 Xin Xu1,2,3,4,5
Xin Xu1,2,3,4,5 Zuliyaer Talifu1,2,3,4,5,6
Zuliyaer Talifu1,2,3,4,5,6 Chun-Jia Zhang1,2,3,4,5
Chun-Jia Zhang1,2,3,4,5 Feng Gao1,2,3,4,5Han Ke1,2,3,4,5,6
Feng Gao1,2,3,4,5Han Ke1,2,3,4,5,6 Yun-Zhu Pan1,2,3,4,5,6
Yun-Zhu Pan1,2,3,4,5,6 Han Gong1,2,3,4,5Hua-Yong Du1,2,3,4,5
Han Gong1,2,3,4,5Hua-Yong Du1,2,3,4,5 Yan Yu1,3,4,5
Yan Yu1,3,4,5 Ying-Li Jing1,3,4,5Liang-Jie Du1,2,3,4,5
Ying-Li Jing1,3,4,5Liang-Jie Du1,2,3,4,5 Jian-Jun Li1,2,3,4,5,6*De-Gang Yang1,2,3,4,5*
Jian-Jun Li1,2,3,4,5,6*De-Gang Yang1,2,3,4,5*- 1School of Rehabilitation, Capital Medical University, Beijing, China
- 2Department of Spinal and Neural Functional Reconstruction, China Rehabilitation Research Center, Beijing, China
- 3Chinese Institute of Rehabilitation Science, Beijing, China
- 4Center of Neural Injury and Repair, Beijing Institute for Brain Disorders, Beijing, China
- 5Beijing Key Laboratory of Neural Injury and Rehabilitation, Beijing, China
- 6School of Rehabilitation Sciences and Engineering, University of Health and Rehabilitation Sciences, Qingdao, Shandong, China
Spinal cord injury leads to loss of innervation of skeletal muscle, decreased motor function, and significantly reduced load on skeletal muscle, resulting in atrophy. Factors such as braking, hormone level fluctuation, inflammation, and oxidative stress damage accelerate skeletal muscle atrophy. The atrophy process can result in skeletal muscle cell apoptosis, protein degradation, fat deposition, and other pathophysiological changes. Skeletal muscle atrophy not only hinders the recovery of motor function but is also closely related to many systemic dysfunctions, affecting the prognosis of patients with spinal cord injury. Extensive research on the mechanism of skeletal muscle atrophy and intervention at the molecular level has shown that inflammation and oxidative stress injury are the main mechanisms of skeletal muscle atrophy after spinal cord injury and that multiple pathways are involved. These may become targets of future clinical intervention. However, most of the experimental studies are still at the basic research stage and still have some limitations in clinical application, and most of the clinical treatments are focused on rehabilitation training, so how to develop more efficient interventions in clinical treatment still needs to be further explored. Therefore, this review focuses mainly on the mechanisms of skeletal muscle atrophy after spinal cord injury and summarizes the cytokines and signaling pathways associated with skeletal muscle atrophy in recent studies, hoping to provide new therapeutic ideas for future clinical work.
Introduction
Spinal cord injury (SCI) is a serious and disabling disease. In recent years, the incidence of spinal cord injury caused by traffic accidents, industrial accidents, and sports injuries has been increasing (1–4). SCI leads to the loss of central regulation of peripheral nerves below the injured segment, resulting in sensory, motor, and autonomic dysfunction, muscle paralysis, and reduced muscle load (5, 6).
Injury to the neuromuscular system and reduced integrity of the musculoskeletal system are important features of SCI (7), and are also significant obstacles to the recovery of motor function. As the main component of human tissue structure, skeletal muscle accounts for about 40% of body weight (8). In addition to maintaining the homeostasis of exercise, skeletal muscle also plays a number of physiological functions such as support, protection, and respiration (9, 10). The muscle type most frequently atrophied after SCI is skeletal muscle, manifesting as loss of mass and strength (8), with 18–46% decreases in cross-sectional area (CSA) of skeletal muscle 6 weeks after injury (7, 11, 12).
After SCI, somatic and visceral nerve functions are affected (4), and these strongly correlate with skeletal muscle atrophy. After somatic nerve function damage, the corresponding skeletal muscle is denervated, and motor dysfunction is severe in patients with higher injury segments. The limbs of these patients maintain long-term braking and lose the nutritional effect of the nerve, causing skeletal muscle physiological, biochemical and biomechanical changes, and many functional reductions (13–15). Visceral nerve function damage leads to disruption of some hormones secretion, many of which are closely related to the maintenance of skeletal muscle mass, such as testosterone, insulin, growth hormone, and others (16–18), with multiple factors coinciding to aggravate skeletal muscle atrophy after SCI (19). Skeletal muscle atrophy can cause systemic secondary metabolic dysfunction, such as glucose intolerance, type 2 diabetes and insulin resistance (6, 20). Although the quality and function of skeletal muscles can be recovered to some extent through voluntary movement, the effect is often very limited (21) and is easily affected by the patient's psychological focus and other factors, and the optimal recovery period may be missed. Muscle atrophy is irreversible even if the axons, endplates, and skeletal muscle can be reconnected later (22). Therefore, maintaining skeletal muscle integrity is crucial for maintaining cell homeostasis and systemic metabolism, and research on the prevention and treatment of skeletal muscle atrophy after SCI is highly significant for optimal patient benefit (23, 24).
In recent years, there have been many studies on skeletal muscle atrophy after spinal cord injury, but most of them are experimental studies aimed at prevention and treatment strategies, but much work is still needed before practical clinical application. There is also a lack of review of the related mechanisms of skeletal muscle atrophy. However, it is important to clarify and organize the factors, pathological changes and related mechanisms of skeletal muscle atrophy after spinal cord injury for subsequent research and treatment.
Factors in skeletal muscle atrophy
SCI often results in upper or lower motor neuron damage or both (7), upper being relatively common, and the muscle atrophy differs between the two (25). When upper motor neurons are injured, the lower motor neurons are often intact. In SCI the conduction process is blocked (26) (Figure 1). Spinal motor neurons become highly excited (27), thereby pathologically activating the antigravity muscles of the lower limbs, such as the quadriceps femoris, gastrocnemius, and others, leading to spasticity. Deep hyperreflexia occurs (28), but moderate spasticity also reduces muscle atrophy to some extent (29, 30).
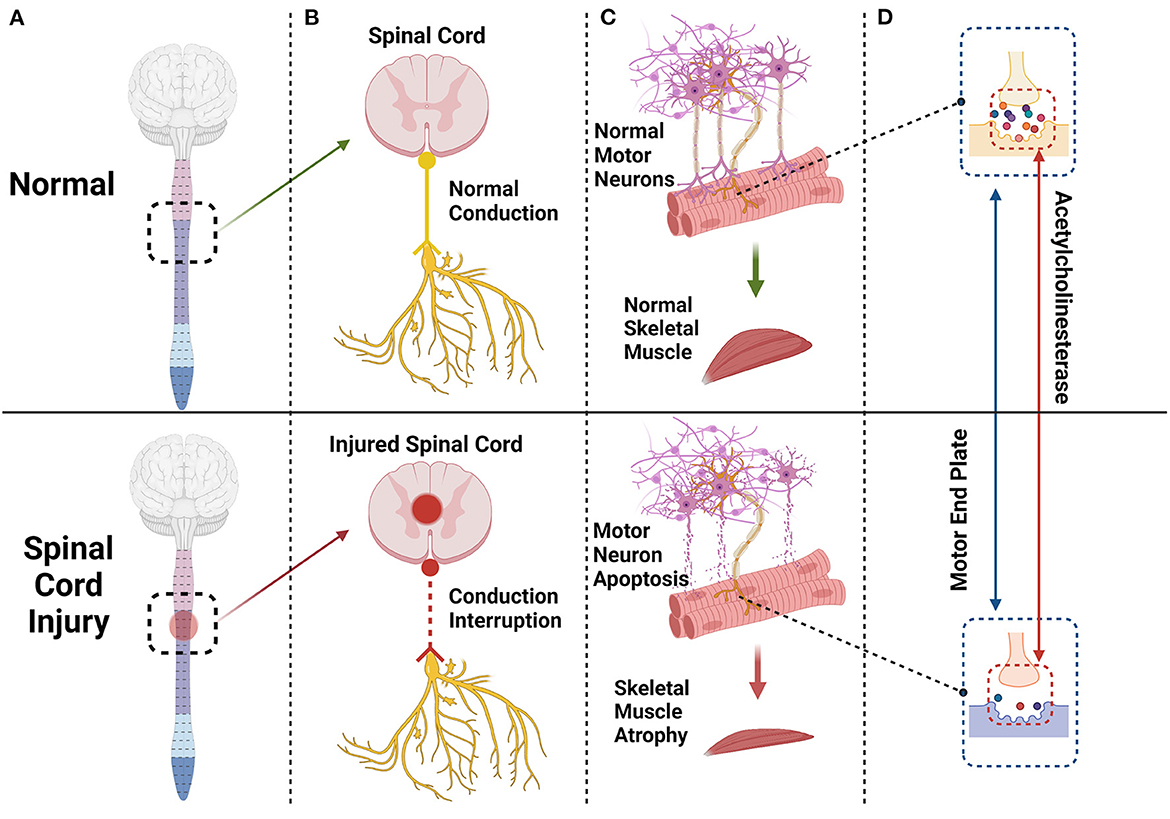
Figure 1. The spinal cord and brain constitute the central nervous system. After a spinal cord injury (A) the conduction of peripheral nerves innervating skeletal muscle will be interrupted or blocked (B) and some motor neurons innervating skeletal muscle will undergo apoptosis (C) Endplates degenerate, and acetylcholinesterase content of synapses will decrease significantly (D) eventually leading to skeletal muscle atrophy (C).
Although the signs of muscle atrophy after SCI are standard in most forms of disuse muscle atrophy, skeletal muscle atrophy is often faster after SCI due to denervation, immobilization, and other factors (23). This is because the γ-loop is damaged after spinal cord injury (31), leading to inhibition of α motor neurons that excite muscles, accompanied by different levels and patterns of muscle atrophy signals and anabolic signals. A combination of factors caused rapid skeletal muscle atrophy after SCI (20). Severe SCI results in impaired neural drive function, accompanied by changes in neuromuscular junctions (8, 20), and leads to impaired skeletal muscle function around and below the injury site. Etzel et al. (32) found in rats that compared with simple immobilization, SCI resulted in a more significant decrease in muscle cross-sectional area, wet muscle weight, and muscle strength within 7–21 days after immobilization. This indicates that neural input and mechanical load have a combined effect on the mass and strength of skeletal muscle after SCI.
Pathophysiological process
The spinal cord establishes a nutritional connection with skeletal muscle through peripheral nerves, and spinal motor neurons also trigger the contraction of skeletal muscle by transmitting action potentials to the motor endplate (33). When the spinal cord is injured, some motor neurons undergo apoptosis. The morphology and function of the remaining motor neurons also change; synapses are shortened (34), and the skeletal muscles innervated by these motor neurons and synapses undergo some level of atrophy and fibrosis (35–37). The motor endplate, also known as the neuromuscular junction, is the chemical synapse between motor neurons and skeletal muscle, consisting of motor nerve endings, the synaptic cleft, acetylcholine-containing synaptic vesicles, and a postsynaptic membrane (38). The motor endplate degenerates after injury, and its acetylcholinesterase (AchE) content decreases significantly (Figure 1). Acetylcholine cannot be removed in time, and excess calcium flows into the postsynaptic membrane through acetylcholine receptor channels. Intracellular proteases are activated in skeletal muscle cells, triggering protein degradation and apoptosis (33, 39, 40) (Figure 2).
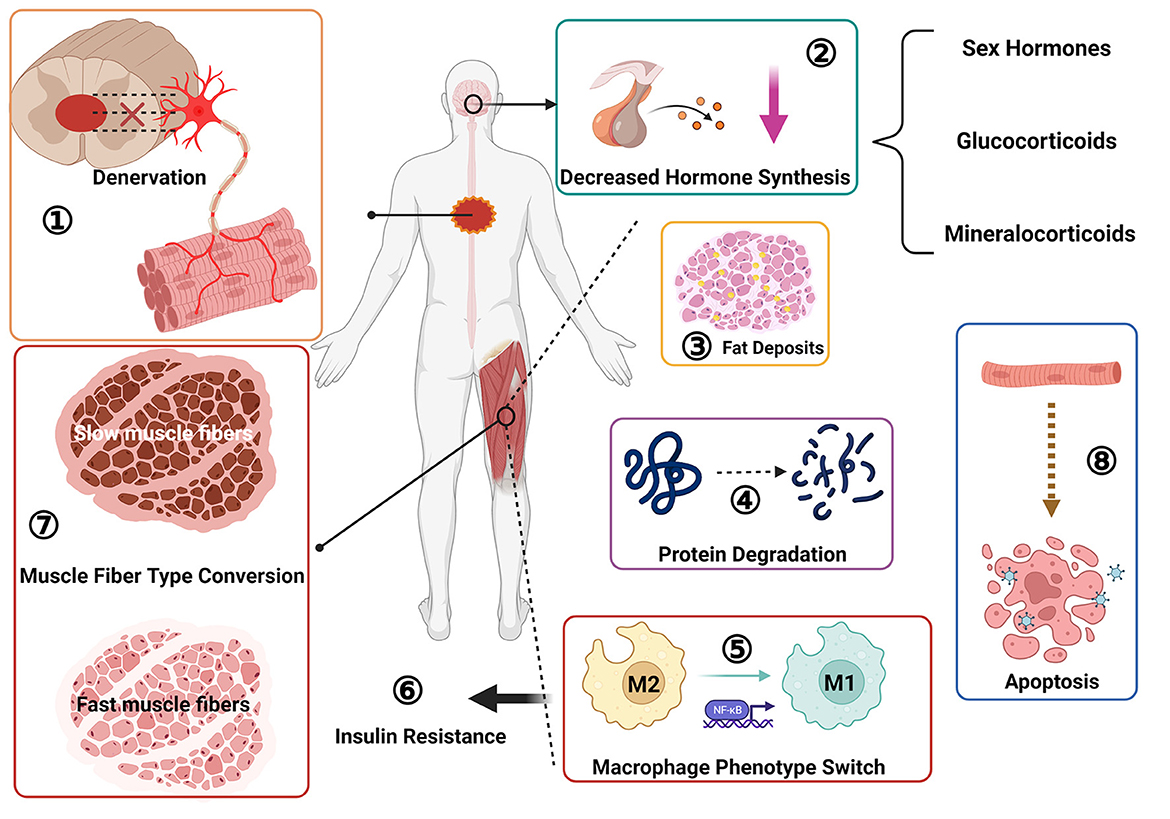
Figure 2. Pathological changes of skeletal muscle after spinal cord injury: ① muscle denervation, neuromuscular junction degeneration; ② decreased sex hormone secretion; ③ fat deposition; ④ protein degradation; ⑤ macrophage phenotype switch, from M2 to M1 type transition; ⑥ insulin resistance; ⑦ muscle fiber type transition, from slow oxidation type to fast glycolysis type; ⑧ muscle cell apoptosis.
