
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr. , 20 January 2023
Sec. Nutritional Epidemiology
Volume 9 - 2022 | https://doi.org/10.3389/fnut.2022.1054089
 Kinga Skoracka1,2*
Kinga Skoracka1,2* Szymon Hryhorowicz3
Szymon Hryhorowicz3 Anna Maria Rychter1,2
Anna Maria Rychter1,2 Alicja Ewa Ratajczak1,2
Alicja Ewa Ratajczak1,2 Aleksandra Szymczak-Tomczak1Agnieszka Zawada1
Aleksandra Szymczak-Tomczak1Agnieszka Zawada1 Ryszard Słomski3Agnieszka Dobrowolska1
Ryszard Słomski3Agnieszka Dobrowolska1 Iwona Krela-Kaźmierczak1
Iwona Krela-Kaźmierczak1The prevalence of celiac disease increased in recent years. In addition to the genetic and immunological factors, it appears that environmental determinants are also involved in the pathophysiology of celiac disease. Gastrointestinal infections impact the development of celiac disease. Current research does not directly confirm the protective effect of natural childbirth and breastfeeding on celiac disease. However, it seems that in genetically predisposed children, the amount of gluten introduced into the diet may have an impact on celiac disease development. Also western lifestyle, including western dietary patterns high in fat, sugar, and gliadin, potentially may increase the risk of celiac disease due to changes in intestinal microbiota, intestinal permeability, or mucosal inflammation. Further research is needed to expand the knowledge of the relationship between environmental factors and the development of celiac disease to define evidence-based preventive interventions against the development of celiac disease. The manuscript summarizes current knowledge on factors predisposing to the development of celiac disease including factors associated with the western lifestyle.
Celiac disease (CD) is a systemic, T-cell mediated, autoimmune disorder that is triggered by exposure to dietary gluten in genetically predisposed individuals. This chronic disease is characterized by specific serum autoantibody response in IgG and IgA class: anti-transglutaminase IgA and anti-endomysial antibodies IgA and deamidated gliadin-related peptide IgA and IgG (1). Today, the only accepted treatment for CD is a strict gluten-free diet (2).
CD presents in about 1% of the population. Singh et al. in meta-analysis and systemic review reported that worldwide seroprevalence and prevalence of CD are 1.4 and 0.7% respectively. According to the authors, the prevalence of CD varies with sex, age, and country and it increased from 0.6% in 1991–2000 to 0.8% between 2001 and 2016 (3). It is supposed that the increase in diagnosed cases of CD is partly the result of better diagnostic tools and more frequent screening tests in individuals at risk. However, it seems that also environmental factors can contribute to that phenomenon (4). There are scientific reports, that besides genetic and immunological features, there are environmental factors that could trigger the development of CD (5). These factors (Figure 1) will be discussed in this paper.
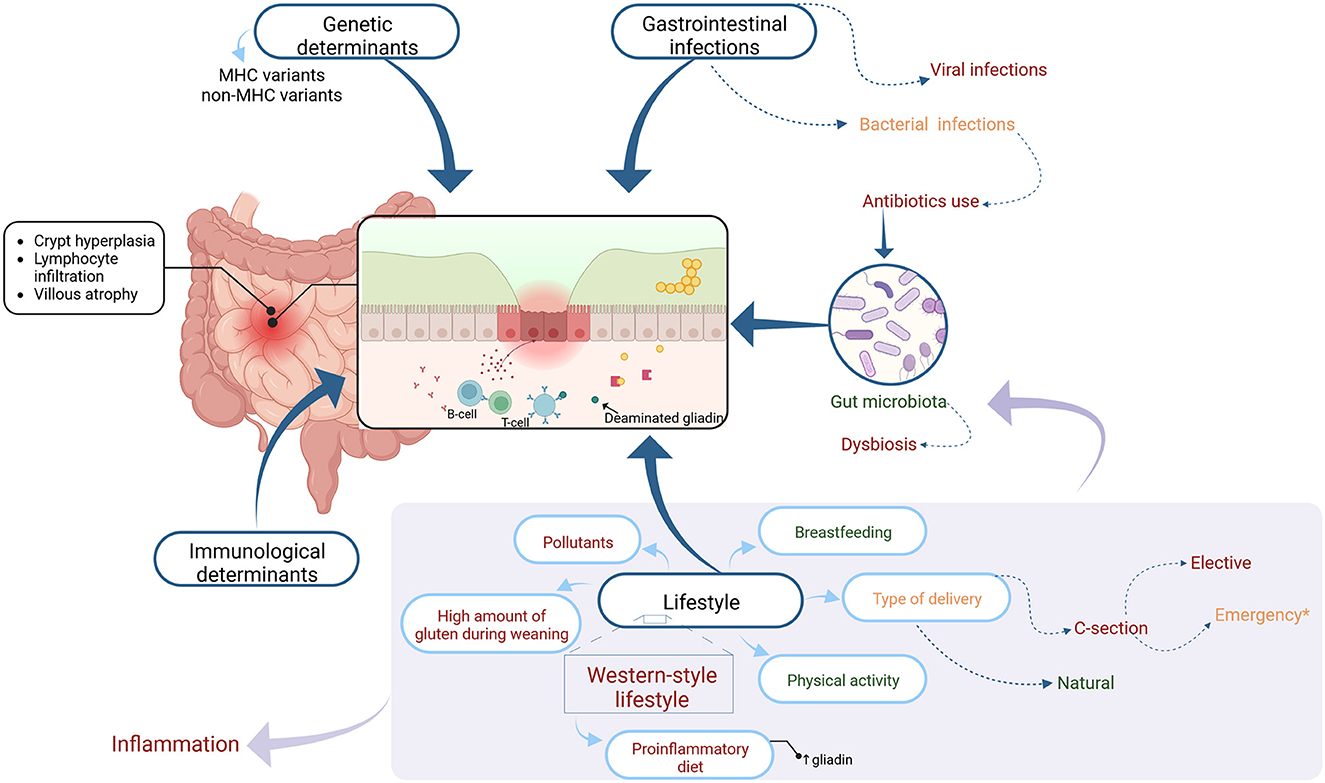
Figure 1. The figure shows the factors influencing and potentially influencing the development of celiac disease marked in color as appropriate: general factors-black, factors showing a potentially protective effect-green, factors negatively influencing the development of celiac disease-red, and factors that may have a dual effect-orange. Each group of factors is discussed in further detail in the text.
The data presented in this study are openly available in Medline and PubMed databases and on the publisher's website. The keywords that were used for the search: celiac disease; celiac disease genetics; celiac disease risk factors; celiac disease prevention; western lifestyle; western diet; autoimmunity; inflammation; Mediterranean diet; proinflammatory diet; viruses; bacteria; infection; gut microbiota; dysbiosis; antibiotics use; breastfeeding; physical activity; c-section; gluten proteins; gluten; gliadin.
Gluten is a complex of storage proteins found in grains such as wheat, rye, and barley. In wheat, gluten constitutes mainly alcohol-soluble gliadins and insoluble glutenins. It constitutes about 85% of total grain protein mass (6). In barley, the major storage proteins are alcohol-soluble hordeins, and in the rye—secalins (7). These proteins belong to a fraction called prolamins, and they are the main endosperm storage proteins in grains (7). Wheat is the most concentrated source of gluten. The wheat kernel is one of the major cereal crops worldwide, occupying 17% of the cultivated land and constituting a staple food for 35% of the population worldwide (8). Wheat constitutes a relevant part of the contemporary diet. According to WHO, it provides 19% of the total caloric input in the population. Global wheat production amounts to an average of around 750 million tons a year and the major producers of wheat are Europe, North America, and Asia (9).
Among prolamins, the two types of proteins in comparable amounts can be distinguished: gliadins and glutenins. Gliadin, due to the high content of proline residues, is resistant to gastric-pancreatic and intestinal proteases. As result, long gliadin fragments can reach high concentrations in the gut epithelium (10). Some of the undigested peptides derived from gliadin have been reported to activate T cell response while others have pleiotropic biological activity (11). α-gliadin 33-mer peptide, P57–89 is one of the most immunogenic gluten peptides (12). It has been suggested that α-gliadin 33-mer peptide could reach lamina propria, and presented by HLA-DQ2 or 8 to T cells, can be an activator of the adaptive immune response, playing a major role in the pathogenic cascade of CD (10). Another, relatively less immunogenic peptides are 25-mer peptide, P31–43, which is not presented by HLA-DQ to T cells, or P57–73, and P111–130 (13). Undigested peptides are active in vivo in the celiac intestine after gluten ingestion and induce an immune response with the production of INF-alpha and IL-15 in CD biopsies and fibroblasts (14, 15).
Even though the inheritance of celiac disease is still unknown, it has long been known that genetics is involved in the triggering and later development of the disease (16, 17). Celiac disease, like most autoimmune diseases, is strongly associated with human leukocyte antigens (HLA) regions. The role of the HLA region as a susceptibility factor was understood in 2008 with the first genome-wide association study of celiac disease (18). However, the first reports that a major histocompatibility complex (MHC) was related to CD came 50 years earlier with the identification of the disease's association with HLA-B8 (19, 20) accounting for about 40% of CD genetic variation.
In the following years, disease associations were proven with HLA-A1, HLA-DR3, and HLA-DR7, as well as with HLA-DQ2 (21–25) which are due to the strong coupling imbalance and the fact that these alleles are part of the conservative A1-B8-DR-DQ2 haplotype.
A crucial point in the diagnosis of celiac disease was the identification of MHC class II alleles: HLA-DQ2.5, HLA-DQ8, and HLA-DQ2.2 (26). It is estimated that about 90–95% of patients with celiac disease are carriers of HLA-DQ2.5 (DQA1*05:01, DQB1*02:01). Patients who do not express HLA-DQ2.5 usually express HLA-DQ2.2 (DQA1*02:01, DQB1*02:01) or HLA-DQ8 (DQA1*03, DQB1*03:02) (27, 28). For HLA-DQ2.5, the risk of celiac disease is particularly high by the fact that carriers of the DR3DQ2 haplotype encoded by DQA1 and DQB1 have both alleles on the same chromosome-cis configuration, while heterozygote carriers of the DR5DQ7/DR7DQ2 haplotype have alleles on opposite chromosomes, which is trans configuration (28, 29).
