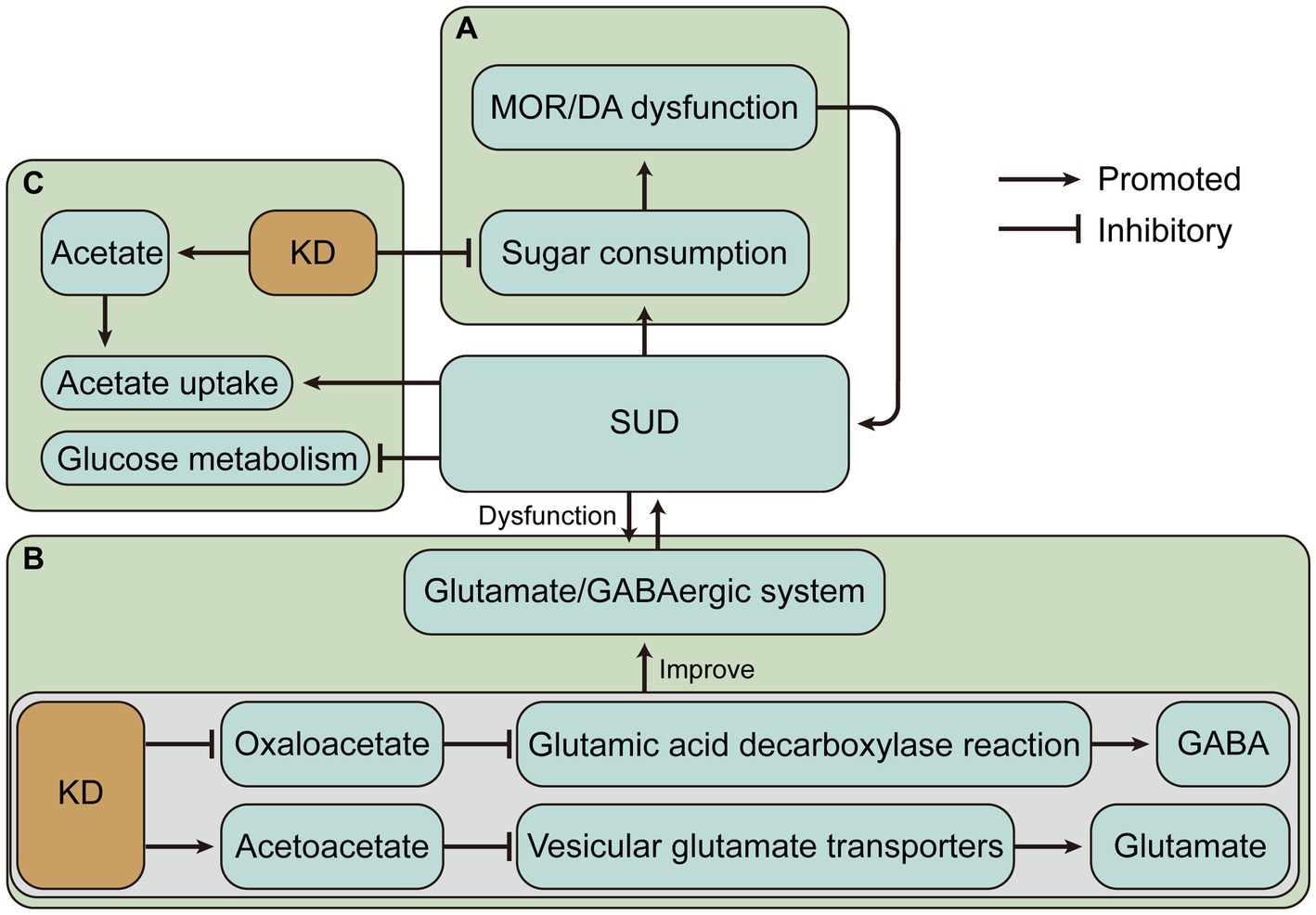
Figure 2. The ketogenic diet (KD) may have beneficial effects on substance use disorders (SUD) by altering dietary intake and changing metabolic status. (A) KD may alleviate the vicious cycle caused by SUD and sugar intake. (B) KD may alleviate SUD-induced glutamate/GABAergic disorders by decreasing glutamate and increasing GABA. (C) Ketone bodies produced by KD may alleviate impaired glucose metabolism and increased ketone body requirements due to SUD. MOR, mu-opioid receptor; DA, dopamine; GABA, γ-aminobutyric acid.
3. Ketogenic diet and glutamate/GABAergic system
DA is critical for acute reward and the onset of addiction, but the later stages of addiction are primarily related to glutamatergic projections. Dysregulation of glutamate, an excitatory neurotransmitter, leads to changes in neuroplasticity, which reduces the value of natural rewards, decreases cognitive control, and promotes compulsive drug-seeking (80–82). Imbalance in glutamate homeostasis is a key neurometabolic feature of SUD. By a proton magnetic resonance spectroscopy protocol, it was found that basal glutamate concentrations in the NAc were significantly lower in cocaine addicts and that glutamate levels increased during cue-induced cravings in cocaine-addicted individuals compared to baseline (83). Changes in relapse behavior induced by activation or inhibition of glutamate receptors suggest that glutamate may be the major mediator of substance-seeking behavior recovery. For example, stimulation of group II metabotropic glutamate receptor and blockade of group I metabotropic glutamate receptor could prevent relapse of cocaine, heroin, nicotine, and alcohol (84–90).
The rapid elevation and release of DA in the voxel nucleus are important causes of the reward response in addiction which can be antagonized by increasing γ-aminobutyric acid (GABA) levels (91). Diminished GABA signaling leads to increased susceptibility to SUD. For example, the excitatory signal from the entopeduncular nucleus to the lateral habenula is limited by GABAergic cotransmission, but during cocaine withdrawal, the presynaptic vesicular GABA transporter is reduced, resulting in a decrease in this inhibitory component and causing relapse. And stress-induced relapse can be prevented by restoring GABAergic neurotransmission (92). GABA release in DA neurons is dependent on the GABA synthesis pathway mediated by aldehyde dehydrogenase 1a1 and decreased it leads to increased alcohol consumption and preference (93). In addition, exposure to morphine blocks the long-term potentiation of GABA-mediated synaptic transmission and may contribute to the development of addiction (94).
KD may alleviate SUD by decreasing glutamate and increasing GABA. A similar example is that KD significantly increased the level of GABA in cerebrospinal fluid in children with refractory epilepsy (95). Previous studies have shown that KD treatment increases GABA levels in the hippocampus of the rat brain (96). The possible mechanism is that KD may limit the availability of oxaloacetate in the aspartate aminotransferase reaction, allowing more glutamate to enter the glutamic acid decarboxylase reaction and produce GABA (97). KD also produces ketone body carbon which is metabolized to glutamine, the basic precursor of GABA, in addition to the increased consumption of acetate in ketosis, which is converted to glutamine via astrocytes in the brain (97). Relatively, the glutamate level in the brain of rats receiving KD was reduced (98). It has also been proposed that KD intervention for anxiety and depression in Alzheimer’s Disease may related to the modulation of the glutamatergic neurotransmission system (99). Acetone and BHB act as glutamate inhibitors in the N-methyl-D-aspartic acid receptor receptor (99, 100). Evidence suggests that acetoacetate inhibits glutamate release and another mechanism by which KD reduces glutamate is that Cl-activates vesicular glutamate transporters, while increased ketone bodies, especially acetoacetate, inhibit vesicular glutamate transporters by competing with Cl−, thereby inhibiting glutamate release (101) (Figure 2B). Overall, KD leads to an enhanced conversion of glutamate to glutamine, which permits a more efficient removal of glutamate and conversion of glutamine to GABA (102). These evidences provide rationale for the treatment of SUD by the glutamate/GABAergic system.
4. Ketogenic diet increases ketone metabolism
The KD creates a special metabolic state that changes the energy metabolism, which is dominated by glucose metabolism in the brain, to ketone body metabolism. Because of this particular state of energy metabolism that develops, the KD may be more effective in the treatment of AUD. The effects of alcoholism on brain glucose and acetate metabolism were assessed by positron emission tomography, and brain glucose metabolism was found to be reduced and acetate uptake was increased during alcoholism, suggesting that increasing acetate concentration through a KD may have therapeutic benefits for AUD (103). And several preclinical and clinical studies have provided a partial basis for this. KD decreased the rigidity and irritability symptoms and reduced convulsions and anxiety-like behaviors during the alcohol withdrawal (31, 32). And KD reduces alcohol consumption in alcohol-dependent rats (28). Mice exposed to a ketogenic diet exhibited an overall reduction in ethanol consumption during stable ketosis, while gene expression analysis revealed several changes in the DA, adenosine, and cannabinoid systems (30). In addition, ketogenic diet-induced fibroblast growth factor 21 administration significantly reduced sweet taste and alcohol preference in mice and sweet taste preference in cynomolgus monkeys, and was associated with reduced DA concentrations in the NAc (104). Human studies on AUD have shown that patients who received KD experienced decreased withdrawal symptoms, required less benzodiazepines, had reduced cravings for alcohol, and exhibited increased reactivity to alcohol cues in the dorsal anterior cingulate cortex (28). However, a case report showed that prolonged KD combined with alcohol intake can disrupt glucose homeostasis and lead to significant hypoglycemia (105), suggesting that counseling patients about alcohol intake during the KD is necessary.
During alcohol abuse, the demand for ketone bodies in the brain increases, and adaptation to repeated alcohol intake occurs, while acetate levels decline during withdrawal, and supplementation of ketone bodies through a KD may alleviate withdrawal symptoms due to the sudden shift in metabolic status (106). These may be the mechanisms by which the KD alleviates AUD. It is worth noting that several studies have shown that other substance use disorders also exist with impaired brain glucose metabolism. For example, regional cerebral glucose metabolism was lower in both opioid withdrawal and methadone maintenance subjects than in control subjects (107, 108). Cocaine and nicotine reduce brain glucose metabolism (109, 110). During methamphetamine abuse and withdrawal, glucose metabolism is reduced in several brain regions including the thalamus, striatum, and frontal lobes (111–113). The prevalence of impaired cerebral glucose metabolism suggests that KD may have beneficial effects by changing the major energy metabolism of the brain in SUD (Figure 2C).
5. Neuroprotective effects of the ketogenic diet
Mitochondria is an important organelle for energy production and is also involved in a series of processes of signaling and cell death. During oxidative metabolism and various cellular reactions, mitochondria produce reactive oxygen species (ROS), associated with the function of the mitochondrial electron transport chain. Due to the lack of adequate antioxidant systems, the brain and nervous system are easily affected by oxidative stress (114). Impaired mitochondrial redox homeostasis, such as oxidative stress due to excessive ROS production and impairment of antioxidant function, can cause mitochondrial dysfunction and the onset of the cell death cascade, leading to neuronal damage (115, 116). And oxidative stress can cause free radicals to attack nerve cells, leading to neurodegeneration (117). Substance abuse causes widespread neurotoxicity, mainly related to mitochondrial dysfunction and oxidative stress (118). In response to the dopaminergic neural activation induced by substances of abuse, such as heroin (119, 120), methamphetamine (121, 122), and cocaine (123), the mitochondrial respiratory chain is rapidly activated, leading to mitochondrial dysfunction, such as decreased mitochondrial membrane potential, mitochondrial DNA, and mitochondrial proteins, and increased cytochrome c release, resulting in increased ROS and oxidative stress (124).
KD is neuroprotective and, importantly, its primary activity may be associated with improved mitochondrial function and reduced oxidative stress (125). KD, especially BHBA, inhibits dynamin-related protein 1-mediated mitochondrial fission (126). Dynamin-related protein 1 is a GTPase that interacts with the mitochondrial fission protein Fis1 to induce mitochondrial fission, which produces ROS and activates nucleotide-binding domain-like receptor protein 3 (NLRP3) inflammasome (127, 128). Detection of mRNA and protein levels showed that KD or ketone bodies can improve mitochondrial biogenesis and bioenergetics through the peroxisome proliferator-activated receptor γ-coactivator-1α – sirtuin 3 – uncoupling proteins 2 axis, and uncoupling proteins upregulated by mitochondrial respiration can reduce the production of ROS and oxidative stress (129, 130). Similarly, enhanced mitochondrial biogenesis was verified by define of gene expression patterns in rat hippocampus at 3 weeks of KD (131). 3-week KD increased glutathione biosynthesis in rat hippocampal mitochondria, enhanced mitochondrial antioxidant status, and protected mitochondrial DNA from oxidant-induced damage (132). BHBA and acetoacetate reduce glutamate-induced free radical formation by increasing the nicotinamide adenine dinucleotide (+)/ nicotinamide adenine dinucleotide hydrogen ratio and strengthening mitochondrial respiration (133), and prevent mitochondrial permeability transition and oxidative injury (134, 135). In addition, KD reduces the release of mitochondrial cytochrome c, thus achieving a neuroprotective effect (136). These findings provide a wealth of evidence for a possible neuroprotective role of KD in SUD (Figure 3A).
