 Manisha Aswal
Manisha Aswal Neelja Singhal
Neelja Singhal Manish Kumar
Manish Kumar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Nutr. , 04 April 2023
Sec. Nutrition and Microbes
Volume 10 - 2023 | https://doi.org/10.3389/fnut.2023.1082566
This article is part of the Research Topic Emerging Perspectives on Probiotics, Prebiotics, and Synbiotics for Prevention and Management of Chronic Disease View all 9 articles
Hypercholesterolemia is a major risk factor for cardiovascular diseases (CVDs). Chemotherapeutic agents for CVDs exhibit several side effects. Specific probiotics with hypocholesterolemic effects can be safe and effective alternatives to chemotherapeutics. Here, we have analyzed and compared the genome of a novel rhizospheric Enterococcus faecium LR13 cholesterol-assimilating probiotic with other probiotic/pathogenic E. faecium strains to discern genetic factors underlying probiotic efficacy and cholesterol-assimilation. Genomic analyses of E. faecium probiotic strains revealed that LR13 and WEFA23 (cholesterol-assimilating probiotics) harbored 21 unique proteins absent in non-cholesterol-assimilating probiotics. Of these, 14 proteins could directly help in cholesterol-assimilation by producing short chain fatty acids, lipid (sterol) transport and membrane stabilization, and bile salt hydrolase activity. This suggests that cholesterol-assimilation is an intrinsic, strain-specific trait exhibited by probiotics with a specific genetic constitution. Moreover, the unique proteins identified in this study can serve as biomarkers for discerning/characterizing cholesterol-assimilating probiotics as novel biotherapeutics against CVDs.
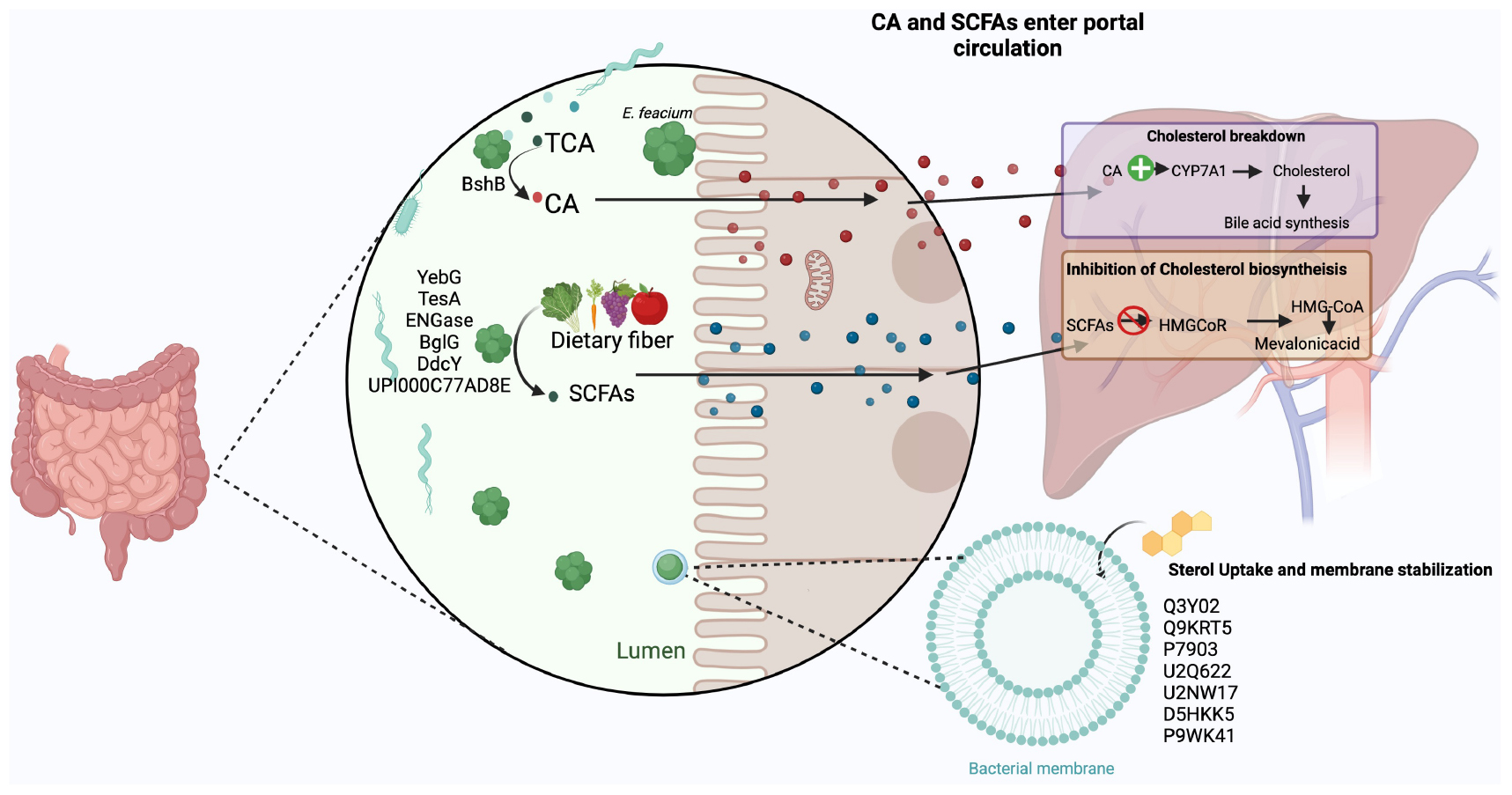
Graphical Abstract. The three probable mechanisms by which the unique proteins discerned in hypo-cholesterolemic E. faecium LR13 and WEFA23 probiotic strains might aid in cholesterol-assimilation : (i) bile salt hydrolysis: bacterial bile salt hydrolases (BshB) convert taurine/glycine cholic acid (T/GCA) to cholic acid (CA) that aids in the activation of cholesterol 7α-hydroxylase (Cyp7a) in the liver resulting in degradation of cholesterol into bile acids (110), (ii) production of short chain fatty acids (SCFAs): the bacterial proteins YebG, TesA, ENGase, BglG, DdcY, and UPI000C77AD8E are involved in the uptake and breakdown of dietary fibers into short chain fatty acids (SCFAs) like propionate, acetate and butyrate. In the liver, these inhibit HMG-CoA reductase enzyme (3-hydroxy-3-methylglutaryl-CoA) which converts HMG-CoA to mevalonic acid resulting in inhibition of cholesterol biosynthesis, and (iii) lipid (sterol) transport, membrane stabilization and binding of cholesterol to the bacterial cell walls: bacterial proteins Q3Y02, Q9KRT5, P7903, U2Q622, U2NW17, D5HKK5, and P9WK41 aid in uptake of sterols (lipids) from the intestinal lumen and incorporation in their cell membranes/walls. Created by BioRender.com.
Elevated serum cholesterol is a major risk factor for cardiovascular diseases (CVDs). Though statins and other drugs are effective in lowering serum cholesterol, they have several side effects. Thus, agents which can exhibit hypocholesterolemic effects without side effects should be discovered. Recently, probiotics have been promoted as important food additives/supplements for improving human health. According to the World Health Organization (WHO) and Food and Agricultural Organization (WHO/FAO) probiotics may be defined as, “live microbes which when administered in a certain amount deliver several health benefits on the host” (1). Probiotics are mainly associated with the maintenance of a healthy gastrointestinal tract (GIT) by restoring the microbial balance of the host gut (2). However, several studies have reported that probiotics might also confer some other health benefits, like prevention of oral/mouth diseases, bacterial/fungal vaginal infections, cancer, osteoporosis, type 2 diabetes, CVDs, antibiotics induced diarrhea, irritable bowel syndrome (IBS), etc. (2, 3). Various researchers have identified many species/strains of the genera Lactobacillus, Lactococcus, Levilactobacillus, Limosilactobacillus, Pediococcus, Streptococcus, Weissella, Enterococcus, etc. with probiotic potential and/or other health benefits (4).
Enterococcus faecium is a common commensal of the human GIT. E. faecium species is composed of both probiotic and pathogenic strains and reportedly due to genomic diversity and plasticity, E. faecium strains might exhibit probiotic or pathogenic traits (5–7). Some pathogenic strains of E. faecium carrying virulence and antibiotic resistance genes, especially vancomycin-resistant genes, were reported to be responsible for severe nosocomial outbreaks (8, 9). These E. faecium strains were known as vancomycin-resistant enterococci (VRE). Also, these E. faecium (VRE) strains were categorized in the WHO “Global Priority List of Antibiotic-resistant Bacteria to Guide Research, Discovery and Development of New Antibiotics” under priority 2 (high) category (10). On the other hand, many E. faecium strains were reportedly associated with health benefits such as regulation of gut microbiota, reduction of IBS symptoms, allergy and diarrhea (2, 11), reduction of obesity, immune system modulation (12), lowering of cholesterol-levels (13), etc. Many strains of E. faecium, E. faecium T110 (14), E. faecium 170M39 (15), E. faecium SP15 (16), E. faecium WEFA23 (17), E. faecium LR13 (18) have been reported as potential probiotics and, E. faecium SF68®, Enterococcus faecalis Symbioflor® 1, etc. (11, 19) are approved commercial probiotics. Reportedly, specific genomic differences were found to exist in the food-grade and nosocomial enterococci strains (20).
Most of the current probiotics available in Indian markets are non-indigenous strains whose efficacy is dubious (21, 22). Due to different genetic compositions and geographical variations, the microbiome of the Indian population is different from the microbiomes of the populations living in other parts of the world (23, 24). Indigenous probiotic strains isolated from animal or environmental niches specific to a particular geographical location might help in identifying population-specific probiotics which can confer better health benefits on the local population than non-indigenous strains.
In an earlier study we had reported a rhizospheric isolate of E. faecium LR13 which exhibited several probiotic attributes and hypo-cholesterolemic potential in vitro (18). Also, this strain was found to be devoid of virulence factors and antibiotic resistance genes which suggested its use as a potential probiotic candidate. In the present study we have performed de novo complete genome analysis of the indigenous E. faecium LR13 strain to unravel the genes/pathways associated with the probiotic traits. Since, mobile genetic elements (MGEs) such as plasmids, insertion sequences, prophages, pathogenicity islands, and transposons are reportedly involved in transmission of traits such as virulence, antimicrobial resistance (AMR), and probiotic attributes (25–29), MGEs were also characterized to understand if the AMR/virulence/probiotic traits depicted by E. faecium LR13 were intrinsic or acquired. Additionally, the genome sequence of E. faecium LR13 was compared with the genome sequences of other probiotic strains such as, E. faecium T110 (14), E. faecium 170M39 (15), E. faecium SP15 (16), and E. faecium WEFA23 (17) to understand its genomic relatedness with E. faecium probiotic strains isolated from other parts of the world. Also, the genome of E. faecium LR13 was compared with the genomes of pathogenic and non-pathogenic non-probiotic E. faecium strains to determine its genomic relatedness/differences with other E. faecium strains.