The protein content of skeletal muscle accounts for about 60% of total body protein (41). Skeletal muscle atrophy after SCI is mainly a process of protein production and degradation imbalance (42). After an injury, protein hydrolysis is activated, the rate of protein degradation is greater than the rate of synthesis (8) and many organelles and muscle contractile proteins are degraded into free amino acids (43–45). Studies have shown that long-term disuse-related muscle atrophy is mainly due to a reduced rate of protein synthesis. In contrast, short-term disuse-related muscle atrophy is accompanied by a reduction in protein synthesis and an increase in decomposition, so the early stage of muscle disuse is manifested as clearly apparent muscle atrophy (46), indicating that short-term disuse-related muscle atrophy is more closely related to muscle mass and volume reduction. A study by Moore et al. (11) showed that muscle CSA in an SCI group decreased compared with that of a control group (8, 11), and the degree of muscle atrophy was closely related to the degree of injury. Muscle atrophy is more pronounced in complete than incomplete SCI (47). Metabolic protein changes have consequences throughout the lifespan, so age can also exacerbate the process of muscle wasting in patients with SCI (46) (Figure 2).
A persistent secondary injury cascade follows SCI (28), including many systemic effects closely related to skeletal muscle atrophy. For example, levels of hormones such as testosterone (a sex hormone), glucocorticoids, mineralocorticoids and others may fluctuate. Studies have found that dihydrotestosterone, the active metabolite of testosterone, may reduce synaptic dissection after nerve injury (35), strengthen afferent central nervous system signals, and promote the recovery of motor function (48). Men are prone to hypotestosteronemia after SCI, and exogenous testosterone treatment can prevent oxidative neural stress damage, effectively preventing SCI skeletal muscle atrophy and maintaining skeletal muscle mass [16]. Studies have found that another sex hormone, estradiol, can improve motor function and has anti-inflammatory effects, effectively reducing apoptosis (35, 49) and preventing further damage to spinal cord tissue. Its main target is the nervous system, but whether estradiol has a direct therapeutic effect on skeletal muscle atrophy after SCI remains unclear. Hormone therapy has shown strong potential in improving motor function and reducing skeletal muscle atrophy. Synthetic steroid hormones are used clinically in treating skeletal muscle atrophy, such as testosterone, insulin-like growth factor, and others. Combating skeletal muscle atrophy by increasing the transcriptional level of myogenic fibronectin DNA and activating the proliferation and differentiation of muscle satellite cells (50) (Figure 2). However, hormone therapy, while having a better therapeutic effect, is also associated with a certain risk of side effects, so it has to be applied appropriately and may not be suitable for all patients, and therefore researchers are currently exploring alternative treatments.
The spinal cord mainly relies on α motor neurons to regulate skeletal muscle, at the same time according to the regulation of different types of muscle fibers, α motor neurons can be divided into S motor neurons (slow contraction, anti-fatigue), FR motor neurons (fast contraction, fatigue-resistant), FF motor neurons (fast contraction, easy fatigue) three subtypes, so as to coordinate the various functions and effects of skeletal muscle, control type I muscle fibers, IIb muscle fibers and IIx muscle fibers respectively (51). Spinal cord injury can cause slow motor neuron axon conduction speed to slow down (52, 53). Skeletal muscle fiber undergoes a transition after SCI from a slow-oxidative to a fast fatigue and fast glycolysis variety, and at the same time develops toward the direction of muscle fibrosis, that is, the transformation of type II fiber to type I fiber (7, 54–57). which often precedes the deposition of adipose tissue (7), this may attribute to changes in the expression of genes that control myosin subunits, resulting in a significant decrease in the proportion of the slow myosin heavy chain (MHC) isoform and a corresponding increase in the fast MHC isoform (58, 59). However, if spasticity occurs after spinal cord injury, the change in muscle fiber type may not be obvious (56) (Figure 2).
In addition to skeletal muscle fiber changes, specific abnormalities occur in the glucose and lipid metabolism of the skeletal muscle. Intramuscular fat (IMF) comprises fat infiltrated within a single muscle group (intramuscular and extra muscular fat compartments) and intermuscular adipose tissue between different muscle groups. An essential pathological change after skeletal muscle atrophy is the deposition and infiltration of numerous IMFs (11) more extensively in patients with SCI than in healthy individuals. Elder found that the IMF content in the muscles of SCI subjects was more than three times that of control subjects, and the subfascial fat content was about four times that of controls, and this is a cause of decreased strength in SCI patients (60) (Figure 2).
The glucose metabolism of skeletal muscle becomes disordered after SCI. Skeletal muscle, as an important consumer of glucose (61), develops insulin resistance (60) and initiates an inflammatory response in skeletal muscle after SCI, with an increase in macrophages within the muscle. The phenotype of muscle macrophages can affect the insulin sensitivity, and macrophages will polarize from the M2 phenotype to the M1 phenotype (62, 63), inducing insulin resistance. Macrophages may also affect the uptake and metabolism of glucose in skeletal muscle by affecting the secretion of factors related to glucose homeostasis (64), also resulting in insulin resistance in the muscle. These changes account for the occurrence of type 2 diabetes mellitus after SCI (65, 66) (Figure 2).
Cytokines associated with skeletal muscle atrophy after SCI
Skeletal muscle atrophy is often closely related to the severity of spinal cord injury. Incomplete spinal cord injury tends to cause skeletal muscle atrophy within the first 6 weeks of injury (6), while complete spinal cord injury continues to cause skeletal muscle atrophy within 24 weeks after injury (7). Chronic inflammatory response and oxidative stress in skeletal muscle after SCI may be the mechanisms leading to atrophy (9, 67–69). At present, the known factors and related proteins involved in skeletal muscle atrophy include: tumor necrosis factor-alpha (TNF-α) and its receptor (70–72), human tumor necrosis factor-related weak apoptosis-inducing factor (TWEAK) and its receptor (56, 67, 73), interleukin-1β (IL-1β) (72, 74), interleukin-6 (IL-6) and its receptor (75, 76), growth factor (IGF-1) (77, 78), human dystrophin (Fbox-1, also known as Atrogin-1) (79, 80), muscle-specific RING finger protein 1 (MuRF1) (79, 80), peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) (20, 58, 81), fibroblast growth factor-inducible receptor 14 (Fn14) (73), reactive oxygen species (ROS) (20, 68, 82) and others. The discovery of these skeletal dystrophins has provided a deeper understanding of skeletal muscle atrophy at the molecular level and suggest the possibility of intervening via corresponding signaling pathways and factors to delay the atrophy process or promote skeletal muscle regeneration.
Inflammation-mediated skeletal muscle atrophy after spinal cord injury
Tumor necrosis factor-alpha
As a pro-inflammatory cytokine, TNF-α is a potent inducer of skeletal muscle atrophy after SCI. It was found that the expression levels of TNF-α and TNF-α receptors were elevated in the atrophied skeletal muscle during the chronic phase of spinal cord injury patients, which played a very important role in mediating skeletal muscle atrophy during the chronic phase of SCI (5, 67, 72). TNF-α binding to tumor necrosis factor receptor 1 (TNFR1) induces atrophy and autophagy of C2C12 myotubes in skeletal muscle (9, 42), while leading to ROS accumulation and activating the inflammatory response pathway. The expression level of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is also increased, and the degradation rate of skeletal muscle protein is accelerated (83, 84). TNF-α is closely related to the occurrence of energy disorder and stress as well as abnormal glucose and lipid metabolism in the process of skeletal muscle atrophy (42). Consistent with this, its levels are significantly positively correlated with muscle strength (85). The MAPK signaling pathway is activated to varying degrees after SCI due to various factors such as oxidative stress and inflammation (86, 87). TNF-α as an inflammatory factor activates the MAPK signaling pathway mediated by extracellular regulated protein kinas (ERK), p38 MAPK, c-Jun aminoterminal kinase (JNK) mediated MAPK signaling pathway (83, 88) (Figure 3) and enhances the expression of Atrogin-1 and MuRF1.
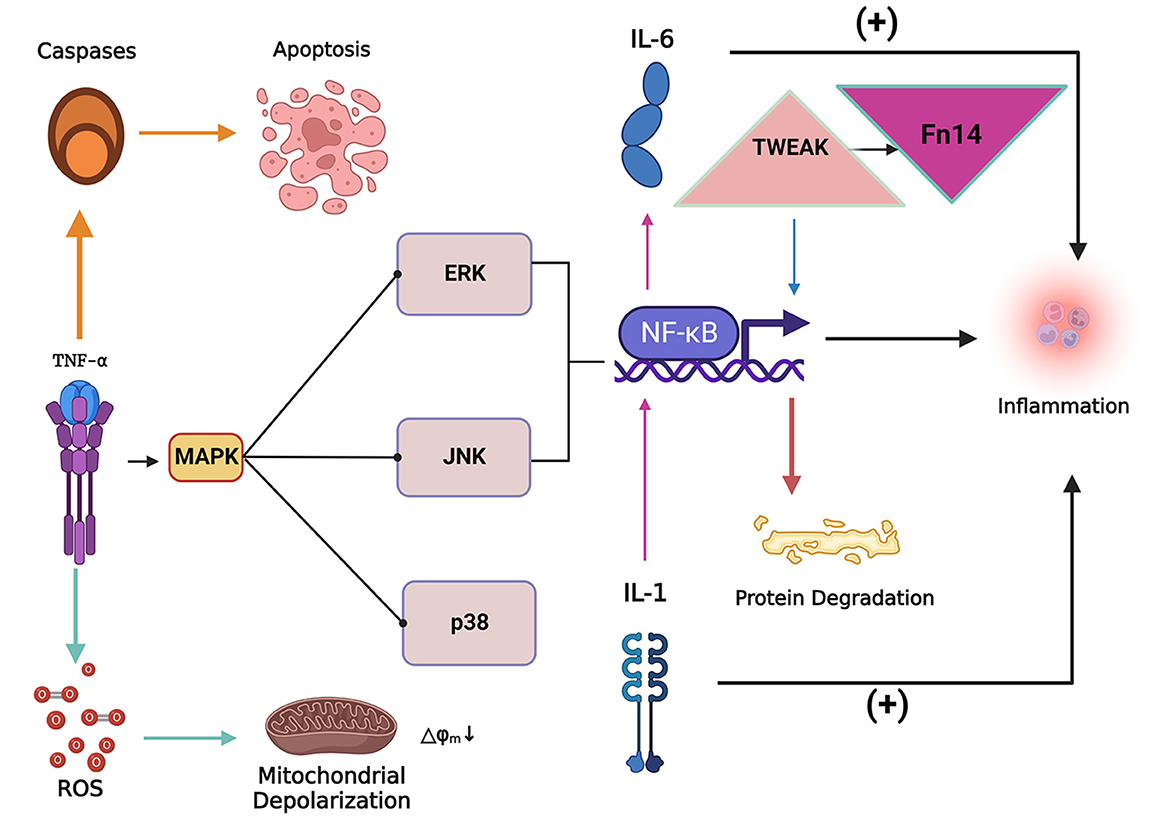
Figure 3. The inflammatory factor tumor necrosis factor-alpha (TNF-α) can induce the apoptosis of skeletal muscle cells and lead to the accumulation of reactive oxygen species (ROS). The collection of ROS can lead to mitochondrial dysfunction and depolarization, while TNF-α can activate the MAPK signaling pathway and cause nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). NF-κB expression levels to increase, inducing inflammation and protein degradation. Tumor necrosis factor-related weak apoptosis-inducing factor (TWEAK) can activate NF-κB signaling to promote proteolysis and simultaneously increase the expression level of fibroblast growth factor-inducing factor 14 (Fn14), both of which act synergistically in the process of skeletal muscle atrophy. IL-1 (Interleukin 1) and IL-6 (Interleukin 6) are critical inflammatory factors that induce skeletal muscle atrophy. IL-1 stimulates the expression of IL-6 by stimulating the expression of NF-κB. Overexpression of IL-1 and IL-6 induces an inflammatory response in the body and thus induces skeletal muscle atrophy.