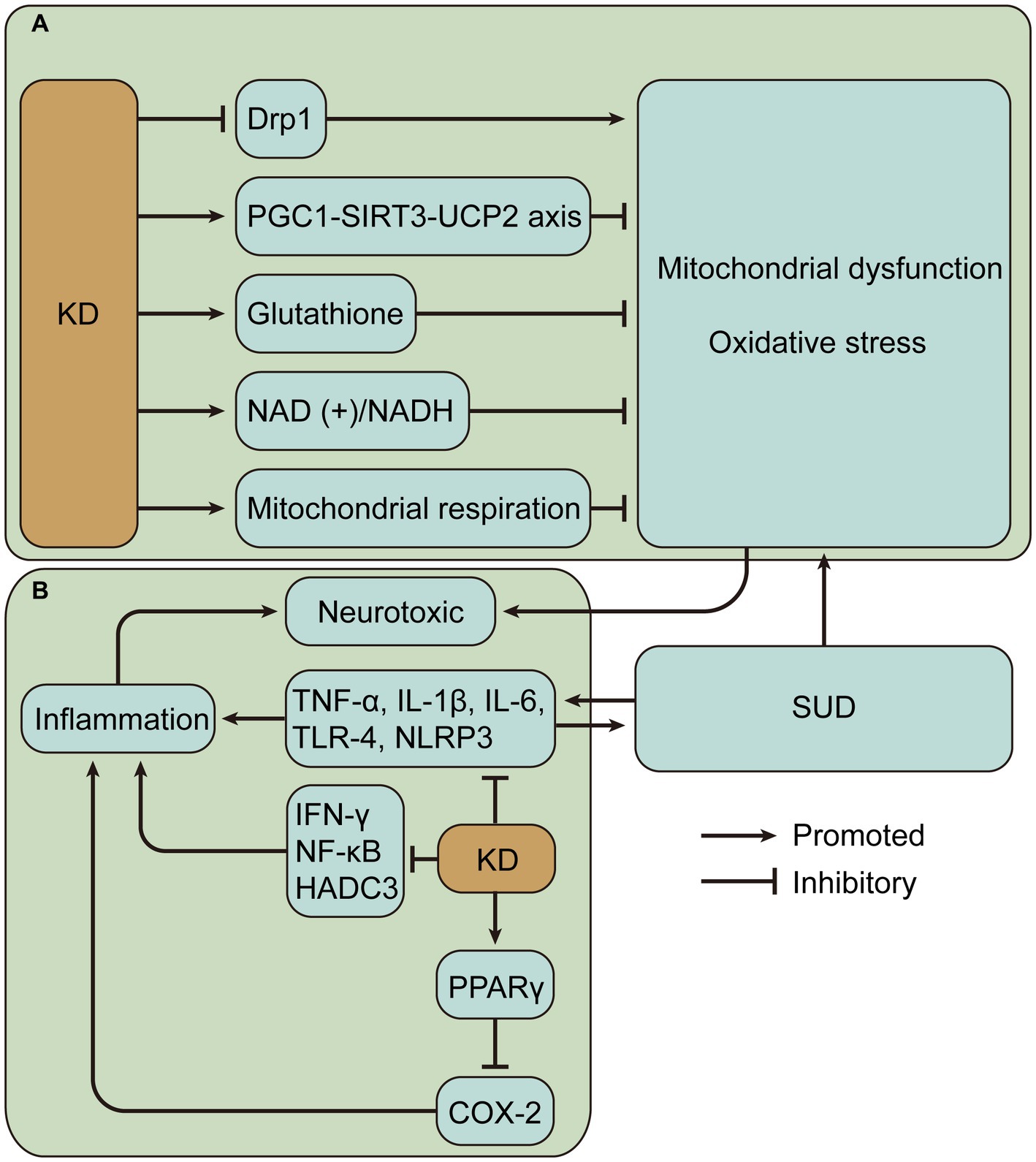
Figure 3. Potential mechanisms of neuroprotective effects of ketogenic diet (KD) in substance use disorders (SUD). (A) KD may alleviate mitochondrial dysfunction and oxidative stress caused by SUD. (B) KD may alleviate inflammation caused by SUD. Drp1, dynamin-related protein 1; PGC1-SIRT3-UCP2, peroxisome proliferator-activated receptor γ-coactivator-1α – sirtuin 3 – uncoupling proteins 2; NAD (+)/NADH, nicotinamide adenine dinucleotide (+)/nicotinamide adenine dinucleotide hydrogen; TNF, tumor necrosis factor; IL, interleukin; NLRP3, nucleotide-binding domain-like receptor protein 3; IFN, interferon; NF-κB, nuclear factor-kappa B; HADC3, histone deacetylase 3; PPAR γ, peroxisome proliferator-activated receptor γ; COX-2, cyclooxygenase-2.
Inflammation is another important factor in neurodegeneration, which may lead to the production of neurotoxic mediators (137). Many substances of abuse can lead to increased inflammation, mainly characterized by upregulation of pro-inflammatory cytokine levels, such as tumor necrosis factor (TNF)-α, Interleukin (IL)-1, and IL-6 (138–140). In addition, neuroinflammation in SUD can also be induced by the activation of toll-like receptor 4 receptors and NLRP3 inflammasome (141–143). Cytokines have been shown to influence host behavior (144). Inflammation appears to lead to persistent changes in basal ganglia and DA function, as shown by lack of pleasure, fatigue, and psychomotor slowing. And inflammation also leads to reduced neural responses to hedonic rewards, reduced DA metabolites, and increased reuptake and decreased turnover of presynaptic DA (145, 146). In SUD, inflammatory responses may contribute to drug-induced rewards and drug relapse. An interesting example is that cannabidiol may reduce METH relapse by improving cytokine expressions, such as IL-1β, IL-6, IL-10, and TNF-α (147).
Evidence suggests that KD reduces inflammation in a variety of diseases. KD activates peroxisome proliferator-activated receptor gamma to inhibit cyclooxygenase-2-dependent pathways and thereby suppress neuroinflammation in the mouse model of epilepsy (148). Inflammation is a key secondary pathological process in spinal cord injury, and KD attenuates inflammation, including reduced nuclear factor-kappa B pathway and expression levels of TNF-α, IL-1β, and interferon-γ (149). In colorectal tumor-bearing mice, the ketogenic formula suppressed systemic inflammation associated with tumors, such as lowering plasma IL-6 levels (150). In a demyelination mouse model with neuroinflammation, KD treatment inhibits the activation of pro-inflammatory glial cells and decreases the production of pro-inflammatory cytokines, including IL-1β and TNF-α, in addition to significantly reducing histone deacetylase 3 and NLRP3 (151). Histone deacetylases alter chromatin structure and accessibility, impairing memory function and synaptic plasticity (152). In a mouse model of Alzheimer’s disease, a 4-month KD reduced neuroinflammation, decreased microglia activation, and thus improved cognitive function (153). Single-cell RNA sequencing of adipose-resident immune cells showed that KD expanded metabolically protective γδ T cells and suppressed inflammation (154). The results suggest that KD mostly alleviates inflammation by reducing pro-inflammatory cytokine levels and plays a beneficial role in different diseases (Figure 3B). However, there are also contrary evidence suggesting the complexity of KD regulation of inflammation. One study evaluated postprandial responses in 17 men after transitioning from a baseline diet to an isocaloric KD and found increased markers of inflammation (155). Hyperketonemic diabetic patients have significantly higher levels of TNF-α and IL-6 than normoketonemic diabetic patients (156, 157). Short-term use of exogenous ketone supplements increases blood BHBA but leads to increased markers of NLRP3 inflammasome activation including caspase-1 and IL-1β secretion (158). In addition, it was found that high concentrations of BHB induced an increase in pro-inflammatory signals such as TNF-α, IL-6 and IL-1β (159). BHB can also mediate inflammation by promoting neutrophil adhesion through inhibition of autophagy (160). Therefore, it is crucial to measure ketosis levels, such as BHB levels, to properly evaluate the impact of KD.
According to the above results, KD may play an important protective role in SUD-mediated neurological damage mainly by protecting mitochondria and reducing oxidative stress and inflammation. However, different strategies, such as the timing of intervention, need to be considered during the KD.
6. Glial cells in the ketogenic diet
Glial cells account for about half of the central nerve cells, and they influence the formation and function of the neural system (161). Glial cells regulate neurotransmission, synaptic connections, and neural circuits, which can influence brain function and behavior in SUD (162–164). In the past, microglia were usually thought of as phagocytes in the central nervous system; however, in fact, recent studies have revealed that microglia are important participants in central nervous system homeostasis, and their dysregulation causes neurological diseases (165, 166). Microglia may regulate neuronal function and be associated with the development of SUD. Chronic high-dose alcohol intake induces reactive gliosis and may increase the risk of dementia (167). In contrast, depletion of microglia reduced alcohol consumption in mice, decreased anxiety-like behavior, and reduced GABA and glutamate receptor-mediated synaptic transmission in the central nucleus of the amygdala (168). Morphine withdrawal in mice leads to microglia adaptations and reduced glutamatergic transmission, resulting in reduced synaptic excitation and social behavior (169). Many substances of abuse cause neurological dysfunction by activating microglia. For example, cocaine may activate microglia by downregulating miR-124 targeting kruppel-like factor 4 and toll-like receptor 4 signaling, which may contribute to cocaine-induced synaptic plasticity (170, 171). Methamphetamine activates the NLRP3 inflammasome in microglia and promotes the processing and release of IL-1β, resulting in neurotoxicity (142). In addition to microglia, another type of glial cell in the central nervous system, astrocytes, is also important in neurological diseases (172, 173). Interestingly, MOR is highly expressed in astrocytes (174). Activation of astrocyte MOR elevates cytosolic calcium ions, leading to rapid glutamate release, and further regulating neuronal activity (175, 176). The activation of astrocytes may be involved in the addiction process. Studies have shown that glutamate released by astrocyte MOR activation enhances synaptic transmission and can drive conditioned place preference (CPP) (177). In addition, activation of astrocytes is associated with neuroinflammation (178, 179). For example, morphine activates astrocytes and promotes the production of pro-inflammatory cytokines (180). There are also interactions between glial cells. Morphine-mediated release of miR-138 from astrocyte-derived extracellular vesicles promotes microglia activation and further causes neurological dysfunction (181). Thus, glial cell disorders play a key role in SUD, and improving glial cell disorders may facilitate recovery from SUD.
In a study of normal adult rats, KD was able to alter glial morphology, suggesting that KD may have neuroprotective effects by affecting glial cells (182). In a variety of disease models, KD has played a similar role in reducing glial cell activation. Studies have shown that KD reduced the inflammatory activation of microglia in glaucoma, depression, and multiple sclerosis mouse models (183–185). Similarly, activation of reactive astrocytes can be inhibited by KD (185). BHBA produced by KD may play a major role in these effects. In AD mice, BHB reduces microglia proliferation and activation (135, 186). In neuroinflammatory models, BHBA promotes microglia polarization toward M2, which exerts anti-inflammatory effects and prevents depressive-like behaviors (187). In spinal cord injury, BHB inhibits NLRP3 inflammasome and transfers the activation state of microglia from M1 to M2a phenotype, reducing neuroinflammation (188). However, some studies have also raised questions about the central role of ketone bodies. For instance, the medium-chain triglyceride KD is a commonly used variation (189). Medium-chain fatty acids may play a central role in the KD, which may regulate mitochondrial metabolism in astrocytes and have positive effects on the brain such as accelerated glycolysis, enhanced lactate shuttle, and accelerated ketone body production (190). Thus, KD may play a neuroprotective role by reducing the activation of glial cells, which is crucial in the pathogenesis of SUD. However, there is no direct evidence that KD attenuates glial cell activation in SUD, and further relevant studies are necessary.
7. Ketogenic diet modulate gut microbiota
Human microbes are mainly found in the gastrointestinal tract, including bacteria, fungi, viruses, etc. Gut microbes regulate neurological function and behavior and are associated with many neurological diseases and psychiatric disorders, such as multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (191), as well as depression, and anxiety (192).
