
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr., 24 March 2022
Sec. Nutrition and Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fnut.2022.862277
This article is part of the Research TopicFlavonoids and Cardiovascular MetabolismView all 5 articles
 Ruo-Lan Li1†
Ruo-Lan Li1† Ling-Yu Wang1†
Ling-Yu Wang1† Shuqin Liu2Hu-Xinyue Duan1
Shuqin Liu2Hu-Xinyue Duan1 Qing Zhang1Ting Zhang1
Qing Zhang1Ting Zhang1 Wei Peng1*Yongliang Huang2*
Wei Peng1*Yongliang Huang2* Chunjie Wu1*
Chunjie Wu1*
Atherosclerosis, as a chronic inflammatory response, is one of the main causes of cardiovascular diseases. Atherosclerosis is induced by endothelial cell dysfunction, migration and proliferation of smooth muscle cells, accumulation of foam cells and inflammatory response, resulting in plaque accumulation, narrowing and hardening of the artery wall, and ultimately leading to myocardial infarction or sudden death and other serious consequences. Flavonoid is a kind of natural polyphenol compound widely existing in fruits with various structures, mainly including flavonols, flavones, flavanones, flavanols, anthocyanins, isoflavones, and chalcone, etc. Because of its potential health benefits, it is now used in supplements, cosmetics and medicines, and researchers are increasingly paying attention to its role in atherosclerosis. In this paper, we will focus on several important nodes in the development of atherosclerotic disease, including endothelial cell dysfunction, smooth muscle cell migration and proliferation, foam cell accumulation and inflammatory response. At the same time, through the classification of flavonoids from fruits, the role and potential mechanism of flavonoids in atherosclerosis were reviewed, providing a certain direction for the development of fruit flavonoids in the treatment of atherosclerosis drugs.
Cardiovascular disease (CVD) is a kind of disease with extremely high morbidity and mortality. According to relevant investigations, CVD deaths accounted for about 31% of global deaths in 2016, among which atherosclerosis is the main cause of CVD (1). Atherosclerosis is a chronic inflammatory disease, mostly affecting adults and the elderly. It is characterized by plaque accumulation, narrowing and hardening of coronary artery walls, which will directly affect the completion of blood oxygen supply to various organs in the body, resulting in serious consequences such as myocardial infarction, angina pectoris and sudden death (2, 3). The pathogenesis of atherosclerosis is diverse. In current studies, the factors that affect atherosclerosis are mainly hyperlipidemia, diabetes, smoking, high blood pressure, genetic, and other cardiovascular risk factors. These factors can induce dysfunction of endothelial cells through mediating oxidative stress, and then leads to the beginning of the atherosclerotic disease process (4).
At present, pharmacologic treatment with medications, stent-based therapy or coronary artery bypass surgery are commonly used in clinical treatment of atherosclerosis to relieve symptoms, but three methods have certain limitations (5, 6). For example, statin is a widely used drug in clinical practice, which can inhibit the occurrence and development of atherosclerosis by inhibiting cholesterol synthesis. However, due to its poor targeting, oral or intravenous administration can also attack normal tissues and cells, resulting in strong side effects. In addition, when the disease develops to an advanced stage, drug treatment is less effective (7). Although coronary artery bypass surgery can significantly reduce the mortality of patients with atherosclerosis, its prognosis is poor and it is easy to cause various complications (6). Stent-based therapy can also help relieve patients’ related symptoms and have a low incidence of disease complications in the advanced stage of the disease when drugs fail to play a role. But it’s a pity that problems such as artery stenosis, inflammation and thrombosis in patients with stent treatment have not been solved, so the treatment can only relieve their symptoms but not solve their causes (5). Therefore, it is urgent to find new compounds for the treatment of atherosclerosis.
Flavonoids are a kind of natural organic compounds widely present in fruits, which are composed of two aromatic rings and have typical C6-C3-C6 skeleton (8). Previous studies have found that a diet rich in flavonoids can significantly reduce CVD mortality, which is directly related to atherosclerosis. At the same time, the effect is related to the source, dose and bioavailability of flavonoids (9). The current pharmacological studies have showed that a variety of flavonoids from fruits could not only reduce cholesterol transport, but also enhanced the immune function by regulating the level of intracellular inflammatory factors (10). In addition, hydroxyl radicals, which are widely present in flavonoids, also play a role in protecting blood vessels by mediating antioxidant effects (11). Epidemiological studies linking flavonoid intake to a reduced risk of death from CVD have generated considerable interest in this preventive mechanism (12). As fruit is the most important component in the source of flavonoids, the treatment of atherosclerosis by flavonoids derived from fruit will be reviewed in this paper.
Due to the different components, the arteries can be classified as elastic arteries, muscular arteries, and transitional regions between the two kinds of arteries. The artery wall has three layers of tissue structure, of which the most inward layer is composed of endothelial cells, known as the intima. The outermost layer is composed of connective tissues, collagen, and elastic fibers, while the medial membrane is composed of vascular smooth muscle cells (VSMCs) (13). As a chronic inflammatory response, atherosclerosis is at increased risk for environmental and genetic factors. In the early stage of atherosclerotic disease, hypercholesterolemia induces the entry of low-density lipoprotein (LDL) into the subcutaneous space of intima and promotes the oxidation of LDL under enzymatic or non-enzymatic modification, thereby activating endothelial cells and causing endothelial dysfunction (14). In particular, activated endothelial cells attract monocytes and other white blood cells by upregulation of adhesion molecules and secretion of chemokines, which ultimately lead to chronic inflammatory responses (15, 16). During the development of atherosclerosis, monocytes differentiate into macrophages and phagocytose oxidized low-density lipoprotein (ox-LDL) to form foam cells. Subsequently, foam cells can attract VSMCs to migrate to the subcutaneous space and proliferate, resulting in the formation of new intima in the arterial lumen and leading to arterial narrowing (17). As the inflammatory response within the arterial vasculature continues to occur and the lumen becomes progressively narrower, the arterial vasculature is highly susceptible to rupture and subsequent thrombosis, which can lead to more serious clinical complications. Therefore, in this section, we will focus on an overview of several important points in the development of atherosclerotic disease, namely, endothelial dysfunction, foam cell formation, migration, and proliferation of VSMCs and inflammatory response.
Vascular endothelial cells are epithelial cells arranged in a single layer on the inner side of blood vessels with a large surface area and at a critical location where blood circulation and tissue intersection (18). They have multiple physiological functions. Functioning endothelial cells can effectively regulate vascular permeability and vascular tension, and also be used as active signal transducers for circulating influences that modify the vessel wall phenotype (19). However, when endothelial cells encounter shear stress, dyslipidemia, hyperglycemia, aging and other factors, endothelial cell dysfunction and vascular homeostasis disorders, which then lead to a series of consequences such as vasoconstriction, leukocyte adherence, platelet activation, and promotion of oxidation, and ultimately lead to atherosclerosis (20). Continuous DNA replication, oxidative stress, and mitochondrial dysfunction may exert pressure on cells to permanently inhibit proliferation and lead to cell senescence (21). In response to this stress, cells secrete a variety of proteins named senescence-associated secretory phenotype (SASP), including pro-inflammatory cytokines (interleukin-6, interleukin-8, macrophage inflammatory proteins, etc.), chemokines, growth factors, matrix metalloproteinases, and other signaling molecules. There is no doubt that the transient expression of these proteins will repair the damaged tissue, but when the body is exposed to this environment for a long time, it will accelerate endothelial dysfunction (22–24). In addition to aging, dyslipidemia is another important cause of endothelial cell dysfunction. When the level of serum high-density lipoprotein (HDL) decreases and the level of total cholesterol (TC), triglyceride (TG), and low-density lipoprotein cholesterol (LDL-C) increases, LDLs will accumulate in the subcutaneous space of the artery wall and oxidize to form oxLDL under enzymatic or non-enzymatic modification (25). It further promoted the expression of monocyte chemotactic protein 1 (MCP-1), vascular cell adhesion molecule-1 (VCAM-1), endothelial leukocyte adhesion molecule (E-selectin), and finally induced inflammatory response (26, 27).
Endothelial dysfunction is characterized by endothelium-dependent vasodilation injury and endothelial activation marked by proinflammatory, proliferative, and procoagulant states, in which disruption of nitric oxide (NO) bioavailability is central (28). NO is a major vasodilator. Due to its small molecular weight, NO can diffuse to VSMC to activate guanylate cyclase, leading to cGMP-mediated vasodilation. At the same time, it can also spread to vascular lumen to inhibit platelet aggregation and adhesion, thus achieving anti-thrombotic effect (29, 30). In addition, as shown in Figure 1, NO can inhibit vascular smooth muscle proliferation by inhibiting platelet and leukocyte activation. However, when endothelial cells are activated, the production of endothelial NO synthase (eNOS) from L-arginine is reduced and tetrahydrobiopterin is absent, leading to NOS uncoupling and the generation of reactive oxygen species (ROS) such as superoxide and hydrogen peroxide (31, 32). Thus, endothelial cells switch from NO signal to ROS-mediated oxidative stress signal, activating the nuclear transcription factor kappaB (NF-κB) and other signaling pathways (33). In atherosclerosis, ROS production is associated with NADPH oxidase (NOX), myeloperoxidase (MPO), eNOS, and lipoxygenase. Of course, in addition to NO, prostacyclin prostacyclin (PGI2) and endothelium-derived hyperpolarizing factor (EDHF) and other vasodilators maintain vascular motility together with endothelin-1 (ET-1) and angiotensin II (AngII) (34–36).
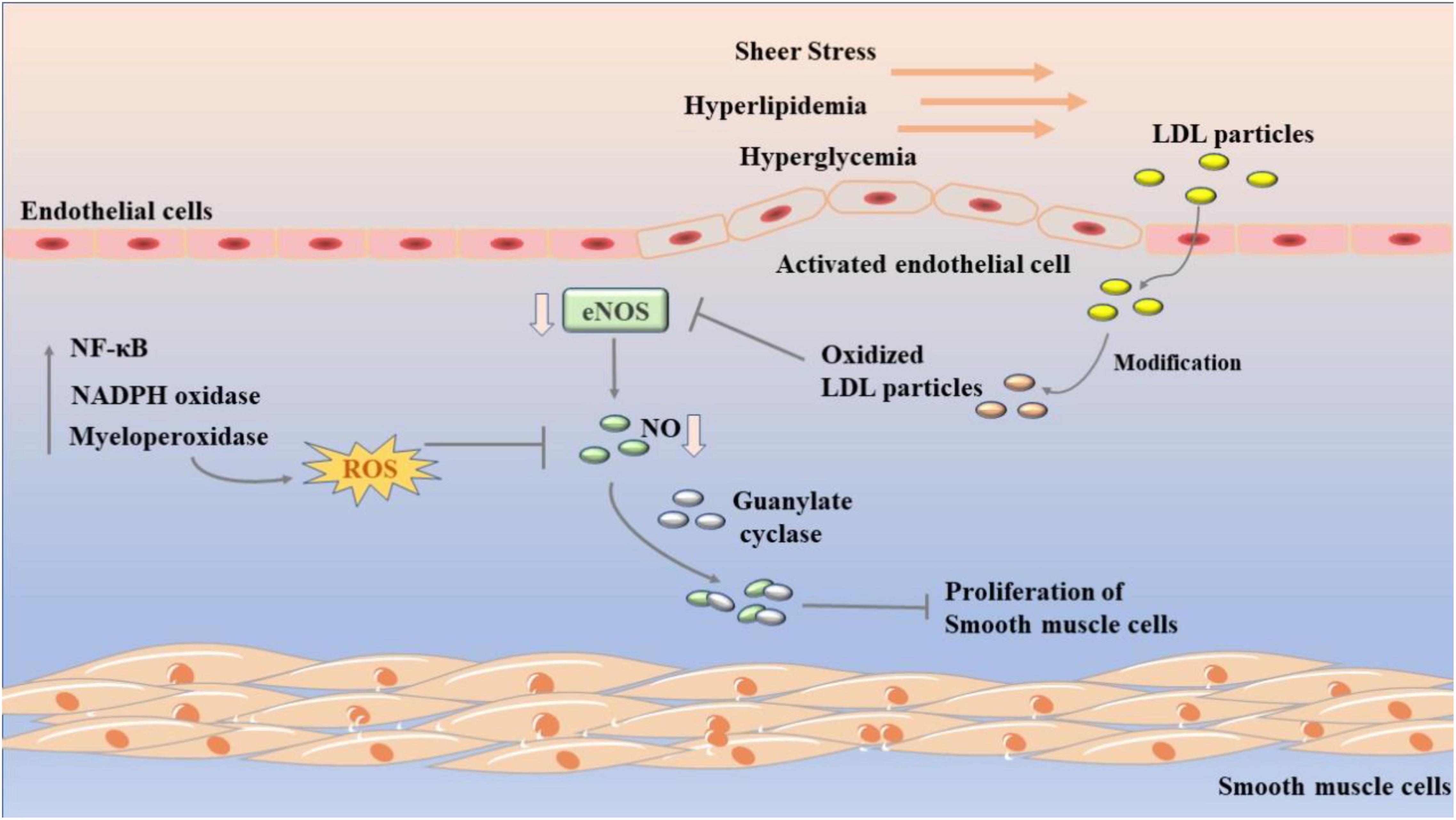
Figure 1. Development of endothelial dysfunction in atherosclerosis. Sheer stress, hyperlipidemia, and hyperglycemia leads to endothelial dysfunction. LDLs will accumulate in the subcutaneous space of the artery wall and oxidize to form oxLDL. It further decreases the activity of eNOS, which in turn reduce the content of NO. The nuclear transcription factor kappa B (NF-κB), NADPH oxidase (NOX), and myeloperoxidase (MPO) are related to its process.