Genome-wide association studies have so far identified 43 genetic risk factors, in addition to MHC, that explain about 50% of the genetic variation in celiac disease (17). Currently we know relatively well what role HLA-DQ2 and -DQ8 play in celiac disease (28–30) but the involvement of other loci as the celiac disease determinants, not related to HLA, is still largely undiscovered. Only three of these loci have single nucleotide polymorphisms (SNPs) located in protein-coding regions (31). Most of them—more than 90% are SNPs located in non-coding—intergenic regions, where in T and B cells they are most probably responsible for deregulating the expression of essential genes involved in the pathogenesis of celiac disease (17).
Activation of gluten-specific CD4 T lymphocytes, which recognize gluten peptides, is the first factor initiating an immune response in celiac disease. Molbergu et al. showed that gliadin-specific CD4+ T cells are present only in the mucosa of patients with celiac disease (32). This supports the concept that most T cells recognize gliadin peptide(s) when present by binding to DQ2 (33). Villi atrophy in patients after gluten ingestion can occur even after years on a gluten-free diet, proving that activation of gluten-specific T cells is crucial to the onset and pathology of celiac disease. It has also been shown that gluten-specific T cells secrete many signaling molecules upon stimulation, including interleukin IL-2, IL-4, IL-6, IL-8, IL-10, IL-21, CD40LG, IFNγ, and TNF (34–40) with IL-4, IL-21, CD40LG, and CXCL13 (C-X-C motif chemokine 13), being important in the differentiation and activation of T cells and B cells (33, 41–43). From GWAS studies, we also know that genes SH2B3, TAGAP, PTPN2, CD28, CTLA4, CIITA, and IL2/IL21 are involved in T-cell activation. Interleukins IRAK1, IL12A, IL18RAP/IL18R1, IL1RL1/IL1RL2 (44) are in turn responsible for the differentiation of inflammatory T cells CD4, while genes IL21/IL2, RGS1, MAP3K7, CCR1,2,3, CCR4, UBASH3 are responsible for the migration of cytolytic effector T cells (44). It has also been documented that cytokines secreted by gluten-specific T cells are important for the activation and proliferation of CD8+ intraepithelial lymphocytes (IELs) in celiac disease. Thus, it follows that gluten-specific T cells play a key role in the response to gluten peptides, thereby leading to inflammation, anti-TG2 antibodies production, and atrophy of intestinal villi (45–47).
Alteration analyses of regulatory regions suggest that adaptor cells (gluten-specific T CD4 + cells and B cells) are strongly associated with celiac disease and that specific genes may contribute to the development of celiac disease through innate immune cells, epithelial cells, and D8+ TCRαβ intraepithelial cytotoxic lymphocytes (48, 49). In addition, mapping of expression quantitative trait loci (eQTL), signaling pathway analyses, and functional SNP analyses have established associations between celiac disease and T cell receptor (TCR), NFκB, and interferon (IFN) signaling pathways and several candidate genes like UBASH3A, CD28, CSK, CD274, SH2B3, and STAT4 (49–51). These reports confirm the role of CD4+ T cells in celiac disease but do not, however, define how single nucleotide changes associated with CD affect gluten-specific CD4 T cells upon activation, most likely due to an incompletely understood mechanism of regulation of the stimulation response in gluten-specific T cells. In a 2018 study, Harley et al. showed that transcription factors (TFs) occupy multiple loci associated with individual complex genetic disorders, including celiac disease encompassing STAT4, STAT5A, STAT5B, T-BET, and transcription factors from the NFκB pathway. While many of these are involved in the regulation of CD4+ T-cell activation (52–55), the role of TFs as well as the transcriptional and epigenetic response in gluten-specific T cells activation has not been described, nor has the role of CD-associated genetic variants in these dynamic processes.
Infections are among the environmental factors that may underline the etiopathogenesis of autoimmune diseases. The interdependencies between the influence of an infectious agent and the disease occurrence are usually complex and multidirectional. According to Lerner et al. infectious agents that could induce CD autoimmunity include Cytomegalovirus, Rotavirus, Enterovirus, Pneumococcus, Bacteroides species, Campylobacter Jejuni, Helicobacter Pylori, Mycobacterium tuberculosis, hepatitis B virus. Contrary to this, some studies show potential protective effects of Cytomegalovirus, Toxoplasma gondii, H. Pylori, Rubella, and Epstein-Barr virus. It should be noted that the results of the research on Cytomegalovirus and H. Pylori infections are contradictory (56).
Jiang et al. in a meta-analysis showed that infections are associated with a 37% increase in the odds of CD, particularly among hospitalized patients. The risk was not modulated by the time of infection, type of infectious agent, and site of infection (57). Moreover, patients with CD have an increased risk of infections. Possible mechanisms for this phenomenon include hyposplenism, malnutrition, vitamin D deficiency, increased mucosal permeability, and altered immune response determined by genetic risk factors (58, 59).
Given the role of IFN secreted by host cells upon viral infection (60) in the pathophysiology of celiac disease—this cytokine has been suggested to promote T helper type 1 (Th1) responses in CD and its high levels can be observed in the duodenal mucosa from celiac patients (61)—it is speculated that viral infections are greater than bacterial risk factors for CD.
The first observations on the relationship between viral infection and CD come from the 1980s. Kagnoff et al. postulated that viruses may play a role in the pathogenesis of CD via an immunological cross-reaction between antigenic determinants common to the viral protein and gluten. However, this mechanism has not been confirmed (62). That observation was the beginning of further research. Beyerlein et al. analyzed data on nearly 300,000 infants from Germany. They revealed that infections of the digestive tract in the first year of life correlate with a higher risk of CD later in life (63). Also, a prospective cohort study based on The Norwegian Mother and Child Cohort Study including 72,921 children suggests that early-life infections may have a role in CD development (64).
Kemppainen et al. made similar observations after analyzing the infection data from 6,327 children from USA and Europe that carry HLA risk genotypes for CD. The researchers showed an increased risk of CD in children with recurrent infections of the digestive and respiratory tracts. The risk was modified by HLA genotype, gluten consumption, breastfeeding, and rotavirus vaccination status, and what is important, the risk was lower in children vaccinated against rotavirus (65).
Moreover, interesting are observations of Lindfors et al. who investigated the correlation between viral exposures alone or together with gluten and the risk of CD autoimmunity in 83 genetically predisposed children whose stool samples were collected monthly up to 2 years of age. The authors observed an interaction between enteroviral exposure and gluten intake suggesting that infections early in life and high gluten intake may trigger CD development in genetically predisposed children. Furthermore, they observed that children consuming bigger amounts of gluten had a higher risk of CD autoimmunity. This indicates the cumulative effect of gluten consumption and enterovirus infections in children at-risk of CD (66).
The mechanism of action of viruses in the context of gluten intolerance is not fully understood. One of the hypotheses is that gastrointestinal infections can lead to changes in the permeability of the intestinal barrier, thus leading to increased gluten penetration. The structure of the virus may be similar to gluten, which in turn may lead to an initiated anti-gluten response. Kagnoff et al. and Lähdeaho et al. in their researches demonstrated the presence of antibodies to viral peptides (62, 67). Viruses from the Reoviridae family are segmented, double-stranded RNA viruses (dsRNA) that cause infections in humans of all ages (68). These infections are often asymptomatic. Specific reovirus infections stimulate characteristic inflammatory pathways, thus leading to T-cell responses to food antigens, which in turn impairs the development of tolerance to the food antigen (69). Reovirus infection stimulates interferon type 1 signaling and increases the expression of the interferon regulating factor 1 (IRF1) transcription factor. Then it blocks the transition of T cells to regulatory T cells and stimulates the Th1 response to food antigens which may lead to the development of celiac disease (70, 71).
Moreover, Assa et al. observed that individuals born in May and June present a higher risk of CD (72). It may be associated with rotavirus infection, which peak occurs in early winter-the same time as exposure to gluten in children born in May and June (72). Additionally, the risk of CD is higher in children, which had a gastrointestinal infection in the first year of life (63).
However, more research is needed to fully understand the importance of viral host interactions and infection in CD development.
Compared to the extensive, but constantly updated, knowledge of viral risk factors for CD, less is known about bacterial infections. The results of various studies on H. pylori infections both in children and adults are contradictory. In the meta-analysis Amlashi et al. observed a negative association between H. pylori and CD autoimmunity, implying a mild protective role of H. pylori against celiac disease (73). On the other hand, there are indications that Campylobacter can increase the risk of CD (74–76). The mechanism that could explain the relation between bacterial infections and CD is molecular mimicry (77).
However, antibiotics seem to increase the risk of CD through their negative effect on gut microbiota (78). Microbiome evolves during the first years of children's lives to stabilize at 3 years of age and present microbial composition similar to adult microbiota with the dominance of Firmicutes and Bacteroidetes phyla (79). Healthy gut microbiota allows for the maturation of the immune system and plays a role in the degradation of gluten in the gastrointestinal tract, affecting this way the immunogenicity of gluten peptides (78).
In meta-analysis higher doses of antibiotics correlated with a higher risk of CD. Yet, after subgroup analysis, only penicillin V was found to increase the risk of CD (57). An observational study of two nationwide cohorts including more than 1.7 million children from Denmark and Norway, among which 3,346 were diagnosed with CD, showed a positive association between antibiotic use in the first year of life with a diagnosis of CD. This relation was dose-dependent (78). Contrary to these outcomes, a systematic review analyzing data from six studies, showed no evidence of an association between prenatal or postnatal antibiotic exposure and CD (80).