Although not yet clear, there is growing evidence of a strong link between SUD and gut microbes (193–195). A study of the gut microbiota composition of patients with more than 10 years of alcohol overconsumption showed a higher relative abundance of bacteria from phylum Proteobacteria and higher levels of the genera Sutterella, Holdemania, and Clostridium, and a lower relative abundance of bacteria from genus Faecalibacterium compared to control patients with no or low alcohol intake history (196). Decreased fecal microbial alpha diversity in people with AUD who actively drink alcohol, characterized by a decrease in Akkermansia and an increase in Bacteroides (197). Chronic vapor alcohol exposure mice significant increases in genus Alistipes and significant reductions in genra Clostridium IV and XIVb, Dorea, and Coprococcus (198). However, another study showed that alcohol led to an opposite significant increase in gut microbial diversity in mice, and the abundance of phylum Firmicutes and class Clostridiales were elevated (167). Alcohol overconsumption leads to changes in the composition of the gut microbiota and appears to cause pro-inflammatory effects. Alcohol-induced neuroinflammation and gut inflammation in mice can be attenuated by reducing the bacterial load in the gut with antibiotics (138), suggesting that alcohol-induced inflammation may be related to gut bacteria. Methadone maintenance therapy leads to an imbalance of key bacterial communities, mainly Akkermansia muciniphila, required for the production of short-chain fatty acids, mucus degradation, and maintenance of barrier integrity. And Bifidobacteriaceae was significantly increased (199). Chronic morphine treatment significantly alters the gut microbial composition, decreases the Bacteroidetes/Firmicutes ratio, and induces amplification of gram-positive pathogenic bacteria (200). The heroin-dependent mouse model had reduced gut microbial alpha diversity with higher levels of Bifidobacterium and Sutterella and reduced levels of Akkermansia compared to controls (201). Cocaine causes alterations in the gut microbiota of mice and is associated with the upregulation of pro-inflammatory mediators, such as nuclear factor-kappa B and IL-1β (202). In addition, methamphetamine use disorders resulted in altered gut microbes, increased the relative abundance of pathogenic gut bacteria, and decreased the relative abundance of probiotic bacteria in mice, which was associated with systemic inflammation (203, 204). Based on the above studies, it appears that different types of SUD lead to some similar trends in gut microbes. Typically, Sutterella is significantly increased in alcohol and heroin use (196, 201), and Bifidobacterium is generally increased in opioid use (199, 201), and Akkermansia is generally decreased in alcohol and opioid use (197, 199, 201). Interestingly, an increased abundance of Sutterella may be associated with autism spectrum disorder (205). Bifidobacterium has been shown to induce the accumulation of gut pro-inflammatory Th17 cells (206). Akkermansia is associated with improved glucose homeostasis, modulation of the immune response, and protection of barrier function (207–209). Through mucus protein degradation, Akkermansia has beneficial effects on the gut barrier (210). Synbiotic treatment reduced the escalation and relapse to alcohol intake, possibly related to Akkermansia abundance restoration (211). However, it has also been shown that in a methamphetamine-induced CPP rat model, increased Akkermansia is associated with higher CPP scores (212). In addition, chronic alcohol feeding may lead to an increase in Akkermansia muciniphila in mice (213, 214). The opposite results seem to be related to the different types of substances of abuse and experimental subjects. Even so, it is clear that SUD causes widespread gut microbial disorders, resulting in a range of adverse consequences.
Gut microbial disorders resulting from SUD may also lead to behavior changes. Some studies have shown a close correlation between SUD-related behaviors and gut microbes. For example, dysbiosis of the gut microbiota during chronic alcohol exposure was strongly associated with alcohol-induced behaviors, and a decrease in the Adlercreutzia spp. was positively associated with alcohol preference and negatively associated with anxiety-like behavior (215). Methamphetamine-altered gut microbial composition is associated with depressive-like behavior (216). There is growing interest in using fecal microbiota to treat people with SUD. Through the treatment of fecal microbiota transplantation (FMT), the important role of gut microbes is revealed. In a double-blind randomized clinical trial, FMT from a donor enriched in Lachnospiraceae and Ruminococcaceae caused favorable gut microbial changes in patients with AUD-related cirrhosis and reduced alcohol craving and consumption (217). And a variety of addiction-related behaviors were shown to be associated with Lachnospiraceae and Ruminococcaceae (218). Gut microbes influence the development of morphine dependence, while FMT may reduce opioid withdrawal responses (219). In addition, transplantation by specific probiotics may also affect SUD-induced behavioral responses. For example, Lactobacillus rhamnosus probiotic can reduce cocaine-induced behavioral responses (220). Interestingly, transplantation of SUD-associated gut microbes into normal controls would result in behavioral changes similar to those associated with SUD. For instance, transplantation of gut microbiota from AUD patients into mice induced a decrease in BHBA metabolism that may be associated with social impairment and depression in AUD (221). Similarly, transplantation of gut microbes from alcohol-fed mice to healthy controls triggered anxiety behaviors similar to those induced by alcohol withdrawal (222). Thus, SUD alters gut microbes and induces related behaviors, suggesting that the gut microbiota may be an important regulator of SUD susceptibility.
Changes in gut microbiota associated with KD have been demonstrated to play a beneficial role in a variety of diseases. Importantly, the general decrease in Akkermansia in SUD may be reversed by KD. For example, in epilepsy, KD provides epilepsy protection by increasing the relative abundance of Akkermansia and Parabacteroides and by increasing the ratio of Bacteroidetes/Firmicutes, similarly, transplantation of KD-associated gut microbiota and treatment with Akkermansia and Parabacteroides also provide epilepsy protection in control mice (223, 224). In AD, KD also increased the relative abundance of beneficial gut microbiota (such as Akkermansia muciniphila and Lactobacillus) and decreased the relative abundance of pro-inflammatory microbiota (such as Desulfovibrio and Turicibacter) and improved metabolic conditions. They reduced the risk of Alzheimer’s disease and improved Alzheimer’s disease biomarkers in cerebrospinal fluid (225, 226). In colitis, 16 weeks of KD increases Akkermansia abundance, protects the gut barrier, and alleviates inflammation (227). Moreover, KD was able to reduce gut pro-inflammatory Th17 cell accumulation by inhibiting the growth of Bifidobacterium (206, 228). This may be part of the anti-inflammatory mechanism of KD.
However, some researchers have also raised concerns about the negative impact of KD on gut microbiota and health. In children with severe epilepsy, KD resulted in a significant decrease in the relative abundance of Bifidobacteria as well as E. rectale and Dialister, and an increase in the relative abundance of E. coli, indicating a decrease in the abundance of health-promoting fiber-consuming bacteria (229). Similarly, KD pre-treatment for 1 month resulted in an increase in gut pathogenic bacteria and a decrease in beneficial bacteria in inflammatory bowel disease mice, which increased gut and systemic inflammation, disrupted the gut barrier, and exacerbated colitis (230). These different outcomes resulting from KD may be attributed to different populations, diseases, and treatment protocols. The exact reasons need to be further explored, given that different gut microbial alterations have been observed in different diseases. However, certainly, KD may partially regulate gut microbial dysbiosis caused by SUD, such as Akkermansia dysbiosis.
8. Limitations
Various effects of KD may be beneficial for recovery from SUD. However, there are few reports of KD being used for the treatment of SUD besides AUD. Also, some researchers have shown concerns during the application of KD in neurological diseases, such as decreased appetite, increased risk of malnutrition, and the occurrence of some adverse effects (231). Common KD adverse effects include metabolic abnormalities, gastrointestinal symptoms, kidney stones, and slow growth in children (232). However, most of these adverse effects were evaluated in children. Even so, KD should be applied with caution in people with SUD, as this particular population often suffers from multisystem disorders such as the increased risk of malnutrition (36) and gastrointestinal symptoms (233).
9. Conclusion
The successful exploration of the KD in other neurological disorders suggests a positive role in SUD that still cannot be ignored. It may play a positive role in the improvement of various disorders caused by SUD, especially the reduction of sugar intake, alteration of metabolic processes, improvement of neural circuits associated with substance addiction, neuroprotective effects, improvement of glial cell activation, and modulation of gut microbiota (Table 2). In conclusion, there are potential therapeutic implications of the KD in SUD, but many problems remain. Further preclinical studies and clinical randomized controlled trials are needed to investigate and improve strategies for KD, such as optimization of the timing of interventions and nutrient composition, to assess the suitability, effectiveness, and safety of KD in the treatment of SUD.
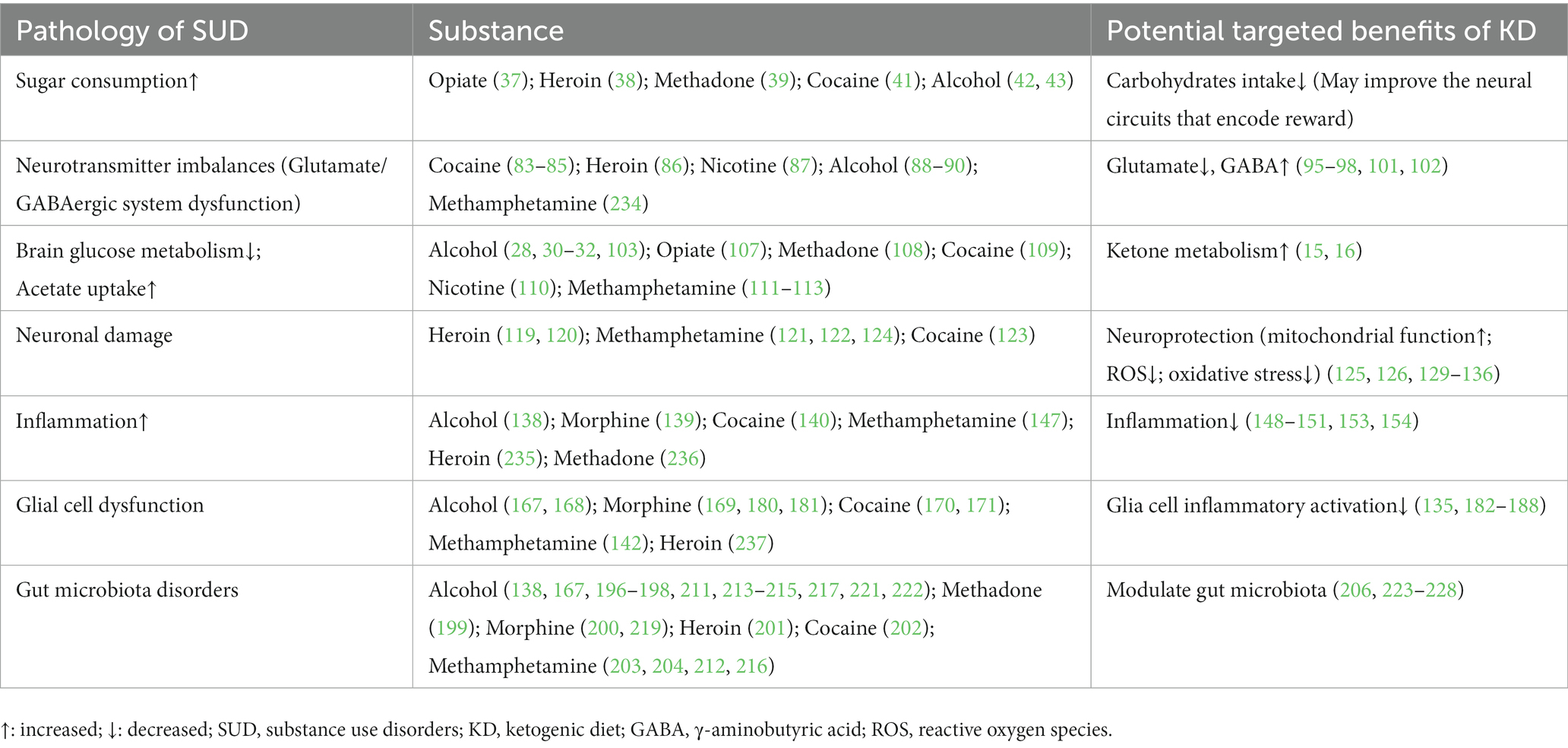
Table 2. Pathology of SUD and potential targeted benefits of KD.
Author contributions
DK, J-xS, and J-qY: conceptualization, original draft preparation, review, and editing. Y-sL, KB, Z-yZ, and K-hW: conceptualization, review, and editing. H-yL, MZ, and YX: review and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was partly supported by the Yunnan Technological Innovation Centre of Drug Addiction Medicine (202305AK340001); the Biomedical Science Resource Database of Kunming Medical University (Drug Abuse Population Cohort; Digitalization, Development and Application of Biotic Resource) (No. 202002AA100007); the National Natural Science Foundation of China (No. 81860100); the high-level health technology talent reserve in Yunnan Province (No. H-2018062); the Yunnan Province “high-level talent training support program” training program (Nos. YNWR-QNBJ-2019-243 and RLMY20200019); the Reserve Talent Project for Young and Middle-aged Academic and Technical Leaders of Yunnan Province Science and Technology Department (No. 202005AC160057); the Huang Chang-ming Expert Workstation of Yunnan Province (No. 202005AF150090); and the Yunnan Province and the Education Department of Yunnan Province (No. 2019J1226).
