 Amir Mohammad Malvandi
Amir Mohammad Malvandi Sara Shahba
Sara Shahba Jalil Mehrzad
Jalil Mehrzad Giovanni Lombardi
Giovanni Lombardi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr., 24 June 2022
Sec. Food Chemistry
Volume 9 - 2022 | https://doi.org/10.3389/fnut.2022.915681
This article is part of the Research TopicRisk Assessment of Mycotoxins in FoodView all 5 articles
Naturally occurring food/feed contaminants have become a significant global issue due to animal and human health implications. Despite risk assessments and legislation setpoints on the mycotoxins' levels, exposure to lower amounts occurs, and it might affect cell homeostasis. However, the inflammatory consequences of this possible everyday exposure to toxins on the vascular microenvironment and arterial dysfunction are unexplored in detail. Circulation is the most accessible path for food-borne toxins, and the consequent metabolic and immune shifts affect systemic health, both on vascular apparatus and bone homeostasis. Their oxidative nature makes mycotoxins a plausible underlying source of low-level toxicity in the bone marrow microenvironment and arterial dysfunction. Mycotoxins could also influence the function of cardiomyocytes with possible injury to the heart. Co-occurrence of mycotoxins can modulate the metabolic pathways favoring osteoblast dysfunction and bone health losses. This review provides a novel insight into understanding the complex events of coexposure to mixed (low levels) mycotoxicosis and subsequent metabolic/immune disruptions contributing to chronic alterations in circulation.
Mycotoxins are small, highly stable toxic molecules contaminating feed and food chains (1). They are classified into the main categories of carcinogenic substances, including aflatoxins (AFs), ochratoxins, fumonisins, trichothecenes, and zearalenone (ZON) (2). Each mycotoxin is considered a secondary metabolite naturally produced by various fungi genera, Aspergillus, Penicillium, Fusarium, Alternaria, and Claviceps. The lack of quality control, climate change, intensive farm production, and storage technologies might favor a more frequent mycotoxin occurrence even in Europe/western countries (3). Although regulatory limits on significant levels are established in many countries, they remain a concerning issue due to the continuous exposure, although at very low concentrations.
Both acute and chronic exposure to mycotoxins can lead to teratogenic, mutagenic, carcinogenic, nephrotoxic, hepatotoxic, and immunotoxic effects on different organs (4). We recently reviewed the impact of naturally occurring mycotoxins dysregulating the immune system in the brain (5). However, knowledge regarding the consequences of exposure to low-level mycotoxins on circulation is modest. Circulating cells, bone marrow niche, and vascular tissue are easy targets of mycotoxins' toxicity (Table 1). There is no safe level; synergistic interaction of nanomolar concentrations of common mycotoxins could cause serious problems compared to higher concentrations (39).

Table 1. The metabolic and immune shifts induced by mycotoxins (focusing on low concentrations).
Generation of reactive oxygen species (ROS) occurs with normal cellular metabolism; however, it can also result from exposure to naturally occurring mycotoxins (40). ROS leads to alteration of cell components, including DNA breaks, protein cross-links, lipid peroxidation, and consequent impairment of cell function and hemostasis (41, 42). Although cells are endowed with endogenous antioxidant agents [e.g., superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx)], the deleterious effects of mycotoxin-mediated ROS could overcome these defense mechanisms. Oxidative stress is an imbalance between oxidants-free radicals and other reactive species and antioxidants (41). The accumulation of oxidized molecules also accelerates reactive oxygen intermediates' generation; this would be the overall manifestations of disturbance to redox potential and the main cellular processes.
Accordingly, oxidative damage is assumed to be the underlying mechanism of various molecular injuries, such as immune and metabolic disruptions contributing to cancer's chronic inflammatory phenotype profile, neurodegeneration, and vascular diseases. Indeed, this damage would explain how mycotoxins toxicity at low levels may determine harmful effects on the vasculature. Considering the contribution of low levels of mycotoxins to oxidative stress, a series of activated immune cells, endothelial dysfunctions, oxidized lipoproteins, and recruiting additional reinforcements as crucial players in crosstalk between oxidative imbalance and vascular inflammation can be pretty predictable. Paralleling the evidence, the dysregulated lipid metabolism and the lowered availability of lipid-soluble vitamins with exposure to mycotoxins direct the pressure on osteocytes to release the calcium into the circulation, thus moving toward osteoporosis in a long-term perspective (43). Besides, the high mitochondrial activity in the cells (i.e., cardiomyocytes, hematopoietic cells) makes them more fragile to redox imbalance (44, 45). Therefore, cardio-specific disruptions can be another target of mycotoxins-mediated oxidative damage. This review provides a novel perspective on the oxidative aspects of relatively low-dose mycotoxins in the framework of systemic risk and vascular function.
Obstruction of the arteries (supplying blood to different tissues), defined as arterial dysfunction, is mainly the buildup of oxidative reactions and calcium and inflammatory deposits restoring each other (46). Tumor necrosis factor (TNF) inhibitors, the most common biologics used in treating rheumatological diseases, would exert favorable effects on the pathophysiology of the blood vessels (47). However, sustained and chronic inflammation is quickly becoming a controversial issue. Recent research has revealed a rich field for further investigation: a dual challenge of integrated oxidative and immune disruptions in circulating cells. There is a fundamental question: how repeated episodes of oxidative stress can induce a persistent inflammatory phenotype?
A significant consequence of the enhanced ROS formation and prolonged oxidative stress exposure is endothelial dysfunction via scavenging of NO by superoxide. The vascular microenvironment and the associated endothelial dysfunction look to predict the prognosis of the lesions. Endothelial cells (ECs) are a source of production contributing to chronic inflammatory states (48). EC's phenotype becomes activated through interaction with leukocytes and platelets, the expression of several proinflammatory cytokines [TNFα and interleukin-1 (IL-1)], and adhesion molecules. NF-κB is the pathway regulating endothelium-leukocyte interactions by adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin (49). Activation of the NF-κB pathway leads to the inflammatory EC responses by chemokines release, such as macrophage chemoattractant protein-1 (MCP-1) and IL-8 (50). That is, the activated ECs modulate inflammatory cells to overexpress scavenger receptors (CD36) and TLRs, essential for vascular oxidative stress (51).
In addition, ECs are a primary target of oxidative signals in circulation, importantly oxidizing lipoproteins. In the vessel wall, impairment of lipid metabolism could cause pathologic lipids accumulation (52). Malondialdehyde (MDA) represents an important factor in atherosclerosis because it principally exists in low-density lipoprotein cholesterol (LDL-C). Hence, excessive oxidation can act on lipids determining lipid peroxidation products. Indeed, MDA levels could precisely indicate the severity of lipid peroxidation injury, one of the initiating links of endothelial dysfunction in atherosclerosis (53). The oxidative modification of low-density lipoprotein (LDL) is an important inducer for atherogenic and proinflammatory pathways. In the atherosclerotic lesions, the primary source of chronic inflammatory responses is represented by lipid-laden foam cells caused by excessive lipid accumulation, ROS generation, intramural retention of oxidized LDLs (oxLDLs), and minimally oxLDLs, with many of these responses mediated by toll-like receptor-4 (TLR-4, by recognition and assembly of multi-molecular complexes, reviewed elsewhere) (54).
The oxidative imbalance may be inevitably enhanced during the atherosclerotic plaque progression as long as the modified lipoproteins and reinforcements possess proinflammatory properties (Figure 1). Of note, environmental triggers are a part of the story when the imbalance between cellular oxidative and antioxidant status becomes a critical underlying factor. Many genes, biomolecules, and immune cells associated with vascular dysfunction are considerably oxidative status-sensitive, the stress enhanced by relatively low mycotoxin levels (Table 2); thereby, a mixture of mycotoxins (Table 3) is an emerging issue in modern toxicological science contributing to different organs' pathobiology (Figure 2).
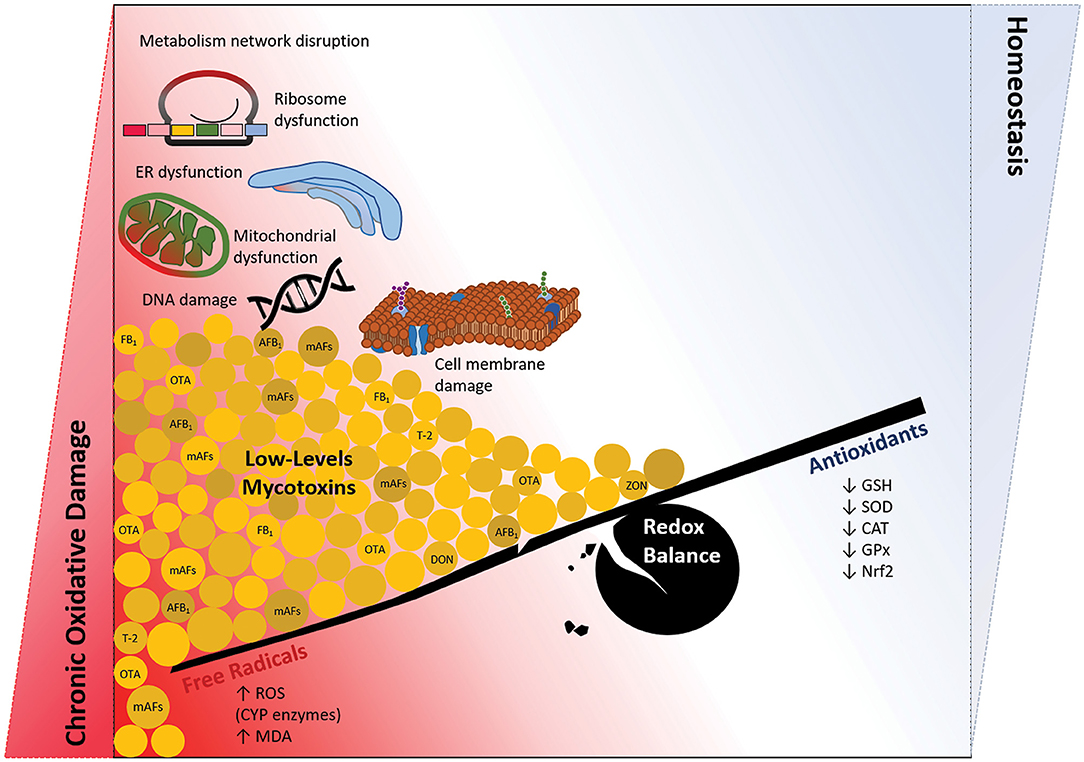
Figure 1. Long-term exposure to foods and feed contaminated by low doses of mycotoxins; the chronic oxidative damage vs. hemostasis. Coexposure to the mixture of mycotoxins, especially at nanomolar doses below the allowable levels (40), induces a chronic condition of oxidative disturbance in the cells. There would not be the context of redox balance regarding a continuous production of free radicals and loss of antioxidants defense system. Then, significant damage to macromolecules (DNA, protein, and lipid), organelles (mitochondria, ER, and ribosome), and metabolism network in the cell may orchestrate several pathophysiological conditions, including cardiovascular damages.
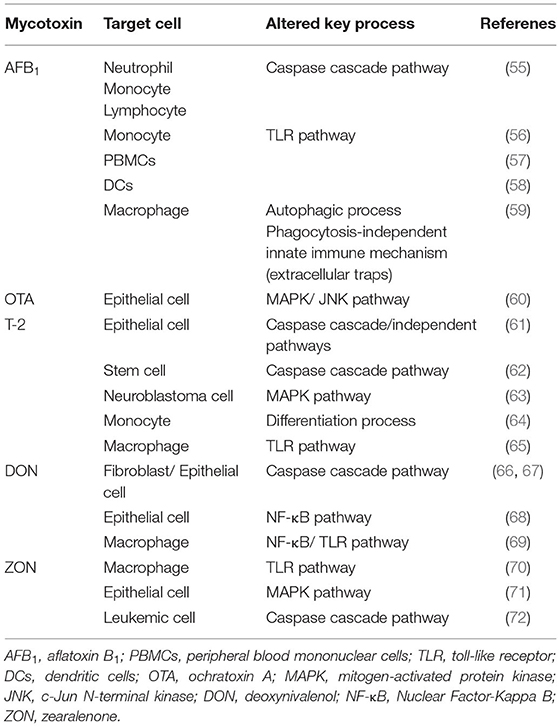
Table 2. The summarized list of stress-related immune pathways modulated by mycotoxins at very low doses; with possibly toxic action at the vascular level.