Previous studies have found that the differentiation of C2C12 myoblasts in the early stage of skeletal muscle injury is strongly correlated with the concentration of TNF-α (89), which is closely related to the tissue repair effect of TNF-α in the acute phase, while in the chronic phase TNF-α is related to tissue damage (90). Low doses of TNF-α can induce the proliferation of C2C12 myoblasts, promote muscle production in skeletal muscle cells, while high doses cause skeletal muscle atrophy by interfering with the ability of muscle cells to differentiate into muscle fibers. Therefore, TNF-α exhibits a time- and dose-dependent induction of skeletal muscle atrophy (42). It was found that functional electrical stimulation therapy and endurance exercise training can effectively reduce the level of TNF-α in SCI patients and animals, thus acting as a treatment against skeletal muscle atrophy (91, 92). Therefore, suppressing elevated levels of inflammatory factors in the acute phase of SCI or fighting chronic inflammation in the chronic phase are also effective strategies to treat skeletal muscle atrophy.
Tumor necrosis factor-like weak inducer of apoptosis /fibroblast growth factor-inducing factor 14
TWEAK may be an essential mediator of chronic inflammation and fibrotic changes in skeletal muscle after SCI, and higher levels of TWEAK and TWEAK R in SCI may affect the oxidative metabolism of the body. On the one hand, TWEAK inhibits the normal oxidative metabolic process in skeletal muscle by activating NF-κb, thus causing oxidative stress injury (67, 93). On the other hand, TWEAK can activate NF-κB signaling and other proteolytic pathways (94) (Figure 3), activate the autophagy pathway, induce muscle proteolysis, and inhibit the proliferation of myoblasts, thereby inhibiting the regeneration of skeletal muscle fibers. In addition, TWEAK reduces the number of mitochondria in skeletal muscle cells, weakens the ability of skeletal muscle cells to resist oxidative stress (93), and causes metabolic dysfunction in skeletal muscle cells (93, 95).
Fn14 is stimulated and up-regulated after SCI, and can combine with TWEAK to synergistically affect the atrophy of skeletal muscle (93) (Figure 3). It may also be involved in the transition process of skeletal muscle fiber types (93), but at higher levels Fn14 has a TWEAK-independent effect, promoting the expression of critical factors in skeletal muscle regeneration, thereby promoting skeletal muscle regeneration (94). Activation of TWEAK/Fn14 is also coupled to TNF-α-TNFR1 signaling and can sensitize skeletal muscle to TNF-α signaling (96).
In the persistent state of injury after SCI, TWEAK-TWEAK R activation may lead to pathological remodeling of muscle, activating proliferation and activation of fibroblasts and causing muscle fibrosis. Yarar-Fisher et al. (67) found that expression levels of TWEAK R, Fn14, and NK-κB in the skeletal muscle of patients with SCI were significantly increased, and the degree of muscle fibrosis was also significantly increased, suggesting a close association. In addition, TWEAK is also an important regulator of the skeletal muscle fiber type transition process after SCI (67). Mittal (55) and others compared mice with overexpression or knockout of the TWEAK gene and found that skeletal muscle atrophy and fast muscle fibers increased significantly after 4 to 5 months of overexpression, with increased Fn14 expression level and the opposite finding in the knockout group. These studies have confirmed that the TWEAK/TWEAK R/NF-κB signaling pathway and Fn14 play essential roles in skeletal muscle atrophy after SCI.
Interleukin 1/6
SCI activates the body's immune and inflammatory responses, causes an increase in interleukin levels, such as IL-1β, IL-6, and IL-1β causes secondary spinal cord injury, which further leads to loss of central regulation of skeletal muscle, also IL-1β has a role in promoting neurogenic heterotopic ossification (NHO) in skeletal muscle after SCI, which limits joint movement and affects the normal life of patients (97–100). IL-6 is mainly produced during skeletal muscle contraction (101). High levels of IL-6 are also an important influence on skeletal muscle atrophy after SCI (102).
Satellite cells are the power source for skeletal muscle cell proliferation and regeneration, maintain skeletal muscle quality, and endow skeletal muscle with a degree of plasticity (103). IL-1 and IL-6 have pro-inflammatory effects, activating satellite cells in skeletal muscle, promoting muscle cell proliferation to some extent, and have a particularly positive impact on promoting muscle regeneration in the early stage of injury (99, 104). However, high levels of IL-1 and IL-6 combined with TNF-α can inhibit the synthesis and metabolism of IGF-1 (69). At an appropriate level, IL-1β can promote the expression of cyclooxygenase 2 (COX-2) in muscle and reduce myostatin levels (103). A study (105) showed that the proliferation and differentiation of myoblasts was reduced in IL-1 knockout mice. With low concentrations of exogenous IL-1 introduced to the body, the MAPK signaling pathway was activated, and NF-κB stimulated the secretion of chemokines and IL-6 (Figure 3). Low concentrations of IL-6 have a positive effect on the promotion of muscle cell proliferation by IL-1β. However, when spinal cord injury enters the chronic inflammatory phase, it has the opposite effect, mainly by activating the ubiquitin-proteasome system, inducing skeletal muscle atrophy (9, 106), and simultaneously generating the production of myostatin and causing cell damage. The accumulation of mitochondrial ROS (84, 107) and other factors induce skeletal muscle atrophy.
Skeletal muscle is an integral producer of IL-1 and IL-6 (97), healthy individuals can promote IL-6 levels through exercise, but under short-term exercise SCI patients lack such a regulatory mechanism due to skeletal muscle atrophy (101), and studies have found that endurance exercise can regulate the expression of IL-1 and IL-6 in skeletal muscle (103, 108), which may explain the positive effect of endurance training on skeletal muscle function and muscle strength after SCI (75, 109).
Oxidative stress-mediated skeletal muscle atrophy after spinal cord injury
Peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α
PGC-1α is a regulator of mitochondrial bioenergetics and abundantly expressed in skeletal muscle, and plays an essential role in skeletal muscle repair after SCI via transformation of fast-twitch to slow-twitch muscle fibers (77, 81, 110, 111). It can induce mitochondrial biogenesis, and by up-regulating nuclear respiratory factors 1,2 (Nrf1, 2) and mitochondrial transcription factors expressed against oxidative stress in skeletal muscle and spinal cord (81, 112) it can increase myoglobin activity and promote energy metabolism in skeletal muscle. After SCI, PGC-1α expression decreases (110), and expression of myosin heavy chain protein also decreases accordingly (31). PGC-1α has an inhibitory effect on FoxO3 associated with muscle atrophy and mass loss (110)whose expression is increased after SCI, indirectly causing the expression of Atrogin-1 and MuRF1, resulting in muscle atrophy (113). It was found that exercise training promotes the expression of PGC-1α and the transformation of spinal cord injury patients' muscle fibers from fatigue-prone fast muscle fibers to more endurance-prone slow muscle fibers, while improving muscle endurance (111).Therefore, PGC-1α plays a vital role in the repair of nerve and skeletal muscle structure and function after SCI.
Reactive oxygen species
Changes in mitochondrial structure and function in skeletal muscle cells play a vital role in regulating overall skeletal muscle mass and function (93). Denervation of skeletal muscle after SCI can lead to mitochondrial toxicity (68, 114), this leads to oxidative stress damage, mainly in the form of an imbalance between ROS production and detoxification (68). After the mitochondrial function is damaged, intracellular ROS cannot be effectively and promptly removed. The antioxidant capacity of skeletal muscle cells decreases, resulting in oxidative stress and ROS accumulation, causing oxidative stress damage, apoptosis and mitochondrial depolarization of myoblasts (Figures 3, 4) (115, 116), so scavenging ROS and alleviating oxidative stress damage is an integral part of the treatment of skeletal muscle atrophy after SCI. Under normal circumstances, ROS scavenging mainly depends on antioxidant-related enzymes and factors in vivo, such as catalase, glutathione, superoxide dismutase, and others (41, 117, 118). The antioxidant capacity of the spinal cord and skeletal muscle is impaired after SCI, so exogenous interventions are often required to reduce or reverse oxidative stress injury (119). Studies have found that many vitamins, proteins, and other nutrients have antioxidant capacity. For example, vitamin D, especially its 1,25-(OH)2D form, is generally considered to be an antioxidant and its supplementation can reduce skeletal muscle, illustrating the potential for ROS generation to combat oxidative stress (120).
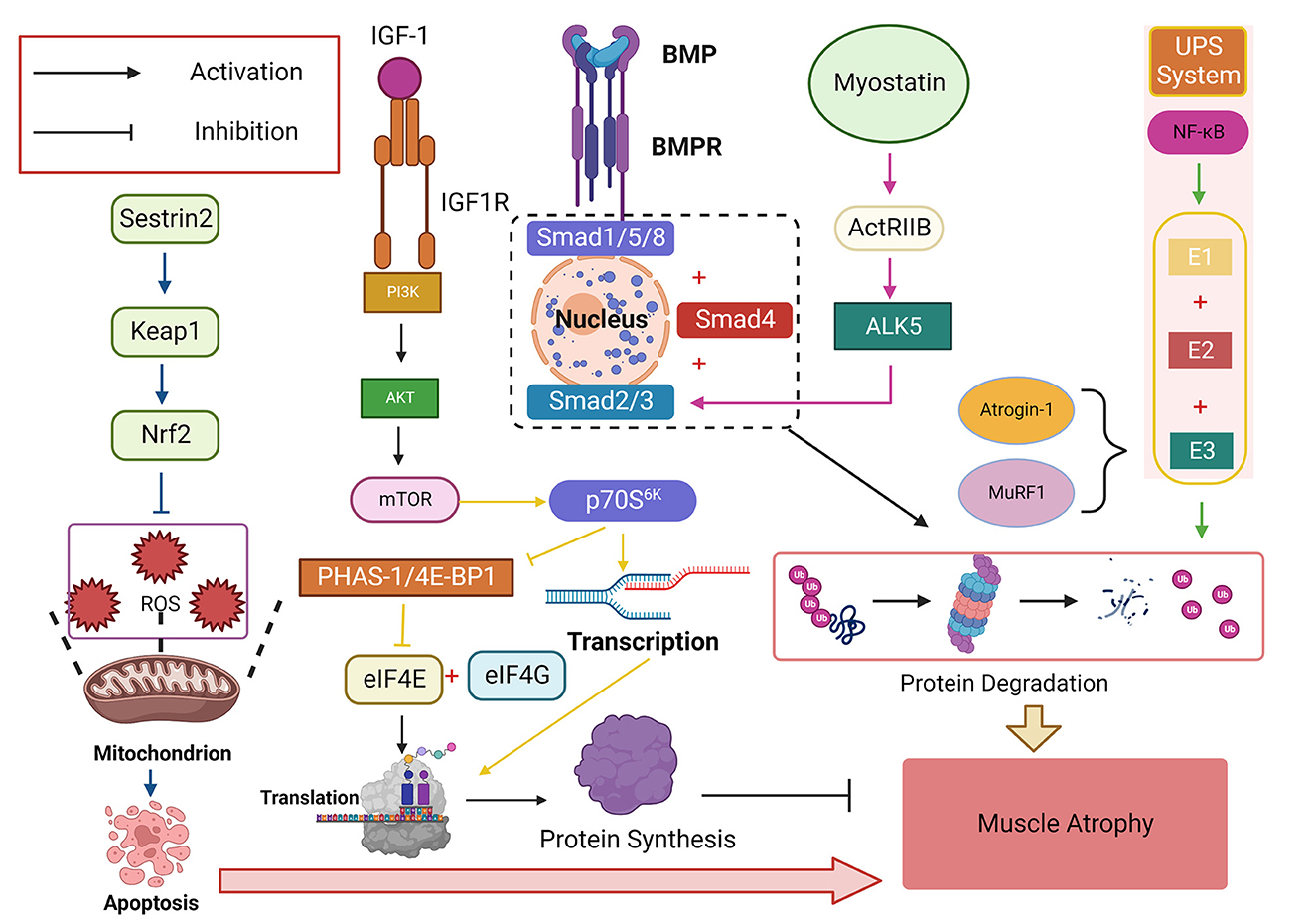
Figure 4. The insulin-like growth factor-1 (IGF-1)/phosphatidylinositol trikinase (PI3K)/threonine kinase (Akt)/mammalian target of rapamycin (mTOR) signaling pathway is a critical signaling pathway that promotes protein synthesis and plays a vital role in regulating skeletal muscle mass. Its downstream target p70S6K can promote protein transcription while inhibiting eIF4E, a negative regulator of PHAS-1/4E-BP1, which binds eIF4E to eIF4G and initiates translation. NF-κB activates ubiquitin-proteasome System (UPS) and degrades proteins by binding E1, E2, and E3, with Atrogin-1 and muscle-specific RING finger protein 1 (MuRF1) being the two most common E3s Ligase, bone morphogenetic protein (BMP) affects the signaling pathway of Smad1, Smad5, and Smad8 (Smad1/5/8) protein phosphorylation and finally converges to Smad4. Myostatin-ActRIIB-ALK5-Smad2/3 is another signaling pathway that finally combines to Smad4 to cause protein degradation in skeletal muscle. Sestrin2 can activate the Sestrin2-Keap1-Nrf2 pathway, increase the expression level of Nrf2, resist ROS-induced apoptosis, and play an antioxidant role.
Sestrins (Sesns)
Sestrins is a highly conserved stress-inducible protein family, which has attracted the attention of researchers in recent years due to its anti-aging and muscle atrophy effects. It can activate AKT and then autophagy, restoring neuronal autophagy flux. It can also activate the MAPK signaling pathway to combat apoptosis and oxidative stress, can affect the homeostasis of stem cells in skeletal muscle (121–125), combat pathological changes such as insulin resistance and fat accumulation (126), and coordinate skeletal muscle synthesis and catabolism to delay the disuse of muscle atrophy (127).