Acknowledgments
The authors thank all the authors who participated in the writing and revision of this review article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AUD, alcohol use disorders; BHB, β-hydroxybutyric; CPP, conditioned place preference; DA, dopamine; DUD, drug use disorders; GABA, γ-aminobutyric acid; IL, interleukin; KD, ketogenic diet; MOR, mu-opioid receptor; NAc, nucleus accumbens; NLRP3, nucleotide-binding domain-like receptor protein 3; ROS, reactive oxygen species; SUD, substance use disorders; TNF, tumor necrosis factor.
References
1. Volkow, ND, Koob, GF, and McLellan, AT. Neurobiologic advances from the brain disease model of addiction. N Engl J Med. (2016) 374:363–71. doi: 10.1056/NEJMra1511480
2. Substance Abuse and Mental Health Services Administration. Key substance use and mental health indicators in the United States: Results from the 2021 National Survey on Drug Use and Health (HHS Publication No. PEP22-07-01-005, NSDUH Series H-57). Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration. (2022). Available at: https://www.samhsa.gov/data/report/2021-nsduh-annual-national-report.
3. Castaldelli-Maia, JM, and Bhugra, D. Analysis of global prevalence of mental and substance use disorders within countries: focus on sociodemographic characteristics and income levels. Int Rev Psychiatry. (2022) 34:6–15. doi: 10.1080/09540261.2022.2040450
4. Degenhardt, L, Whiteford, HA, Ferrari, AJ, Baxter, AJ, Charlson, FJ, Hall, WD, et al. Global burden of disease attributable to illicit drug use and dependence: findings from the global burden of disease study 2010. Lancet. (2013) 382:1564–74. doi: 10.1016/S0140-6736(13)61530-5
5. Alcohol, GBD, and Drug, UC. The global burden of disease attributable to alcohol and drug use in 195 countries and territories, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Psychiatry. (2018) 5:987–1012. doi: 10.1016/S2215-0366(18)30337-7
6. Roth, GA, Abate, D, Abate, KH, Abay, SM, Abbafati, C, Abbasi, N, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2018) 392:1736–88. doi: 10.1016/s0140-6736(18)32203-7
7. Volkow, ND, and Morales, M. The brain on drugs: from reward to addiction. Cells. (2015) 162:712–25. doi: 10.1016/j.cell.2015.07.046
8. Friedman, H, Newton, C, and Klein, TW. Microbial infections, immunomodulation, and drugs of abuse. Clin Microbiol Rev. (2003) 16:209–19. doi: 10.1128/CMR.16.2.209-219.2003
9. Patnaik Kuppili, P, Vengadavaradan, A, and Bharadwaj, B. Metabolic syndrome and substance use: a narrative review. Asian J Psychiatr. (2019) 43:111–20. doi: 10.1016/j.ajp.2019.05.022
10. Bathla, M, Singh, M, Anjum, S, Kulhara, P, and Jangli, SI. Metabolic syndrome in drug naive patients with substance use disorder. Diabetes Metab Syndr. (2017) 11:167–71. doi: 10.1016/j.dsx.2016.08.022
11. Tran, BX, Moir, M, Latkin, CA, Hall, BJ, Nguyen, CT, Ha, GH, et al. Global research mapping of substance use disorder and treatment 1971-2017: implications for priority setting. Subst Abuse Treat Prev Policy. (2019) 14:21. doi: 10.1186/s13011-019-0204-7
12. NIDA. Treatment and recovery. (2023). Available at: https://nida.nih.gov/publications/drugs-brains-behavior-science-addiction/treatment-recovery (Accessed April, 24, 2023).
13. Kalin, NH. Substance use disorders and addiction: mechanisms, trends, and treatment implications. Am J Psychiatry. (2020) 177:1015–8. doi: 10.1176/appi.ajp.2020.20091382
14. Veech, RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. (2004) 70:309–19. doi: 10.1016/j.plefa.2003.09.007
15. Owen, OE, Morgan, AP, Kemp, HG, Sullivan, JM, Herrera, MG, and Cahill, GF Jr. Brain metabolism during fasting. J Clin Invest. (1967) 46:1589–95. doi: 10.1172/JCI105650
16. Fukao, T, Lopaschuk, GD, and Mitchell, GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids. (2004) 70:243–51. doi: 10.1016/j.plefa.2003.11.001
17. Paoli, A, Rubini, A, Volek, JS, and Grimaldi, KA. Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur J Clin Nutr. (2013) 67:789–96. doi: 10.1038/ejcn.2013.116
18. Neal, EG, Chaffe, H, Schwartz, RH, Lawson, MS, Edwards, N, Fitzsimmons, G, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. (2008) 7:500–6. doi: 10.1016/S1474-4422(08)70092-9
19. Yancy, WS Jr, Olsen, MK, Guyton, JR, Bakst, RP, and Westman, EC. A low-carbohydrate, ketogenic diet versus a low-fat diet to treat obesity and hyperlipidemia: a randomized, controlled trial. Ann Intern Med. (2004) 140:769–77. doi: 10.7326/0003-4819-140-10-200405180-00006
20. Feinman, RD, Pogozelski, WK, Astrup, A, Bernstein, RK, Fine, EJ, Westman, EC, et al. Dietary carbohydrate restriction as the first approach in diabetes management: critical review and evidence base. Nutrition. (2015) 31:1–13. doi: 10.1016/j.nut.2014.06.011
21. Henderson, ST, Vogel, JL, Barr, LJ, Garvin, F, Jones, JJ, and Costantini, LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab. (2009) 6:31. doi: 10.1186/1743-7075-6-31
22. Vanitallie, TB, Nonas, C, Di Rocco, A, Boyar, K, Hyams, K, and Heymsfield, SB. Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study. Neurology. (2005) 64:728–30. doi: 10.1212/01.WNL.0000152046.11390.45
23. Tennant, DA, Duran, RV, and Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. (2010) 10:267–77. doi: 10.1038/nrc2817
24. Evangeliou, A, Vlachonikolis, I, Mihailidou, H, Spilioti, M, Skarpalezou, A, Makaronas, N, et al. Application of a ketogenic diet in children with autistic behavior: pilot study. J Child Neurol. (2003) 18:113–8. doi: 10.1177/08830738030180020501
25. Choi, IY, Piccio, L, Childress, P, Bollman, B, Ghosh, A, Brandhorst, S, et al. A diet mimicking fasting promotes regeneration and reduces autoimmunity and multiple sclerosis symptoms. Cell Rep. (2016) 15:2136–46. doi: 10.1016/j.celrep.2016.05.009
26. Di Lorenzo, C, Coppola, G, Sirianni, G, Di Lorenzo, G, Bracaglia, M, Di Lenola, D, et al. Migraine improvement during short lasting ketogenesis: a proof-of-concept study. Eur J Neurol. (2015) 22:170–7. doi: 10.1111/ene.12550
27. Stafstrom, CE, and Rho, JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. (2012) 3:59. doi: 10.3389/fphar.2012.00059
28. Wiers, CE, Vendruscolo, LF, van der Veen, JW, Manza, P, Shokri-Kojori, E, Kroll, DS, et al. Ketogenic diet reduces alcohol withdrawal symptoms in humans and alcohol intake in rodents. Sci Adv. (2021) 7:eabf6780. doi: 10.1126/sciadv.abf6780
29. Martinez, LA, Lees, ME, Ruskin, DN, and Masino, SA. A ketogenic diet diminishes behavioral responses to cocaine in young adult male and female rats. Neuropharmacology. (2019) 149:27–34. doi: 10.1016/j.neuropharm.2019.02.001
30. Blanco-Gandia, MDC, Rodenas-Gonzalez, F, Pascual, M, Reguilon, MD, Guerri, C, Minarro, J, et al. Ketogenic diet decreases alcohol intake in adult male mice. Nutrients. (2021) 13:2167. doi: 10.3390/nu13072167
31. Dencker, D, Molander, A, Thomsen, M, Schlumberger, C, Wortwein, G, Weikop, P, et al. Ketogenic diet suppresses alcohol withdrawal syndrome in rats. Alcohol Clin Exp Res. (2018) 42:270–7. doi: 10.1111/acer.13560
32. Bornebusch, AB, Mason, GF, Tonetto, S, Damsgaard, J, Gjedde, A, Fink-Jensen, A, et al. Effects of ketogenic diet and ketone monoester supplement on acute alcohol withdrawal symptoms in male mice. Psychopharmacology. (2021) 238:833–44. doi: 10.1007/s00213-020-05735-1
33. Ródenas-González, F, Blanco-Gandía, MC, Miñarro, J, and Rodríguez-Arias, M. Effects of ketosis on cocaine-induced reinstatement in male mice. Neurosci Lett. (2022) 778:136619. doi: 10.1016/j.neulet.2022.136619
34. Li, H, Wan, X, Wu, Z, Zhou, Y, Chen, R, Xu, W, et al. β-hydroxybutyrate reduces reinstatement of cocaine conditioned place preference through hippocampal CaMKII-α β-hydroxybutyrylation. Cell Rep. (2022) 41:111724. doi: 10.1016/j.celrep.2022.111724
35. Trinko, R, Diaz, DM, Foscue, E, Thompson, SL, Taylor, JR, and DiLeone, RJ. Ketogenic diet enhances the effects of oxycodone in mice. Sci Rep. (2023) 13:7507. doi: 10.1038/s41598-023-33458-8
36. Jeynes, KD, and Gibson, EL. The importance of nutrition in aiding recovery from substance use disorders: a review. Drug Alcohol Depend. (2017) 179:229–39. doi: 10.1016/j.drugalcdep.2017.07.006
37. Morabia, A, Fabre, J, Chee, E, Zeger, S, Orsat, E, and Robert, A. Diet and opiate addiction: a quantitative assessment of the diet of non-institutionalized opiate addicts. Br J Addict. (1989) 84:173–80. doi: 10.1111/j.1360-0443.1989.tb00566.x
38. Neale, J, Nettleton, S, Pickering, L, and Fischer, J. Eating patterns among heroin users: a qualitative study with implications for nutritional interventions. Addiction. (2012) 107:635–41. doi: 10.1111/j.1360-0443.2011.03660.x
39. Nolan, LJ, and Scagnelli, LM. Preference for sweet foods and higher body mass index in patients being treated in long-term methadone maintenance. Subst Use Misuse. (2007) 42:1555–66. doi: 10.1080/10826080701517727
40. Zador, D, Lyons Wall, PM, and Webster, I. High sugar intake in a group of women on methadone maintenance in south western Sydney. Australia Addict. (1996) 91:1053–61. doi: 10.1111/j.1360-0443.1996.tb03601.x
41. Janowsky, DS, Pucilowski, O, and Buyinza, M. Preference for higher sucrose concentrations in cocaine abusing-dependent patients. J Psychiatr Res. (2003) 37:35–41. doi: 10.1016/s0022-3956(02)00063-8
42. Kampov-Polevoy, AB, Tsoi, MV, Zvartau, EE, Neznanov, NG, and Khalitov, E. Sweet liking and family history of alcoholism in hospitalized alcoholic and non-alcoholic patients. Alcohol Alcohol. (2001) 36:165–70. doi: 10.1093/alcalc/36.2.165
43. Krahn, D, Grossman, J, Henk, H, Mussey, M, Crosby, R, and Gosnell, B. Sweet intake, sweet-liking, urges to eat, and weight change: relationship to alcohol dependence and abstinence. Addict Behav. (2006) 31:622–31. doi: 10.1016/j.addbeh.2005.05.056
44. Kroemer, G, Lopez-Otin, C, Madeo, F, and de Cabo, R. Carbotoxicity-noxious effects of carbohydrates. Cells. (2018) 175:605–14. doi: 10.1016/j.cell.2018.07.044
45. Hoerr, J, Fogel, J, and Van Voorhees, B. Ecological correlations of dietary food intake and mental health disorders. J Epidemiol Glob Health. (2017) 7:81–9. doi: 10.1016/j.jegh.2016.12.001
46. Fowler, L, Ivezaj, V, and Saules, KK. Problematic intake of high-sugar/low-fat and high glycemic index foods by bariatric patients is associated with development of post-surgical new onset substance use disorders. Eat Behav. (2014) 15:505–8. doi: 10.1016/j.eatbeh.2014.06.009
47. Lange, LA, Kampov-Polevoy, AB, and Garbutt, JC. Sweet liking and high novelty seeking: independent phenotypes associated with alcohol-related problems. Alcohol Alcohol. (2010) 45:431–6. doi: 10.1093/alcalc/agq040