In the past 10 years, the global rate of cesarean section has increased to 21%, and every year it increases by 4%. In some regions of Africa, the rate of cesarean section is 4%, in Canada, the c-section rate is 27.1%, and in some countries of Latin America almost 60% (81, 82).
Children delivered by cesarean are exposed to different microbial ligands and bacterial colonization when compared to neonates born vaginally (83). Here, it is worth recalling the Danish, population-based cohort study on two million infants. The analysis showed that there is no association between cesarean delivery and CD but at the same time, cesarean delivery was positively correlated with such diseases as asthma, inflammatory bowel disease, or immune deficiencies (84). Moreover, Koletzko et al. reported a lack of association between cesarean delivery and the risk of CD or CD autoimmunity in 6,087 genetically predisposed children (85). On the other hand, a multicentre, case-control study showed, that the type of delivery may affect gut microbiota and enhance the risk of CD (86). Finally, in 2022 Yang et al. performed a meta-analysis of 11 observational studies and found that C-section is not associated with CD in offspring (87).
However, it is worth noting the study by Mårild et al. who observed that elective, but not an emergency, cesarean delivery was associated with a higher risk of CD (88). In this type of cesarean section, the birth must occur before the onset of labor thus children do not have initial contact with maternal vaginal microbiota in the birth canal. Moreover, mothers undergoing elective cesarean birth and those with an indication for scheduled deliveries are also more likely to receive antibiotic prophylaxis compared with mothers undergoing emergency C-sections. So perhaps it would be more valuable to study the impact of the antibiotic therapy used at C-section and the type of C-section rather than the impact of C-section overall on the development of CD.
Low- and middle-income countries generally have high rates of breastfeeding initiation of over 90%, while in high-income countries, there is a wide variation of breastfeeding initiation—countries such as Italy, Australia, or Nordic countries report rates of over 90%, while France, UK, USA, Republic of Ireland report lower rates (89). Breast milk is a complete source of nutrients for newborns, but also contains immunogenic molecules, which are immune active and affect the immune response of mucosal. Therefore, for a long time, there were suggestions, that breastfeeding may protect against CD—a meta-analyse performed in the 2006 year showed, that breastfeeding during gluten exposure reduced the risk of CD when compared with the exposition to gluten after stopping breastfeeding (90). Yet actual data do not show evidence for the preventing impact of breastfeeding. In a study performed by Lionetti et al. breastfeeding did not affect the risk of CD among children at-risk (91). Similar observations had Vriezinga et al. in the multicenter study (92). According to a systematic review by Silano et al., there is no evidence that breastfeeding duration can impact CD development in predisposed children (93). Finally, Szajewska et al. in a meta-analysis including 21 publications, showed that breastfeeding does not affect the development of CD (94).
Besides, it is worth considering the quantity and timing of gluten introduction. Andrén Aronsson et al. also showed that the age of first gluten exposition is not an independent risk factor of CD (95). Similarly, in an earlier mentioned study by Lionetti et al. delayed introduction of gluten did not affect the risk of CD (91). Vriezinga et al. observed that the introduction of gluten between 16 and 24 weeks of age did not decrease the risk of CD by 3 years of age (92). In a meta-analysis performed by Szajewska et al. time of gluten introduction also did not impact CD development (94).
However, greater amounts of gluten seem to increase the risk of CD in genetically predisposed individuals. This evidence brought three separate cohort studies published in 2019 by Andrén-Aronsson et al. (96), Lund-Blix et al. (97), Mårild et al. (98). Andrén-Aronsson et al. followed for about 9 years 6,605 children. They observed that in genetically predisposed children, 20.7% of those who consumed the reference amount of gluten developed CD at 3 years. Among the group of children that consumed daily 1 g more of gluten, CD developed in 27.9% of children (96). Mårild et al. revealed that 1-year-olds with the highest gluten intake had a 2-fold greater hazard of CD, and the incidence of CD increased with higher cumulative gluten intake throughout childhood (98). Finally, Lund-Blix et al. conducted population-based research on 67,608 children independent of HLA. Increased gluten intake at 18 months was associated with a modestly increased risk of CD later in childhood. The association with gluten amount was independent of the age at the introduction of gluten. Gluten introduction ≥6 months was also an independent risk factor for CD (97). Also, the latest study by Aurrichio et al. showed that among 83 children predisposed to CD, 27 children who developed CD had eaten significantly more gluten in the second year of life than the remaining children who did not develop CD by 6 years of age (15).
Some of the studies cited in the article indicate the cumulative effects of individual factors. For example, in the study by Lindfors et al. there is an interaction between enteroviral exposures and higher gluten intake early in life indicating cumulative effects of these factors in CD development (66). Herein, we can find potential relation between increased gut permeability as an outcome of viral infection and subsequent gluten penetration. A similar relationship was observed in another incident case-referent study—it was observed that frequent infectious episodes in early life increase the risk of later celiac disease. Furthermore, there was a synergistic effect between gastrointestinal infections and the amount of daily gluten consumption, especially among infants for whom breastfeeding had been discontinued prior to gluten introduction (99).
It seems that viruses and gluten consumption may share a common pathway—a vesicular pathway regulating the innate/inflammatory response to viral ligands and bioactive dietary peptides—and trigger CD autoimmunity. The mechanism of this relation could be explained by gliadin peptide P31–43 activation of the INF alpha-mediated immune response to viruses in intestinal cells and an enterocyte cell line, CaCo-2 (14). The P31–43 peptide shows similar sequence homology with hepatocyte growth factor-regulated tyrosine kinase (HRS), which is a major regulator of endocytic vesicle maturation. Silencing or mutation of this protein contributes to delayed vesicle transport and activation of several different pathways. P31–43, therefore, delays endocytic movement in the same way as “altered” HRS leading to the same biological effects, including activation of inflammatory pathways (100).
To conclude it is worth recalling the ESPGHAN recommendations published in the 2016 year (101). Experts state that there is no evidence that neither breastfeeding nor breastfeeding during gluten introduction reduces the risk of CD. It is recommended to introduce gluten into infants' diet between 4 and 12 completed months of age—the time of gluten introduction to diet does not have an impact on overall CD development, yet the earlier introduction of gluten is associated with earlier development of CD autoimmunity. There is no recommendation regarding the type and the amount of gluten to be used at introduction but ESPGHAN suggests avoiding large quantities of gluten during the first month after gluten introduction and during infancy (101).
Western-style diet's (WD) characteristic (Figure 2) is strongly associated with calorie-dense foods, rich in saturated fatty acids, simple carbohydrates, and animal proteins (102, 103). The term is commonly used but there is no scientifically accepted definition of WD (104). Its relationship with non-communicable diseases (NCDs), e.g., obesity or diabetes is highly discussed (105). However, there is generally little known about the impact of a western-style diet on CD. Although several causal links could be suggested, it has not been widely investigated. The possible influence of WD on CD could be associated with changes in intestinal microbiota, mucosal inflammation, and an increase in intestinal permeability further leading to bacterial translocation, endotoxemia, and systemic low-grade inflammation (106–108). WD's influence on CD seems important, as—according to the current data—its role in the pathogenesis and clinical course of other intestinal diseases, such as inflammatory bowel disease (IBD), is also suggested and investigated (109). Moreover, diet and general lifestyle are major factors of chronic NCDs and can harm immune responses and stimulate the development of a variety of inflammatory diseases (100, 110). According to the Christ et al. study, NLRP3 mediates trained immunity following a western-style diet and thus could mediate the potentially deleterious effects of trained immunity in inflammatory diseases (111).
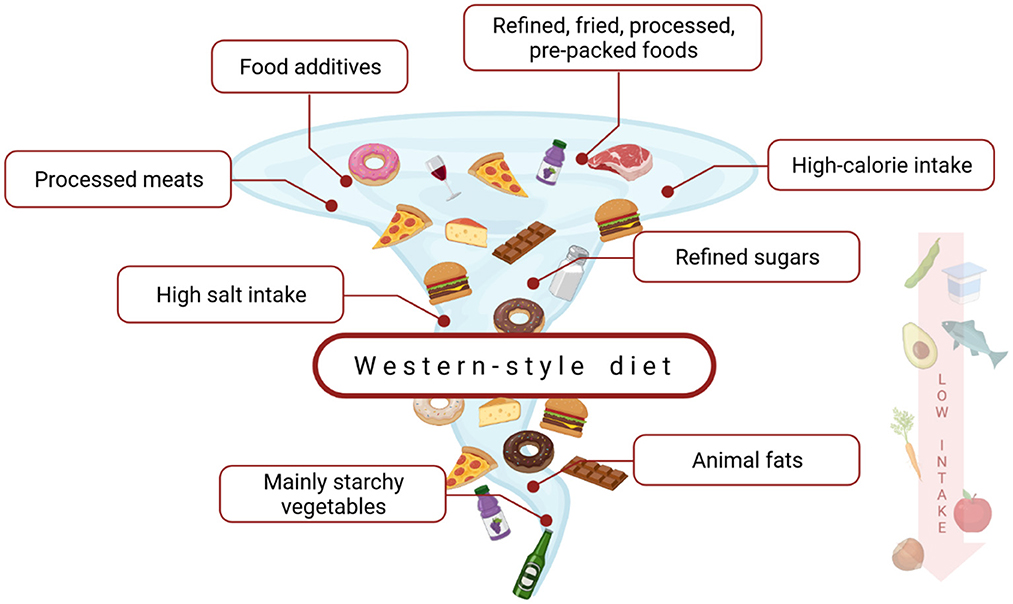
Figure 2. The characterization of a western-style diet. This dietary model is a high-caloric diet, rich in saturated fats, refined carbohydrates, animal protein, salt, and food additives. At the same time, it is low in fiber, vitamins, and minerals (102, 103).
