 Mengyao Zhao
Mengyao Zhao Boya Zhang
Boya Zhang Linlin Deng1
Linlin Deng1
- 1State Key Laboratory of Bioreactor Engineering, School of Biotechnology, East China University of Science and Technology, Shanghai, China
- 2Shanghai Collaborative Innovation Center for Biomanufacturing Technology (SCICBT), Shanghai, China
Acrylamide (ACR), a potential neurotoxin, is produced by the Maillard reaction between reducing sugars and free amino acids during food processing. Over the past decade, the neurotoxicity of ACR has caused increasing concern, prompting many related studies. This review summarized the relevant literature published in recent years and discussed the exposure to occupational, environmental, and daily ACR contamination in food. Moreover, ACR metabolism and the potential mechanism of ACR-induced neurotoxicity were discussed, with particular focus on the axonal degeneration of the nervous system, nerve cell apoptosis, oxidative stress, inflammatory response, and gut-brain axis homeostasis. Additionally, the limitations of existing knowledge, as well as new perspectives, were examined, specifically regarding the connection between the neurotoxicity caused by ACR and neurodegenerative diseases, NOD-like receptor protein 3 (NLRP3) inflammasome-related neuroinflammation, and microbiota-gut-brain axis signaling. This review might provide systematic information for developing an alternative pathway approach to assess ACR risk.
Introduction
For decades, acrylamide (ACR) has been widely used in the paper industry, as well as for wastewater treatment and soil conditioning. It is also used in medicine and the textile industry, mainly to synthesize high molecular polymers, such as poly-ACR (1). In 1994, ACR was listed as a class 2A substance by the International Agency for Research on Cancer (IARC) (“most likely carcinogenic to humans”) (2). In 2002, Swedish scientists first discovered the presence of ACR in heat-processed foods rich in asparagine, raising widespread concern (3, 4). In the same year, two articles on the formation mechanism of ACR were published in Nature, confirming that the Maillard reaction was primarily responsible for ACR formation in heat-processed foods, causing concern regarding ACR exposure via normal dietary intake (5, 6). In 2015, the European Food Safety Authority (EFSA) issued a survey report on the ACR content in 43,419 food products. The results showed that the average ACR content in fried potato products was as high as 1.0 mg/kg, while the highest ACR content in coffee was 4.5 mg/kg (7). Moreover, the European Commission set the residue limits of ACR in potato crisps at 750 μg/kg and roast coffee and coffee substances at 400–850 μg/kg (8). Therefore, ACR toxicity and its risk to human health require urgent attention due to its abundance in food products and the environment (9).
Acrylamide is responsible for developmental genotoxicity, neurotoxicity, and potentially carcinogenicity, of which neurotoxicity has been confirmed via human and animal experiments (10). Therefore, neurotoxicity is closely connected with human health. For decades, results showed similar phenotypical neurotoxicity in various laboratory animals, including dogs, cats, guinea pigs, rabbits, and rodents when repeatedly exposed to ACR levels ranging between 0.5 and 50 mg/kg/day (11–13). These neurological disorders may be caused by covalent adduct formation between highly nucleophilic cysteine and ACR at the active location of the presynaptic neuron. This process deactivates neurons and impacts neurotransmitter transfer, leading to neurotoxicity (14). Moreover, oxidative stress serves as a biochemical and physiological activation signal and is, directly and indirectly, related to the neurotoxicity caused by ACR (15, 16). Although the potential molecular mechanism reported in recent years as underlying ACR-related neurotoxicity is multifaceted, a complete characterization and summary of the comprehensive mechanical system and its impact are still required.
Therefore, this review summarizes the relevant literature published in recent years, discusses the primary potential mechanisms, and provides insight into the axonal degeneration of the nervous system, nerve cell apoptosis, oxidative stress, inflammatory response, and gut-brain axis homeostasis. Furthermore, the existing challenges and research aspects showing potential for examining ACR-induced neurotoxicity are discussed. This review may provide a more complete theoretical foundation to uncover the ACR toxicity mechanism while improving its subsequent adverse impact. This paper offers a fresh perspective on neurotoxicity while furnishing guidance regarding health safety development.
Exposure to Acrylamide
Occupational and Environmental Exposure
As a raw material used during industrial production, ACR often enters the water, soil, atmosphere, and other environmental media, damaging human health and severely affecting the nervous system (17). Occupational exposure is one of the main ACR exposure pathways. As early as the 1950s, it was reported that several workers exposed to ACR showed symptoms of poisoning, such as weakness, numbness, leg weakness, and unsteady gait. In recent years, occasional studies have examined ACR neurotoxicity, primarily acute occupational ACR poisoning during construction, coal mining, flocculator manufacturing, and tunnel construction (18). In addition to occupational exposure, ACR exposure can occur via drinking water, especially when public drinking water sources are treated using polyacrylamide as a flocculant (19), leading to high ACR concentrations in tap water (20, 21). Moreover, residual ACR is present in cosmetics, packaging materials, and cigarettes, while exposure can also occur via direct skin contact and alimentary contact (22–24). Table 1 summarizes the environmental and occupational characteristics of ACR exposure, including routes, estimated doses, typical cases, and threshold values. In summary, extensive research has been conducted involving the potentially harmful effect of ACR in occupational settings and daily life. However, additional studies are necessary to fully understand the chronic toxic effect of ACR and improve risk assessment to protect public health.
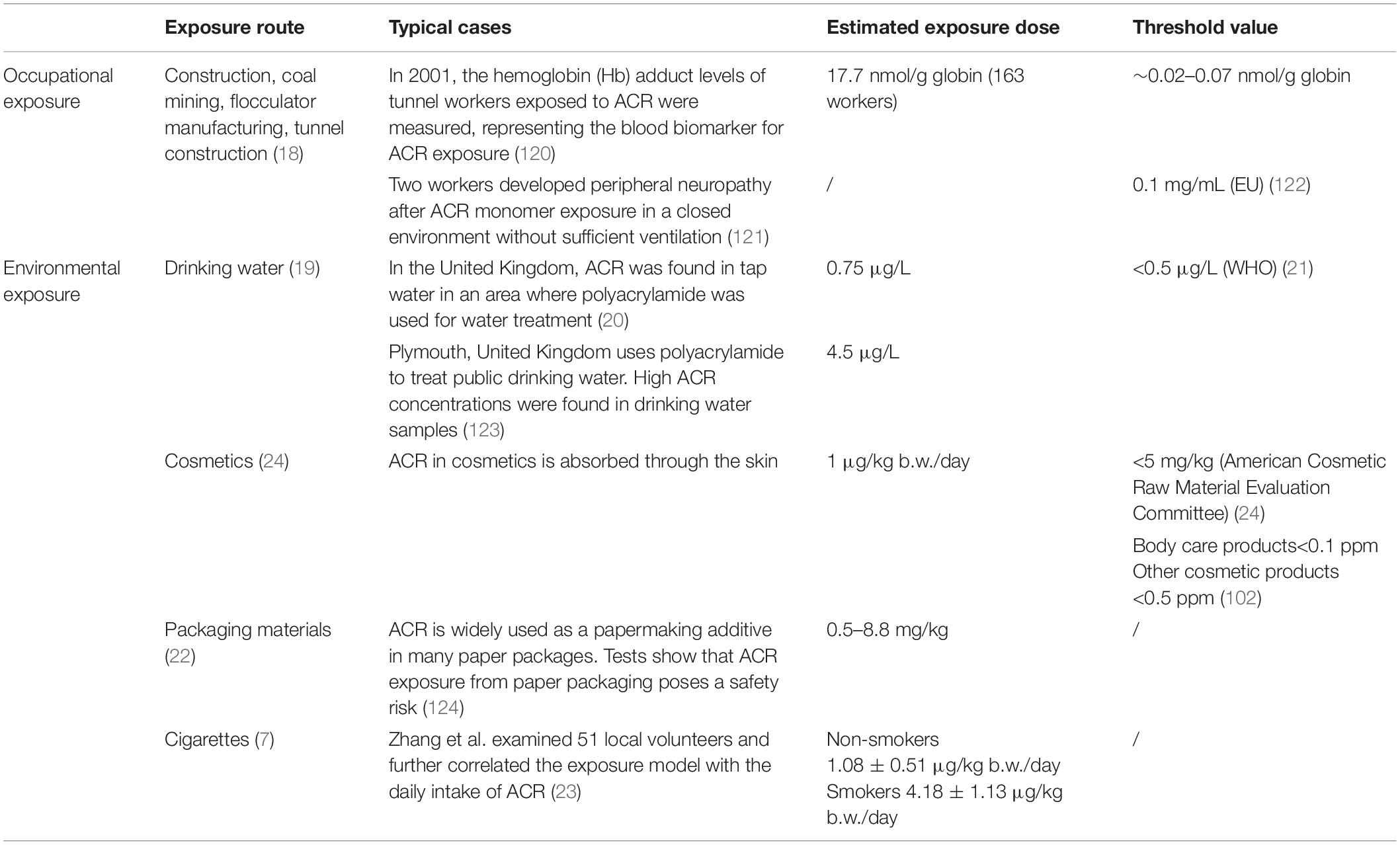
Table 1. A summary of the occupational and environmental exposure to ACR.
Foodborne Exposure
Consumers can be directly exposed to ACR via the oral intake of high-carbohydrate foods, such as potato chips, baked cereals, and bread (18). As early as 2010, the JECFA presented data regarding the ACR detected in 12,582 food samples from 31 countries, which included fried potatoes, bread, biscuits, coffee, and other food products. The results indicated the presence of ACR in almost all the analyzed food samples, with the highest levels in fried potato products and coffee (21). In 2011, the JECFA evaluated the dietary intake of ACR in eight representative countries. The findings showed that the average daily intake of the general population was about 1 μg/kg b.w., with the highest consumption at about 4 μg/kg b.w. (25). Moreover, young people tend to consume food products high in carbohydrates more frequently. The average exposure of adolescents aged 10–18 is 0.4–0.9 μg/kg b.w./day, and the high-level exposure is 0.9–2.0 μg/kg b.w./day. For children aged 3–10 the average exposure is 0.9–1.6 μg/kg b.w./day, while the high-level exposure can reach 1.4–3.2 μg/kg b.w./day. Based on body weight, the intake of ACR in children is two to three times higher than that of adults (7). In 2015–2017, a survey report by EFSA showed that the average ACR content in fried potatoes and coffee products remained high at 1 and 5 mg/kg, respectively (7). In 2017, the EU set the benchmark levels for the presence of ACR in food products, with that of French fries (ready-to-eat) at 500 μg/kg and potato dough products at 750 μg/kg. The benchmark level for roasted coffee was 400 μg/kg, while that of instant coffee was 850 μg/kg (26). Therefore, ACR toxicity and its risk to human health require urgent attention due to its abundance in food products and high exposure frequency.
The Metabolic Pathway of Acrylamide
Acrylamide is a small-molecule hydrophilic substance. It is absorbed via the gastrointestinal tracts of humans and animals and passively diffused to the entire body. ACR can also pass through the blood-brain barrier to directly exert its toxic effect on the nervous system (27). ACR follows two main metabolic pathways in the body (Figure 1). (1) Briefly, an enzymatic reaction occurs when catalyzed by the cytochrome P450 enzyme system, CYP2E1, converting ACR into glycidamide (GA) (28). Studies have found that GA can combine with purine bases on deoxyribonucleic acid (DNA) molecules to form DNA adducts, inhibit the release of neurotransmitters, cause nerve terminal degeneration, damage nerve structures, and display distinct cumulative effects. In addition, the ability of GA to form Hb and DNA adducts is more significant than ACR. Therefore, it is believed that this pathway is the main route of ACR-induced neurotoxicity (29, 30). (2) ACR undergoes biotransformation and is catalyzed by glutathione S-transferase in the liver, combining with glutathione to generate N-acetyl-S-cysteine. It is further degraded into mercapturic ACR acids, which are excreted in the urine. This pathway is mainly responsible for ACR detoxification (31). Glutathione consumption reduces antioxidant levels, leading to excessive active oxygen accumulation and causing oxidative stress and neurotoxicity (32). Various reviews elaborate on the metabolic pathways of ACR. Those published by Koszucka et al. (27), Rifai et al. (28), and Fang et al. (30) are recommended for more details.