Sestrin1 has the highest expression level in skeletal muscle, and decreases rapidly after SCI, while overexpression of the Sestrin1 gene may reduce atrophy of skeletal muscle (127, 128). SCI can also activate the Sestrin2-Keap1-Nrf2 pathway, increase the expression level of Nrf2, enhancing its antioxidant effect (126) (Figure 4). Elevated sestrin2 levels were found to improve functional recovery and neuronal survival after spinal cord injury through activation of autophagy (122). Therefore, the Sestrins protein family significantly improves SCI skeletal muscle function and promotes structural recovery, and should be further explored as a potential therapeutic target.
Other related factors regulating skeletal muscle atrophy after spinal cord injury
Insulin-like growth factor-1
IGF-1 is expressed in skeletal muscle where it plays a vital role in muscle and nerve metabolism. IGF-1 can regulate the proliferation and differentiation of muscle satellite cells by activating PI3K/Akt (77, 129, 130) and promote collagen formation, in turn promoting skeletal muscle hypertrophy (131). It also has a specific nutritional effect on the nervous system, enabling the protein synthesis of neurons and glial cells, inhibiting apoptosis, promoting nerve regeneration and myelination (132), and strengthening the nervous system and skeletal muscle connection. However, IGF-1 is highly sensitive to the inflammatory response. Cheng et al. (133) found that the inflammatory response after SCI resulted in decreased IGF-1 levels in skeletal muscle, which caused diminished anabolism of muscle tissue and atrophy of skeletal muscle (23, 134).
Hypothalamus-growth hormone (GH)-IGF-1 axis
GH plays a role in denervated muscle regeneration after SCI by inducing the production of IGF-1 in the liver (132). In chronic SCI patients, the (GH)-IGF-1 axis is inhibited (77, 135). The main reason may be that chronic inflammation caused by SCI leads to increased expression of pro-inflammatory cytokines, stimulates the hypothalamus to secrete growth Inhibitors, inhibits the hypothalamus-GH-IGF-1 axis, and increases GH resistance. Some studies have proposed that inflammatory factors work by inhibiting GH signal transduction (132, 134). For example, TNF-α and IL-1β can inhibit the abundance of growth hormone receptors, while IL-6 plays a role in promoting the expression of suppressor of cytokine signaling 3 (SOCS3), which has an enhancing effect on GH resistance (134).
IGF-1/PI3K/Akt/mTOR signaling pathway
IGF-1/phosphatidylinositol trikinase (PI3K)/threonine kinase (Akt)/mammalian target of rapamycin (mTOR) is a signaling pathway that promotes protein synthesis and is involved in the regulation of bone. It plays a vital role in muscle mass (77, 131, 136) (Figure 4). This signaling pathway is inhibited after SCI, and reactivation can resist the resulting skeletal muscle atrophy (137–139) and inflammatory response (133). The PI3K/Akt/mTORC1 pathway plays an essential role in the recovery of denervated skeletal muscle (107).
PI3K is located in the hypothalamus and is anti-flammatory after SCI, and activation of this kinase can be used primarily to counteract the inflammatory response, which has a protective effect in preventing the development of insulin resistance after SCI (133). Akt/mTOR regulates skeletal muscle cell proliferation and growth, normal protein metabolism, and prevents skeletal muscle atrophy. This signaling pathway is down-regulated in skeletal muscle atrophy (41, 137). After SCI, the PI3K/Akt/mTOR pathway is inhibited, and the skeletal muscle protein degradation program is initiated (50). IGF-1 creates this signaling pathway through PI3K and Akt kinases in vivo, activating mTOR and phosphorylating it. mTOR is sensitive to changes in amino acids and is a critical protein turnover regulator that integrates nutrition signals, growth factors, energy status, and stress (19, 140). Amino acid-sensitive signals converge on GTPases, which are immobilized on the surface of lysosomes by the Ragator (RAG) complex (141). mTOR is inhibited in amino acid deficiency. Supplementation of amino acids to regulate the mTOR signaling pathway and protein metabolism has received considerable attention as a potential treatment strategy for muscle atrophy after SCI (142–145). Amino acids activate the RAG complex by regulating the guanine dissociation and binding states of RAG. Activation of the RAG complex promotes the translocation and activation of mTORC1 to the lysosome (141, 146), thereby promoting the activation of mTORC1 and its downstream targets p70S6K and PHAS-1/4E-BP1, and thus enhancing synthesis of skeletal muscle protein (141).
The downstream target p70S6K of the PI3K/Akt/mTOR pathway can positively promote protein transcription: p70S6K is phosphorylated after activation and inhibits the negative regulator PHAS-1/4E-BP1 of the eukaryotic transcription initiation factor eIF4E (147). Release of this factor from the inhibitory complex allows it to bind to eIF4G and initiate translation, simultaneously promoting protein synthesis by prolonging the translation process (147) (Figure 4). Research has shown that 95% of the compensatory hypertrophic changes in muscle were blocked after rapamycin's specific inhibition of mTOR (137). In contrast, in unloaded muscles, the muscles showed a marked atrophic state, with significantly decreased phosphorylation levels of Akt protein and its downstream targets, and recovery of these when the load was restored. This suggests that phosphorylation of AKT and its downstream targets and activation of mTOR are required during muscle hypertrophy. Signaling pathways play a crucial role in mass recovery and hypertrophy of skeletal muscle atrophy caused by SCI (137, 148). The current study found that androgens and β2-adrenergic agonists have better therapeutic effects in activating IGF-1/PI3K/AKT/mTOC signaling pathway for skeletal muscle atrophy after spinal cord injury (19).
Ubiquitin-proteasome system
The main component of skeletal muscle is myogenic fibers, and a characteristic of muscle atrophy is the rapid degradation of myogenic fibers (149). UPS in skeletal muscle is activated after SCI, the breakdown of proteins in skeletal muscle after SCI is mainly accomplished through the ubiquitin-proteasome pathway (50, 150), resulting in greater catabolism than anabolism in skeletal muscle (151), in which the degraded proteins are covalently linked to ubiquitin molecules via the ubiquitin ligase complex, which is then processed by the 26S protease. It has been shown that immobilization can up-regulate the mRNA expression of rat 26S proteasome (25, 50, 152, 153). As a critical pro-inflammatory factor, NF-κB, plays an important role between the inflammatory response to skeletal muscle atrophy after SCI and the balance of apoptotic and anti-apoptotic signaling, on the one hand, and determines whether cells will undergo apoptosis in response to other apoptosis-inducing-related factors such as TNF-α and TWEAK, affecting the proliferation and survival of skeletal muscle cells (83). However, the targets of NF-κB are ubiquitin-proteasome members, which can trigger ubiquitin molecular markers. The target protein is ultimately degraded by the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) (107, 154). Two E3 ligases, Atrogin-1 and MuRF1 play a vital role in the protein degradation process that mediates skeletal muscle atrophy (7, 107) (Figure 4), and their expression levels are upregulated in skeletal muscle after SCI (155). While the UPS is also key to the neural recovery of SCI (153). Gonzalez-Ruiz et al. found that the level of protein ubiquitination was significantly reduced after the application of epicatechin [(-)-epicatechi] in clinical SCI patients, the CSA of skeletal muscle was significantly increased; indicating that using UPS as a target of intervention can prevent skeletal muscle atrophy after SCI (156).
Calpain
Calpain is the key enzyme in regulating apoptosis by mediating the degradation of cytoskeletal and membrane proteins, calpain is activated thus causing apoptosis of neural cells in the spinal cord and skeletal muscle cells in skeletal muscle after SCI (149, 157, 158), usually acting in conjunction with caspase-3 in the apoptosis of skeletal muscle, this is usually a key step in skeletal muscle atrophy (68, 151, 158, 159). There are studies that attenuate apoptosis after spinal cord injury from inhibition of calpain activation, and the current study found that SJA6017 (159), calpeptin, MDL-28170 could inhibit apoptosis by inhibiting calpain activity and have neuroprotective effects (158), but the effect on inhibiting skeletal muscle atrophy still needs to be further investigated.
Autophagic lysosome system
Moderately activated autophagy helps protect cell structure and maintain normal cellular energy metabolism, and autophagic lysosomes are overactivated in skeletal muscle after SCI (160, 161). Beclin-1, a protein specific for autophagy, was found to be significantly elevated after SCI, resulting in the degradation of muscle proteins in skeletal muscle (160). Autophagy may depend on the regulation of mTOR signaling pathway (162), so it was found that regulation of mTOR may affect the autophagic process after injury, such as the application of AMPK inhibitors (163) or activators (164); also the autophagic process may not depend on mTOR, and inosito also have the effect of inhibiting autophagy (163). Exercise therapy is currently a very effective mainstream treatment for skeletal muscle atrophy, and studies have found that exercise training can slow down skeletal muscle atrophy by reducing autophagosome levels in skeletal muscle and thereby regulating autophagy (162).
Myostatin
Myostatin is an essential member of the TGFβ family and acts as a negative regulator of skeletal muscle mass (20, 165). Some studies have found that its expression level may gradually increase with the inhibitory level of the PI3K-AKT signal after SCI, resulting in the loss of skeletal muscle mass (136, 166). It is a crucial muscle growth regulator, its inhibition increasing the mass of denervated skeletal muscle. In previous studies, myostatin-ActRIIB-ALK5-Smad2/3 was found to be an essential pathway affecting muscle mass (167) (Figure 4), and may be targeted at the molecular level. Previous studies have used synthetic myostatin inhibitors to interfere with Smad2/3, inhibiting the myostatin pathway (165) and delaying the skeletal muscle atrophy and metabolism caused by SCI (107, 168, 169) to treat post-SCI patients. The inhibition of myostatin has not been found to alleviate skeletal muscle atrophy of denervated limbs in experimental animals but has a specific therapeutic effect on disuse muscle atrophy, indicating that myostatin may be effective only in the presence of innervation (20). Studies have found (170) that the level of myostatin in the serum of patients with aerobic exercise, commonly used in the clinical treatment of SCI, increases to some extent over time. However, the mechanism underlying this increase needs to be further explored.
Bone morphogenetic protein
BMP is a member of the TGFβ family and acts on a signaling pathway that affects the phosphorylation of Smad1, Smad5, and Smad8 proteins and ultimately converges on Smad4, thereby affecting muscle mass (Figure 4). BMP can inhibit muscle atrophy by binding to BMP-type receptor (ALK3). It can also negatively regulate the Fbxo30 (Musa1) gene, which plays a vital role in the regulation of the muscle atrophy-related ubiquitin-protease system (169, 171–173). Normal BMP signaling protects the neuromuscular junction (NMJ) and prevents excessive denervation of muscle fibers, and disturbed BMP signaling accelerates muscle atrophy (171, 174); BMP is expressed at low levels in motor neurons in a functionally intact state, and the expression of BMP2/4/7 and the corresponding ligands and receptors, phosphorylated Smad, are significantly upregulated in the spinal cord after SCI (175), thereby participating in the inflammatory response and neuronal apoptosis in the spinal cord, leading to abnormal neuromuscular signaling between the spinal cord and skeletal muscle, thereby affecting subsequent neurological recovery and skeletal muscle atrophy (173).
Discussion
Skeletal muscle atrophy after SCI is a more complex and difficult clinical problem that requires multidisciplinary intervention. In recent years, little literature has outlined and analyzed the mechanisms of skeletal muscle atrophy after SCI. Therefore, this paper summarizes the pathophysiological changes of skeletal muscle atrophy after SCI. It analyzes the related factors and atrophy mechanisms, which can provide a specific theoretical basis and research direction for future clinical treatment to delay of skeletal muscle atrophy and enhance recovery of the patient's motor function. The current mainstream prevention and treatment methods are mostly exercise and hormone therapy. As described above in the article, exercise training and hormone therapy have shown relatively good therapeutic effects through various molecular pathways. The current clinical treatment for skeletal muscle atrophy after SCI is based on exercise training. For example, activity-based physical rehabilitation therapies (ABTs) (176–178) have shown sound therapeutic effects in improving neuromuscular plasticity and are widely used in many SCI patients for motor function training. They can enhance residual muscle strength (179), delay skeletal muscle atrophy, maintain and improve residual motor function, and improve patients' quality of life (180). More common forms of treatment include body weight-supported treadmill training, robot-assisted mobility training (8), heavy load strength training (HLT) (181), resistance training (RT) (182), but the therapeutic effect of ABTs decreases with the severity of the patient's injury. ABTs alone may therefore not be enough to promote muscle regeneration and motor function reconstruction in patients, and a combination of other adjuvant therapies may be needed (8, 176). In a review by Alvaro Megía García (183) et al. found that non-invasive transcutaneous spinal cord stimulation (tSCS) was effective in activating lower limb muscles, increasing muscle strength and improving muscle function; several studies (184, 185) found that the combination of N-3 unsaturated fatty acids and appropriate training delayed muscle atrophy and improved physical function. In addition, testosterone and androgen therapy can also significantly delay skeletal muscle atrophy (82).