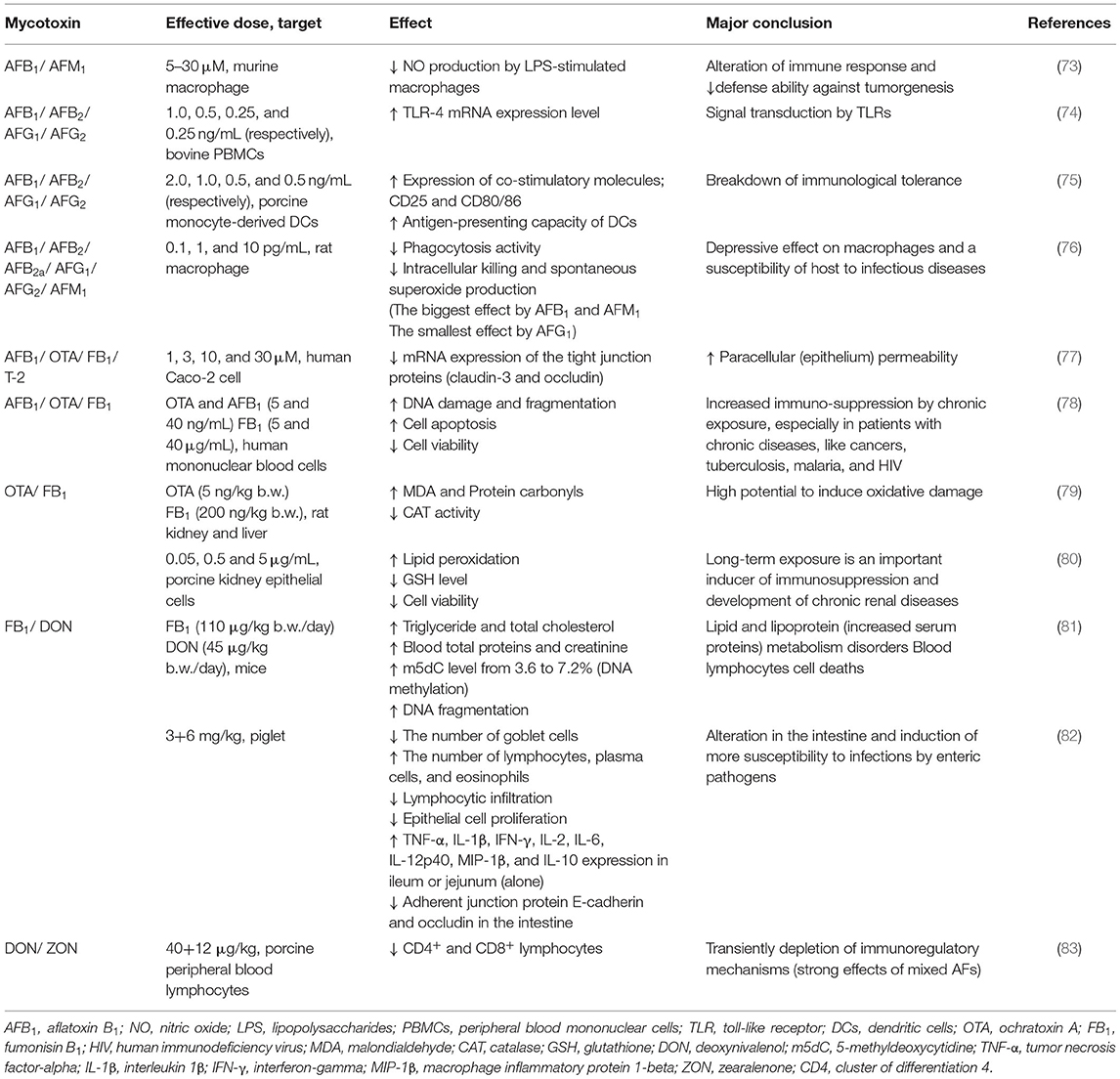
Table 3. Examples of combined mycotoxins effects disturbing metabolic and immune reactions (focusing on low concentrations).
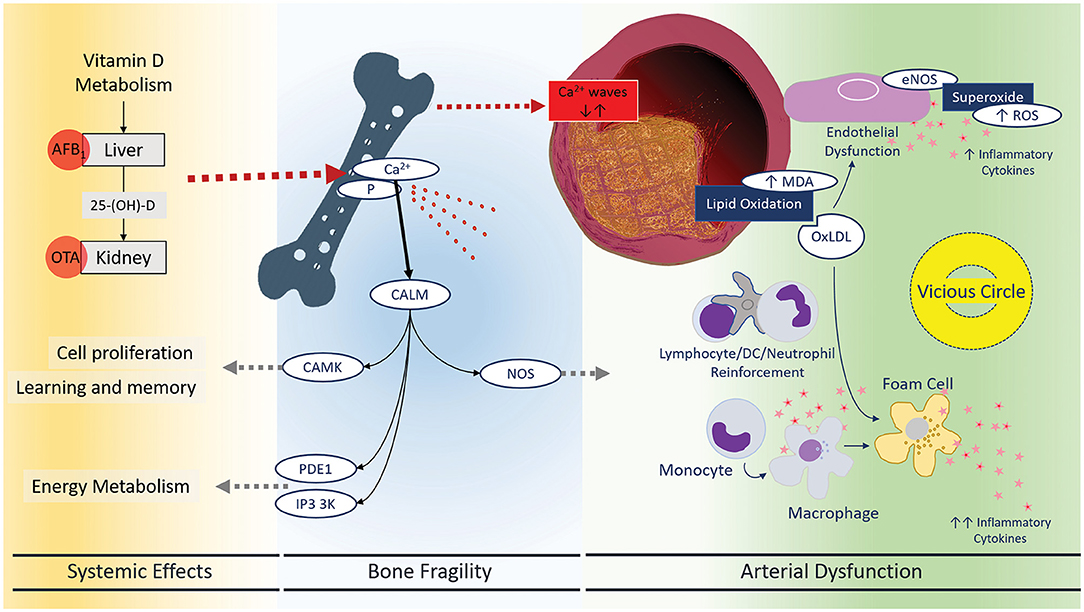
Figure 2. Systemic effects of mycotoxins leading to bone fragility and arterial dysfunction. There has been a meaningful connection between oxidative status and atherosclerotic vascular diseases. Now, mycotoxins are assumed to be an important environmental factor disrupting and or modulating oxidative and immune mechanisms in the blood vessels. Endothelial dysfunction, lipid peroxidation, and foam cell formation are three main characteristics of vascular inflammation: inflammatory responses and immune cell reinforcement. Mycotoxins can restore this vicious circle that may explain the high risk of stroke, heart attack, and PAD. CALM, Calmodulin 1–6, CAMK, calcium/calmodulin-dependent protein kinases, PDE1, phosphodiesterases, IP3 3K, inositol-trisphosphate 3-kinases, NOS, nitric oxide synthase.
Altering redox balance toward oxidation is a pivotal effect of prolonged exposure to mycotoxins. Circulation is the way by which oxidative agents reach different targets, from endothelial cells to bone marrow/structural cells. Therefore, prolonged exposure to low mycotoxins concentrations activates metabolic feedback between different tissues, affecting, finally, the whole metabolic homeostasis in a fragile condition. Reducing the general antioxidant capacity plus local inflammatory responses can result from exposure to low doses of different mycotoxins.
The earliest evidence show ROS system impairment as the underlying mechanism of the AFB1 toxic effects at the nanomolar levels (10–1000 nM) (84, 85). The toxic effects of nanomolar AFB1 metabolites on oxidative status and immune cells (neutrophils, monocytes, lymphocytes, and dendritic cells [DCs]) in mammals are represented by increased free radicals, inflammatory molecules, and mitochondrial pathways of cell apoptosis (via caspase-3/7/9 activation and ATP depletion) (9, 55, 86–88). The induction of apoptosis caused by 20 ng AFB1/mL is due to free radical production (Table 2), the most common sign of intercellular oxidative stress (42, 89). This, in turn, could be considered a potential risk for susceptibility to many chronic diseases. Nonetheless, the knowledge of low-level AFs' oxidative effects increasing the risk of infectious and non-infectious diseases is limited.
The cytochrome P450 (CYP) isoforms, which have key roles in metabolic pathways of AFB1, are stimulated via environmentally relevant levels of AFB1 (10 and 100 ng/mL) in circulating lymphocytes and monocytes (56). AFB1-epoxide strongly binds to the cell macromolecules, triggering more oxidative conditions (90). There is a positive correlation between the induction of CYPs and TLR-4, an essential inflammatory mediator. Highly induced CYPs (CYP1A1 and CYP1B1) by low doses of AFB1 in monocytes may lead to a 10-fold upregulation of TLR-4 (Table 2) (56). Thus, ROS production, caused by AFs, drives immune cells to prooxidant/inflammation status, such as autophagy and extracellular trap formation in macrophages, through TLRs (59). The sequential status of oxidative stress is concluded by several folds overexpression of myeloid differentiation primary response 88 (MyD88), TLR-2/4, and CD14 genes in nanomolar concentration of mixed AFs-exposed leukocytes (Table 2) (57), exacerbating immune and metabolic alterations.
Low AFB1 levels (50 μg/kg DM) can also induce substantial metabolic alterations involving cell membrane-associated metabolism, the tricarboxylic acid cycle, lipids, glycolysis, amino acid metabolism, serum lipoproteins, and N-acetyl glycoproteins. These are attributable to Cori and Krebs cycle disruptions, energy metabolism and mitochondrial function deregulations, decreased β-hydroxybutyrate and acetate, and increased acetoacetate and acetone production, suggesting the point of low-level AFB1-induced β-oxidation of fatty acids and suppressed tricarboxylic acid cycle (91). Together, AFB1 is a potent biosynthetic inhibitor affecting the metabolic processes toward the prooxidant status.
We recently showed that AFB1 and AFM1 at picomolar concentrations could further affect the metabolic pathways by inhibiting insulin production in pancreatic islets in vitro (92). In this work, a new framework for the toxicity of mycotoxins has opened. The Aflatoxins, and their metabolites, modulate the endocrine system, either directly or indirectly, via alteration in redox homeostasis. Further studies will scope low and biologically relevant levels of AFB1-induced oxidative imbalance translating to infection, metabolic alterations, and chronic inflammation.
Ochratoxin A (OTA) is a well-known potent inducer of ROS and lipid peroxidation in the mammalian system (93). OTA stably accumulates in many tissues and detection of nanomolar OTA (from 0.15 to 9.15 ng/mL) in plasma/serum is not surprising; hence it may be a risk factor for humans, especially following long-term exposure (94). In this context, the oxidative nature of low OTA levels in immune and metabolic processes should be more noticed.
A specific effect of low-level OTA, which is disturbing normal cell homeostasis (Figure 1), is based on OTA's interaction with secondary messengers and carriers, such as mitogen-activated protein kinases (MAPKs) (95). The oxidative stress amplifies the consequences of direct damage to DNA structure and interferes with the DNA repair system in cytotoxic T lymphocyte activity at a low concentration of OTA (96). OTA leads to a time- and dose-dependent induction of apoptosis through disruption in mitochondrial function and B-cell lymphoma extra-large (Bcl-xL) expression in leukocytes and Kit 225 cells. Bcl-xL, a transmembrane molecule in the mitochondria, would inhibit apoptosis by preventing mitochondria oxidative contents, such as cytochrome c efflux to the cytosol and activating caspases. At a low dose (0.5 μM), OTA exposes the cells to mitochondrial transmembrane potential loss and apoptosis (97). Since this toxin has a long half-life, chronic exposure to low-concentration OTA (98) may cause immunosuppression and severe vascular effects.