The intestinal barrier comprises the mucus layer, intestinal epithelial cells, immune cells, gut microbiota, tight junctions located in the epithelial layer of the intestinal wall—which regulates the intestinal integrity—and gut-associated lymphoid tissue (GALT) (112). Any disruption in the components of the intestinal barrier—the physical barrier to luminal inflammatory molecules—increases its permeability, which can further implicate, e.g., inflammation in various tissues, including intestinal tissues (102, 106, 113, 114). Dietary fats expose the intestinal barrier and enhance the permeation of luminal contents into the mucosal and submucosal layers in proximity to resident immune cells, which promotes mucosal inflammation (115, 116). Moreover, high-fat diets (HFD) can enhance lipopolysaccharide (LPS) uptake and increase concentrations of bacteria-derived LPS in the circulatory system (117). Furthermore, increased permeability can also be driven by HFD-dysbiosis (118). Impaired mucosal integrity, which results in microbial translocation and increased concentrations of LPS in the bloodstream (LPS can also enter the circulation after incorporation into bile acid micelles) can lead to endotoxemia, and further low-grade systemic inflammation (119). Further, receptors such as NOD-like, C-type lectin, or toll-like receptors (TLR4) activate the NF-κB pathway increasing pro-inflammatory response and increasing concentrations of pro-inflammatory cytokines and chemokines (120). Within a short time after ingestion of saturated fats, TLR4 is downregulated and LPS translocation is increased (121).
Besides a high amount of fat, WD is also rich in simple carbohydrates, which can potentially and negatively affect intestinal permeability, possibly due to intestinal dysbiosis (122). In the study of Fajstova et al., mice fed with a high-sucrose diet had increased intestinal permeability (without damaged colon mucosa) and a higher state of immune system activation [increased proportion of neutrophils and T helper 17 cells (Th17) in the spleen, and increased proportion of Ly6Clow, Lyc6Chigh, and macrophages in colon tissue] when compared with mice fed with control diet (120). As the authors highlighted, the negative effect of a high-sucrose diet on intestinal inflammation was not only associated with transferred microbiota but was also dependent on TLR4 signaling. Similar results were obtained by Laffin et al. where mice fed with a high-sugar diet (50% of sucrose) had increased permeability and pro-inflammatory cytokines concentrations, impaired gut microbiota, and reduced short-chain fatty acids (SCFA), and increased susceptibility to colitis (123). Frequent and high intake of animal protein is also specific for WD. However, it remains unknown how and if WD through high-protein content can affect intestinal permeability. Oddly enough, an animal study by Zhu et al. suggests that low protein intake can negatively affect the intestinal barrier (124). Possibly, animal protein can even have a positive influence on intestinal permeability, due to the high content of glutamine and tryptophan (125, 126). However, WD-derived meat is usually highly processed, and therefore, high content of saturated and trans fatty acids can shift the balance in favor of the negative influence of WD-derived meat. Further, WD is also generally low in fresh fruits and vegetables and therefore is low in anti-inflammatory and antioxidant nutrients (e.g., polyphenols, minerals, or vitamins), which additionally predisposes to low-grade chronic inflammation and possibly increased intestinal permeability (127, 128). For example, in in vitro study by Amasheh et al., quercetin restored TNF-α-induced intestinal permeability by downregulating claudin-2 concentrations (129, 130). A low intake of fruits and vegetables may result in a low intake of dietary fiber, which, further, predisposes to impaired intestinal barrier through reduced SCFA production and negative changes in intestinal microbiota (131).
It has been proven that anti-inflammatory diets based on the Mediterranean diet (MD) models can reduce the risk of inflammatory diseases, but still there are a lot of questions about this relation (132–134). Barroso et al. in the prospective analysis of data from the Generation R Study including 1997 children, examined associations between dietary patterns of children at 1 year with the occurrence of CD autoimmunity at 6 years. There was a correlation between the consumption of a diet that based on vegetables and grains and low amounts of refined cereals and sweet beverages, with lower odds of CD autoimmunity. These outcomes suggest that early-life dietary patterns can have an impact on CD development (135). Furthermore, Auricchio et al. conducted a study on a cohort of 239 CD genetically predisposed infants and suggested that not only the amount of gluten consumed is an important factor influencing CD development, but also dietary patterns modulate this risk. Twenty-seven children who developed CD by the age of 6 years had different dietary patterns than the remaining 56 controls, i.e., predisposed children who did not develop the disease. CD children consumed greater amounts of carbohydrates, particularly starch and sugars, and lower amounts of legumes, vegetables, fruits, and milk products. This study suggests that children who developed CD consumed a diet that was more similar to the pro-inflammatory WD than to the anti-inflammatory Mediterranean diet (15).
Important for the health of the immune system is physical activity, which may affect the production of pro-inflammatory cytokines and inflammation that underline the development of many diseases (136). The animal study showed that physical activity increased the Th17 level, which plays important role in autoimmune diseases, including CD (137, 138). Moreover, physical activity decreases the risk of autoimmune diseases. The study showed that physical activity increases Th1 production and interleukin 6 (Il-6) in muscle, which induces an anti-inflammatory response (139). However, about 31% of the worldwide population is inactive (140), which also may be a factor responsible for the increasing incidence of autoimmune diseases. Furthermore, low levels of physical activity increase the risk of being overweight and obese. The prevalence of overweight and obesity in patients with CD increases and varies from 6 to 39% and from 3 to 13%, respectively (141–145). Obesity promotes chronic, systemic low-grade inflammation and is linked to increased production of pro-inflammatory cytokines and chemokines through endocrinal active white adipose tissue (146). Considering the increasing prevalence of both CD and obesity, studies contemplating obesity as one of the risk factors for developing CD would be interesting.
Gaylord et al. analyzed persistent organic pollutant (POP) exposure in a cohort of 30 children aged 3–12 years with CD and 58 non-celiac patients aged 5–11 years presenting with gastrointestinal complaints. Authors observed the statistically significant association of POPs, especially p,p′-dichlorodiphenyldichloroethylene (DDE) with the manifestation and development of celiac disease (147). What is interesting, the meta-analysis by Wijarnpreecha et al. shows that current smokers have a significantly decreased risk of developing CD compared with never-smokers (148).
WD changes intestinal microbiota composition and increases the risk of gut dysbiosis which conversely is linked to a higher risk of CD (108, 114). The intestinal microbiota of people consuming WD is characterized by increased Firmicutes: Bacteroidetes ratio (105).
What is interesting, it seems that the HLA genotype impacts gut colonization (149). Herein, Olivares et al. observed in a cohort of healthy infants with at least one first-degree relative with CD, that infants carrying the HLA-DQ-2 haplotype have different microbiota composition than non-HLA-DQ-2/8-carriers. Children with a high risk of CD had increased Firmicutes and Proteobacteria proportions but reduced Actinobacteria proportions compared to those with low genetic risk (150). HLA-DQ-2 carriers had significantly less Bifidobacterium and unclassified Bifidobacteriaceae proportions and more Corynebacterium, Gemella, Clostridium, unclassified Clostridiaceae, unclassified Enterobacteriaceae and Raoultella proportions (150). On the other hand, gut microbiota alterations seem to contribute to the development of CD (108). In the same cohort, Olivares et al. observed that the diversity of gut microbiota in healthy children increased over time—increases in Firmicutes families. Those who remained healthy showed increases in TNF-α correlated to Bifidobacterium spp. and increased relative abundance of Bifidobacterium longum. Increased proportions of Bifidobacterium breve and Enterococcus spp. and reduction in secretory IgA (sIgA) levels were associated with CD development (151). Ou et al. characterized the microbiota of the small intestine in children with CD and controls. They observed that a significant fraction constituted rod-shaped bacteria—Clostridium spp., Prevotella spp., and Actinomyces spp.,—suggesting that it could be an important risk factor for CD, contributing to the increase in disease incidence in children below 2 years of age during Swedish CD epidemic (152).
Actual data are insufficient to compose evidence-based recommendations for CD prevention. Yet, considering the latest findings, we can present the following directions.
First, we underline the role of the Mediterranean diet as a possible preventive approach to CD (15, 135). MD is characterized by a high intake of vegetables including legumes, olive oil, fresh, seasonal fruits, daily intake of whole grains, and regular consumption of nuts and seeds. Furthermore, according to MD's dietary pattern, fish should be consumed 2–3 times a week, dairy several times a week in moderate amounts, and meat should be consumed in moderate amounts. In addition to dishes should be used herbs and spices, and sweets should be limited. Three to four eggs per week and a moderate intake of wine can also be part of the MD diet (153). MD has the potential of a gut microbiota modulator, since consumption of MD increases the production of favorable bacteria and their metabolites, whereas dysbiosis and LPS levels decrease. In contrast to occidental dietary patterns, MD is associated with greater gut microbiota diversity and better function and integrity of the gut barrier (105).
Furthermore, taking into account the outcomes of several studies indicating the impact of viral infections on CD development, we suggest considering the preventive role of vaccination (65). Finally, it seems that in the prevention of CD bigger quantities of gluten should be avoided (101). Moreover, the latest studies indicate the cumulative effect of gluten and enterovirus infections, so it seems that especially during gastrointestinal infections gluten intake should be limited (66).
In conclusion, apart from genetic and immunological factors, environmental determinants play an important role in the pathogenesis of CD. Viral infections in early life seem to be important agents predisposing to CD development. On the other hand, current research does not confirm the protective effect of natural childbirth and breastfeeding on celiac disease. Regarding gluten introduction to an infant's diet—evidence shows that the time of gluten introduction does not have an impact on overall CD development but it seems that greater amounts of gluten can increase the risk of CD in genetically predisposed individuals. Furthermore, a western lifestyle, including a high-fat and high-sugar diet and a diet high in gluten, particularly gliadin, may be associated with a higher prevalence of CD. The predisposing effect of a WD on the development of CD may be related to changes in intestinal microbiota, intestinal permeability, or mucosal inflammation. Further research is needed to expand the knowledge of the relationship between environmental factors and the development of CD. Further validated studies are needed to define evidence-based preventive interventions against the development of CD.