Foam cells are a group of cells with multiple lipid inclusions in the cytoplasm. Most of them exist in the lipid rich endothelial space beneath the arteries. The appearance of foam cells is often regarded as one of the early manifestations of atherosclerosis (37). In the current study, it is generally believed that foam cells are mainly derived from macrophages, endothelial cells and VSMCs, and are mostly combined with modified LDL and cholesteryl ester (CE) after macrophages pass through the endothelial barrier (38). According to relevant data, 90% of macrophages in the artery are located in the adventitial layer, only 10% are located in the intima. Besides this, macrophages in the intima are almost formed only after birth (39, 40). Hypercholesterolemia is often accompanied by persistent inflammation, endothelial cell activation and secretion of chemokines such as CCL2/MCP-1, CX3CL1, and CCL5. This phenomenon will cause a large number of monocytes recruit to the area of LDL modification and promote the differentiation of monocytes into macrophages, which can quickly recognize and absorb modified LDL into foam cells (41–43). Foam cell formation is a complex process which is affected by many factors. Although the accumulation of lipid in macrophages is mainly derived from modified LDL, unmodified LDL in blood does not induce foam cell formation under normal physiological conditions. During the development of atherosclerosis, LDL will undergo a variety of modifications such as oxidation, carbamylation, and glycosylation to change its characteristics, so that it can be recognized and absorbed by macrophages (44).
Besides the modified LDL, the disorder of lipid metabolism in macrophages is another important factor of foam cell production (45). As shown in Figure 2, the homeostasis of lipid metabolism in macrophages is mainly coordinated by three main processes, including cholesterol uptake, cholesterol esterification, and cholesterol efflux (45). The imbalance of any of the three processes may lead to the increase of foam cells. Cholesterol uptake in macrophages mainly recognizes and absorbs modified LDL through a variety of scavenging receptors. CD36 is a glycoprotein that can promote cholesterol uptake. Its high expression often follows the emergence of ox-LDL (46, 47). Therefore, CD36 is often used as a biomarker of atherosclerosis in modern diagnosis and treatment. In macrophages, the expression of CD36 is often activated by peroxisome proliferator-activated receptors-γ (PPAR γ), nuclear erythroid-related factor 2 (Nrf2), signal transducer and activator of transcription (STAT) 1, and activator protein-1 (AP-1) are regulated (48, 49). When the expression of CD36 was inhibited, cholesterol uptake was significantly reduced and the symptoms of atherosclerosis were alleviated. In addition to CD36, scavenging receptor A1 (SR-A1) and lectin like ox-LDL receptor-1 (LOX-1) can also promote the recognition and absorption of modified LDL, while LOX-1 is the main receptor for endothelial cells to bind ox-LDL (50). What’s more, in macrophages, the expression of SR-A1 is regulated by NF-κB, AP-1, and PPAR γ, while the expression of LOX-1 is mainly regulated by NF- κB, AP-1, and POU-domain transcription factors (45, 51).
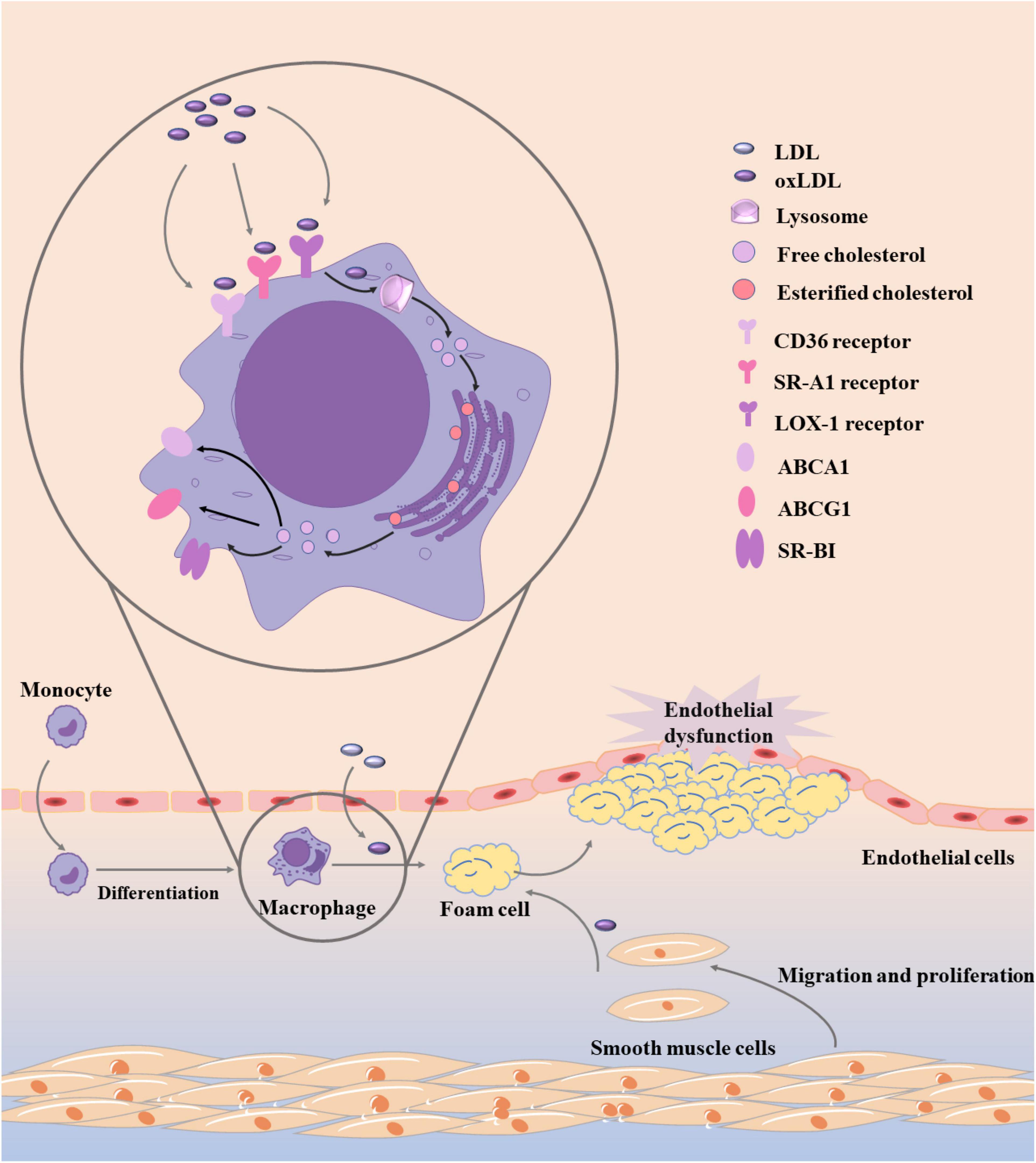
Figure 2. Development of foam cells in atherosclerosis. Monocytes recruit to the area of LDL modification and differentiate into macrophages, which can quickly recognize and absorb modified LDL into foam cells. Besides the modified LDL, the disorder of lipid metabolism in macrophages is another important factor of foam cell production, and the homeostasis of lipid metabolism in macrophages is mainly coordinated by three main processes, including cholesterol uptake, cholesterol esterification, and cholesterol efflux.
After the modified LDL was recognized and absorbed by macrophages, it was first transformed into free cholesterol by lysosomal acid lipase (LAL) in lysosome. When cholesterol accumulates excessively, cholesterol acyltransferases-1 (ACAT1) and –2 (ACAT2) in the endoplasmic reticulum will esterificate free cholesterol again. Subsequently, cholesteryl ester hydrolases (CEH) such as hormone sensitive lipase (HSL), carboxyl ester lipase (CEL), and neutral cholesterol ester hydrolase 1 (NCEH1) can hydrolyze esterified cholesterol again (52–54). During this process, the esterification and hydrolysis of cholesterol should be balanced. If the balance is broken, the generation rate of foam cells will be accelerated. The re-esterification of cholesterol in macrophages can prevent the accumulation of free cholesterol from damaging cells, but this process has a certain limit. When the cholesterol exceeds a certain range after re-esterification, a large number of lipid droplets will be generated in cells (55).
Of course, the content of free cholesterol in macrophages should not exceed the limit. In addition to the re-esterification mentioned above, free cholesterol can also maintain intracellular metabolic balance through cholesterol efflux process. ATP-binding cassette transporter A1 (ABCA1), ATP-binding cassette transporter G1 (ABCG1), and scavenger receptor class B type 1 (SR-BI) are mainly involved in the process of cholesterol efflux, which can bind to free cholesterol and transport out of cells (56). Subsequently, ABCA1 carrying cholesterol preferentially bind to apolipoprotein A1 (apoA1) to produce HDL particles, ABCG1 preferentially interacts with mature HDL particles, and SR-BI interacts with a variety of lipoproteins (45, 57). PPAR γ, liver X receptor (LXR), retinoid X receptor, and some miRNAs can regulate the expression of ABCA1, ABCG1, and SR-BI (58, 59).
Vascular smooth muscle cells are the most abundant cell type in the arterial wall, and have phenotypic plasticity. They can show different phenotypes in different arteries or different diseases (60). In healthy blood vessels, VSMCs can maintain homeostasis by adjusting their phenotypes to adapt to changes in blood flow when hemodynamics changes. Conversely, when arteries become diseased, this ability was reduced and homeostasis was broken, which could exacerbate the disease (61). Mature VSMCs, for example, have a low proliferation rate and can respond to changes of NO and ET-1 from endothelium and regulate blood flow by regulating blood vessel diameter through contraction (62). Unfortunately, because VSMCs are not in a final differentiation state, when atherosclerosis occurs, the expression of specific markers of mature VSMCs under biochemical and biomechanical stimulation is inhibited, a large number of VSMCs differentiate into synthetic phenotypes and migrate to the intima of arterial wall under the guidance of platelet-derived growth factor B (PDGF-B) (63). On the one hand, activated VSMCs proliferate in the intima and narrow the arterial lumen, which are regarded as the main features of atherosclerosis. On the other hand, VSMCs produce collagen fibers and elastic fibers under the stimulation of transformational growth factor-β (TGF-β), change the composition of extracellular matrix and envelops lipids by fiber caps to form typical atherosclerotic plaques (64). In addition, the latest research also shows that fibro-myocytes differentiated by VSMC can stabilize the plaque, and when they differentiate into cartilage, osteoblasts or inflammatory cells, they can aggravate the development of atherosclerosis (65, 66). Thus, the phenotypic transformation of VSMCs is crucial in atherosclerosis.
In the past few decades, increasing experiments have focused on the process of controlling VSMC phenotypic conversion, but the key molecular mechanism has not been clearly clarified (67). Subsequently, there are growing evidences that epigenetic mechanisms provide transcriptional control that can directly cause phenotypic switch in VSMC, which is shown in Figure 3 (68). Theoretically, epigenetic mechanism is to change gene expression through three main epigenetic modifications, DNA methylation, histone modification, and non-coding RNA (ncRNA) modification without changing the genome (69, 70). In atherosclerosis, DNA methylation can regulate a variety of genes that define VSMC phenotypic transformation, such as serum response factor (SRF), PDGF-B, and TAGLN (64). DNA methyltransferase 1 (DMNT1) and Ten-eleven translocated methylcytosine deoxygenase 2 (TET2), two major enzymes that control DNA methylation, also play an important role (71). It was found that knockdown of TET2 suppressed the expression of key VSMC genes such as MYOCD and SRF, while transcriptional upregulation of KLF4 initiated VSMC phenotypic transition (72). When TET2 was overexpressed, VSMC phenotype conversion was inhibited and intimal hyperplasia was significantly improved. In contrast, when DNMT1 was repressed, MYOCD expression was increased and VSMC phenotypic conversion was inhibited (73). Histone modification mainly includes methylation, acetylation, and ubiquitination. The role of histone methylation and acetylation in atherosclerosis and VSMC phenotype transformation cannot be ignored, and most of them appear in a combination form (74). For example, a significant decrease in H3K9 and H3K27 methylation and a significant increase in H3K9 and H3K27 acetylation levels were observed in atherosclerotic plaques (75, 76). Besides, VSMC phenotypic transition can be regulated by microRNAs (miRNAs) and long-stranded non-coding RNA (lncRNAs) (77).