Surprisingly, in a sub-chronic intoxication model (547.2, 752.5, and 930.3 ng OTA/g kidney tissue in rats within 10, 30, and 60 days), OTA concentrations were higher in serum than in kidneys. Thereafter, oxidative stress and apoptosis are evident through increased MDA formation, lipid peroxidation, and decreased SOD. Increased free radicals level and subsequent mitochondrial membrane damage may accelerate the apoptosis process within 60 days of exposure to OTA (99). OTA-induced loss of Matrix metalloproteinases (MMPs) and antioxidant enzymes demonstrates activation of c-Jun N-terminal kinase (JNK)-mediated caspase3-dependent apoptosis and potentiating inflammatory mediators, such as TNFα (60). The 30-day exposure to low concentrations of OTA (0.05 mg/kg) significantly decreases nuclear factor erythroid 2-related factor 2 (Nrf2) expression, further confirming OTA-mediated prooxidant micro/macroenvironment, exacerbating inflammation (100).
A significant correlation between circulating inflammatory markers, such as C-reactive protein (CRP), and OTA level may be derived from systemic inflammation or mild tissue damage in chronic terms. The hypothesis of OTA-dependent inflammation comes from lipid peroxidation, ROS production, and apoptosis. Concerning the long-term exposure to OTA allied with oxidative subcellular structures (DNA, protein, and lipid), chronic inflammatory conditions, such as autoimmunity, rheumatoid sclerosis, musculoskeletal disorders, and cancer, are a new perspective to warrant further investigations.
There is little evidence about the induction of oxidative and inflammatory processes by prolonged exposure to low levels of FB1. The progress in FB1 research on oxidative imbalance as the plausible mechanism for pleiotropic toxicities has not been addressed at nanomolar concentrations. The proposed mechanism of action is the inhibition of ceramide synthase by occupying sphingosine and fatty acyl-CoA interactions. Following the inhibition of ceramide biosynthesis, several mechanisms would be correlated with oxidative stress and apoptosis due to immunolocalization of the lower doses of FB1 in specific targets, such as mitochondria and nucleus (Figure 1) (101). The lowest FB1 concentration (0.5 μM) may raise the rate of cytosolic ROS production and inhibit mitochondrial respiration (102). At the low levels of FB1, apoptosis is likely the mechanism of cell death initiating uncontrolled pathways.
The oxidative nature of FB1 mediates acute lipid metabolism. The lowest FB1 doses (10 and 50 mg/kg) in rats, for a minimum period, changes the main cellular polar lipid fractions and the fatty acid (FA) composition of the hepatic mitochondrial phospholipids. A decreased phosphatidylcholine and phosphatidylethanolamine decreased the polyunsaturated/saturated fatty acids (FA) ratio which suggests that FB1 impairs the membrane fluidity (103). Also, the proportion of fatty acids decreased in the phospholipid fraction, modifying the hepatocellular membrane lipids, indicates the acute membrane-damaging effect of FB1. C22 N3 polyunsaturated FA (PUFA) shows a proportional decline because of very high sensitivity to oxidative stress and lipid peroxidation reflected by MDA and glutathione (GSH). The hepatic GSH level, a well-known marker of antioxidative capacity, decreases under FB1 exposure. And one might hypothesize that FB1 targets and modifies the membrane phospholipids to generate hydrogen peroxides. Therefore, lipid peroxidation in the FB1-induced stress is probably reflected by increased lipid-associated parameters, including hepatic lipoprotein secretion, total cholesterol, and MDA and GSH levels in blood plasma (103).
Exposure to even permissible levels of FB1 would induce lipid peroxidation and redox imbalance due to mitochondria involvement, which could lead to sequential proinflammatory micro/macroenvironment in various cells and organs (104). The effects, naturally, will be more pronounced in muscles and neurons due to their significant dependence on mitochondria (Figure 3). It is needed to do more on the lower or nanomolar levels of FB1 to elucidate the molecular mechanism.
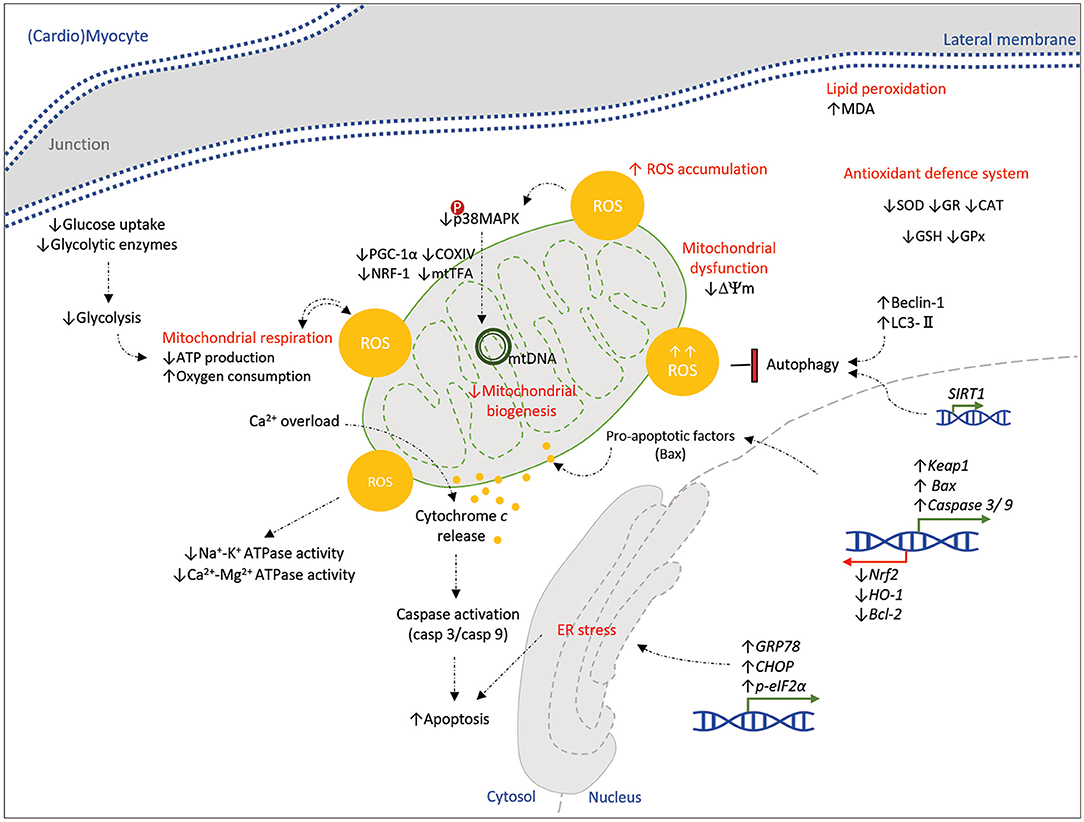
Figure 3. Schematic representation of the molecular pathways affected by mycotoxins in a (cardio)myocyte. Mycotoxins (AFB1, OTA, T-2, DON, and ZON) show toxicity on a (cardio)myocyte, principally by excessive oxidative stress and ROS accumulation. Alterations in mitochondrial bioenergetics, mitochondrial dysfunction, transcription of stress/apoptotic genes, and ER stress orchestrate the cell damage and apoptosis. Dysregulation of the antioxidative system and protective pathways (autophagy) would enhance these mechanisms of toxic myopathy. Down- and upstream signaling pathways that might be affected by mycotoxins in a myocyte are still unknown to be represented here; hence it is highly suggested to be determined by further studies. Green and red arrows (blocked line) indicate activation and inhibition, respectively.
T-2 toxin is lipophilic and easily absorbed through the skin, gut, and pulmonary mucosa. Exposure to a sub-lethal dose of T-2 toxin refers to several hematologic and immunotoxic effects targeting immune cells, macrophages, and peripheral blood B and T lymphocytes (105). Hematopoietic and lymphoid tissues are susceptible to T-2 toxin (106). It causes apoptotic death in highly proliferating cells, such as lymphocytes (107) and intestinal crypt epithelial cells (108). Nanomolar T-2 and its major metabolite (HT-2) may account for redox imbalance in the circulation (Figure 1) and intracellular spaces underlying apoptosis induction (109).
Therefore, oxidative stress is the suggested mechanism of T-2 toxicity as LC50 of 10 ng/mL induces apoptosis in HeLa cells through caspases dependent and independent pathways (61). Even at a concentration <2 ng/mL, DNA fragmentation and apoptosis occur through the involvement of mitochondrial apoptogenic factors, Bcl-2-associated X (Bax), B-cell lymphoma 2 (Bcl-2), cytochrome c, apoptosis-inducing factor (AIF), and caspases 9, 3, and 7 (Table 2) (42, 62). Then, increased ROS and MDA levels, associated with loss of intracellular GSH, SOD, and catalase (CAT) activities, prove the evidence of oxidative stress as the main mechanisms of 1 and 2 ng T-2 toxin/mL-induced apoptosis (110).
T-2 toxin activates apoptotic processes through the ROS-mediated MAPK signaling pathway (Table 2) (63). Exposure to high nanomolar levels (40 nM) causes a significant time-dependent increase in ROS and MMP loss in GH3 cells (111). The implication of T-2 toxicity at nanomolar concentrations can be significantly reduced by protecting the mitochondrial activity (112).
Monocytes and macrophages are privileged targets of T-2, enough to control the principal mediators of inflammation (64). The potential mechanism of this disturbance in monocyte differentiation has not been explored.
A pre-exposure to nanomolar T-2 concentrations reduces nitric oxide (NO) production and cell viability in the porcine alveolar macrophages activated by TLR-4 and TLR-2/6 agonists (Table 2) (65). Then, TLRs signal inflammatory pathways and metabolic inflammation. It can explain how metabolic imbalance underlying low-level inflammatory processes resulting from T-2 would be the common characteristic of chronic conditions such as vascular dysfunction and sarcopenia.
DON or vomitoxin disturbs oxidative status in rainbow trout at concentrations below the limit fixed by the European Commission (113). In animals fed with low-dose DON, prooxidant and antioxidant parameters analyses indicate changes in SOD, GPx, and CAT activities in erythrocytes and the blood (114).
The metabolomics data can confirm the role of oxidative stress and metabolic alterations in cellular processes, including redox homeostasis, energy balance, membrane integrity, and lipid metabolism by DON (Figure 1) (115). Peroxidation of phospholipids and perturbation of membrane integrity are expected in exposed cells. It triggers lipid peroxidation (high MDA levels), responsible for membrane and DNA damage in human lymphocytes (116). Nanomolar of DON causes antioxidants (GSH) depletion and oxidative DNA damage, leading to reduced expression of heme oxygenase-1 (HO-1) (a homeostatic and protective agent against cellular stress) and loss of DNA reparative potential. Thus, it potentially induces genotoxicity and apoptosis in human peripheral blood lymphocytes (42, 116).
The intracellular apoptotic signals of DON are also explored on epithelial cells. Activation of the caspase cascade system, such as caspase-3, caspase-8, caspase-9, and poly (ADP-ribose) polymerase, and the increased level of P53, ROS, and the ratio of Bax/Bcl-2 contribute to loss of MMP in epithelial cells (66). Following 250 ng/mL DON exposure, the intracellular ROS and ribotoxic stress drive the expression of inflammatory genes, NF-κB and cyclooxygenase-2 (COX-2) (Table 2) (68). In macrophages, this DON-mediated effect on NF-κB is stimulated by MyD88-dependent TLR signals. Hence, DON poses a risk of an oxidative imbalance in the organelles, such as ribosome and endoplasmic reticulum (ER), leading to T-cell activation and apoptosis (117). Characterization of the modes of action of DON, therefore, formulates the potential mechanisms of DON toxicity on immune and endothelial cells in the blood.
ZON is known for its robust estrogenic activity because it competes with 17β-estradiol to bind to cytosolic estrogen receptors (118). But, oxidative damage has been recently documented as an initiating event responsible for different genotoxic and cytotoxic effects of ZON (Figure 1) (119). Non-cytotoxic concentrations impairs cell viability through oxidative DNA damage, GSH depletion, and stress-responsive genes (Hsp70 and Hsp90) induction (119). Mitochondrial oxidative stress, plasma membrane permeabilization, and apoptosis are induced by 3 and 100 nanomolar (120).