Enterococcus faecium LR13 preserved as glycerol stock (50% v/v) at −80°C in our laboratory was revived by incubating overnight in deMan, Rogosa and Sharpe (MRS) broth at 37°C, 200 rpm. Bacteria were grown up to the exponential phase (OD600 = 0.8) and harvested by centrifugation at 8,000 rpm for 8 min at 4°C. Genomic DNA was extracted using Nucleospin Microbial DNA kit (Takara Bio, San Jose, CA, USA) following the manufacturer’s instructions. DNA concentrations were determined using Qubit 4 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
The sequencing DNA library was prepared by QIASeq FX DNA Library Kit (Qiagen, USA). Quantitative and qualitative library QC was done by Qubit 4 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and tapestation 4150 (Agilent technologies, Santa Clara, CA, USA), respectively. The libraries were sequenced on Novaseq 6000 (Illumina, USA) by using 2 × 150 bp paired end sequencing chemistry. The quality of the raw reads was assessed using FastQC v0.11.8 (30) and BLAST against NCBI Nucleotide Database (NT) at e-value 1e−6. The reads were trimmed using Trimmomatic v0.39 (31) at default parameters to remove Illumina adaptors, low-quality bases and/or reads less than 36 bp. The trimmed short overlapped paired-end reads were merged using Flash (v1.2.11) (32) at default parameters to create longer reads (single-end). The merged single-end reads along with the remaining trimmed pair-end reads were used to perform de novo genome assembly using SPAdes v3.13.0 (33) and Unicycler v0.4.8 (34). From the assembled contigs, all contigs with ≤200 bp length were discarded using SeqKit v0.16.1 (35). The quality of the assembled genome was further assessed using QUAST v5.0.2 (36) and checkM v1.1.3 (37).
The assembled genome was annotated using prokaryotic genome annotation pipeline PGAP (2021-07-01) (38), Prokka v1.14.6 (39), KAAS server with BBH (bi-directional best hit) (40) and PATRIC RAST tool kit (RASTtk) v3.6.12 (41). The protein coding genes, tRNA and rRNA genes were predicted using Glimmer version v3.02 (42), tRNAscan-SE v2.0.9 (43), and RNAmmer v1.2 (44), respectively.
The presence of essential probiotic genes in E. faecium LR13 was confirmed by ascertaining the presence of genes essential for survival in the human gut such as dltA (D-alanine D-alanyl carrier protein ligase), dltB (teichoic acid D-alanyltransferase), copA (copper-importing P-type ATPase A), gadC (glutamate/gamma-aminobutyrate antiporter), dps (DNA protection during starvation), clpC (ATP-dependent Clp protease ATP-binding subunit ClpC), clpE (ATP-dependent Clp protease ATP-binding subunit ClpE), msrB (peptide methionine sulfoxide reductase), treC (tellurium resistance protein), and bsh (bile salt hydrolase) (14).
Presence of genes for desirable probiotic attributes such as biosynthesis of essential amino acids and vitamins was determined by complete genome classification of E. faecium LR13 using the PANTHER database (45) and mapping of biosynthetic pathways on the KEGG database (46). The presence of phosphoenolpyruvate-dependent sugar phosphotransferase (sugar PTS) genes such as, ptsG_1 (PTS system glucose-specific EIICB component), ptsG_2 (PTS system glucose-specific EIICB component), ptsG_3 (PTS system glucose-specific EIICB component), ptsH (phosphocarrier protein HPr), ptsI (phosphoenolpyruvate protein phosphotransferase), and ptsP (phosphoenolpyruvate protein phosphotransferase) (47) and biofilm formation such as, ebpB (endocarditis and biofilm-associated pili), srtC (Sortase C), efaA (endocarditis specific antigen), slrA (transcriptional regulator control initiation of biofilm formation), and bopD (biofilm on plastic surface/a putative sugar-binding transcriptional regulator) were also confirmed (48). The presence of antimicrobial peptides (AMPs) was checked by tBLASTn search against the Antimicrobial Peptide Database (APD3) (49) at e-value 100, alignment identity >85% and query coverage of 100%. The antimicrobial mechanisms of the AMPs were predicted using antiSMASH v5.0 (50) and BAGEL v4.0 (51) web servers.
The presence of MGEs was confirmed using the CRISPRFinder v4.2.20 (52). Prediction and annotation of the prophage sequences was done using the PHAST server1 (53) and of insertion sequence elements (ISs) was done using the server ISfinder2 (54). The Islandviewer tool was used to detect horizontal transfer of genes.
The presence of virulence factors was verified using the VFDB server3 (55) and of AMR genes using AMR databases such as CARD v3.1.4 (56), ResFinder v4.1 (57), and CBMAR (58). The presence of bacterial drug efflux pump genes was confirmed using BacEffluxPred (59).
The genome of E. faecium LR13 was compared with the genomes of other reported probiotics such as E. faecium T110, E. faecium 170M39, E. faecium SP15 E. faecium, and a cholesterol-lowering probiotic strain E. faecium WEFA23 which assimilated 85% cholesterol in vitro; (17) by in silico DNA–DNA Hybridization (DDH) using the web server Genome-to-Genome Distance Calculator (version 2.1)4 (60). The average nucleotide identity (ANI) was calculated using the ANI Calculator at EZ-Biocloud server (61). The proteomes were compared by clustering and annotating protein sequences using OrthoFinder tool (62) and webMGA server (63), respectively. The clustered ortholog proteins were further divided into three categories using in-house Perl script as: (i) core (protein sequences present in all the five probiotic strains), (ii) accessory (protein sequences present in different strains but not present in all the five strains), and (iii) unique (protein sequences exclusively present in a particular strain). Functional annotation was performed using NCBI’s COG (cluster of orthologous genes) database to calculate abundance of genes pertaining to a COG family/pathway present in a particular cluster using WebMGA server in (i) core, (ii) accessory, and (iii) unique protein cluster. The statistical analysis was performed using WebMGA server to calculate:
Total number of genes in COG family/pathway in database.
The genome of E. faecium LR13 was compared with the genomes of five pathogenic strains: E. faecium Aus0085, E. faecium 6E6, E. faecium DO, E. faecium Aus0004, E. faecium ATCC70021, and E. faecium E39; two non-pathogenic non-probiotic strains: E. faecium 64/3 and E. faecium ATCC8459 and four probiotic strains: E. faecium T110, E. faecium 170M39, E. faecium SP15, and E. faecium WEFA23. The genome sequences of these strains were retrieved from NCBI and were compared using the genome comparator tool available at PubMLST (64) which identifies the core-genome based multiple locus typing (cgMLST) pattern and builds a Neighbor-Net plot.
Also, the genome of E. faecium LR13 was compared with the genomes of 317 E. faecium strains reported in an earlier study (65) using Average Nucleotide Index (ANI) (66). These 317 strains represented foodborne, gut commensal, pathogenic, non-pathogenic non-probiotic, and probiotic E. faecium strains (65). The ANI-based comparison was performed using the FASTANI program (67) that creates a distance matrix of ANI values, followed by divergence calculation (100-ANI data). The divergence values were used to build a dendrogram with the MEGAX software (68) using the neighbor joining (NJ) algorithm (69).
The genome sequencing of E. faecium LR13 was conducted with a sequencing depth of ∼400× which was sufficient for de novo assembly. Quality assessment of the reads revealed that Illumina sequencing data was optimal for assembly (Supplementary Links 1, 2) since ∼95% of the reads aligned with the E. faecium genome (Table S1 in Supplementary File 1). After trimming, the Phred value of ∼98% reads was >33 indicating a high quality (Table S2 in Supplementary File 1). The overlapping reads were further merged for a better assembly (Table S3 in Supplementary File 1). The final draft assembly showed 97 contigs of varying lengths. Nodes with length 200 bp were discarded and 97 contigs with a length >200 bp were selected for further analysis. Finally, the assembled genome showed 97 contigs, 37.79% GC content and a genome size of 2.66 Mbp. The N50 value of 123,181 bp (Table 1 and Table S4 in Supplementary File 1). Thus, the assembled genome showed 99.63% completeness after marker gene analysis (Table S5 in Supplementary File 1). Within the genome, 2,522 protein-coding genes were identified, of which putative functions were annotated for 1,558 genes while 964 genes were annotated as hypothetical. Also, 18 pseudogenes, 1 tmRNA, 3 rRNAs, and 56 tRNAs were annotated (Table 1 and Figure 1). Genes encoding for proteins residing inside the cellular machinery were analyzed through the RAST subsystem which revealed that 74% of the genes coded for metabolic proteins, protein processing, stress response, DNA and RNA processing and membrane transport, while 26% of the genes were poorly characterized (Fig. S1 in Supplementary Figure 1). The KEGG Automatic Annotation Server (KAAS) revealed enrichment of proteins involved in carbohydrate metabolic pathways in E. faecium LR13.
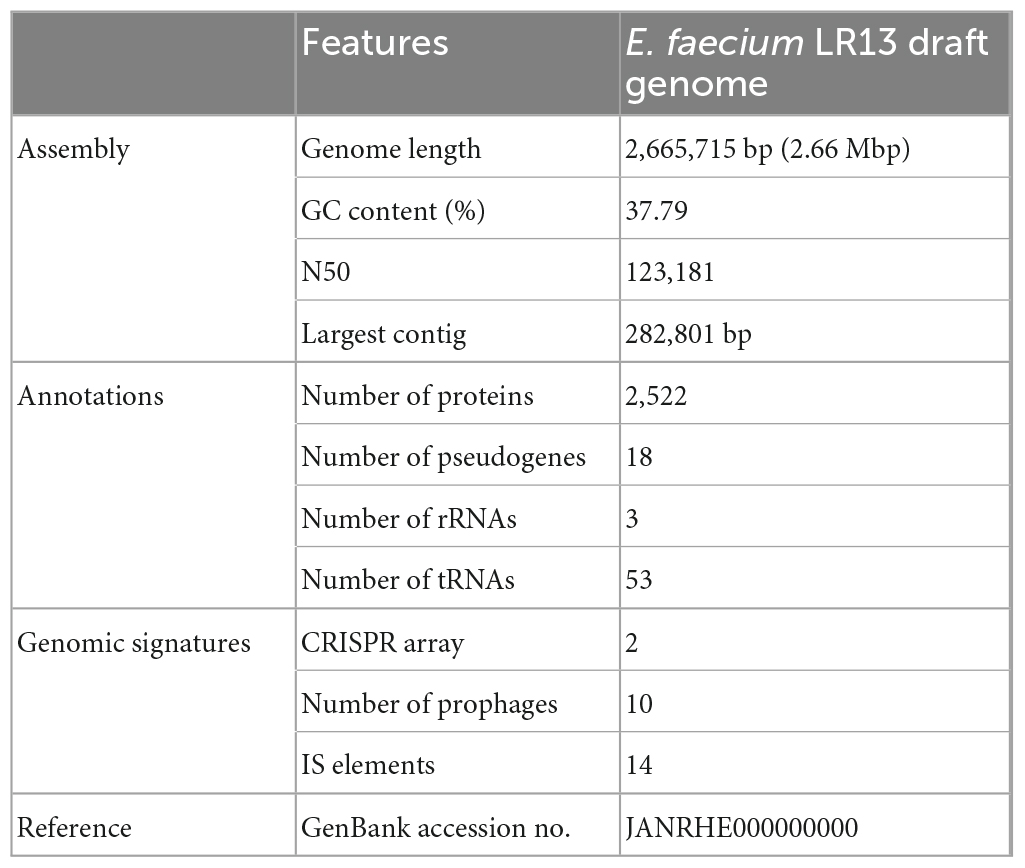
Table 1. Genomic features of E. faecium LR13.

Figure 1. Circular representation of Enterococcus faecium LR13 draft genome. The circles were generated using a CG view server.
