 Yuanyuan Song
Yuanyuan Song Kai Chen3
Kai Chen3
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Nutr. , 26 September 2022
Sec. Nutrition and Microbes
Volume 9 - 2022 | https://doi.org/10.3389/fnut.2022.1003763
This article is part of the Research Topic Gut Microbial Response to Host Metabolic Phenotypes, Volume 2 View all 19 articles
The gut microbiota is a complex ecosystem that is essential for the metabolism, immunity and health of the host. The gut microbiota also plays a critical role in nutrient absorption and metabolism, and nutrients can influence the growth and composition of the gut microbiota. To gain a better understanding of the relationship between the gut microbial composition and nutrient metabolism, we used a pig model by collecting the contents of the different intestinal locations from six pigs to investigate microbial composition in different intestinal locations based on 16S rRNA gene sequencing and the concentrations of short-chain fatty acids (SCFAs), amino acids, fat, and crude ash in different intestinal locations using gas chromatography and chemical analysis. The results showed that the richness and diversity of intestinal microbial communities gradually increased from the small intestine to the large intestine. The relative abundance of Proteobacteria was higher in the jejunum and ileum, whereas the proportion of Firmicutes was higher in the cecum and colon. The concentrations of SCFAs were higher in the cecum and colon (P < 0.05). The concentrations of amino acids were higher in the small intestine than in the large intestine, while the amino acid content was significantly higher in the ascending colon than in the transverse colon and descending colon. The correlation analysis revealed that Ruminococcaceae UCG-005, Coriobacteriaceae_uncultured, [Eubacterium] hallii group, Mogibacterium and Lachnospiraceae AC2044 group had a higher positive correlation with SCFAs, crude ash and fat but had a negative correlation with amino acids in different gut locations of pigs. These findings may serve as fundamental data for using nutrient metabolism to regulate human and animal gut microbes and health and provide guidance for exploring host-microbe bidirectional interaction mechanisms and driving pathways.
The gastrointestinal tract (GIT) is the largest interface between the external and internal environments and contains the largest amount and the greatest diversity of microorganisms. Humans and mammals are colonized with a vast and complex microbial community, which is known to play an important role in host metabolism, including digestion and absorption of nutrients, energy acquisition, carbohydrate metabolism and the immune system (1–4). Along the longitudinal axis of the intestinal lumen, distinct microhabitats selectively harbor characteristic microbes (5, 6).
The gut microbiota can directly affect the utilization and absorption of nutrients, including carbohydrates, lipids, proteins and minerals, and are also regulated by the host nutrition (6–9). SCFAs are produced by gut saccharolytic microbes through fermenting and degrading indigestible carbohydrate (10, 11), which can provide energy for intestinal epithelial cells and serve as an energy source for the growth of anaerobic bacteria (12, 13). Lachnospiraceae_uncultured may be a potential beneficial bacterium involved in the metabolism of a variety of carbohydrates, and the fermentation product acetic acid is the main source of bacteria that provides energy for the host. The fermentation metabolites of Ruminococcaceae (CG-004, 05, 013, 014) are mainly acetic acid and formic acid, and bacteria in the Prevotella-dominated enterotype mainly absorb monosaccharides and degrade mucins to obtain energy (14, 15). Some species of the genus [Eubacterium] rectale group can ferment carbohydrates in food and produce large amounts of acetic acid and butyric acid (16). In addition, acetate produced by Bacteroides, Bifidobacterium, Prausnitzii, and Ruminococcus acts on the brain through the blood–brain barrier (17). The gut microbiota may play important roles in the host amino acid balance and health through multiple pathways (18, 19). Bacteria have various metabolic pathways for amino acid metabolism, such as the synthesis of cellular proteins and amino acid catabolism (20). Studies have shown that small intestinal bacteria may mainly utilize amino acids for the synthesis of bacterial proteins, while large intestinal bacteria mainly use them for catabolism (21). Bacteria in the rumen and large intestine of animals and humans can degrade amino acids in large quantities. Clostridium can degrade amino acids through the Stickland reaction, and the main metabolites are branched-chain fatty acids and ammonia (22). Some studies have shown that amino acid metabolism by intestinal bacteria is ultimately performed for their survival and growth in the complex intestinal environment. For example, Lactobacillus can synthesize bacterial proteins by utilizing exogenous amino acids, which have an important role in host nutrition and physiology (23). The gut microbiota also synthesizes many essential amino acids, such as lysine, threonine, and arginine. For example, some intestinal microorganisms are able to bind NH3 from the breakdown of amino acids and resynthesize essential amino acids or proteins (24).
In recent years, numerous studies have shown a correlation between the gut microbiota and host metabolism (25–30), but the impact of the gut microbiota on nutrient metabolism remains unclear, and few studies have examined the metabolic compartmentalization of nutrients in different locations of the intestinal tract. Pigs are very similar to humans in terms of gastrointestinal tract development, physiology, digestive function and composition, the gut microbiota of pigs is 96% similar to humans in functional pathway (31–34). The minimum nutritional requirement of pigs is equivalent to the daily nutritional requirement of humans and the common physiological structure leads to a similar digestive tract transit time and nutrient absorption process, the pigs also can collect repeated measurement data (35). So pigs are an ideal model for studying human gut microbiota and nutrient metabolism. Herein, we investigated the intestinal microbiota structure in different parts of pigs and analyzed its association with the concentrations of SCFAs, amino acids, fat, and crude ash in different intestinal locations. The present findings could provide insights into the association between the microbial community and nutrient metabolism with gut homeostasis and whole-body health in humans and mammals.
All animal experiments were approved by the Institutional Animal Care of the Zhejiang Academy of Agricultural Sciences in accordance with the relevant rules and regulations (ZAAS-2017-009).
Six female Jinhua pigs were randomly selected from the pig farm of Jinhua Academy of Agricultural Sciences, reared in the same environment and fed the same diet daily and water ad libitum. The detailed ingredients and nutrient contents of the diet are presented in Table 1. The pigs (71.90 ± 0.82 kg) were killed at the age of 250 days under anesthesia, and then the contents of the jejunum, ileum, cecum, ascending colon, transverse colon, and intermediate descending colon were collected immediately. We collected samples in sterile tubes and quickly frozen them in liquid nitrogen, and then transferred to a refrigerator at−80 °C for future chemical and microbial analyses.
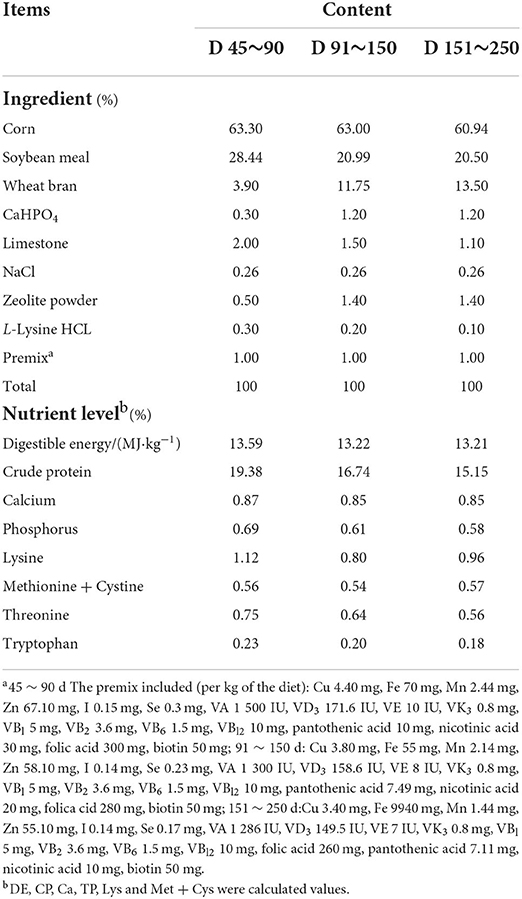
Table 1. Composition and nutrient levels of basal diet.
The CTAB/SDS method was used to extract genomic DNA from 36 samples. DNA concentration and purity were monitored on a 1% agarose gel and DNA was diluted to 1 ng/μl using sterile water according to the concentration. The 16S rRNA gene V4-V5 region was amplified using primers (515F: 5′- GTGCCAGCMGCCGCGGTAA-3′; 907R: 5′-CCGTCAATTCCTTTGAGTTT-3′) with barcodes. All PCRs were conducted in 30 μL reactions with 15 μL of Phusion®High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM forward and reverse primers, and approximately 10 ng of template DNA. Initially, the samples were denaturated at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, elongation at 72 °C for 60 s and finally 72 °C for 5 min. We used A GeneJET Gel Extraction Kit (Thermo Scientific) to purify the PCR mixture and NEB Next®UltraTM DNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) to prepare sequencing libraries following the manufacturer's recommendations, and index codes were added. The Illumina paired-end reads were filtered and demultiplexed (36), merged into tags using FLASH, and assorted to each sample according to the attached barcode (37).
The RDP classifier was used to performs taxonomy assignment of the OTUs (38). We calculated and visualized the alpha diversity (Chao 1 estimator, Shannon, and Simpson indices) using GraphPad Prism 8 (GraphPad software, San Diego, CA, USA). The differences in microbial communities in different intestinal segments using principal coordinate analysis (PCoA). An analysis of linear discriminant analysis (LDA) coupled with effect size (LEfSe) were carried out to identify differentially abundant features between groups (39).