Low levels of ZON lead to an imbalance in immune cells and pro-/anti-inflammatory cytokines. It changes blood lymphocyte populations and proliferative capacity, with a significant increase in IFN-ɤ levels (83). Co-exposure to ZON metabolites might cause more substantial adverse effects on the viability and expression of inflammatory factors, TLR ligand stimulation, IL-1β, TNFα, IL-6, and IL-8 (121). MAPK pathways involving extracellular signal-regulated kinases (ERKs), p38, and JNK signaling are essential in transducing extracellular stimuli; that is, oxidative signals derived from ZON (71). However, the exact molecular mechanisms of ZON toxicity inducing inflammatory reactions have not been investigated. The cytotoxic effects of ZON are, in part, thought to be correlated with its apoptotic potential by activation of mitochondrial cytochrome c, ROS production, induction of caspase-3/8, and ER stress in human leukemia cells (Table 2) (72). Regarding changes in blood metabolic profile associated with long-term exposure to low doses of ZON, it would represent a probable reason behind vascular damage.
Among mycotoxins, AFs are more studied due to their higher potential to disrupt. Mycotoxins share many mechanisms of action; therefore, here, the discussion will follow with AFs. The acute effect of AFs on the cardiovascular system is vascular fragility and thus hemorrhage and teratogenicity. Metabolic alterations and inflammatory conditions might occur due to AFs exposure to oxidative stress leading to lipid peroxidation and oxidative DNA damage (Figure 1) (122). Lipid peroxidation is the most destructive process in cells associated with excessive ROS production. The high or moderate increase in ROS seems detrimental for myocardial cells, while its physiological level is necessary for normal cell signaling (123).
Intraperitoneal administration of AFB1 as a single dose of 2 mg or 17 μg/kg increases markers of oxidative damage in the heart, such as thiobarbituric reactive substances, like MDA, and calcium (Ca)2+ levels (124). Cardiomyocytes, and more generally all muscle cells, are specifically susceptible to oxidative modifications and free radicals because the antioxidant defense mechanisms are very limited in these cells compared to other types of cells in the body (125). Indeed, the susceptibility of cardiomyocytes and vascular muscles to oxidative damage under aflatoxicosis is due to the significant reduction in total antioxidant capacity, ATPase, and antioxidant enzymatic and non-enzymatic activities, including SOD, CAT, glutathione reductase (GR), GPx, GSH, and vitamins C and E (123).
In addition, AFB1 interferes with cellular energy supply in myocardial cells (126). Glycolysis is also one of the most important metabolic processes participating directly in ATP production to meet cellular energy demands in the heart. Myocytes need continuous ATP production as it cannot be restored (127, 128). AFB1 alters such bioenergetic sources, increasing myocytes' susceptibility to metabolic stress and affecting myocardial contractile performance (126). A single dose of 1 mg AFB1/kg can inhibit glycogen synthesis, glucose uptake (a decrease in glucose contents of myocardial cells), and cardiac glycolysis in rats. Then, glycolytic enzymes, hexokinase, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), phosphoglucose isomerase (PGI), and lactate dehydrogenase (LDH), as well as the rate of glycolysis, are declined by AF exposure (126).
As energy producers (90% of ATP), mitochondria have attracted much interest regarding impairment of mitochondrial activity which may result in cardiomyopathy. In rat cardiomyocytes, low doses of AFB1 result in mitochondrial damage through disruption in mitochondrial membrane and cristae (129). Mitochondrial dysfunction and expression of apoptotic proteins, such as active caspase-3, Bax, and Bcl-2 caused by low levels (0.75 mg/kg) of AFB1 can finally cause severe cardiomyocyte dysfunction (129). It could be a hidden danger because cardiomyocytes' renewal is extremely low in humans (130). However, the precise mechanisms of AFB1 and cardiomyocytes' apoptotic pathways are not precise.
High NO concentration and pro-inflammatory cytokines, TNFα and IL-1α by AFB1 ingestion (131), can also inhibit mitochondrial ATP production and stimulate cardiomyocytes apoptosis (132). In the low range of concentrations, 0.5, 1.0, 2.5, and 5.0 μmol/L of AFB1, mitochondrial functions are determined by glutamate/malate and succinate-driven respiration. In a concentration-dependent manner, glutamate/malate-driven respiration is significantly uncoupled, increasing the state of respiration significantly vs. a decrease in broiler cardiomyocytes (BCMs) (133). Hence, AFB1 may uncouple the mitochondrial respiration and oxidative phosphorylation in BCMs. AFB1 results in a significant and concentration-dependent increase in intracellular ROS production in BCMs because a main source of ROS can be mitochondrial respiratory chain uncoupling (133). High levels of mitochondrial ROS and membrane lipids peroxidation exhibit mitochondrial oxidative stress-induced apoptosis in BCMs. On the other hand, this toxicity response to AFB1 is proven by a significant increase in LDH and cardiac troponins leakage from BCMs, as sensitive markers for acute cardiac damage (133). It is well-known that the antioxidant defense system, including antioxidant enzymes and sensitive transcription factors, tries to preserve the redox balance (134). Considering this condition, there is a significant decrease and increase in mRNA expression of SOD and Nrf2 genes, respectively, in BCMs by exposure to AFB1 (133). So, the myocardium of rats indicates patchy necrosis with a surrounding inflammatory reaction within the first 4 days of AFB1 exposure.
The histopathological examination of ventricular muscle sections in rabbits orally administered at a dose of 30 μg AFB1/kg revealed nuclear pyknosis and peripheralization, sarcoplasmic vacuolation, and myofibrils degeneration with the use of minimization of disrupted intercalated disks and congestion and dilatation blood vessels, in addition to mononuclear cellular infiltration. Edema distribution among myocardial fibers illustrates severe cardiotoxicity and subsequent heart damage by AFs. Figure 3 summarizes mycotoxins toxicity in a (cardio)myocyte at the molecular level. According to similar data in Table 4 for cardiotoxicity induced by other common mycotoxins at low and high levels, the chronic oxidative stress and damage would be concluded by long-term exposure to different levels of mycotoxins. Although this condition represents cardiac cell damage and heart failure in vitro and in vivo, there is still a need to do large-scale investigations.
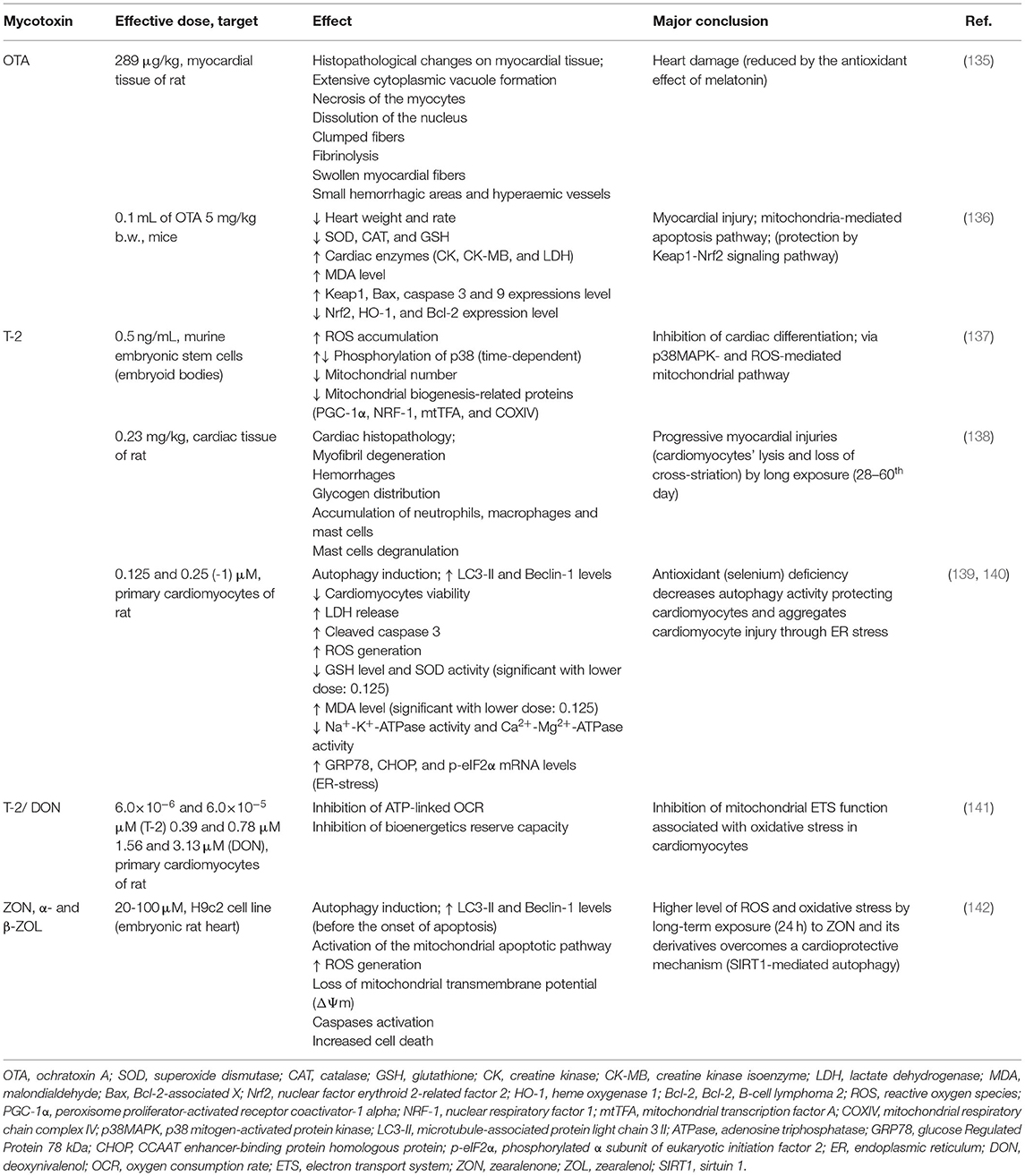
Table 4. The cardiotoxicity of mycotoxins.
Mycotoxins affect different cells throughout their travel in circulation. As aforementioned, the effects on redox balance favoring more oxidation can reduce the antioxidant capacity in the circulation and damage protective mechanisms. Bone homeostasis strictly depends on the correct function of metabolic pathways and the endocrine system, particularly vitamin D. There is experimental evidence that low-level AFs supposed to change vitamin D and parathyroid hormone (PTH) metabolism (143).
Also, vitamin D metabolism is consistently associated with the modulation of vascular tone. It is correlated with atherogenic blood lipid profiles, such as total cholesterol, triglycerides, and LDL/HDL-cholesterol, as risk factors for cardiovascular diseases (144). Supplementation studies evident the vitamin D deficiency in the development of atherosclerosis at the site of the blood vessels (145). It is an atherogenic factor influencing adhesion molecules, ECs activation, and VSMC proliferation. Simultaneously, vitamin D deficiency is associated with oxidative stress and inflammation involving immune cells, particularly monocytes and macrophages (144). The 22-oxacalcitriol is a vitamin D analog that can suppress the high expression levels of p22phox and NADPH oxidase enzyme (generating superoxide) and improve eNOS coupling, thereby reducing ROS production oxidative stress in the vasculature (146).
Considering the mechanism of action, vitamin D, in the active form of 1,25-(OH)2-D, binds to nuclear vitamin D receptor (VDR), membrane-bound and cytoplasmic receptors. These receptors are considerably expressed in all cells playing in atherosclerosis, such as ECs, VSMCs, and immune cells. Hence, vitamin D might regulate a wide range of physiological and pathological processes, including vascular cell growth, migration, and differentiation, immune response modulation, and inflammatory pathways in the blood vessels (147).
Naturally occurring mycotoxins might indirectly affect bone and skeletal apparatus hemostasis via interference with vitamin D metabolism. Increasing low-level AF intake significantly decreases bone mineralization parameters, such as tibia breaking strength (TBS) and the percentages of Ca and P in the tibia, associated with increased PTH and decreased PTH 1,25-dihydroxycholecalciferol. These adverse effects on broilers' P metabolism and bone mineralization might relate to the vitamin D metabolism (143).
It has been reviewed elsewhere that certain mycotoxins, singly and severally, including AFB1, OTA, T-2 toxin, and FB1, can affect bone growth strength and cause bone fragility (148). As a crucial hormone in Ca bone's homeostasis and metabolism, vitamin D has been essential for bone health (149). Mycotoxins cause a metabolic situation, such as reducing circulating Ca and P content in the blood, eventually reducing bone strength (148).