48. Radke, AK, Zlebnik, NE, Holtz, NA, and Carroll, ME. Cocaine-induced reward enhancement measured with intracranial self-stimulation in rats bred for low versus high saccharin intake. Behav Pharmacol. (2016) 27:133–6. doi: 10.1097/FBP.0000000000000182
49. Radke, AK, Holtz, NA, Gewirtz, JC, and Carroll, ME. Reduced emotional signs of opiate withdrawal in rats selectively bred for low (LoS) versus high (HiS) saccharin intake. Psychopharmacology. (2013) 227:117–26. doi: 10.1007/s00213-012-2945-0
50. Smail-Crevier, RL, Maracle, AC, Wash, SIJ, and Olmstead, MC. Binge-like intake of sucrose reduces the rewarding value of sucrose in adult rats. Physiol Behav. (2018) 194:420–9. doi: 10.1016/j.physbeh.2018.06.027
51. Collins, GT, Chen, Y, Tschumi, C, Rush, EL, Mensah, A, Koek, W, et al. Effects of consuming a diet high in fat and/or sugar on the locomotor effects of acute and repeated cocaine in male and female C57BL/6J mice. Exp Clin Psychopharmacol. (2015) 23:228–37. doi: 10.1037/pha0000019
52. Bogucka-Bonikowska, A, Baran-Furga, H, Chmielewska, K, Habrat, B, Scinska, A, Kukwa, A, et al. Taste function in methadone-maintained opioid-dependent men. Drug Alcohol Depend. (2002) 68:113–7. doi: 10.1016/s0376-8716(02)00186-2
53. Green, A, Kaul, A, O’Shea, J, Sharma, E, Bennett, L, Mullings, EL, et al. Opiate agonists and antagonists modulate taste perception in opiate-maintained and recently detoxified subjects. J Psychopharmacol. (2013) 27:265–75. doi: 10.1177/0269881112472567
54. Garfield, JBB, and Lubman, DI. Associations between opioid dependence and sweet taste preference. Psychopharmacology. (2021) 238:1473–84. doi: 10.1007/s00213-021-05774-2
55. Charbogne, P, Kieffer, BL, and Befort, K. 15 years of genetic approaches in vivo for addiction research: opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology. (2014) 76:204–17. doi: 10.1016/j.neuropharm.2013.08.028
56. Herz, A. Endogenous opioid systems and alcohol addiction. Psychopharmacology. (1997) 129:99–111. doi: 10.1007/s002130050169
57. Ostlund, SB, Kosheleff, A, Maidment, NT, and Murphy, NP. Decreased consumption of sweet fluids in mu opioid receptor knockout mice: a microstructural analysis of licking behavior. Psychopharmacology. (2013) 229:105–13. doi: 10.1007/s00213-013-3077-x
58. Awad, G, Roeckel, LA, Massotte, D, Olmstead, MC, and Befort, K. Deletion of mu opioid receptors reduces palatable solution intake in a mouse model of binge eating. Behav Pharmacol. (2020) 31:249–55. doi: 10.1097/FBP.0000000000000496
59. Katsuura, Y, and Taha, SA. Mu opioid receptor antagonism in the nucleus accumbens shell blocks consumption of a preferred sucrose solution in an anticipatory contrast paradigm. Neuroscience. (2014) 261:144–52. doi: 10.1016/j.neuroscience.2013.12.004
60. Eikemo, M, Loseth, GE, Johnstone, T, Gjerstad, J, Willoch, F, and Leknes, S. Sweet taste pleasantness is modulated by morphine and naltrexone. Psychopharmacology. (2016) 233:3711–23. doi: 10.1007/s00213-016-4403-x
61. Nathan, PJ, O’Neill, BV, Bush, MA, Koch, A, Tao, WX, Maltby, K, et al. Opioid receptor modulation of hedonic taste preference and food intake: a single-dose safety, pharmacokinetic, and pharmacodynamic investigation with GSK1521498, a novel mu-opioid receptor inverse agonist. J Clin Pharmacol. (2012) 52:464–74. doi: 10.1177/0091270011399577
62. Hakim, JD, Chami, J, and Keay, KA. Mu-opioid and dopamine-D2 receptor expression in the nucleus accumbens of male Sprague-Dawley rats whose sucrose consumption, but not preference, decreases after nerve injury. Behav Brain Res. (2020) 381:112416. doi: 10.1016/j.bbr.2019.112416
63. Castro, DC, and Berridge, KC. Opioid hedonic hotspot in nucleus accumbens shell: mu, delta, and kappa maps for enhancement of sweetness “liking” and “wanting”. J Neurosci. (2014) 34:4239–50. doi: 10.1523/JNEUROSCI.4458-13.2014
64. Mitchell, MR, Berridge, KC, and Mahler, SV. Endocannabinoid-enhanced “liking” in nucleus accumbens shell hedonic hotspot requires endogenous opioid signals. Cannabis Cannabinoid Res. (2018) 3:166–70. doi: 10.1089/can.2018.0021
65. Di Chiara, G. Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav Brain Res. (2002) 137:75–114. doi: 10.1016/s0166-4328(02)00286-3
66. Koob, GF. Neural mechanisms of drug reinforcement. Ann N Y Acad Sci. (1992) 654:171–91. doi: 10.1111/j.1749-6632.1992.tb25966.x
67. Poisson, CL, Engel, L, and Saunders, BT. Dopamine circuit mechanisms of addiction-like behaviors. Front Neural Circuits. (2021) 15:752420. doi: 10.3389/fncir.2021.752420
68. Nestler, EJ, and Luscher, C. The molecular basis of drug addiction: linking epigenetic to synaptic and circuit mechanisms. Neuron. (2019) 102:48–59. doi: 10.1016/j.neuron.2019.01.016
69. Volkow, ND, Michaelides, M, and Baler, R. The neuroscience of drug reward and addiction. Physiol Rev. (2019) 99:2115–40. doi: 10.1152/physrev.00014.2018
70. Rada, P, Avena, NM, and Hoebel, BG. Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience. (2005) 134:737–44. doi: 10.1016/j.neuroscience.2005.04.043
71. Hajnal, A, Smith, GP, and Norgren, R. Oral sucrose stimulation increases accumbens dopamine in the rat. Am J Physiol Regul Integr Comp Physiol. (2004) 286:R31–7. doi: 10.1152/ajpregu.00282.2003
72. Jacques, A, Chaaya, N, Beecher, K, Ali, SA, Belmer, A, and Bartlett, S. The impact of sugar consumption on stress driven, emotional and addictive behaviors. Neurosci Biobehav Rev. (2019) 103:178–99. doi: 10.1016/j.neubiorev.2019.05.021
73. Fritz, BM, Munoz, B, Yin, F, Bauchle, C, and Atwood, BK. A high-fat, high-sugar ‘Western’ diet alters dorsal striatal glutamate, opioid, and dopamine transmission in mice. Neuroscience. (2018) 372:1–15. doi: 10.1016/j.neuroscience.2017.12.036
74. Winterdahl, M, Noer, O, Orlowski, D, Schacht, AC, Jakobsen, S, Alstrup, AKO, et al. Sucrose intake lowers mu-opioid and dopamine D2/3 receptor availability in porcine brain. Sci Rep. (2019) 9:16918. doi: 10.1038/s41598-019-53430-9
75. Hakim, JD, and Keay, KA. Prolonged ad libitum access to low-concentration sucrose changes the neurochemistry of the nucleus accumbens in male Sprague-Dawley rats. Physiol Behav. (2019) 201:95–103. doi: 10.1016/j.physbeh.2018.12.016
76. Bello, NT, Lucas, LR, and Hajnal, A. Repeated sucrose access influences dopamine D2 receptor density in the striatum. Neuroreport. (2002) 13:1575–8. doi: 10.1097/00001756-200208270-00017
77. Ferrario, CR, and Robinson, TE. Amphetamine pretreatment accelerates the subsequent escalation of cocaine self-administration behavior. Eur Neuropsychopharmacol. (2007) 17:352–7. doi: 10.1016/j.euroneuro.2006.08.005
78. Blomqvist, O, Ericson, M, Johnson, DH, Engel, JA, and Soderpalm, B. Voluntary ethanol intake in the rat: effects of nicotinic acetylcholine receptor blockade or subchronic nicotine treatment. Eur J Pharmacol. (1996) 314:257–67. doi: 10.1016/s0014-2999(96)00583-3
79. Hu, S, Wang, L, Yang, D, Li, L, Togo, J, Wu, Y, et al. Dietary fat, but not protein or carbohydrate, regulates energy intake and causes adiposity in mice. Cell Metab. (2018) 28:415–31 e4. doi: 10.1016/j.cmet.2018.06.010
80. Kalivas, PW, and Volkow, ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. (2005) 162:1403–13. doi: 10.1176/appi.ajp.162.8.1403
81. Kalivas, PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. (2009) 10:561–72. doi: 10.1038/nrn2515
82. Scofield, MD, Heinsbroek, JA, Gipson, CD, Kupchik, YM, Spencer, S, Smith, AC, et al. The nucleus accumbens: mechanisms of addiction across drug classes reflect the importance of glutamate homeostasis. Pharmacol Rev. (2016) 68:816–71. doi: 10.1124/pr.116.012484
83. Engeli, EJE, Zoelch, N, Hock, A, Nordt, C, Hulka, LM, Kirschner, M, et al. Impaired glutamate homeostasis in the nucleus accumbens in human cocaine addiction. Mol Psychiatry. (2021) 26:5277–85. doi: 10.1038/s41380-020-0828-z
84. Cornish, JL, and Kalivas, PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. (2000) 20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000
85. Moran, MM, McFarland, K, Melendez, RI, Kalivas, PW, and Seamans, JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. (2005) 25:6389–93. doi: 10.1523/JNEUROSCI.1007-05.2005
86. Bossert, JM, Gray, SM, Lu, L, and Shaham, Y. Activation of group II metabotropic glutamate receptors in the nucleus accumbens shell attenuates context-induced relapse to heroin seeking. Neuropsychopharmacology. (2006) 31:2197–209. doi: 10.1038/sj.npp.1300977
87. Liechti, ME, Lhuillier, L, Kaupmann, K, and Markou, A. Metabotropic glutamate 2/3 receptors in the ventral tegmental area and the nucleus accumbens shell are involved in behaviors relating to nicotine dependence. J Neurosci. (2007) 27:9077–85. doi: 10.1523/JNEUROSCI.1766-07.2007
88. Kapasova, Z, and Szumlinski, KK. Strain differences in alcohol-induced neurochemical plasticity: a role for accumbens glutamate in alcohol intake. Alcohol Clin Exp Res. (2008) 32:617–31. doi: 10.1111/j.1530-0277.2008.00620.x
89. Besheer, J, Grondin, JJM, Cannady, R, Sharko, AC, Faccidomo, S, and Hodge, CW. Metabotropic glutamate receptor 5 activity in the nucleus accumbens is required for the maintenance of ethanol self-administration in a rat genetic model of high alcohol intake. Biol Psychiatry. (2010) 67:812–22. doi: 10.1016/j.biopsych.2009.09.016
90. Zhou, Z, Karlsson, C, Liang, T, Xiong, W, Kimura, M, Tapocik, JD, et al. Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proc Natl Acad Sci U S A. (2013) 110:16963–8. doi: 10.1073/pnas.1309839110
91. Silverman, RB. Design and mechanism of GABA aminotransferase inactivators. Treatments for epilepsies and addictions. Chem Rev. (2018) 118:4037–70. doi: 10.1021/acs.chemrev.8b00009
92. Meye, FJ, Soiza-Reilly, M, Smit, T, Diana, MA, Schwarz, MK, and Mameli, M. Shifted pallidal co-release of GABA and glutamate in habenula drives cocaine withdrawal and relapse. Nat Neurosci. (2016) 19:1019–24. doi: 10.1038/nn.4334
93. Kim, JI, Ganesan, S, Luo, SX, Wu, YW, Park, E, Huang, EJ, et al. Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science. (2015) 350:102–6. doi: 10.1126/science.aac4690
94. Nugent, FS, Penick, EC, and Kauer, JA. Opioids block long-term potentiation of inhibitory synapses. Nature. (2007) 446:1086–90. doi: 10.1038/nature05726
95. Dahlin, M, Elfving, A, Ungerstedt, U, and Amark, P. The ketogenic diet influences the levels of excitatory and inhibitory amino acids in the CSF in children with refractory epilepsy. Epilepsy Res. (2005) 64:115–25. doi: 10.1016/j.eplepsyres.2005.03.008
