 Donghui Luo1,2
Donghui Luo1,2 Hao Dong
Hao Dong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Nutr. , 28 September 2022
Sec. Food Chemistry
Volume 9 - 2022 | https://doi.org/10.3389/fnut.2022.1001671
This article is part of the Research Topic Novel Chemical, Microbiological and Physical Approaches In Food Safety Control, volume II View all 11 articles
A solid phase extraction-high-performance liquid chromatography-tandem Orbitrap high resolution mass spectrometry (HPLC-Orbitrap HRMS) method was established for the determination of 12 mycotoxins (ochratoxin A, ochratoxin B, aflatoxin B1, aflatoxin B2, aflatoxin G1, aflatoxin G2, HT-2 toxin, sterigmatocystin, diacetoxysciroenol, penicillic acid, mycophenolic acid, and citreoviridin) in edible oil, soy sauce, and bean sauce. Samples were extracted by 80:20 (v:v) acetonitrile-water solution, purified by PRiME HLB column, separated by aQ C18 column with mobile phase consisting of 0.5 mmol/L ammonium acetate-0.1% formic acid aqueous solution and methanol. The results showed that the limits of detection (LODs) and limits of quantification (LOQs) of 12 mycotoxins were 0.12–1.2 μg/L and 0.40–4.0 μg/L, respectively. The determination coefficients of 12 mycotoxins in the range of 0.20–100 μg/L were > 0.998. The average recoveries in soy sauce and bean sauce were 78.4–106.8%, and the relative standard deviations (RSDs) were 1.2–9.7% under three levels, including LOQ, 2× LOQ and 10 × LOQ. The average recoveries in edible oil were 78.3–115.6%, and the precision RSD (n = 6) was 0.9–8.6%. A total of 24 edible oils, soy sauce and bean sauce samples were analyzed by this method. AFB1, AFB2, sterigmatocystin and mycophenolic acid were detected in several samples at concentrations ranging from 1.0 to 22.1 μg/kg. The method is simple, sensitive, and rapid and can be used for screening and quantitative analysis of mycotoxin contamination in edible oil, soy sauce, and bean sauce.
Mycotoxins are toxic secondary metabolites produced by mycotoxin-producing fungi under suitable environmental conditions (1–3). Currently, there are more than 400 known mycotoxins. Edible oils such as rapeseed oil, peanut oil, corn oil, sesame oil, soy sauce and bean sauce are easily contaminated by mycotoxins because most cereals are used as raw materials in the preparation process (4, 5). It has been reported that ~25% of wheat, corn, sorghum and rice produce toxic and harmful mycotoxins due to mildew during production, processing, transportation and storage every year (6). At present, aflatoxin (AFT), ochratoxin A (OTA) and zearalenone (ZEN) have a great influence on human health, which will damage human liver function, cause cancer and teratogenicity and induce immunosuppressive diseases after exceeding a certain intake (7, 8). The World Health Organization has included mycotoxins in the key monitoring objects of the food safety system (9). In China, GB2761-2017 “National Standard for Food Safety Limits of Mycotoxins in Food” also has strict regulations on the limit indicators of some mycotoxins in food. Under natural conditions, edible oils and condiments may be contaminated by various mycotoxins. According to the current national standards, it is necessary to use several detection methods for multiple experimental analyses to determine the content of different mycotoxins (10–12). The determination methods are not only cumbersome but also inefficient. Therefore, it is urgent to establish a synchronous detection method for multiple mycotoxins.
Anastassiades et al. (13) proposed the QuEChERS (Quick, Easy, Cheap, Effective, Rugged, Safe) method, namely, dispersive solid phase extraction, but it is still insufficient to extract many toxins from complex substrates. Oasis PRiME HLB is a type of reversed-solid phase extraction (SPE) adsorbent that can simplify and accelerate the SPE process and can obtain cleaner extracts compared with other sample pretreatment methods. Compared with other SPE products, it can also remove more than 90% of endogenous phospholipids and is widely used in the detection of food organic pollutants (14). At present, colloidal gold immunochromatography, enzyme-linked immunosorbent assay (ELISA) kits, immunoaffinity column-high-performance liquid chromatography and isotope dilution liquid chromatography tandem mass spectrometry are commonly used to detect mycotoxins (3, 15, 16). Liquid chromatography-tandem mass spectrometry (LC–MS/MS) has higher selectivity and sensitivity and has gradually become the main means for the simultaneous detection of various mycotoxins (9, 17, 18). Orbitrap high-resolution mass spectrometry (Orbitrap HRMS) has higher selectivity and resolution than ordinary mass spectrometry and can effectively reduce the interference of impurities in complex matrices (19–22).