Notably, the expression of VDRs was significantly downregulated by the natural occurrence of AFB1 (at nM concentrations) in osteosarcoma cell line SaOs-2. It could suggest that naturally occurring mycotoxins that are toxic toward VDRs can potentially interfere with vitamin D's action on Ca-binding gene expression in tissues (150). Therefore, the unexpected toxicity of the AFB1 toward the VDRs proposes indirect effects of combined mycotoxins on the cardiovascular system.
T-2 toxin targets bone and exposure to T2 may associate with several bone diseases. Indeed, the C57BL/6 T-2 toxin suppresses Wnt/β-catenin signaling and expression of downstream target genes and increases autophagy and apoptosis (151).
Bone marrow can be a sensible target for mycotoxins action. For instance, mesenchymal stem cells (MSCs) are susceptible to oxidative stress that can induce DNA breaks and genomic instability (152). Exposure of pregnant rats to FBs impairs bone metabolism of the offspring via alteration of the osteoprotegerin (OPG)-to-RANκL (receptor activator of nuclear factor κB ligand), possibly driven by immunological alterations (153), and by affecting the expression of remodeling enzymes [MMPs and their tissue inhibitors (TIMPs)] and angiogenic factors [vascular endothelial growth factor (VEGF)] (154).
Besides this evidence, calcium-phosphorous metabolism, vitamin D action, and bone turnover emerge as important targets of mycotoxins' effects. Therefore, it may be of interest to enhance the knowledge of these aspects.
Acknowledging potential mechanisms responsible for releasing the peroxidative decomposition products, for example, MDA, in the arteria would open new perceptions of biochemical and cellular events in arterial dysfunction and strategies for intervention. Oxidative stress represents a long-term reaction against some stimuli, which is characterized by a long-lasting inflammatory condition. Therefore, it becomes a unifier of various chronic diseases, such as artery diseases, considering the key facts. Activating mitochondrial permeability transition leads to potential loss and the release of cytochrome c upregulates the caspase-3 and−9 (62). Excessive ROS production under the pathological condition cannot be efficiently inhibited by the protective antioxidant mechanisms, leading to a state of lipid and DNA peroxidation.
Mycotoxins-assisted oxidative imbalance, mitochondrial dysfunction, and cytotoxicity are unusual mechanisms of action in biological systems. Due to the widespread presence of mycotoxins as contaminants frequently occurring in feed and food, it is likely that the oxidative aspect of mycotoxins individually could also be enhanced by subchronic coexposure to all of them. As represented in Table 3, chronic exposure to combined mycotoxins may have additive or synergistic impacts on the oxidative and immune players. It is necessary to fully understand to what extent the oxidative status in this microenvironment, involving an essential component of the human cardiovascular system, may be responsible for priming immune disruptions, artery diseases, and cardiomyopathies in individuals exposed to the combined mycotoxins. Future studies may also indicate the importance of antioxidant applications in human health to benefit from the promising therapeutic and preventive strategies in people at risk of diseases.
Apart from the oxidative nature of mycotoxins, their intracellular molecular hits remain inconclusive. There is little knowledge regarding the putative proteins that can be targeted directly with these small toxins and/or their metabolites. The lack of knowledge is more remarkable considering the other groups of mycotoxins, such as masked and minor mycotoxins, all of which are unregulated. Further research is needed to address this critical piece of understanding.
Mycotoxins' indirect systemic effects throughout the unfavorable influence on calcium signaling and VitD metabolism (Figure 2) involve key organs responsible for maintaining homeostasis. In conditions with lowering the VitD precursors due to mycotoxins' effects on intestinal cells or hepatocytes, the bone will release the calcium into the circulation. Calcium release and reabsorption are cycles that dynamically keep the general homeostasis; however, dysregulation of this dynamic can create a pulse of high Ca concentration in circulation promoting calculations in arterial walls. Hence, lowering the Ca content of the bone leads to osteoporosis susceptibility and/or bone fragility. The unusual release of Ca from the bone activates downstream Ca/calmodulin-dependent cascades, affecting cell survival, inflammation, and energy metabolism. The discovery of proteins affected by most and/or the possible more susceptible protein isoforms can be the next step in this research field.
AM and GL: conceptualization, writing, editing, and final approval. SS: writing, editing, and final approval. JM: conceptualization, editing, and final approval. All authors contributed to the article and approved the submitted version.
The works of JM and AM, cited in this review, have been supported by grants from the Ferdowsi University of Mashhad, the University of Tehran, and the Iranian National Elite Foundation. AM has received support from Italian Ministry of Health, Ricerca Corrente Program, to the IRCCS Istituto Ortopedico Galeazzi.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Alshannaq A, Yu J-H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int J Environ Res Public Health. (2017) 14:632. doi: 10.3390/ijerph14060632
2. EFSA. Risk assessment of aflatoxins in food. EFSA J. (2020) 18:112. doi: 10.2903/j.efsa.2020.6040
3. Marin S, Ramos A, Cano-Sancho G, Sanchis V. Mycotoxins: Occurrence, toxicology, exposure assessment. Food Chem Toxicol. (2013) 60:218–37. doi: 10.1016/j.fct.2013.07.047
4. Taheur FB, Kouidhi B, Al Qurashi YMA, Salah-Abbès JB, Chaieb K. Biotechnology of mycotoxins detoxification using microorganisms and enzymes. Toxicon. (2019) 160:12–22. doi: 10.1016/j.toxicon.2019.02.001
5. Shahba S, Mehrzad J, Malvandi AM. Neuroimmune disruptions from naturally occurring levels of mycotoxins. Environ Sci Pollut Res. (2021) 28:32156–76. doi: 10.1007/s11356-021-14146-4
6. Qian G, Tang L, Guo X, Wang F, Massey ME, Su J, et al. Aflatoxin B1 modulates the expression of phenotypic markers and cytokines by splenic lymphocytes of male F344 rats. J Appl Toxicol. (2014) 34:241–9. doi: 10.1002/jat.2866
7. Reddy R, Sharma R. Effects of aflatoxin B1 on murine lymphocytic functions. Toxicology. (1989) 54:31–44. doi: 10.1016/0300-483X(89)90076-0
8. Raisuddin S, Singh K, Zaidi S, Paul B, Ray P. Immunosuppressive effects of aflatoxin in growing rats. Mycopathologia. (1993) 124:189–94. doi: 10.1007/BF01103737
9. Mehrzad J, Hosseinkhani S, Malvandi AM. Human microglial cells undergo proapoptotic induction and inflammatory activation upon in vitro exposure to a naturally occurring level of aflatoxin B1. Neuroimmunomodulation. (2018) 25:176–83. doi: 10.1159/000493528
10. Mehrzad J, Devriendt B, Baert K, Cox E. Aflatoxin B1 interferes with the antigen-presenting capacity of porcine dendritic cells. Toxicol In Vitro. (2014) 28:531–7. doi: 10.1016/j.tiv.2013.11.015
11. Peng X, Bai S, Ding X, Zhang K. Pathological impairment, cell cycle arrest and apoptosis of thymus and bursa of fabricius induced by aflatoxin-contaminated corn in Broilers. Int J Environ Res Public Health. (2017) 14:77. doi: 10.3390/ijerph14010077
12. Wang F, Zuo Z, Chen K, Gao C, Yang Z, Zhao S, et al. Histopathological injuries, ultrastructural changes, and depressed TLR expression in the small intestine of broiler chickens with aflatoxin B1. Toxins. (2018) 10:131. doi: 10.3390/toxins10040131
13. Jiang M, Peng X, Fang J, Cui H, Yu Z, Chen Z. Effects of aflatoxin B1 on T-cell subsets and mRNA expression of cytokines in the intestine of broilers. Int J Mol Sci. (2015) 16:6945–59. doi: 10.3390/ijms16046945
14. Cusumano V, Rossano F, Merendino R, Arena A, Costa G, Mancuso G, et al. Immunobiological activities of mould products: functional impairment of human monocytes exposed to aflatoxin B1. Res Microbiol. (1996) 147:385–91. doi: 10.1016/0923-2508(96)84713-9
15. Methenitou G, Maravelias C, Athanaselis S, Dona A, Koutselinis A. Immunomodulative effects of aflatoxins and selenium on human natural killer cells. Vet Hum Toxicol. (2001) 43:232–4.
16. Chen X, Horn N, Cotter P, Applegate T. Growth, serum biochemistry, complement activity, and liver gene expression responses of Pekin ducklings to graded levels of cultured aflatoxin B1. Poult Sci. (2014) 93:2028–36. doi: 10.3382/ps.2014-03904
17. Marin D, Taranu I, Bunaciu R, Pascale F, Tudor D, Avram N, et al. Changes in performance, blood parameters, humoral and cellular immune responses in weanling piglets exposed to low doses of aflatoxin. J Anim Sci. (2002) 80:1250–7. doi: 10.2527/2002.8051250x
18. Yang X, Zhang Z, Wang X, Wang Y, Zhang X, Lu H, et al. Cytochrome P450 2A13 enhances the sensitivity of human bronchial epithelial cells to aflatoxin B1-induced DNA damage. Toxicol Appl Pharmacol. (2013) 270:114–21. doi: 10.1016/j.taap.2013.04.005
19. Caloni F. Transport of aflatoxin M1 in human intestinal Caco-2/TC7 cells. Front Pharmacol. (2012) 3:111. doi: 10.3389/fphar.2012.00111
20. Shirani K, Zanjani BR, Mahmoudi M, Jafarian AH, Hassani FV, Giesy JP, et al. Immunotoxicity of aflatoxin M1: as a potent suppressor of innate and acquired immune systems in a subacute study. J Sci Food Agric. (2018) 98:5884–92. doi: 10.1002/jsfa.9240
21. Ferrante M, Raso GM, Bilancione M, Esposito E, Iacono A, Meli R. Differential modification of inflammatory enzymes in J774A.1 macrophages by ochratoxin A alone or in combination with lipopolysaccharide. Toxicol Lett. (2008) 181:40–6. doi: 10.1016/j.toxlet.2008.06.866
22. Lautert C, Ferreiro L, Wolkmer P, Paim FC, Da Silva CB, Jaques JA, et al. Individual in vitro effects of ochratoxin A, deoxynivalenol and zearalenone on oxidative stress and acetylcholinesterase in lymphocytes of broiler chickens. Springerplus. (2014) 3:1–7. doi: 10.1186/2193-1801-3-506
23. Marin DE, Taranu I, Pascale F, Lionide A, Burlacu R, Bailly J-D, et al. Sex-related differences in the immune response of weanling piglets exposed to low doses of fumonisin extract. Br J Nutr. (2006) 95:1185–92. doi: 10.1079/BJN20061773
24. Obremski K, Podlasz P, Zmigrodzka M, Winnicka A, Wozny M, Brzuzan P, et al. The effect of T-2 toxin on percentages of CD4+, CD8+, CD4+ CD8+ and CD21+ lymphocytes, and mRNA expression levels of selected cytokines in porcine ileal Peyer's patches. Pol J Vet Sci. (2013) 16:341–9. doi: 10.2478/pjvs-2013-0046
25. Yang G-H, Jarvis BB, Chung Y-J, Pestka JJ. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: relationship to ERK, p38 MAPK, SAPK/JNK activation. Toxicol Appl Pharmacol. (2000) 164:149–60. doi: 10.1006/taap.1999.8888
26. Sugita-KONISHI Y, Hara-Kudo Y, KASUGA F, KUMAGAI S. The effects of trichothecenes on host defense against infectious diseases. JSM Mycotoxins. (1998) 1998:19–23. doi: 10.2520/myco1975.1998.47_19
27. A.N. da Costa, Keen JN, Wild CP, Findlay JB. An analysis of the phosphoproteome of immune cell lines exposed to the immunomodulatory mycotoxin deoxynivalenol. Biochim Biophys Acta. (2011) 1814:850–7. doi: 10.1016/j.bbapap.2011.04.001
28. Pinton P, Accensi F, Beauchamp E, Cossalter A-M, Callu P, Grosjean F, et al. Ingestion of deoxynivalenol (DON) contaminated feed alters the pig vaccinal immune responses. Toxicol Lett. (2008) 177:215–22. doi: 10.1016/j.toxlet.2008.01.015