However, these treatments often have their own limitations, such as hormone therapy has certain side effects, and exercise training is often difficult to achieve the desired results due to the lack of patient's endurance. SCI patients have energy and substance metabolism disorders (186), so a good nutritional intervention program may be able to improve the treatment effect of SCI skeletal muscle atrophy and optimize the treatment plan. Glutathione (GSH) is a small-molecule antioxidant substance that can counteract oxidative stress damage after SCI, thereby reducing skeletal muscle damage and protecting skeletal muscle from atrophy (68). Glycine is a non-essential amino acid as well as one of the main components of GSH. Glycine levels in serum, spinal cord and skeletal muscle tissue are decreased in SCI (187, 188), however glycine has been found to increase skeletal muscle mass, protect skeletal muscle functions under pathological conditions (189, 190), and fight against inflammatory response after disease (191). Leucine has also been found to be a potent amino acid effective in reducing skeletal muscle catabolism. The body's perception of leucine is impaired after injury due to inflammation and other factors, but glycine can restore the role of leucine in the body (192) and activates GSH metabolism (189). Glycine can be supplemented orally (193), therefore glycine therapy may be a safe, effective and promising dietary treatment.
Vitamin D has also been shown to be strongly associated with skeletal muscle health. Vitamin D can activate the vitamin D receptor in skeletal muscle cells by affecting the balance of calcium and phosphate, which promotes the proliferation and differentiation of skeletal muscle cells (194), and also affects the strength of skeletal muscle to a certain extent (195). The vitamin D content in the skeletal muscle of SCI patients is decreased (196), therefore, how to improve the skeletal muscle function of SCI patients through vitamin D supplementation has received a lot of attention from researchers in recent years (196, 197).
The metabolism of nutrients in the skeletal muscle of SCI patients is disturbed after the injury, and timely supplementation is needed. Therefore, appropriate dietary treatment and nutritional therapy are necessary to perhaps compensate for some of the drawbacks of medication and exercise training, and to improve the physical function of SCI patients by means of dietary intake, so that the therapeutic effects of multiple treatments can be maximized. However, how to carry out multiple nutrient supplementation, i.e., the ratio and content of each component nutrient, still needs to be clarified through further investigation of safe and effective nutritional intervention programs and the development of strict guidelines.
Limitation
In order to write this review, we conducted a literature search in PUBMED, ISI Web of Science, and MEDLINE and Google Scholar databases before April 2022, searching “spinal cord injury,” “skeletal muscle,” and “atrophy” and so on as keywords. Although we conducted as extensive a literature search as possible and cited relevant and high-quality literature in our field whenever possible, there are still some relevant literature that may have been overlooked, as well as some ongoing studies and recent results that may not have been included.
Conclusions
Multiple factors cause skeletal muscle atrophy after SCI, and the mechanism of atrophy is complex. The current clinical treatment methods involving drugs or exercise training are often insufficiently effective, especially for patients with more severe injuries. This review reveals that future therapeutic modalities may be explored and investigated at the cellular and molecular levels to optimize current clinical treatment options and improve the therapeutic effect of skeletal muscle atrophy after SCI, thus effectively promoting functional recovery after SCI. Besides, many regulatory factors related to skeletal muscle atrophy have a certain value in theory, but the relationship and influence between various regulatory factors also need to be further explored. Meanwhile, the development of regulatory means with clinical translational significance and how to carry out appropriate regulation are still an academic problem that needs to be further studied.
Author contributions
Design and concepts: J-JL, D-GY, and XX. Definition of intellectual content: XX and ZT. Literature search: C-JZ, HK, HG, H-YD, and Y-ZP. Manuscript preparation: YY and Y-LJ. Manuscript editing: Y-LJ, L-JD, D-GY, and FG. Manuscript review: J-JL, D-GY, and FG. All authors approved the final version of the manuscript.
Funding
This work was supported by the National Key R&D Program of China, No. 2018YFF0301104 (to J-JL) and China Rehabilitation Research Center Project, No. 2022ZX-02 (to J-JL).
Acknowledgments
We thank Charlesworth Author Services (https://www.cwauthors.com.cn/) for its linguistic assistance during the preparation of this manuscript. We also thank https://Biorender.com for the support of the diagram drawing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
SCI, spinal cord injury; CSA, cross-sectional area; AchE, acetylcholinesterase; IMF, intramuscular fat; TNF-α, tumor necrosis factor; TNFR1, tumor necrosis factor receptor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; TWEAK, human tumor necrosis factor-related weak apoptosis-inducing factor; Fn14, fibroblast growth factor-inducible receptor; TWEAK R, TWEAK receptor; ERK, extracellular regulated protein kinase; JNK, c-Jun N-terminal kinase; MuRF1, muscle-specific RING finger protein 1; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; ROS, Reactive Oxygen Species; IL-1, interleukin 1; IL-6, interleukin 6; NHO, neurogenic heterotopic ossification; COX-2, cyclooxygenase 2; Nrf-1,2, nuclear respiratory factors 1,2; Sesns, Sestrins; IGF-1, Insulin-like growth factor-1; GH, growth hormone; SOCS3, suppressor of signaling 3; PI3K, phosphatidylinositol trikinase; Akt, threonine kinase; mTOR, mammalian target of rapamycin; UPS, Ubiquitin–proteasome System; FGF6, Fibroblast growth factor 6; FGFR, fibroblast growth factor receptor; BMP, Bone morphogenetic protein; ABTs, Activity-based physical rehabilitation therapies; GSH, Glutathione.
References
1. Eckert MJ, Martin MJ. Trauma: spinal cord injury. Surg Clin North Am. (2017) 97:1031–45. doi: 10.1016/j.suc.2017.06.008
2. Karsy M, Hawryluk G. Modern medical management of spinal cord injury. Curr Neurol Neurosci Rep. (2019) 19:65. doi: 10.1007/s11910-019-0984-1
3. Quadri SA, Farooqui M, Ikram A, Zafar A, Khan MA, Suriya SS, et al. Recent update on basic mechanisms of spinal cord injury. Neurosurg Rev. (2020) 43:425–41. doi: 10.1007/s10143-018-1008-3
4. McDonald J W, Sadowsky C. Spinal-cord injury. Lancet. (2002) 359:417–25. doi: 10.1016/S0140-6736(02)07603-1
5. Graham ZA, Collier L, Peng Y, Saéz JC, Bauman WA, Qin W, et al. A soluble activin receptor IIB fails to prevent muscle atrophy in a mouse model of spinal cord injury. J Neurotrauma. (2016) 33:1128–35. doi: 10.1089/neu.2015.4058
6. Gorgey AS, Dudley GA. Skeletal muscle atrophy and increased intramuscular fat after incomplete spinal cord injury. Spinal Cord. (2007) 45:304–9. doi: 10.1038/sj.sc.3101968
7. Otzel DM, Lee J, Ye F, Borst SE, Yarrow JF. Activity-based physical rehabilitation with adjuvant testosterone to promote neuromuscular recovery after spinal cord injury. Int J Mol Sci. (2018) 19:701. doi: 10.3390/ijms19061701
8. Yin L, Li N, Jia W, Wang N, Liang M, Yang X, et al. Skeletal muscle atrophy: from mechanisms to treatments. Pharmacol Res. (2021) 172:105807. doi: 10.1016/j.phrs.2021.105807
9. Abrigo J, Simon F, Cabrera D, Vilos C, Cabello-Verrugio C. Mitochondrial dysfunction in skeletal muscle pathologies. Curr Protein Pept Sci. (2019) 20:536–46. doi: 10.2174/1389203720666190402100902
10. Zhang S, Chen N. Regulatory role of microRNAs in muscle atrophy during exercise intervention. Int J Mol Sci. (2018) 19:405. doi: 10.3390/ijms19020405
11. Moore CD, Craven BC, Thabane L, Laing AC, Frank-Wilson AW, Kontulainen SA, et al. Lower-extremity muscle atrophy and fat infiltration after chronic spinal cord injury. J Musculoskelet Neuronal Interact. (2015) 15:32–41. Available online at: https://pubmed.ncbi.nlm.nih.gov/25730650/
12. Shah PK, Stevens JE, Gregory CM, Pathare NC, Jayaraman A, Bickel SC, et al. Lower-extremity muscle cross-sectional area after incomplete spinal cord injury. Arch Phys Med Rehabil. (2006) 87:772–8. doi: 10.1016/j.apmr.2006.02.028
13. Liu M, Bose P, Walter GA, Thompson FJ, Vandenborne K. A longitudinal study of skeletal muscle following spinal cord injury and locomotor training. Spinal Cord. (2008) 46:488–93. doi: 10.1038/sj.sc.3102169
14. Baligand C, Chen Y-W, Ye F, Pandey SN, Lai S-H, Liu M, et al. Transcriptional pathways associated with skeletal muscle changes after spinal cord injury and treadmill locomotor training. Biomed Res Int. (2015) 2015:387090. doi: 10.1155/2015/387090
15. Cho KH, Nam JH. Evaluation of stiffness of the spastic lower extremity muscles in early spinal cord injury by acoustic radiation force impulse imaging. Ann Rehabil Med. (2015) 39:393–400. doi: 10.5535/arm.2015.39.3.393
16. Byers JS, Huguenard AL, Kuruppu D, Liu N-K, Xu X-M, Sengelaub DR. Neuroprotective effects of testosterone on motoneuron and muscle morphology following spinal cord injury. J Comp Neurol. (2012) 520:2683–96. doi: 10.1002/cne.23066
17. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. (2009) 32:S157–163. doi: 10.2337/dc09-S302
18. Kraemer WJ, Ratamess NA, Hymer WC, Nindl BC, Fragala MS. Growth hormone(s), testosterone, insulin-like growth factors, and cortisol: roles and integration for cellular development and growth with exercise. Front Endocrinol. (2020) 11:33. doi: 10.3389/fendo.2020.00033
19. Otzel DM, Kok HJ, Graham ZA, Barton ER, Yarrow JF. Pharmacologic approaches to prevent skeletal muscle atrophy after spinal cord injury. Curr Opin Pharmacol. (2021) 60:193–9. doi: 10.1016/j.coph.2021.07.023
20. O'Brien LC, Gorgey AS. Skeletal muscle mitochondrial health and spinal cord injury. World J Orthop. (2016) 7:628–37. doi: 10.5312/wjo.v7.i10.628
21. Battistuzzo CR, Callister RJ, Callister R, Galea MP. A systematic review of exercise training to promote locomotor recovery in animal models of spinal cord injury. J Neurotrauma. (2012) 29:1600–13. doi: 10.1089/neu.2011.2199
22. Gordon T, Tyreman N, Raji MA. The basis for diminished functional recovery after delayed peripheral nerve repair. J Neurosci. (2011) 31:5325–34. doi: 10.1523/JNEUROSCI.6156-10.2011
23. Chandrasekaran S, Davis J, Bersch I, Goldberg G, Gorgey AS. Electrical stimulation and denervated muscles after spinal cord injury. Neural Regen Res. (2020) 15:1397–407. doi: 10.4103/1673-5374.274326
24. Yang X, Xue P, Chen H, Yuan M, Kang Y, Duscher D, et al. Denervation drives skeletal muscle atrophy and induces mitochondrial dysfunction, mitophagy and apoptosis via miR-142a-5p/MFN1 axis. Theranostics. (2020) 10:1415–32. doi: 10.7150/thno.40857
25. Zeman RJ, Zhao J, Zhang Y, Zhao W, Wen X, Wu Y, et al. Differential skeletal muscle gene expression after upper or lower motor neuron transection. Pflugers Arch. (2009) 458:525–35. doi: 10.1007/s00424-009-0643-5
26. Bryden AM, Hoyen HA, Keith MW, Mejia M, Kilgore KL, Nemunaitis GA. Upper extremity assessment in tetraplegia: the importance of differentiating between upper and lower motor neuron paralysis. Arch Phys Med Rehabil. (2016) 97:S97–s104. doi: 10.1016/j.apmr.2015.11.021
27. Sangari S, Lundell H, Kirshblum S, Perez MA. Residual descending motor pathways influence spasticity after spinal cord injury. Ann Neurol. (2019) 86:28–41. doi: 10.1002/ana.25505