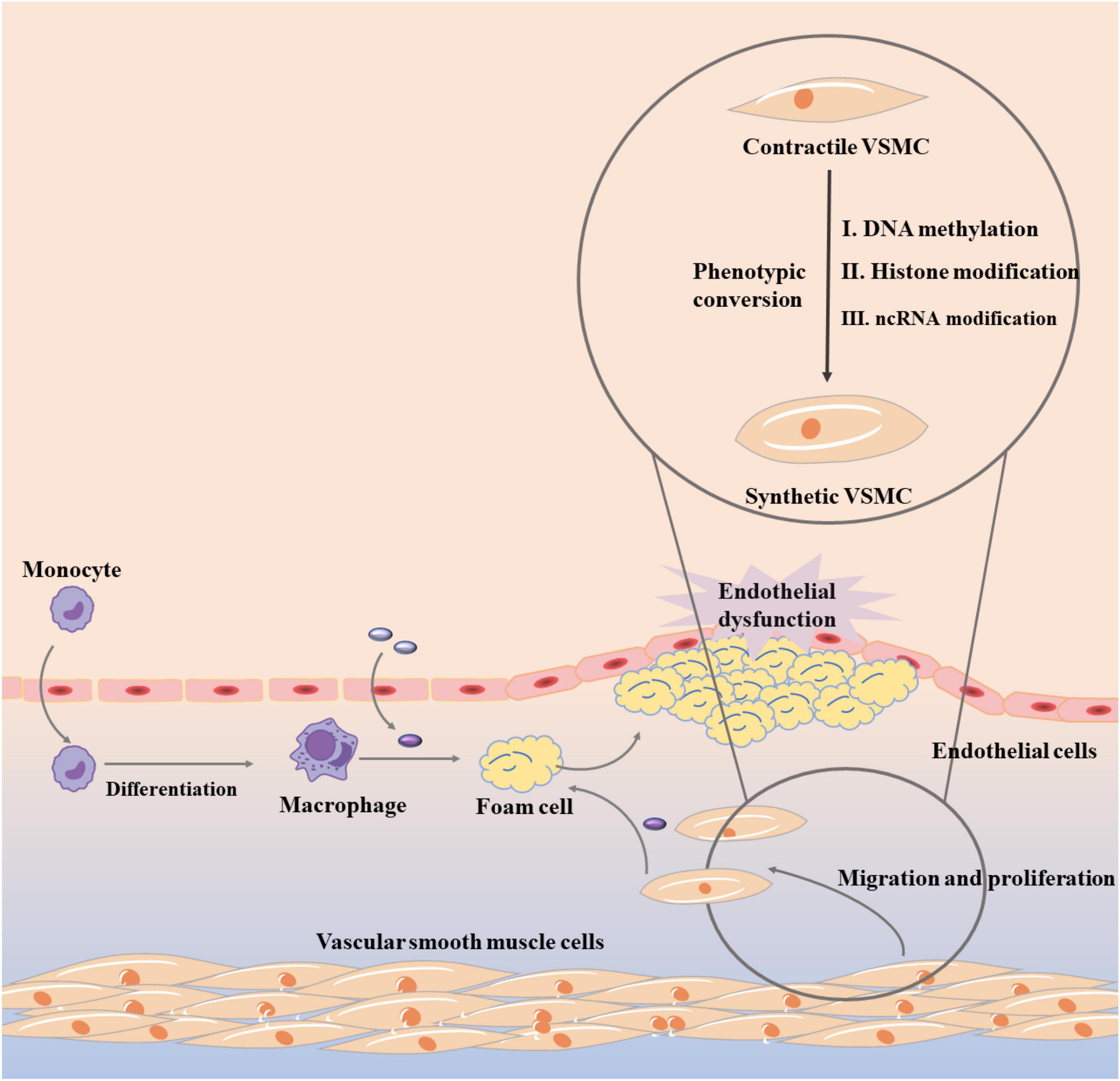
Figure 3. Vascular smooth muscle cell phenotypic conversion in atherosclerosis.
As we all know, atherosclerosis is a chronic inflammatory disease. Inflammation is accompanied by the initiation and development of the whole disease. After decades of extensive research, we have preliminarily elucidated that the related inflammatory response in atherosclerosis is mediated by proinflammatory cytokines, adhesion molecules, inflammatory signaling pathways and bioactive lipids (78). In general, healthy endothelial cells are able to effectively resist leukocyte adhesion, and acute inflammation can restore normal tissue structure through leukocyte infiltration and subsequent clearance mechanism (79). However, when related events such as hypertension and hyperglycemia occur, endothelial cells activate and consequently express monocyte chemoattractant protein-1, interleukin (IL)-8, intercellular adhesion molecule-1 (ICAM-1), vascular adhesion molecule-1 (VCAM-1), E-selectin, P-selectin, and other inflammatory factors, resulting in monocyte retention and triggering chronic inflammatory injury (80). This view was further proved in vitro experiments. Pro-inflammatory monocytes with high expression of Ly6C preferentially adhere to cytokine-stimulated endothelial cells, and dendritic cells, T cells and neutrophils are also involved in this inflammatory response (81). With the development of atherosclerosis, macrophages, VSMC, and endothelial cells in arteries can secrete a variety of matrix metalloproteinases (MMPs). MMP-9 can increase macrophage infiltration and collagen deposition, while MMP-2 can promote extracellular matrix degradation and VSMC migration (82, 83). Both of them work together to form an arterial pro-inflammatory environment and aggravate the inflammatory reaction in atherosclerosis.
In fact, although inflammatory response is involved in all processes of atherosclerosis, there is no practical evidence to support the inflammatory hypothesis in early studies until the discovery of inflammatory markers (84). In addition to MMPs, IL-6, C-Reactive protein (CRP), and adhesion molecules are inflammatory markers. When the inflammatory response is turned on in arteries, macrophages and adipocytes release large amounts of IL-6 and TNF- α,inducing a downstream inflammatory cascade to occur (85). In another experiment, it was also found that the risk of coronary heart disease increased with the upregulating of IL-6 level in plasma and was positively correlated with the severity of the disease (86). At the same time, the release of IL-6, IL-1β, and TNF-α stimulated the synthesis of CRP in the liver and adipose tissue, prevents the proliferation and repair of vascular endothelial cells (87). Selectin family, immunoglobulin superfamily (IgSF) and integrin family of adhesion molecules are involved in the development of atherosclerosis (88). Among them, vascular cell adhesion molecule-1 (VCAM-1) and ICAM-1 play an important role. As shown in Figure 4, VCAM-1 can activate endothelial cells by upregulating the transcription factor nuclear factor-κB (NF-κB), which causes endothelial cells to release various pro-inflammatory cytokines such as IL-1, TNF-a, IL-6, and IL-8 (89). The high expression of VCAM-1 and ICAM-1 can promote the proliferation of macrophages, lead to the excessive accumulation of macrophages in the plaque and reduce the stability of the plaque (90). At the same time, VCAM-1 and ICAM-1 can also promote the formation of high permeability and fragile neovascularization (91).
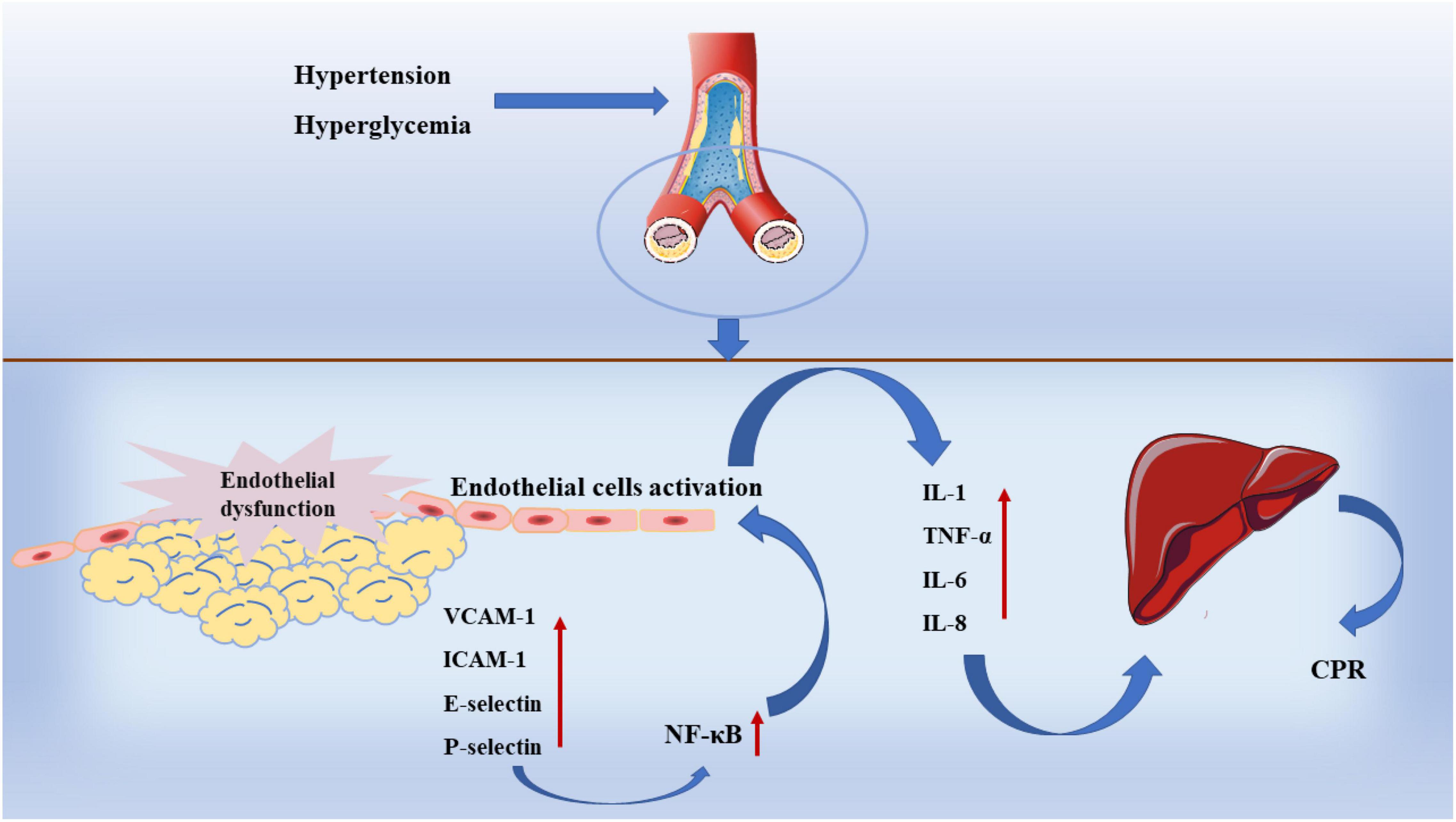
Figure 4. Inflammation in atherosclerosis.
As researchers have progressively studied atherosclerosis, the inflammatory signaling pathways associated with the disease continue to attract more attention. From the known studies, toll like receptor 4 (TLR4), NF- κB, Janus kinase (JAK) signal transducers and activators of transcription (STAT) have been identified as major signaling pathways. ABCG1, a key gene linking lipid accumulation and inflammation, can be regulated by TLR4 in the organism (92). After the onset of atherosclerotic process, TLR4 activates the peroxisome proliferator-activated receptor γ (PPAR-γ)/liver X receptor α (LXRα) signaling pathway, which in turn downregulates ABCG1 expression (93). Meanwhile, TLR4 can also promote the release of MCP-1, IL-1α, and IL-6 by activating NF-κB, which induces lipid accumulation in the arterial vasculature and the development of inflammation (94). In atherosclerotic, JAK/STAT is mainly activated by cytokines of JAK kinases (JAK1, JAK2, JAK3) and tyrosine kinase (Tyk)2. In experiments, it was found that activation of p-STAT3 was often accompanied by elevated levels of IL-6 and TNF-α, while activation of STAT4 similarly caused secretion of IFN-γ and TNF-α, which activated macrophages and made arterial plaques larger (95, 96).
Flavones are a kind of compounds existing in nature and fruits, which play an important role in fruit growth, development, and antibacterial activities. In modern pharmacological research, it is found that the flavones are inseparable from the anti-atherosclerotic effect of fruits (97). Apigenin is a kind of typical flavones named 4′,5,7,-trihydroxyflavone. It is widely found in oranges, grapefruit, and other fruits. Apigenin has high biological activity, can play neuroprotection, antioxidant, anti-tumor, and other effects (98). In addition, it has been found in recent studies that apigenin could participate in all stages of atherosclerosis through a variety of mechanisms, so as to play an anti-atherosclerotic role. As mentioned earlier, hyperlipidemia caused by high-fat diet may be an important factor in inducing atherosclerosis. In SD rats fed with high-fat diet, 8.0 g/kg apigenin was given by gavage for 2 weeks. The results showed that after apigenin treatment, the thickening of aortic intima was alleviated, the contents of TC, TG, and LDL-C decreased, and the content of HDL-C increased, indicating that apigenin could reduce the possibility of atherosclerosis by improving hyperlipidemia (99). However, when hyperlipidemia occurred in vivo, the expression of LOX-1 in endothelial cells increased, which promoted the binding of endothelial cells to oxLDL, resulting in endothelial dysfunction. Surprisingly, in HUVECs activated by oxLDL, apigenin could alleviate endothelial cell dysfunction by reducing the expression of LOX-1, VCAM-1, and E-selectin (100). Subsequently, glucose-induced HUVECs and HAECs and trimethylamine-N-oxide-induced has cells were used to study the underlying mechanism (101–103). The results showed that apigenin could protect endothelial cells through a variety of signal pathways. For example, apigenin could inhibit endothelial cell apoptosis by decreasing the expression of PKCβII and phosphorylation of NF-κB through ROS/caspase-3 and NO signaling pathway (101). Furthermore, apigenin could also improve the uncontrolled vasodilation and enhance the antioxidant activity of endothelial cells by up-regulating the activity of eNOS and the content of NO and SOD (102, 103). Apigenin also plays an important role in the formation of foam cells. For example, in vitro experiments, apigenin could enhance the expression of ABCA1 by inhibiting miR-33, promote the cholesterol efflux in macrophages, and effectively reduce the content of TC, FC and CE in foam cells (104). In vivo, apigenin was used to treat LPS-induced ApoE–/– mice, and the same results were obtained. That is, apigenin affected the expression of miR-33, ABCA1, NF-κB p65, and TLR-4, promoting cholesterol efflux and reducing the number of macrophages and smooth muscle cells in atherogenesis, which leads to the decrease in foam cells as well (104). Of course, in addition to affecting lipid metabolism, apigenin could also down-regulate the expression of PAI-2 by inhibiting the phosphorylation of Akt at ser473 site, increase the expression of Bax and cleaved caspase-3 in oxLDL-induced macrophages, and promote macrophage apoptosis (105). In further studies, it was also found that after apigenin treatment, autophagy mediated by ATG5/Atg7 was enhanced in oxLDL-induced macrophages. The role of apoptosis and autophagy accelerates the attenuation of macrophages and relieves the formation of foam cells (106). Simultaneously, apigenin not only inhibited the activation of Caspase-1 by destroying NLRP3 inflammasome assembly, but also reduced mRNA stability by inhibiting ERK1/2 activation in response to inflammation throughout the development of atherosclerosis. The combination of two effects inhibited the secretion of IL-6, IL-1β, and TNF-α, thereby inhibiting the activation of NF-κB in LPS-induced macrophages (107). In addition to apigenin, a variety of flavones derived from fruits in Supplementary Table 1, such as luteolin, tangeretin and chrysoeriol, can inhibit the development of atherosclerosis.