29. Becker C, Reiter M, Pfaffl MW, Meyer HH, Bauer J, Meyer KH. Expression of immune relevant genes in pigs under the influence of low doses of deoxynivalenol (DON). Mycotoxin Res. (2011) 27:287. doi: 10.1007/s12550-011-0106-7
30. Wu X, Kohut M, Cunnick J, Bailey T, Hendrich S. Deoxynivalenol suppresses circulating and splenic leukocyte subpopulations in BALB/c mice: dose response, time course and sex differences. Food Addit Contam. (2009) 26:1070–80. doi: 10.1080/02652030902832959
31. Maresca M, Mahfoud R, Garmy N, Fantini J. The mycotoxin deoxynivalenol affects nutrient absorption in human intestinal epithelial cells. J Nutr. (2002) 132:2723–31. doi: 10.1093/jn/132.9.2723
32. Tardivel C, Airault C, Djelloul M, Guillebaud F, Barbouche R, Troadec J-D, et al. The food born mycotoxin deoxynivalenol induces low-grade inflammation in mice in the absence of observed-adverse effects. Toxicol Lett. (2015) 232:601–11. doi: 10.1016/j.toxlet.2014.12.017
33. Meky F, Hardie L, Evans S, Wild C. Deoxynivalenol-induced immunomodulation of human lymphocyte proliferation and cytokine production. Food Chem Toxicol. (2001) 39:827–36. doi: 10.1016/S0278-6915(01)00029-1
34. Zhou H-R, Islam Z, Pestka JJ. Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol Sci. (2003) 72:130–42. doi: 10.1093/toxsci/kfg006
35. J. Van De Walle, Romier B, Larondelle Y, Schneider Y-J. Influence of deoxynivalenol on NF-κB activation and IL-8 secretion in human intestinal Caco-2 cells. Toxicol Lett. (2008) 177:205–14. doi: 10.1016/j.toxlet.2008.01.018
36. Maresca M, Yahi N, Younès-Sakr L, Boyron M, Caporiccio B, Fantini J. Both direct and indirect effects account for the proinflammatory activity of enteropathogenic mycotoxins on the human intestinal epithelium: Stimulation of interleukin-8 secretion, potentiation of interleukin-1β effect and increase in the transepithelial passage of commensal bacteria. Toxicol Appl Pharmacol. (2008) 228:84–92. doi: 10.1016/j.taap.2007.11.013
37. Obremski K. The effect of in vivo exposure to zearalenone on cytokine secretion by Th1 Th2 lymphocytes in porcine Peyer's patches after in vitro stimulation with LPS. Pol J Vet Sci. (2014) 17:625–32. doi: 10.2478/pjvs-2014-0093
38. Obremski K. Changes in Th1 and Th2 cytokine concentrations in ileal Peyer's patches in gilts exposed to zearalenone. Pol J Vet Sci. (2014) 17:53–9. doi: 10.2478/pjvs-2014-0007
39. Alassane-Kpembi I, Kolf-Clauw M, Gauthier T, Abrami R, Abiola FA, Oswald IP, et al. New insights into mycotoxin mixtures: the toxicity of low doses of Type B trichothecenes on intestinal epithelial cells is synergistic. Toxicol Appl Pharmacol. (2013) 272:191–8. doi: 10.1016/j.taap.2013.05.023
40. Mehrzad J, Klein G, Kamphues J, Wolf P, Grabowski N, Schuberth H. In vitro effects of very low levels of aflatoxin B1 on free radicals production and bactericidal activity of bovine blood neutrophils. Vet Immunol Immunopathol. (2011) 141:16–25. doi: 10.1016/j.vetimm.2011.01.010
41. Forman HJ, Zhang H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat Rev Drug Discov. (2021) 20:689–709. doi: 10.1038/s41573-021-00233-1
42. Chatzidoukaki O, Stratigi K, Goulielmaki E, Niotis G, Akalestou-Clocher A, Gkirtzimanaki K, et al. R-loops trigger the release of cytoplasmic ssDNAs leading to chronic inflammation upon DNA damage. Sci Adv. (2021) 7:eabj5769. doi: 10.1126/sciadv.abj5769
43. Bolland MJ, Grey A, Avenell A. Effects of vitamin D supplementation on musculoskeletal health: a systematic review, meta-analysis, and trial sequential analysis. Lancet Diabetes Endocrinol. (2018) 6:847–58. doi: 10.1016/S2213-8587(18)30265-1
44. Pohjoismäki JL, Goffart S. The role of mitochondria in cardiac development and protection. Free Radic Biol Med. (2017) 106:345–54. doi: 10.1016/j.freeradbiomed.2017.02.032
45. Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. (2015) 347:1374–7. doi: 10.1126/science.aaa2361
46. Shokr H, Dias IH, Gherghel D. Microvascular function and oxidative stress in adult individuals with early onset of cardiovascular disease. Sci Rep. (2020) 10:1–8. doi: 10.1038/s41598-020-60766-0
47. van Sijl AM, van Eijk IC, Peters MJ, Serné EH, van der Horst-Bruinsma IE, Smulders YM, et al. Tumour necrosis factor blocking agents and progression of subclinical atherosclerosis in patients with ankylosing spondylitis. Ann Rheum Dis. (2015) 74:119–123. doi: 10.1136/annrheumdis-2013-203934
48. Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med. (2014) 24:165–9. doi: 10.1016/j.tcm.2013.12.001
49. Zhong L, Simard MJ, Huot J. Endothelial microRNAs regulating the NF-κB pathway and cell adhesion molecules during inflammation. FASEB J. (2018) 32:4070–84. doi: 10.1096/fj.201701536R
50. Bhaskar S, Sudhakaran P, Helen A. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-κB signaling pathway. Cell Immunol. (2016) 310:131–40. doi: 10.1016/j.cellimm.2016.08.011
51. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. (2014) 46:e99–e99. doi: 10.1038/emm.2014.38
52. Zhao L, Varghese Z, Moorhead J, Chen Y, Ruan XZ. CD36 and lipid metabolism in the evolution of atherosclerosis. Br Med Bull. (2018) 126:101–12. doi: 10.1093/bmb/ldy006
53. Yamada T, Ogawa K, Tanaka TD, Nagoshi T, Minai K, Ogawa T, et al. Increase in oxidized low-density lipoprotein level according to hyperglycemia in patients with cardiovascular disease: a study by structure equation modeling. Diabetes Res Clin Pract. (2020) 161:108036. doi: 10.1016/j.diabres.2020.108036
54. Falck-Hansen M, Kassiteridi C, Monaco C. Toll-like receptors in atherosclerosis. Int J Mol Sci. (2013) 14:14008–23. doi: 10.3390/ijms140714008
55. Mehrzad J, Fazel F, Pouyamehr N, Hosseinkhani S, Dehghani H. Naturally occurring level of Aflatoxin B1 injures human, canine and bovine leukocytes through ATP depletion and caspase activation. Int J Toxicol. (2020) 39:30–8. doi: 10.1177/1091581819892613
56. Bahari A, Mehrzad J, Mahmoudi M, Bassami MR, Dehghani H. Cytochrome P450 isoforms are differently upregulated in aflatoxin B1-exposed human lymphocytes and monocytes. Immunopharmacol Immunotoxicol. (2014) 36:1–10. doi: 10.3109/08923973.2013.850506
57. Malvandi AM, Mehrzad J, Saleh-moghaddam M. Biologically relevant doses of mixed aflatoxins B and G upregulate MyD88, TLR2, TLR4 and CD14 transcripts in human PBMCs. Immunopharmacol Immunotoxicol. (2013) 35:528–32. doi: 10.3109/08923973.2013.803572
58. Mohammadi A, Mehrzad J, Mahmoudi M, Schneider M. Environmentally relevant level of aflatoxin B1 dysregulates human dendritic cells through signaling on key toll-like receptors. Int J Toxicol. (2014) 33:175–86. doi: 10.1177/1091581814526890
59. An Y, Shi X, Tang X, Wang Y, Shen F, Zhang Q, et al. Aflatoxin B1 induces reactive oxygen species-mediated autophagy and extracellular trap formation in macrophages. Front Cell Infect Microbiol. (2017) 7:53. doi: 10.3389/fcimb.2017.00053
60. Gekle M, Schwerdt G, Freudinger R, Mildenberger S, Wilflingseder D, Pollack V, et al. Ochratoxin A induces JNK activation and apoptosis in MDCK-C7 cells at nanomolar concentrations. J Pharmacol Exp Ther. (2000) 293:837–44.
61. Chaudhari M, Jayaraj R, Bhaskar A, Rao PL. Oxidative stress induction by T-2 toxin causes DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology. (2009) 262:153–61. doi: 10.1016/j.tox.2009.06.002
62. Fang H, Wu Y, Guo J, Rong J, Ma L, Zhao Z, et al. T-2 toxin induces apoptosis in differentiated murine embryonic stem cells through reactive oxygen species-mediated mitochondrial pathway. Apoptosis. (2012) 17:895–907. doi: 10.1007/s10495-012-0724-3
63. Agrawal M, Bhaskar A, Rao PL. Involvement of mitogen-activated protein kinase pathway in T-2 toxin-induced cell cycle alteration and apoptosis in human neuroblastoma cells. Mol Neurobiol. (2015) 51:1379–94. doi: 10.1007/s12035-014-8816-4
64. Hymery N, Léon Léon K, Carpentier F-G, Jung J-L, Parent-Massin D. T-2 toxin inhibits the differentiation of human monocytes into dendritic cells and macrophages. Toxicol In Vitro. (2009) 23:509–19. doi: 10.1016/j.tiv.2009.01.003
65. Seeboth J, Solinhac R, Oswald IP, Guzylack-Piriou L. The fungal T-2 toxin alters the activation of primary macrophages induced by TLR-agonists resulting in a decrease of the inflammatory response in the pig. Vet Res. (2012) 43:35. doi: 10.1186/1297-9716-43-35
66. Li D, Ye Y, Lin S, Deng L, Fan X, Zhang Y, et al. Evaluation of deoxynivalenol-induced toxic effects on DF-1 cells in vitro: cell-cycle arrest, oxidative stress, and apoptosis. Environ Toxicol Pharmacol. (2014) 37:141–9. doi: 10.1016/j.etap.2013.11.015
67. Li D, Ma H, Ye Y, Ji C, Tang X, Ouyang D, et al. Deoxynivalenol induces apoptosis in mouse thymic epithelial cells through mitochondria-mediated pathway. Environ Toxicol Pharmacol. (2014) 38:163–71. doi: 10.1016/j.etap.2014.05.015
68. Krishnaswamy R, Devaraj SN, Padma VV. Lutein protects HT-29 cells against Deoxynivalenol-induced oxidative stress and apoptosis: prevention of NF-κB nuclear localization and down regulation of NF-κB and Cyclo-Oxygenase−2 expression. Free Radical Biol Med. (2010) 49:50–60. doi: 10.1016/j.freeradbiomed.2010.03.016
69. Sugiyama K-i, Muroi M, Kinoshita M, Hamada O, Minai Y, Sugita-Konishi Y, et al. NF-κB activation via MyD88-dependent Toll-like receptor signaling is inhibited by trichothecene mycotoxin deoxynivalenol. J Toxicol Sci. (2016) 41:273–9. doi: 10.2131/jts.41.273
70. Islam MR, Kim JW, Roh Y-S, Kim J-H, Han KM, Kwon H-J, et al. Evaluation of immunomodulatory effects of zearalenone in mice. J Immunotoxicol. (2017) 14:125–36. doi: 10.1080/1547691X.2017.1340371
71. Shen T, Miao Y, Ding C, Fan W, Liu S, Lv Y, et al. Activation of the p38/MAPK pathway regulates autophagy in response to the CYPOR-dependent oxidative stress induced by zearalenone in porcine intestinal epithelial cells. Food Chem Toxicol. (2019) 131:110527. doi: 10.1016/j.fct.2019.05.035
72. Banjerdpongchai R, Kongtawelert P, Khantamat O, Srisomsap C, Chokchaichamnankit D, Subhasitanont P, et al. Mitochondrial and endoplasmic reticulum stress pathways cooperate in zearalenone-induced apoptosis of human leukemic cells. J Hematol Oncol. (2010) 3:1–16. doi: 10.1186/1756-8722-3-50
73. Bianco G, Russo R, Marzocco S, Velotto S, Autore G, Severino L. Modulation of macrophage activity by aflatoxins B1 and B2 and their metabolites aflatoxins M1 and M2. Toxicon. (2012) 59:644–50. doi: 10.1016/j.toxicon.2012.02.010
74. Mehrzad J, Milani M, Mahmoudi M. Naturally occurring level of mixed aflatoxins B and G stimulate toll-like receptor-4 in bovine mononuclear cells. Vet Q. (2013) 33:186–90. doi: 10.1080/01652176.2013.873960
75. Mehrzad J, Devriendt B, Baert K, Cox E. Aflatoxins of type B and G affect porcine dendritic cell maturation in vitro. J Immunotoxicol. (2015) 12:174–80. doi: 10.3109/1547691X.2014.916366
76. Cusumano V, Costa G, Seminara S. Effect of aflatoxins on rat peritoneal macrophages. Appl Environ Microbiol. (1990) 56:3482–4. doi: 10.1128/aem.56.11.3482-3484.1990
77. Romero A, Ares I, Ramos E, Castellano V, Martínez M, Martínez-Larrañaga M-R, et al. Mycotoxins modify the barrier function of Caco-2 cells through differential gene expression of specific claudin isoforms: Protective effect of illite mineral clay. Toxicology. (2016) 353:21–33. doi: 10.1016/j.tox.2016.05.003
78. Mulunda M, Dutton MF. A study of single and combined cytotoxic effects of fumonisin B1, aflatoxin B1 and ochratoxin A on human mononuclear blood cells using different cytotoxic methods. Glob J Med Res. (2014) 14:1–19.