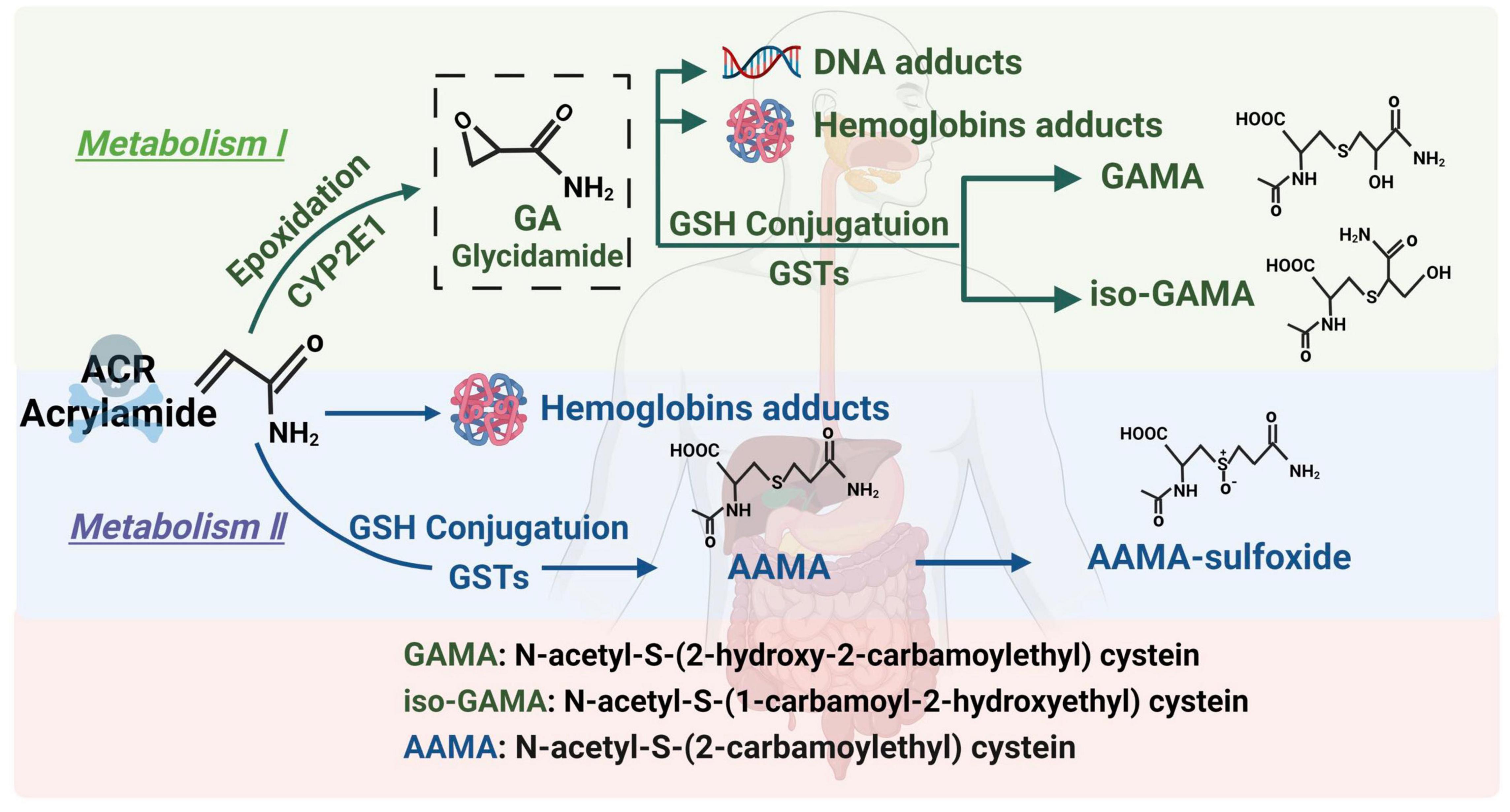
Figure 1. The metabolic pathway of ACR.
Model and Dosage
Previous studies involving ACR neurotoxicity mostly used a dose of 5–50 mg/kg/day during animal experiments in the in vivo model, with an average dose range of 1–5 mM in various cell lines. Toxic relationship and mechanism investigation mainly established between exposure to high concentrations (>2 mM in vitro or 20 mg/kg body weight in vivo) of ACR and neurotoxicity.
Barber and LoPachin exposed Sprague Dawley (SD) rats to a dose of 50 mg/kg/day for 28 days to explore ACR neurotoxicity, revealing that the mice displayed significant weight loss and abnormal gait (33). Yu et al. exposed male Wistar rats to doses of 20 and 40 mg/kg/days for 8 weeks, revealing the same phenomenon (34). In 2011, another study exposed adult male SD rats to 50 mg/kg/day ACR for 10 days. The results showed that ACR toxicity was associated with selective nerve terminal damage in the central and peripheral nervous systems (35). Santhanasabapathy and Vasudevan exposed adult Swiss albino male mice to a dose of 20 mg/kg/day for 4 weeks, indicating that ACR promoted microglial and astrocyte activation (36). Recently Guo and Cao exposed adult male SD rats to 40 mg/kg/day ACR for 4 weeks, showing the death of hippocampal neurons and neurotoxicity caused by oxidative stress (37). Moreover, the ACR exposure dose is relatively high in most in vitro studies. Human embryonic stem cells (H1hESC) were exposed to 2.5 mM and 5 mM for 24 h, demonstrating that ACR inhibited neuron differentiation based on the oxidative stress response (38). Similarly, Li and Sun exposed rat adrenal pheochromocytoma cells (PC12) to 5 mM ACR for 24 h, revealing oxidative stress-induced cytotoxicity (39). Triningsih and Yang used 5 mM 24 h ACR treatment to explore the relationship between ACR neurotoxicity and autophagy in PC12 cells (40).
Exposure to high ACR concentrations can produce central-peripheral signal transduction obstacles and influence neural development in occupationally exposed humans and laboratory animals (41–43). However, dietary intake is the main source of ACR exposure in humans (4). Several in vivo studies established models with a dose below 5 mg/kg/day and exposure times exceeding 60 days to investigate the neurotoxic effect and mechanism of foodborne ACR exposure. For example, SD rats provided with treated drinking water containing ACR dosages of 0, 0.5, or 5 mg/kg/day for 12 months displayed gait abnormality and cognitive dysfunction (44). Zhao et al. orally exposed C57BL/6 male mice to a low ACR dose of 5 mg/kg/day for 60 day (sub-chronic toxicity) to explore its neurotoxicity. This group displayed a slight gait abnormality at the end of the intonation period (45). Similarly, doses (about 1–5 μM) closer to chronic foodborne exposure are relatively rare in in vitro exposure models. Zong et al. demonstrated that ACR exposure increased the expression of cytokines and inflammatory markers in BV-2 microglial cells after treatment with a low ACR dose of 5 μM (46). Currently, extensive research has focused on the high-dose neurotoxicity, while chronic foodborne exposure of ACR has not received enough attention. In addition, health risk assessments require information regarding the relationship between exposure, internal brain dosage, and the observed toxic effects. Therefore, further research is needed to clarify and assess the risk of foodborne ACR neurotoxicity.
Mechanisms of Acrylamide-Induced Neurotoxicity
Axonal Degeneration
Early research suggests that ACR-induced neurotoxicity may be related to nerve-ending damage in the peripheral and central nervous systems (47). Studies have shown that ACR can change the β-actin, β-tubulin, and other cytoskeletal proteins, destroying the neuron structures to cause neurotoxicity. Subsequent morphological, electrophysiological, and electrochemical research shows that nerve endings represent the primary target of ACR toxicity (48). With the passage of exposure time, the damage is gradually aggravated and eventually leads to axonal degeneration. Chronic ACR poisoning can cause selective peripheral and central nerve fiber degeneration, which initially occurs at the ends of long and large nerve fibers, followed by progressive, continuous proximal axon degeneration (49). Further studies show that ACR affects the levels of actin, motor proteins, and other neuronal proteins, resulting in an insufficient supply of adenosine triphosphate, impairing axonal transport functionality (50).
With the development of mass spectrometry and nuclear magnetic resonance technology, researchers found that ACR, as an electrophilic reagent, can quickly attack the sulfhydryl group on proteins and react with molecular DNA while adduct formation may be a reason for its neurotoxicity (51). In addition, ACR can also attack protein sites containing thiols, interfere with the presynaptic nitric oxide (NO) signaling pathway, damage presynaptic nerve endings, disturb nerve signal transmission, and produce neurotoxicity (52). Moreover, phosphorylated Tau agglomeration causes cytoskeletal instability and neuronal malfunction or even death (53). According to recent research, ACR induces Tau tubulin hyperphosphorylation and brain-derived neurotrophic factor (BDNF) reduction in rat hippocampi, resulting in synaptic damage and spatial cognitive impairment (54). Furthermore, chronic ACR exposure leads to motor dysfunction by significantly degenerating dopaminergic and acetyl cholinergic neurons (55). Kopańska et al. believed that the cholinergic anti-inflammatory pathway was closely related to ACR-induced neurotoxicity. ACR decreased cholinergic conduction and acetylcholine secretion, inhibiting the cholinergic anti-inflammatory pathway and causing a potential systemic inflammatory reaction (56). Moreover, it is worth noting that the cognitive impairment caused by neuron degeneration is a precursor of Alzheimer’s disease (57), while the loss of dopaminergic neurons in the brain is related to Parkinson’s disease, indicating that ACR may be a risk factor for the pathological development of neurodegenerative diseases (58).
Apoptosis and Autophagy
As a common form of programmed cell death, apoptosis plays a vital role in neurodegenerative diseases (59). Neurocytes, including neurons, astrocytes, and microglia, are essential in maintaining brain function (60). Astrocytes mainly support neurotrophic functionality (61). As the resident immune cells in the central nervous system (62), microglial activation is crucial for collective immune monitoring, but excessive activation leads to the release of pro-inflammatory factors (63). Neurons represent the main executors of nerve impulses and, if damaged, cause cognitive impairment and motor dysfunction (64). Early studies have indicated that ACR can induce the apoptosis of various neurocytes, such as human neuroblastoma SH-SY5Y cells, astrocytoma U1240-MG cells, and rat stellate cells (65, 66). Therefore, ACR may damage brain homeostasis and cause neurotoxicity by inducing nerve cell apoptosis.