Several genes responsible for essential probiotic attributes were discerned in the genome of E. faecium LR13 genes for acid tolerance: gadC, dltA and dltB, copA, Dps, clpC, clpE, msrB, and terC; for adhesion and colonization of the gastro-intestinal (GI) tract: ebpB, srtC, efaA, slrA, and bopD; for hydrolysis of bile salts: bsh; for anti-phagocytosis and host-evasion: cpsA, cpsB, rgpG, and epsE. The complete list of essential probiotic genes discerned in LR13 is presented in Table 2.
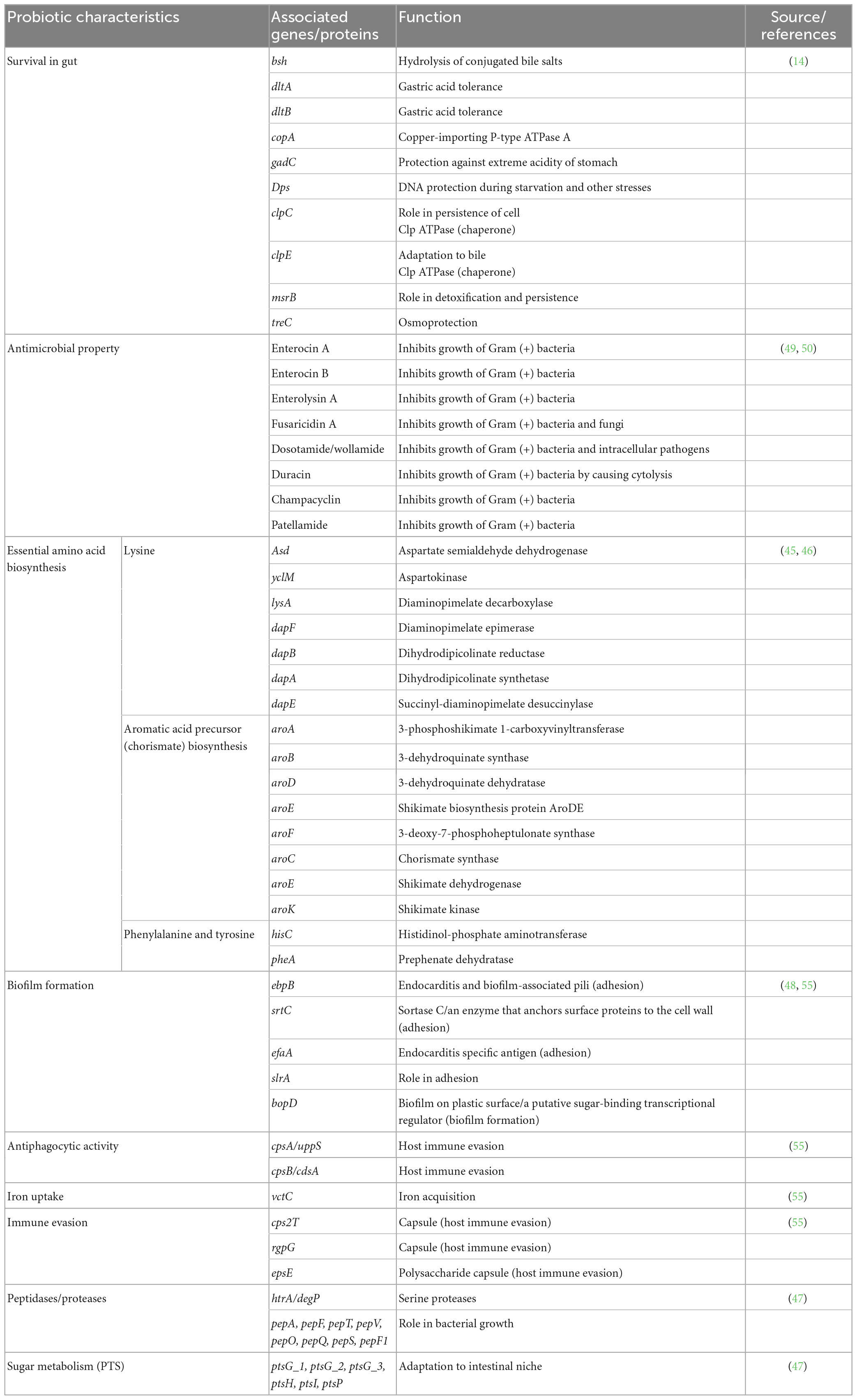
Table 2. Essential and desirable probiotic genes discerned in E. faecium LR13.
Genes that confer additional metabolic benefits on the host, genes for essential amino acid biosynthesis, iron acquisition, carbohydrate metabolism and transport were discerned in E. faecium LR13 have been discussed below:
Genes encoding the enzymes involved in biosynthesis of host essential amino acids such as lysine, phenylalanine and tyrosine were discerned in LR13. Genes encoding for enzymes involved in biosynthesis of lysine such as asd (encodes for aspartate semialdehyde dehydrogenase), yclM (encodes for aspartokinase), lysA (encodes for diaminopimelate decarboxylase), dapF (encodes for diaminopimelate epimerase), dapB (encodes for dihydrodipicolinate reductase, dapA (encodes for dihydrodipicolinate synthase), and dapE (encodes for succinyl-diaminopimelate desuccinylase) were discerned. Also, precursors for biosynthesis of aromatic amino acids (phenylalanine and tyrosine) aroA (encodes for 3-phosphoshikimate 1-carboxyvinyltransferase), aroB (encodes for 3-dehydroquinate synthase), aroC (encodes for chorismate synthase), aroD (encodes for 3-dehydroquinate dehydratase), aroE (encodes for shikimate dehydrogenase), aroF (encodes for 3-deoxy-7-phosphoheptulonate synthase), aroK (for shikimate kinase), hisC (encodes for histidinol-phosphate aminotransferase, and pheA (prephenate dehydratase) were discerned. The complete biosynthetic pathways for lysine and aromatic amino acids depicting the role of these genes are shown in Figures 2A, B.
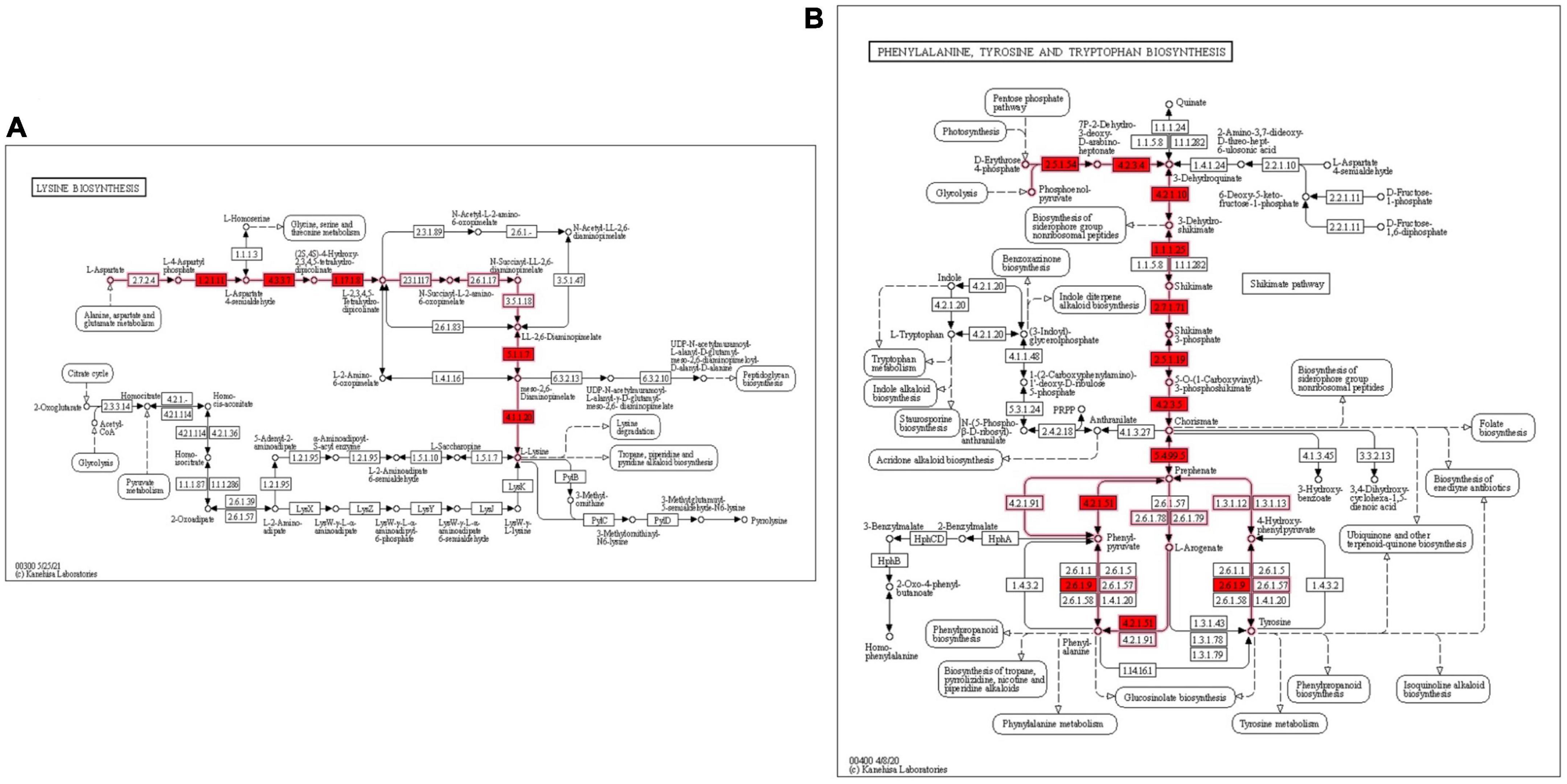
Figure 2. KEGG metabolic pathway for biosynthesis of essential amino acids lysine and aromatic amino acids in E. faecium LR13 (A) lysine biosynthesis genes are highlighted in red (B) phenylalanine and tyrosine biosynthesis genes are highlighted in red. The pathway maps were created using the KEGG database.
KEGG Automatic Annotation Server annotations revealed an abundance of genes related to carbohydrate metabolism in LR13. Also, a variety of genes related to sugar metabolism such as di-, mono-, and oligosaccharides were observed. RAST, PROKKA, and PGAP annotations revealed genes related to glycolysis, pentose phosphate, pyruvate metabolism pathways, etc. Also, genes encoding for carbohydrate transport systems such as phosphoenolpyruvate-dependent sugar phosphotransferase (sugar PTS) genes: ptsG_1, ptsG_2, ptsG_3, gene for phosphocarrier protein (ptsH), phosphoenolpyruvate-protein phosphotransferase genes ptsI and ptsP were discerned in LR13 (Table 2).
The gene vctC which encodes for the iron chelate ABC transporter ATP-binding protein was discerned in LR13. Also, genes for serine proteases and metallopeptidases such pepA, pepF, pepT, pepV, pepO, pepQ, pepS, and pepF1 were discerned in LR13 (Table 2).
Genes encoding stress response such as heat shock proteins of the chaperone class III Clp proteases ClpC and E were discerned in LR13. Also, dps which encodes a DNA-binding protein that helps in DNA protection and in universal stress; msrB which encodes a protein that plays role in detoxification and treC which encodes for proteins mainly involved in osmoprotection were discerned (Table 2).