As described in our previous study, the concentrations of SCFAs in intestinal contents were detected by gas chromatography (40, 41). Briefly, 0.1 g intestinal contents of each bowel segment were weighed into a 1.5–mL centrifuge tube and suspended in Milli-Q water (9 volumes). After centrifugation (12 000 rpm, 10 min), the supernatant (0.5 mL) was collected and mixed with 0.1 mL of 25% (w/v) metaphosphoric acid and crotonic acid solution and stored at −20 °C overnight. The samples were filtered with a microporous membrane (0.22 μm), and the continuous filtrate was collected for analysis. The levels of SCFAs in each bowel content were determined using gas chromatography (GC) (Shimadzu, Kyoto, Japan), and the chromatographic column was a capillary column (InertCap FAPF). The chromatographic conditions were as follows: FID detector operating at 180 °C, column at 110 °C, vaporization chamber at 180 °C, and carrier gas nitrogen at 0.06 MPa, while the auxiliary gas consisted of hydrogen and air at pressure of 0.05 MPa and 0.05 MPa, respectively.
Samples of intestinal contents were freeze-dried. The procedure set by the Association of Official Analytical Chemists (AOAC, 2000) was used to determine the concentrations of moisture, crude fat and crude ash. We used a Hitachi L-8900 amino acid analyzer (Hitachi, Tokyo, Japan) to determine the contents of all the amino acids following acid hydrolysis as described by AOAC (2000) (42).
Based on Spearman's correlation matrices, correlation networks were analyzed between the relative abundance of bacterial abundance at the genus level and the content of SCFAs, amino acids, fat, crush ash, and moisture at different gut locations in pigs. An analysis of network structures was performed using Gephi v0.9.2 software (43).
We used GraphPad Prism 8 software to perform one-way analysis of variance (ANOVA). Data were expressed as means ± standard deviations (SD), and a P value < 0.05 was set as the level of statistical significance.
A total of 1,492,547 high-quality reads were generated from the 36 samples and were classified into 16,191 bacterial OTUs. Alpha diversity was observed in different gut locations. The samples from the large intestine (cecum and colon) had significantly higher Chao and Shannon indices than samples from the small intestine (jejunum and ileum) (Table 2, P < 0.05), suggesting that the richness and diversity of intestinal microbiota communities in pigs gradually increased from the small intestine to the large intestine in the digestive tract.
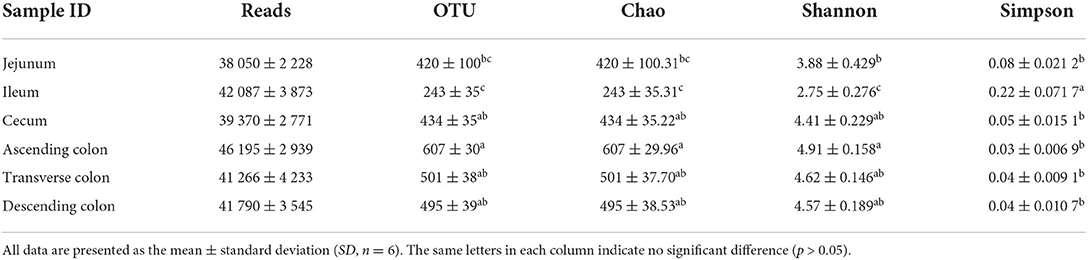
Table 2. Diversity of sequencing data of pig intestinal contents from different intestinal segments.
To measure the similarity between microbiota communities, a principal coordinate analysis (PCoA) was performed and revealed distinct clustering of the microbiota compositions of the different gut locations in pig (Figure 1). The community structures observed in the large intestine were significantly different from those detected in the small intestine.
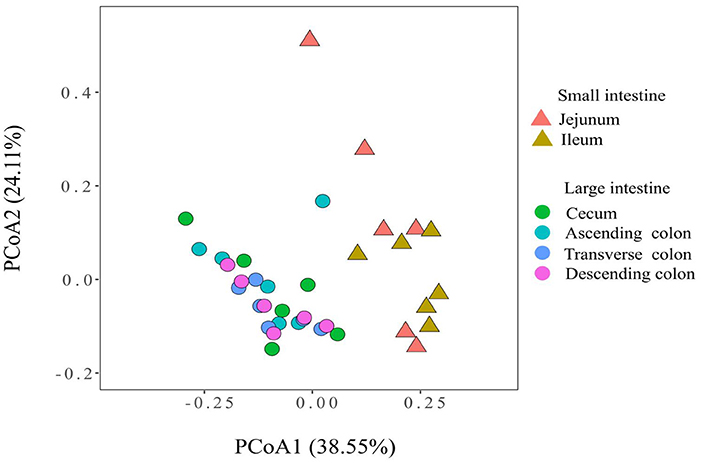
Figure 1. Principal coordinate analysis of the microbial communities based on the weighted UniFrac distance.
To reveal the microbial composition in different intestinal locations, we calculated the abundances of the top 6 phyla and the top 10 genera in all pig samples. The dominant phyla were Firmicutes, Bacteroidetes, and Proteobacteria. Firmicutes was the most dominant phylum in different intestinal segments, representing 64.36 to 82.81% of the total microbial population in pigs (Figure 2). However, the abundance of Firmicutes (64.36%) in the jejunum was lower than that in other intestinal segments of pigs (> 70%). Firmicutes and Tenericutes were predominant in the cecum and colon of pigs and made up a smaller percentage in the jejunum and ileum. Otherwise, the abundance of Proteobacteria in the small intestine was significantly higher, ranging from 13.62 to 16.32% in the jejunum and ileum. At the genus level, the most abundant genera appeared to be very diverse in different guts. Lactobacillus, Clostridium sensu stricto1, Bacteroidales S24-7 group norank, and Ruminococcaceae UCG-005 were the dominant genera. Lactobacillus was present at higher levels in the small intestine than in the large intestine, and Ruminococcaceae UCG-005 and Mollicutes RF9_norank were more abundant in the large intestine. The ileum had a higher abundance of Lactobacillus (46.30%) than the jejunum, cecum and colon.
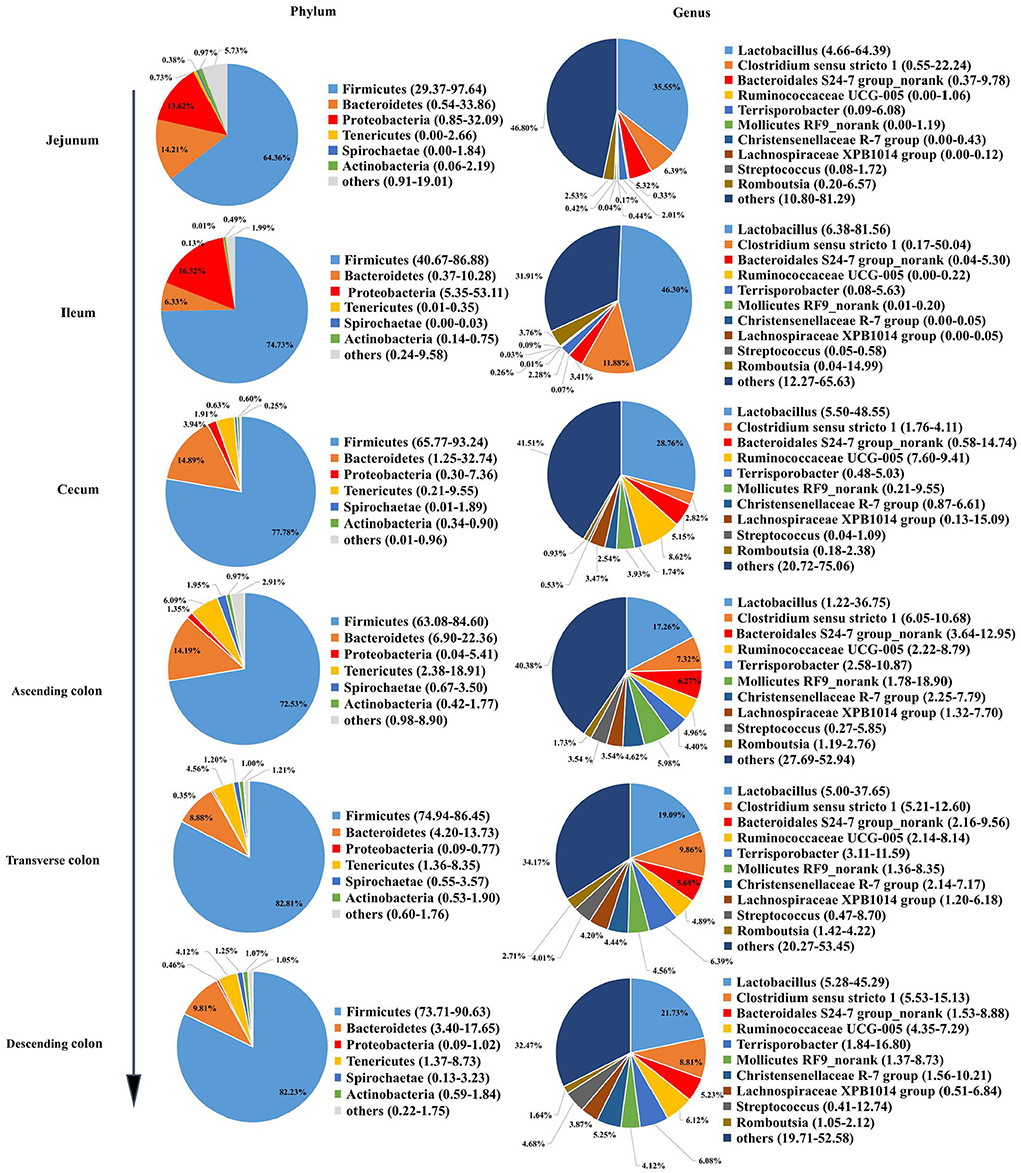
Figure 2. Microbial community structure in different intestinal locations of pigs at the phylum and genus levels.