Liu et al. conducted a mechanism investigation and found that ACR caused mitochondrial malfunctioning in human astrocytoma cells and BV-2 mouse microglia, activated caspase-9 and its downstream pathway, up-regulated the BCL2-associated X protein (Bax)/B-cell lymphoma-2 (Bcl-2) ratio, and induced mitochondrial-dependent apoptosis and neurotoxicity (67). Moreover, other signaling pathways are also involved in ACR-induced apoptosis. Mitogen-activated protein kinase (MAPK), a serine-threonine protein kinase, can control many cell activities, such as apoptosis and the proliferation and expression of signal-regulated protein kinase [extracellular regulated protein kinases (ERK), c-Jun N-terminal kinase (JNK), and p38 protein] genes (68, 69). The inactivation of ERK and activation of JNK and p38 are essential for inducing apoptosis. Tabeshpour et al. found that intraperitoneal ACR injection reduced the p-ERK/ERK ratio while increasing the Bax/Bcl-2, p-JNK/JNK, and p-p38/p38 ratios in the cerebral cortexes of rats, indicating that the MAPK signaling pathway promoted ACR-induced apoptosis (70). Additionally, nuclear factor-κB (NF-κB) regulates a variety of target genes, such as proliferation and apoptosis, while the NF-κB signal is prone to crosstalk and affects several signaling pathways (71). ACR can promote apoptosis via the MAPK-driven NF-κB signaling pathway, ultimately resulting in cytotoxicity (72). The nuclear factor E2-related factor-2 (Nrf2) family represents a transcriptional factor that regulates the redox state of cells and participates in the coordination of adaptive responses to various stimuli (73). Pan et al. found that ACR activated the Nrf2 and MAPK pathways in PC12 cells. The MAPK served as an upstream regulator to control the nuclear translocation of Nrf2, exhibiting an antioxidative effect (74). It is worth noting that MAPK could control both the Nrf2 and NF-κB cascades as an upstream factor and facilitate dual-direction regulation (75). Consequently, crosstalk between the NF-κB, MAPK, and Nrf2 signaling pathways is vital during ACR-induced apoptosis and neurotoxicity.
Lysosome-mediated protective autophagy is essential for maintaining intracellular redox balance and is crucial for clearing damaged cell proteins and organelles, as well as regulating cell death and survival (76). However, high ACR doses may inhibit autophagy. For example, Liu et al. revealed that sub-chronic exposure to ACR could block autophagy flow by inhibiting lysosomal protease D in the hippocampus, decreasing the inflammasome clearance, and inducing local inflammation (44), which might be a decisive factor in ACR neurotoxicity. Song et al. reported that ACR caused autophagic marker microtubule-associated protein 1 light chain 3-II (LC3-II) and p62 accumulation, suggesting that ACR might inhibit cellular autophagy (77). However, autophagy flow is a finely regulated process and includes (1) autophagosome formation, (2) autophagosome and lysosome fusion, and (3) autolysosome degradation (78). Moreover, the crosstalk affiliation between apoptosis and autophagy is critical for brain health maintenance and central nervous system pathogenesis (79, 80). A study by Deng et al. confirmed the immunofluorescence and co-localization positions and co-expressed intensity of key autophagic markers during the three-autophagic process. The ACR-induced autophagosome accumulation could probably be ascribed to blocked autophagic flux, preventing the autophagosomes from combining with lysosomes. Additional information substantiating the connection between cellular apoptosis and autophagic flux showed that restricting the protective autophagy caused by ACR further promoted the initiation of apoptosis (Figure 2)(81).
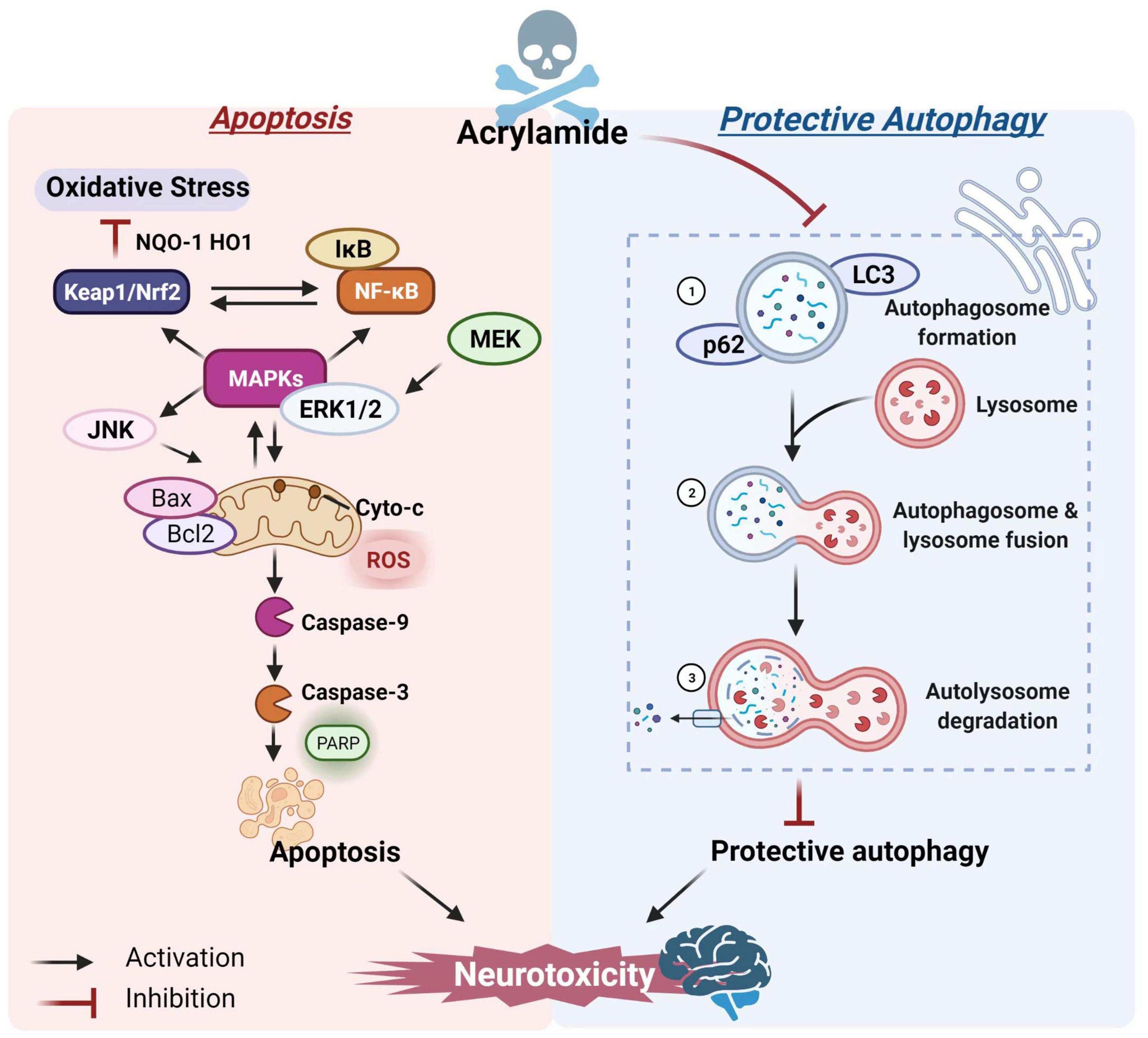
Figure 2. The potential mechanism of ACR-induced apoptosis and autophagy.
Oxidative Stress
The brain is more vulnerable to oxidative stress than other organs (82, 83), and most neurocyte components can be oxidized and damaged (84). Recent studies have shown that oxidative stress may be involved in the occurrence and development of neurodegenerative and chronic diseases (such as stroke, diabetes, Parkinson’s, Alzheimer’s, and other diseases) (85). Although reactive oxygen species (ROS) are produced during the normal physiological process, excessive production inhibits antioxidant reductase activity and disrupts redox balance, leading to oxidative stress damage (36). ACR-induced neurotoxicity typically manifests as intracellular glutathione depletion (86), indicating that oxidative stress may play a significant role in ACR neurotoxicity.
Mitochondria are essential organelles for intracellular energy metabolism and redox system regulation. Zhao et al. indicated that ACR disrupted the activity of the mitochondrial electron transport chain complexes, I, III, IV, and V, resulting in mitochondrial membrane swelling, the collapse of inner membrane potential, obvious electron transfer disorder, and electron leakage, leading to ROS accumulation (87). Liu et al. also found that ACR significantly increased the ROS level in the BV-2 microglial cell line, leading to oxidative stress, mitochondrial damage, and functionality loss while increasing the expression of caspase-dependent apoptosis-related genes (67). Damage to the mitochondrial structure and functional loss are hallmarks of endogenous apoptosis, activating the Bcl-2 protein family (88, 89). These results suggest that ACR-mediated mitochondrial dysfunction and oxidative stress injury are crucial factors leading to caspase-cascade activation and cellular apoptosis. Moreover, subsequent studies have revealed ACR-induced crosstalk in multiple signaling pathways during oxidative stress response and mitochondrial dysfunction. The Nrf2/NF-κB pathway in astrocytes and microglia was sequentially activated, gradually causing glutathione consumption, ROS accumulation, mitochondrial damage, and neuroinflammation (16).
Oxidative stress is deemed a significant predisposing factor in the pathogenesis of ACR-induced neurotoxicity, of which an increase in ROS accumulation or induced oxidative stress in the brain is only phenotypical. Of fundamental importance is linking these redox shifts to various signaling pathways and explaining how the changes occur. For example, research should explore (1) how oxidative stress arises via ACR in the brain, (2) how it is neutralized, (3) which typical signaling or genetic factors may trigger ACR-induced neurotoxicity development, and (4) how this impacts inflammation or autoimmunity in the central nervous system. Consequently, novel, more precise, and effective therapies can then be established to better understand how oxidative stress drives the underlying neurotoxic mechanisms.
Inflammation
The immune system recognizes external stimuli like pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) via pattern recognition receptors (PRRs). Furthermore, cellular PRRs, such as the nucleotide oligomerization domain (NOD)-like receptor (NLR), can activate the downstream immune-inflammatory response. Of these, NOD-like receptor protein 3 (NLRP3) in the NOD subfamily has been widely studied due to its unique response mechanism to a variety of stimuli (90). Research has shown that misfolded proteins like α-synuclein and β-amyloid can activate NLRP3 inflammasomes in the microglia, which may participate in neurodegenerative disease development, such as Parkinson’s and Alzheimer’s diseases (91). Mild cognitive impairment and the early symptoms of Alzheimer’s disease are similar to that of chronic neurotoxicity induced by ACR, suggesting the involvement of NLRP3 inflammasomes.
The primary manifestation of immune inflammation involves inflammatory factor secretion, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-18 (IL-18), and interleukin-1β (IL-1β) (92). Previous research by Zhao et al. found increased TNF-α, IL-6, and IL-1β levels in the primary microglia during the later stage of ACR exposure, further confirming the involvement of the immune-inflammatory response in ACR-induced neurotoxicity (16). Recent studies have shown that the NLRP3 inflammasome level was up-regulated in the hippocampi of rats after oral ACR administration, further increasing the expression levels of pro-inflammatory cytokines, IL-1β, IL-6, and IL-18 (46). Liu reported that inflammatory factors were released via the microglial activation facilitated by persistent ACR exposure. NLRP3 inflammasome activation increased the level of IL-1β, raising the levels of other inflammatory factors directly responsible for neuronal injury in the cerebrums of rats (44). Moreover, treatment with the NLRP3 inhibitor (MCC950) or NLRP3 siRNA safeguarded BV-2 microglial cells against the cytotoxicity produced by ACR while reversing NLRP3 inflammasome activation and the subsequent inflammatory response (93). Similarly, in mice exposed to MCC950, NLRP3 knockout intervention provided protection against the neurotoxic damage caused by ACR by restricting Nrf2 antioxidant pathway activation and neuroinflammation (93). Therefore, NLRP3 inflammasomes participate in the neurotoxicity facilitated by ACR and show promise as a target to improve therapeutic strategies.
Microbiota-Gut-Brain Axis Homeostasis
The brain-gut axis, which is linked by the immune system, is closely associated with mental disease and neurodevelopmental disorders (94). The mechanism underlying the influence of the intestinal barrier and intestinal flora on brain function may include synapse formation regulation (95), neuronal signaling activation, ROS formation inhibition (96), and blood-brain barrier function regulation via secondary metabolites (97).