This manuscript summarizes current knowledge of the environmental factors predisposing to celiac disease development.
IK-K: conceptualization. KS, SH, AMR, AER, AS-T, and AZ: writing—original draft. AMR: graphic design. IK-K, RS, AD, and KS: revision and edition of the whole manuscript. All authors have read and agreed to the present version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
CD, celiac disease; dsRNA, double-stranded RNA viruses; eQTL, expression quantitative trait loci; FAO, Food and Agriculture Organization; GALT, gut-associated lymphoid tissue; GWAS, genome-wide association studies; HFD, high-fat diet; HLA, human leukocyte antigen; HMW-GS, high molecular weight glutenin subunits; IBD, inflammatory bowel disease; IL-2, interleukin 2; IL-6, interleukin 6; IL-10, interleukin 20; IFN, interferon; IRF1, interferon regulating factor 1; LMW-GS, low molecular weight glutenin subunits; LPS, lipopolysaccharide; lncRNA, long non-coding RNA; miRNAs, microRNAs; MHC, major histocompatibility complex; MD, Mediterranean diet; NCDs, non-communicable diseases; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SCFA, short-chain fatty acids; sIgA, secretory IgA; SNPs, single nucleotide polymorphisms; TCR, and T cell receptor; TFs, transcription factors; Th1, T helper 1 cells; Th17, T helper 17 cells; TLR4, toll-like receptors; TNF, tumor necrosis factor; WD, Western-style diet; WHO, World Health Organization.
1. Lebwohl B, Sanders DS, Green PHR. Coeliac disease. Lancet Lond Engl. (2018) 391:70–81. doi: 10.1016/S0140-6736(17)31796-8
2. Caio G, Volta U, Sapone A, Leffler DA, De Giorgio R, Catassi C, et al. Celiac disease: a comprehensive current review. BMC Med. (2019) 17:142. doi: 10.1186/s12916-019-1380-z
3. Singh P, Arora A, Strand TA, Leffler DA, Catassi C, Green PH, et al. Global prevalence of celiac disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol. (2018) 16:823–36.e2. doi: 10.1016/j.cgh.2017.06.037
4. King JA, Jeong J, Underwood FE, Quan J, Panaccione N, Windsor JW, et al. Incidence of celiac disease is increasing over time: a systematic review and meta-analysis. Am J Gastroenterol. (2020) 115:507–25. doi: 10.14309/ajg.0000000000000523
5. Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol. (2013) 13:294–302. doi: 10.1038/nri3407
6. Wheat Gluten Protein Its Impacts on Wheat Processing Quality (n.d.). Available online at: https://journal.hep.com.cn/fase/EN/10.15302/J-FASE-2019267 (accessed February 28, 2022).
7. Balakireva AV, Zamyatnin AA. Properties of gluten intolerance: gluten structure, evolution, pathogenicity and detoxification capabilities. Nutrients. (2016) 8:644. doi: 10.3390/nu8100644
8. IDRC. Facts & Figures on Food and Biodiversity. International Development Research Centre (n.d.). Available online at: https://www.idrc.ca/en/research-in-action/facts-figures-food-and-biodiversity (accessed December 6, 2021).
9. Grote U, Fasse A, Nguyen TT, Erenstein O. Food security and the dynamics of wheat and maize value chains in Africa and Asia. Front Sustain Food Syst. (2021) 4:317. doi: 10.3389/fsufs.2020.617009
10. Barone MV, Zimmer KP. Endocytosis and transcytosis of gliadin peptides. Mol Cell Pediatr. (2016) 3:8. doi: 10.1186/s40348-015-0029-z
11. Barone MV, Salvatore A. Pro-inflammatory nutrient: focus on gliadin and celiac disease. Int J Mol Sci. (2022) 23:5577. doi: 10.3390/ijms23105577
12. Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, et al. Structural basis for gluten intolerance in celiac sprue. Science. (2002) 297:2275–9. doi: 10.1126/science.1074129
13. Yoosuf S, Makharia GK. Evolving therapy for celiac disease. Front Pediatr. (2019) 7:193. doi: 10.3389/fped.2019.00193
14. Nanayakkara M, Lania G, Maglio M, Auricchio R, De Musis C, Discepolo V, et al. P31–43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: a role in celiac disease. Sci Rep. (2018) 8:10821. doi: 10.1038/s41598-018-28830-y
15. Auricchio R, Calabrese I, Galatola M, Cielo D, Carbone F, Mancuso M, et al. Gluten consumption and inflammation affect the development of celiac disease in at-risk children. Sci Rep. (2022) 12:5396. doi: 10.1038/s41598-022-09232-7
16. Hunt KA, van Heel DA. Recent advances in coeliac disease genetics. Gut. (2009) 58:473–6. doi: 10.1136/gut.2008.155879
17. Withoff S, Li Y, Jonkers I, Wijmenga C. Understanding celiac disease by genomics. Trends Genet. (2016) 32:295–308. doi: 10.1016/j.tig.2016.02.003
18. Hunt KA, Zhernakova A, Turner G, Heap GAR, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. (2008) 40:395–402. doi: 10.1038/ng.102
19. Falchuk ZM, Rogentine GN, Strober W. Predominance of histocompatibility antigen HL-A8 in patients with gluten-sensitive enteropathy. J Clin Invest. (1972) 51:1602–5. doi: 10.1172/JCI106958
20. Stokes PL, Holmes GKT, Asquith P, Mackintosh P, Cooke WT. Histocompatibility antigens associated with adult cœliac. disease. Lancet. (1972) 300:162–4. doi: 10.1016/S0140-6736(72)91330-X
21. Betuel H, Gebuhrer L, Descos L, Percebois H, Minaire Y, Bertrand J. Adult celiac disease associated with HLA-DRw3 and -DRw7. Tissue Antigens. (1980) 15:231–8. doi: 10.1111/j.1399-0039.1980.tb00912.x
22. DeMarchi M, Borelli I, Olivetti E, Richiardi P, Wright P, Ansaldi N, et al. Two HLA-D and DR alleles are associated with coeliac disease. Tissue Antigens. (1979) 14:309–16. doi: 10.1111/j.1399-0039.1979.tb00854.x
23. Keuning JJ, Peña AS, van Leeuwen A, van Hooff JP, va Rood JJ. HLA-DW3 associated with coeliac disease. Lancet Lond Engl. (1976) 1:506–8. doi: 10.1016/S0140-6736(76)90294-4
24. Solheim BG, Albrechtsen D, Thorsby E, Thune P. Strong association between an HLA-Dw3 associated B cell alloantigen and dermatitis herpetiformis. Tissue Antigens. (1977) 10:114–8. doi: 10.1111/j.1399-0039.1977.tb01126.x
25. Tosi R, Vismara D, Tanigaki N, Ferrara GB, Cicimarra F, Buffolano W, et al. Evidence that celiac disease is primarily associated with a DC locus allelic specificity. Clin Immunol Immunopathol. (1983) 28:395–404. doi: 10.1016/0090-1229(83)90106-X
26. Sollid LM. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics. (2017) 69:605–16. doi: 10.1007/s00251-017-0985-7
27. Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, et al. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol. (2003) 64:469–77. doi: 10.1016/S0198-8859(03)00027-2
28. Sollid LM, Qiao S-W, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics. (2012) 64:455–60. doi: 10.1007/s00251-012-0599-z
29. Kim C-Y, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci USA. (2004) 101:4175–9. doi: 10.1073/pnas.0306885101
30. Petersen J, Montserrat V, Mujico JR, Loh KL, Beringer DX, van Lummel M, et al. T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat Struct Mol Biol. (2014) 21:480–8. doi: 10.1038/nsmb.2817
31. Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. (2011) 43:1193–201. doi: 10.1038/ng.998
32. Molberg O, Kett K, Scott H, Thorsby E, Sollid LM, Lundin KE. Gliadin specific, HLA DQ2-restricted T cells are commonly found in small intestinal biopsies from coeliac disease patients, but not from controls. Scand J Immunol. (1997) 46:103–9. doi: 10.1046/j.1365-3083.1997.d01-93.x-i2
33. Christophersen A, Risnes LF, Bergseng E, Lundin KEA, Sollid LM, Qiao S-W. Healthy HLA-DQ2.5+ subjects lack regulatory and memory T cells specific for immunodominant gluten epitopes of celiac disease. J Immunol. (2016) 196:2819–26. doi: 10.4049/jimmunol.1501152
34. Gianfrani C, Levings MK, Sartirana C, Mazzarella G, Barba G, Zanzi D, et al. Gliadin-specific type 1 regulatory T cells from the intestinal mucosa of treated celiac patients inhibit pathogenic T cells. J Immunol. (2006) 177:4178–86. doi: 10.4049/jimmunol.177.6.4178
35. Bodd M, Ráki M, Tollefsen S, Fallang LE, Bergseng E, Lundin KEA, et al. HLA-DQ2-restricted gluten-reactive T cells produce IL-21 but not IL-17 or IL-22. Mucosal Immunol. (2010) 3:594–601. doi: 10.1038/mi.2010.36
36. Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut. (1995) 37:766–76. doi: 10.1136/gut.37.6.766
37. Brottveit M, Beitnes A-CR, Tollefsen S, Bratlie JE, Jahnsen FL, Johansen F-E, et al. Mucosal cytokine response after short-term gluten challenge in celiac disease and non-celiac gluten sensitivity. Am J Gastroenterol. (2013) 108:842–50. doi: 10.1038/ajg.2013.91