A clustered heatmap based on the 50 most dominant genera is shown in Figure 3. The most abundant genera in the jejunum were Sarcina, Bacteroides, and Faecalibacterium, and the proportions of Escherichia-Shigella, Lactobacillus, and Veillonella were notably increased in the ileum. Alloprevotella and Prevotellaceae were the predominant genera in the cecum. For the different colon segments, the genera Prevotellaceae NK3B31 group and Prevotella 1 were considerably more abundant in the ascending colon; Blautia, Marvinbryantia, and Mogibacterium were enriched in the transverse colon; and Christensenellaceae R-7 group, Coriobacteriaceae_uncultured, and Ruminococcaceae UCG-013 had a relatively high relative abundance of bacteria in the descending colon.
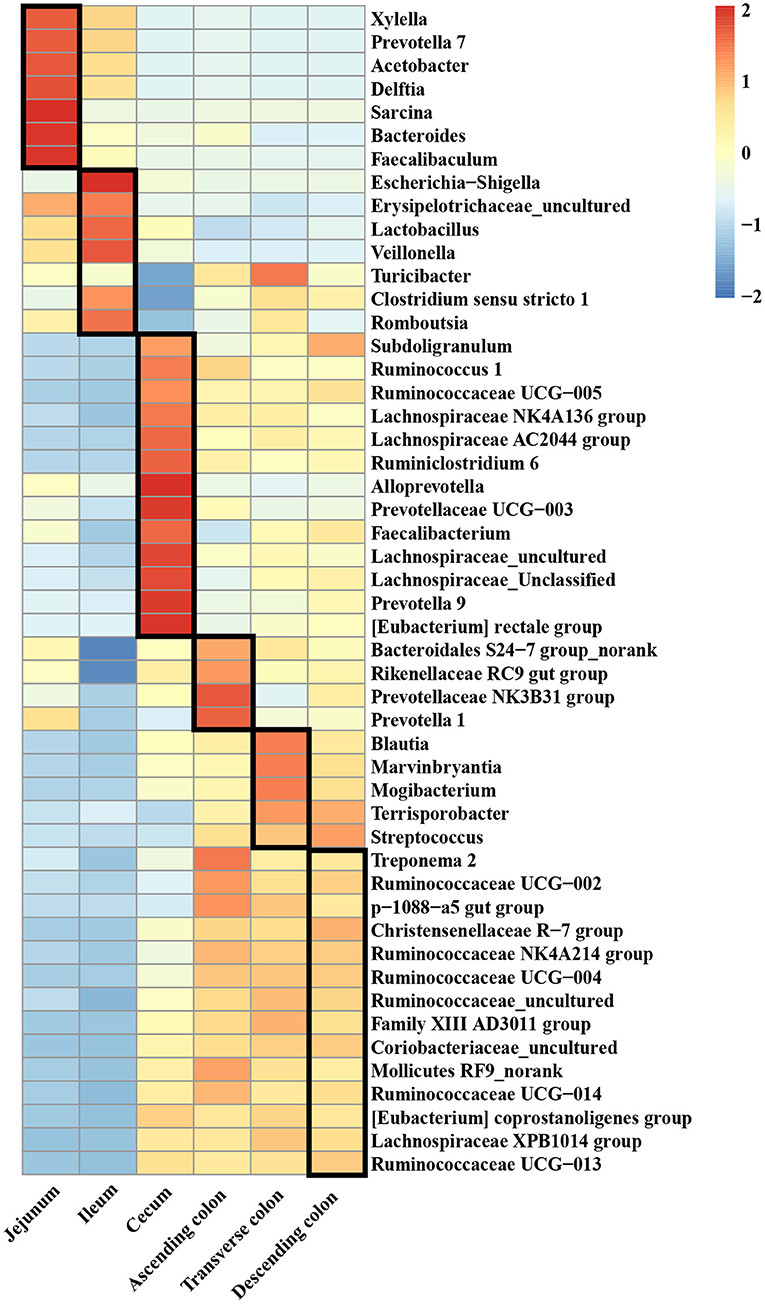
Figure 3. The top 50 genera in different intestinal locations of pigs.
To further explore the specificity of the distribution of microflora in each intestinal segment, LEfSe analysis was used to identify the genera with a differential abundance between the small and large intestine, and then the analysis identified differentially abundant genera in different colon segments. In the large intestine, a total of 57 genera were significantly enriched, while only 22 genera were enriched in the small intestine (Figure 4A). The relative abundances of Acetobacter, Veillonella, Bacteroides, Erysipelotrichaceae_uncultured, Xylella and Faecalibaculum were more abundant in the small intestine, while the abundances of Ruminococcaceae UCG-005, Mollicutes RF9_norank, Christensenellaceae R-7 group, Lachnospiraceae XPB1014 group, Streptococcus, Terrisporobacter and Ruminococcaceae UCG-014 were significantly higher in the large intestine. Concerning the different colon segments, the descending colon had higher proportions of Ruminococcaceae UCG-005, whereas Terrisporobacter, Clostridium sensu stricto 1, Romboutsia, Turicibacter, Ruminococcaceae UCG-008, Coprococcus 1, and Lachnospiraceae AC2044 group were enriched in the transverse colon. In addition, Prevotellaceae NK3B31 group, Treponema 2, Ruminiclostridium 5, Prevotellaceae UCG-001, Bacteroides, and Prevotella 1 were more abundant in the ascending colon (Figure 4B).
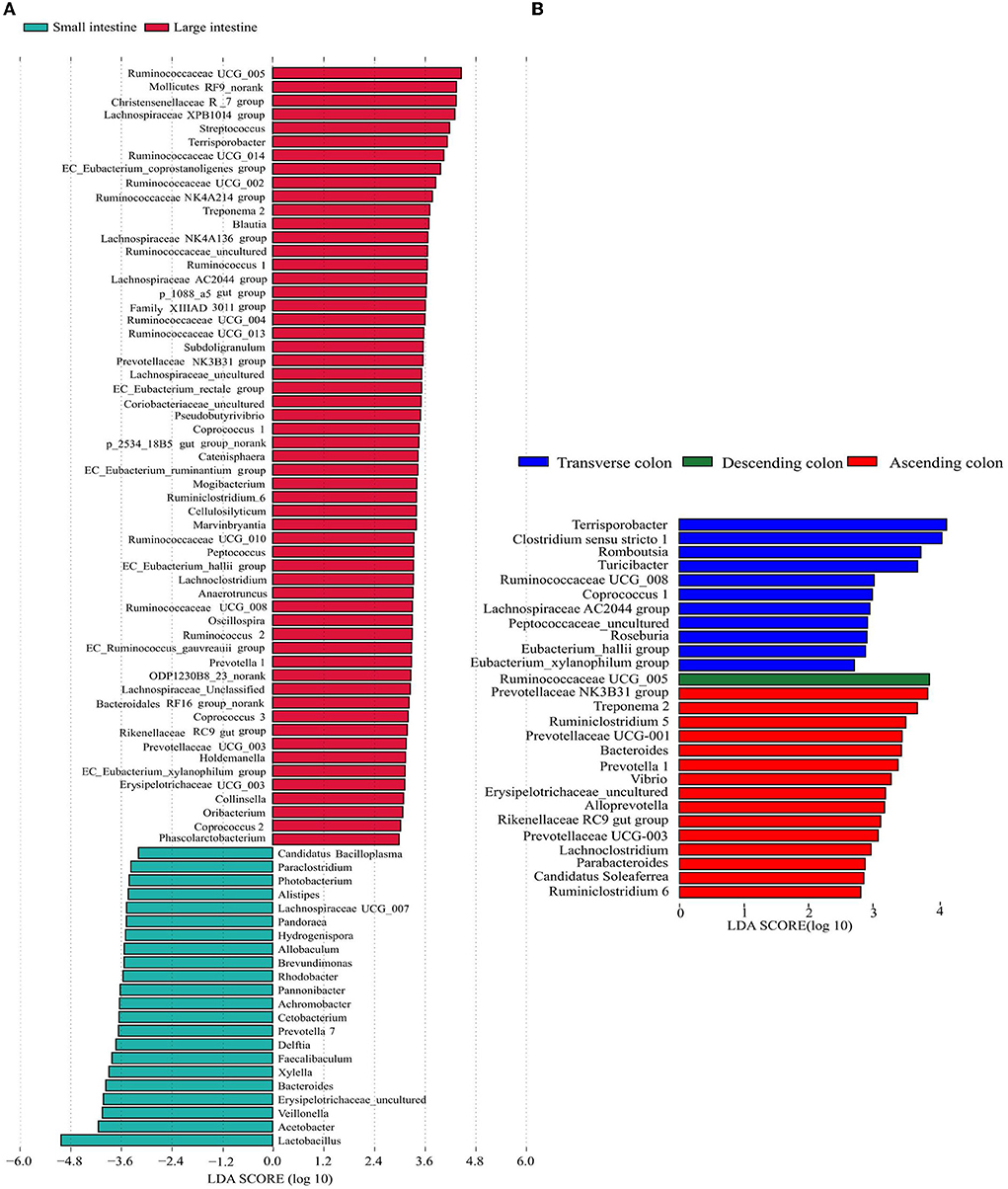
Figure 4. Differentially represented genera in different intestinal locations of pigs identified by LEFSe using an LDA score threshold of > 2.5. (A) plots the discriminative genera enriched in the small and large intestine, (B) plots the discriminative genera enriched in different colon segments. The vertical axis represents the names of different genera, and the colors correspond to different intestinal locations.