96. Calderon, N, Betancourt, L, Hernandez, L, and Rada, P. A ketogenic diet modifies glutamate, gamma-aminobutyric acid and agmatine levels in the hippocampus of rats: a microdialysis study. Neurosci Lett. (2017) 642:158–62. doi: 10.1016/j.neulet.2017.02.014
97. Yudkoff, M, Daikhin, Y, Melo, TM, Nissim, I, Sonnewald, U, and Nissim, I. The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu Rev Nutr. (2007) 27:415–30. doi: 10.1146/annurev.nutr.27.061406.093722
98. Melø, TM, Nehlig, A, and Sonnewald, U. Neuronal-glial interactions in rats fed a ketogenic diet. Neurochem Int. (2006) 48:498–507. doi: 10.1016/j.neuint.2005.12.037
99. de la Rubia Ortí, JE, Fernández, D, Platero, F, and García-Pardo, MP. Can ketogenic diet improve Alzheimer’s disease? Association with anxiety, depression, and glutamate system. Front Nutr. (2021) 8:744398. doi: 10.3389/fnut.2021.744398
100. Pflanz, NC, Daszkowski, AW, James, KA, and Mihic, SJ. Ketone body modulation of ligand-gated ion channels. Neuropharmacology. (2019) 148:21–30. doi: 10.1016/j.neuropharm.2018.12.013
101. Juge, N, Gray, JA, Omote, H, Miyaji, T, Inoue, T, Hara, C, et al. Metabolic control of vesicular glutamate transport and release. Neuron. (2010) 68:99–112. doi: 10.1016/j.neuron.2010.09.002
102. Yudkoff, M, Daikhin, Y, Horyn, O, Nissim, I, and Nissim, I. Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia. (2008) 49:73–5. doi: 10.1111/j.1528-1167.2008.01841.x
103. Volkow, ND, Kim, SW, Wang, GJ, Alexoff, D, Logan, J, Muench, L, et al. Acute alcohol intoxication decreases glucose metabolism but increases acetate uptake in the human brain. Neuroimage. (2013) 64:277–83. doi: 10.1016/j.neuroimage.2012.08.057
104. Talukdar, S, Owen, BM, Song, P, Hernandez, G, Zhang, Y, Zhou, Y, et al. FGF21 regulates sweet and Alcohol preference. Cell Metab. (2016) 23:344–9. doi: 10.1016/j.cmet.2015.12.008
105. Malaeb, S, and Spoke, C. A case of hypoglycemia associated with the ketogenic diet and alcohol use. J Endocr Soc. (2020) 4:bvaa045. doi: 10.1210/jendso/bvaa045
106. Mahajan, VR, Elvig, SK, Vendruscolo, LF, Koob, GF, Darcey, VL, King, MT, et al. Nutritional ketosis as a potential treatment for alcohol use disorder. Front Psych. (2021) 12:781668. doi: 10.3389/fpsyt.2021.781668
107. Galynker, II, Eisenberg, D, Matochik, JA, Gertmenian-King, E, Cohen, L, Kimes, AS, et al. Cerebral metabolism and mood in remitted opiate dependence. Drug Alcohol Depend. (2007) 90:166–74. doi: 10.1016/j.drugalcdep.2007.03.015
108. Prosser, J, London, ED, and Galynker, II. Sustained attention in patients receiving and abstinent following methadone maintenance treatment for opiate dependence: performance and neuroimaging results. Drug Alcohol Depend. (2009) 104:228–40. doi: 10.1016/j.drugalcdep.2009.04.022
109. Thornton, C, Grad, E, and Yaka, R. The role of mitochondria in cocaine addiction. Biochem J. (2021) 478:749–64. doi: 10.1042/BCJ20200615
110. Stapleton, JM, Gilson, SF, Wong, DF, Villemagne, VL, Dannals, RF, Grayson, RF, et al. Intravenous nicotine reduces cerebral glucose metabolism: a preliminary study. Neuropsychopharmacology. (2003) 28:765–72. doi: 10.1038/sj.npp.1300106
111. Volkow, ND, Chang, L, Wang, GJ, Fowler, JS, Franceschi, D, Sedler, MJ, et al. Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am J Psychiatry. (2001) 158:383–9. doi: 10.1176/appi.ajp.158.3.383
112. Wang, GJ, Volkow, ND, Chang, L, Miller, E, Sedler, M, Hitzemann, R, et al. Partial recovery of brain metabolism in methamphetamine abusers after protracted abstinence. Am J Psychiatry. (2004) 161:242–8. doi: 10.1176/appi.ajp.161.2.242
113. Kim, SJ, Lyoo, IK, Hwang, J, Sung, YH, Lee, HY, Lee, DS, et al. Frontal glucose hypometabolism in abstinent methamphetamine users. Neuropsychopharmacology. (2005) 30:1383–91. doi: 10.1038/sj.npp.1300699
114. Halliwell, B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. (2006) 97:1634–58. doi: 10.1111/j.1471-4159.2006.03907.x
115. Ray, PD, Huang, BW, and Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
116. Angelova, PR, and Abramov, AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. (2018) 592:692–702. doi: 10.1002/1873-3468.12964
117. Uttara, B, Singh, AV, Zamboni, P, and Mahajan, RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. (2009) 7:65–74. doi: 10.2174/157015909787602823
118. Cunha-Oliveira, T, Rego, AC, and Oliveira, CR. Cellular and molecular mechanisms involved in the neurotoxicity of opioid and psychostimulant drugs. Brain Res Rev. (2008) 58:192–208. doi: 10.1016/j.brainresrev.2008.03.002
119. Xu, B, Wang, Z, Li, G, Li, B, Lin, H, Zheng, R, et al. Heroin-administered mice involved in oxidative stress and exogenous antioxidant-alleviated withdrawal syndrome. Basic Clin Pharmacol Toxicol. (2006) 99:153–61. doi: 10.1111/j.1742-7843.2006.pto_461.x
120. Cunha-Oliveira, T, Rego, AC, Garrido, J, Borges, F, Macedo, T, and Oliveira, CR. Street heroin induces mitochondrial dysfunction and apoptosis in rat cortical neurons. J Neurochem. (2007) 101:543–54. doi: 10.1111/j.1471-4159.2006.04406.x
121. Xiong, Q, Ru, Q, Tian, X, Zhou, M, Chen, L, Li, Y, et al. Krill oil protects PC12 cells against methamphetamine-induced neurotoxicity by inhibiting apoptotic response and oxidative stress. Nutr Res. (2018) 58:84–94. doi: 10.1016/j.nutres.2018.07.006
122. Wu, CW, Ping, YH, Yen, JC, Chang, CY, Wang, SF, Yeh, CL, et al. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol Appl Pharmacol. (2007) 220:243–51. doi: 10.1016/j.taap.2007.01.011
123. Cunha-Oliveira, T, Rego, AC, Garrido, J, Borges, F, Macedo, T, and Oliveira, CR. Neurotoxicity of heroin-cocaine combinations in rat cortical neurons. Toxicology. (2010) 276:11–7. doi: 10.1016/j.tox.2010.06.009
124. Shiba, T, Yamato, M, Kudo, W, Watanabe, T, Utsumi, H, and Yamada, K. In vivo imaging of mitochondrial function in methamphetamine-treated rats. Neuroimage. (2011) 57:866–72. doi: 10.1016/j.neuroimage.2011.05.041
125. Pinto, A, Bonucci, A, Maggi, E, Corsi, M, and Businaro, R. Anti-oxidant and anti-inflammatory activity of ketogenic diet: new perspectives for neuroprotection in Alzheimer’s disease. Antioxidants. (2018) 7:63. doi: 10.3390/antiox7050063
126. Guo, M, Wang, X, Zhao, Y, Yang, Q, Ding, H, Dong, Q, et al. Ketogenic diet improves brain ischemic tolerance and inhibits NLRP3 inflammasome activation by preventing Drp1-mediated mitochondrial fission and endoplasmic reticulum stress. Front Mol Neurosci. (2018) 11:86. doi: 10.3389/fnmol.2018.00086
127. Archer, SL. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med. (2013) 369:2236–51. doi: 10.1056/NEJMra1215233
128. Knott, AB, Perkins, G, Schwarzenbacher, R, and Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. (2008) 9:505–18. doi: 10.1038/nrn2417
129. Hasan-Olive, MM, Lauritzen, KH, Ali, M, Rasmussen, LJ, Storm-Mathisen, J, and Bergersen, LH. A ketogenic diet improves mitochondrial biogenesis and bioenergetics via the PGC1alpha-SIRT3-UCP2 Axis. Neurochem Res. (2019) 44:22–37. doi: 10.1007/s11064-018-2588-6
130. Sullivan, PG, Rippy, NA, Dorenbos, K, Concepcion, RC, Agarwal, AK, and Rho, JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. (2004) 55:576–80. doi: 10.1002/ana.20062
131. Bough, KJ, Wetherington, J, Hassel, B, Pare, JF, Gawryluk, JW, Greene, JG, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. (2006) 60:223–35. doi: 10.1002/ana.20899
132. Jarrett, SG, Milder, JB, Liang, LP, and Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J Neurochem. (2008) 106:1044–51. doi: 10.1111/j.1471-4159.2008.05460.x
133. Maalouf, M, Sullivan, PG, Davis, L, Kim, DY, and Rho, JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. (2007) 145:256–64. doi: 10.1016/j.neuroscience.2006.11.065
134. Kim, DY, Davis, LM, Sullivan, PG, Maalouf, M, Simeone, TA, van Brederode, J, et al. Ketone bodies are protective against oxidative stress in neocortical neurons. J Neurochem. (2007) 101:1316–26. doi: 10.1111/j.1471-4159.2007.04483.x
135. Wu, Y, Gong, Y, Luan, Y, Li, Y, Liu, J, Yue, Z, et al. BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer’s disease. FASEB J. (2020) 34:1412–29. doi: 10.1096/fj.201901984R
136. Wang, BH, Hou, Q, Lu, YQ, Jia, MM, Qiu, T, Wang, XH, et al. Ketogenic diet attenuates neuronal injury via autophagy and mitochondrial pathways in pentylenetetrazol-kindled seizures. Brain Res. (2018) 1678:106–15. doi: 10.1016/j.brainres.2017.10.009
137. Glass, CK, Saijo, K, Winner, B, Marchetto, MC, and Gage, FH. Mechanisms underlying inflammation in neurodegeneration. Cells. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
138. Lowe, PP, Gyongyosi, B, Satishchandran, A, Iracheta-Vellve, A, Cho, Y, Ambade, A, et al. Reduced gut microbiome protects from alcohol-induced neuroinflammation and alters intestinal and brain inflammasome expression. J Neuroinflammation. (2018) 15:298. doi: 10.1186/s12974-018-1328-9
139. Osmanlioglu, HO, Yildirim, MK, Akyuva, Y, Yildizhan, K, and Naziroglu, M. Morphine induces apoptosis, inflammation, and mitochondrial oxidative stress via activation of TRPM2 channel and nitric oxide signaling pathways in the hippocampus. Mol Neurobiol. (2020) 57:3376–89. doi: 10.1007/s12035-020-01975-6
140. Sil, S, Niu, F, Tom, E, Liao, K, Periyasamy, P, and Buch, S. Cocaine mediated neuroinflammation: role of dysregulated autophagy in pericytes. Mol Neurobiol. (2019) 56:3576–90. doi: 10.1007/s12035-018-1325-0
141. Carranza-Aguilar, CJ, Hernandez-Mendoza, A, Mejias-Aponte, C, Rice, KC, Morales, M, Gonzalez-Espinosa, C, et al. Morphine and fentanyl repeated administration induces different levels of NLRP3-dependent pyroptosis in the dorsal raphe nucleus of male rats via cell-specific activation of TLR4 and opioid receptors. Cell Mol Neurobiol. (2022) 42:677–94. doi: 10.1007/s10571-020-00957-5
142. Xu, E, Liu, J, Liu, H, Wang, X, and Xiong, H. Inflammasome activation by methamphetamine potentiates lipopolysaccharide stimulation of IL-1beta production in microglia. J Neuroimmune Pharmacol. (2018) 13:237–53. doi: 10.1007/s11481-018-9780-y
143. Wang, X, Northcutt, AL, Cochran, TA, Zhang, X, Fabisiak, TJ, Haas, ME, et al. Methamphetamine activates toll-like receptor 4 to induce central immune signaling within the ventral tegmental area and contributes to extracellular dopamine increase in the nucleus accumbens shell. ACS Chem Neurosci. (2019) 10:3622–34. doi: 10.1021/acschemneuro.9b00225
144. Salvador, AF, de Lima, KA, and Kipnis, J. Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol. (2021) 21:526–41. doi: 10.1038/s41577-021-00508-z