Flavonols refer to a class of compounds containing 2-phenyl-3-hydroxy (or oxygen-substituted) benzo-γ-pyrone (2-phenyl-3-hydroxy-chromone). They are the most abundant flavonoids, and there are more than 1,700 kinds of flavonols have been found. Quercetin, one of the most abundant flavonols in fruits, has been widely shown to be useful in the prevention and treatment of atherosclerosis. First, quercetin was used to treat Caco-2 cells and human embryonic kidney 293T cells who expressing NPC1L1, and it was found that quercetin inhibited cellular cholesterol uptake by reducing NPC1L1 mRNA levels (132). Subsequently, quercetin was administered to ApoE–/– mice induced by high-fat diet. The results showed that quercetin regulated lipid metabolism by up-regulating the expressions of PPARγ, LXR-α, ABCA1, and down-regulating the expressions of PCSK9 and CD36, reducing the content of TC, LDL-C, oxLDL, and lipid droplets in the cytoplasm, and alleviated the symptoms of atherosclerosis (133). At the same time, quercetin could also reduce the content of TNF-α and IL-6 in the serum of mice, increase the content of IL-10 (133). In another oxLDL-induced RAW264.7 macrophage, quercetin promoted the expression of LC3-II/I and Beclin 1 by reducing the expression of MST1. Simultaneously, quercetin also inhibited the expression of Bcl-2, P21, and P16, which ultimately triggered autophagy in macrophages and reduced foam cell formation (134). Without doubt, quercetin also has excellent efficacy in inhibiting inflammation. In high-glucose-induced human THP-1 monocytic cells, quercetin inhibited the expression of pro-inflammatory genes and related proteins, including TNF-α, IL-1β, COX-2, etc., through the MAPK signaling pathway (115). Correspondingly, in ApoE–/– mice fed on a high-fat diet, quercetin reduced inflammatory by up-regulating Sirt1 and down-regulating Slcam-1 and VCAM-1 expression. All the above results suggest that quercetin is a potential natural compound for the treatment of atherosclerosis (135).
Kaempferol is another flavonol with broad bioactivity that has been shown to reduce the risk of atherosclerosis. Kaempferol was initially confirmed to play a synergistic role with urate in plasma to jointly exert antioxidant effect and reduce oxidative modification of LDL, which preliminarily suggested that kaempferol may have an anti-atherosclerosis effect (136). Subsequently, kaempferol was applied to rabbits fed with a high cholesterol diet. The results showed that after kaempferol treatment, the levels of TNF-α, IL-1β, and MDA in aorta decreased significantly, and the activity of SOD in serum increased. Meanwhile, the expression of genes and proteins related to inflammation, such as E-sel, ICAM-1, VCAM-1, and MCP-1, decreased significantly, which inhibited the occurrence of inflammation (137). In ox-LDL-induced endothelial cells, kaempferol not only inhibited the PI3K/Akt/mTOR pathway, but also upregulated LC3-II/I and Beclin-1, which reduced endothelial cell apoptosis (138). Nevertheless, in a recent study, researchers applied kaempferol to atherosclerotic mice. It was a surprise to everyone that kaempferol inhibited inflammation and apoptosis by activating the membrane G-protein conjugated estrogen receptor (GPER), thereby activating the PI3K/AKT/Nrf2 signaling pathway (139). It’s revealed that when kaempferol acts on different models, its mechanism of action is different, but its preventive and therapeutic effects on atherosclerosis cannot be neglected. Besides, there are a variety of flavonols in fruit that have the same effect, as shown in Table 1.

Table 1. Flavonols derived from fruits are potential agents against Atherosclerosis.
As an important component of flavonoids, the role of flavone glycoside in atherosclerosis has been gradually concerned. Rutin, a typical flavonoid glycoside found in apples, green tea and other sources, has antioxidant and anti-inflammatory activities and multiple therapeutic effects in atherosclerosis. In HUVEC cells induced by H2O2, rutin could enhance the expression and activity of eNOS by up-regulating the expression of basic fibroblast growth factor (bFGF), thereby increasing the production of NO and improving endothelial function (150). In high glucose induced VSMCs, rutin inhibited the migration and proliferation of VSMCs by inhibiting the MAPK (ERK1/2), BMK1, PI3K, and NF-κB signaling pathways (151). In vivo, when rutin was applied to streptozotocin (STZ)-induced ApoE–/– mice, a significant reduction of atherosclerotic plaque in aorta was observed, accompanied by an increased proportion of VSMCs and enhanced plaque stability (152).
Naringin is the main compound of tomato, grapefruit, and related citrus. It is a flavanone glycoside with a disaccharide neohesperidose linked at C7 of the C6 (A ring)-C3 (C ring)-C6 (B ring) flavanone skeleton. It was first found in mice on a high-fat/high-cholesterol diet that treatment with naringin reduced plasma non-HDL cholesterol concentrations and ICAM-1, a biomarker of endothelial dysfunction. Transcriptome analysis of potential molecular targets suggested that the therapeutic effect of naringin may be related to its ability to reduce the adhesion of monocytes to endothelial cells and the proliferation of smooth muscle cells (153). In the following experiments, TNF-α-induced HUVECs were used as an in vitro model to further study the anti-atherosclerosis effect of naringin. The results showed that naringin inhibited the expression of adhesion molecules and chemokines, including VCAM-1, ICAM-1, and E-selectin, by inhibiting the activation of IKK/NF-κB signaling pathway (154). In addition, ox-LDL was used as a model drug to induce HUVECs. After naringin administration, VE-cadherin decomposition and F-actin remodeling were inhibited, and endothelial function was protected. At the same time, IL-1β, IL-6, IL-18, and other pro-inflammatory factors were decreased, and this protective effect was directly related to the YAP signaling pathway (155). In addition to these effects, naringin also has an ideal effect on regulating atherosclerosis through gut microbiota. Previous reports have found that naringin is highly hydrophilic and lacks the corresponding hydrolase in the body. This property protects naringin against digestion and absorption in the small intestine. Therefore, naringin can reach the colon and affect the composition of the gut microbiota after oral intake. The results showed that after naringin reached the colon, the relative abundance of g_Bacteroides, g_Bifidobacterium, and g_Lactococcus in the colon was decreased, and the content of bile salt hydrolyase was decreased. In contrast, the abundance of 7α-dehydroxylase producing bacteria-Eubacterium_fissicatena, Eubacterium_coprostanoligenes, and Eubacterium_brachy increased. The changes of the gut microbial community structure could directly promote the degradation of free bile acid, regulate the metabolism of cholesterol in the body, and increase the excretion of bile acid and neutral sterol by 1.6-fold and 4.3-fold, respectively. Cholesterol levels in serum and liver were also decreased to different degrees. These results suggested that naringin could alleviate atherosclerosis through the gut microbiome–liver–cholesterol axis (156). Other flavonoid glycosides are shown in Supplementary Table 2.
In addition to the above classification, there are many other classes of natural flavonoid compounds in fruits that can be used to treat atherosclerosis. For example, dihydromyricetin, derived from actinidia arguta, is structurally classified as a flavanones. In recent years, previous studies have found that dihydromyricetin can significantly improve hyperlipidemia in mice, reduce the levels of ox-LDL, IL-6, and TNF-α in serum, and restore inflammation to normal levels (173). Meanwhile, the protein expression of PPARα, LXRα, and ABCA1 was increased to promote lipid efflux and prevent lipid accumulation (173). In addition, in vitro cell models, dihydromyricetin could protect endothelial cell function, inhibit endothelial cell apoptosis, and prevent monocyte adhesion by activating Nrf2 or mir-21 signaling pathways (174, 175). Catechins derived from apples and peaches are flavanols that could reduce high glucose induced inflammation in human THP-1 cells through MAPK signaling pathway, reduce the expression of pro-inflammatory genes and proteins, including TNF-α, IL-1β, and COX-2, and reduce monocyte adhesion (115). Epicatechin, which also belongs to flavanols, is also distributed in apples, and has a good effect on severe atherosclerosis. Epicatechin attenuates the inflammatory process of atherosclerosis by inhibiting NF-κB signaling, reducing neutrophils and chemokines, and slowing stromal remodeling (176). In addition to these, anthocyanins such as pelargonidin, delphinidin, petunidin, xanthohumol, and phloretin in chalcone can also be used for the treatment of atherosclerosis, see Table 2 for details.
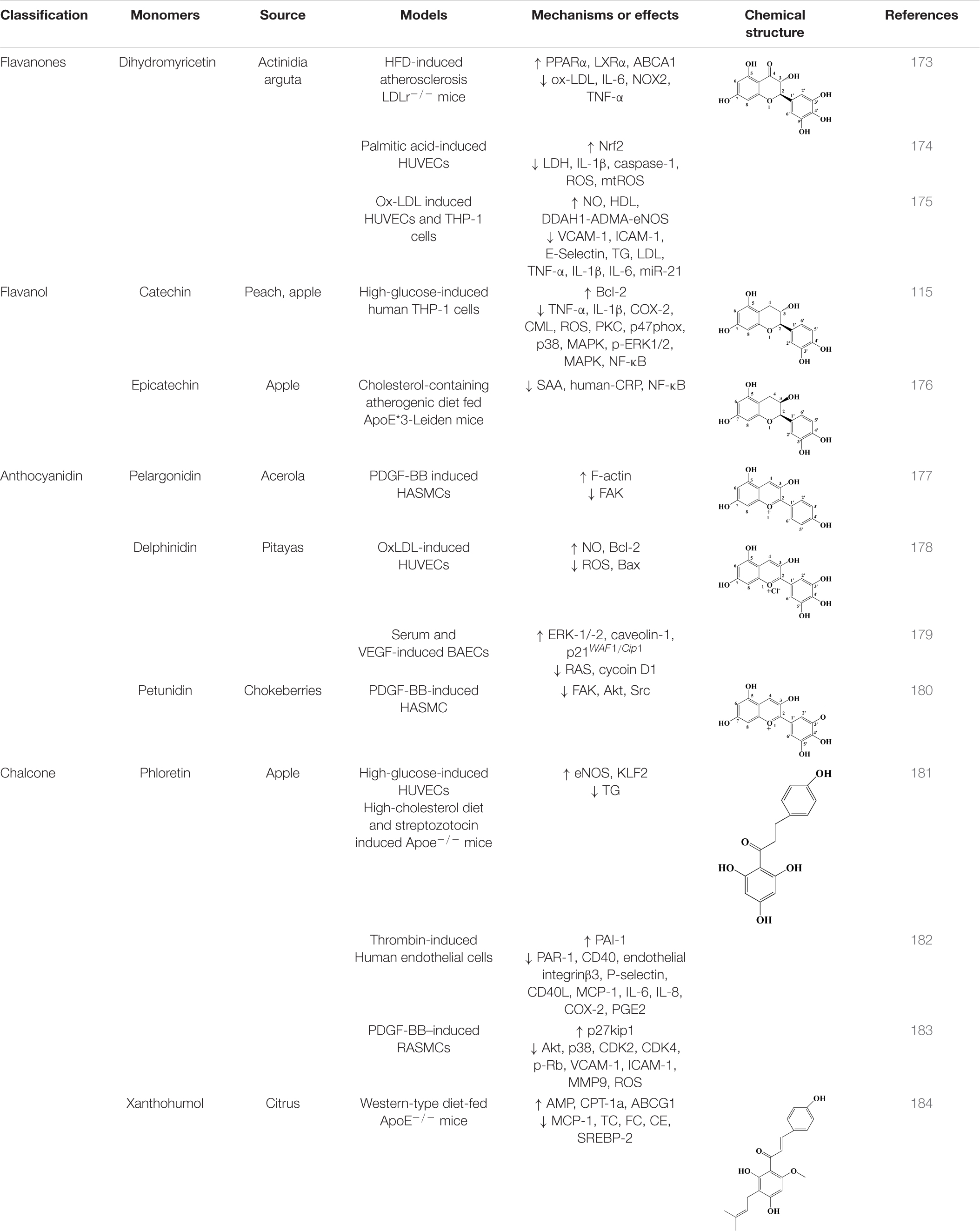
Table 2. Other flavonoids derived from fruits are potential agents against Atherosclerosis.