The total levels of SCFAs were significantly higher in the cecum and colon (Figure 5) (P < 0.05), and acetic acid, propionic acid, and butyric acid were the main metabolic products of the microbiota. To better understand the interaction between the microbiota and SCFAs, Spearman's correlation analysis was used to assess the association between the top 100 genera and SCFAs in different intestinal segments (Figure 6). The genera [Eubacterium] hallii group, Mogibacterium, Ruminococcaceae UCG-005, Ruminococcaceae UCG-004, Pseudobutyrivibrio, Marvinbryantia, Catenisphaera, [Eubacterium] coprostanoligenes group, and Lachnospiraceae AC2044 group were positively correlated with a variety of SCFAs. The genera Ruminococcaceae UCG-014, Ruminococcaceae UCG-013 and Coriobacteriaceae_uncultured also had a positive correlation with valeric acid, isovaleric acid, isobutyric acid, butyric acid, and propionic acid. Furthermore, Brevundimonas and Pandoraea showed a negative correlation with the contents of all kinds of SCFAs.
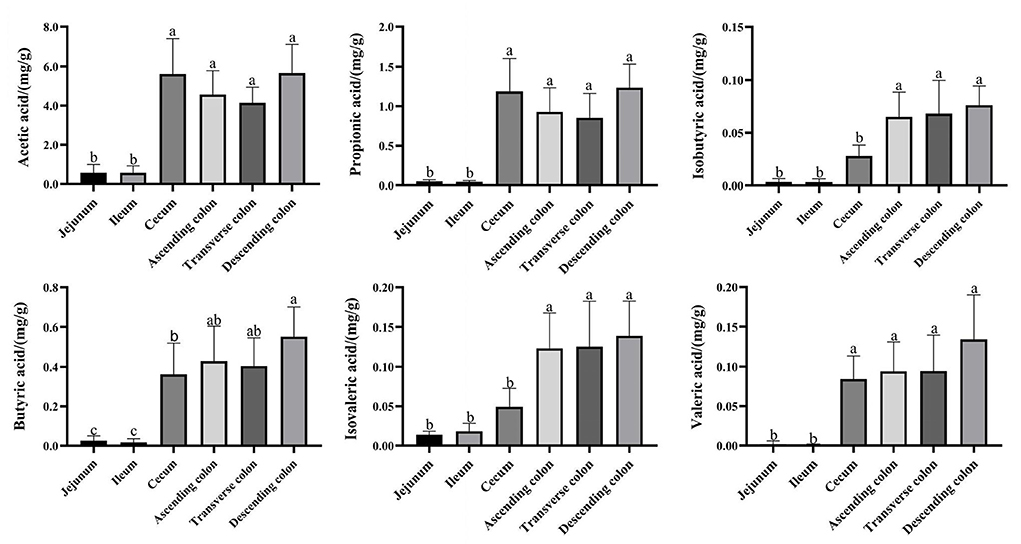
Figure 5. Concentrations of SCFA in different intestinal locations of pigs. Data are shown as mean ± SD (n = 6). The same letter within each column indicates no significant difference (p > 0.05).
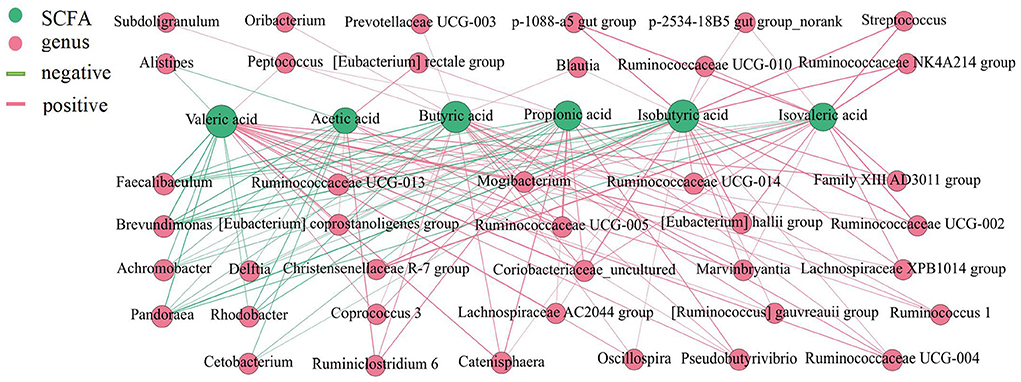
Figure 6. Correlation analysis between the microbial community and the concentrations of SCFAs in the intestine of pigs. The association between the top 100 genera and SCFAs were analyzed using Spearman's correlation method. The node size is proportional to the number of connections.
The proportion of crude ash in the large intestine was significantly greater than that in the small intestine, and the colon had the highest content of crude ash (Figure 7) (P < 0.05). Additionally, the colon had a lower moisture content and approximately the same fat content in each intestinal segment. Next, we performed a network correlation (Figure 8) to analyze the association of the top 100 genera with nutrients in different intestinal segments of pigs. The genera [Eubacterium] hallii group, Lachnospiraceae AC2044 group, Ruminococcaceae UCG-005, Coriobacteriaceae_uncultured, Mogibacterium, Ruminococcaceae UCG-013, Terrisporobacter, Lachnospiraceae NK4A136 group, Peptococcus, Turicibacter, Subdoligranulum and Marvinbryantia showed a positive correlation with the contents of fat and crude ash, whereas the genera Brevundimonas, Veillonella, and Rhodobacter had a negative correlation.
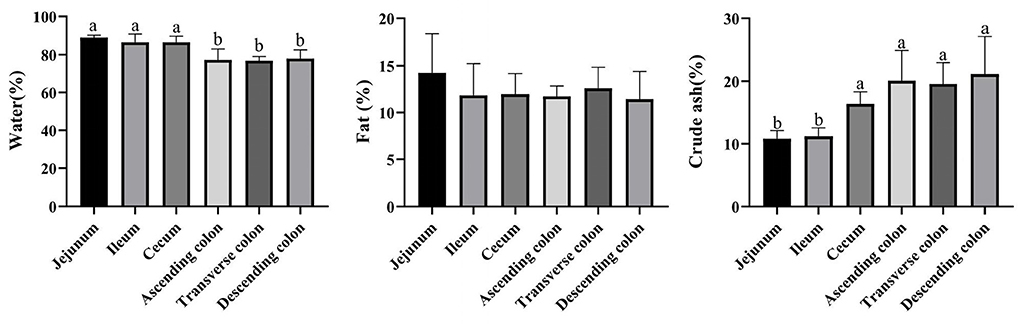
Figure 7. Concentrations of moisture, fat, and crude ash in different intestinal locations of pigs. The concentration of fat and crude ash was based on the dry matter of the intestinal contents. Data are shown as mean ± SD (n = 6). The same letter within each column indicates no significant difference (p > 0.05).
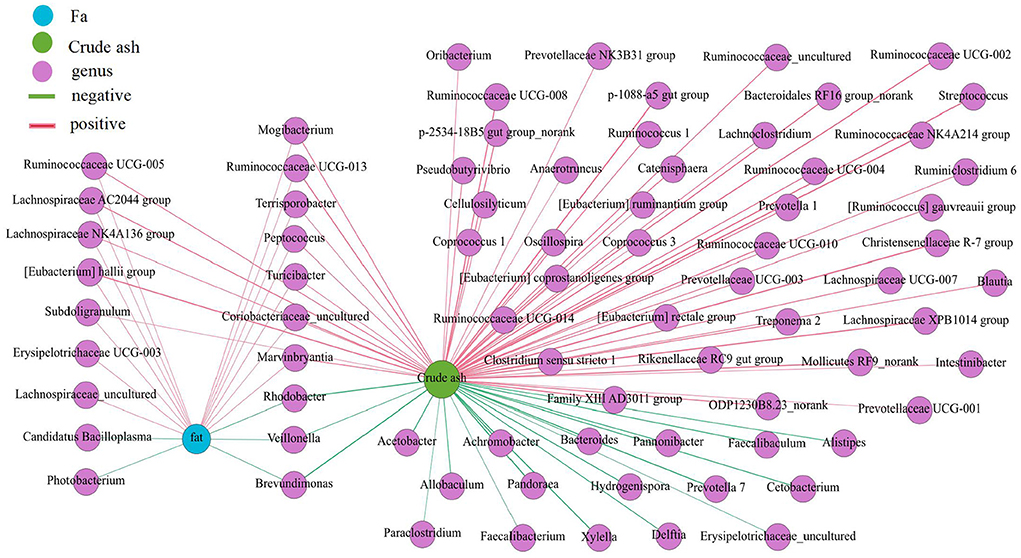
Figure 8. Correlation analysis between the microbial community and the concentration of fat and crude ash in intestine of pigs. The association between the top 100 genera and the fat and crude ash were analyzed using Spearman's correlation method. The node size is proportional to the number of connections.