38. Lahat N, Shapiro S, Karban A, Gerstein R, Kinarty A, Lerner A. Cytokine profile in coeliac disease. Scand J Immunol. (1999) 49:441–6. doi: 10.1046/j.1365-3083.1999.00523.x
39. Sjöberg V, Sandström O, Hedberg M, Hammarström S, Hernell O, Hammarström M-L. Intestinal T-cell responses in celiac disease - impact of celiac disease associated bacteria. PLoS ONE. (2013) 8:e53414. doi: 10.1371/journal.pone.0053414
40. Kooy-Winkelaar YMC, Bouwer D, Janssen GMC, Thompson A, Brugman MH, Schmitz F, et al. CD4 T-cell cytokines synergize to induce proliferation of malignant and nonmalignant innate intraepithelial lymphocytes. Proc Natl Acad Sci USA. (2017) 114:E980–9. doi: 10.1073/pnas.1620036114
41. Tangye S. Cytokine-mediated regulation of plasma cell generation: IL-21 takes center stage. Front Immunol. (2014) 5:65. doi: 10.3389/fimmu.2014.00065
42. Vasquez H, Mazure R, Gonzalez D, Flores D, Pedreira S, Niveloni S, et al. Risk of fractures in celiac disease patients: a cross-sectional, case-control study. Am J Gastroenterol. (2000) 95:183–9. doi: 10.1111/j.1572-0241.2000.01682.x
43. Mesin L, Sollid LM, Di Niro R. The intestinal B-cell response in celiac disease. Front Immunol. (2012) 3:313. doi: 10.3389/fimmu.2012.00313
44. Jabri B, Sollid LM. T cells in celiac disease. J Immunol. (2017) 198:3005–14. doi: 10.4049/jimmunol.1601693
45. Meresse B, Malamut G, Cerf-Bensussan N. Celiac disease: an immunological jigsaw. Immunity. (2012) 36:907–19. doi: 10.1016/j.immuni.2012.06.006
46. Korneychuk N, Ramiro-Puig E, Ettersperger J, Schulthess J, Montcuquet N, Kiyono H, et al. Interleukin 15 and CD4+ T cells cooperate to promote small intestinal enteropathy in response to dietary antigen. Gastroenterology. (2014) 146:1017–27. doi: 10.1053/j.gastro.2013.12.023
47. Zorro MM, Aguirre-Gamboa R, Mayassi T, Ciszewski C, Barisani D, Hu S, et al. Tissue alarmins and adaptive cytokine induce dynamic and distinct transcriptional responses in tissue-resident intraepithelial cytotoxic T lymphocytes. J Autoimmun. (2020) 108:102422. doi: 10.1016/j.jaut.2020.102422
48. Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. (2015) 518:337–43. doi: 10.1038/nature13835
49. van der Graaf A, Zorro MM, Claringbould A, Võsa U, Aguirre-Gamboa R, Li C, et al. Systematic prioritization of candidate genes in disease loci identifies TRAFD1 as a master regulator of IFNγ signaling in celiac disease. Front Genet. (2020) 11:562434. doi: 10.3389/fgene.2020.562434
50. Kumar V, Wijmenga C, Xavier RJ. Genetics of immune-mediated disorders: from genome-wide association to molecular mechanism. Curr Opin Immunol. (2014) 31:51–7. doi: 10.1016/j.coi.2014.09.007
51. Ricaño-Ponce I, Zhernakova DV, Deelen P, Luo O, Li X, Isaacs A, et al. Refined mapping of autoimmune disease associated genetic variants with gene expression suggests an important role for non-coding RNAs. J Autoimmun. (2016) 68:62–74. doi: 10.1016/j.jaut.2016.01.002
52. Monteleone G, Mann J, Monteleone I, Vavassori P, Bremner R, Fantini M, et al. A failure of transforming growth factor-beta1 negative regulation maintains sustained NF-kappaB activation in gut inflammation. J Biol Chem. (2004) 279:3925–32. doi: 10.1074/jbc.M303654200
53. Fernandez-Jimenez N, Castellanos-Rubio A, Plaza-Izurieta L, Irastorza I, Elcoroaristizabal X, Jauregi-Miguel A, et al. Coregulation and modulation of NFκB-related genes in celiac disease: uncovered aspects of gut mucosal inflammation. Hum Mol Genet. (2014) 23:1298–310. doi: 10.1093/hmg/ddt520
54. Maiuri MC, De Stefano D, Mele G, Fecarotta S, Greco L, Troncone R, et al. Nuclear factor kappa B is activated in small intestinal mucosa of celiac patients. J Mol Med Berl Ger. (2003) 81:373–9. doi: 10.1007/s00109-003-0440-0
55. Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. (2012) 490:543–6. doi: 10.1038/nature11530
56. Lerner A, Arleevskaya M, Schmiedl A, Matthias T. Microbes and viruses are bugging the gut in celiac disease. Are they friends or foes? Front Microbiol. (2017) 8:1392. doi: 10.3389/fmicb.2017.01392
57. Jiang H, Zhang X, Zhou Y, Jiang C, Shi Y. Infection, antibiotic exposure, and risk of celiac disease: a systematic review and meta-analysis. J Gastroenterol Hepatol. (2020) 35:557–66. doi: 10.1111/jgh.14928
58. Lebwohl B, Nobel YR, Green PHR, Blaser MJ, Ludvigsson JF. Risk of clostridium difficile infection in patients with celiac disease: a population-based study. Am J Gastroenterol. (2017) 112:1878–84. doi: 10.1038/ajg.2017.400
59. Pes GM, Bibbò S, Dore MP. Coeliac disease: beyond genetic susceptibility and gluten. A narrative review. Ann Med. (2019) 51:1–16. doi: 10.1080/07853890.2019.1569254
60. Wirusanti NI, Baldridge MT, Harris VC. Microbiota regulation of viral infections through interferon signaling. Trends Microbiol. (2022) 30:778–92. doi: 10.1016/j.tim.2022.01.007
61. Monteleone G, Pender SL, Alstead E, Hauer AC, Lionetti P, McKenzie C, et al. Role of interferon alpha in promoting T helper cell type 1 responses in the small intestine in coeliac disease. Gut. (2001) 48:425–9. doi: 10.1136/gut.48.3.425
62. Kagnoff MF, Austin RK, Hubert JJ, Bernardin JE, Kasarda DD. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med. (1984) 160:1544–57. doi: 10.1084/jem.160.5.1544
63. Beyerlein A, Donnachie E, Ziegler A-G. Infections in early life and development of celiac disease. Am J Epidemiol. (2017) 186:1277–80. doi: 10.1093/aje/kwx190
64. Mårild K, Kahrs CR, Tapia G, Stene LC, Størdal K. Infections and risk of celiac disease in childhood: a prospective nationwide cohort study. Am J Gastroenterol. (2015) 110:1475–84. doi: 10.1038/ajg.2015.287
65. Kemppainen KM, Lynch KF, Liu E, Lönnrot M, Simell V, Briese T, et al. factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin Gastroenterol Hepatol. (2017) 15:694–702.e5. doi: 10.1016/j.cgh.2016.10.033
66. Lindfors K, Lin J, Lee H-S, Hyöty H, Nykter M, Kurppa K, et al. Metagenomics of the faecal virome indicate a cumulative effect of enterovirus and gluten amount on the risk of coeliac disease autoimmunity in genetically at risk children: the TEDDY study. Gut. (2020) 69:1416–22. doi: 10.1136/gutjnl-2019-319809
67. Lähdeaho ML, Parkkonen P, Reunala T, Mäki M, Lehtinen M. Antibodies to E1b protein-derived peptides of enteric adenovirus type 40 are associated with celiac disease and dermatitis herpetiformis. Clin Immunol Immunopathol. (1993) 69:300–5. doi: 10.1006/clin.1993.1184
68. Bouziat R, Hinterleitner R, Brown JJ, Stencel-Baerenwald JE, Ikizler M, Mayassi T, et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science. (2017) 356:44–50. doi: 10.1126/science.aah5298
69. Caminero A, Verdu EF. Celiac disease: should we care about microbes? Am J Physiol Gastrointest Liver Physiol. (2019) 317:G161–70. doi: 10.1152/ajpgi.00099.2019
70. Tye-Din JA, Galipeau HJ, Agardh D. Celiac disease: a review of current concepts in pathogenesis, prevention, and novel therapies. Front Pediatr. (2018) 6:350. doi: 10.3389/fped.2018.00350
71. Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol. (2006) 101:2333–40. doi: 10.1111/j.1572-0241.2006.00741.x
72. Assa A, Waisbourd-Zinman O, Daher S, Shamir R. Birth month as a risk factor for the diagnosis of celiac disease later in life: a population-based study. J Pediatr Gastroenterol Nutr. (2018) 67:367–70. doi: 10.1097/MPG.0000000000002001
73. Amlashi FI, Norouzi Z, Sohrabi A, Shirzad-Aski H, Norouzi A, Ashkbari A, et al. A systematic review and meta-analysis for association of Helicobacter pylori colonization and celiac disease. PLoS ONE. (2021) 16:e0241156. doi: 10.1371/journal.pone.0241156
74. Riddle MS, Murray JA, Cash BD, Pimentel M, Porter CK. Pathogen-specific risk of celiac disease following bacterial causes of foodborne illness: a retrospective cohort study. Dig Dis Sci. (2013) 58:3242–5. doi: 10.1007/s10620-013-2733-7
75. Verdu EF, Mauro M, Bourgeois J, Armstrong D. Clinical onset of celiac disease after an episode of Campylobacter jejuni enteritis. Can J Gastroenterol. (2007) 21:453–5. doi: 10.1155/2007/169591
76. Sabayan B, Foroughinia F, Imanieh M-H. Can Campylobacter jejuni play a role in development of celiac disease? A hypothesis. World J Gastroenterol. (2007) 13:4784–5. doi: 10.3748/wjg.v13.i35.4784
77. Petersen J, Ciacchi L, Tran MT, Loh KL, Kooy-Winkelaar Y, Croft NP, et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat Struct Mol Biol. (2020) 27:49–61. doi: 10.1038/s41594-019-0353-4