Recent studies suggest that brain-gut axis homeostasis may be another important cause of ACR-induced neurotoxicity. ACR can facilitate circadian cognitive damage and spatial memory impairment by downregulating the protein expression of the circadian rhythm protein (Clock) and BDNF in mice (98). Moreover, ACR increases intestinal permeability, decreases tight junction protein (Occludin) expression, and increases the lipopolysaccharide (LPS) content in the intestine and serum, while the expression of pro-inflammatory cytokines, IL-6, and IL-1β are significantly up-regulated (98). Excessive LPS and inflammatory factors in the blood can induce an immune-inflammatory response, destroy the close blood-brain barrier connection, and aggravate neurogenic inflammation, damaging the neurons and leading to memory impairment (98). Therefore, ACR may interfere with the communication between the intestine and brain via the brain-gut axis, leading to circadian rhythm dysfunction, further promoting neurotoxicity development. In addition, ACR can change the diversity of flora in rat feces, reduce the abundance of some beneficial bacteria, and significantly increase the abundance of pathogenic bacteria, resulting in an intestinal flora imbalance and reduced short-chain fatty acids (SCFA) production, further stimulating neurotoxicity (99).
The brain-gut axis attracts increasing attention for the physiological and biological exploration of neurodegenerative, age-related, and neurodevelopmental diseases. Considering the close relationship between ACR toxicity and induced neurodegenerative disorders, the underlying neurotoxic mechanism via brain-gut axis homeostasis requires further investigation.
Conclusion: The Role of Multiple Signaling Pathways in Acrylamide-Induced Neurotoxicity
Acrylamide neurotoxicity has attracted extensive research attention globally. These reports confirmed that exposure to ACR can lead to neurological disorders like gait abnormality, cognitive impairment, and learning deficiencies. The representative effects and signaling pathways of the neurotoxicity caused by ACR are listed in Table 2. Developing research has gradually proposed a hypothesis regarding the neurotoxic mechanism of ACR and can be summarized as follows (Figure 3): (1) Axonal degeneration, neuronal deficits, and DNA-protein adduct formation are primarily responsible for ACR-induced neurological disorders. (2) Oxidative stress is a typical response caused by ACR and may be associated with mitochondrial malfunction resulting from ACR in conjunction with calcium dyshomeostasis. (3) Multiple neural pathways involving apoptosis, autophagy, and inflammation are attributed to ACR-induced neurotoxicity. (4) Brain-intestinal axis homeostasis and circadian rhythms also participate in ACR-induced neurotoxicity.
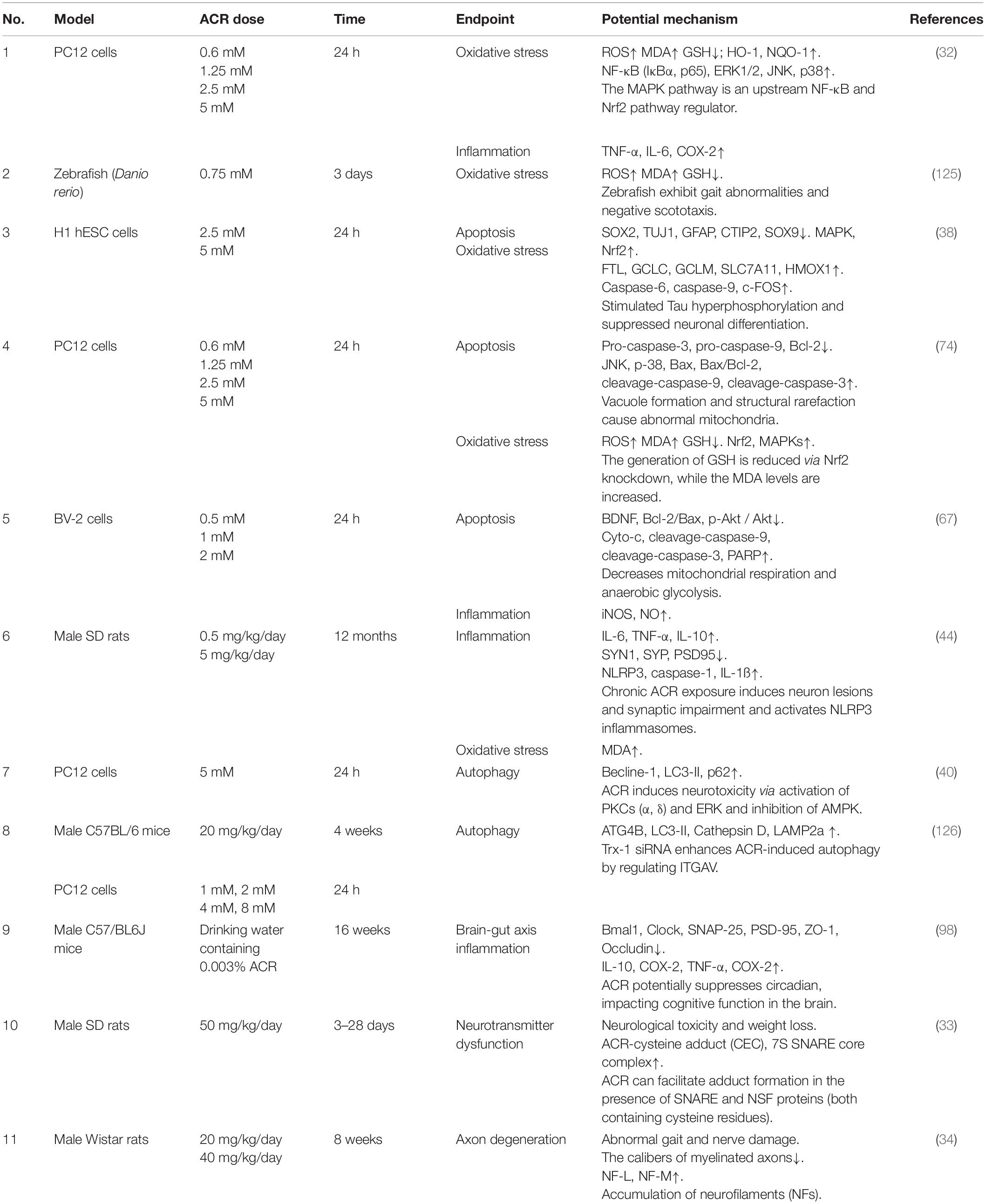
Table 2. A summary of the representative neurotoxic effect of ACR in different models.
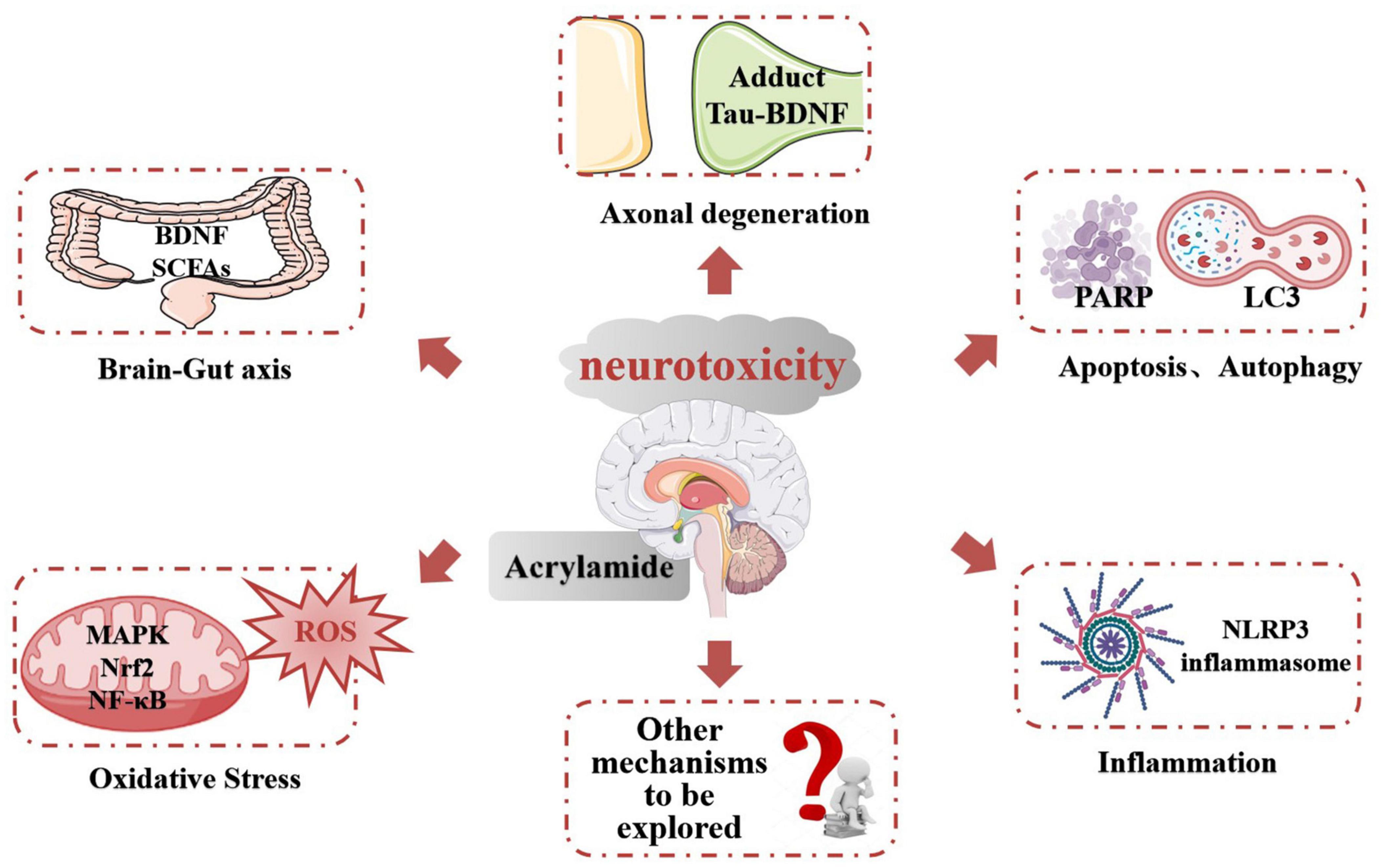
Figure 3. The potential mechanism of ACR-induced neurotoxicity.
Future Perspectives
Studies are increasingly concentrating on the neurological impact and various underlying mechanisms of ACR. However, despite extensive research involving multiple models, fundamental questions persist, requiring resolution. This section discusses future research from three perspectives, requiring further attention. These insights may further prompt the development of alternative approaches examining ACR risk factors.
Acrylamide and Neurodegenerative Diseases
As early as 2008, Lopachin et al. suggested that the onset and progression of some neuropathogenic processes, like Parkinson’s and Alzheimer’s diseases and amyotrophic lateral sclerosis, are accelerated by environmental exposure to some type-2 alkenes, such as ACR (100). Moreover, a recent epidemiological study involving 2,534 elderly non-smoking Chinese men indicated that ACR exposure was related to a mild cognitive decrease and a higher risk of poor cognition in four years (101). Extensive research has shown that the various neurotoxic syndromes due to ACR exposure and neurodegenerative diseases, including biochemical changes and neuropathic events in the brain and spinal cord, are similar (102, 103). The inflammatory response and oxidative stress, as well as the related activated signaling pathways present in both neurodegenerative diseases and ACR-induced neurotoxicity, were investigated to clarify the underlying mechanism (13, 104, 105). Therefore, a close connection may exist between the neurotoxicity caused by ACR and neurodegenerative diseases.