79. Domijan AM, Peraica M, Vrdoljak AL, Radić B, Žlender V, Fuchs R. The involvement of oxidative stress in ochratoxin A and fumonisin B1 toxicity in rats. Mol Nutr Food Res. (2007) 51:1147–51. doi: 10.1002/mnfr.200700079
80. Klarić MŠ, Pepeljnjak S, Domijan AM, Petrik J. Lipid peroxidation and glutathione levels in porcine kidney PK15 cells after individual and combined treatment with fumonisin B1, beauvericin and ochratoxin A. Basic Clin Pharmacol Toxicol. (2007) 100:157–64. doi: 10.1111/j.1742-7843.2006.00019.x
81. Kouadio J, Moukha S, Brou K, Gnakri D. Lipid metabolism disorders, lymphocytes cells death, and renal toxicity induced by very low levels of deoxynivalenol and fumonisin B1 alone or in combination following 7 days oral administration to mice. Toxicol Int. (2013) 20:218. doi: 10.4103/0971-6580.121673
82. A.-Bracarense PF, Lucioli J, Grenier B, Pacheco GD, Moll W-D, Schatzmayr G, et al. Chronic ingestion of deoxynivalenol and fumonisin, alone or in interaction, induces morphological and immunological changes in the intestine of piglets. Br J Nutr. (2012) 107:1776–86. doi: 10.1017/S0007114511004946
83. Dabrowski M, Obremski K, Gajecka M, Gajecki MT, Zielonka Ł. Changes in the subpopulations of porcine peripheral blood lymphocytes induced by exposure to low doses of zearalenone (ZEN) and deoxynivalenol (DON). Molecules. (2016) 21:557. doi: 10.3390/molecules21050557
84. Shen H-M, Shi C-Y, Shen Y, Ong C-N. Detection of elevated reactive oxygen species level in cultured rat hepatocytes treated with aflatoxin B1. Free Radic Biol Med. (1996) 21:139–46. doi: 10.1016/0891-5849(96)00019-6
85. Murcia HW, Diaz GJ. In vitro hepatic aflatoxicol production is related to a higher resistance to aflatoxin B 1 in poultry. Sci Rep. (2020) 10:1–8. doi: 10.1038/s41598-020-62415-y
86. Vahidi-Ferdowsi P, Mehrzad J, Malvandi A, Hosseinkhani S. Bioluminescence-based detection of astrocytes apoptosis and ATP depletion induced by biologically relevant level aflatoxin B1. World Mycotox J. (2018) 11:589–98. doi: 10.3920/WMJ2017.2275
87. Mehrzad J, Bahari A, Bassami MR, Mahmoudi M, Dehghani H. Immunobiologically relevant level of aflatoxin B1 alters transcription of key functional immune genes, phagocytosis and survival of human dendritic cells. Immunol Lett. (2018) 197:44–52. doi: 10.1016/j.imlet.2018.03.008
88. Mehrzad J, Bahari A, Bassami MR, Mahmoudi M, Dehghani H. Data on environmentally relevant level of aflatoxin B1-induced human dendritic cells' functional alteration. Data Brief. (2018) 18:1576–80. doi: 10.1016/j.dib.2018.04.104
89. Mehrzad J, Malvandi AM, Alipour M, Hosseinkhani S. Environmentally relevant level of aflatoxin B1 elicits toxic proinflammatory response in murine CNS-derived cells. Toxicol Lett. (2017) 279:96–106. doi: 10.1016/j.toxlet.2017.07.902
90. Dohnal V, Wu Q, Kuča K. Metabolism of aflatoxins: key enzymes and interindividual as well as interspecies differences. Arch Toxicol. (2014) 88:1635–44. doi: 10.1007/s00204-014-1312-9
91. Cheng J, Huang S, Fan C, Zheng N, Zhang Y, Li S, et al. Metabolomic analysis of alterations in lipid oxidation, carbohydrate and amino acid metabolism in dairy goats caused by exposure to Aflotoxin B1. J Dairy Res. (2017) 84:401–6. doi: 10.1017/S0022029917000590
92. Mehrzad J, Zahraei Salehi T, Khosravi A, Hosseinkhani S, Tahamtani Y, Hajizadeh-Saffar E, et al. Environmentally occurring aflatoxins B1 and M1 notifyably harms pancreatic islets. Toxin Rev. (2021) 1–10. doi: 10.1080/15569543.2021.2010758
93. EFSA. Outcome of a public consultation on the risk assessment of ochratoxin A in food. EFSA Support Pub. (2020) 17:1845E. doi: 10.2903/sp.efsa.2020.EN-1845
94. R. Di Giuseppe, Bertuzzi T, Rossi F, Rastelli S, Mulazzi A, Capraro J, et al. Plasma ochratoxin A levels, food consumption, and risk biomarkers of a representative sample of men and women from the Molise region in Italy. Eur J Nutr. (2012) 51:851–60. doi: 10.1007/s00394-011-0265-5
95. Sauvant C, Silbernagl S, Gekle M. Exposure to ochratoxin A impairs organic anion transport in proximal-tubule-derived opossum kidney cells. J Pharmacol Exp Ther. (1998) 287:13–20.
96. González-Arias CA, Benitez-Trinidad AB, Sordo M, Robledo-Marenco L, Medina-Díaz IM, Barrón-Vivanco BS, et al. Low doses of ochratoxin A induce micronucleus formation and delay DNA repair in human lymphocytes. Food Chem Toxicol. (2014) 74:249–54. doi: 10.1016/j.fct.2014.10.006
97. Assaf H, Azouri H, Pallardy M. Ochratoxin A induces apoptosis in human lymphocytes through down regulation of Bcl-xL. Toxicol Sci. (2004) 79:335–44. doi: 10.1093/toxsci/kfh123
98. Studer-Rohr I, Schlatter J, Dietrich DR. Kinetic parameters and intraindividual fluctuations of ochratoxin A plasma levels in humans. Arch Toxicol. (2000) 74:499–510. doi: 10.1007/s002040000157
99. Rutigliano L, Valentini L, Martino NA, Pizzi F, Zanghì A, Dell'Aquila ME, et al. Ochratoxin A at low concentrations inhibits in vitro growth of canine umbilical cord matrix mesenchymal stem cells through oxidative chromatin and DNA damage. Reprod Toxicol. (2015) 57:121–9. doi: 10.1016/j.reprotox.2015.05.017
100. Marin DE, Pistol GC, Gras MA, Palade ML, Taranu I. Comparative effect of ochratoxin A on inflammation and oxidative stress parameters in gut and kidney of piglets. Regul Toxicol Pharmacol. (2017) 89:224–31. doi: 10.1016/j.yrtph.2017.07.031
101. Kouadio JH, Mobio TA, Baudrimont I, Moukha S, Dano SD, Creppy EE. Comparative study of cytotoxicity and oxidative stress induced by deoxynivalenol, zearalenone or fumonisin B1 in human intestinal cell line Caco-2. Toxicology. (2005) 213:56–65. doi: 10.1016/j.tox.2005.05.010
102. Domijan A-M, Abramov AY. Fumonisin B1 inhibits mitochondrial respiration and deregulates calcium homeostasis—implication to mechanism of cell toxicity. Int J Biochem Cell Biol. (2011) 43:897–904. doi: 10.1016/j.biocel.2011.03.003
103. Szabó A, Szabó-Fodor J, Kachlek M, Mézes M, Balogh K, Glávits R, et al. Dose and exposure time-dependent renal and hepatic effects of intraperitoneally administered fumonisin B1 in rats. Toxins. (2018) 10:465. doi: 10.3390/toxins10110465
104. Minervini F, Garbetta A, D'Antuono I, Cardinali A, Martino NA, Debellis L, et al. Toxic mechanisms induced by fumonisin B 1 mycotoxin on human intestinal cell line. Arch Environ Contam Toxicol. (2014) 67:115–23. doi: 10.1007/s00244-014-0004-z
105. Ahmadi A, Poursasan N, Amani J, Salimian J. Adverse effect of T-2 toxin and the protective role of selenium and vitamin E on peripheral blood B lymphocytes. Iran J Immunol. (2015) 12:64–9.
106. Mezes M, Barta M, Nagy G. Comparative investigation on the effect of T-2 mycotoxin on lipid peroxidation and antioxidant status in different poultry species. Res Vet Sci. (1999) 66:19–23. doi: 10.1053/rvsc.1998.0233
107. Shinozuka J, Li G, Kiatipattanasakul W, Uetsuka K, Nakayama H, Doi K. T-2 toxin-induced apoptosis in lymphoid organs of mice. Exp Toxicol Pathol. (1997) 49:387–92. doi: 10.1016/S0940-2993(97)80124-8
108. Li G, Shinozuka J, Uetsuka K, Nakayama H, Doi K. T-2 toxin-induced apoptosis in intestinal crypt epithelial cells of mice. Exp Toxicol Pathol. (1997) 49:447–50. doi: 10.1016/S0940-2993(97)80132-7
109. Zhang Y, Han J, Zhu C-C, Tang F, Cui X-S, Kim N-H, et al. Exposure to HT-2 toxin causes oxidative stress induced apoptosis/autophagy in porcine oocytes. Sci Rep. (2016) 6:1–8. doi: 10.1038/srep33904
110. Wu J, Tu D, Yuan L-Y, Yuan H, Wen L-X. T-2 toxin exposure induces apoptosis in rat ovarian granulosa cells through oxidative stress. Environ Toxicol Pharmacol. (2013) 36:493–500. doi: 10.1016/j.etap.2013.03.017
111. Fatima Z, Guo P, Huang D, Lu Q, Wu Q, Dai M, et al. The critical role of p16/Rb pathway in the inhibition of GH3 cell cycle induced by T-2 toxin. Toxicology. (2018) 400:28–39. doi: 10.1016/j.tox.2018.03.006
112. Deyu H, Luqing C, Xianglian L, Pu G, Qirong L, Xu W, et al. Protective mechanisms involving enhanced mitochondrial functions and mitophagy against T-2 toxin-induced toxicities in GH3 cells. Toxicol Lett. (2018) 295:41–53. doi: 10.1016/j.toxlet.2018.05.041
113. Commission E. Commission Recommendation of 17 August 2006 on the presence of deoxynivalenol, zearalenone, ochratoxin A, T-2 HT-2 fumonisins in products intended for animal feeding (2006/576/EC). J Eur Union. (2006) 229:7–9.