145. Felger, JC, and Miller, AH. Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol. (2012) 33:315–27. doi: 10.1016/j.yfrne.2012.09.003
146. Felger, JC, and Treadway, MT. Inflammation effects on motivation and motor activity: role of dopamine. Neuropsychopharmacology. (2017) 42:216–41. doi: 10.1038/npp.2016.143
147. Karimi-Haghighi, S, Dargahi, L, and Haghparast, A. Cannabidiol modulates the expression of neuroinflammatory factors in stress-and drug-induced reinstatement of methamphetamine in extinguished rats. Addict Biol. (2020) 25:e12740. doi: 10.1111/adb.12740
148. Jeong, EA, Jeon, BT, Shin, HJ, Kim, N, Lee, DH, Kim, HJ, et al. Ketogenic diet-induced peroxisome proliferator-activated receptor-gamma activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp Neurol. (2011) 232:195–202. doi: 10.1016/j.expneurol.2011.09.001
149. Lu, Y, Yang, YY, Zhou, MW, Liu, N, Xing, HY, Liu, XX, et al. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-kappaB signaling pathways. Neurosci Lett. (2018) 683:13–8. doi: 10.1016/j.neulet.2018.06.016
150. Nakamura, K, Tonouchi, H, Sasayama, A, and Ashida, K. A ketogenic formula prevents tumor progression and cancer cachexia by attenuating systemic inflammation in colon 26 tumor-bearing mice. Nutrients. (2018) 10:206. doi: 10.3390/nu10020206
151. Zhang, N, Liu, C, Zhang, R, Jin, L, Yin, X, Zheng, X, et al. Amelioration of clinical course and demyelination in the cuprizone mouse model in relation to ketogenic diet. Food Funct. (2020) 11:5647–63. doi: 10.1039/c9fo02944c
152. Peixoto, L, and Abel, T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology. (2013) 38:62–76. doi: 10.1038/npp.2012.86
153. Xu, Y, Jiang, C, Wu, J, Liu, P, Deng, X, Zhang, Y, et al. Ketogenic diet ameliorates cognitive impairment and neuroinflammation in a mouse model of Alzheimer’s disease. CNS Neurosci Ther. (2022) 28:580–92. doi: 10.1111/cns.13779
154. Goldberg, EL, Shchukina, I, Asher, JL, Sidorov, S, Artyomov, MN, and Dixit, VD. Ketogenesis activates metabolically protective gammadelta T cells in visceral adipose tissue. Nat Metab. (2020) 2:50–61. doi: 10.1038/s42255-019-0160-6
155. Rosenbaum, M, Hall, KD, Guo, J, Ravussin, E, Mayer, LS, Reitman, ML, et al. Glucose and lipid homeostasis and inflammation in humans following an isocaloric ketogenic diet. Obesity. (2019) 27:971–81. doi: 10.1002/oby.22468
156. Jain, SK, Kannan, K, Lim, G, McVie, R, and Bocchini, JA Jr. Hyperketonemia increases tumor necrosis factor-alpha secretion in cultured U937 monocytes and type 1 diabetic patients and is apparently mediated by oxidative stress and cAMP deficiency. Diabetes. (2002) 51:2287–93. doi: 10.2337/diabetes.51.7.2287
157. Jain, SK, Kannan, K, Lim, G, Matthews-Greer, J, McVie, R, and Bocchini, JA Jr. Elevated blood interleukin-6 levels in hyperketonemic type 1 diabetic patients and secretion by acetoacetate-treated cultured U937 monocytes. Diabetes Care. (2003) 26:2139–43. doi: 10.2337/diacare.26.7.2139
158. Neudorf, H, Durrer, C, Myette-Cote, E, Makins, C, O’Malley, T, and Little, JP. Oral ketone supplementation acutely increases markers of NLRP3 inflammasome activation in human monocytes. Mol Nutr Food Res. (2019) 63:e1801171. doi: 10.1002/mnfr.201801171
159. Shi, X, Li, X, Li, D, Li, Y, Song, Y, Deng, Q, et al. β-Hydroxybutyrate activates the NF-κB signaling pathway to promote the expression of pro-inflammatory factors in calf hepatocytes. Cell Physiol Biochem. (2014) 33:920–32. doi: 10.1159/000358664
160. He, J, Wang, K, Liu, M, Zeng, W, Li, D, Majigsuren, Z, et al. β-hydroxybutyrate enhances bovine neutrophil adhesion by inhibiting autophagy. Front Immunol. (2022) 13:1096813. doi: 10.3389/fimmu.2022.1096813
161. Allen, NJ, and Lyons, DA. Glia as architects of central nervous system formation and function. Science. (2018) 362:181–5. doi: 10.1126/science.aat0473
162. Linker, KE, Cross, SJ, and Leslie, FM. Glial mechanisms underlying substance use disorders. Eur J Neurosci. (2019) 50:2574–89. doi: 10.1111/ejn.14163
163. Stellwagen, D, Kemp, GM, Valade, S, and Chambon, J. Glial regulation of synaptic function in models of addiction. Curr Opin Neurobiol. (2019) 57:179–85. doi: 10.1016/j.conb.2019.02.010
164. Jones, JD. Potential of glial cell modulators in the management of substance use disorders. CNS Drugs. (2020) 34:697–722. doi: 10.1007/s40263-020-00721-9
165. Prinz, M, Jung, S, and Priller, J. Microglia biology: one century of evolving concepts. Cells. (2019) 179:292–311. doi: 10.1016/j.cell.2019.08.053
166. Nayak, D, Roth, TL, and McGavern, DB. Microglia development and function. Annu Rev Immunol. (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
167. Lundgaard, I, Wang, W, Eberhardt, A, Vinitsky, HS, Reeves, BC, Peng, S, et al. Beneficial effects of low alcohol exposure, but adverse effects of high alcohol intake on glymphatic function. Sci Rep. (2018) 8:2246. doi: 10.1038/s41598-018-20424-y
168. Warden, AS, Wolfe, SA, Khom, S, Varodayan, FP, Patel, RR, Steinman, MQ, et al. Microglia control escalation of drinking in alcohol-dependent mice: genomic and synaptic drivers. Biol Psychiatry. (2020) 88:910–21. doi: 10.1016/j.biopsych.2020.05.011
169. Valentinova, K, Tchenio, A, Trusel, M, Clerke, JA, Lalive, AL, Tzanoulinou, S, et al. Morphine withdrawal recruits lateral habenula cytokine signaling to reduce synaptic excitation and sociability. Nat Neurosci. (2019) 22:1053–6. doi: 10.1038/s41593-019-0421-4
170. Periyasamy, P, Liao, K, Kook, YH, Niu, F, Callen, SE, Guo, ML, et al. Cocaine-mediated downregulation of miR-124 activates microglia by targeting KLF4 and TLR4 signaling. Mol Neurobiol. (2018) 55:3196–210. doi: 10.1007/s12035-017-0584-5
171. Cotto, B, Li, H, Tuma, RF, Ward, SJ, and Langford, D. Cocaine-mediated activation of microglia and microglial MeCP2 and BDNF production. Neurobiol Dis. (2018) 117:28–41. doi: 10.1016/j.nbd.2018.05.017
172. Sofroniew, MV. Astrocyte reactivity: subtypes, states, and functions in CNS innate immunity. Trends Immunol. (2020) 41:758–70. doi: 10.1016/j.it.2020.07.004
173. Liddelow, SA, and Barres, BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
174. Nam, MH, Han, KS, Lee, J, Bae, JY, An, H, Park, S, et al. Expression of micro-opioid receptor in CA1 hippocampal astrocytes. Exp Neurobiol. (2018) 27:120–8. doi: 10.5607/en.2018.27.2.120
175. Woo, DH, Bae, JY, Nam, MH, An, H, Ju, YH, Won, J, et al. Activation of astrocytic mu-opioid receptor elicits fast glutamate release through TREK-1-containing K2P channel in hippocampal astrocytes. Front Cell Neurosci. (2018) 12:319. doi: 10.3389/fncel.2018.00319
176. Corkrum, M, Rothwell, PE, Thomas, MJ, Kofuji, P, and Araque, A. Opioid-mediated astrocyte-neuron signaling in the nucleus accumbens. Cells. (2019) 8:586. doi: 10.3390/cells8060586
177. Nam, MH, Han, KS, Lee, J, Won, W, Koh, W, Bae, JY, et al. Activation of astrocytic mu-opioid receptor causes conditioned place preference. Cell Rep. (2019) 28:1154–1166.e5. doi: 10.1016/j.celrep.2019.06.071
178. Giovannoni, F, and Quintana, FJ. The role of astrocytes in CNS inflammation. Trends Immunol. (2020) 41:805–19. doi: 10.1016/j.it.2020.07.007
179. Linnerbauer, M, Wheeler, MA, and Quintana, FJ. Astrocyte crosstalk in CNS inflammation. Neuron. (2020) 108:608–22. doi: 10.1016/j.neuron.2020.08.012
180. Sil, S, Periyasamy, P, Guo, ML, Callen, S, and Buch, S. Morphine-mediated brain region-specific astrocytosis involves the ER stress-autophagy axis. Mol Neurobiol. (2018) 55:6713–33. doi: 10.1007/s12035-018-0878-2
181. Liao, K, Niu, F, Hu, G, Yang, L, Dallon, B, Villarreal, D, et al. Morphine-mediated release of miR-138 in astrocyte-derived extracellular vesicles promotes microglial activation. J Extracell Vesicles. (2020) 10:e12027. doi: 10.1002/jev2.12027
182. Gzielo, K, Soltys, Z, Rajfur, Z, and Setkowicz, ZK. The impact of the ketogenic diet on glial cells morphology. A quantitative morphological analysis. Neuroscience. (2019) 413:239–51. doi: 10.1016/j.neuroscience.2019.06.009
183. Harun-Or-Rashid, M, and Inman, DM. Reduced AMPK activation and increased HCAR activation drive anti-inflammatory response and neuroprotection in glaucoma. J Neuroinflammation. (2018) 15:313. doi: 10.1186/s12974-018-1346-7
184. Guan, YF, Huang, GB, Xu, MD, Gao, F, Lin, S, Huang, J, et al. Anti-depression effects of ketogenic diet are mediated via the restoration of microglial activation and neuronal excitability in the lateral habenula. Brain Behav Immun. (2020) 88:748–62. doi: 10.1016/j.bbi.2020.05.032
185. Liu, C, Zhang, N, Zhang, R, Jin, L, Petridis, AK, Loers, G, et al. Cuprizone-induced demyelination in mouse hippocampus is alleviated by ketogenic diet. J Agric Food Chem. (2020) 68:11215–28. doi: 10.1021/acs.jafc.0c04604
186. Shippy, DC, Wilhelm, C, Viharkumar, PA, Raife, TJ, and Ulland, TK. beta-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J Neuroinflammation. (2020) 17:280. doi: 10.1186/s12974-020-01948-5
187. Huang, C, Wang, P, Xu, X, Zhang, Y, Gong, Y, Hu, W, et al. The ketone body metabolite beta-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia. (2018) 66:256–78. doi: 10.1002/glia.23241
188. Kong, G, Liu, J, Li, R, Lin, J, Huang, Z, Yang, Z, et al. Ketone metabolite beta-hydroxybutyrate ameliorates inflammation after spinal cord injury by inhibiting the NLRP3 inflammasome. Neurochem Res. (2021) 46:213–29. doi: 10.1007/s11064-020-03156-2
189. Augustin, K, Khabbush, A, Williams, S, Eaton, S, Orford, M, Cross, JH, et al. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. (2018) 17:84–93. doi: 10.1016/S1474-4422(17)30408-8
190. Thevenet, J, De Marchi, U, Domingo, JS, Christinat, N, Bultot, L, Lefebvre, G, et al. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. (2016) 30:1913–26. doi: 10.1096/fj.201500182
191. Tremlett, H, Bauer, KC, Appel-Cresswell, S, Finlay, BB, and Waubant, E. The gut microbiome in human neurological disease: a review. Ann Neurol. (2017) 81:369–82. doi: 10.1002/ana.24901
192. Foster, JA, and McVey Neufeld, KA. Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. (2013) 36:305–12. doi: 10.1016/j.tins.2013.01.005
193. Angoa-Perez, M, and Kuhn, DM. Evidence for modulation of substance use disorders by the gut microbiome: hidden in plain sight. Pharmacol Rev. (2021) 73:571–96. doi: 10.1124/pharmrev.120.000144
194. Russell, JT, Zhou, Y, Weinstock, GM, and Bubier, JA. The gut microbiome and substance use disorder. Front Neurosci. (2021) 15:725500. doi: 10.3389/fnins.2021.725500
195. Meckel, KR, and Kiraly, DD. A potential role for the gut microbiome in substance use disorders. Psychopharmacology. (2019) 236:1513–30. doi: 10.1007/s00213-019-05232-0