The amino acid content in the small intestine was higher than that in the large intestine, while the amino acid content in the ascending colon was significantly higher than that in the transverse colon and descending colon (Figure 9) (P < 0.05). Then, a network correlation (Figure 10) was used to analyze the association of the top 100 genera with amino acids in the colon. The genera Dorea, Marvinbryantia, Terrisporobacter, Romboutsia, [Eubacterium] rectale group, and [Eubacterium] hallii group showed a negative correlation with the content of a variety of amino acids, indicating a robust contribution to the synthesis of amino acids. The genus Ruminococcaceae UCG-005 was negatively correlated with histidine, Lachnospiraceae AC2044 group, Coriobacteriaceae_uncultured and Mogibacterium and had a negative correlation with isoleucine and phenylalanine. The genera Lachnoclostridium, Vibrio, Parabacteroides, and Treponema 2 showed a positive correlation with the contents of various amino acids.
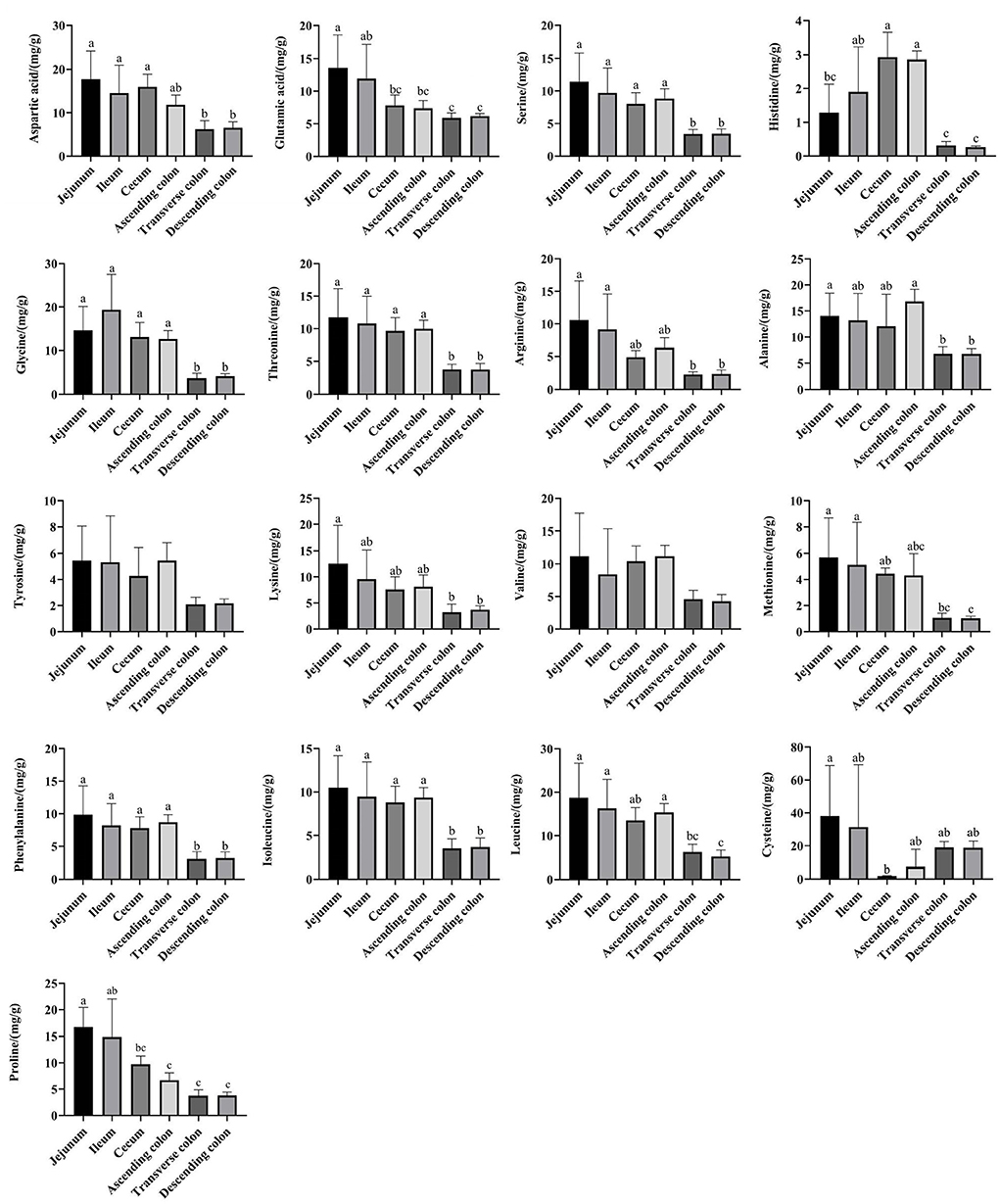
Figure 9. The concentrations of amino acids in different intestinal locations of pigs. The concentration was based on the dry matter of the intestinal contents. Data are shown as mean ± SD (n = 6). The same letter within each column indicates no significant difference (p > 0.05).
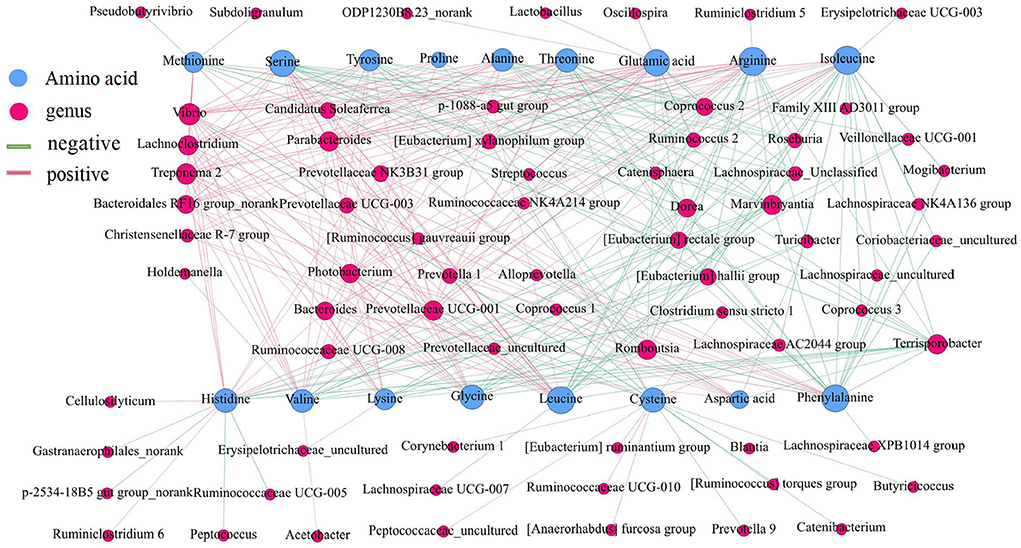
Figure 10. Correlation analysis between the microbial community and the concentrations of amino acids in the intestine of pigs. The association between the top 100 genera in the colon and the amino acids was analyzed using Spearman's correlation method. The node size is proportional to the number of connections.
The genera Ruminococcaceae UCG-005, [Eubacterium] hallii group, Lachnospiraceae AC2044 group, Coriobacteriaceae_uncultured and Mogibacterium clearly showed a positive correlation with the contents of SCFA, fat, and crude ash and a negative correlation with amino acids. Then, we analyzed the relative abundance of these genera in different intestinal locations. The results showed that Ruminococcaceae UCG-005, [Eubacterium] hallii group, Lachnospiraceae AC2044 group, Coriobacteriaceae_uncultured and Mogibacterium were more abundant in the colon and cecum than in the jejunum and ileum (Figure 11) (P < 0.05).