Enterococcus faecium LR13 harbored genes encoding for three bacteriocins and five antimicrobial peptides (AMPs). The three bacteriocin genes exhibited 100% identity with the reported enterocin A (Figure 3), 97% identity with the enterocin B (Supplementary Figure 5), and 42% identity with enterolysin A (Supplementary Figure 4) gene sequences, respectively. The genes encoding for the five AMPs, fusaricidin A, dosotamide/wollamide, duracin, champacyclin, and patellamide exhibited 100% query coverage with Antimicrobial Peptide Database (APD3) records. The identity with APD3 records was 96.42% for fusaricidin A and dosotamide/wollamide, 87.50% for champacyclin and patellamide and 96.42% for duracin (Table 2).

Figure 3. Multiple sequence analysis (MSA) of E. faecium LR13 enterocin A (red) with other enterocin A using Muscle tool (111).
Several MGEs such as plasmids, bacteriophages, and transposons were found in E. faecium. The identification of the mobilome in the chromosomal genome was based on screening for CRISPR-Cas elements. No CRISPR and cas gene clusters were identified. A total of four prophage regions were detected, of which two of 42.8 and 33.4 kb were intact (PHASTER score ≥100), while two of 20.4 and 15.7 kb were incomplete (PHASTER score ≤50), comprising 4% of the chromosomal genome (Fig. S2 in Supplementary Figure 1). In total, 14 Insertion sequences (ISs) were detected (ISCac2, ISSsu9, ISAac3, ISEfa4, IS21, ISEf1, ISEfa13, IS1485, ISEfa10, ISLca1, IS30, IS6, ISAhy2, and ISBs2) with no associated virulence or AMR gene.
When compared with a commercially available probiotic strain E. faecium T110, three Genomic islands (GIs) were identified in the genome of LR13, representing 4% of the aligned genome (the chromosomal region). Annotation of GIs revealed that GI1 was composed mainly of ribosomal proteins and transporter genes, GI2 of ISEfa13 element and GI3 of hypothetical proteins (Supplementary Table 2). None of the probiotic, bile salt hydrolase, virulence or AMR genes were found on the GIs of LR13. The MGEs discerned in LR13 are depicted in Figure 4.
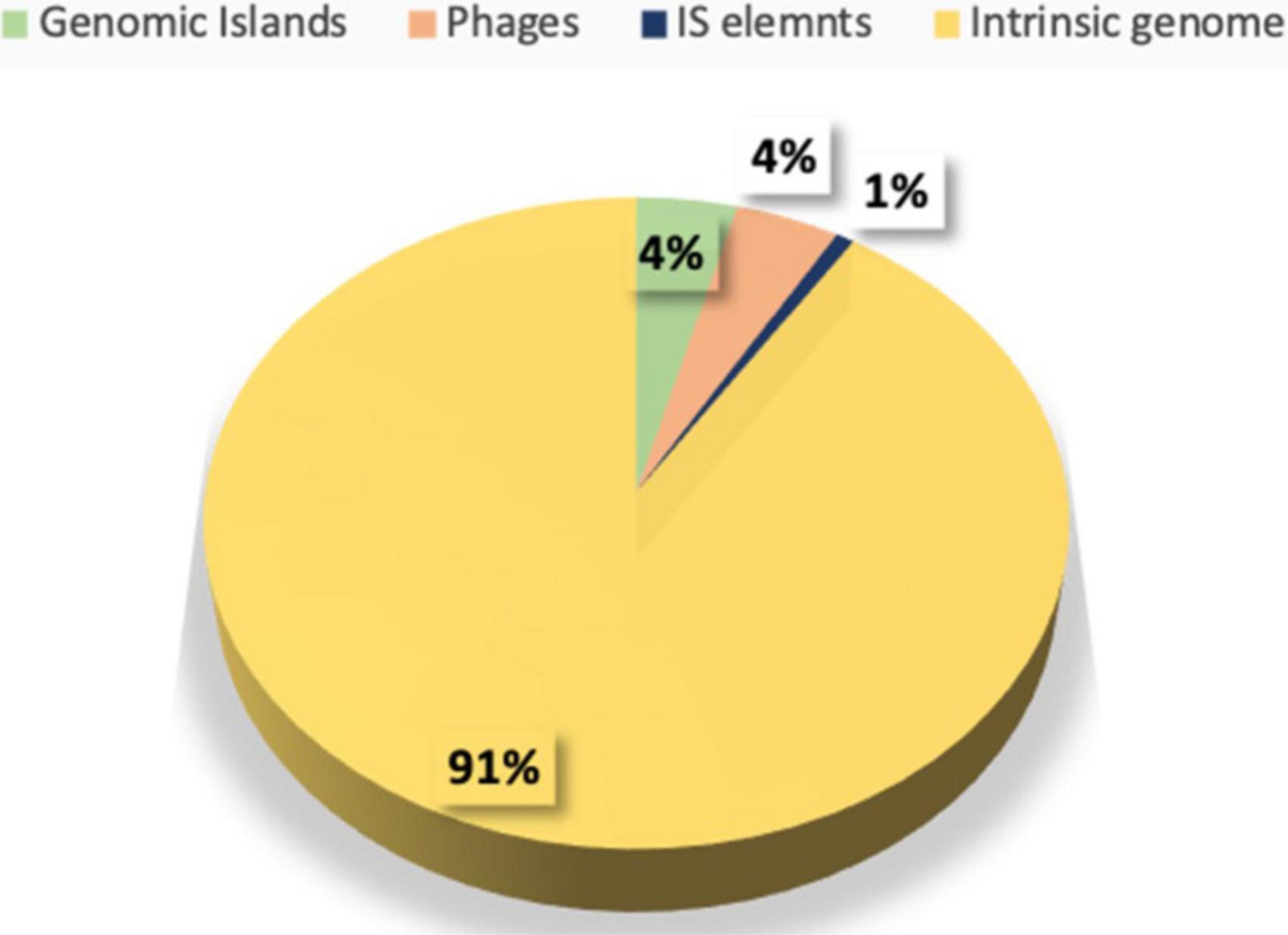
Figure 4. Distribution of various mobile genetic elements like insertion sequences (ISs), genomic islands (GIs), phage (Φ) elements in the genome of E. faecium LR13.
None of the genes for virulence factors (VFs), antibiotic resistance or efflux of antibiotics were discerned in LR13. However, two AMR genes were found, msrC which confers resistance for macrolides and, aac (6′)-Ii which confers resistance for fluoroquinolones and aminoglycosides. ResFinder revealed that none of the AMR gene was an acquired resistance gene or present on the plasmids.
The genome comparison of LR13 with other probiotic strains revealed its genomic proximity with the general probiotic strains in the order: DDH = 60.10% and ANI = 94.85% with strain T110, DDH = 59.90% and ANI = 98.91% with strain SP15, DDH = 56.60% and ANI = 94.95% with strain 170M39 (Fig. S3 in Supplementary Figure 1). However, LR13 exhibited high genomic relatedness with the cholesterol lowering strain WEFA23 with DDH = 91.70% and ANI = 98.91% (Fig. S3 in Supplementary Figure 1).
Comparison of the proteome of LR13 with the proteomes of the strains WEFA23, T110, 170M39, and SP15 revealed a total of 3,741 Cluster of Orthologous Genes (COG), of which 1,807 (48.3%) clusters belonged to the core proteome, 863 (23%) to the accessory, and 1,070 (28.6%) to the unique proteome. Overall representation of the COG clusters present in the core, accessory and unique proteome is shown in Fig. S4 in Supplementary Figure 1. Further analysis of the COG clusters revealed that 1,897 proteins were core proteins, 2,475 were accessory proteins, and 1,070 were unique proteins. The strain specific proteins in LR13, WEFA23, T110, 170M39, and SP15 were 716, 48, 83, 131, and 92, respectively. OrthoFinder revealed that 13.7 % of the core, 10.6% of the accessory, and 14.3% of the unique proteins were hypothetical and uncharacterized.
OrthoFinder revealed that in the core proteome of all the five probiotic strains, genes corresponding to 1,654 proteins were present as a single copy and 243 proteins as multiple copies. Functional annotation of the core proteins revealed presence of various functional categories within each COG class viz, cell growth, DNA replication, recombination and repair, transcription, translation, ribosomal structure and biogenesis, carbohydrate, nucleotide, lipid and amino acid metabolism and transport, defense mechanism, signal transduction, and various transporters. The highest number of core proteins of all the probiotic strains belonged to the COG cluster carbohydrate metabolism and transport (11.4% proteins), followed by translation, ribosomal structure and biogenesis (9% proteins), amino acid transport (7% proteins), nucleotide transport (4% proteins), and coenzyme transport and metabolism (2% proteins). The complete list of core proteins and their corresponding genes is provided in Supplementary Table 3.
Overall functional annotation of the accessory genes/proteins of all the five probiotic strains revealed two important COG subsystems: (a) carbohydrate metabolism and transport with 30% abundance, (b) DNA transcription with 8% abundance. The complete list of accessory proteins and their corresponding genes is provided in Supplementary Table 4.
In the unique proteome of all the five probiotic strains 18% abundance was observed for DNA replication, recombination and repair proteins, especially the plasmid maintenance proteins, helicases, and transcriptional regulators. This was followed by hypothetical proteins or proteins with unknown functions (14.3% abundance), followed by proteins of the COG category cell wall/membrane/envelope biogenesis ubiquinone, cytochromes, and Phosphotransferase system PTS proteins (12.7% abundance). The unique proteins and their corresponding genes in the probiotic strains are enlisted in Supplementary Table 5. The overall representation of COG in core, accessory and unique proteome is depicted in Supplementary Figure 4.
Comparison of the proteomes of cholesterol lowering (LR13 and WEFA23) and general probiotic strains (T110, 170M39, and SP15) revealed that 52% of the proteins of the hypo-cholesterolemic strains were abundant in the COG category carbohydrate metabolism and transport while 17% of the proteins of the general probiotic strains were abundant in DNA transcription (Figure 5). In hypo-cholesterolemic strains, 3% of the proteins were involved in the COG category cell wall/membrane/envelope biogenesis, while in the general probiotic strains only 0.7% of the proteins were involved in this COG category. Twenty-one proteins were found to be exclusively present in hypo-cholesterolemic strains (LR13 and WEAF3) which were absent in general probiotic strains (T110, SP15, and 17OM9). The details of these proteins are presented in Table 3.