196. Bjorkhaug, ST, Aanes, H, Neupane, SP, Bramness, JG, Malvik, S, Henriksen, C, et al. Characterization of gut microbiota composition and functions in patients with chronic alcohol overconsumption. Gut Microbes. (2019) 10:663–75. doi: 10.1080/19490976.2019.1580097
197. Addolorato, G, Ponziani, FR, Dionisi, T, Mosoni, C, Vassallo, GA, Sestito, L, et al. Gut microbiota compositional and functional fingerprint in patients with alcohol use disorder and alcohol-associated liver disease. Liver Int. (2020) 40:878–88. doi: 10.1111/liv.14383
198. Peterson, VL, Jury, NJ, Cabrera-Rubio, R, Draper, LA, Crispie, F, Cotter, PD, et al. Drunk bugs: chronic vapour alcohol exposure induces marked changes in the gut microbiome in mice. Behav Brain Res. (2017) 323:172–6. doi: 10.1016/j.bbr.2017.01.049
199. Cruz-Lebron, A, Johnson, R, Mazahery, C, Troyer, Z, Joussef-Pina, S, Quinones-Mateu, ME, et al. Chronic opioid use modulates human enteric microbiota and intestinal barrier integrity. Gut Microbes. (2021) 13:1946368. doi: 10.1080/19490976.2021.1946368
200. Banerjee, S, Sindberg, G, Wang, F, Meng, J, Sharma, U, Zhang, L, et al. Opioid-induced gut microbial disruption and bile dysregulation leads to gut barrier compromise and sustained systemic inflammation. Mucosal Immunol. (2016) 9:1418–28. doi: 10.1038/mi.2016.9
201. Yang, J, Xiong, P, Bai, L, Zhang, Z, Zhou, Y, Chen, C, et al. The association of altered gut microbiota and intestinal mucosal barrier integrity in mice with heroin dependence. Front Nutr. (2021) 8:765414. doi: 10.3389/fnut.2021.765414
202. Chivero, ET, Ahmad, R, Thangaraj, A, Periyasamy, P, Kumar, B, Kroeger, E, et al. Cocaine induces inflammatory gut milieu by compromising the mucosal barrier integrity and altering the gut microbiota colonization. Sci Rep. (2019) 9:12187. doi: 10.1038/s41598-019-48428-2
203. Deng, D, Su, H, Song, Y, Chen, T, Sun, Q, Jiang, H, et al. Altered fecal microbiota correlated with systemic inflammation in male subjects with methamphetamine use disorder. Front Cell Infect Microbiol. (2021) 11:783917. doi: 10.3389/fcimb.2021.783917
204. Chen, LJ, Zhi, X, Zhang, KK, Wang, LB, Li, JH, Liu, JL, et al. Escalating dose-multiple binge methamphetamine treatment elicits neurotoxicity, altering gut microbiota and fecal metabolites in mice. Food Chem Toxicol. (2021) 148:111946. doi: 10.1016/j.fct.2020.111946
205. Wang, L, Christophersen, CT, Sorich, MJ, Gerber, JP, Angley, MT, and Conlon, MA. Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Mol Autism. (2013) 4:42. doi: 10.1186/2040-2392-4-42
206. Tan, TG, Sefik, E, Geva-Zatorsky, N, Kua, L, Naskar, D, Teng, F, et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A. (2016) 113:E8141–50. doi: 10.1073/pnas.1617460113
207. Shin, NR, Lee, JC, Lee, HY, Kim, MS, Whon, TW, Lee, MS, et al. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. (2014) 63:727–35. doi: 10.1136/gutjnl-2012-303839
208. Ottman, N, Reunanen, J, Meijerink, M, Pietila, TE, Kainulainen, V, Klievink, J, et al. Pili-like proteins of Akkermansia muciniphila modulate host immune responses and gut barrier function. PLoS One. (2017) 12:e0173004. doi: 10.1371/journal.pone.0173004
209. Ottman, N, Geerlings, SY, Aalvink, S, de Vos, WM, and Belzer, C. Action and function of Akkermansia muciniphila in microbiome ecology, health and disease. Best Pract Res Clin Gastroenterol. (2017) 31:637–42. doi: 10.1016/j.bpg.2017.10.001
210. Belzer, C, and de Vos, WM. Microbes inside--from diversity to function: the case of Akkermansia. ISME J. (2012) 6:1449–58. doi: 10.1038/ismej.2012.6
211. Pizarro, N, Kossatz, E, González, P, Gamero, A, Veza, E, Fernández, C, et al. Sex-specific effects of synbiotic exposure in mice on addictive-like behavioral alterations induced by chronic alcohol intake are associated with changes in specific gut bacterial taxa and brain tryptophan metabolism. Front Nutr. (2021) 8:750333. doi: 10.3389/fnut.2021.750333
212. Yang, C, Fu, X, Hao, W, Xiang, X, Liu, T, Yang, BZ, et al. Gut dysbiosis associated with the rats’ responses in methamphetamine-induced conditioned place preference. Addict Biol. (2021) 26:e12975. doi: 10.1111/adb.12975
213. Hartmann, P, Chen, P, Wang, HJ, Wang, L, McCole, DF, Brandl, K, et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology. (2013) 58:108–19. doi: 10.1002/hep.26321
214. Yan, AW, Fouts, DE, Brandl, J, Starkel, P, Torralba, M, Schott, E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. (2011) 53:96–105. doi: 10.1002/hep.24018
215. Xu, Z, Wang, C, Dong, X, Hu, T, Wang, L, Zhao, W, et al. Chronic alcohol exposure induced gut microbiota dysbiosis and its correlations with neuropsychic behaviors and brain BDNF/Gabra1 changes in mice. Biofactors. (2019) 45:187–99. doi: 10.1002/biof.1469
216. Forouzan, S, Hoffman, KL, and Kosten, TA. Methamphetamine exposure and its cessation alter gut microbiota and induce depressive-like behavioral effects on rats. Psychopharmacology. (2021) 238:281–92. doi: 10.1007/s00213-020-05681-y
217. Bajaj, JS, Gavis, EA, Fagan, A, Wade, JB, Thacker, LR, Fuchs, M, et al. A randomized clinical trial of fecal microbiota transplant for alcohol use disorder. Hepatology. (2021) 73:1688–700. doi: 10.1002/hep.31496
218. Peterson, VL, Richards, JB, Meyer, PJ, Cabrera-Rubio, R, Tripi, JA, King, CP, et al. Sex-dependent associations between addiction-related behaviors and the microbiome in outbred rats. EBioMedicine. (2020) 55:102769. doi: 10.1016/j.ebiom.2020.102769
219. Thomaz, AC, Iyer, V, Woodward, TJ, and Hohmann, AG. Fecal microbiota transplantation and antibiotic treatment attenuate naloxone-precipitated opioid withdrawal in morphine-dependent mice. Exp Neurol. (2021) 343:113787. doi: 10.1016/j.expneurol.2021.113787
220. Chivero, ET, Sil, S, Singh, S, Thangaraj, A, Gordon, L, Evah-Nzoughe, GB, et al. Protective role of Lactobacillus rhamnosus probiotic in reversing cocaine-induced oxidative stress, glial activation and locomotion in mice. J Neuroimmune Pharmacol. (2021) 17:62–75. doi: 10.1007/s11481-021-10020-9
221. Leclercq, S, Le Roy, T, Furgiuele, S, Coste, V, Bindels, LB, Leyrolle, Q, et al. Gut microbiota-induced changes in beta-hydroxybutyrate metabolism are linked to altered sociability and depression in alcohol use disorder. Cell Rep. (2020) 33:108238. doi: 10.1016/j.celrep.2020.108238
222. Xiao, HW, Ge, C, Feng, GX, Li, Y, Luo, D, Dong, JL, et al. Gut microbiota modulates alcohol withdrawal-induced anxiety in mice. Toxicol Lett. (2018) 287:23–30. doi: 10.1016/j.toxlet.2018.01.021
223. Olson, CA, Vuong, HE, Yano, JM, Liang, QY, Nusbaum, DJ, and Hsiao, EY. The gut microbiota mediates the anti-seizure effects of the ketogenic diet. Cells. (2018) 173:1728–1741. e13. doi: 10.1016/j.cell.2018.04.027
224. Zhang, Y, Zhou, S, Zhou, Y, Yu, L, Zhang, L, and Wang, Y. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. (2018) 145:163–8. doi: 10.1016/j.eplepsyres.2018.06.015
225. Ma, D, Wang, AC, Parikh, I, Green, SJ, Hoffman, JD, Chlipala, G, et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci Rep. (2018) 8:6670. doi: 10.1038/s41598-018-25190-5
226. Nagpal, R, Neth, BJ, Wang, S, Craft, S, and Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine. (2019) 47:529–42. doi: 10.1016/j.ebiom.2019.08.032
227. Kong, C, Yan, X, Liu, Y, Huang, L, Zhu, Y, He, J, et al. Ketogenic diet alleviates colitis by reduction of colonic group 3 innate lymphoid cells through altering gut microbiome. Signal Transduct Target Ther. (2021) 6:154. doi: 10.1038/s41392-021-00549-9
228. Ang, QY, Alexander, M, Newman, JC, Tian, Y, Cai, J, Upadhyay, V, et al. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cells. (2020) 181:1263–75 e16. doi: 10.1016/j.cell.2020.04.027
229. Lindefeldt, M, Eng, A, Darban, H, Bjerkner, A, Zetterstrom, CK, Allander, T, et al. The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy. NPJ Biofilms Microbiomes. (2019) 5:5. doi: 10.1038/s41522-018-0073-2
230. Li, S, Zhuge, A, Wang, K, Lv, L, Bian, X, Yang, L, et al. Ketogenic diet aggravates colitis, impairs intestinal barrier and alters gut microbiota and metabolism in DSS-induced mice. Food Funct. (2021) 12:10210–25. doi: 10.1039/d1fo02288a
231. Wlodarek, D. Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). Nutrients. (2019) 11:169. doi: 10.3390/nu11010169
232. Kossoff, EH, Zupec-Kania, BA, Amark, PE, Ballaban-Gil, KR, Christina Bergqvist, AG, Blackford, R, et al. Optimal clinical management of children receiving the ketogenic diet: recommendations of the international ketogenic diet study group. Epilepsia. (2009) 50:304–17. doi: 10.1111/j.1528-1167.2008.01765.x
233. Bell, TJ, Panchal, SJ, Miaskowski, C, Bolge, SC, Milanova, T, and Williamson, R. The prevalence, severity, and impact of opioid-induced bowel dysfunction: results of a US and European patient survey (PROBE 1). Pain Med. (2009) 10:35–42. doi: 10.1111/j.1526-4637.2008.00495.x
234. Su, H, Chen, T, Zhong, N, Jiang, H, Du, J, Xiao, K, et al. Decreased GABA concentrations in left prefrontal cortex of methamphetamine dependent patients: a proton magnetic resonance spectroscopy study. J Clin Neurosci. (2020) 71:15–20. doi: 10.1016/j.jocn.2019.11.021
235. Neri, M, Panata, L, Bacci, M, Fiore, C, Riezzo, I, Turillazzi, E, et al. Cytokines, chaperones and neuroinflammatory responses in heroin-related death: what can we learn from different patterns of cellular expression? Int J Mol Sci. (2013) 14:19831–45. doi: 10.3390/ijms141019831
236. Chan, YY, Yang, SN, Lin, JC, Chang, JL, Lin, JG, and Lo, WY. Inflammatory response in heroin addicts undergoing methadone maintenance treatment. Psychiatry Res. (2015) 226:230–4. doi: 10.1016/j.psychres.2014.12.053
Keywords: ketogenic diet, substance use disorders, addiction, metabolism, neuroprotection, gut microbiota
Citation: Kong D, Sun J-x, Yang J-q, Li Y-s, Bi K, Zhang Z-y, Wang K-h, Luo H-y, Zhu M and Xu Y (2023) Ketogenic diet: a potential adjunctive treatment for substance use disorders. Front. Nutr. 10:1191903. doi: 10.3389/fnut.2023.1191903
Edited by:
Jose Enrique De La Rubia Ortí, Catholic University of Valencia San Vicente Mártir, SpainReviewed by:
Alina Arulsamy, Monash University, MalaysiaAurelijus Burokas, Vilnius University, Lithuania
Copyright © 2023 Kong, Sun, Yang, Li, Bi, Zhang, Wang, Luo, Zhu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua-you Luo, MTY3NzU0NjI5NkBxcS5jb20=; Mei Zhu, emh1bWVpc0AxNjMuY29t; Yu Xu, eGlhb3l1ZXI1MjA2N0BxcS5jb20=
†These authors have contributed equally to this work