In recent years, atherosclerosis is increasingly threatening to human life, and the existing drugs or surgical treatments have certain limitations, so it is urgent to develop new drugs or treatment methods. Flavonoids are important bioactive components in fruits and are widely used in various nutritional products, cosmetics and medicines. At the same time, flavonoids from fruit have been shown to be effective in various stages of atherosclerosis development in recent studies. Based on our conclusion, current evidence suggests that fruit flavones have therapeutic effects on atherosclerosis by protecting endothelial cells, inhibiting foam cell formation, regulating lipid metabolism, and anti-inflammation, and the underlying molecular mechanisms are gradually being elucidated in more specific ways.
However, there are limitations and controversies that prevent the generalization of these results. It can be seen from Tables 1, 2 and Supplementary Tables 1, 2 that there are multiple in vivo and in vitro models to choose from in the study of flavonoids against atherosclerosis. When a compound acts on the same cell or animal stimulated by different modeling agents, its efficacy and mechanism of action will also be different, and an appropriate research model is an important prerequisite to ensure the accuracy of mechanism exploration and the reliability of results. At present, human or mouse cell lines are mostly used in vitro studies. For example, when studying the effect of flavonoids on inflammation in atherosclerosis, mouse leukemia cell lines RAW264.7 and J774 and human leukemia monocyte cell line THP-1 are mostly used, and THP-1 can differentiate into macrophages after the intervention of a variety of factors. However, in modern studies, it is generally believed that the atherosclerotic lesions in mice have been affected by microenvironmental factors, and the results of immortal cell lines do not reflect the in vivo process. Therefore, primary macrophages and peritoneal macrophages derived directly from animals have been applied in the study of flavonoid anti-atherosclerosis. In vivo models, researchers mostly use mouse and rabbit models as the main experimental platform. However, the major sites of atherosclerosis in humans are the coronary and carotid arteries, whereas in mice, the major sinus and innominate arteries are predominant. The in vivo model is also limited by the difference of lesion location.
All flavones from fruits have been studied in many aspects in clinical trials, but there is still a lack of clinical trials on atherosclerosis. At present, the research trend of flavonoids in fruits still remains to study their mechanism of action and molecular target, so as to explore their medicinal potential in atherosclerosis. However, whether a compound is suitable for development into a drug is also related to its bioavailability, metabolism, distribution, etc. As mentioned earlier, quercetin is an excellent potential drug with multiple therapeutic effects on atherosclerosis. When the solubility of quercetin was studied, it was found that the solubility of quercetin was 2.1 mg/L in water and 2 g/L in ethanol. This physical property directly limits the absorption of quercetin in the body (185). Pharmacokinetic results in human showed that the bioavailability of quercetin was very poor after a single oral administration. In addition, dietary quercetin is usually present in the form of glycosylation, which can be hydrolyzed by β-glucosidase in the digestive system and absorbed in the intestinal mucosa. Subsequently, quercetin can be transported to the liver via the portal vein and metabolized by glucuronidation, methylation, or sulfonylation (186). However, in recent studies, it was found that quercetin glucuronides, a major circulating metabolite, was rapidly eliminated in the human body, and the short elimination half-life was also an important reason for limiting the development of quercetin drugs (187). To solve the existing problems, the preparation of different delivery systems using nanotechnology has been widely accepted. For example, quercetin was encapsulated in nano-polymeric micelles, and then relevant pharmacokinetic experiments were performed in beagle dogs. The results showed that compared with free quercetin, the half-life of nano-quercetin was prolonged by 2.19 times after the application of nanotechnology, and its relative oral bioavailability was increased by 286%. Therefore, nanotechnology also has high potential in the treatment of atherosclerosis (188). In addition to polymer nanomaterials, inorganic nanomaterials, lipid-based nanomaterials, and biomimetic nanomaterials have been involved in the development of effective drugs for the treatment of atherosclerosis. Unfortunately, most drug development is still in the pre-clinical stage and has not been widely studied. In addition, the design of nanomaterials for dual therapy is also an important direction in future research.
Interest in the interaction of gut microbiota with flavonoids has increased in recent years. Under the action of intestinal flora, flavonoids can be hydrolyzed into aglycones in intestinal tract, and then reduced by hydrogenation of C ring. Finally, O-C2 bonds on C-ring are cleaved to form phenolic ketones and phenolic acids. In this process, the metabolic transformation of flavonoids enables them to be better absorbed by the small intestine and improve the bioavailability of flavonoids through systemic and local anti-atherosclerosis effects (189). At the same time, flavonoids in the intestinal tract can also affect the structure and function of gut microbiota, affecting the balance of gut microbiota (190). The interaction between flavonoids and gut microbiota provides a new perspective for understanding the effect mechanism of flavonoids on atherosclerosis. It is worth noting that up to now, the study on absorption, distribution and metabolism of flavonoids in the gut microbiota is still in its infancy. The therapeutic effect of flavonoid on atherosclerosis under the action of gut microbiota is not stable, and the underlying mechanism needs to be further explored.
Therefore, in future experiments, based on existing studies, we should increase the study of its pharmacokinetic, metabolic and pharmacodynamic characteristics in vivo, and find better flavonoid compounds and nanomaterials for the treatment of atherosclerosis, so as to find more reliable drugs for the treatment of disease.
R-LL and L-YW participated in the whole work. SL and H-XD participated in manuscript design, literature acquisition, and analysis. QZ and TZ participated in the drafting and revision of the manuscript. WP, YH, and CW gave final approval to the forthcoming edition, agreed on the journal to which the manuscript was submitted, and agreed to be responsible for all aspects of the work. All authors made a significant contribution to the work reported.
This work was supported by Sichuan Science and Technology Program (no. 2020YFS0523), Research Promotion Plan for Xinglin Scholars in Chengdu University of Traditional Chinese Medicine (no. QNXZ2019018), and Science and Technology Development Fund of Hospital of Chengdu University of Traditional Chinese Medicine (nos. Y2019128 and Y2019114).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2022.862277/full#supplementary-material
1. Otreba M, Kosmider L, Rzepecka-Stojko A. Polyphenols’ cardioprotective potential: review of rat fibroblasts as well as rat and human cardiomyocyte cell lines research. Molecules. (2021) 26:774. doi: 10.3390/molecules26040774
2. Palasubramaniam J, Wang XW, Peter K. Myocardial infarction-from atherosclerosis to thrombosis uncovering new diagnostic and therapeutic approaches. Arter Throm Vas. (2019) 39:E176–85. doi: 10.1161/ATVBAHA.119.312578
3. Chen J, Zhang X, Millican R, Sherwood J, Martin S, Jo H, et al. Recent advances in nanomaterials for therapy and diagnosis for atherosclerosis. Adv Drug Deliv Rev. (2021) 170:142–99. doi: 10.1016/j.addr.2021.01.005
4. Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392:1736–88. doi: 10.1016/s0140-6736(18)32203-7
5. d’SOUZA J, Giri J, Kobayashi T. Stent-based revascularization for complex lesions in PAD. J Cardiovasc Surg. (2017) 58:715–21. doi: 10.23736/S0021-9509.17.09994-3
6. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114:1852–66. doi: 10.1161/CIRCRESAHA.114.302721
7. Davies JT, Delfino SF, Feinberg CE, Johnson MF, Nappi VL, Olinger JT, et al. Current and emerging uses of statins in clinical therapeutics: a review. Lipid Insights. (2016) 9:13–29. doi: 10.4137/LPI.S37450
8. Beecher GR. Overview of dietary flavonoids: nomenclature, occurrence and intake. J Nutr. (2003) 133:3248S–54S. doi: 10.1093/jn/133.10.3248S
9. Bohn T. Dietary factors affecting polyphenol bioavailability. Nutr Rev. (2014) 72:429–52. doi: 10.1111/nure.12114
10. Millar CL, Duclos Q, Blesso CN. Effects of Dietary Flavonoids on reverse cholesterol transport, HDL metabolism, and HDL function. Adv Nutr. (2017) 8:226–39. doi: 10.3945/an.116.014050
11. Kumari D, Bandyopadhyay P, Suryaprakash N. Discrimination of a-amino acids using green tea flavonoid (–)-epigallocatechin gallate as a chiral solvating agent. J Org Chem. (2013) 78:2373–8. doi: 10.1021/jo3025016
12. Graf BA, Milbury PE, Blumberg JB. Flavonols, flavones, flavanones, and human health: epidemiological evidence. J Med Food. (2005) 8:281–90. doi: 10.1089/jmf.2005.8.281
13. Wilting J. Integrated vascular anatomy. In: P Lanzer, EJ Topol editors. Pan Vascular Medicine: Integrated Clinical Management. Berlin: Springer (2002). p. 50–75. doi: 10.1007/978-3-642-56225-9_4
14. Goikuria H, Vandenbroeck K, Alloza I. Inflammation in human carotid atheroma plaques. Cytokine Growth Factor Rev. (2018) 39:62–70. doi: 10.1016/j.cytogfr.2018.01.006
15. Jaminon A, Reesink K, Kroon A, Schurgers L. The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci. (2019) 20:5694. doi: 10.3390/ijms20225694
16. Libby P, Ridker PM, Hansson GK Leducq Transatlantic Network on Atherothrombosis. Inflammation in Atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. (2009) 54:2129–38. doi: 10.1016/j.jacc.2009.09.009
17. Owens G, Meena SK, Wamhoff B. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Rev. (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
18. Sumpio BE, Timothy Riley J, Dardik A. Cells in focus: endothelial cell. Int Biochem Cell Biol. (2002) 34:1508–12. doi: 10.1016/s1357-2725(02)00075-4
19. Vita JA, Keaney JF. Endothelial function: a barometer for cardiovascular risk. Circulation. (2002) 106:640–2. doi: 10.1161/01.cir.0000028581.07992.56
20. Verma S, Anderson TJ. Fundamentals of endothelial function for the clinical cardiologist. Circulation. (2002) 105:546–9. doi: 10.1161/hc0502.104540
21. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
22. Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. (2008) 133:1006–18. doi: 10.1016/j.cell.2008.03.038
23. Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn hR, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. (2008) 133:1019–31. doi: 10.1016/j.cell.2008.03.039
24. Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, Ferbeyre G. The senescence-associated secretory phenotype and its regulation. Cytokine. (2019) 117:15–22. doi: 10.1016/j.cyto.2019.01.013
25. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxidative Med Cell Longevity. (2019) 2019:1–32. doi: 10.1155/2019/8563845
26. Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. (2013) 123:540–1. doi: 10.1172/JCI66843
27. Martin-Ventura JL, Rodrigues-Diez R, Martinez-Lopez D, Salaices M, Blanco-Colio LM, Briones AM. Oxidative stress in human atherothrombosis: sources, markers and therapeutic targets. Int J Mol Sci. (2017) 18:2315. doi: 10.3390/ijms18112315
28. Konukoglu D, Uzun H. Endothelial dysfunction and hypertension. Adv Exp Med Biol. (2017) 956:511–40. doi: 10.1007/5584_2016_90
29. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. (1980) 288:373–6. doi: 10.1038/288373a0
30. Radomski MW, Palmer RM, Moncada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Commun. (1987) 148:1482–9. doi: 10.1016/s0006-291x(87)80299-1
31. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. (2006) 113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532
32. Anderson TJ. Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. (1999) 34:631–8. doi: 10.1016/s0735-1097(99)00259-4
33. Gimbrone MA Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
34. Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. (1987) 84:9265–9. doi: 10.1073/pnas.84.24.9265
35. Violi F, Loffredo L, Carnevale R, Pignatelli P, Pastori D. Atherothrombosis and oxidative stress: mechanisms and management in elderly. Antioxid Redox Signal. (2017) 27:1083–124. doi: 10.1089/ars.2016.6963
36. Farbstein D, Soloveichik YZ, Levy NS, Levy AP. Genetics of redox systems and their relationship with cardiovascular disease. Curr Atheroscler Rep. (2011) 13:215–24. doi: 10.1007/s11883-011-0170-7
37. Orekhov AN. LDL and foam cell formation as the basis of atherogenesis. Curr Opin Lipidol. (2018) 29:279–84. doi: 10.1097/mol.0000000000000525
38. Chistiakov A, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med. (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8
39. Weinberger T, Esfandyari D, Messerer D, Percin G, Schleifer C, Thaler R, et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat Commun. (2020) 11:4549. doi: 10.1038/s41467-020-18287-x
40. Williams JW, Zaitsev K, Kim K-W, Ivanov S, Saunders BT, Schrank PR, et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol. (2020) 21:1–11. doi: 10.1038/s41590-020-0768-4
41. Marchini T, Mitre LS, Wolf D. Inflammatory cell recruitment in cardiovascular disease. Front Cell Dev Biol. (2021) 9:635527. doi: 10.3389/fcell.2021.635527
42. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. (2007) 117:195–205. doi: 10.1172/jci29950
43. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117:185–94. doi: 10.1172/jci28549
44. Summerhill VI, Grechko AV, Yet SF, Sobenin IA, Orekhov AN. The atherogenic role of circulating modified lipids in atherosclerosis. Int J Mol Sci. (2019) 20:3561. doi: 10.3390/ijms20143561
45. Hutchins PH, Heinecke JW. Cholesterol efflux capacity, macrophage reverse cholesterol transport and cardioprotective HDL. Curr Opin Lipidol. (2015) 26:388–93. doi: 10.1097/mol.0000000000000209
46. Zhao L, Varghese Z, Moorhead JF, Chen Y, Ruan XZ. CD36 and lipid metabolism in the evolution of atherosclerosis. Br Med Bull. (2018) 126:101–12. doi: 10.1093/bmb/ldy006
47. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. (2014) 46:e99. doi: 10.1038/emm.2014.38
48. Chistiakov DA, Bobryshev YV, Orekhov AN. Macrophage-mediated cholesterol handling in atherosclerosis. J Cell Mol Med. (2016) 20:17–28. doi: 10.1111/jcmm.12689
49. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. (2013) 19:557–66.