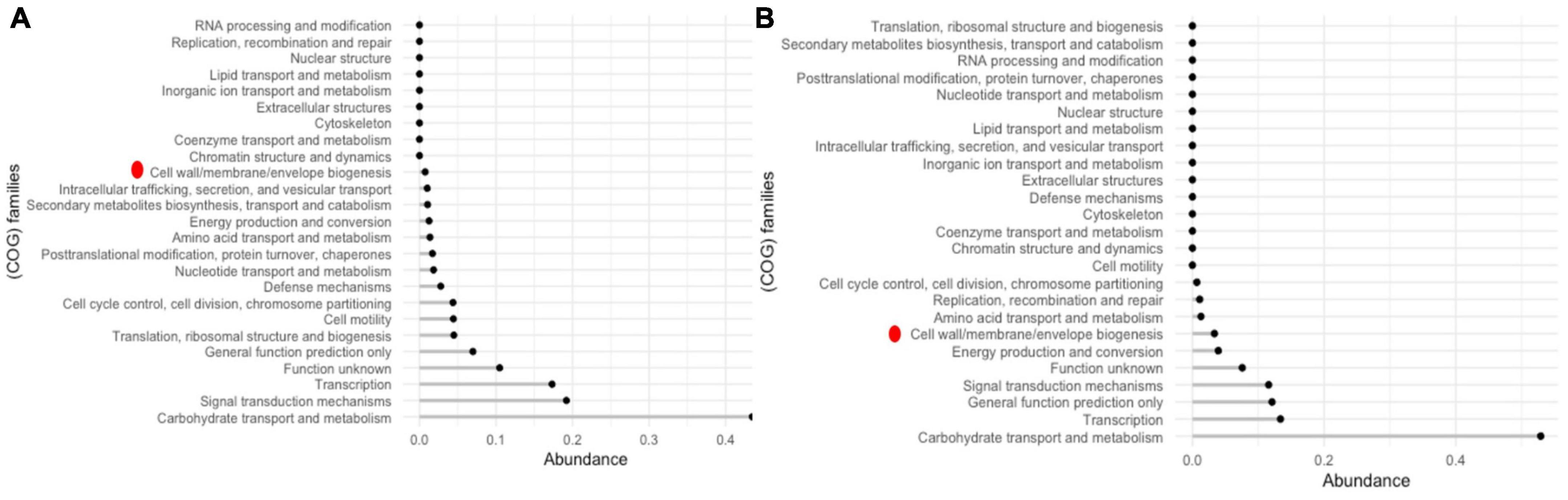
Figure 5. COG families of accessory proteins (A) general E. faecium probiotic strains – T110, 170M39, and SP15LR13 and (B) cholesterol-lowering E. faecium probiotic strains LR 13 and WEFA23. Red dot represents the cell wall/membrane/envelope biogenesis COG family proteins which exhibited a fourfold abundance in LR13 and WEFA23.
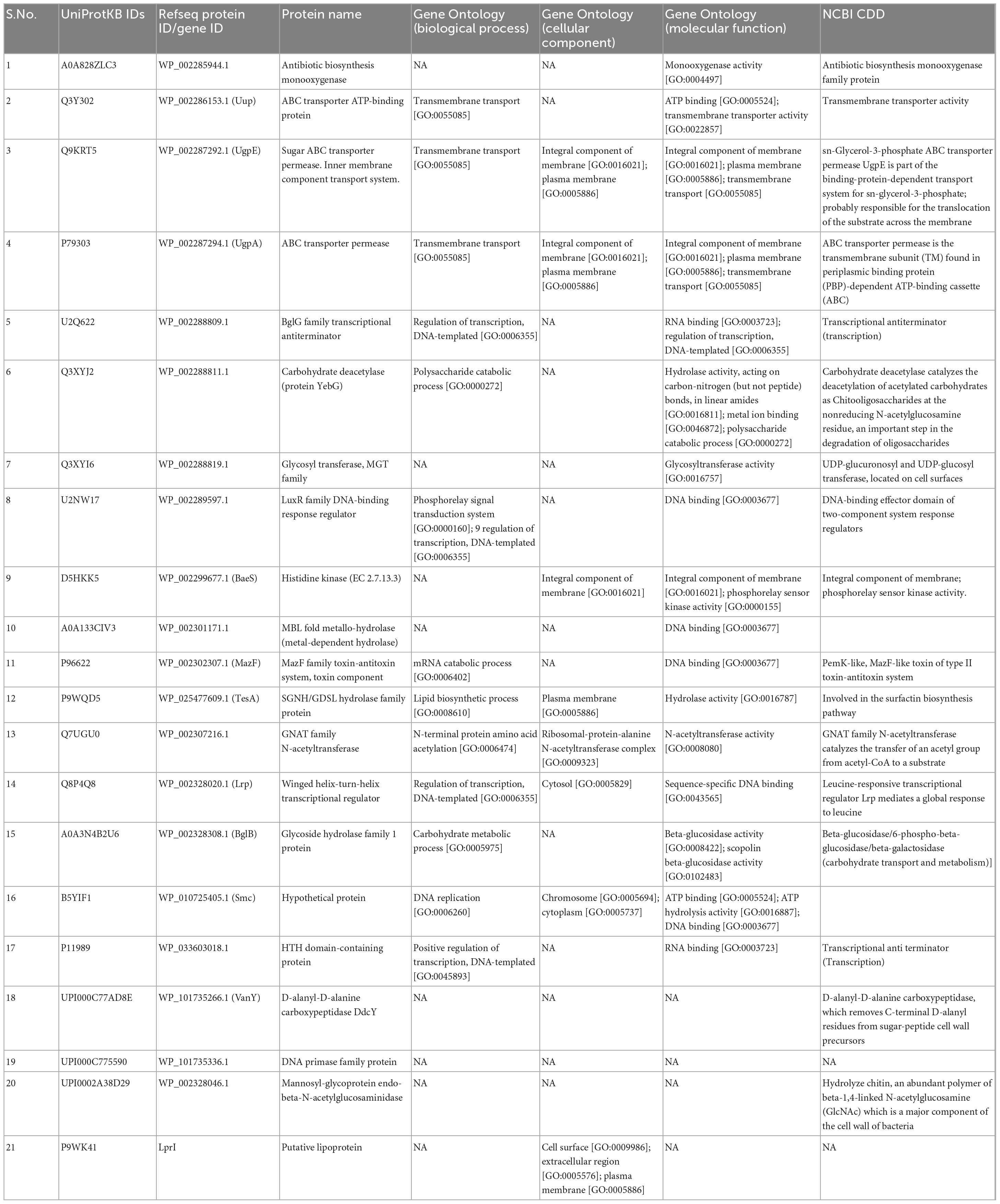
Table 3. Details of the proteins discerned through COG-clustering as exclusive to hypo-cholesterolemic probiotics E. faecium LR13 and WEAF3.
The cgMLST of the 12 E. faecium strains comprising pathogenic, non-pathogenic non-probiotic, and probiotic strains revealed three clusters, (i) pathogenic strains, (ii) non-pathogenic, non-probiotic strains and, (iii) probiotic strains. E. faecium LR13 belonged to the probiotic cluster (Supplementary Figure 2 and Table S6 in Supplementary File 1).
Calculation of the nucleotide-level genomic similarity of LR13 with 317 E. faecium strains reported in an earlier study (65) revealed that E. faecium LR13 belonged to the cluster containing gut-associated strains with the ANI value above 99.5 (Supplementary Figure 3).
Most of the probiotic E. faecium strains are isolated from animal/human gut and fermented foods (70, 71). E. faecium LR13 was the first rhizospheric isolate which not only exhibited several probiotic attributes but also bile salt hydrolase activity and hypocholesterolemic activity in vitro (18). Thus, a complete understanding of the genomic features of this strain and comparison with other probiotic and pathogenic E. faecium strains can help in understanding the genomic relatedness/differences in probiotic/non-probiotic environmental or animal isolates.
The essential attributes possessed by a probiotic strain included the genes that helps in adherence, competence, survival, tolerance, and persistence in harsh conditions of the GI-tract. Genome sequencing and computational analysis revealed that the probiotic attributes exhibited phenotypically by E. faecium LR13 co-related exactly with the presence of the respective genes. Genes encoding for essential probiotic attributes such as acid tolerance, (dltA, dltB, and gadC), bile resistance (clpC, clpE, and dps), self-aggregation (bopD), adhesion and colonization (ebpB, srtC, efaA, and slrA) (48) hydrolysis of conjugated bile salts (bsh), were discerned in LR13.
Also, several other genes beneficial in populating the host GI-tract were discerned in LR13 such as genes for colonization and host immune evasion (cpsA, cpsB, cps2T, rgpG, and epsE), persistence and detoxification (msrB) and, growth and osmoprotection (treC) (14). Also, several genes encoding serine proteases and metallopeptidases such as pepA, pepF, pepT, pepV, pepO, pepQ, pepS, and pepF1 were discerned in LR13. The peptidases/proteases helps in bacterial growth inside the host (47). A variety of genes related to sugar metabolism, glycolysis, pentose phosphate, and pyruvate metabolism pathways were discerned. An earlier study also reported that genes for carbohydrate metabolism are more abundant in probiotic strains than the pathogenic strains (72). Genes encoding carbohydrate transport systems including phosphoenolpyruvate-dependent sugar phosphotransferases (sugar PTS), PTS system glucose-specific EIICB component (ptsG_1, ptsG_2, and ptsG_3), phosphocarrier protein (ptsH), phosphoenolpyruvate-protein phosphotransferase (ptsI) and (ptsP) were identified in LR13. The PTS component genes are essential for adaptation in the intestinal niche and their presence is frequently reported in gut/commensal bacteria (47). The presence of these genes suggests that LR13 might fairly survive and transit in the GI-tract without the need for any protective encapsulation.
Besides, genes whose products might confer some additional metabolic benefits on the host were discerned in LR13. These included genes for biosynthesis of amino acids, lysine, phenylalanine, and tyrosine which are essential amino acids. Essential amino acids cannot be synthesized by the human body and, since they are necessary for the normal functioning of the cells they have to be taken with the diet. Besides, these amino acids are precursors for short-chain fatty acids which play diverse physiological roles in human health, production of vitamins, lipids, and energy (73). The presence of iron-chelate ABC transporter ATP-binding protein, vctC in the genome of E. faecium LR13 might also have metabolic implications for the host because the host iron is used by several pathogenic bacteria for colonization and pathogenesis (74, 75). The presence of iron uptake genes in probiotics can pose competition to the pathogens for the host-iron and thus reduce the pathogen population inside the host. As was also observed in the case of probiotic Escherichia coli Nissle, which helped in reducing the host colonization by pathogenic Salmonella typhimurium (76).
Enterococcus faecium LR13 harbored genes encoding for three bacteriocins and five AMPs. The three bacteriocin genes belonged to the bacteriocin-families, enterocin A, enterocin B, and enterolysin A, respectively. Interestingly, the enterocin A of LR13 was identical with an earlier reported enterocin from E. faecium 170M39 which exhibited antimicrobial activity against Listeria monocytogenes (15). L. monocytogenes is a pathogenic food-borne bacteria associated with a serious infection called listeriosis. This suggests that E. faecium LR13 might also protect against listeriosis. Besides, the presence of genes encoding AMPs, Fusaricidin A, Dosotamide/wollamide, Duracin, Champacyclin, and Patellamide can play an important role in the development of a healthy microbiome inside the host because AMPs reportedly inhibit the growth of Gram (+) positive bacteria (77).
Enterococcus faecium is composed of both pathogenic and non-pathogenic strains (78, 79) and many strains can acquire virulence genes over the course of time. Thus, identification of virulence genes is pivotal before categorizing an E. faecium strain as a probiotic. None of the virulence genes prevalent in enterococci, genes encoding for cytolysins, hemotoxins, aggregation, and chemotaxis were discerned in LR13 indicating that it was safe for human consumption. VRE are notorious nosocomial pathogens, hence, potential probiotic E. faecium strains should not harbor vancomycin resistance genes (11). Our analysis revealed that LR13 neither harbored any AMR genes reported for pathogenic enterococci (80) nor vancomycin resistance genes.