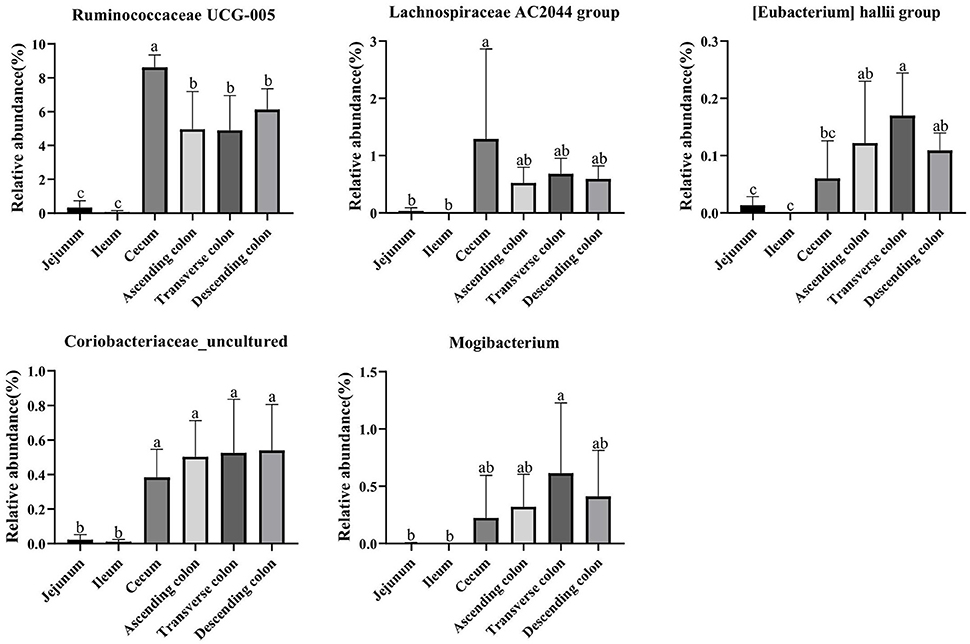
Figure 11. The relative abundance of Ruminococcaceae UCG-005, [Eubacterium] hallii group, Lachnospiraceae AC2044 group, Coriobacteriaceae_uncultured and Mogibacterium in different intestinal locations of pigs. Data are shown as mean ± SD (n = 6). The same letter within each column indicates no significant difference (p > 0.05).
The intestinal tract of humans and mammals is colonized by a dense and highly complex microbial community composed mainly of bacteria, which form a relatively dynamic and stable microecosystem in the intestinal environment and play an important role in the growth and health of the host (44–46). The intestinal microbiota is involved in nutrient absorption, metabolism and storage (47). Previous studies have shown that the intestinal microbiota contribute a lot in regulating the nutrient metabolism of hosts and maintaining energy balance (28, 48, 49). Due to the similarity between the human and pig microbiota and the knowledge that approximately 96% similarity of the functional pathways between the human and pig gut microbiota (34, 50), in this study, we used the pig as a model to analyze the microbial composition and contents of SCFA, amino acids, fat, and crude ash in different intestinal locations to explore the correlation between the intestinal microbiota and the metabolism and absorption of nutrients.
The diversity and abundance of the microbiota were higher in the large intestine than in the small intestine; simultaneously, the microbiota in the colon was the richest and the most diverse, which was consistent with the results of a previous study showing that the richness and diversity of the microbiota gradually increased along the digestive tract from the duodenum to the colon (51). PCoA showed that the microbiota in the small intestine clearly differed from that in the large intestine, which might be closely related to the physiological functions of each intestinal segment. Additionally, each region harbor specific microbial community (52). Proteobacteria were predominant in the jejunum and ileum and made up a smaller percentage in the cecum and colon, whereas the proportion of Firmicutes was higher than in the cecum and colon, which was consistent with the findings of several previous studies on the distribution of different intestinal microbiota in pigs (1) (53, 54). In contrast, the relative abundance of Acetobacter, Veillonella, Bacteroides, Erysipelotrichaceae_uncultured, Xylella and Faecalibaculum were higher in the small intestine of pigs, while [Eubacterium], Ruminococcaceae, Mollicutes, Christensenellaceae, Lachnospiraceae, Streptococcus, Terrisporobacter and Ruminococcaceae were enriched in the large intestine. The main reason for the unique microbiota in the difficult intestine is that from the proximal small intestine to the large intestine, a strong pH gradient and a large oxygen gradient can be observed (55, 56). The small intestine is a harsh microenvironment formicrobial life because of the shorter transit time and lower pH values and there is an increased proportion of aerobic or facultative anaerobic bacteria. In contrast, the large intestine dominantly hosts a number of anaerobic bacteria (57). Moreover, within the same intestinal segment, the distribution of bacteria in different spaces was also distinct. In this study, diversity of the microbial composition was found in different colonic locations, with the descending colon containing higher proportions of the Ruminococcaceae UCG-005 while the transverse colon was enriched with Terrisporobacter, Clostridium sensu stricto 1, Romboutsia and Turicibacter, and the ascending colon had greater abundances of Prevotellaceae NK3B31 group, Treponema 2, Ruminiclostridium 5. Numerous investigations have been conducted to examine the intestinal microbiota in different human intestinal segments and have shown that the jejunum is mainly composed of gram-positive aerobic bacteria, including Lactobacillus, Streptococcus and Staphylococcus; the number of ileal anaerobic bacteria exceeds aerobic bacteria; and more than 98% of the colon contains obligate anaerobic bacteria, mainly Bacteroides, Bifidobacterium, and Eubacterium (56, 58, 59). The similarity of bacterial structure leads to the similar nutrient absorption process between pigs and human sand the colon is the main place of bacterial fermentation between pigs and humans (35, 60), therefore the application of pigs in human nutrition model models is increasingly gaining traction.
SCFAs are considered indirect microbial metabolism substrates and involved in the regulation of energy metabolism, immunity, adipose tissue expansion and modulation of cancer cell development (61, 62). In our study, the concentrations of SCFA were higher in the large intestine than in the small intestine due to the slower rate of flow in the large intestine and metabolism favoring fermentation of indigestible carbohydrates; additionally, the anaerobic state of the colon provides an ideal environment for anaerobic digestion (56, 63). Previous studies have reported that anaerobic bacteria in the colon ferment undigested carbohydrates from the small intestine to produce SCFAs. Propionic acid is the main product of Bacteroidetes fermentation, and butyric acid is mainly produced by the metabolism of Firmicutes (64), in accordance with our findings. The genera Ruminococcaceae (UCG-004, UCG-005, UCG-014, UCG-013), Coriobacteriaceae_uncultured, [Eubacterium] (hallii group, coprostanoligenes group), Mogibacterium, Pseudobutyrivibrio, Marvinbryantia, Catenisphaera, and Lachnospiraceae AC2044 group had a positive correlation with a variety of SCFAs based on the network analysis. Simultaneously, these genera were enriched in the large intestine, which suggested that the large intestine was the main region of SCFA production and that these genera were the core bacteria of fiber fermentation.
The crude ash and fat are important indicators to reflect nutrients. The content of crude ash is related to the absorption and utilization of minerals, the gut microbiota can affect the host mineral metabolism and participate in the metabolism of calcium, iron, magnesium, selenium, copper, and zinc (65–67). The fat content is involved in metabolism and body energy reserves and is an important index affecting human health (1, 68). Due to different digestive enzymes in the jejunum, ileum, cecum and colon, there are differences in digestion, absorption and utilization of nutrients in different intestinal locations (45, 56, 69). In our study, the proportion of crude ash in the large intestine was significantly greater than that in the small intestine. The genera [Eubacterium] hallii group, Lachnospiraceae AC2044 group, Ruminococcaceae UCG-005, Coriobacteriaceae_uncultured, and Mogibacterium showed a positive correlation with fat and crude ash. The utilization of amino acids is widely distributed among bacteria residing in the digestive tract of humans and animals (18, 70), and the microbial community structure in different intestinal segments affects compartmentalized amino acid metabolism (54). In our study, the amino acid content was higher in the small intestine than the large intestine, while it was significantly higher in the ascending colon than in the transverse colon and descending colon, which is consistent with a previous study showing faster transit and a preference for amino acid metabolism in the small intestine, where the community primarily consisted of rapidly dividing facultative anaerobes such as Proteobacteria and Lactobacillales (71). In contrast, bacteria in the large intestine use amino acids mainly for catabolism. Bacteria in the large intestine of humans and animals can degrade a large number of amino acids, and the products of fermented amino acids, including short-chain and branched-chain fatty acids, can also participate in the energy metabolism of the host (72). The genera Dorea, Marvinbryantia, Terrisporobacter, Romboutsia, [Eubacterium] rectale group, [Eubacterium] hallii group, Ruminococcaceae UCG-005, Lachnospiraceae AC2044 group, Coriobacteriaceae_uncultured, and Mogibacterium showed a negative correlation with the contents of a variety of amino acids in the colon. Moreover, most of these bacteria were enriched in the transverse colon and descending colon, making a robust contribution to the degradation of amino acids. The genera Ruminococcaceae UCG-005, [Eubacterium] hallii group, Lachnospiraceae AC2044 group, Coriobacteriaceae_uncultured and Mogibacterium showed a positive correlation with the contents of SCFA, fat, and crude ash and a negative correlation with amino acids and these genera were mainly distributed in the colon and cecum.
This work presents the first overview of the microbial community and nutrient metabolism in different intestinal locations using a pig model. The richness and diversity of intestinal microbial communities gradually increased from the small intestine to the large intestine. The concentrations of SCFA were higher in the large intestine than in the small intestine, and the concentrations of amino acids in the small intestine were higher than in the large intestine, while the amino acid content in the ascending colon was significantly higher than those in the transverse colon and descending colon. The Ruminococcaceae UCG-005, Coriobacteriaceae_uncultured, [Eubacterium] hallii, Mogibacterium, and Lachnospiraceae AC2044 group had a positive correlation with the contents of SCFA, fat, and crude ash and a negative correlation with amino acids in different gut locations of pigs. Collectively, these correlation outcomes prompted us to further understand the relationship between the microbiota and nutrient metabolism in different gut environments, which is important for gut health and whole-body homeostasis.