50. Zhang Z, Zhang D, Du B, Chen Z. Hyperoside inhibits the effects induced by oxidized low-density lipoprotein in vascular smooth muscle cells via oxLDL-LOX-1-ERK pathway. Mol Cell Biochem. (2017) 433:169–76. doi: 10.1007/s11010-017-3025-x
51. Hermonat PL, Zhu H, Cao M, Mehta JL. LOX-1 transcription, cardiovasc. Drugs Ther. (2011) 25:393–400. doi: 10.1007/s10557-011-6322-8
52. Korber M, Klein I, Daum G. Steryl ester synthesis, storage and hydrolysis: a contribution to sterol homeostasis, Biochimica et Biophysica Acta. Mol Cell Biol Lipids. (2017) 1862:1534–45. doi: 10.1016/j.bbalip.2017.09.002
53. Sekiya M, Osuga J, Igarashi M, Okazaki H, Ishibashi S. The role of neutral cholesterol ester hydrolysis in macrophage foam cells. J Atheroscler Thromb. (2011) 18:359–64. doi: 10.5551/jat.7013
54. Ghosh S. Early steps in reverse cholesterol transport: cholesteryl ester hydrolase and other hydrolases, Current Opinion in Endocrinology. Diabetes Obesity. (2012) 19:136–41. doi: 10.1097/MED.0b013e3283507836
55. Ghosh S. Macrophage cholesterol homeostasis and metabolic diseases: critical role of cholesteryl ester mobilization. Expert Rev Cardiovasc Ther. (2011) 9:329–40. doi: 10.1586/erc.11.16
56. Favari A, Chroni UJ, Tietge I, Zanotti JC, Escolà-Gil BF. Cholesterol efflux and reverse cholesterol transport. Handb Exp Pharmacol. (2015) 224:181–206. doi: 10.1007/978-3-319-09665-0_4
57. Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. (2014) 289:24020–9. doi: 10.1074/jbc.R114.583658
58. Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis, nature reviews. Endocrinology. (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
59. Majdalawieh A, Ro HS. PPARgamma1 and LXRalpha face a new regulator of macrophage cholesterol homeostasis and inflammatory responsiveness, AEBP1. Nucl Recept Signal. (2010) 8:e004. doi: 10.1621/nrs.08004
60. Quintavalle M, Condorelli G, Elia L. Arterial remodeling and atherosclerosis: miRNAs involvement. Vascul Pharmacol. (2011) 55:106–10. doi: 10.1016/j.vph.2011.08.216
61. Leung DY, Glagov S, Mathews MB. Cyclic stretching stimulates synthesis of matrix components by arterial smooth muscle cells in vitro. Science. (1976) 191:475–7. doi: 10.1126/science.128820
62. Haga JH, Li YS, Chien S. Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech. (2007) 40:947–60. doi: 10.1016/j.jbiomech.2006.04.011
63. Wang D, Wang Z, Zhang L, Wang Y. Roles of cells from the arterial vessel wall in atherosclerosis. Med Inflamm. (2017) 2017:8135934. doi: 10.1155/2017/8135934
64. Montes de Oca A, Madueño JA, Martinez-Moreno JM, Guerrero F, Muñoz-Castañeda J, Rodriguez-Ortiz ME. High-phosphate-induced calcification is related to SM22α promoter methylation in vascular smooth muscle cells. J Bone Miner Res. (2010) 25:1996–2005. doi: 10.1002/jbmr.93
65. Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. (2019) 25:1280–9. doi: 10.1038/s41591-019-0512-5
66. Miano JM, Fisher EA, Majesky MW. Fate and State of Vascular smooth muscle cells in atherosclerosis. Circulation. (2021) 143:2110–6. doi: 10.1161/CIRCULATIONAHA.120.049922
67. Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. (2018) 52:48–64. doi: 10.1016/j.cellsig.2018.08.019
68. Findeisen HM, Kahles FK, Bruemmer D. Epigenetic regulation of vascular smooth muscle cell function in atherosclerosis. Curr Atheroscler Rep. (2013) 15:319. doi: 10.1007/s11883-013-0319-7
69. Al-Hasani K, Mathiyalagan P, El-Osta A. Epigenetics, cardiovascular disease, and cellular reprogramming. J Mol Cell Cardiol. (2019) 128:129–33. doi: 10.1016/j.yjmcc.2019.01.019
70. Ming X, Zhu B, Li Y. Mitotic inheritance of DNA methylation: more than just copy and paste. J Genet Genomics. (2021) 48:1–13. doi: 10.1016/j.jgg.2021.01.006
71. Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. (2011) 12:206–22. doi: 10.1002/cbic.201000195
72. Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. (2013) 128:2047–57. doi: 10.1161/circulationaha.113.002887
73. Zhuang J, Luan P, Li H, Wang K, Zhang P, Xu Y, et al. The Yin-Yang Dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler Thromb Vasc Biol. (2017) 37:84–97. doi: 10.1161/atvbaha.116.307923
74. Lawrence M, Daujat S, Schneider R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet. (2016) 32:42–56. doi: 10.1016/j.tig.2015.10.007
75. Greißel A, Culmes M, Napieralski R, Wagner E, Gebhard H, Schmitt M, et al. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb Haemost. (2015) 114:390–402. doi: 10.1160/th14-10-0852
76. Greißel A, Culmes M, Burgkart R, Zimmermann A, Eckstein HH, Zernecke A, et al. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol. (2016) 25:79–86. doi: 10.1016/j.carpath.2015.11.001
77. Panni S, Lovering RC, Porras P, Orchard S. Non-coding RNA regulatory networks. Biochim Biophys Acta Gene Regul Mech. (2020) 1863:194417. doi: 10.1016/j.bbagrm.2019.194417
78. Ross R. Atherosclerosis—An inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
79. Jones SA. Directing transition from innate to acquired immunity:defining a role for IL-6. J Immunol. (2005) 175:3463–8. doi: 10.4049/jimmunol.175.6.3463
80. Chistiakov DA, Melnichenko AA, Grechko AV, Myasoedova VA, Orekhov AN. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp Mol Pathol. (2018) 104:114–24. doi: 10.1016/j.yexmp.2018.01.008
81. Swirski FK, Nahrendorf M, Libby P. Mechanisms of myeloid cell modulation of atherosclerosis. Microbiol Spectr. (2016) 4:26. doi: 10.1128/microbiolspec.MCHD-0026-2015
82. Luttun A, Lutgens E, Manderveld A, Maris K, Collen D, Carmeliet P, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. (2004) 109:1408–14. doi: 10.1161/01.CIR.0000121728.14930.DE
83. Kuwahara S, Fukuoka M, Koan Y, Miyake H, Ono Y, Moriki A, et al. Subdural hyperintense band on diffusion-weighted imaging of chronic subdural hematoma indicates bleeding from the outer membrane. Neurol Med Chir. (2005) 45:125–31. doi: 10.2176/nmc.45.125
84. Kottoor SJ, Arora RR. The utility of anti-inflammatory agents in cardiovascular disease: a novel perspective on the treatment of atherosclerosis. J Cardiovasc Pharmacol Ther. (2018) 23:483–93. doi: 10.1177/1074248418778548
85. Hakobyan S, Harri CL, van den Berg CW, Fernandez-Alonso MC, de Jorge EG, de Cordoba SR, et al. Complement factor H binds to denatured rather than to native pentameric C-reactive protein. J Biol Chem. (2008) 283:30451–60. doi: 10.1074/jbc.M803648200
86. Aker S, Bantis C, Reis P, Kuhr N, Schwandt C, Grabensee B, et al. Influence of interleukin-6 G-174C gene polymorphism on coronary artery disease, cardiovascular complications and mortality in dialysis patients. Nephrol Dial Transplant. (2009) 24:2847–51. doi: 10.1093/ndt/gfp141
87. Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Investig. (2003) 111:1805–12. doi: 10.1172/JCI18921
88. Price DT, Loscalzo J. Cellular adhesion molecules and atherogenesis. Am J Med. (1999) 107:85–97. doi: 10.1016/s0002-9343(99)00153-9
89. Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. (2009) 29:1419–23. doi: 10.1161/ATVBAHA.108.180497
90. Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, et al. Atherosclerotic plaque progression and vulnerability to rupture: Angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. (2005) 25:2054–61. doi: 10.1161/01.ATV.0000178991.71605.18
91. Fotis L, Agrogiannis G, Vlachos IS, Pantopoulou A, Margoni A, Kostaki M, et al. Intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 at the early stages of atherosclerosis in a rat model. In Vivo. (2012) 26:243–50.