The MGEs including plasmids, ISs, transposons, bacteriophages, and GIs play an important role in horizontal gene transfer and transmission of virulence/antibiotic resistance genes (81). Reportedly, the MGEs of probiotic enterococci are different from pathogenic enterococci (72). ISs, ISEfa11, and ISEfa5 have been reportedly associated with vancomycin resistant genes vanS, vanX, and vanY of E. faecium (82). Bacteriophages or prophages provide new genetic characteristics to the host and increase virulence of pathogenic bacteria, Vibrio cholerae (83), E. coli (84), and Corynebacterium diphtheriae (85). Genes present on GIs are usually mobile genes representing the acquired traits (26). Thus, it was necessary to ascertain if the antibiotic resistance, virulence and/or probiotic traits of LR13 were intrinsic or acquired traits. Our results revealed that LR13 did not carry plasmid, IS element, transposon, bacteriophage or GI-linked antibiotic resistance or virulence genes. Moreover, none of the genes for probiotic attributes were associated with MGEs suggesting that the probiotic attributes exhibited by LR13 were intrinsic traits and not acquired.
The genomic comparison of LR13 with the genomes of foodborne, gut commensal, pathogenic, non-pathogenic non-probiotic, and probiotic E. faecium strains revealed that it lies within the cluster of gut commensal strains. The cgMLST based genotyping further strengthened the fact that LR13 was quite distinct from pathogenic and laboratory strains. These findings are in concordance with an earlier study which also reported that food-grade and nosocomial enterococci strains are genetically distinct (20).
Cholesterol-assimilation capability has been reported for only a few probiotic Lactobacillus plantarum and E. faecium strains (17, 18, 72, 86, 87). Comparative genomic and proteomic analysis of the cholesterol-lowering probiotic E. faecium strains (LR13 and WEFA23) and general E. faecium probiotic strains (T110, 170M39, and SP15) revealed that LR13 was genetically more related to the cholesterol-lowering strain WEFA23 (DDH = 91.70% and ANI = 98.91%), than other probiotic strains.
Comparison of the COG categories revealed at least a 4.28-fold abundance of cell wall/membrane/envelope biogenesis COG category proteins in cholesterol lowering probiotics than general probiotic strains. GO-based functional annotation and literature survey of the proteins exclusively discerned in hypo-cholesterolemic probiotic strains (LR13 and WEAF3) revealed that of the 21 proteins, 17 proteins might be directly/indirectly related to cholesterol assimilation. The four proteins for which a correlation with cholesterol-assimilation could not be discerned were, A0A133CIV3, P96622, B5YIF1, and UPI000C775590. A0A133CIV3 was a MBL fold metallo-hydrolase (metal-dependent hydrolase), P96622 – a MazF family toxin-antitoxin system protein, B5YIF1 – a hypothetical protein and UPI000C775590 – a DNA primase family protein. Interestingly of the remaining 17 proteins, 14 proteins might help directly in cholesterol-assimilation by employing either of the three mechanisms, (i) production of short chain fatty acids (SCFAs), (ii) lipid (sterol) transport and membrane stabilization, and (iii) bile salt hydrolase (bsh) activity (Graphical Abstract).
Short chain fatty acids are fatty acids with less than 6 carbon atoms. These are produced in the human gut by the gut microbes as a by-product of breakdown/fermentation of dietary fibers (88). SCFAs are transported from the gut to the liver where they inhibit the HMG-CoA reductase enzyme (3-hydroxy-3-methylglutaryl-CoA) which converts HMG-CoA to mevalonic acid (89). Interestingly, this step is the rate limiting step of the cholesterol biosynthesis pathway and also the target of statins (90). GO annotations, NCBI Conserved Domain Database (CDD) search and literature evidences revealed that six proteins, Q3XYJ2 (yebG), Q3XYI6, P9WQD5 (TesA), UPI0002A38D29 (ENGase), P1198 (BglG), and UPI000C77AD8E (DdcY) were involved in sugar uptake and breakdown/fermentation into SCFAs (Table 3). The protein Q3XYJ2 was a carbohydrate deacetylase, Q3XYI6, a glycosyltransferase, P9WQD5 was a SGNH/GDSL hydrolase family protein, UPI0002A38D29 a mannosyl-glycoprotein endo-beta-N-acetylglucosaminidase, P1198 an RNA binding protein that regulates the expression of genes involved in sugar utilization and UPI000C77AD8E removes D-alanyl residue from sugars. These proteins participate in uptake and breakdown of complex sugars which are fermented to produce SCFAs (91–94).
Several studies have indicated that gut bacteria might assimilate cholesterol by binding/attaching to the cell walls and incorporating in their membranes (87, 95). Recently, cholesterol-assimilation was related to the metabolic state of the bacteria and live cells reportedly accumulated more cholesterol than the dead cells (95). GO annotations, NCBI-CDD and literature evidences revealed that Q3Y02, Q9KRT5, P7903, and U2Q622 (membrane transporter/importers) and, U2NW17, D5HKK5, and P9WK41 (membrane stabilization proteins) might be involved in cholesterol uptake and stabilization of bacterial membrane. Q3Y302 was an ABC transporter ATP-binding protein, and Q9KRT5 and P79303 were ABC transporter permeases. ABC transporters are involved in transport of lipids, sterols (96) while Q9KRT5 and P79303 are bacterial importers also known as permeases which transport sugars, ions, and lipids (97). U2Q622 was a BglG family transcriptional anti-terminator which regulates oppA that encodes an oligopeptide transporter (98). U2NW17 was a LuxR family DNA-binding response regulator while D5HKK5 was a histidine kinase. Both U2NW17 and D5HKK5 are putative stress response proteins involved in cell wall homeostasis and regulation of peptidoglycan plasticity (99–101). P9WK41 was a putative lipoprotein that plays an important role in cell wall biosynthesis (102). A recent report has suggested that lipoproteins are important for cholesterol uptake (103).
GO annotations and NCBI-CDD revealed that Q7UGU0 (bshB) was a homolog of the bile salt hydrolase, bshA. Most of the gut microbes usually have one, but some probiotics might harbor multiple BSH homologs (104). Q7UGU0 (bshB) is a GNAT family N-acetyl transferase which deconjugates conjugated bile salts. Since deconjugated bile salts are not as efficiently absorbed in the intestine, they are excreted out in the feces as free bile acids. Thus, deconjugation of bile salts can potentially reduce the serum cholesterol levels by increasing the use of host cholesterol for bile acid synthesis (105). Also, deconjugated bile salts do not efficiently solubilize and absorb lipids resulting in reduced cholesterol solubility and absorption by the intestinal lumen (104).
The other three proteins, A0A828ZLC3, A0A3N4B2U6, and Q8P4Q8 did not exhibit a direct correlation with cholesterol-assimilation but might help in bacteria in other ways. A0A828ZLC3, was an antibiotic biosynthesis monooxygenase protein which initiates degradation of sterol side chain in bacteria (106). A0A3N4B2U6 is a glycoside hydrolase family 1 protein which helps in auto-aggregation of bacterium by linking cell and extracellular molecules of glucan (107). Q8P4Q8 was a winged helix-turn-helix transcriptional regulator which plays an important role in bacterial DNA stabilization (108).
To summarize, a comprehensive genome analysis of E. faecium LR13 revealed that it was genetically related to food grade enterococci and was safe for human consumption. Additionally, besides the usual probiotic benefits it can bestow several other additional metabolic benefits on the host as the genome of LR13 harbored genes for biosynthesis of essential amino acids, carbohydrate metabolism, iron acquisition, and production of bacteriocins and antimicrobial peptides. Cholesterol-assimilation capability has been reported for only a few probiotic L. plantarum and two E. faecium strains. Among all the probiotic strains tested, LR13 was genetically more related to WEFA23 – another cholesterol assimilating probiotic. Both LR13 and WEFA23 harbored 21 unique proteins which were absent in other probiotic strains. Of these, three proteins were indirectly related to cholesterol-assimilation while 14 proteins might directly help in cholesterol assimilation. However, in vivo and mutation-based studies are required to establish the mechanism and identify the precise role of these proteins in cholesterol assimilation. The fact that the general probiotic strains were devoid of proteins directly/indirectly related with cholesterol assimilation, suggests that cholesterol-assimilation might be an inherent, strain specific trait present in a few probiotics with a specific genetic constitution. A recent study identified many cholesterol-interacting microbes suggesting that a specific microbiome can precisely regulate the host-cholesterol homeostasis (109). They also identified a novel mechanism for microbial transformation of dietary cholesterol enabled by the presence of a cholesterol-responsive sulfotransferase in Bacteroides (109). Moreover, the unique proteins identified in this study can serve as biomarkers for discerning/characterizing cholesterol-assimilating probiotics as novel biotherapeutics for treating CVDs. However, more cholesterol assimilating probiotics need to be studied to arrive at some definite conclusion.
The datasets presented in this study can be found in online repositories. The names of the repositories and accession number(s) can be found below: National Center for Biotechnology Information (NCBI) GenBank, https://www.ncbi.nlm.nih.gov/genbank/, JANRHE000000000 NCBI Sequence Reads Archive (SRA), https://www.ncbi.nlm.nih.gov/sra, SRR20852288.
NS designed the experiment. MA performed the bioinformatics analysis. NS and MA analyzed the data and prepared the manuscript. MK conceived the idea. All authors reviewed the manuscript.
MA was supported by the CSIR-JRF scheme (Grant Number: 09/045/(1637)/2019-EMR-1). NS was supported by CSIR Senior Research Associateship (Scientists’ Pool Scheme) (Grant Number: 13(9089-A)/2019-Pool).
The authors acknowledge University of Delhi for providing excellent research support. The authors would like to acknowledge lab members Ms. Aakriti Jain and Govind Rao Dabburu for their meaningful suggestions and inputs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2023.1082566/full#supplementary-material
Supplementary Figure 1 | Phages present in LR13 genome. Figure S2: RAST subsystem analysis. Figure S3: DDH and ANI calculation of other strains represented with LR13 genome. Figure S4: Overall COG representation of core, accessory and unique present in LR13, WEFA23, T110, 170M39, and SP15 probiotic strains.
Supplementary Figure 2 | An overview of genome comparison of E. faecium LR13 with 317 published genomes of foodborne, gut commensal, pathogenic, non-pathogenic non-probiotic and probiotic E. faecium strains. The comparison was performed with an ANI-based approach using FASTANI. E. faecium LR13 was found to be related to the gut-associated strains (marked in black color).
Supplementary Figure 3 | A neighbor-net view of the core-genome comparative analysis of E. faecium LR13 with five pathogenic E. faecium strains (Aus0085, 6E6, DO, Aus0004, ATCC70021 and E39), two non-pathogenic non-probiotic strains (64/3, ATCC8459) and four probiotic strains (T110, 170M39, SP15 and WEFA23). The comparison was performed using a genome comparator tool available at PubMLST to discern the cgMLST. E. faecium LR13 showed more proximity with the probiotic strains (green color) than non-probiotic non-pathogenic (yellow color) and pathogenic strains (orange color).
Supplementary Figure 4 | Multiple sequence analysis (MSA) of E. faecium LR13 enterolysin A (Red) with other enterolysin A using Muscle tool.
Supplementary Figure 5 | Multiple sequence analysis (MSA) of E. faecium LR13 enterolysin B (Red) with other enterolysin B using Muscle tool.