28. Ahuja CS, Nori S, Tetreault L, Wilson J, Kwon B, Harrop J, et al. Traumatic spinal cord injury. Nat Rev Dis Primers. (2017) 3:17018. doi: 10.1038/nrdp.2017.18
29. Yoshizaki S, Yokota K, Kubota K, Saito T, Tanaka M, Konno D-J, et al. The beneficial aspects of spasticity in relation to ambulatory ability in mice with spinal cord injury. Spinal Cord. (2020) 58:537–43. doi: 10.1038/s41393-019-0395-9
30. Hidler JM, Harvey RL, Rymer WZ. Frequency response characteristics of ankle plantar flexors in humans following spinal cord injury: relation to degree of spasticity. Ann Biomed Eng. (2002) 30:969–81. doi: 10.1114/1.1500409
31. Higashino K, Matsuura T, Suganuma K, Yukata K, Nishisho T, Yasui N. Early changes in muscle atrophy and muscle fiber type conversion after spinal cord transection and peripheral nerve transection in rats. J Neuroeng Rehabil. (2013) 10:46. doi: 10.1186/1743-0003-10-46
32. Ye F, Baligand C, Keener JE, Vohra R, Lim W, Ruhella A, et al. Hindlimb muscle morphology and function in a new atrophy model combining spinal cord injury and cast immobilization. J Neurotrauma. (2013) 30:227–35. doi: 10.1089/neu.2012.2504
33. Tintignac LA, Brenner HR, Rüegg MA. Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol Rev. (2015) 95:809–52. doi: 10.1152/physrev.00033.2014
34. Lin CS-Y, Macefield VG, Elam M, Wallin BG, Engel S, Kiernan MC. Axonal changes in spinal cord injured patients distal to the site of injury. Brain. (2007) 130: 985–94. doi: 10.1093/brain/awl339
35. Sengelaub DR, Han Q, Liu N-K, Maczuga MA, Szalavari V, Valencia SA, et al. Protective effects of estradiol and dihydrotestosterone following spinal cord injury. J Neurotrauma. (2018) 35:825–41. doi: 10.1089/neu.2017.5329
36. Pallafacchina G, Blaauw B, Schiaffino S. Role of satellite cells in muscle growth and maintenance of muscle mass. Nutr Metab Cardiovasc Dis. (2013) 23:S12–18. doi: 10.1016/j.numecd.2012.02.002
37. Moon L, Bunge MB. From animal models to humans: strategies for promoting CNS axon regeneration and recovery of limb function after spinal cord injury. J Neurol Phys Ther. (2005) 29:55–69. doi: 10.1097/01.NPT.0000282512.16964.94
38. Iyer SR, Shah SB, Lovering RM. The neuromuscular junction: roles in aging and neuromuscular disease. Int J Mol Sci. (2021) 22:58. doi: 10.3390/ijms22158058
39. Wang J, Sun J, Tang Y, Guo G, Zhou X, Chen Y, et al. Basic fibroblast growth factor attenuates the degeneration of injured spinal cord motor endplates. Neural Regen Res. (2013) 8:2213–24. doi: 10.3969/j.issn.1673-5374.2013.24.001
40. Legay C. Why so many forms of acetylcholinesterase? Microsc Res Tech. (2000) 49:56–72. doi: 10.1002/(SICI)1097-0029(20000401)49:1<56::AID-JEMT7>3.0.CO;2-R
41. Brown LA, Guzman SD, Brooks SV. Emerging molecular mediators and targets for age-related skeletal muscle atrophy. Transl Res. (2020) 221:44–57. doi: 10.1016/j.trsl.2020.03.001
42. Bernacchioni C, Ghini V, Squecco R, Idrizaj E, Garella R, Puliti E, et al. Role of sphingosine 1-phosphate signalling axis in muscle atrophy induced by TNFα in C2C12 myotubes. Int J Mol Sci. (2021) 22:80. doi: 10.3390/ijms22031280
43. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. (2013) 6:25–39. doi: 10.1242/dmm.010389
44. Bhatnagar S, Kumar A. The TWEAK-Fn14 system: breaking the silence of cytokine-induced skeletal muscle wasting. Curr Mol Med. (2012) 12:3–13. doi: 10.2174/156652412798376107
45. Cabello-Verrugio C, Córdova G, Salas JD. Angiotensin II: role in skeletal muscle atrophy. Curr Protein Pept Sci. (2012) 13:560–9. doi: 10.2174/138920312803582933
46. Wall BT, Dirks ML, van Loon LJ. Skeletal muscle atrophy during short-term disuse: implications for age-related sarcopenia. Ageing Res Rev. (2013) 12:898–906. doi: 10.1016/j.arr.2013.07.003
47. Battistuzzo CR, Rank MM, Flynn JR, Morgan DL, Callister R, Callister RJ, et al. Effects Of treadmill training on hindlimb muscles of spinal cord-injured mice. Muscle Nerve. (2017) 55:232–42. doi: 10.1002/mus.25211
48. Yarrow JF, Kok HJ, Phillips EG, Conover CF, Lee J, Bassett TE, et al. Locomotor training with adjuvant testosterone preserves cancellous bone and promotes muscle plasticity in male rats after severe spinal cord injury. J Neurosci Res. (2020) 98:843–68. doi: 10.1002/jnr.24564
49. Namjoo Z, Moradi F, Aryanpour R, Piryaei A, Joghataei MT, Abbasi Y, et al. Combined effects of rat Schwann cells and 17β-estradiol in a spinal cord injury model. Metab Brain Dis. (2018) 33:1229–42. doi: 10.1007/s11011-018-0220-8
50. Urso ML. Disuse atrophy of human skeletal muscle: cell signaling and potential interventions. Med Sci Sports Exerc. (2009) 41:1860–8. doi: 10.1249/MSS.0b013e3181a6458a
51. Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci. (2010) 33:409–40. doi: 10.1146/annurev.neuro.051508.135722
52. Thomas CK, Bakels R, Klein CS, Zijdewind I. Human spinal cord injury: motor unit properties and behaviour. Acta Physiol. (2014) 210:5–19. doi: 10.1111/apha.12153
53. Celichowski J, Kryściak K, Krutki P, Majczyński H, Górska T, Sławińska U, et al. Time-related changes of motor unit properties in the rat medial gastrocnemius muscle after the spinal cord injury. II Effects of a spinal cord hemisection. J Electromyogr Kinesiol. (2010) 20:532–41. doi: 10.1016/j.jelekin.2009.07.003
54. Gorgey AS, Witt O, O'Brien L, Cardozo C, Chen Q, Lesnefsky EJ, et al. Mitochondrial health and muscle plasticity after spinal cord injury. Eur J Appl Physiol. (2019) 119:315–31. doi: 10.1007/s00421-018-4039-0
55. Mittal A, Bhatnagar S, Kumar A, Lach-Trifilieff E, Wauters S, Li H, et al. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J Cell Biol. (2010) 188:833–49. doi: 10.1083/jcb.200909117
56. Biering-Sørensen B, Kristensen IB, Kjaer M, Biering-Sørensen F. Muscle after spinal cord injury. Muscle Nerve. (2009) 40:499–519. doi: 10.1002/mus.21391
57. Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol. (2013) 45:2191–9. doi: 10.1016/j.biocel.2013.05.016
58. Wu Y, Zhao J, Zhao W, Pan J, Bauman WA, Cardozo CP. Nandrolone normalizes determinants of muscle mass and fiber type after spinal cord injury. J Neurotrauma. (2012) 29:1663–75. doi: 10.1089/neu.2011.2203
59. Negredo P, Rivero JL, González B, Ramón-Cueto A, Manso R. Slow- and fast-twitch rat hind limb skeletal muscle phenotypes 8 months after spinal cord transection and olfactory ensheathing glia transplantation. J Physiol. (2008) 586:2593–610. doi: 10.1113/jphysiol.2007.149120
60. Elder CP, Apple DF, Bickel CS, Meyer RA, Dudley GA. Intramuscular fat and glucose tolerance after spinal cord injury–a cross-sectional study. Spinal Cord. (2004) 42:711–6. doi: 10.1038/sj.sc.3101652
61. Hernandez-Carretero A, Weber N, LaBarge SA, Peterka V, Doan NY, Schenk S. Cysteine- and glycine-rich protein 3 regulates glucose homeostasis in skeletal muscle. Am J Physiol Endocrinol Metab. (2018) 315:E267–78. doi: 10.1152/ajpendo.00435.2017
62. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
63. Paccoud R, Saint-Laurent C, Piccolo E, Tajan M, Dortignac A, Pereira O, et al. SHP2 drives inflammation-triggered insulin resistance by reshaping tissue macrophage populations. Sci Transl Med. (2021) 13:591. doi: 10.1126/scitranslmed.abe2587
64. Fink LN, Oberbach A, Costford SR, Chan KL, Sams A, Blüher M, et al. Expression of anti-inflammatory macrophage genes within skeletal muscle correlates with insulin sensitivity in human obesity and type 2 diabetes. Diabetologia. (2013) 56:1623–8. doi: 10.1007/s00125-013-2897-x
65. Lai Y-J, Lin C-L, Chang Y-J, Lin M-C, Lee S-T, Sung F-C, et al. Spinal cord injury increases the risk of type 2 diabetes: a population-based cohort study. Spine J. (2014) 14:1957–64. doi: 10.1016/j.spinee.2013.12.011
66. Cragg JJ, Noonan VK, Dvorak M, Krassioukov A, Mancini GBJ, Borisoff JF. Spinal cord injury and type 2 diabetes: results from a population health survey. Neurology. (2013) 81:1864–8. doi: 10.1212/01.wnl.0000436074.98534.6e
67. Yarar-Fisher C, Bickel CS, Kelly NA, Stec MJ, Windham ST, McLain AB, et al. Heightened TWEAK-NF-κB signaling and inflammation-associated fibrosis in paralyzed muscles of men with chronic spinal cord injury. Am J Physiol Endocrinol Metab. (2016) 310:E754–761. doi: 10.1152/ajpendo.00240.2015
68. Savikj M, Kostovski E, Lundell LS, Iversen PO, Massart J, Widegren U. Altered oxidative stress and antioxidant defence in skeletal muscle during the first year following spinal cord injury. Physiol Rep. (2019) 7:e14218. doi: 10.14814/phy2.14218
69. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. (2018) 15:505–22. doi: 10.1038/s41569-018-0064-2
70. Lin S, Zhou Z, Zhao H, Xu C, Guo Y, Gao S, et al. TNF promotes M1 polarization through mitochondrial metabolism in injured spinal cord. Free Radic Biol Med. (2021) 172:622–32. doi: 10.1016/j.freeradbiomed.2021.07.014
71. Stratos I, Behrendt A-K, Anselm C, Gonzalez A, Mittlmeier T, Vollmar B. Inhibition of TNF-α restores muscle force, inhibits inflammation, and reduces apoptosis of traumatized skeletal muscles. Cells. (2022) 11:397. doi: 10.3390/cells11152397
72. Howard EE, Pasiakos SM, Blesso CN, Fussell MA, Rodriguez NR. Divergent roles of inflammation in skeletal muscle recovery from injury. Front Physiol. (2020) 11:87. doi: 10.3389/fphys.2020.00087
73. Meijboom KE, Sutton ER, McCallion E, McFall E, Anthony D, Edwards B, et al. Dysregulation of tweak and Fn14 in skeletal muscle of spinal muscular atrophy mice. Skelet Muscle. (2022) 12:18. doi: 10.1186/s13395-022-00301-z
74. Gensel J C, Zhang B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. (2015) 1619:1–11. doi: 10.1016/j.brainres.2014.12.045
75. Drasites K, Shams R, Zaman V, Matzelle D, Shields D, Garner D, et al. Pathophysiology, biomarkers, and therapeutic modalities associated with skeletal muscle loss following spinal cord injury. Brain Sci. (2020) 10:933. doi: 10.3390/brainsci10120933
76. Paulson TAW, Goosey-Tolfrey VL, Lenton JP, Leicht CA, Bishop NC. Spinal cord injury level and the circulating cytokine response to strenuous exercise. Med Sci Sports Exerc. (2013) 45:1649–55. doi: 10.1249/MSS.0b013e31828f9bbb
77. Qin W, Bauman W A, Cardozo C. Bone and muscle loss after spinal cord injury: organ interactions. Ann N Y Acad Sci. (2010) 1211:66–84. doi: 10.1111/j.1749-6632.2010.05806.x
78. Gorgey A S, Gater D R. Insulin growth factors may explain relationship between spasticity and skeletal muscle size in men with spinal cord injury. J Rehabil Res Dev. (2012) 49:373–80. doi: 10.1682/JRRD.2011.04.0076
79. Aravena J, Abrigo J, Gonzalez F, Aguirre F, Gonzalez A, Simon F. Angiotensin (1-7) decreases myostatin-induced Nf-κb signaling and skeletal muscle atrophy. Int J Mol Sci. (2020) 21:1167. doi: 10.3390/ijms21031167
80. Léger B, Senese R, Al-Khodairy AW, Dériaz O, Gobelet C, Giacobino JP, et al. Atrogin-1, MuRF1, and FoXO, as well as phosphorylated GSK-3beta and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord-injured patients. Muscle Nerve. (2009) 40:69–78. doi: 10.1002/mus.21293
81. Zhu Z, Wang X, Song Z, Zuo X, Ma Y, Zhang Z, et al. Photobiomodulation promotes repair following spinal cord injury by restoring neuronal mitochondrial bioenergetics via AMPK/PGC-1α/TFAM pathway. Front Pharmacol. (2022) 13:991421. doi: 10.3389/fphar.2022.991421
82. Invernizzi M, de Sire A, Renò F, Cisari C, Runza L, Baricich A, et al. Spinal cord injury as a model of bone-muscle interactions: therapeutic implications from in vitro and in vivo studies. Front Endocrinol. (2020) 11:204. doi: 10.3389/fendo.2020.00204
83. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. (2003) 3:745–56. doi: 10.1038/nri1184
84. Ren H, Chen X, Tian M, Zhou J, Ouyang H, Zhang Z. Regulation of inflammatory cytokines for spinal cord injury repair through local delivery of therapeutic agents. Adv Sci. (2018) 5:1800529. doi: 10.1002/advs.201800529
85. Tuttle CSL, Thang LAN, Maier AB. Markers of inflammation and their association with muscle strength and mass: a systematic review and meta-analysis. Ageing Res Rev. (2020) 64:101185. doi: 10.1016/j.arr.2020.101185