114. Pietsch C, Michel C, Kersten S, Valenta H, Dänicke S, Schulz C, et al. In vivo effects of deoxynivalenol (DON) on innate immune responses of carp (Cyprinus carpio L). Food Chem Toxicol. (2014) 68:44–52. doi: 10.1016/j.fct.2014.03.012
115. Liu Y, Ran R, Hu C, Cui B, Xu Y, Liu H, et al. The metabolic responses of HepG2 cells to the exposure of mycotoxin deoxynivalenol. World Mycotox J. (2016) 9:577–86. doi: 10.3920/WMJ2015.1981
116. Yang W, Yu M, Fu J, Bao W, Wang D, Hao L, et al. Deoxynivalenol induced oxidative stress and genotoxicity in human peripheral blood lymphocytes. Food Chem Toxicol. (2014) 64:383–96. doi: 10.1016/j.fct.2013.12.012
117. Katika MR, Hendriksen PJ, van Loveren HA. ACM Peijnenburg, Characterization of the modes of action of deoxynivalenol (DON) in the human Jurkat T-cell line. J Immunotoxicol. (2015) 12:206–16. doi: 10.3109/1547691X.2014.925995
118. Liu J, Applegate T. Zearalenone (ZEN) in Livestock and Poultry: Dose, toxicokinetics, toxicity and estrogenicity. Toxins. (2020) 12:377. doi: 10.3390/toxins12060377
119. Hassen W, Ayed-Boussema I, Oscoz AA, Lopez ADC, Bacha H. The role of oxidative stress in zearalenone-mediated toxicity in Hep G2 cells: Oxidative DNA damage, gluthatione depletion and stress proteins induction. Toxicology. (2007) 232:294–302. doi: 10.1016/j.tox.2007.01.015
120. Kowalska K, Habrowska-Górczyńska DE, Domińska K, Piastowska-Ciesielska AW. The dose-dependent effect of zearalenone on mitochondrial metabolism, plasma membrane permeabilization and cell cycle in human prostate cancer cell lines. Chemosphere. (2017) 180:455–66. doi: 10.1016/j.chemosphere.2017.04.027
121. Marin D, Pistol G, Bulgaru C, Taranu I. Cytotoxic and inflammatory effects of individual and combined exposure of HepG2 cells to zearalenone and its metabolites. Naunyn Schmiedebergs Arch Pharmacol. (2019) 392:937–47. doi: 10.1007/s00210-019-01644-z
122. Bbosa GS, Kitya D, Lubega A, Ogwal-Okeng J, Anokbonggo WW, Kyegombe DB. Review of the biological and health effects of aflatoxins on body organs and body systems. Aflatoxins Recent Adv Future Prospect. (2013) 12:239–65. doi: 10.5772/51201
123. Yilmaz S, Kaya E, Karaca A, Karatas O. Aflatoxin B1 induced renal and cardiac damage in rats: protective effect of lycopene. Res Vet Sci. (2018) 119:268–75. doi: 10.1016/j.rvsc.2018.07.007
124. Mannaa FA, Abdel-Wahhab KG, Abdel-Wahhab MA. Prevention of cardiotoxicity of aflatoxin B 1 via dietary supplementation of papaya fruit extracts in rats. Cytotechnology. (2014) 66:327–34. doi: 10.1007/s10616-013-9579-x
125. Singal P, Kirshenbaum L. A relative deficit in antioxidant reserve may contribute in cardiac failure. Can J Cardiol. (1990) 6:47–9.
126. Mohamed AM, Metvvally NS. Antiaflatoxigenic activities of some plant aqueous extracts against Aflatoxin-B1 induced renal and cardiac damage. J Pharmacol Toxicol. (2009) 4:1–16. doi: 10.3923/jpt.2009.1.16
127. Diakos NA, Navankasattusas S, Abel ED, Rutter J, McCreath L, Ferrin P, et al. Evidence of glycolysis up-regulation and pyruvate mitochondrial oxidation mismatch during mechanical unloading of the failing human heart: implications for cardiac reloading and conditioning. JACC Basic Transl Sci. (2016) 1:432–44. doi: 10.1016/j.jacbts.2016.06.009
128. Malvandi AM, Loretelli C, Nasr MB, Zuccotti GV, Fiorina P. Sitagliptin favorably modulates immune-relevant pathways in human beta cells. Pharmacol Res. (2019) 148:104405. doi: 10.1016/j.phrs.2019.104405
129. Ge J, Yu H, Li J, Lian Z, Zhang H, Fang H, et al. Assessment of aflatoxin B1 myocardial toxicity in rats: mitochondrial damage and cellular apoptosis in cardiomyocytes induced by aflatoxin B1. J Int Med Res. (2017) 45:1015–23. doi: 10.1177/0300060517706579
130. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, et al. Evidence for cardiomyocyte renewal in humans. Science. (2009) 324:98–102. doi: 10.1126/science.1164680
131. Abdel-Wahhab MA, Ahmed HH, Hagazi MM. Prevention of aflatoxin B1-initiated hepatotoxicity in rat by marine algae extracts. J Appl Toxicol. (2006) 26:229–38. doi: 10.1002/jat.1127
132. Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. (1996) 98:2854–65. doi: 10.1172/JCI119114
133. Wang WJ, Xu ZL Yu C, Xu XH. Effects of aflatoxin B1 on mitochondrial respiration, ROS generation and apoptosis in broiler cardiomyocytes. Anim Sci J. (2017) 88:1561–8. doi: 10.1111/asj.12796
134. Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic Res. (2010) 44:1267–88. doi: 10.3109/10715762.2010.507670
135. Okutan H, Aydin G, Ozcelik N. Protective role of melatonin in ochratoxin a toxicity in rat heart and lung. J Appl Toxicol. (2004) 24:505–12. doi: 10.1002/jat.1010
136. Cui G, Li L, Xu W, Wang M, Jiao D, Yao B, et al. Astaxanthin protects ochratoxin A-induced oxidative stress and apoptosis in the heart via the Nrf2 pathway. Oxid Med Cell Longev. (2020) 2020:7639109. doi: 10.1155/2020/7639109
137. Fang H, Cong L, Zhi Y, Xu H, Jia X, Peng S. T-2 toxin inhibits murine ES cells cardiac differentiation and mitochondrial biogenesis by ROS and p-38 MAPK-mediated pathway. Toxicol Lett. (2016) 258:259–66. doi: 10.1016/j.toxlet.2016.06.2103
138. Jaćević V, Wu Q, Nepovimova E, Kuča K. Cardiomyopathy induced by T-2 toxin in rats. Food Chem Toxicol. (2020) 137:111138. doi: 10.1016/j.fct.2020.111138
139. Chen X, Xu J, Liu D, Sun Y, Qian G, Xu S, et al. The aggravating effect of selenium deficiency on T-2 toxin-induced damage on primary cardiomyocyte results from a reduction of protective autophagy. Chem Biol Interact. (2019) 300:27–34. doi: 10.1016/j.cbi.2019.01.009
140. Xu J, Pan S, Gan F, Hao S, Liu D, Xu H, et al. Selenium deficiency aggravates T-2 toxin-induced injury of primary neonatal rat cardiomyocytes through ER stress. Chem Biol Interact. (2018) 285:96–105. doi: 10.1016/j.cbi.2018.01.021
141. Ngampongsa S, Hanafusa M, Ando K, Ito K, Kuwahara M, Yamamoto Y, et al. Toxic effects of T-2 toxin and deoxynivalenol on the mitochondrial electron transport system of cardiomyocytes in rats. J Toxicol Sci. (2013) 38:495–502. doi: 10.2131/jts.38.495
142. Salem IB, Boussabbeh M, Da Silva JP, Guilbert A, Bacha H, Abid-Essefi S, et al. SIRT1 protects cardiac cells against apoptosis induced by zearalenone or its metabolites α-and β-zearalenol through an autophagy-dependent pathway. Toxicol Appl Pharmacol. (2017) 314:82–90. doi: 10.1016/j.taap.2016.11.012
143. Bai S, Wang L, Luo Y, Ding X, Yang J, Bai J, et al. Effects of corn naturally contaminated with aflatoxins on performance, calcium and phosphorus metabolism, and bone mineralization of broiler chicks. J Poult Sci. (2013) 51:157–64. doi: 10.2141/jpsa.0130053
144. Surdu AM, Pînzariu O, Ciobanu D-M, Negru A-G, Căinap S-S, Lazea C, et al. Vitamin D and its role in the lipid metabolism and the development of atherosclerosis. Biomedicines. (2021) 9:172. doi: 10.3390/biomedicines9020172
145. Mirhosseini N, Rainsbury J, Kimball SM. Vitamin D supplementation, serum 25 (OH) D concentrations and cardiovascular disease risk factors: a systematic review and meta-analysis. Frontiers Cardiovasc Med. (2018) 5:87. doi: 10.3389/fcvm.2018.00087
146. Hirata M, Serizawa K-i, Aizawa K, Yogo K, Tashiro Y, Takeda S, et al. 22-Oxacalcitriol prevents progression of endothelial dysfunction through antioxidative effects in rats with type 2 diabetes and early-stage nephropathy. Nephrol Dial Transplant. (2013) 28:1166–74. doi: 10.1093/ndt/gfs536
147. Kassi E, Adamopoulos C, Basdra EK, Papavassiliou AG. Role of vitamin D in atherosclerosis. Circulation. (2013) 128:2517–31. doi: 10.1161/CIRCULATIONAHA.113.002654
148. Devegowda G, Ravikiran D. Mycotoxins and skeletal problems in poultry. World Mycotoxin J. (2009) 2:331–7. doi: 10.3920/WMJ2008.1085
149. LeBoff MS, Chou SH, Murata EM, Donlon CM, Cook NR, Mora S, et al. Effects of supplemental vitamin D on bone health outcomes in women and men in the VITamin D and OmegA-3 TriaL (VITAL). J Bone Miner Res. (2020) 35:883–93. doi: 10.1002/jbmr.3958
150. Costanzo P, Santini A, Fattore L, Novellino E, Ritieni A. Toxicity of aflatoxin B1 towards the vitamin D receptor (VDR). Food Chem Toxicol. (2015) 76:77–9. doi: 10.1016/j.fct.2014.11.025
151. Zhang J, Song M, Cui Y, Shao B, Zhang X, Cao Z, et al. T-2 toxin-induced femur lesion is accompanied by autophagy and apoptosis associated with Wnt/beta-catenin signaling in mice. Environ Toxicol. (2022) 37:1653–61. doi: 10.1002/tox.23514
152. Hladik D, Hofig I, Oestreicher U, Beckers J, Matjanovski M, Bao X, et al. Long-term culture of mesenchymal stem cells impairs ATM-dependent recognition of DNA breaks and increases genetic instability. Stem Cell Res Ther. (2019) 10:218. doi: 10.1186/s13287-019-1334-6
153. Tomaszewska E, Rudyk H, Swietlicka I, Hulas-Stasiak M, Donaldson J, Arczewska M, et al. The influence of prenatal fumonisin exposure on bone properties, as well as OPG and RANKL expression and immunolocalization, in newborn offspring is sex and dose dependent. Int J Molecul Sci. (2021) 22:13234. doi: 10.3390/ijms222413234
154. Tomaszewska E, Rudyk H, Swietlicka I, Hulas-Stasiak M, Donaldson J, Arczewska M, et al. Trabecular bone parameters, TIMP-2, MMP-8, MMP-13, VEGF expression and immunolocalization in bone and cartilage in newborn offspring prenatally exposed to fumonisins. Int J Molecul Sci. (2021) 22:12528. doi: 10.3390/ijms222212528
Keywords: mycotoxins, low dose toxicity, oxidative stress, metabolic and immune alterations, bone homeostasis, cardiovascular diseases
Citation: Malvandi AM, Shahba S, Mehrzad J and Lombardi G (2022) Metabolic Disruption by Naturally Occurring Mycotoxins in Circulation: A Focus on Vascular and Bone Homeostasis Dysfunction. Front. Nutr. 9:915681. doi: 10.3389/fnut.2022.915681
Received: 08 April 2022; Accepted: 30 May 2022;
Published: 24 June 2022.
Edited by:
Nada El Darra, Beirut Arab University, LebanonReviewed by:
Otniel Freitas-Silva, Embrapa Food Agroindustry, BrazilCopyright © 2022 Malvandi, Shahba, Mehrzad and Lombardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amir Mohammad Malvandi, YW1pcm1vaGFtbWFkLm1hbHZhbmRpQGdydXBwb3NhbmRvbmF0by5pdA==; orcid.org/0000-0003-1243-2372
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.