Supplementary File 1 | Table S1: BLAST analysis of pair-end reads of E. faecium LR13. Table S2: After trimmomatic analysis the reads left for further analysis. Table S3: Reads after merge of overlapping reads. Table S4: N50, GC%, and NG50 value when aligned with the reference E. faecium genome. Table S5: Completeness of assembly. Table S6: cgMLST output.
Supplementary File 2 | Detailed information of the core genes in the studied genomes.
Supplementary File 3 | Detailed information of the accessory genes in the studied genomes.
Supplementary File 4 | Detailed information of the unique genes in the studied genomes.
Supplementary File 5 | Detailed information of the Genomic Islands in E. faecium LR13.
Supplementary Link 1 | FastQC output html file (LR13_HHH23DSXX_L3_1_fastqc.html).
Supplementary Link 2 | FastQC output html file (LR13_HHH23DSXX_L3_2_fastqc.html).
1. FAO, WHO. Guidelines for the Evaluation of Probiotics in Foods. Geneva: World Health Organization (2002).
2. Nagpal R, Kumar A, Kumar M, Behare P, Jain S, Yadav H. Probiotics, their health benefits and applications for developing healthier foods: a review. FEMS Microbiol Lett. (2012) 334:1–15. doi: 10.1111/j.1574-6968.2012.02593.x
3. Blaabjerg S, Artzi D, Aabenhus R. Probiotics for the prevention of antibiotic-associated diarrhea in outpatients—a systematic review and meta-analysis. Antibiotics (Basel). (2017) 6:21. doi: 10.3390/antibiotics6040021
4. Ehrmann MA, Kurzak P, Bauer J, Vogel RF. Characterization of lactobacilli towards their use as probiotic adjuncts in poultry. J Appl Microbiol. (2002) 92:966–75. doi: 10.1046/j.1365-2672.2002.01608.x
5. Abeijón M, Medina R, Katz M, González S. Technological properties of Enterococcus faecium isolated from ewe’s milk and cheese with importance for flavour development. Can J Microbiol. (2006) 52:237–45. doi: 10.1139/w05-136
6. Hassanzadazar H, Ehsani A, Mardani K. Antibacterial activity of Enterococcus faecium derived from koopeh cheese against Listeria monocytogenes in probiotic ultra-filtrated cheese. Vet Res Forum Int Q J. (2014) 5:169–75.
7. Satish Kumar R, Kanmani P, Yuvaraj N, Paari K, Pattukumar V, Arul V. Purification and characterization of enterocin MC13 produced by a potential aquaculture probiont Enterococcus faecium MC13 isolated from the gut of Mugil cephalus. Can J Microbiol. (2011) 57:993–1001. doi: 10.1139/w11-092
8. Rosenthal V, Maki D, Mehta A, Alvarez-Moreno C, Leblebicioglu H, Higuera F, et al. International nosocomial infection control consortium report, data summary for 2002-2007, issued january 2008. Am J Infect Control. (2008) 36:627–37. doi: 10.1016/j.ajic.2008.03.003
9. Arias C, Murray B. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. (2012) 10:266–78.
10. WHO. WHO Publishes List of Bacteria for Which New Antibiotics are Urgently Needed. Geneva: World Health Organization (2017).
11. Franz C, Huch M, Abriouel H, Holzapfel W, Gálvez A. Enterococci as probiotics and their implications in food safety. Int J Food Microbiol. (2011) 151:125–40.
12. Nissen L, Chingwaru W, Sgorbati B, Biavati B, Cencic A. Gut health promoting activity of new putative probiotic/protective Lactobacillus spp. strains: a functional study in the small intestinal cell model. Int J Food Microbiol. (2009) 135:288–94. doi: 10.1016/j.ijfoodmicro.2009.08.027
13. Ejtahed H, Mohtadi-Nia J, Homayouni-Rad A, Niafar M, Asghari-Jafarabadi M, Mofid V, et al. Effect of probiotic yogurt containing Lactobacillus acidophilus and Bifidobacterium lactis on lipid profile in individuals with type 2 diabetes mellitus. J Dairy Sci. (2011) 94:3288–94. doi: 10.3168/jds.2010-4128
14. Natarajan P, Parani M. First complete genome sequence of a probiotic Enterococcus faecium strain T-110 and its comparative genome analysis with pathogenic and non-pathogenic Enterococcus faecium genomes. J Genet Genomics. (2015) 42:43–6. doi: 10.1016/j.jgg.2014.07.002
15. Ghattargi V, Nimonkar Y, Burse S, Davray D, Kumbhare S, Shetty S, et al. Genomic and physiological analyses of an indigenous strain, Enterococcus faecium 17OM39. Funct Integr Genomics. (2018) 18:385–99. doi: 10.1007/s10142-018-0596-x
16. Aziz F, Khan M, Ahmed S, Andrews S. Draft genome sequence of Enterococcus faecium SP15, a potential probiotic strain isolated from spring water. BMC Res Notes. (2019) 12:99. doi: 10.1186/s13104-019-4136-0
17. Zhang F, Qiu L, Xu X, Liu Z, Zhan H, Tao X, et al. Bene?cial effects of probiotic cholesterol-lowering strain of Enterococcus faecium WEFA23 from infants on diet-induced metabolic syndrome in rats. J Dairy Sci. (2017) 100:1618–28. doi: 10.3168/jds.2016-11870
18. Singhal N, Maurya A, Mohanty S, Kumar M, Virdi J. Evaluation of bile salt hydrolases, cholesterol-lowering capabilities, and probiotic potential of Enterococcus faecium isolated from rhizosphere. Front Microbiol. (2019) 10:1567. doi: 10.3389/fmicb.2019.01567
19. Serio A, Chaves-López C, Paparella A, Suzzi G. Evaluation of metabolic activities of enterococci isolated from pecorino Abruzzese cheese. Int Dairy J. (2010) 20:459–64. doi: 10.1016/j.idairyj.2010.02.005
20. Montealegre M, Singh K, Murray B. Gastrointestinal tract colonization dynamics by different Enterococcus faecium clades. J Infect Dis. (2016) 213:1914–22. doi: 10.1093/infdis/jiv597
21. Singhal N, Singh N, Mohanty S, Singh P, Virdi J. Evaluation of probiotic characteristics of lactic acid bacteria isolated from two commercial preparations available in Indian market. Indian J Microbiol. (2019) 59:112–5. doi: 10.1007/s12088-018-0762-9
22. Raghuwanshi S, Misra S, Sharma R, Bisen P. Indian perspective for probiotics: a review. Indian J Dairy Sci. (2015) 63:195–205.
23. Shetty S, Marathe N, Shouche Y. Opportunities and challenges for gut microbiome studies in the Indian population. Microbiome. (2013) 1:24. doi: 10.1186/2049-2618-1-24
24. Bhute S, Pande P, Shetty S, Shelar R, Mane S, Kumbhare S, et al. Molecular characterization and meta-analysis of gut microbial communities illustrate enrichment of Prevotella and Megasphaera in Indian subjects. Front Microbiol. (2016) 7:660. doi: 10.3389/fmicb.2016.00660
25. Hollenbeck B, Rice L. Intrinsic and acquired resistance mechanisms in Enterococcus. Virulence. (2012) 3:421–33. doi: 10.4161/viru.21282
26. Dobrindt U, Hochhut B, Hentschel U, Hacker J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol. (2004) 2:414–24.
27. Juhas M, van der Meer J, Gaillard M, Harding R, Hood D, Crook D. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev. (2009) 33:376–93.
28. Marothi Y, Agnihotri H, Dubey D. Enterococcal resistance–an overview. Indian J Med Microbiol. (2005) 23:214–9.
29. Gold H. Vancomycin-resistant enterococci: mechanisms and clinical observations. Clin Infect Dis. (2001) 33:210–9.
30. Andrews, S. FastQC: a quality control tool for high throughput sequence data. ScienceOpen. (2010). Available online at: http://www.bioinformatics.babrwaham.ac.uk/projects/fastqc
31. Bolger A, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. (2014) 30:2114–20.
32. Magoè T, Salzberg S. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. (2011) 27:2957–63. doi: 10.1093/bioinformatics/btr507
33. Bankevich A, Nurk S, Antipov D, Gurevich A, Dvorkin M, Kulikov A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77.
34. Wick R, Judd L, Gorrie C, Holt K. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. (2017) 13:e1005595. doi: 10.1371/journal.pcbi.1005595
35. Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. (2016) 11:e0163962. doi: 10.1371/journal.pone.0163962
36. Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinforma Oxf Engl. (2013) 29:1072–5.
37. Parks D, Imelfort M, Skennerton C, Hugenholtz P, Tyson G. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. (2015) 25:1043–55. doi: 10.1101/gr.186072.114
38. Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. PPGAP: pan-genomes analysis pipeline. Bioinformatics. (2012) 28:416–8. doi: 10.1093/bioinformatics/btr655
39. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
40. Moriya Y, Itoh M, Okuda S, Yoshizawa A, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. (2007) 35:W182–5.
41. Brettin T, Davis J, Disz T, Edwards R, Gerdes S, Olsen G, et al. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep. (2015) 5:8365. doi: 10.1038/srep08365
42. Kelley D, Liu B, Delcher A, Pop M, Salzberg S. Gene prediction with glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res. (2012) 40:e9. doi: 10.1093/nar/gkr1067
43. Chan P, Lowe T. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. (2019) 1962:1–14. doi: 10.1007/978-1-4939-9173-0_1
44. Lagesen K, Hallin P, Rødland E, Stærfeldt H, Rognes T, Ussery D. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. (2007) 35:3100–8. doi: 10.1093/nar/gkm160
45. Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, Vandergriff J, Rabkin S, et al. The panther database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. (2005) 33:D284–8.
46. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. (2017) 45:D353–61. doi: 10.1093/nar/gkw1092
47. O’Sullivan O, O’Callaghan J, Sangrador-Vegas A, McAuliffe O, Slattery L, Kaleta P, et al. Comparative genomics of lactic acid bacteria reveals a niche-specific gene set. BMC Microbiol. (2009) 9:50. doi: 10.1186/1471-2180-9-50
49. Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. (2016) 44:D1087–93. doi: 10.1093/nar/gkv1278
50. Medema M, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach M, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. (2011) 39:W339–46. doi: 10.1093/nar/gkr466
51. van Heel A, de Jong A, Song C, Viel J, Kok J, Kuipers O. BAGEL4: a user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. (2018) 46:W278–81. doi: 10.1093/nar/gky383
52. Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. (2007) 35:W52–7.
53. Arndt D, Grant J, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. (2016) 44:W16–21. doi: 10.1093/nar/gkw387
54. Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. (2006) 34:D32–6.
55. Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. (2005) 33:D325–8.
56. McArthur A, Waglechner N, Nizam F, Yan A, Azad M, Baylay A, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. (2013) 57:3348–57.
57. Bortolaia V, Kaas R, Ruppe E, Roberts M, Schwarz S, Cattoir V, et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J Antimicrob Chemother. (2020) 75:3491–500.