86. Yarar-Fisher C, Bickel CS, Windham ST, McLain AB, Bamman MM. Skeletal muscle signaling associated with impaired glucose tolerance in spinal cord-injured men and the effects of contractile activity. J Appl Physiol. (2013) 115:756–64. doi: 10.1152/japplphysiol.00122.2013
87. Huang H, Xue J, Zheng J, Tian H, Fang Y, Wang W, et al. Bioinformatic analysis of the gene expression profile in muscle atrophy after spinal cord injury. Sci Rep. (2021) 11:21903. doi: 10.1038/s41598-021-01302-6
88. Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. (2006) 25:409–16. doi: 10.1007/s10555-006-9005-3
89. Levitt DE, Yeh AY, Prendergast MJ, Jr RGB, Adler KA, Cook G, et al. Chronic alcohol dysregulates skeletal muscle myogenic gene expression after hind limb immobilization in female rats. Biomolecules. (2020) 10:441. doi: 10.3390/biom10030441
90. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
91. Rosety-Rodriguez M, Camacho A, Rosety I, Fornieles G, Rosety MA, Diaz AJ, et al. Low-grade systemic inflammation and leptin levels were improved by arm cranking exercise in adults with chronic spinal cord injury. Arch Phys Med Rehabil. (2014) 95:297–302. doi: 10.1016/j.apmr.2013.08.246
92. Griffin L, Decker MJ, Hwang JY, Wang B, Kitchen K, Ding Z, et al. Functional electrical stimulation cycling improves body composition, metabolic and neural factors in persons with spinal cord injury. J Electromyogr Kinesiol. (2009) 19:614–22. doi: 10.1016/j.jelekin.2008.03.002
93. Sato S, Ogura Y, Kumar A. TWEAK/Fn14 signaling axis mediates skeletal muscle atrophy and metabolic dysfunction. Front Immunol. (2014) 5:18. doi: 10.3389/fimmu.2014.00018
94. Pascoe AL, Johnston AJ, Murphy RM. Controversies in TWEAK-Fn14 signaling in skeletal muscle atrophy and regeneration. Cell Mol Life Sci. (2020) 77:3369–81. doi: 10.1007/s00018-020-03495-x
95. Sato S, Ogura Y, Mishra V, Shin J, Bhatnagar S, Hill BG, et al. TWEAK promotes exercise intolerance by decreasing skeletal muscle oxidative phosphorylation capacity. Skelet Muscle. (2013) 3:18. doi: 10.1186/2044-5040-3-18
96. Vince JE, Chau D, Callus B. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol. (2008) 182:171–84. doi: 10.1083/jcb.200801010
97. Luo G, Hershko DD, Robb BW, Wray CJ, Hasselgren PO. IL-1beta stimulates IL-6 production in cultured skeletal muscle cells through activation of MAP kinase signaling pathway and NF-kappa B. Am J Physiol Regul Integr Comp Physiol. (2003) 284:R1249–1254. doi: 10.1152/ajpregu.00490.2002
98. Tsuchiya M, Sekiai S, Hatakeyama H, Koide M, Chaweewannakorn C, Yaoita F, et al. Neutrophils provide a favorable IL-1-mediated immunometabolic niche that primes GLUT4 translocation and performance in skeletal muscles. Cell Rep. (2018) 23:2354–64. doi: 10.1016/j.celrep.2018.04.067
99. Tseng HW, Kulina I, Girard D, Gueguen J, Vaquette C, Salga M, et al. Interleukin-1 is overexpressed in injured muscles following spinal cord injury and promotes neurogenic heterotopic ossification. J Bone Miner Res. (2022) 37:531–46. doi: 10.1002/jbmr.4482
100. Zhang X, Xu H, Zhu L, Huang D, Kong L, Wang Z, et al. Thoracic Jia-Ji electro-acupuncture mitigates low skeletal muscle atrophy and improves motor function recovery following thoracic spinal cord injury in rats. Am J Transl Res. (2022) 14:8103–16. Available online at: https://pubmed.ncbi.nlm.nih.gov/36505337/
101. Kouda K, Furusawa K, Sugiyama H, Sumiya T, Ito T, Tajima F, et al. Does 20-min arm crank ergometer exercise increase plasma interleukin-6 in individuals with cervical spinal cord injury? Eur J Appl Physiol. (2012) 112:597–604. doi: 10.1007/s00421-011-2004-2
102. Haddad F, Zaldivar F, Cooper DM. IL-6-induced skeletal muscle atrophy. J Appl Physiol. (2005) 98:911–7. doi: 10.1152/japplphysiol.01026.2004
103. Alvarez AM, DeOcesano-Pereira C, Teixeira C, Moreira V. IL-1β and TNF-α modulation of proliferated and committed myoblasts: IL-6 and COX-2-derived prostaglandins as key actors in the mechanisms involved. Cells. (2020) 9:205. doi: 10.3390/cells9092005
104. Otis JS, Niccoli S, Hawdon N, Sarvas JL, Frye MA, Chicco AJ, et al. Pro-inflammatory mediation of myoblast proliferation. PLoS ONE. (2014) 9:e92363. doi: 10.1371/journal.pone.0092363
105. Chaweewannakorn C, Tsuchiya M, Koide M, Hatakeyama H, Tanaka Y, Yoshida S, et al. Roles of IL-1α/β in regeneration of cardiotoxin-injured muscle and satellite cell function. Am J Physiol Regul Integr Comp Physiol. (2018) 315:R90–r103. doi: 10.1152/ajpregu.00310.2017
106. Silva KAS, Dong J, Dong Y, Dong Y, Schor N, Tweardy DJ, et al. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J Biol Chem. (2015) 290:11177–87. doi: 10.1074/jbc.M115.641514
107. Peris-Moreno D, Cussonneau L, Combaret L, Polge C, Taillandier D. Ubiquitin ligases at the heart of skeletal muscle atrophy control. Molecules. (2021) 26:407. doi: 10.3390/molecules26020407
108. Peake JM, Gatta PD, Suzuki K, Nieman DC. Cytokine expression and secretion by skeletal muscle cells: regulatory mechanisms and exercise effects. Exerc Immunol Rev. (2015) 21:8–25. Available online at: https://pubmed.ncbi.nlm.nih.gov/25826432/
109. Ogawa T, Nakamura T, Banno M, Sasaki Y, Umemoto Y, Kouda K, et al. Elevation of interleukin-6 and attenuation of tumor necrosis factor-α during wheelchair half marathon in athletes with cervical spinal cord injuries. Spinal Cord. (2014) 52:601–5. doi: 10.1038/sc.2014.88
110. Sandri M, Lin J, Handschin C, et al. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A. (2006) 103:16260–5. doi: 10.1073/pnas.0607795103
111. Petrie MA, Sharma A, Taylor EB, Suneja M, Shields RK. Impact of short- and long-term electrically induced muscle exercise on gene signaling pathways, gene expression, and PGC1a methylation in men with spinal cord injury. Physiol Genomics. (2020) 52:71–80. doi: 10.1152/physiolgenomics.00064.2019
112. Kang C, Li L. Role of PGC-1α signaling in skeletal muscle health and disease. Ann N Y Acad Sci. (2012) 1271:110–7. doi: 10.1111/j.1749-6632.2012.06738.x
113. Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ, et al. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol. (2013) 45:2288–301. doi: 10.1016/j.biocel.2013.06.024
114. Scalabrin M, Pollock N, Staunton CA, Brooks SV, McArdle A, Jackson MJ, et al. Redox responses in skeletal muscle following denervation. Redox Biol. (2019) 26:101294. doi: 10.1016/j.redox.2019.101294
115. Wang D, Yang Y, Zou X, Zhang J, Zheng Z, Wang Z. Antioxidant apigenin relieves age-related muscle atrophy by inhibiting oxidative stress and hyperactive mitophagy and apoptosis in skeletal muscle of mice. J Gerontol A Biol Sci Med Sci. (2020) 75:2081–8. doi: 10.1093/gerona/glaa214
116. Ji LL, Yeo D, Kang C, Zhang T. The role of mitochondria in redox signaling of muscle homeostasis. J Sport Health Sci. (2020) 9:386–93. doi: 10.1016/j.jshs.2020.01.001
117. Liu T, Sun L, Zhang Y, Wang Y, Zheng J. Imbalanced GSH/ROS and sequential cell death. J Biochem Mol Toxicol. (2022) 36:e22942. doi: 10.1002/jbt.22942
118. Diaz-Vivancos P, de Simone A, Kiddle G, Foyer CH. Glutathione–linking cell proliferation to oxidative stress. Free Radic Biol Med. (2015) 89:1154–64. doi: 10.1016/j.freeradbiomed.2015.09.023
119. Rosales-Antequera C, Viscor G, Araneda OF. Inflammation and oxidative stress as common mechanisms of pulmonary, autonomic and musculoskeletal dysfunction after spinal cord injury. Biology. (2022) 11:550. doi: 10.3390/biology11040550
120. Raiteri T, Zaggia I, Reano S, Scircoli A, Salvadori L, Prodam F, et al. The atrophic effect of 1,25(OH)(2) vitamin D(3) (Calcitriol) on C2C12 myotubes depends on oxidative stress. Antioxidants. (2021) 10:980. doi: 10.3390/antiox10121980
121. Yang BA, Castor-Macias J, Fraczek P, Cornett A, Brown LA, Kim M, et al. Sestrins regulate muscle stem cell metabolic homeostasis. Stem Cell Reports. (2021) 16:2078–88. doi: 10.1016/j.stemcr.2021.07.014
122. Li Y, Zhang J, Zhou K, Xie L, Xiang G, Fang M, et al. Elevating sestrin2 attenuates endoplasmic reticulum stress and improves functional recovery through autophagy activation after spinal cord injury. Cell Biol Toxicol. (2021) 37:401–19. doi: 10.1007/s10565-020-09550-4
123. Gombos Z, Koltai E, Torma F, Bakonyi P, Kolonics A, Aczel D, et al. Hypertrophy of rat skeletal muscle is associated with increased SIRT1/Akt/mTOR/S6 and suppressed sestrin2/SIRT3/FOXO1 levels. Int J Mol Sci. (2021) 22:14. doi: 10.3390/ijms22147588
124. Lee JH, Cho US. Karin M. Sestrin regulation of TORC1: is sestrin a leucine sensor? Sci Signal. (2016) 9:re5. doi: 10.1126/scisignal.aaf2885
125. Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. (2010) 327:1223–8. doi: 10.1126/science.1182228
126. Ho A, Cho C-S, Namkoong S, Cho U-S, Lee JH. biochemical basis of sestrin physiological activities. Trends Biochem Sci. (2016) 41:621–32. doi: 10.1016/j.tibs.2016.04.005
127. Segalés J, Perdiguero E, Serrano AL, Sousa-Victor P, Ortet L, Jardí M, et al. Sestrin prevents atrophy of disused and aging muscles by integrating anabolic and catabolic signals. Nat Commun. (2020) 11:189. doi: 10.1038/s41467-019-13832-9
128. Chen Y, Huang T, Yu Z, Yu Q, Wang Y, Hu J, et al. The functions and roles of sestrins in regulating human diseases. Cell Mol Biol Lett. (2022) 27:2. doi: 10.1186/s11658-021-00302-8
129. Yan J, Herzog JW, Tsang K, Brennan CA, Bower MA, Garrett WS, et al. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc Natl Acad Sci U S A. (2016) 113:E7554–63. doi: 10.1073/pnas.1607235113
130. Day CS, Riano F, Tomaino MM, Buranatanitkit B, Somogyi G, Sotereanos D, et al. Growth factor may decrease muscle atrophy secondary to denervation. J Reconstr Microsurg. (2001) 17:51–7. doi: 10.1055/s-2001-12689
131. Yan B, Zeng C, Chen Y, Huang M, Yao N, Zhang J, et al. Mechanical stress-induced IGF-1 facilitates col-I and col-III synthesis via the IGF-1R/AKT/mTORC1 signaling pathway. Stem Cells Int. (2021) 2021:5553676. doi: 10.1155/2021/5553676
132. Bianchi VE, Locatelli V, Rizzi L. Neurotrophic and neuroregenerative effects of GH/IGF1. Int J Mol Sci. (2017) 18:441. doi: 10.3390/ijms18112441
133. Cheng R-D, Ren W, Sun P, Tian L, Zhang L, Zhang J, et al. Spinal cord injury causes insulin resistance associated with PI3K signaling pathway in hypothalamus. Neurochem Int. (2020) 140:104839. doi: 10.1016/j.neuint.2020.104839
134. Martín AI, Priego T, Moreno-Ruperez Á. IGF-1 and IGFBP-3 in Inflammatory cachexia. Int J Mol Sci. (2021) 22:469. doi: 10.3390/ijms22179469
135. Kostovski E, Iversen PO, Birkeland K, Torjesen PA, Hjeltnes N. Decreased levels of testosterone and gonadotrophins in men with long-standing tetraplegia. Spinal Cord. (2008) 46:559–64. doi: 10.1038/sc.2008.3
136. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. (2015) 14:58–74. doi: 10.1038/nrd4467
137. Bodine S C, Stitt T N, Gonzalez M. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. (2001) 3:1014–9. doi: 10.1038/ncb1101-1014
138. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. (2001) 3:1009–13. doi: 10.1038/ncb1101-1009
139. Feng L, Li B, Xi Y, Cai M, Tian Z. Aerobic exercise and resistance exercise alleviate skeletal muscle atrophy through IGF-1/IGF-1R-PI3K/Akt pathway in mice with myocardial infarction. Am J Physiol Cell Physiol. (2022) 322:C164–c176. doi: 10.1152/ajpcell.00344.2021
140. Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. (2019) 12:71. doi: 10.1186/s13045-019-0754-1
141. Ham DJ, Lynch GS, Koopman R. Amino acid sensing and activation of mechanistic target of rapamycin complex 1: implications for skeletal muscle. Curr Opin Clin Nutr Metab Care. (2016) 19:67–73. doi: 10.1097/MCO.0000000000000240
142. Kinoshita H, Orita S, Inage K, Yamauchi K, Abe K, Inoue M, et al. Skeletal muscle cell oxidative stress as a possible therapeutic target in a denervation-induced experimental sarcopenic model. Spine. (2019) 44:E446–55. doi: 10.1097/BRS.0000000000002891