92. O’Shea JJ, Pesu M, Borie DC, Changelian PS. A new modality for immunosuppression: Targeting the JAK/STAT pathway. Nat Rev Drug Discov. (2004) 3:555–64. doi: 10.1038/nrd1441
93. Hollestelle SC, de Vries MR, van Keulen JK, Schoneveld AH, Vink A, Strijder CF, et al. Toll-like receptor 4 is involved in outward arterial remodeling. Circulation. (2004) 109:393–8. doi: 10.1161/01.CIR.0000109140.51366.72
94. Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. (1996) 380:630–3. doi: 10.1038/380630a0
95. Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. Circulation. (1999) 100:230–5. doi: 10.1161/01.cir.100.3.230
96. Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, et al. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules. (2018) 8:80. doi: 10.3390/biom8030080
97. Micha R, Peñalvo JL, Cudhea F, Imamura F, Rehm CD, Mozaffarian D. Association between dietary factors and mortality from heart disease, stroke, and type 2 diabetes in the United States. J Am Med Assoc. (2017) 317:912–24. doi: 10.1001/jama.2017.0947
98. Shukla S, Gupta S. Apigenin: a promising molecule for cancer prevention. Pharmaceut Res. (2010) 27:962–78. doi: 10.1007/s11095-010-0089-7
99. Xu Q, Li YC, Du C, Wang LN, Xiao YH. Effects of Apigenin on the expression of LOX-1. Bcl-2, and Bax in hyperlipidemia rats. Chem Biodivers. (2021) 18:e2100049. doi: 10.1002/cbdv.202100049
100. Jeong YJ, Choi YJ, Choi JS, Kwon HM, Kang SW, Bae JY, et al. Attenuation of monocyte adhesion and oxidised LDL uptake in luteolin-treated human endothelial cells exposed to oxidised LDL. Br J Nutr. (2007) 97:447–57. doi: 10.1017/S0007114507657894
101. Qin W, Ren B, Wang S, Liang S, He B, Shi X, et al. Apigenin and naringenin ameliorate PKCβII-associated endothelial dysfunction via regulating ROS/caspase-3 and NO pathway in endothelial cells exposed to high glucose. Vascul Pharmacol. (2016) 85:39–49. doi: 10.1016/j.vph.2016.07.006
102. Yamagata K, Hashiguchi K, Yamamoto H, Tagami M. Dietary apigenin reduces induction of LOX-1 and NLRP3 expression, leukocyte adhesion, and acetylated low-density lipoprotein uptake in human endothelial cells exposed to trimethylamine-N-oxide. J Cardiovasc Pharmacol. (2019) 74:558–65. doi: 10.1097/FJC.0000000000000747
103. Zhang K, Song W, Li D, Jin X. Apigenin in the regulation of cholesterol metabolism and protection of blood vessels. Exp Ther Med. (2017) 13:1719–24. doi: 10.3892/etm.2017.4165
104. Ren K, Jiang T, Zhou HF, Liang Y, Zhao GJ. Apigenin Retards Atherogenesis by Promoting ABCA1-Mediated Cholesterol Efflux and Suppressing Inflammation. Cell Physiol Biochem. (2018) 47:2170–84. doi: 10.1159/000491528
105. Zeng P, Liu B, Wang Q, Fan Q, Diao JX, Tang J, et al. Apigenin attenuates atherogenesis through inducing macrophage apoptosis via inhibition of AKT Ser473 phosphorylation and downregulation of plasminogen activator inhibitor-2. Oxid Med Cell Longev. (2015) 2015:379538. doi: 10.1155/2015/379538
106. Wang Q, Zeng P, Liu Y, Wen G, Fu X, Sun X. Inhibition of autophagy ameliorates atherogenic inflammation by augmenting apigenin-induced macrophage apoptosis. Int Immunopharmacol. (2015) 27:24–31. doi: 10.1016/j.intimp.2015.04.018
107. Zhang X, Wang G, Gurley EC, Zhou H. Flavonoid apigenin inhibits lipopolysaccharide-induced inflammatory response through multiple mechanisms in macrophages. PLoS One. (2014) 9:e107072. doi: 10.1371/journal.pone.0107072
108. Ding X, Zheng L, Yang B, Wang X, Ying Y. Luteolin attenuates atherosclerosis via modulating signal transducer and activator of transcription 3-mediated inflammatory response. Drug Des Devel Ther. (2019) 13:3899–911. doi: 10.2147/DDDT.S207185
109. Wu YT, Chen L, Tan ZB, Fan HJ, Xie LP, Zhang WT, et al. Luteolin inhibits vascular smooth muscle cell proliferation and migration by inhibiting TGFBR1 signaling. Front Pharmacol. (2018) 9:1059. doi: 10.3389/fphar.2018.01059
110. Li J, Dong JZ, Ren YL, Zhu JJ, Cao JN, Zhang J, et al. Luteolin decreases atherosclerosis in LDL receptor-deficient mice via a mechanism including decreasing AMPK-SIRT1 signaling in macrophages. Exp Ther Med. (2018) 16:2593–9. doi: 10.3892/etm.2018.6499
111. Kim MS, Kim DS, Kim HS, Kang SW, Kang YH. Inhibitory effects of luteolin on transendothelial migration of monocytes and formation of lipid-laden macrophages. Nutrition. (2012) 28:1044–54. doi: 10.1016/j.nut.2011.12.003
112. Lang Y, Chen D, Li D, Zhu M, Xu T, Zhang T, et al. Luteolin inhibited hydrogen peroxide-induced vascular smooth muscle cells proliferation and migration by suppressing the Src and Akt signalling pathways. J Pharm Pharmacol. (2012) 64:597–603. doi: 10.1111/j.2042-7158.2011.01438.x
113. Zhu M, Chen D, Li D, Ding H, Zhang T, Xu T, et al. Luteolin inhibits angiotensin II-induced human umbilical vein endothelial cell proliferation and migration through downregulation of Src and Akt phosphorylation. Circ J. (2013) 77:772–9. doi: 10.1253/circj.cj-12-0310
114. Xia F, Wang C, Jin Y, Liu Q, Meng Q, Liu K, et al. Luteolin protects HUVECs from TNF-α-induced oxidative stress and inflammation via its effects on the Nox4/ROS-NF-κB and MAPK pathways. J Atheroscler Thromb. (2014) 8:768–83. doi: 10.5551/jat.23697
115. Wu CH, Wu CF, Huang HW, Jao YC, Yen GC. Naturally occurring flavonoids attenuate high glucose-induced expression of proinflammatory cytokines in human monocytic THP-1 cells. Mol Nutr Food Res. (2009) 53:984–95. doi: 10.1002/mnfr.200800495
116. Jiang Q, Pan D, Yang Y, Hu Y, Fang L, Shang P, et al. Luteolin regulates macrophage polarization via the PI3K/Akt pathway to inhibit the apoptosis stimulated by angiotensin II. Curr Pharm Biotechnol. (2018) 19:428–37. doi: 10.2174/1389201019666180629143251
117. Jia Z, Nallasamy P, Liu D, Shah H, Li JZ, Chitrakar R, et al. Luteolin protects against vascular inflammation in mice and TNF-alpha-induced monocyte adhesion to endothelial cells via suppressing IK Bα/NF-κB signaling pathway. J Nutr Biochem. (2015) 26:293–302. doi: 10.1016/j.jnutbio.2014.11.008
118. Pangestika I, Oksal E, Tengku Muhammad TS, Amir H, Syamsumir DF, Wahid MEA, et al. Inhibitory effects of tangeretin and trans-ethyl caffeate on the HMG-CoA reductase activity: Potential agents for reducing cholesterol levels. Saudi J Biol Sci. (2020) 27:1947–60. doi: 10.1016/j.sjbs.2020.06.010
119. Seo J, Lee HS, Ryoo S, Seo JH, Min BS, Lee JH. Tangeretin, a citrus flavonoid, inhibits PGDF-BB-induced proliferation and migration of aortic smooth muscle cells by blocking AKT activation. Eur J Pharmacol. (2011) 673:56–64. doi: 10.1016/j.ejphar.2011.10.011
120. Cha BY, Shi WL, Yonezawa T, Teruya T, Nagai K, Woo JT. An inhibitory effect of chrysoeriol on platelet-derived growth factor (PDGF)-induced proliferation and PDGF receptor signaling in human aortic smooth muscle cells. J Pharmacol Sci. (2009) 110:105–10. doi: 10.1254/jphs.08282fp
121. Whitman SC, Kurowska EM, Manthey JA, Daugherty A. Nobiletin, a citrus flavonoid isolated from tangerines, selectively inhibits class A scavenger receptor-mediated metabolism of acetylated LDL by mouse macrophages. Atherosclerosis. (2005) 178:25–32. doi: 10.1016/j.atherosclerosis.2004.07.034
122. Eguchi A, Murakami A, Ohigashi H. Nobiletin, a citrus flavonoid, suppresses phorbol ester-induced expression of multiple scavenger receptor genes in THP-1 human monocytic cells. FEBS Lett. (2006) 580:3321–8. doi: 10.1016/j.febslet.2006.04.077
123. Mulvihill EE, Assini JM, Lee JK, Allister EM, Sutherland BG, Koppes JB, et al. Nobiletin attenuates VLDL overproduction, dyslipidemia, and atherosclerosis in mice with diet-induced insulin resistance. Diabetes. (2011) 60:1446–57. doi: 10.2337/db10-0589
124. He PP, Shen QQ, Wen M, Zou JQ, Wang Y, Yang JX, et al. Nobiletin reduces LPL-mediated lipid accumulation and pro-inflammatory cytokine secretion through upregulation of miR-590 expression. Biochem Biophys Res Commun. (2019) 508:97–101. doi: 10.1016/j.bbrc.2018.11.075
125. Wang S, Zhang X, Liu M, Luan H, Ji Y, Guo P, et al. Chrysin inhibits foam cell formation through promoting cholesterol efflux from RAW264.7 macrophages. Pharm Biol. (2015) 53:1481–7. doi: 10.3109/13880209.2014.986688
126. Lin CM, Wang BW, Pan CM, Fang WJ, Chua SK, Cheng WP, et al. Chrysin boosts KLF2 expression through suppression of endothelial cell-derived exosomal microRNA-92a in the model of atheroprotection. Eur J Nutr. (2021) 60:4345–55. doi: 10.1007/s00394-021-02593-1
127. Anandhi R, Thomas PA, Geraldine P. Evaluation of the anti-atherogenic potential of chrysin in Wistar rats. Mol Cell Biochem. (2014) 385:103–13. doi: 10.1007/s11010-013-1819-z
128. Ramírez-Espinosa JJ, Saldaña-Ríos J, García-Jiménez S, Villalobos-Molina R, Ávila-Villarreal G, Rodríguez-Ocampo AN, et al. Chrysin induces antidiabetic, antidyslipidemic and anti-inflammatory effects in athymic nude diabetic mice. Molecules. (2017) 23:67. doi: 10.3390/molecules23010067
129. Han WM, Chen XC, Li GR, Wang Y. Acacetin protects against high glucose-induced endothelial cells injury by preserving mitochondrial function via activating Sirt1/Sirt3/AMPK Signals. Front Pharmacol. (2020) 11:607796. doi: 10.3389/fphar.2020.607796
130. Wu Y, Song F, Li Y, Li J, Cui Y, Hong Y, et al. Acacetin exerts antioxidant potential against atherosclerosis through Nrf2 pathway in apoE-/- Mice. J Cell Mol Med. (2021) 25:521–34. doi: 10.1111/jcmm.16106
131. Tsuboi T, Lu R, Yonezawa T, Watanabe A, Woo JT, Abe-Dohmae S, et al. Molecular mechanism for nobiletin to enhance ABCA1/G1 expression in mouse macrophages. Atherosclerosis. (2020) 297:32–9. doi: 10.1016/j.atherosclerosis.2020.01.024
132. Nekohashi M, Ogawa M, Ogihara T, Nakazawa K, Kato H, Misaka T, et al. Luteolin and quercetin affect the cholesterol absorption mediated by epithelial cholesterol transporter niemann-pick c1-like 1 in caco-2 cells and rats. PLoS One. (2014) 9:e97901. doi: 10.1371/journal.pone.0097901
133. Jia Q, Cao H, Shen D, Li S, Yan L, Chen C, et al. Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARγ, LXRα and ABCA1. Int J Mol Med. (2019) 44:893–902. doi: 10.3892/ijmm.2019.4263
134. Cao H, Jia Q, Yan L, Chen C, Xing S, Shen D. Quercetin suppresses the progression of atherosclerosis by regulating MST1-mediated autophagy in ox-LDL-induced RAW264.7 macrophage foam cells. Int J Mol Sci. (2019) 20:6093. doi: 10.3390/ijms20236093
135. Jiang YH, Jiang LY, Wang YC, Ma DF, Li X. Quercetin attenuates atherosclerosis via modulating oxidized LDL-induced endothelial cellular senescence. Front Pharmacol. (2020) 11:512. doi: 10.3389/fphar.2020.00512
136. Filipe P, Lança V, Silva JN, Morlière P, Santus R, Fernandes A. Flavonoids and urate antioxidant interplay in plasma oxidative stress. Mol Cell Biochem. (2001) 221:79–87. doi: 10.1023/a:1010944919952
137. Kong L, Luo C, Li X, Zhou Y, He H. The anti-inflammatory effect of kaempferol on early atherosclerosis in high cholesterol fed rabbits. Lipids Health Dis. (2013) 12:115. doi: 10.1186/1476-511X-12-115
138. Che J, Liang B, Zhang Y, Wang Y, Tang J, Shi G. Kaempferol alleviates ox-LDL-induced apoptosis by up-regulation of autophagy via inhibiting PI3K/Akt/mTOR pathway in human endothelial cells. Cardiovasc Pathol. (2017) 31:57–62. doi: 10.1016/j.carpath.2017.08.001
139. Feng Z, Wang C, Yue J, Meng Q, Wu J, Sun H. Kaempferol-induced GPER upregulation attenuates atherosclerosis via the PI3K/AKT/Nrf2 pathway. Pharm Biol. (2021) 59:1106–16. doi: 10.1080/13880209.2021.1961823
140. Meng Z, Wang M, Xing J, Liu Y, Li H. Myricetin ameliorates atherosclerosis in the low-density-lipoprotein receptor knockout mice by suppression of cholesterol accumulation in macrophage foam cells. Nutr Metab (Lond). (2019) 16:25. doi: 10.1186/s12986-019-0354-7
141. Chen G, Xu H, Wu Y, Han X, Xie L, Zhang G, et al. Myricetin suppresses the proliferation and migration of vascular smooth muscle cells and inhibits neointimal hyperplasia via suppressing TGFBR1 signaling pathways. Phytomedicine. (2021) 92:153719. doi: 10.1016/j.phymed.2021.153719
142. Bai Y, Liu X, Chen Q, Chen T, Jiang N, Guo Z. Myricetin ameliorates ox-LDL-induced HUVECs apoptosis and inflammation via lncRNA GAS5 upregulating the expression of miR-29a-3p. Sci Rep. (2021) 11:19637. doi: 10.1038/s41598-021-98916-7