58. Srivastava A, Singhal N, Goel M, Virdi J, Kumar M. CBMAR: a comprehensive β-lactamase molecular annotation resource. Database. (2014) 2014:bau111. doi: 10.1093/database/bau111
59. Pandey D, Kumari B, Singhal N, Kumar M. BacEffluxPred: a two-tier system to predict and categorize bacterial efflux mediated antibiotic resistance proteins. Sci Rep. (2020) 10:9287. doi: 10.1038/s41598-020-65981-3
60. Auch A, Klenk H, Göker M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci. (2010) 2:142–8. doi: 10.4056/sigs.541628
61. Yoon S, Ha S, Kwon S, Lim J, Kim Y, Seo H, et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Microbiol Soc. (2017) 67:1613–7. doi: 10.1099/ijsem.0.001755
62. Emms D, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. (2019) 20:238. doi: 10.1186/s13059-019-1832-y
63. Wu S, Zhu Z, Fu L, Niu B, Li W. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics. (2011) 12:444. doi: 10.1186/1471-2164-12-444
64. Jolley K, Bray J, Maiden M. Open-access bacterial population genomics: BIGSdb software, the website and their applications. Wellcome Open Res. (2018) 3:124. doi: 10.12688/wellcomeopenres.14826.1
65. Aka S, Dridi B, Bolotin A, Yapo E, Koussemon-Camara M, Bonfoh B, et al. Characterization of lactic acid bacteria isolated from a traditional Ivoirian beer process to develop starter cultures for safe sorghum-based beverages. Int J Food Microbiol. (2020) 322:108547. doi: 10.1016/j.ijfoodmicro.2020
66. ANI. Average Nucleotide Identity (ANI). (2022). Available online at: https://img.jgi.doe.gov/docs/ANI.pdf (accessed February 22, 2022).
67. Jain C, Rodriguez-R L, Phillippy A, Konstantinidis K, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. (2018) 9:5114.
68. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. (2018) 35:1547–9. doi: 10.1093/molbev/msy096
69. Saitou N, M Nei. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. (1987) 4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454
70. Archana C, Beena AK, Bhagya S, Rathish RL, Rahila MP. Probiotic characterisation of Enterococcus faecali strain isolated from a household Dahi sample of Wayanad district, Kerala. J Vet Anim Sci. (2022) 53:70–8.
71. Nami Y, Vaseghi Bakhshayesh R, Mohammadzadeh Jalaly H, Lotfi H, Eslami S, Hejazi M. Probiotic properties of Enterococcus isolated from artisanal dairy products. Front Microbiol. (2019) 10:300. doi: 10.3389/fmicb.2019.00300
72. Ghattargi V, Gaikwad M, Meti B, Nimonkar Y, Dixit K, Prakash O, et al. Comparative genome analysis reveals key genetic factors associated with probiotic property in Enterococcus faecium strains. BMC Genomics. (2018) 19:652. doi: 10.1186/s12864-018-5043-9
73. Neis E, Dejong C, Rensen S. The role of microbial amino acid metabolism in host metabolism. Nutrients. (2015) 7:2930–46.
74. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. (2014) 19:164–74.
75. Rusu I, Suharoschi R, Vodnar D, Pop C, Socaci S, Vulturar R, et al. Iron supplementation influence on the gut microbiota and probiotic intake effect in iron deficiency—a literature-based review. Nutrients. (2020) 12:1993. doi: 10.3390/nu12071993
76. Deriu E, Liu J, Pezeshki M, Edwards R, Ochoa R, Contreras H, et al. Probiotic bacteria reduce Salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe. (2013) 14:26–37. doi: 10.1016/j.chom.2013.06.007
77. Malanovic N, Lohner K. Antimicrobial peptides targeting gram-positive bacteria. Pharmaceuticals. (2016) 9:59.
78. Semedo T, Almeida Santos M, Martins P, Silva Lopes M, Figueiredo Marques J, Tenreiro R, et al. Comparative study using type strains and clinical and food isolates to examine hemolytic activity and occurrence of the CYL operon in enterococci. J Clin Microbiol. (2003) 41:2569–76. doi: 10.1128/JCM.41.6.2569-2576.2003
79. Gilmore M, Clewell D, Ike Y, Shankar N. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Boston, MA: Massachusetts Eye and Ear Infirmary (2014).
80. Lebreton F, Willems R, Gilmore M. Enterococcus diversity, origins in nature, and gut colonization. In: Gilmore M, Clewell D, Ike Y, Shankar N editors. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. (Boston, MA: Massachusetts Eye and Ear Infirmary) (2014).
81. Schmidt H, Hensel M. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev. (2004) 17:14–56.
82. Mikalsen T, Pedersen T, Willems R, Coque T, Werner G, Sadowy E, et al. Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis. BMC Genomics. (2015) 16:282. doi: 10.1186/s12864-015-1407-6
83. Davis B, Waldor M. Filamentous phages linked to virulence of Vibrio cholerae. Curr Opin Microbiol. (2003) 6:35–42. doi: 10.1016/s1369-5274(02)00005-x
84. O’Brien A, Newland J, Miller S, Holmes R, Smith H, Formal S. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science. (1984) 226:694–6.
85. Freeman V. Studies on the virulence of bacteriophage-infected strains of Corynebacterium diphtheriae. J Bacteriol. (1951) 61:675–88. doi: 10.1128/jb.61.6.675-688.1951
86. Singhal N, Singh N, Mohanty S, Kumar M, Virdi J. Rhizospheric Lactobacillus plantarum (Lactiplantibacillus plantarum) strains exhibit bile salt hydrolysis, hypocholestrolemic and probiotic capabilities in vitro. Sci Rep. (2021) 11:15288. doi: 10.1038/s41598-021-94776-3
87. Noh D, Kim S, Gilliland S. Incorporation of cholesterol into the cellular membrane of Lactobacillus acidophilus ATCC 43121. J Dairy Sci. (1997) 80:3107–13. doi: 10.3168/jds.S0022-0302(97)76281-7
88. Canfora E, Jocken J, Blaak E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol. (2015) 11:577–91.
89. Alvaro A, Solà R, Rosales R, Ribalta J, Anguera A, Masana L, et al. Gene expression analysis of a human enterocyte cell line reveals downregulation of cholesterol biosynthesis in response to short-chain fatty acids. IUBMB Life. (2008) 60:757–64. doi: 10.1002/iub.110
90. Istvan E. Statin inhibition of HMG-CoA reductase: a 3-dimensional view. Atheroscler Suppl. (2003) 4:3–8. doi: 10.1016/s1567-5688(03)00003-5
91. Inman M. How bacteria turn fiber into food. PLoS Biol. (2011) 9:e1001227. doi: 10.1371/journal.pbio.1001227
92. Abriouel H, Pérez Montoro B, Casimiro-Soriguer C, Pérez Pulido A, Knapp C, Caballero Gómez N, et al. Insight into potential probiotic markers predicted in Lactobacillus pentosus MP-10 genome sequence. Front Microbiol. (2017) 8:891. doi: 10.3389/fmicb.2017.00891
93. Duman H, Kaplan M, Arslan A, Sahutoglu A, Kayili H, Frese S, et al. Potential applications of Endo-β-N-acetylglucosaminidases from Bifidobacterium longum subspecies infantis in designing value-added. Next-generation infant formulas. Front Nutr. (2021) 8:646275. doi: 10.3389/fnut.2021.646275
94. Zhao C, Dong H, Zhang Y, Li Y. Discovery of potential genes contributing to the biosynthesis of short-chain fatty acids and lactate in gut microbiota from systematic investigation in E. coli. Npj Biofilms Microbiomes. (2019) 5:1–8. doi: 10.1038/s41522-019-0092-7
95. Tjandrawinata R, Kartawijaya M, Hartanti A. In vitro evaluation of the anti-hypercholesterolemic effect of Lactobacillus isolates from various sources. Front Microbiol. (2022) 13:825251. doi: 10.3389/fmicb.2022.825251
96. Borst P, Zelcer N, van Helvoort A. ABC transporters in lipid transport. Biochim Biophys Acta. (2000) 1486:128–44.
97. Garmory H, Titball R. ATP-binding cassette transporters are targets for the development of antibacterial vaccines and therapies. Infect Immun. (2004) 72:6757–63.
98. Harwani D, Zangoui P, Mahadevan S. The β-glucoside (bgl) operon of Escherichia coli is involved in the regulation of oppA, encoding an oligopeptide transporter. J Bacteriol. (2012) 194:90–9. doi: 10.1128/JB.05837-11
99. Senan S, Prajapati J, Joshi C. Whole-genome based validation of the adaptive properties of Indian origin probiotic Lactobacillus helveticus MTCC 5463. J Sci Food Agric. (2015) 95:321–8. doi: 10.1002/jsfa.6721
100. Dubrac S, Bisicchia P, Devine K, Msadek T. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol Microbiol. (2008) 70:1307–22. doi: 10.1111/j.1365-2958.2008.06483.x
101. Dobihal G, Brunet Y, Flores-Kim J, Rudner D. Homeostatic control of cell wall hydrolysis by the WalRK two-component signaling pathway in Bacillus subtilis. Elife. (2019) 8:e52088. doi: 10.7554/eLife.52088
102. Jayashree S, Sivakumar R, Karthikeyan R, Gunasekaran P, Rajendhran J. Genome-wide identification of probiotic Escherichia coli Nissle 1917 (EcN) fitness genes during adhesion to the intestinal epithelial cells Caco-2. Gene. (2021) 803:145890. doi: 10.1016/j.gene.2021.145890
103. Nakayama H, Kurokawa K, Lee B. Lipoproteins in bacteria: structures and biosynthetic pathways. FEBS J. (2012) 279:4247–68.
104. Begley M, Hill C, Gahan C. Bile salt hydrolase activity in probiotics. Appl Environ Microbiol. (2006) 72:1729–38.
105. Choi E, Chang H. Cholesterol-lowering effects of a putative probiotic strain Lactobacillus plantarum EM isolated from kimchi. LWT Food Sci Technol. (2015) 62:210–7.
106. Frank D, Zhao Y, Wong S, Basudhar D, De Voss J, Ortiz de Montellano P. Cholesterol analogs with degradation-resistant alkyl side chains are effective mycobacterium tuberculosis growth inhibitors. J Biol Chem. (2016) 291:7325–33. doi: 10.1074/jbc.M115.708172
107. Couvigny B, Kulakauskas S, Pons N, Quinquis B, Abraham A, Meylheuc T, et al. Identification of new factors modulating adhesion abilities of the pioneer commensal bacterium Streptococcus salivarius. Front Microbiol. (2018) 9:273. doi: 10.3389/fmicb.2018.00273
109. Le H, Lee M, Besler K, Comrie J, Johnson E. Characterization of interactions of dietary cholesterol with the murine and human gut microbiome. Nat Microbiol. (2022) 7:1390–403.
110. Wang H, Chen J, Hollister K, Sowers L, Forman B. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. (1999) 3:543–53. doi: 10.1016/s1097-2765(00)80348-2
Keywords: cholesterol-assimilation, mobile genetic elements, probiotics, metabolic effect, hypercholesterolemia
Citation: Aswal M, Singhal N and Kumar M (2023) Comprehensive genomic analysis of hypocholesterolemic probiotic Enterococcus faecium LR13 reveals unique proteins involved in cholesterol-assimilation. Front. Nutr. 10:1082566. doi: 10.3389/fnut.2023.1082566
Received: 28 October 2022; Accepted: 14 March 2023;
Published: 04 April 2023.
Edited by:
Alberto Finamore, Council for Agricultural Research and Economics (CREA), ItalyReviewed by:
Mónica Sánchez-Tapia, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoCopyright © 2023 Aswal, Singhal and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Neelja Singhal, bmVlbGphQHNvdXRoLmR1LmFjLmlu; Manish Kumar, bWFuaXNoQHNvdXRoLmR1LmFjLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.