The sequencedata of this study have been submitted to the NCBI Sequence Read Archive (SRA) database under the study accession number: PRJNA853391.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA853391.
The animal study was reviewed and approved by (ZAAS-2017-009).
Experiment design: KC, LL, YX, and YS. Animal experiments: YS, XZ, and GZ. Data analysis and visualization: YS, XD, and YX. Roles, writing—original draft, writing—review and editing: YS and YX. All authors contributed to the article and approved the submitted the manuscript.
This work was supported by the National Natural Science Foundation of China (31972999), Development Center of Animial Husbandry and Agricultural Machinery in Jinhua City (OBANG2021-FW030-ZFCG0321), State Key Laboratory for Managing Biotic and Chemical Threats to the Quality and Safety of Agro-products, Zhejiang Academy of Agricultural Sciences (2010DS700124-ZZ1905).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Armet A, Deehan E, O'Sullivan A, Mota J, Field C, Prado C, et al. Rethinking healthy eating in light of the gut microbiome. Cell Host Microbe. (2022) 30:764–85. doi: 10.1016/j.chom.2022.04.016
2. Li M, Zhou H, Pan X, Xu T, Zhang Z, Zi X, et al. Corrigendum: cassava foliage affects the microbial diversity of Chinese indigenous geese caecum using 16s Rrna Sequencing. Sci Rep. (2017) 7:46837. doi: 10.1038/srep46837
3. Miao Z, Du W, Xiao C, Su C, Gou W, Shen L, et al. Gut microbiota signatures of long-term and short-term plant-based dietary pattern and cardiometabolic health: a prospective cohort study. BMC Med. (2022) 20:204. doi: 10.1186/s12916-022-02402-4
4. Corrigan A, Leeuw Mde, Penaud-Frézet S, Dimova D, Murphy RA. Phylogenetic and functional alterations in bacterial community compositions in broiler ceca as a result of mannan oligosaccharide supplementation. Applied Environ Microbiol. (2015) 81:3460–70. doi: 10.1128/AEM.04194-14
5. Yang H, Wu J, Huang X, Zhou Y, Zhang Y, Liu M, et al. Abo genotype alters the gut microbiota by regulating galnac levels in pigs. Nature. (2022) 606:358–67. doi: 10.1038/s41586-022-04769-z
6. Forgie A, Drall K, Bourque S, Field C, Kozyrskyj A, Willing B. The impact of maternal and early life malnutrition on health: a diet-microbe perspective. BMC Med. (2020) 18:135. doi: 10.1186/s12916-020-01584-z
7. Murakami M, Tognini P, Liu Y, Eckel-Mahan K, Baldi P, Sassone-Corsi P. Gut Microbiota directs Pparγ-driven reprogramming of the liver circadian clock by nutritional challenge. EMBO Rep. (2016) 17:1292–303. doi: 10.15252/embr.201642463
8. Pifer R, Russell R, Kumar A, Curtis M, Sperandio V. Redox, amino acid, and fatty acid metabolism intersect with bacterial virulence in the gut. Proc Natl Acad Sci U S A. (2018) 115:E10712–E9. doi: 10.1073/pnas.1813451115
9. Hua Y, Yun X, Robinson K, Wang J, Xiao Y. Gut microbiota is a major contributor to adiposity in pigs. Front Microbiol. (2018) 9:3045. doi: 10.3389/fmicb.2018.03045
10. Alou MT, Lagier JC, Raoult D. Diet influence on the gut microbiota and dysbiosis related to nutritional disorders. Human Microbiome J. (2016) 1:3–11. doi: 10.1016/j.humic.2016.09.001
11. Yao Y, Cai X, Fei W, Ye Y, Zhao M, Zheng C. The role of short-chain fatty acids in immunity, inflammation and metabolism. Critical Rev Food Sci Nutr. (2022) 62:1–12. doi: 10.1080/10408398.2020.1854675
12. Stinson L, Geddes D. Microbial metabolites: the next frontier in human milk. Trends Microbiol. (2022) 30:408–10. doi: 10.1016/j.tim.2022.02.007
13. Yu H, Kim SH, Noh MY, Lee S, Park Y. Relationship between dietary fiber intake and the prognosis of amytrophic lateral sclerosis in Korea. Nutrients. (2020) 12:3420. doi: 10.3390/nu12113420
14. Zhang YJ, Liu Q, Zhang WM, Zhang ZJ, Wang WL, Zhuang S. Gastrointestinal microbial diversity and short-chain fatty acid production in pigs fed different fibrous diets with or without cell wall-degrading enzyme supplementation. Livestock Ence. (2018) 207:105–16. doi: 10.1016/j.livsci.2017.11.017
15. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. (2014) 12:661–72. doi: 10.1038/nrmicro3344
16. Sheridan OP, Martin JC, Lawley TD, Browne HP, Harris HMB, Bernalier-Donadille A, et al. Polysaccharide utilization loci and nutritional specialization in a dominant group of butyrate-producing human colonic firmicutes. Microb Genom. (2016) 2: e000043. doi: 10.1099/mgen.0.000043
17. Kaiko G, Ryu S, Koues O, Collins P, Solnica-Krezel L, Pearce E, et al. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell. (2016) 16:1708–20. doi: 10.1016/j.cell.2016.05.018
18. Dai Z, Wu G, Zhu W. Amino acid metabolism in intestinal bacteria: links between gut ecology and host health. Frontiers Biosci. (2011) 16:1768–86. doi: 10.2741/3820
19. Zhang Z, Mu X, Shi Y, Zheng H. Distinct roles of honeybee gut bacteria on host metabolism and neurological processes. Microbiol Spect. (2022) 10:e0243821. doi: 10.1128/spectrum.02438-21
20. Liu Y, Wang X, Hu CA. Therapeutic potential of amino acids in inflammatory bowel disease. Nutrients. (2017) 9:920. doi: 10.3390/nu9090920
21. Duca F, Waise T, Peppler W, Lam T. The metabolic impact of small intestinal nutrient sensing. Nat Commun. (2021) 12:903. doi: 10.1038/s41467-021-21235-y
22. Agus A, Planchais J, Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease - sciencedirect. Cell Host Microbe. (2018) 23:716. doi: 10.1016/j.chom.2018.05.003
23. Jochems P, Garssen J, van Keulen A, Masereeuw R, Jeurink P. Evaluating human intestinal cell lines for studying dietary protein absorption. Nutrients. (2018) 10:3022. doi: 10.3390/nu10030322
24. Metges CC. Contribution of microbial amino acids to amino acid homeostasis of the host. J Nutr. (2000) 130:1857S. doi: 10.1093/jn/130.7.1857S
25. Wu C, Lyu W, Hong Q, Zhang X, Yang H, Xiao Y. Gut microbiota influence lipid metabolism of skeletal muscle in pigs. Front Nutr. (2021) 8:675445. doi: 10.3389/fnut.2021.675445
26. Zhao G, Xiang Y, Wang X, Dai B, Zhang X, Ma L, et al. Exploring the possible link between the gut microbiome and fat deposition in pigs. Oxid Med Cell Longev. (2022) 2022:1098892. doi: 10.1155/2022/1098892
27. Lyu W, Xiang Y, Wang X, Li J, Yang C, Yang H, et al. Differentially expressed hepatic genes revealed by transcriptomics in pigs with different liver lipid contents. Oxid Med Cell Longev. (2022) 2022:2315575. doi: 10.1155/2022/2315575
28. Yang H, Huang X, Fang S, He M, Zhao Y, Wu Z, et al. Unraveling the fecal microbiota and metagenomic functional capacity associated with feed efficiency in pigs. Front Microbiol. (2017) 8:1555. doi: 10.3389/fmicb.2017.01555
29. Berry SE, Valdes AM, Long HN, Segata N. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals nature. Medicine. (2021) 27:321–32. doi: 10.1038/s41591-020-01183-8
30. Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med. (2017) 15:73. doi: 10.1186/s12967-017-1175-y
31. Wang X, Tsai T, Zuo B, Wei X, Deng F, Li Y, et al. Donor age and body weight determine the effects of fecal microbiota transplantation on growth performance, and fecal microbiota development in recipient pigs. J Anim Sci Biotechnol. (2022) 13:49. doi: 10.1186/s40104-022-00696-1
32. Tilocca B, Burbach K, Heyer C, Hoelzle L, Mosenthin R, Stefanski V, et al. Dietary changes in nutritional studies shape the structural and functional composition of the pigs' fecal microbiome-from days to weeks. Microbiome. (2017) 5:144. doi: 10.1186/s40168-017-0362-7
33. Lee S, Kang K. Omega-3 fatty acids modulate cyclophosphamide induced markers of immunosuppression and oxidative stress in pigs. Sci Rep. (2019) 9:2684. doi: 10.1038/s41598-019-39458-x
34. Chang HW, Mcnulty NP, Hibberd MC, O'Donnell D, Gordon JI. Gut microbiome contributions to altered metabolism in a pig model of undernutrition. Proc Natl Acad Sci U S A. (2021) 118:e2024446118. doi: 10.1073/pnas.2024446118
35. Lunney J, Van Goor A, Walker K, Hailstock T, Franklin J, Dai C. Importance of the pig as a human biomedical model. Sci Trans Med. (2021) 13:eabd5758. doi: 10.1126/scitranslmed.abd5758
36. Lawley B, Tannock GW. Analysis of 16s Rrna gene amplicon sequences using the qiime software package. Methods Molecular Biol. (2017) 1537:153. doi: 10.1007/978-1-4939-6685-1_9
37. Tanja M. Steven, Salzberg. Flash: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. (2011) 27:2957–63. doi: 10.1093/bioinformatics/btr507
38. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve bayesian classifier for rapid assignment of Rrna sequences into the new bacterial taxonomy. Appl Environ Microbiol. (2007) 73:5261–7. doi: 10.1128/AEM.00062-07
39. Segata N, Izard J, Waldron L, Gevers D. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
40. Xiao Y, Li K, Yun X, Zhou W, Hua Y. The fecal microbiota composition of boar duroc, yorkshire, landrace and hampshire pigs. Asian-Australas J Anim Sci. (2017) 30:1456–63. doi: 10.5713/ajas.16.0746
41. Du X, Xiang Y, Lou F, Tu P, Zhang X, Hu X, et al. Microbial community and short-chain fatty acid mapping in the intestinal tract of quail. Animals. (2020) 10:1006. doi: 10.3390/ani10061006