However, despite existing comparative studies, some fundamental questions remain. (1) Discoveries regarding ACR-related neurotoxic mechanisms require further investigation based on the neuropathological process of neurodegenerative diseases. (2) How ACR promotes the neuropathological process of neurodegenerative diseases and whether ACR can induce the expression of neurodegenerative pathogenic factors like β-amyloid protein aggregation in the brain remain unclear. However, this presents a fascinating research topic for the future. (3) Clarification is required regarding the existence of a shared target or signaling connection during ACR-induced neurotoxicity and pathological neurodegenerative processing, such as innate immune receptors, toll-like receptor-4, and NLRP3, as well as their signaling interaction. Therefore, more evidence is required. (4) These challenges necessitate a more in-depth understanding of the mechanisms underlying the neurotoxicity caused by ACR. The knowledge regarding the mechanism can be used to develop novel strategies or inhibitors to reduce and block the targets required to manifest adverse ACR effects.
Nucleotide Oligomerization Domain-Like Receptor Protein 3 Inflammasome-Related Immune Inflammation
Several studies have clarified the role of inflammation in the neurotoxicity caused by ACR (36, 72), revealing a connection with NLRP3 inflammasomes (93). However, the specific regulatory role and mechanism of NLRP3 inflammasomes in ACR-induced neurotoxicity require further clarification (Figure 4).
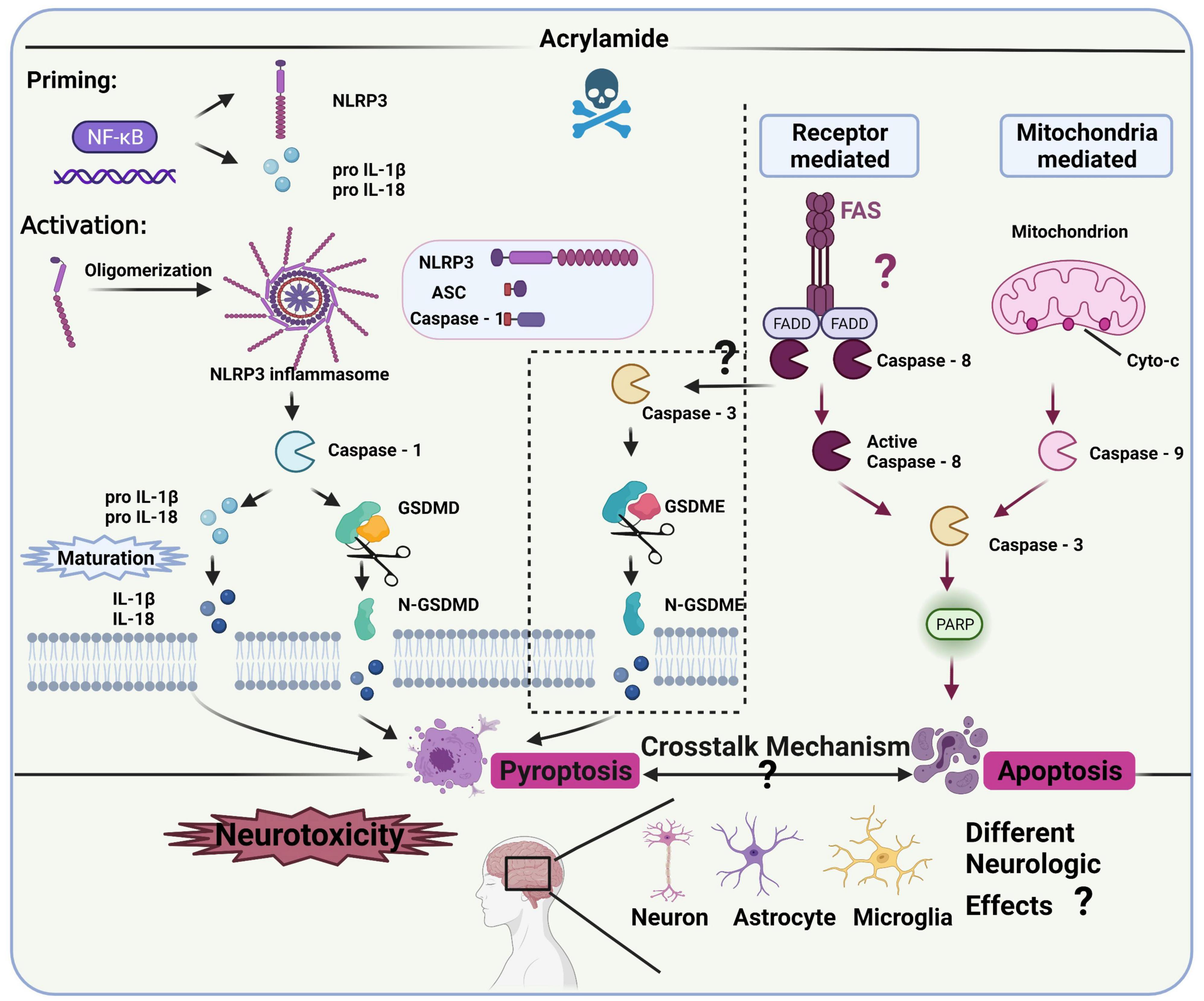
Figure 4. NLRP3 inflammasomes and the related pathway in ACR-induced neurotoxicity.
Canonical NLRP3 inflammasomes contain effector protein caspase-1, adaptor protein ASC (a speck-like protein containing a CARD, associated with adaptor molecule apoptosis), and sensory protein NLRP3 (106), while it requires priming and activation to become operational. The priming process includes pro-IL-18, pro-IL-1β, and NLRP3 expression, the initiation of which occurs via the NF-κB signaling pathway (107, 108). The activation process allows inflammasome complex assembly, facilitating pro-caspase-1 cleavage to obtain active caspase-1. Then, mature IL-18 and IL-1β are released via pro-IL-18 and pro-IL-1β cleavage (44). Additionally, the N-terminal domain of gasdermin D (GSDMD) is released via activated caspase-1 cleavage, forming pores in the plasma membrane to rapidly release mature IL-18 and IL-1β, which may cause pyroptosis (109). Besides the canonical pathway, caspase-3 and gasdermin E (GSDME) can also facilitate the release of mature IL-18 and IL-1β while inducing inflammatory cell death, known as alternative pyroptosis in many neurodegenerative diseases and inflammatory response models (110, 111). However, whether the activated caspase-3-IL-1β/IL-18-GSDME signaling contributes to ACR-induced pyroptosis and neurotoxicity remains unknown. Therefore, the pyroptosis-related pathway representing the main signal cascade (NLRP3-ASC-caspase-1-IL-1β/IL-18-GSDMD or caspase-3-IL-1β/IL-18-GSDME) induced by ACR and its neurological impact requires exploration. Furthermore, future studies should also investigate which cascade is activated first during ACR exposure and whether the activation is time-dependent. Additionally, both pyroptosis and apoptosis are involved in programmed cell death but are differentiated via morphology (112). ACR can induce pyroptosis and apoptosis with phenotypical traits (109), while caspase-3 represents the effector protein in both the mitochondrially mediated apoptosis pathway and caspase-3-IL-1β/IL-18-GSDME cascade. How ACR dosage, period, and various other factors affect the functionality of caspase-3, as well as the synergistic effect of pyroptosis and apoptosis on the neurological mechanism, require further exploration.
Given the role of inflammasome signaling in the ACR-induced neurotoxic conditions mentioned above, researchers should explore the crucial biomarkers in the pathways to develop specific inhibitors against ACR-induced neurotoxicity. In this case, the correlation between oxidative stress and neuroinflammation should be further refined. For example, how the specific modification of cellular oxidative stress impacts neuroinflammation warrants further attention. Clarification is necessary regarding the connection between mitochondrial dysfunction and inflammasome signaling and whether ion efflux and energy metabolism contribute to this association. Moreover, the potential biomarkers in the NLRP3-related pathway like NLRP3, ASC, caspase-1, GSDMD, and the alternative pyroptosis pathway like caspase-3 and GSDME, should be further confirmed in vitro and in vivo. An adequate transgenic model must be employed to further verify the NLRP3-related biomarkers and investigate their mechanistic role.
Microglial cells (such as the BV-2 cell line) are generally considered central neuron system immune cells and are activated during the inflammatory response. However, future studies should compare the functional response diversity of neurons, astrocytes, and microglia using different in vitro neurocyte models to verify the pathways involved in neuroinflammation while assessing their contribution to ACR neurotoxicity. Another challenge involves achieving cell-cell communication between neurons and glia, specifically concerning the microglia-astrocyte-neuron impact on the neuroinflammation produced by ACR. This interaction may be explained using a co-cultural model.
Microbiota-Gut-Brain Axis Signaling
The gut-brain axis is deemed an essential pathway for physiological regulation and communication, while gut microbiota are seemingly crucial in this interactive relationship (113). Several pathways are involved in microbiota-gut-brain axis signaling and include the immune system, neurochemical signaling host recruitment, the vagus nerve and direct enteric nervous system routes, and bacterial metabolite production (114–117).
Altered gut microbial profiles and signaling have been described in various neurological disorders, such as Parkinson’s and Alzheimer’s diseases (118, 119). Although the similar neurological impact has prompted some studies to investigate the connection between ACR-induced neurotoxicity and microbiota-gut-brain axis signaling in recent years, systematic studies involving this topic are still lacking. In general, the hypothetical effect of microbiota-gut-brain axis signaling induced by ACR occurs via the gut bacterial metabolites of a defective gut barrier, leading to a systematic inflammatory response. This process impairs the blood-brain barrier, ultimately leading to neural damage and deterioration (Figure 5).
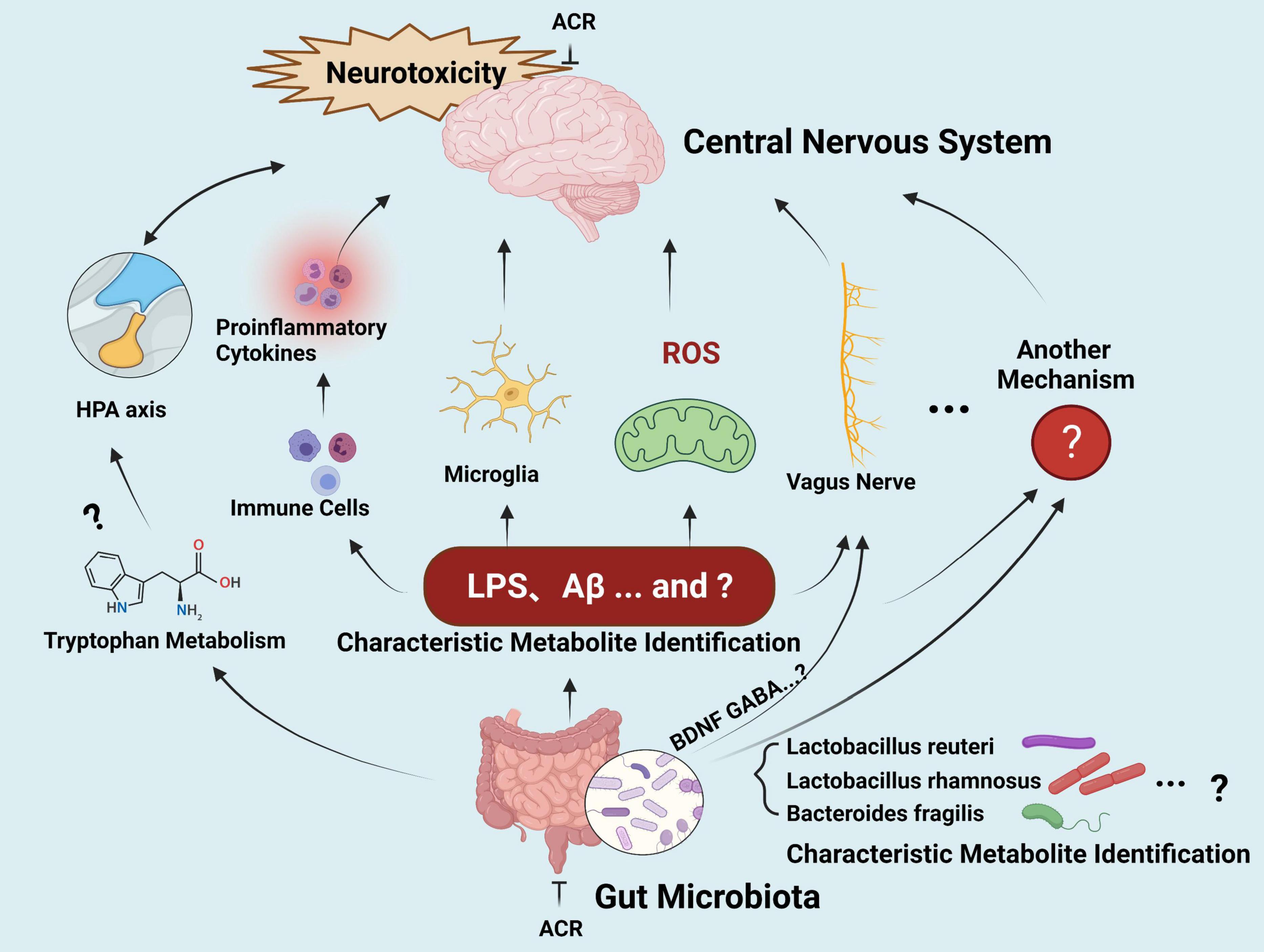
Figure 5. The gut-brain axis in ACR-induced neurotoxicity.