78. Dydensborg Sander S, Nybo Andersen A-M, Murray JA, Karlstad Ø, Husby S, Størdal K. Association between antibiotics in the first year of life and celiac disease. Gastroenterology. (2019) 156:2217–29. doi: 10.1053/j.gastro.2019.02.039
79. Valitutti F, Cucchiara S, Fasano A. Celiac disease and the microbiome. Nutrients. (2019) 11:2403. doi: 10.3390/nu11102403
80. Kołodziej M, Patro-Gołab B, Gieruszczak-Białek D, Skórka A, Pieścik-Lech M, Baron R, et al. Association between early life (prenatal and postnatal) antibiotic administration and coeliac disease: a systematic review. Arch Dis Child. (2019) 104:1083–9. doi: 10.1136/archdischild-2019-317174
81. Nagy S, Papp Z. Global approach of the cesarean section rates. J Perinat Med. (2021) 49:1–4. doi: 10.1515/jpm-2020-0463
82. Hobbs AJ, Mannion CA, McDonald SW, Brockway M, Tough SC. The impact of caesarean section on breastfeeding initiation, duration and difficulties in the first four months postpartum. BMC Pregn Childbirth. (2016) 16:90. doi: 10.1186/s12884-016-0876-1
83. Decker E, Hornef M, Stockinger S. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Gut Microbes. (2011) 2:91–8. doi: 10.4161/gmic.2.2.15414
84. Sevelsted A, Stokholm J, Bønnelykke K, Bisgaard H. Cesarean section and chronic immune disorders. Pediatrics. (2015) 135:e92–98. doi: 10.1542/peds.2014-0596
85. Koletzko S, Lee H-S, Beyerlein A, Aronsson CA, Hummel M, Liu E, et al. Cesarean section on the risk of celiac disease in the offspring: the teddy study. J Pediatr Gastroenterol Nutr. (2018) 66:417–24. doi: 10.1097/MPG.0000000000001682
86. Decker E, Engelmann G, Findeisen A, Gerner P, Laass M, Ney D, et al. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics. (2010) 125:e1433–40. doi: 10.1542/peds.2009-2260
87. Yang XY, Liu YH, Jiang HY, Ying XH. Cesarean section is not associated with increased risk of celiac disease in the offspring: a meta-analysis. J Matern Fetal Neonatal Med. (2022) 35:9570–7. doi: 10.1080/14767058.2022.2048813
88. Mårild K, Stephansson O, Montgomery S, Murray JA, Ludvigsson JF. Pregnancy outcome and risk of celiac disease in offspring: a nationwide case-control study. Gastroenterology. (2012) 142:39–45.e3. doi: 10.1053/j.gastro.2011.09.047
89. Balogun OO, O'Sullivan EJ, McFadden A, Ota E, Gavine A, Garner CD, et al. Interventions for promoting the initiation of breastfeeding. Cochr Database Syst Rev. (2016) 2016:CD001688. doi: 10.1002/14651858.CD001688.pub3
90. Akobeng AK, Ramanan AV, Buchan I, Heller RF. Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies. Arch Dis Child. (2006) 91:39–43. doi: 10.1136/adc.2005.082016
91. Lionetti E, Castellaneta S, Francavilla R, Pulvirenti A, Tonutti E, Amarri S, et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med. (2014) 371:1295–303. doi: 10.1056/NEJMoa1400697
92. Vriezinga SL, Auricchio R, Bravi E, Castillejo G, Chmielewska A, Crespo Escobar P, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med. (2014) 371:1304–15. doi: 10.1056/NEJMoa1404172
93. Silano M, Agostoni C, Sanz Y, Guandalini S. Infant feeding and risk of developing celiac disease: a systematic review. BMJ Open. (2016) 6:e009163. doi: 10.1136/bmjopen-2015-009163
94. Szajewska H, Shamir R, Chmielewska A, Pieścik-Lech M, Auricchio R, Ivarsson A, et al. Systematic review with meta-analysis: early infant feeding and coeliac disease–update 2015. Aliment Pharmacol Ther. (2015) 41:1038–54. doi: 10.1111/apt.13163
95. Aronsson CA, Lee H-S, Liu E, Uusitalo U, Hummel S, Yang J, et al. Age at gluten introduction and risk of celiac disease. Pediatrics. (2015) 135:239–45. doi: 10.1542/peds.2014-1787
96. Andrén Aronsson C, Lee H-S, Hård Af Segerstad EM, Uusitalo U, Yang J, Koletzko S, et al. Association of gluten intake during the first 5 years of life with incidence of celiac disease autoimmunity and celiac disease among children at increased risk. JAMA. (2019) 322:514–23. doi: 10.1001/jama.2019.10329
97. Lund-Blix NA, Mårild K, Tapia G, Norris JM, Stene LC, Størdal K. Gluten intake in early childhood and risk of celiac disease in childhood: a nationwide cohort study. Am J Gastroenterol. (2019) 114:1299–306. doi: 10.14309/ajg.0000000000000331
98. Mårild K, Dong F, Lund-Blix NA, Seifert J, Barón AE, Waugh KC, et al. Gluten intake and risk of celiac disease: long-term follow-up of an at-risk birth cohort. Am J Gastroenterol. (2019) 114:1307–14. doi: 10.14309/ajg.0000000000000255
99. Myléus A, Hernell O, Gothefors L, Hammarström M-L, Persson L-Å, Stenlund H, et al. Early infections are associated with increased risk for celiac disease: an incident case-referent study. BMC Pediatr. (2012) 12:194. doi: 10.1186/1471-2431-12-194
100. Barone MV, Auricchio S, A. Cumulative effect of food and viruses to trigger celiac disease (CD): a commentary on the recent literature. Int J Mol Sci. (2021) 22:2027. doi: 10.3390/ijms22042027
101. ESPGHAN. Gluten Introduction and the Risk of Coeliac Disease (n.d.). Available online at: http://www.espghan.org/knowledge-center/publications/Nutrition/2016_Gluten_Introduction_and_the_Risk_of_Coeliac_Disease (accessed August 21, 2022).
102. Chiba M, Nakane K, Komatsu M. Westernized diet is the most ubiquitous environmental factor in inflammatory bowel disease. Perm J. (2019) 23:18–107. doi: 10.7812/TPP/18-107
103. Zinöcker M, Lindseth I. The western diet–microbiome-host interaction and its role in metabolic disease. Nutrients. (2018) 10:365. doi: 10.3390/nu10030365
104. Malesza IJ, Malesza M, Walkowiak J, Mussin N, Walkowiak D, Aringazina R, et al. High-fat, western-style diet, systemic inflammation, and gut microbiota: a narrative review. Cells. (2021) 10:3164. doi: 10.3390/cells10113164
105. García-Montero C, Fraile-Martínez O, Gómez-Lahoz AM, Pekarek L, Castellanos AJ, Noguerales-Fraguas F, et al. Nutritional components in western diet versus mediterranean diet at the gut microbiota–immune system interplay. Implications for health and disease. Nutrients. (2021) 13:699. doi: 10.3390/nu13020699
106. Thorburn AN, Macia L, Mackay CR. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity. (2014) 40:833–42. doi: 10.1016/j.immuni.2014.05.014
107. Suzuki T. Regulation of the intestinal barrier by nutrients: the role of tight junctions. Anim Sci J. (2020) 91:e13357. doi: 10.1111/asj.13357
108. Girbovan A, Sur G, Samasca G, Lupan I. Dysbiosis a risk factor for celiac disease. Med Microbiol Immunol. (2017) 206:83–91. doi: 10.1007/s00430-017-0496-z
109. Rizzello F, Spisni E, Giovanardi E, Imbesi V, Salice M, Alvisi P, et al. Implications of the westernized diet in the onset and progression of IBD. Nutrients. (2019) 11:1033. doi: 10.3390/nu11051033
110. Statovci D, Aguilera M, MacSharry J, Melgar S. The impact of western diet and nutrients on the microbiota and immune response at mucosal interfaces. Front Immunol. (2017) 8:838. doi: 10.3389/fimmu.2017.00838
111. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. (2018) 172:162–75.e14. doi: 10.1016/j.cell.2017.12.013
112. Binienda A, Twardowska A, Makaro A, Salaga M. Dietary carbohydrates and lipids in the pathogenesis of leaky gut syndrome: an overview. Int J Mol Sci. (2020) 21:8368. doi: 10.3390/ijms21218368
113. Rohr MW, Narasimhulu CA, Rudeski-Rohr TA, Parthasarathy S. Negative effects of a high-fat diet on intestinal permeability: a review. Adv Nutr. (2020) 11:77–91. doi: 10.1093/advances/nmz061
114. Christ A, Lauterbach M, Latz E. Western diet and the immune system: an inflammatory connection. Immunity. (2019) 51:794–811. doi: 10.1016/j.immuni.2019.09.020
115. Mörkl S, Lackner S, Meinitzer A, Mangge H, Lehofer M, Halwachs B, et al. Gut microbiota, dietary intakes and intestinal permeability reflected by serum zonulin in women. Eur J Nutr. (2018) 57:2985–97. doi: 10.1007/s00394-018-1784-0
116. Ji Y, Sakata Y, Tso P. Nutrient-induced inflammation in the intestine. Curr Opin Clin Nutr Metab Care. (2011) 14:315–21. doi: 10.1097/MCO.0b013e3283476e74
117. Moreira AP, Texeira TF, Ferreira AB, Peluzio MD, Alfenas RD. Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr. (2012) 108:801–9. doi: 10.1017/S0007114512001213
118. Tran HQ, Bretin A, Adeshirlarijaney A, Yeoh BS, Vijay-Kumar M, Zou J, et al. “Western diet” -induced adipose inflammation requires a complex gut microbiota. Cell Mol Gastroenterol Hepatol. (2020) 9:313–33. doi: 10.1016/j.jcmgh.2019.09.009