143. Wang C, Nan X, Pei S, Zhao Y, Wang X, Ma S, et al. Salidroside and isorhamnetin attenuate urotensin II-induced inflammatory response in vivo and in vitro: Involvement in regulating the RhoA/ROCK II pathway. Oncol Lett. (2021) 21:292. doi: 10.3892/ol.2021.12553
144. Luo Y, Sun G, Dong X, Wang M, Qin M, Yu Y, et al. Isorhamnetin attenuates atherosclerosis by inhibiting macrophage apoptosis via PI3K/AKT activation and HO-1 induction. PLoS One. (2015) 10:e0120259. doi: 10.1371/journal.pone.0120259
145. Lotito SB, Frei B. Dietary flavonoids attenuate tumor necrosis factor alpha-induced adhesion molecule expression in human aortic endothelial cells. Structure-function relationships and activity after first pass metabolism. J Biol Chem. (2006) 281:37102–10. doi: 10.1074/jbc.M606804200
146. Zhang X, Han X, Zhang P, Zhou T, Chen Y, Jin J, et al. Morin attenuates oxidized low-density lipoprotein-mediated injury by inducing autophagy via activating AMPK signalling in HUVECs. Clin Exp Pharmacol Physiol. (2019) 46:1053–60. doi: 10.1111/1440-1681.13160
147. Shin SS, Ko MC, Noh DH, Hwang B, Park Y, Park SL, et al. Morin inhibits PDGF-induced proliferation, migration, and invasion of vascular smooth muscle cells via modulating p27KIP1, AKT, and MMP-9 activities. Gen Physiol Biophys. (2018) 37:633–45. doi: 10.4149/gpb_2018028
148. Yan L, Jia Q, Cao H, Chen C, Xing S, Huang Y, et al. Fisetin ameliorates atherosclerosis by regulating PCSK9 and LOX-1 in apoE-/- mice. Exp Ther Med. (2021) 21:25. doi: 10.3892/etm.2020.9457
149. Hada Y, Uchida HA, Wada J. Fisetin Attenuates Lipopolysaccharide-Induced Inflammatory Responses in Macrophage. Biomed Res Int. (2021) 2021:5570885. doi: 10.1155/2021/5570885
150. Ugusman A, Zakaria Z, Chua KH, Nordin NA, Abdullah Mahdy Z. Role of rutin on nitric oxide synthesis in human umbilical vein endothelial cells. Scientific World Journal. (2014) 2014:169370. doi: 10.1155/2014/169370
151. Yu SH, Yu JM, Yoo HJ, Lee SJ, Kang DH, Cho YJ, et al. Anti-proliferative effects of rutin on OLETF rat vascular smooth muscle cells stimulated by glucose variability. Yonsei Med J. (2016) 57:373–81. doi: 10.3349/ymj.2016.57.2.373
152. Li Y, Qin R, Yan H, Wang F, Huang S, Zhang Y, et al. Inhibition of vascular smooth muscle cells premature senescence with rutin attenuates and stabilizes diabetic atherosclerosis. J Nutr Biochem. (2018) 51:91–8. doi: 10.1016/j.jnutbio.2017.09.012
153. Chanet A, Milenkovic D, Deval C, Potier M, Constans J, Mazur A, et al. Naringin, the major grapefruit flavonoid, specifically affects atherosclerosis development in diet-induced hypercholesterolemia in mice. J Nutr Biochem. (2012) 23:469–77. doi: 10.1016/j.jnutbio.2011.02.001
154. Hsueh TP, Sheen JM, Pang JH, Bi KW, Huang CC, Wu HT, et al. The anti-atherosclerotic effect of naringin is associated with reduced expressions of cell adhesion molecules and chemokines through NF-κB pathway. Molecules. (2016) 21:195. doi: 10.3390/molecules21020195
155. Zhao H, Liu M, Liu H, Suo R, Lu C. Naringin protects endothelial cells from apoptosis and inflammation by regulating the Hippo-YAP Pathway. Biosci Rep. (2020) 40:BSR20193431. doi: 10.1042/BSR20193431
156. Wang F, Zhao C, Tian G, Wei X, Ma Z, Cui J, et al. Naringin alleviates atherosclerosis in ApoE mice by regulating cholesterol metabolism involved in gut microbiota remodeling. J Agric Food Chem. (2020) 68:12651–60. doi: 10.1021/acs.jafc.0c05800
157. Pardo-Andreu GL, Paim BA, Castilho RF, Velho JA, Delgado R, Vercesi AE, et al. Mangifera indica L. extract (Vimang) and its main polyphenol mangiferin prevent mitochondrial oxidative stress in atherosclerosis-prone hypercholesterolemic mouse. Pharmacol Res. (2008) 57:332–8. doi: 10.1016/j.phrs.2008.03.005
158. Leiro JM, Alvarez E, Arranz JA, Siso IG, Orallo F. In vitro effects of mangiferin on superoxide concentrations and expression of the inducible nitric oxide synthase, tumour necrosis factor-alpha and transforming growth factor-beta genes. Biochem Pharmacol. (2003) 65:1361–71. doi: 10.1016/s0006-2952(03)00041-8
159. Ren K, Li H, Zhou HF, Liang Y, Tong M, Chen L, et al. Mangiferin promotes macrophage cholesterol efflux and protects against atherosclerosis by augmenting the expression of ABCA1 and ABCG1. Aging (Albany N Y). (2019) 11:10992–1009. doi: 10.18632/aging.102498
160. Kim TJ, Kim JH, Jin YR, Yun YP. The inhibitory effect and mechanism of luteolin 7-glucoside on rat aortic vascular smooth muscle cell proliferation. Arch Pharm Res. (2006) 29:67–72. doi: 10.1007/BF02977471
161. Hsuan CF, Hsu HF, Tseng WK, Lee TL, Wei YF, Hsu KL, et al. Glossogyne tenuifolia extract inhibits TNF-α-induced expression of adhesion molecules in human umbilical vein endothelial cells via blocking the NF-kB signaling pathway. Molecules. (2015) 20:16908–23. doi: 10.3390/molecules200916908
162. Sun YZ, Chen JF, Shen LM, Zhou J, Wang CF. Anti-atherosclerotic effect of hesperidin in LDLr-/- mice and its possible mechanism. Eur J Pharmacol. (2017) 815:109–17. doi: 10.1016/j.ejphar.2017.09.010
163. Koga M, Kanaoka Y, Inada K, Omine S, Kataoka Y, Yamauchi A. Hesperidin blocks varenicline-aggravated atherosclerotic plaque formation in apolipoprotein E knockout mice by downregulating net uptake of oxidized low-density lipoprotein in macrophages. J Pharmacol Sci. (2020) 143:106–11. doi: 10.1016/j.jphs.2020.01.012
164. Wang Z, Zhang M, Wang Z, Guo Z, Wang Z, Chen Q. Cyanidin-3-O-glucoside attenuates endothelial cell dysfunction by modulating miR-204-5p/SIRT1-mediated inflammation and apoptosis. Biofactors. (2020) 46:803–12. doi: 10.1002/biof.1660
165. Wang D, Xia M, Gao S, Li D, Zhang Y, Jin T, et al. Cyanidin-3-O-β-glucoside upregulates hepatic cholesterol 7α-hydroxylase expression and reduces hypercholesterolemia in mice. Mol Nutr Food Res. (2012) 56:610–21. doi: 10.1002/mnfr.201100659
166. Yan X, Wu L, Li B, Meng X, Dai H, Zheng Y, et al. Cyanidin-3-O-glucoside induces apoptosis and inhibits Migration of tumor necrosis factor-α-treated rat aortic smooth muscle cells. Cardiovasc Toxicol. (2016) 16:251–9. doi: 10.1007/s12012-015-9333-z
167. Xia M, Ling W, Zhu H, Ma J, Wang Q, Hou M, et al. Anthocyanin attenuates CD40-mediated endothelial cell activation and apoptosis by inhibiting CD40-induced MAPK activation. Atherosclerosis. (2009) 202:41–7. doi: 10.1016/j.atherosclerosis.2008.04.005
168. Jin X, Yi L, Chen ML, Chen CY, Chang H, Zhang T, et al. Delphinidin-3-glucoside protects against oxidized low-density lipoprotein-induced mitochondrial dysfunction in vascular endothelial cells via the sodium-dependent glucose transporter SGLT1. PLoS One. (2013) 8:e68617. doi: 10.1371/journal.pone.0068617
169. Ku SK, Kwak S, Bae JS. Orientin inhibits high glucose-induced vascular inflammation in vitro and in vivo. Inflammation. (2014) 37:2164–73. doi: 10.1007/s10753-014-9950-x
170. Li C, Cai C, Zheng X, Sun J, Ye L. Orientin suppresses oxidized low-density lipoproteins induced inflammation and oxidative stress of macrophages in atherosclerosis. Biosci Biotechnol Biochem. (2020) 84:774–9. doi: 10.1080/09168451.2019.1702871
171. Zhao CR, Yang FF, Cui Q, Wang D, Zhou Y, Li YS, et al. Vitexin inhibits APEX1 to counteract the flow-induced endothelial inflammation. Proc Natl Acad Sci USA. (2021) 118:e2115158118. doi: 10.1073/pnas.2115158118
172. Marino M, Del Bo’ C, Tucci M, Klimis-Zacas D, Riso P, Porrini M. Modulation of adhesion process, E-Selectin and VEGF production by anthocyanins and their metabolites in an in vitro model of atherosclerosis. Nutrients. (2020) 12:655. doi: 10.3390/nu12030655
173. Liu TT, Zeng Y, Tang K, Chen X, Zhang W, Xu XL. Dihydromyricetin ameliorates atherosclerosis in LDL receptor deficient mice. Atherosclerosis. (2017) 262:39–50. doi: 10.1016/j.atherosclerosis.2017.05.003
174. Hu Q, Zhang T, Yi L, Zhou X, Mi M. Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis by activating the Nrf2 signaling pathway in vascular endothelial cells. Biofactors. (2018) 44:123–36. doi: 10.1002/biof.1395
175. Yang D, Yang Z, Chen L, Kuang D, Zou Y, Li J, et al. Dihydromyricetin increases endothelial nitric oxide production and inhibits atherosclerosis through microRNA-21 in apolipoprotein E-deficient mice. J Cell Mol Med. (2020) 24:5911–25. doi: 10.1111/jcmm.15278
176. Morrison M, van der Heijden R, Heeringa P, Kaijzel E, Verschuren L, Blomhoff R, et al. Epicatechin attenuates atherosclerosis and exerts anti-inflammatory effects on diet-induced human-CRP and NFκB in vivo. Atherosclerosis. (2014) 23:149–56. doi: 10.1016/j.atherosclerosis.2013.12.027
177. Son JE, Jeong H, Kim H, Kim YA, Lee E, Lee HJ, et al. Pelargonidin attenuates PDGF-BB-induced aortic smooth muscle cell proliferation and migration by direct inhibition of focal adhesion kinase. Biochem Pharmacol. (2014) 89:236–45. doi: 10.1016/j.bcp.2014.02.015
178. Chen CY, Yi L, Jin X, Mi MT, Zhang T, Ling WH, et al. Delphinidin attenuates stress injury induced by oxidized low-density lipoprotein in human umbilical vein endothelial cells. Chem Biol Interact. (2010) 183:105–12. doi: 10.1016/j.cbi.2009.09.024
179. Martin S, Favot L, Matz R, Lugnier C, Andriantsitohaina R. Delphinidin inhibits endothelial cell proliferation and cell cycle progression through a transient activation of ERK-1/-2. Biochem Pharmacol. (2003) 65:669–75. doi: 10.1016/s0006-2952(02)01568-x
180. Son JE, Lee E, Jung SK, Kim JE, Oak MH, Lee KW, et al. Anthocyanidins, novel FAK inhibitors, attenuate PDGF-BB-induced aortic smooth muscle cell migration and neointima formation. Cardiovasc Res. (2014) 101:503–12. doi: 10.1093/cvr/cvt337
181. Xia Y, Feng H, Li ZW, Tang KX, Gao HQ, Wang WL, et al. Low-dose phloretin alleviates diabetic atherosclerosis through endothelial KLF2 restoration. Biosci Biotechnol Biochem. (2020) 84:815–23. doi: 10.1080/09168451.2019.1699396
182. Kim MS, Park SH, Han SY, Kim YH, Lee EJ, Yoon Park JH, et al. Phloretin suppresses thrombin-mediated leukocyte-platelet-endothelial interactions. Mol Nutr Food Res. (2014) 58:698–708. doi: 10.1002/mnfr.201300267
183. Wang D, Wang Q, Yan G, Qiao Y, Tang C. Phloretin inhibits platelet-derived growth factor-BB-induced rat aortic smooth muscle cell proliferation, migration, and neointimal formation after carotid injury. J Cardiovasc Pharmacol. (2015) 65:444–55. doi: 10.1097/FJC.0000000000000213
184. Doddapattar P, Radović B, Patankar JV, Obrowsky S, Jandl K, Nusshold C, et al. Xanthohumol ameliorates atherosclerotic plaque formation, hypercholesterolemia, and hepatic steatosis in ApoE-deficient mice. Mol Nutr Food Res. (2013) 57:1718–28. doi: 10.1002/mnfr.201200794
185. Srinivas K, King JW, Howard LR, Monrad JK. Solubility and solution thermodynamic properties of quercetin and quercetin dihydrate in subcritical water. J Food Eng. (2010) 100:20818. doi: 10.1016/j.jfoodeng.2010.04.001
186. Zhang L, Dong M, Guangyong X, Yuan T, Tang H, Wang Y. Metabolomics reveals that dietary ferulic acid and quercetin modulate metabolic homeostasis in rats. J Agric Food Chem. (2018) 66:1723–31. doi: 10.1021/acs.jafc.8b00054
187. Dabeek WM, Marra MV. Dietary quercetin and kaempferol: bioavailability and potential cardiovascular-related bioactivity in humans. Nutrients. (2019) 11:2288. doi: 10.3390/nu11102288
188. Wu LD, Enjiang Y, Xiaona C, Xinguo W, Zhengzan Z, Lingzhen Q, et al. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res Lett. (2014) 9:1–11. doi: 10.1186/1556-276X-9-684
189. Zhang BC, Li Z, Xu W, Xiang CH, Ma YF. Luteolin alleviates NLRP3 inflammasome activation and directs macrophage polarization in lipopolysaccharide-stimulated RAW264.7 cells. Am J Transl Res. (2018) 10:265–73.
Keywords: natural flavonoids, fruits, atherosclerosis, cardiovascular diseases, potential mechanism
Citation: Li R-L, Wang L-Y, Liu S, Duan H-X, Zhang Q, Zhang T, Peng W, Huang Y and Wu C (2022) Natural Flavonoids Derived From Fruits Are Potential Agents Against Atherosclerosis. Front. Nutr. 9:862277. doi: 10.3389/fnut.2022.862277
Received: 25 January 2022; Accepted: 17 February 2022;
Published: 24 March 2022.
Edited by:
Shi-Jun Yue, Shaanxi University of Chinese Medicine, ChinaReviewed by:
Kang Xu, Hubei University of Chinese Medicine, ChinaCopyright © 2022 Li, Wang, Liu, Duan, Zhang, Zhang, Peng, Huang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Peng, cGVuZ3dlaUBjZHV0Y20uZWR1LmNu; Yongliang Huang, aHlsQGNkdXRjbS5lZHUuY24=; Chunjie Wu, d3VjamNkdGNtQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.