However, these hypotheses should be investigated further. (1) Screening and identifying the microbiome of a specific type of putative bacterium (biomarkers) related to ACR-induced neurotoxicity deserves further attention. (2) Further exploration is required regarding potential neurotoxic molecule production, such as amyloid proteins, LPS, and additional secondary metabolites that can reach the central neuron system via systemic circulation to activate microglia and ROS signaling. Furthermore, research should clarify whether ACR exposure can modulate the metabolism of normal functional neurotransmitters, such as serotonin and tryptophan, and whether the behavior impacted by ACR-induced gut microbiota and those relying on intact serotonergic neurotransmission overlaps. The underlying crosstalk mechanisms require further elucidation and may be associated with the gut microbial capacity to regulate the metabolism of the host tryptophan in conjunction with the kynurenine pathway. (3) The communication between the brain and the gut during ACR exposure requires clarification. The release of cytokines via the immune response represents the most common assumption during the gut-brain communication mechanism. Other influencing routes include the vagus nerve, hypothalamic-pituitary-adrenal axis, and secondary microbial metabolites, while neuromodulators and neurotransmitters may also be involved. However, the details regarding the signaling pathways and crucial biomarkers for communication require further investigation. (4) Limited epidemiological-related data is available, and it is unclear whether any common changes in the gut microbiota are evident in high-risk individuals during ACR exposure. Moreover, whether common microbiota biomarkers are evident in high-risk exposed individuals and neurodegenerative disease patients requires elucidation. During the extended journey from the gut microbiota to the brain, many connections are still unidentified. If this challenge can be resolved, the microbiota-gut-brain axis can facilitate new therapeutic strategies for ACR-induced neurotoxicity and various neurological conditions.
Author Contributions
MZ: conceptualization, original draft preparation, writing – review and editing, project administration, and funding acquisition. BZ: original draft preparation, especially in summarizing the table and creating the flow chart of the signaling pathways, and writing – review and editing. LD: writing – review and editing and validation. All authors read and approved the final manuscript.
Funding
The National Natural Science Foundation for Young Scientists of China (No. 31801668), The Fundamental Research Funds for the Central Universities (No. 222201814036), The Shanghai PuJiang Program (No. 18J1401900), and The National Key R&D Program of China (No. 2019YFD090180302).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACR, acrylamide; ASC, apoptosis-associated speck-like protein containing CARD; Bax, BCL2-associated X protein; Bcl-2, B-cell lymphoma-2; BDNF, brain-derived neurotrophic factor; Caspase-1, cysteinyl aspartate specific proteinase 1; DAMPs, damage-associated molecular patterns; DNA, deoxyribonucleic acid; EFSA, European Food Safety Authority; ERK, extracellular regulated protein kinases; EU, European Union; GA, glycidamide; GSDMD, gasdermin D; GSDME, gasdermin E; GSH, glutathione; Hb, hemoglobin; IARC, International Agency for Research on Cancer; IL-1 β, interleukin-1 β; IL-6, interleukin-6; IL-18, interleukin-18; JECFA, Joint FAO/WHO Expert Committee on Food Additives; JNK, c-JunN-terminal kinase; LC3-II, microtubule associated protein 1 light chain 3-II; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; Nrf2, nuclear factor E2-related factor-2; NF- κ B, nuclear factor-kappa B; NO, nitric oxide; NLR, nod like receptor; NLRP3, NOD-like receptor protein 3; 8-OHdG, 8-hydroxy-2-deoxyguanosine; PAMPs, pathogen-associated molecular patterns; PRRs, pattern recognition receptors; ROS, reactive oxygen species; SCFA, short-chain fatty acids; SD, Sprague Dawley; TNF- α, tumor necrosis factor- α; WHO, World Health Organization.
References
1. Smith EA, Oehme FW. Acrylamide and polyacrylamide: a review of production, use, environmental fate and neurotoxicity. Rev Environ Health. (1991) 9:215–28. doi: 10.1515/reveh.1991.9.4.215
2. Zhang Y, Zhang Y. Formation and reduction of acrylamide in Maillard reaction: a review based on the current state of knowledge. Crit Rev Food Sci Nutr. (2007) 47:521–42. doi: 10.1080/10408390600920070
3. Capuano E, Fogliano V. Acrylamide and 5-hydroxymethylfurfural (HMF): a review on metabolism, toxicity, occurrence in food and mitigation strategies. LWT Food Sci Technol. (2011) 44:793–810. doi: 10.1016/j.lwt.2010.11.002
4. Tareke E, Rydberg P, Karlsson P, Eriksson S, Tornqvist M. Analysis of acrylamide, a carcinogen formed in heated foodstuffs. J Agric Food Chem. (2002) 50:4998–5006. doi: 10.1021/jf020302f
5. Mottram DS, Wedzicha BL, Dodson AT. Acrylamide is formed in the Maillard reaction. Nature. (2002) 419:448–9. doi: 10.1038/419448a
6. Stadler RH, Blank I, Varga N, Robert F, Hau J, Guy PA, et al. Acrylamide from Maillard reaction products. Nature. (2002) 419:449–50. doi: 10.1038/419449a
7. Panel. Scientific Opinion on acrylamide in food. EFSA J. (2015) 13:4104. doi: 10.2903/j.efsa.2015.4104
8. Powers SJ, Mottram DS, Curtis A, Halford NG. Acrylamide levels in potato crisps in Europe from 2002 to 2016. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. (2017) 34:2085–100. doi: 10.1080/19440049.2017.1379101
9. Pundir CS, Yadav N, Chhillar AK. Occurrence, synthesis, toxicity and detection methods for acrylamide determination in processed foods with special reference to biosensors: a review. Trends Food Sci Technol. (2019) 85:211–25. doi: 10.1016/j.tifs.2019.01.003
10. Exon JH. A review of the toxicology of acrylamide. J Toxicol Environ Health B Crit Rev. (2006) 9:397–412. doi: 10.1080/10937400600681430
11. Barber DS, Stevens S, LoPachin RM. Proteomic analysis of rat striatal synaptosomes during acrylamide intoxication at a low dose rate. Toxicol Sci. (2007) 100:156–67. doi: 10.1093/toxsci/kfm210
12. Baydar T, Erkekoglu P, Sipahi H, Ahin G. Aflatoxin M1 and Ochratoxin contents of infant formulas and baby foods commonly consumed in Ankara. In: Proceedings of the 10th International Congress of Toxicology (2014). Available online at: https://www.researchgate.net/publication/264755252_Aflatoxin_M1_and_Ochratoxin_contents_of_infant_formulas_and_baby_foods_commonly_consumed_in_Ankara
13. LoPachin RM. The changing view of acrylamide neurotoxicity. Neurotoxicology. (2004) 25:617–30. doi: 10.1016/j.neuro.2004.01.004
14. LoPachin RM, Gavin T, DeCaprio A, Barber DS. Application of the hard and soft, acids and bases (HSAB) theory to toxicant–target interactions. Chem Res Toxicol. (2011) 25:239–51. doi: 10.1021/tx2003257
15. Zamani E, Shokrzadeh M, Ziar A, Abedian-Kenari S, Shaki F. Acrylamide attenuated immune tissues’ function via induction of apoptosis and oxidative stress: Protection by l-carnitine. Hum Exp Toxicol. (2018) 37:859–69. doi: 10.1177/0960327117741753
16. Zhao M, Lewis Wang FS, Hu X, Chen F, Chan HM. Acrylamide-induced neurotoxicity in primary astrocytes and microglia: roles of the Nrf2-ARE and NF-kappaB pathways. Food Chem Toxicol. (2017) 106(Pt A):25–35. doi: 10.1016/j.fct.2017.05.007
17. Friedman M. Chemistry, biochemistry, and safety of acrylamide. A review. J Agric Food Chem. (2003) 51:4504–26. doi: 10.1021/jf030204
18. Pennisi M, Malaguarnera G, Puglisi V, Vinciguerra L, Vacante M, Malaguarnera M. Neurotoxicity of acrylamide in exposed workers. Int J Environ Res Public Health. (2013) 10:3843–54. doi: 10.3390/ijerph10093843
19. Backe WJ, Yingling V, Johnson T. The determination of acrylamide in environmental and drinking waters by large-volume injection - hydrophilic-interaction liquid chromatography and tandem mass spectrometry. J Chromatogr A. (2014) 1334:72–8. doi: 10.1016/j.chroma.2014.02.005
20. Brown L, Rhead MM, Hill D, Bancroft KCC. Qualitative and quantitative studies on the in situ adsorption, degradation and toxicity of acrylamide by the spiking of the waters of two sewage works and a river. Water Res. (1982) 16:579–91. doi: 10.1016/0043-135490078-1
21. Who. Evaluation of certain contaminants in food. World Health Organiz Tech Rep Ser. (2011) 959:1–166.
22. Smith EA, Prues SL, Oehme FW. Environmental degradation of polyacrylamides. II. Effects of environmental (outdoor) exposure. Ecotox Environ Saf. (1997) 37:76–91. doi: 10.1006/eesa.1997.1527
23. Zhang Y, Wang Q, Zhang G, Jia W, Ren Y, Wu Y. Biomarker analysis of hemoglobin adducts of acrylamide and glycidamide enantiomers for mid-term internal exposure assessment by isotope dilution ultra-high performance liquid chromatography tandem mass spectrometry. Talanta. (2018) 178:825–33. doi: 10.1016/j.talanta.2017.09.092
24. Lu X. Research progress of acrylamide in cosmetics and its detection. China Med Pharm. (2014). 4:52–55.
25. Viswanath P. Evaluation of certain contaminants in food. In: Proceedings of the 72nd Report of the Joint FAO/WHO Expert Committee on Food Additives. Geneva: Medknow Publications and Media Pvt. Ltd (2012).