119. Netto Candido TL, Bressan J, Alfenas R de CG. Dysbiosis and metabolic endotoxemia induced by high-fat diet. Nutr Hosp. (2018) 35:1432–40. doi: 10.20960/nh.1792
120. Fajstova A, Galanova N, Coufal S, Malkova J, Kostovcik M, Cermakova M, et al. Diet rich in simple sugars promotes pro-inflammatory response via gut microbiota alteration and TLR4 signaling. Cells. (2020) 9:2701. doi: 10.3390/cells9122701
121. Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, Fernandez-Real JM, et al. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care. (2009) 32:2281–7. doi: 10.2337/dc09-0979
122. Do MH, Lee E, Oh M-J, Kim Y, Park H-Y. High-glucose or -fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients. (2018) 10:761. doi: 10.3390/nu10060761
123. Laffin M, Fedorak R, Zalasky A, Park H, Gill A, Agrawal A, et al. A high-sugar diet rapidly enhances susceptibility to colitis via depletion of luminal short-chain fatty acids in mice. Sci Rep. (2019) 9:12294. doi: 10.1038/s41598-019-48749-2
124. Zhu Y, Shi C, Niu Q, Wang J, Zhu W. Dynamic changes in morphology, gene expression and microbiome in the jejunum of compensatory-growth rats induced by protein restriction. Microb Biotechnol. (2018) 11:734–46. doi: 10.1111/1751-7915.13266
125. Zhou Q, Verne ML, Fields JZ, Lefante JJ, Basra S, Salameh H, et al. Randomised placebo-controlled trial of dietary glutamine supplements for postinfectious irritable bowel syndrome. Gut. (2019) 68:996–1002. doi: 10.1136/gutjnl-2017-315136
126. Chen M, Liu Y, Xiong S, Wu M, Li B, Ruan Z, et al. Dietary l-tryptophan alleviated LPS-induced intestinal barrier injury by regulating tight junctions in a Caco-2 cell monolayer model. Food Funct. (2019) 10:2390–8. doi: 10.1039/C9FO00123A
127. Noda S, Tanabe S, Suzuki T. Differential effects of flavonoids on barrier integrity in human intestinal Caco-2 cells. J Agric Food Chem. (2012) 60:4628–33. doi: 10.1021/jf300382h
128. Suzuki T, Tanabe S, Hara H. Kaempferol enhances intestinal barrier function through the cytoskeletal association and expression of tight junction proteins in Caco-2 cells. J Nutr. (2011) 141:87–94. doi: 10.3945/jn.110.125633
129. Amasheh M, Luettig J, Amasheh S, Zeitz M, Fromm M, Schulzke J-D. Effects of quercetin studied in colonic HT-29/B6 cells and rat intestine in vitro. Ann N Y Acad Sci. (2012) 1258:100–7. doi: 10.1111/j.1749-6632.2012.06609.x
130. Shigeshiro M, Tanabe S, Suzuki T. Dietary polyphenols modulate intestinal barrier defects and inflammation in a murine model of colitis. J Funct Foods. (2013) 5:949–55. doi: 10.1016/j.jff.2013.02.008
131. Farré R, Fiorani M, Abdu Rahiman S, Matteoli G. Intestinal permeability, inflammation and the role of nutrients. Nutrients. (2020) 12:1185. doi: 10.3390/nu12041185
132. Casas R, Sacanella E, Estruch R. The immune protective effect of the Mediterranean diet against chronic low-grade inflammatory diseases. Endocr Metab Immune Disord Drug Targets. (2014) 14:245–54. doi: 10.2174/1871530314666140922153350
133. Barbaresko J, Koch M, Schulze MB, Nöthlings U. Dietary pattern analysis and biomarkers of low-grade inflammation: a systematic literature review. Nutr Rev. (2013) 71:511–27. doi: 10.1111/nure.12035
134. Venter C, Eyerich S, Sarin T, Klatt KC. Nutrition and the immune system: a complicated tango. Nutrients. (2020) 12:E818. doi: 10.3390/nu12030818
135. Barroso M, Beth SA, Voortman T, Jaddoe VWV, van Zelm MC, Moll HA, et al. Dietary patterns after the weaning and lactation period are associated with celiac disease autoimmunity in children. Gastroenterology. (2018) 154:2087–96.e7. doi: 10.1053/j.gastro.2018.02.024
136. Terra R, Alves PJF, Gonçalves da Silva SA, Salerno VP, Dutra PML. Exercise improves the Th1 response by modulating cytokine and NO production in BALB/c mice. Int J Sports Med. (2013) 34:661–6. doi: 10.1055/s-0032-1329992
137. Lowder T, Dugger K, Estell K, Deshane J, Schwiebert L. Exercise increases regulatory T cell function and decreases Th2 and Th17 cytokine production in healthy and asthmatic mice (97.15). J Immunol. (2010) 184:97.15. doi: 10.4049/jimmunol.184.Supp.97.15
138. MedCrave Online. The Role of Th1 and Th17 in the Pathogenesis of Celiac Disease (n.d.). Available online at: https://medcraveonline.com/GHOA/the-role-of-th1-and-th17-in-the-pathogenesis-of-celiac-disease.html (accessed February 28, 2022).
139. Sharif K, Watad A, Bragazzi NL, Lichtbroun M, Amital H, Shoenfeld Y. Physical activity and autoimmune diseases: get moving and manage the disease. Autoimmun Rev. (2018) 17:53–72. doi: 10.1016/j.autrev.2017.11.010
140. Hallal PC, Andersen LB, Bull FC, Guthold R, Haskell W, Ekelund U, et al. Global physical activity levels: surveillance progress, pitfalls, and prospects. Lancet Lond Engl. (2012) 380:247–57. doi: 10.1016/S0140-6736(12)60646-1
141. Dickey W, Kearney N. Overweight in celiac disease: prevalence, clinical characteristics, and effect of a gluten-free diet. Am J Gastroenterol. (2006) 101:2356–9. doi: 10.1111/j.1572-0241.2006.00750.x
142. Singh I, Agnihotri A, Sharma A, Verma AK, Das P, Thakur B, et al. Patients with celiac disease may have normal weight or may even be overweight. Indian J Gastroenterol. (2016) 35:20–4. doi: 10.1007/s12664-016-0620-9
143. Cheng J, Brar PS, Lee AR, Green PHR. Body mass index in celiac disease: beneficial effect of a gluten-free diet. J Clin Gastroenterol. (2010) 44:267–71. doi: 10.1097/MCG.0b013e3181b7ed58
144. Ukkola A, Mäki M, Kurppa K, Collin P, Huhtala H, Kekkonen L, et al. Changes in body mass index on a gluten-free diet in coeliac disease: a nationwide study. Eur J Intern Med. (2012) 23:384–8. doi: 10.1016/j.ejim.2011.12.012
145. Tucker E, Rostami K, Prabhakaran S, Al Dulaimi D. Patients with coeliac disease are increasingly overweight or obese on presentation. J Gastrointest Liver Dis. (2012) 21:11–5.
146. Karczewski J, Sledzińska E, Baturo A, Jończyk I, Maleszko A, Maleszko A, et al. Obesity and inflammation. Eur Cytok Netw. (2018) 29:83–94. doi: 10.1684/ecn.2018.0415
147. Gaylord A, Trasande L, Kannan K, Thomas KM, Lee S, Liu M, et al. Persistent organic pollutant exposure and celiac disease: a pilot study. Environ Res. (2020) 186:109439. doi: 10.1016/j.envres.2020.109439
148. Wijarnpreecha K, Lou S, Panjawatanan P, Cheungpasitporn W, Pungpapong S, Lukens FJ, et al. Cigarette smoking and risk of celiac disease: a systematic review and meta-analysis. United Eur Gastroenterol J. (2018) 6:1285–93. doi: 10.1177/2050640618786790
149. Andeweg SP, Keşmir C, Dutilh BE. Quantifying the impact of human leukocyte antigen on the human gut microbiota. MSphere. (2021) 6:e0047621. doi: 10.1128/mSphere.00476-21
150. Olivares M, Neef A, Castillejo G, Palma GD, Varea V, Capilla A, et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut. (2015) 64:406–17. doi: 10.1136/gutjnl-2014-306931
151. Olivares M, Walker AW, Capilla A, Benítez-Páez A, Palau F, Parkhill J, et al. Gut microbiota trajectory in early life may predict development of celiac disease. Microbiome. (2018) 6:36. doi: 10.1186/s40168-018-0415-6
152. Ou G, Hedberg M, Hörstedt P, Baranov V, Forsberg G, Drobni M, et al. Proximal small intestinal microbiota and identification of rod-shaped bacteria associated with childhood celiac disease. Am J Gastroenterol. (2009) 104:3058–67. doi: 10.1038/ajg.2009.524
Keywords: celiac disease, environmental factors, viruses, western diet, gut microbiota, breastfeeding, c-section, gluten
Citation: Skoracka K, Hryhorowicz S, Rychter AM, Ratajczak AE, Szymczak-Tomczak A, Zawada A, Słomski R, Dobrowolska A and Krela-Kaźmierczak I (2023) Why are western diet and western lifestyle pro-inflammatory risk factors of celiac disease? Front. Nutr. 9:1054089. doi: 10.3389/fnut.2022.1054089
Received: 26 September 2022; Accepted: 31 December 2022;
Published: 20 January 2023.
Edited by:
Alessandra Bordoni, University of Bologna, ItalyReviewed by:
Emma E. Hamilton-Williams, The University of Queensland, AustraliaCopyright © 2023 Skoracka, Hryhorowicz, Rychter, Ratajczak, Szymczak-Tomczak, Zawada, Słomski, Dobrowolska and Krela-Kaźmierczak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kinga Skoracka,  a2luZ3Nrb3JhY2thQGdtYWlsLmNvbQ==
a2luZ3Nrb3JhY2thQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.