143. Valenzuela PL, Morales JS, Emanuele E, Pareja-Galeano H, Lucia A. Supplements with purported effects on muscle mass and strength. Eur J Nutr. (2019) 58:2983–3008. doi: 10.1007/s00394-018-1882-z
144. Kobayashi H. Amino acid nutrition in the prevention and treatment of sarcopenia. Yakugaku Zasshi. (2018) 138:1277–83. doi: 10.1248/yakushi.18-00091-4
145. Owens D.J. Nutritional support to counteract muscle atrophy. Adv Exp Med Biol. (2018) 1088:483–95. doi: 10.1007/978-981-13-1435-3_22
146. Peng M, Yin N, Li M O. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell. (2014) 159:122–33. doi: 10.1016/j.cell.2014.08.038
147. Wang X, Yue P, Chan C-B, Ye K, Ueda T, Watanabe-Fukunaga R, et al. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol. (2007) 27:7405–13. doi: 10.1128/MCB.00760-07
148. Ding Y, Chen Q. mTOR pathway: A potential therapeutic target for spinal cord injury. Biomed Pharmacother. (2022) 145:112430. doi: 10.1016/j.biopha.2021.112430
149. Cohen S. Role of calpains in promoting desmin filaments depolymerization and muscle atrophy. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118788. doi: 10.1016/j.bbamcr.2020.118788
150. Cardaci TD, Machek SB, Wilburn DT, Hwang PS, Willoughby DS. Ubiquitin proteasome system activity is suppressed by curcumin following exercise-induced muscle damage in human skeletal muscle. J Am Coll Nutr. (2021) 40:401–11. doi: 10.1080/07315724.2020.1783721
151. Cardozo C.P. Muscle biology after spinal cord injury: Recent advances and future challenges. Acta Physiol (Oxf). (2018) 223:e13073. doi: 10.1111/apha.13073
152. Jones SW, Hill RJ, Krasney PA, O'Conner B, Peirce N, Greenhaff PL. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J. (2004) 18:1025–7. doi: 10.1096/fj.03-1228fje
153. Gong B, Radulovic M, Figueiredo-Pereira ME, Cardozo C. The ubiquitin-proteasome system: potential therapeutic targets for alzheimer's disease and spinal cord injury. Front Mol Neurosci. (2016) 9:4. doi: 10.3389/fnmol.2016.00004
154. Sakuma K, Aoi W, Yamaguchi A. Molecular mechanism of sarcopenia and cachexia: recent research advances. Pflugers Arch. (2017) 469:573–91. doi: 10.1007/s00424-016-1933-3
155. Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab. (2014) 307:E469–484. doi: 10.1152/ajpendo.00204.2014
156. Seaborne RA, Hughes DC, Turner DC, Owens DJ, Baehr LM, Gorski P, et al. UBR5 is a novel E3 ubiquitin ligase involved in skeletal muscle hypertrophy and recovery from atrophy. J Physiol. (2019) 597:3727–49. doi: 10.1113/JP278073
157. Momeni HR. Role of calpain in apoptosis. Cell J. (2011) 13:65–72. Available online at: https://pubmed.ncbi.nlm.nih.gov/23507938/
158. Ray SK, Hogan EL, Banik NL. Calpain in the pathophysiology of spinal cord injury: neuroprotection with calpain inhibitors. Brain Res Brain Res Rev. (2003) 42:169–85. doi: 10.1016/S0165-0173(03)00152-8
159. Akdemir O, Uçankale M, Karaoglan A, Barut S, Sagmanligil A, Bilguvar K, et al. Therapeutic efficacy of SJA6017, a calpain inhibitor, in rat spinal cord injury. J Clin Neurosci. (2008) 15:1130–6. doi: 10.1016/j.jocn.2007.08.011
160. Fry CS, Drummond MJ, Lujan HL, DiCarlo SE, Rasmussen BB. Paraplegia increases skeletal muscle autophagy. Muscle Nerve. (2012) 46:793–8. doi: 10.1002/mus.23423
161. Lundell LS, Savikj M, Kostovski E, Iversen PO, Zierath JR, Krook A, et al. Protein translation, proteolysis and autophagy in human skeletal muscle atrophy after spinal cord injury. Acta Physiol. (2018) 223:e13051. doi: 10.1111/apha.13051
162. Fritzen AM, Madsen AB, Kleinert M, Treebak JT, Lundsgaard A-M, Jensen TE, et al. Regulation of autophagy in human skeletal muscle: effects of exercise, exercise training and insulin stimulation. J Physiol. (2016) 594:745–61. doi: 10.1113/JP271405
163. Meijer AJ, Codogno P. AMP-Activated Protein Kinase and Autophagy. New York, NY: Taylor & Francis. (2007).
164. Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. (2006) 281:34870–9. doi: 10.1074/jbc.M605488200
165. Sartori R, Romanello V, Sandri M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun. (2021) 12:330. doi: 10.1038/s41467-020-20123-1
166. Invernizzi M, Carda S, Rizzi M, Grana E, Squarzanti DF, Cisari C, et al. Evaluation of serum myostatin and sclerostin levels in chronic spinal cord injured patients. Spinal Cord. (2015) 53:615–20. doi: 10.1038/sc.2015.61
167. Sharma M, McFarlane C, Kambadur R. Myostatin: expanding horizons. IUBMB Life. (2015) 67:589–600. doi: 10.1002/iub.1392
168. Lee SJ. Targeting the myostatin signaling pathway to treat muscle loss and metabolic dysfunction. J Clin Invest. (2021) 131:72. doi: 10.1172/JCI148372
169. Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, et al. BMP signaling controls muscle mass. Nat Genet. (2013) 45:1309–18. doi: 10.1038/ng.2772
170. Han D-S, Hsiao M-Y, Wang T-G, Chen S-Y, Yang W-S. Association of serum myokines and aerobic exercise training in patients with spinal cord injury: an observational study. BMC Neurol. (2016) 16:142. doi: 10.1186/s12883-016-0661-9
171. Sartori R, Hagg A, Zampieri S, Armani A, Winbanks CE, Viana LR, et al. Perturbed BMP signaling and denervation promote muscle wasting in cancer cachexia. Sci Transl Med. (2021) 13:605. doi: 10.1126/scitranslmed.aay9592
172. Lowery J W, Rosen V. The BMP Pathway and Its Inhibitors in the Skeleton. Physiol Rev. (2018) 98:2431–52. doi: 10.1152/physrev.00028.2017
173. Al-Sammarraie N, Ray S K. Bone morphogenic protein signaling in spinal cord injury. Neuroimmunol Neuroinflamm. (2021) 8:53–63. doi: 10.20517/2347-8659.2020.34
174. Xiao Q, Du Y, Wu W, Yip HK. Bone morphogenetic proteins mediate cellular response and, together with Noggin, regulate astrocyte differentiation after spinal cord injury. Exp Neurol. (2010) 221:353–66. doi: 10.1016/j.expneurol.2009.12.003
175. Osses N, Henríquez J P. Bone morphogenetic protein signaling in vertebrate motor neurons and neuromuscular communication. Front Cell Neurosci. (2014) 8:453. doi: 10.3389/fncel.2014.00453
176. Behrman AL, Ardolino EM, Harkema SJ. Activity-based therapy: from basic science to clinical application for recovery after spinal cord injury. J Neurol Phys Ther. (2017) 41:S39–45. doi: 10.1097/NPT.0000000000000184
177. Rademeyer HJ, Gauthier C, Zariffa J, Walden K, Jeji T, McCullum S, et al. Using activity-based therapy for individuals with spinal cord injury or disease: interviews with physical and occupational therapists in rehabilitation hospitals. J Spinal Cord Med. (2022) 30:1–11. doi: 10.1080/10790268.2022.2039855
178. Dolbow DR, Gorgey AS, Recio AC, Stiens SA, Curry AC, Sadowsky CL, et al. Activity-based restorative therapies after spinal cord injury: inter-institutional conceptions and perceptions. Aging Dis. (2015) 6:254–61. doi: 10.14336/AD.2014.1105
179. Sutor TW, Kura J, Mattingly AJ, Otzel DM, Yarrow JF. The effects of exercise and activity-based physical therapy on bone after spinal cord injury. Int J Mol Sci. (2022) 23:608. doi: 10.3390/ijms23020608
180. de Oliveira CQ, Refshauge K, Middleton J, de Jong L, Davis GM. Effects of activity-based therapy interventions on mobility, independence, and quality of life for people with spinal cord injuries: a systematic review and meta-analysis. J Neurotrauma. (2017) 34:1726–43. doi: 10.1089/neu.2016.4558
181. Grønfeldt BM, Lindberg Nielsen J, Mieritz RM, Lund H, Aagaard P. Effect of blood-flow restricted vs heavy-load strength training on muscle strength: Systematic review and meta-analysis. Scand J Med Sci Sports. (2020) 30:837–48. doi: 10.1111/sms.13632
182. Grgic J, Schoenfeld BJ, Orazem J, Sabol F. Effects of resistance training performed to repetition failure or non-failure on muscular strength and hypertrophy: a systematic review and meta-analysis. J Sport Health Sci. (2022) 11:202–11. doi: 10.1016/j.jshs.2021.01.007
183. García AM, Serrano-Muñoz D, Taylor J, Avendaño-Coy J, Gómez-Soriano J. Transcutaneous spinal cord stimulation and motor rehabilitation in spinal cord injury: a systematic review. Neurorehabil Neural Repair. (2020) 34:3–12. doi: 10.1177/1545968319893298
184. Lopez-Seoane J, Martinez-Ferran M, Romero-Morales C, Pareja-Galeano H. N-3 PUFA as an ergogenic supplement modulating muscle hypertrophy and strength: a systematic review. Crit Rev Food Sci Nutr. (2021) 62:1–21. doi: 10.1080/10408398.2021.1939262
185. López-Seoane J, Jiménez SL, Coso JD, Pareja-Galeano H. Muscle hypertrophy induced by N-3 PUFA supplementation in absence of exercise: a systematic review of randomized controlled trials. Crit Rev Food Sci Nutr. (2022) 28:1–11. doi: 10.1080/10408398.2022.2034734
186. Farkas GJ, Sneij A, Gater DR. Dietetics after spinal cord injury: current evidence and future perspectives top spinal cord. Inj Rehabil. (2021) 27:100–8. doi: 10.46292/sci20-00031
187. Cantoria MJ, See PA, Singh H, de Leon RD. Adaptations in glutamate and glycine content within the lumbar spinal cord are associated with the generation of novel gait patterns in rats following neonatal spinal cord transection. J Neurosci. (2011) 31:18598–605. doi: 10.1523/JNEUROSCI.3499-11.2011
188. Miyazato M, Sugaya K, Nishijima S, Ashitomi K, Morozumi M, Ogawa Y. Dietary glycine inhibits bladder activity in normal rats and rats with spinal cord injury. J Urol. (2005) 173:314–7. doi: 10.1097/01.ju.0000141579.91638.a3
189. Koopman R, Caldow MK, Ham DJ, Lynch GS. Glycine metabolism in skeletal muscle: implications for metabolic homeostasis. Curr. Metab. Care. (2017) 20:237–42. doi: 10.1097/MCO.0000000000000383
190. Caldow MK, Ham DJ, Trieu J, Chung JD, Lynch GS, Koopman R. Glycine protects muscle cells from wasting in vitro via MTORC1 signaling. Front Nutr. (2019) 6:172. doi: 10.3389/fnut.2019.00172
191. Ham DJ, Murphy KT, Chee A, Lynch GS, Koopman R. Glycine administration attenuates skeletal muscle wasting in a mouse model of cancer cachexia. Clin Nutr. (2014) 33:448–58. doi: 10.1016/j.clnu.2013.06.013
192. Ham DJ, Caldow MK, Chhen V, Chee A, Wang X, Proud CG, et al. Glycine restores the anabolic response to leucine in a mouse model of acute inflammation. Am J Physiol Endocrinol Metab. (2016) 310:E970–981. doi: 10.1152/ajpendo.00468.2015
193. Wang W, Wu Z, Dai Z, Yang Y, Wang J, Wu G. Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids. (2013) 45:463–77. doi: 10.1007/s00726-013-1493-1
194. Ceglia L. Vitamin D and its role in skeletal muscle. Curr Opin Clin Nutr Metab Care. (2009) 12:628–33. doi: 10.1097/MCO.0b013e328331c707
195. Girgis CM, Clifton-Bligh RJ, Hamrick MW, Holick MF, Gunton JE. The roles of vitamin D in skeletal muscle: form, function, and metabolism. Endocr Rev. (2013) 34:33–83. doi: 10.1210/er.2012-1012
196. Amorim S, Teixeira VH, Corredeira R, Cunha M, Maia B, Margalho P, et al. Creatine or vitamin D supplementation in individuals with a spinal cord injury undergoing resistance training: a double-blinded, randomized pilot trial. J Spinal Cord Med. (2018) 41:471–8. doi: 10.1080/10790268.2017.1372058
Keywords: spinal cord injury, skeletal muscle atrophy, denervation, immobilization, inflammation, oxidative stress
Citation: Xu X, Talifu Z, Zhang C-J, Gao F, Ke H, Pan Y-Z, Gong H, Du H-Y, Yu Y, Jing Y-L, Du L-J, Li J-J and Yang D-G (2023) Mechanism of skeletal muscle atrophy after spinal cord injury: A narrative review. Front. Nutr. 10:1099143. doi: 10.3389/fnut.2023.1099143
Received: 15 November 2022; Accepted: 20 February 2023;
Published: 03 March 2023.
Edited by:
Tao Tong, China Agricultural University, ChinaReviewed by:
Joseph S. Marino, University of North Carolina at Charlotte, United StatesHelong Quan, Northeast Normal University, China
Copyright © 2023 Xu, Talifu, Zhang, Gao, Ke, Pan, Gong, Du, Yu, Jing, Du, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-Jun Li, Y3JyY2xpampAMTYzLmNvbQ==; De-Gang Yang, eXp5ZGcyMDA2QDEyNi5jb20=