26. Mesias M, Nouali A, Delgado-Andrade C, Morales FJ. How far is the spanish snack sector from meeting the acrylamide regulation 2017/2158? Foods. (2020) 9:247. doi: 10.3390/foods9020247
27. Koszucka A, Nowak A, Nowak I, Motyl I. Acrylamide in human diet, its metabolism, toxicity, inactivation and the associated European Union legal regulations in food industry. Crit Rev Food Sci Nutr. (2020) 60:1677–92. doi: 10.1080/10408398.2019.1588222
28. Rifai L, Saleh FAA. Review on acrylamide in food: occurrence, toxicity, and mitigation strategies. Int J Toxicol. (2020) 39:93–102. doi: 10.1177/1091581820902405
29. Mucci LA, Wilson KA. Acrylamide intake through diet and human cancer risk. J Agr Food Chem. (2008) 56:6013–9. doi: 10.1021/jf703747b
30. Fang C, Hu X, Liu Y, Wang P, Li D. Metabolism of Acrylamide: Interindividual and Interspecies Differences as Well as the Application as Biomarkers. (Vol. 17). (2015). 2488645 p. doi: 10.1145/2488608.2488645
31. Kirman CR, Gargas ML, Deskin R, Tonner-Navarro L, Andersen ME. A physiologically based pharmacokinetic model for acrylamide and its metabolite, glycidamide, in the rat. J Toxicol Environ Health A. (2003) 66:253–74. doi: 10.1080/15287390306368
32. Pan X, Wu X, Yan D, Peng C, Rao C, Yan H. Acrylamide-induced oxidative stress and inflammatory response are alleviated by N-acetylcysteine in PC12 cells: Involvement of the crosstalk between Nrf2 and NF-kappaB pathways regulated by MAPKs. Toxicol Lett. (2018) 288:55–64. doi: 10.1016/j.toxlet.2018.02.002
33. Barber DS, LoPachin RM. Proteomic analysis of acrylamide-protein adduct formation in rat brain synaptosomes. Toxicol Appl Pharmacol. (2004) 201:120–36. doi: 10.1016/j.taap.2004.05.008
34. Yu S, Zhao X, Zhang T, Yu L, Li S, Cui N, et al. Acrylamide-induced changes in the neurofilament protein of rat cerebrum fractions. Neurochem Res. (2005) 30:1079–85. doi: 10.1007/s11064-005-7413-3
35. Zhang L, Gavin T, Barber DS, LoPachin RM. Role of the Nrf2-ARE pathway in acrylamide neurotoxicity. Toxicol Lett. (2011) 205:1–7. doi: 10.1016/j.toxlet.2011.04.011
36. Santhanasabapathy R, Vasudevan S, Anupriya K, Pabitha R, Sudhandiran G. Farnesol quells oxidative stress, reactive gliosis and inflammation during acrylamide-induced neurotoxicity: behavioral and biochemical evidence. Neuroscience. (2015) 308:212–27. doi: 10.1016/j.neuroscience.2015.08.067
37. Guo J, Cao X, Hu X, Li S, Wang J. The anti-apoptotic, antioxidant and anti-inflammatory effects of curcumin on acrylamide-induced neurotoxicity in rats. BMC Pharmacol Toxicol. (2020) 21:62. doi: 10.1186/s40360-020-00440-3
38. Bu Q, Huang Y, Li M, Dai Y, Fang X, Chen K, et al. Acrylamide exposure represses neuronal differentiation, induces cell apoptosis and promotes tau hyperphosphorylation in hESC-derived 3D cerebral organoids. Food Chem Toxicol. (2020) 144:111643. doi: 10.1016/j.fct.2020.111643
39. Li L, Sun HY, Liu W, Zhao HY, Shao ML. Silymarin protects against acrylamide-induced neurotoxicity via Nrf2 signalling in PC12 cells. Food Chem Toxicol. (2017) 102:93–101. doi: 10.1016/j.fct.2017.01.021
40. Triningsih D, Yang JH, Sim KH, Lee C, Lee YJ. Acrylamide and its metabolite induce neurotoxicity via modulation of protein kinase C and AMP-activated protein kinase pathways. Toxicol In Vitro. (2021) 72:105105. doi: 10.1016/j.tiv.2021.105105
41. Lakshmi D, Gopinath K, Jayanthy G, Anjum S, Prakash D, Sudhandiran G. Ameliorating effect of fish oil on acrylamide induced oxidative stress and neuronal apoptosis in cerebral cortex. Neurochem Res. (2012) 37:1859–67. doi: 10.1007/s11064-012-0794-1
42. Mehri S, Abnous K, Khooei A, Mousavi SH, Shariaty VM, Hosseinzadeh H. Crocin reduced acrylamide-induced neurotoxicity in Wistar rat through inhibition of oxidative stress. Iran J Basic Med Sci. (2015) 18:902–8.
43. Prasad SN, Muralidhara. Mitigation of acrylamide-induced behavioral deficits, oxidative impairments and neurotoxicity by oral supplements of geraniol (a monoterpene) in a rat model. Chem Biol Interact. (2014) 223:27–37. doi: 10.1016/j.cbi.2014.08.016
44. Liu Y, Zhang X, Yan D, Wang Y, Wang N, Liu Y, et al. Chronic acrylamide exposure induced glia cell activation, NLRP3 infl-ammasome upregulation and cognitive impairment. Toxicol Appl Pharmacol. (2020) 393:114949. doi: 10.1016/j.taap.2020.114949
45. Zhao M, Deng L, Lu X, Fan L, Zhu Y, Zhao L. The involvement of oxidative stress, neuronal lesions, neurotransmission impairment, and neuroinflammation in acrylamide-induced neurotoxicity in C57/BL6 mice. Environ Sci Pollut Res Int. (2022). doi: 10.1007/s11356-021-18146-2
46. Zong C, Hasegawa R, Urushitani M, Zhang L, Nagashima D, Sakurai T, et al. Role of microglial activation and neuroinflammation in neurotoxicity of acrylamide in vivo and in vitro. Arch Toxicol. (2019) 93:2007–19. doi: 10.1007/s00204-019-02471-0
47. LoPachin RM Jr., Lehning EJ. Acrylamide-induced distal axon degeneration: a proposed mechanism of action. Neurotoxicology. (1994) 15:247–59. doi: 10.1016/0028-390890123-6
48. Von Burg R, Penney DP, Conroy PJ. Acrylamide neurotoxicity in the mouse: a behavioral, electrophysiological and morphological study. J Appl Toxicol. (1981) 1:227–33. doi: 10.1002/jat.2550010409
49. Spencer PS, Schaumburg HH. Nervous system degeneration produced by acrylamide monomer. Environ Health Perspect. (1975) 11:129–33. doi: 10.1289/ehp.7511129
50. An L, Li G, Si J, Zhang C, Han X, Wang S, et al. Acrylamide retards the slow axonal transport of neurofilaments in rat cultured dorsal root ganglia neurons and the corresponding mechanisms. Neurochem Res. (2016) 41:1000–9. doi: 10.1007/s11064-015-1782-z
51. LoPachin RM, Barber DS, He D, Das S. Acrylamide inhibits dopamine uptake in rat striatal synaptic vesicles. Toxicol Sci. (2006) 89:224–34. doi: 10.1093/toxsci/kfj005
52. Mohr S, Stamler JS, Brüne B. Mechanism of covalent modification of glyceraldehyde-3-phosphate dehydrogenase at its active site thiol by nitric oxide, peroxynitrite and related nitrosating agents. FEBS Lett. (1994) 348:223–7. doi: 10.1016/0014-579300596-6
53. L’Episcopo F, Drouin-Ouellet J, Tirolo C, Pulvirenti A, Giugno R, Testa N, et al. GSK-3beta-induced Tau pathology drives hippocampal neuronal cell death in Huntington’s disease: involvement of astrocyte-neuron interactions. Cell Death Dis. (2016) 7:e2206. doi: 10.1038/cddis.2016.104
54. Yan D, Yao J, Liu Y, Zhang X, Wang Y, Chen X, et al. Tau hyperphosphorylation and P-CREB reduction are involved in acrylamide-induced spatial memory impairment: Suppression by curcumin. Brain Behav Immun. (2018) 71:66–80. doi: 10.1016/j.bbi.2018.04.014
55. Murray SM, Waddell BM, Wu CW. Neuron-specific toxicity of chronic acrylamide exposure in C. elegans. Neurotoxicol Teratol. (2020) 77:106848. doi: 10.1016/j.ntt.2019.106848
56. Kopańska M, Łagowska A, Kuduk B, Banaś-Ząbczyk A. Acrylamide neurotoxicity as a possible factor responsible for inflammation in the cholinergic nervous system. Int J Mol Sci. (2022) 23:2030. doi: 10.3390/ijms23042030
57. Janoutova J, Sery O, Hosak L, Janout V. Is mild cognitive impairment a precursor of alzheimer’s disease? Short Review. Cent Eur J Public Health. (2015) 23:365–7. doi: 10.21101/cejph.a4414
58. Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. Extracellular dopamine potentiates mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet. (2010) 6:182–8. doi: 10.1371/journal.pgen.1001084
59. Unver-Saraydin S, Saraydin D, Sahin Inan ZD. A study of digital image analysis on the acrylamide derivative monomers induced apoptosis in rat cerebrum. Microsc Res Tech. (2020) 83:436–45. doi: 10.1002/jemt.23431
60. Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. (2008) 60:430–40. doi: 10.1016/j.neuron.2008.10.013
61. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
62. Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. (2010) 119:89–105. doi: 10.1007/s00401-009-0622-0
63. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. (2007) 8:57–69. doi: 10.1038/nrn2038
64. Xiao JW, Guo CL, Niu KL, Zhong-Sheng LI, Meng HL, Bin LI. Effects of Schwan cells on damage and repair of motor neuron induced by acrylamide. J J Toxicol. (2011) 25:328–31.
65. Lee JG, Wang YS, Chou CC. Acrylamide-induced apoptosis in rat primary astrocytes and human astrocytoma cell lines. Toxicol In Vitro. (2014) 28:562–70. doi: 10.1016/j.tiv.2014.01.005
66. Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX. (2009) 26:83–94. doi: 10.14573/altex.2009.2.83
67. Liu Z, Song G, Zou C, Liu G, Wu W, Yuan T, et al. Acrylamide induces mitochondrial dysfunction and apoptosis in BV-2 microglial cells. Free Radic Biol Med. (2015) 84:42–53. doi: 10.1016/j.freeradbiomed.2015.03.013
68. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. (2010) 429:403–17. doi: 10.1042/BJ20100323
69. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. (2009) 9:537–49. doi: 10.1038/nrc2694
70. Tabeshpour J, Mehri S, Abnous K, Hosseinzadeh H. Role of oxidative stress, MAPKinase and apoptosis pathways in the protective effects of thymoquinone against acrylamide-induced central nervous system toxicity in rat. Neurochem Res. (2020) 45:254–67. doi: 10.1007/s11064-019-02908-z
71. Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
72. Yan D, Pan X, Yao J, Wang D, Wu X, Chen X, et al. MAPKs and NF-kappaB-mediated acrylamide-induced neuropathy in rat striatum and human neuroblastoma cells SY5Y. J Cell Biochem. (2019) 120:3898–910. doi: 10.1002/jcb.27671
73. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. (2014) 39:199–218. doi: 10.1016/j.tibs.2014.02.002
74. Pan X, Yan D, Wang D, Wu X, Zhao W, Lu Q, et al. Mitochondrion-Mediated Apoptosis Induced by Acrylamide is Regulated by a Balance Between Nrf2 Antioxidant and MAPK Signaling Pathways in PC12 Cells. Mol Neurobiol. (2017) 54:4781–94. doi: 10.1007/s12035-016-0021-1
75. Pan X. The role and Mechanism of MAPKs, Nrf2 and NF-κB Signaling pathWay in Acrylamide Neurotoxicity. Master’s thesis. Wuhan: Huazhong University of Science and Technology (2016).