42. AOAC. Official Methods of Analysis. Arlington, VA: Association of Official Analytical Chemists, Inc (2000).
43. Sarita A, Sushma D. Tracking the spread of Covid-19 in India via social networks in the early phase of the pandemic. J Travel Med. (2020) 27:130. doi: 10.1093/jtm/taaa130
44. Dai D, Yang Y, Yu J, Dang T, Qin W, Teng L, et al. Interactions between gastric microbiota and metabolites in gastric cancer. Cell Death Dis. (2021) 12:1104. doi: 10.1038/s41419-021-04396-y
45. Xiao Y, Kong F, Xiang Y, Zhou W, Wang J, Yang H, et al. Comparative biogeography of the gut microbiome between Jinhua and landrace pigs. Rep. (2018) 8:5985. doi: 10.1038/s41598-018-24289-z
46. Ma L, Shen Q, Lyu W, Lv L, Wang W, Yu M, et al. Clostridium Butyricum and its derived extracellular vesicles modulate gut homeostasis and ameliorate acute experimental colitis. Microbiol Spectrum. (2022) 10:e0136822. doi: 10.1128/spectrum.01368-22
47. Cignarella F, Cantoni C, Ghezzi L, Salter A, Dorsett Y, Chen L, et al. Intermittent fasting confers protection in Cns autoimmunity by altering the gut microbiota. Cell Metabolism. (2018) 27:1222–35.e6. doi: 10.1016/j.cmet.2018.05.006
48. Xiao Y, Zou H, Li J, Song T, Lv W, Wang W, et al. Impact of quorum sensing signaling molecules in gram-negative bacteria on host cells: current understanding and future perspectives. Gut Microbes. (2022) 14:2039048. doi: 10.1080/19490976.2022.2039048
49. Vigors S, Sweeney T, O'Shea C, Kelly A, O'Doherty J. Pigs that are divergent in feed efficiency, differ in intestinal enzyme and nutrient transporter gene expression, nutrient digestibility and microbial activity. Animal. (2016) 10:1848–55. doi: 10.1017/S1751731116000847
50. Xiao L, Estellé J, Kiilerich P, Ramayo-Caldas Y, Xia Z, Feng Q, et al. A reference gene catalogue of the pig gut microbiome. Nature Microbiol. (2016) 1:16161. doi: 10.1038/nmicrobiol.2016.161
51. Yang H, Huang X, Fang S, Xin W, Huang L, Chen C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci Rep. (2016) 6:27427. doi: 10.1038/srep27427
52. Tropini C, Earle K, Huang K, Sonnenburg J. The gut microbiome: connecting spatial organization to function. Cell Host Microbe. (2017) 21:433–42. doi: 10.1016/j.chom.2017.03.010
53. Wang C, Wei S, Chen N, Xiang Y, Wang Y, Jin M. Characteristics of gut microbiota in pigs with different breeds, growth periods and genders. Microb Biotechnol. (2022) 15:793–804. doi: 10.1111/1751-7915.13755
54. Li N, Zuo B, Huang S, Zeng B, Han D, Li T, et al. Spatial heterogeneity of bacterial colonization across different gut segments following inter-species microbiota transplantation. Microbiome. (2020) 8:161. doi: 10.1186/s40168-020-00917-7
55. Albenberg L, Esipova TV, Judge CP, Bittinger K, Wu GD. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota in humans and mice. Gastroenterology. (2014) 147:1055–63.e8. doi: 10.1053/j.gastro.2014.07.020
56. Wang Y, Song W, Wang J, Wang T, Xiong X, Qi Z, et al. Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J Experiment Med. (2020) 217(2). doi: 10.1084/jem.20191130
57. Spiga L, Winter M, Furtado de Carvalho T, Zhu W, Hughes E, Gillis C, et al. An oxidative Central metabolism enables salmonella to utilize microbiota-derived succinate. Cell Host Microbe. (2017) 22:291–301.e6. doi: 10.1016/j.chom.2017.07.018
58. Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The long-term stability of the human gut microbiota. Science. (2013) 341:1237439. doi: 10.1126/science.1237439
59. James KR, Gomes T, Elmentaite R, Kumar N, Teichmann SA. Distinct microbial and immune niches of the human colon. Nat Immunol. (2020) 21:343–53. doi: 10.1038/s41590-020-0602-z
60. Roura E, Koopmans S, Lallès J, Le Huerou-Luron I, de Jager N, Schuurman T, et al. Critical review evaluating the pig as a model for human nutritional physiology. Nutr Res Rev. (2016) 29:60–90. doi: 10.1017/S0954422416000020
61. Zhang J, Sun J, Chen X, Nie C, Zhao J, Guan W, et al. Clostridium butyricumcombination of and corn bran optimized intestinal microbial fermentation using a weaned pig model. Front Microbiol. (2018) 9:3091. doi: 10.3389/fmicb.2018.03091
62. Xu E, Yang H, Ren M, Wang Y, Xiao Y. Identification of enterotype and its effects on intestinal butyrate production in pigs. Animals. (2021) 11:730. doi: 10.3390/ani11030730
63. Tan FPY, Beltranena ET, Zijlstra R. Resistant starch: implications of dietary inclusion on gut health and growth in pigs: a review. J Animal Sci Biotechnol. (2022) 13:613–27. doi: 10.1186/s40104-021-00644-5
64. Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proceedings Nutr Soc. (2003) 62:67–72. doi: 10.1079/PNS2002207
65. Li W, Liu Y, Jiang W, Yan X. Proximate composition and nutritional profile of rainbow trout (oncorhynchus mykiss) heads and skipjack Tuna (katsuwonus pelamis) heads. Molecules. (2019) 24:3189. doi: 10.3390/molecules24173189
66. Pakroo S, Tarrah A, Duarte V, Corich V, Giacomini A. Microbial diversity and nutritional properties of persian “yellow curd” (Kashk Zard), a promising functional fermented food. Microorganisms. (2020) 8:1658. doi: 10.3390/microorganisms8111658
67. Skrypnik K, Suliburska J. Association between the gut microbiota and mineral metabolism. J Sci Food Agric. (2018) 98:2449–60. doi: 10.1002/jsfa.8724
68. Tsukuda N, Yahagi K, Hara T, Watanabe Y, Matsuki T. Key bacterial taxa and metabolic pathways affecting gut short-chain fatty acid profiles in early life. ISME J. (2021) 15:1–17. doi: 10.1038/s41396-021-00937-7
69. Kelly J, Daly, Kristian, Moran, Andrew W, et al. Composition and diversity of mucosa-associated microbiota along the entire length of the pig gastrointestinal tract; dietary influences. Environ Microbiol. (2017) 19:1425–38. doi: 10.1111/1462-2920.13619
70. Mehta S, Huey SL, McDonald D, Knight R, Finkelstein JL. Nutritional interventions and the gut microbiome in children. Annu Rev Nutr. (2021) 41:479–510. doi: 10.1146/annurev-nutr-021020-025755
71. Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch M, Ojeda P, et al. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe. (2018) 23:458–69.e5. doi: 10.1016/j.chom.2018.03.011
Keywords: gut microbiota, short-chain fatty acids, nutrients, amino acid, intestinal tract
Citation: Song Y, Chen K, Lv L, Xiang Y, Du X, Zhang X, Zhao G and Xiao Y (2022) Uncovering the biogeography of the microbial commmunity and its association with nutrient metabolism in the intestinal tract using a pig model. Front. Nutr. 9:1003763. doi: 10.3389/fnut.2022.1003763
Received: 26 July 2022; Accepted: 01 September 2022;
Published: 26 September 2022.
Edited by:
Jie Yin, Hunan Agricultural University, ChinaReviewed by:
Gong Yujie, Henan Agricultural University, ChinaCopyright © 2022 Song, Chen, Lv, Xiang, Du, Zhang, Zhao and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingping Xiao, eGlhb3lwQHphYXMuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.