76. Aldawood N, Alrezaki A, Alanazi S, Amor N, Alwasel S, Sirotkin A, et al. Acrylamide impairs ovarian function by promoting apoptosis and affecting reproductive hormone release, steroidogenesis and autophagy-related genes: An in vivo study. Ecotoxicol Environ Saf. (2020) 197:110595. doi: 10.1016/j.ecoenv.2020.110595
77. Song D, Xu C, Holck AL, Liu R. Acrylamide inhibits autophagy, induces apoptosis and alters cellular metabolic profiles. Ecotoxicol Environ Saf. (2021) 208:111543. doi: 10.1016/j.ecoenv.2020.111543
78. Yorimitsu T, He C, Wang K, Klionsky DJ. Tap42-associated protein phosphatase type 2A negatively regulates induction of autophagy. Autophagy. (2009) 5:616–24. doi: 10.4161/auto.5.5.8091
79. Ghavami S, Sharma P, Yeganeh B, Ojo OO, Jha A, Mutawe MM, et al. Airway mesenchymal cell death by mevalonate cascade inhibition: integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim Biophys Acta. (2014) 1843:1259–71. doi: 10.1016/j.bbamcr.2014.03.006
80. Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A, et al. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci. (2014) 10:1072–83. doi: 10.7150/ijbs.9719
81. Deng L, Zhao M, Cui Y, Xia Q, Jiang L, Yin H, et al. Acrylamide induces intrinsic apoptosis and inhibits protective autophagy via the ROS mediated mitochondrial dysfunction pathway in U87-MG cells. Drug Chem Toxicol. (2021) 21:1–12. doi: 10.1080/01480545.2021.1979030
82. Foyet HS, Tchinda Deffo S, Koagne Yewo P, Antioch I, Zingue S, Asongalem EA, et al. Ficus sycomorus extract reversed behavioral impairment and brain oxidative stress induced by unpredictable chronic mild stress in rats. BMC Complement Altern Med. (2017) 17:502. doi: 10.1186/s12906-017-2012-9
83. da Silva AI, Monteiro Galindo LC, Nascimento L, Moura Freitas C, Manhaes-de-Castro R, Lagranha CJ, et al. Fluoxetine treatment of rat neonates significantly reduces oxidative stress in the hippocampus and in behavioral indicators of anxiety later in postnatal life. Can J Physiol Pharmacol. (2014) 92:330–7. doi: 10.1139/cjpp-2013-0321
84. Chen Z, Zhong C. Oxidative stress in Alzheimer’s disease. Neurosci Bull. (2014) 30:271–81. doi: 10.1007/s12264-013-1423-y
85. Patil SP, Jain PD, Sancheti JS, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective and neurotrophic effects of Apigenin and Luteolin in MPTP induced parkinsonism in mice. Neuropharmacology. (2014) 86:192–202. doi: 10.1016/j.neuropharm.2014.07.012
86. Kopańska M, Lukáč N, Kapusta E, Formicki G. Acrylamide influence on activity of acetylcholinesterase, thiol groups, and malondialdehyde content in the brain of swiss mice. J Biochem Mol Toxicol. (2015) 29:472–8. doi: 10.1002/jbt.21717
87. Zhao M, Wang P, Zhu Y, Liu X, Hu X, Chen F. The chemoprotection of a blueberry anthocyanin extract against the acrylamide-induced oxidative stress in mitochondria: unequivocal evidence in mice liver. Food Funct. (2015) 6:3006–12. doi: 10.1039/c5fo00408j
88. Zhang P, Pan H, Wang J, Liu X, Hu X. Telomerase activity-independent function of telomerase reverse transcriptase is involved in acrylamide-induced neuron damage. Biotech Histochem. (2014) 89:327–35. doi: 10.3109/10520295.2013.855323
89. Cosentino K, Garcia-Saez AJ. Mitochondrial alterations in apoptosis. Chem Phys Lipids. (2014) 181:62–75. doi: 10.1016/j.chemphyslip.2014.04.001
90. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
91. Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. (2014) 8:315. doi: 10.3389/fnins.2014.00315
92. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. (2010) 10:210–5. doi: 10.1038/nri2725
93. Sui X, Yang J, Zhang G, Yuan X, Li W, Long J, et al. NLRP3 inflammasome inhibition attenuates subacute neurotoxicity induced by acrylamide in vitro and in vivo. Toxicology. (2020) 432:152392. doi: 10.1016/j.tox.2020.152392
94. Luna RA, Foster JA. Gut brain axis: diet microbiota interactions and implications for modulation of anxiety and depression. Curr Opin Biotechnol. (2015) 32:35–41. doi: 10.1016/j.copbio.2014.10.007
95. Cryan JF, O’Mahony SM. The microbiome-gut-brain axis: from bowel to behavior. Neurogastroenterol Motil. (2011) 23:187–92. doi: 10.1111/j.1365-2982.2010.01664.x
96. Jiang Y, Li L, Liu B, Zhang Y, Chen Q, Li C. Vagus nerve stimulation attenuates cerebral ischemia and reperfusion injury via endogenous cholinergic pathway in rat. PLoS One. (2014) 9:e102342. doi: 10.1371/journal.pone.0102342
97. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. (2014) 6:263ra158. doi: 10.1126/scitranslmed.3009759
98. Tan X, Ye J, Liu W, Zhao B, Shi X, Zhang C, et al. Acrylamide aggravates cognitive deficits at night period via the gut-brain axis by reprogramming the brain circadian clock. Arch Toxicol. (2019) 93:467–86. doi: 10.1007/s00204-018-2340-7
99. Quigley EMM. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr Neurol Neurosci Rep. (2017) 17:94. doi: 10.1007/s11910-017-0802-6
100. Lopachin RM, Gavin T, Barber DS. Type-2 alkenes mediate synaptotoxicity in neurodegenerative diseases. Neurotoxicology. (2008) 29:871–82. doi: 10.1016/j.neuro.2008.04.016
101. Liu ZM, Tse LA, Chen B, Wu S, Chan D, Kowk T, et al. Dietary acrylamide exposure was associated with mild cognition decline among non-smoking Chinese elderly men. Sci Rep. (2017) 7:6395. doi: 10.1038/s41598-017-06813-9
102. Erkekoglu P, Baydar T. Acrylamide neurotoxicity. Nutr Neurosci. (2014) 17:49–57. doi: 10.1179/1476830513Y.0000000065
103. LoPachin RM, Gavin T. Molecular mechanism of acrylamide neurotoxicity: lessons learned from organic chemistry. Environ Health Perspect. (2012) 120:1650–7. doi: 10.1289/ehp.1205432
104. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. (2014) 14:463–77. doi: 10.1038/nri3705
105. Libby P, Everett BM. Novel Antiatherosclerotic Therapies. Arterioscler Thromb Vasc Biol. (2019) 39:538–45. doi: 10.1161/ATVBAHA.118.310958
106. Hanamsagar R, Torres V, Kielian T. Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem. (2011) 119:736–48. doi: 10.1111/j.1471-4159.2011.07481.x
107. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol. (2017) 18:861–9. doi: 10.1038/ni.3772
108. Wu AG, Zhou XG, Qiao G, Yu L, Tang Y, Yan L, et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev. (2021) 65:101202. doi: 10.1016/j.arr.2020.101202
109. Deng L. Study on the Mechanism Of Acrylamide-Induced Neurotoxicity. [Master’s thesis]. Shanghai: East China University of Science and Technology (2021).
110. Wang Y, Yin B, Li D, Wang G, Han X, Sun X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun. (2018) 495:1418–25. doi: 10.1016/j.bbrc.2017.11.156
111. Xia W, Li Y, Wu M, Jin Q, Wang Q, Li S, et al. Gasdermin E deficiency attenuates acute kidney injury by inhibiting pyroptosis and inflammation. Cell Death Dis. (2021) 12:139. doi: 10.1038/s41419-021-03431-2
112. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. (2005) 73:1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005
113. Foster JA, McVey-Neufeld KA. Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. (2013) 36:305–12. doi: 10.1016/j.tins.2013.01.005
114. Chen J, Li Y, Tian Y, Huang C, Li D, Zhong Q, et al. Interaction between Microbes and host intestinal health: modulation by dietary nutrients and gut-brain-endocrine-immune axis. Curr Protein Pept Sci. (2015) 16:592–603. doi: 10.2174/1389203716666150630135720
115. El Aidy S, Dinan TG, Cryan JF. Immune modulation of the brain-gut-microbe axis. Front Microbiol. (2014) 5:146. doi: 10.3389/fmicb.2014.00146
116. Forsythe P, Bienenstock J, Kunze WA. Vagal pathways for microbiome-brain-gut axis communication. Adv Exp Med Biol. (2014) 817:115–33. doi: 10.1007/978-1-4939-0897-4_5
117. Kuwahara A, Matsuda K, Kuwahara Y, Asano S, Inui T, Marunaka Y. Microbiota-gut-brain axis: enteroendocrine cells and the enteric nervous system form an interface between the microbiota and the central nervous system. Biomed Res. (2020) 41:199–216. doi: 10.2220/biomedres.41.199
118. Hu X, Wang T, Jin F. Alzheimer’s disease and gut microbiota. Sci China Life Sci. (2016) 59:1006–23. doi: 10.1007/s11427-016-5083-9
119. Sun MF, Shen YQ. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s Disease. Ageing Res Rev. (2018) 45:53–61. doi: 10.1016/j.arr.2018.04.004
120. Hagmar L, Tornqvist M, Nordander C, Rosen I, Bruze M, Kautiainen A, et al. Health effects of occupational exposure to acrylamide using hemoglobin adducts as biomarkers of internal dose. Scand J Work Environ Health. (2001) 27:219–26. doi: 10.5271/sjweh.608
121. Mulloy KB. Two case reports of neurological disease in coal mine preparation plant workers. Am J Industr Med. (1996) 30:56–61. doi: 10.1002/(sici)1097-0274(199607)30:13.0.Co;2-q
122. Skowron J. [Priority: safe working conditions]. Med Pr. (2019) 70:497–509. doi: 10.13075/mp.5893.00832
123. Brown L, Rhead M. Liquid chromatographic determination of acrylamide monomer in natural and polluted aqueous environments. The Analyst. (1979) 104:391–9. doi: 10.1039/an9790400391
124. Yang F, Li Z, Bian Z, Tang G, Fan Z, Wang Y, et al. Environmentally friendly method for the determination of acrylamide and trimethylolpropane in paper packaging materials by liquid chromatography with tandem mass spectrometry. J Sep Sci. (2014) 37:3625–31. doi: 10.1002/jssc.201400504
125. Faria M, Valls A, Prats E, Bedrossiantz J, Orozco M, Porta JM, et al. Further characterization of the zebrafish model of acrylamide acute neurotoxicity: gait abnormalities and oxidative stress. Sci Rep. (2019) 9:7075. doi: 10.1038/s41598-019-43647-z
Keywords: acrylamide, neurotoxicity, oxidative stress, apoptosis and autophagy, inflammation, gut-brain axis
Citation: Zhao M, Zhang B and Deng L (2022) The Mechanism of Acrylamide-Induced Neurotoxicity: Current Status and Future Perspectives. Front. Nutr. 9:859189. doi: 10.3389/fnut.2022.859189
Received: 21 January 2022; Accepted: 28 February 2022;
Published: 25 March 2022.
Edited by:
Shuai Chen, Wuhan University, ChinaReviewed by:
Marta Kopańska, University of Rzeszów, PolandYuchen Zhu, China Agricultural University, China
Yuan Yuan, Jilin University, China
Yahui Guo, Jiangnan University, China
Copyright © 2022 Zhao, Zhang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mengyao Zhao, bXl6aGFvQGVjdXN0LmVkdS5jbg==