Therefore, the samples were simply extracted and purified by a PRiME HLB solid phase extraction column in this study, and the conditions of liquid chromatography and mass spectrometry were optimized. An HPLC-Orbitrap HRMS method for the determination of mycotoxins in edible oil, soy sauce, and bean sauce was established. The method has advantages such as simplicity, rapidity and high flux, which is suitable for the screening and detection of mycotoxins in edible oil, soy sauce and bean sauce and reduce food safety problems caused by mycotoxin residues.
The Thermo Q Exactive Focus High Performance Liquid Chromatography–Mass Spectrometry System includes a Dionex Ultimate 3000 Liquid Phase Pump, Autosampler, Column Oven and Orbitrap High Resolution Mass Spectrometry Section (Thermo Fisher Scientific, Massachusetts, USA). XCalibur 4.0 software (Thermo Fisher Scientific, Massachusetts, USA) was used for mass spectrometer control and data processing. A Thermo Accucore aQ C18 (2.1 × 150 mm, 2.6 μm) was used as the chromatographic column. The samples were vortexed with an MS3 basic vortex mixer (IKA GmbH, Staufen, Germany). A KQ-250DV CNC ultrasonic cleaning device (Kunshan, Jiangsu, China) was used for supersonic-assisted extraction. Ultrapure water (18.2 MΩ·cm) prepared from a Milli-Q ultrapure system (Millipore, Bedford, MA, USA) was used in the whole experiment.
Twelve mycotoxins, as shown in Table 1, were purchased from Shanghai Anpu Scientific Instruments Co., Ltd. and Adamas Reagent Company of Switzerland. These standards all have a purity of or higher than 98.0%. Acetonitrile, methanol, ammonium acetate, and formic acid were obtained from CNW Technologies GmbH (CNW, Düsseldorf, Germany). A PRiME HLB solid phase extraction column was purchased from Waters Company (60 mg/3 cc, Waters, Beverly, MA, USA).
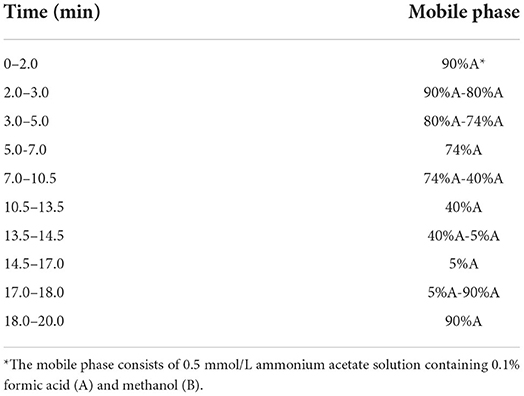
Table 1. The gradient elution procedure of HPLC.
Preparation of standard solutions: The standard reference materials of each mycotoxin were carefully measured and prepared into a 1.00 mg/L mixed standard reserve solution with acetonitrile, which was stored in a refrigerator at 4 °C. Then, an appropriate amount of standard reserve solution was transferred and prepared with acetonitrile to form a series of mixed standard curves with concentrations of 100, 50, 20, 10, 5, 2, 1, 0.5, and 0.2 μg/L.
Thirteen kinds of edible oil samples and 11 soy sauce and bean sauce samples were purchased from local supermarkets and online shopping malls in Guangzhou. A 2.0 g sample was weighed and placed in a 50 mL centrifuge tube, and 20 mL acetonitrile-water 80:20 (v/v) solution was added. After mixing, the sample was extracted by oscillation for 10 min and centrifuged at 500 × g for 5 min at room temperature. Ten milliliters of supernatant was transferred into a 50 mL centrifuge tube, and 10 mL of n-hexane was added and vortexed for 1 min. Then, the mixture was centrifuged at 500 × g for 3 min at room temperature. After that, a PRiME HLB column and HLB solid phase extraction column were adopted for purification. Finally, the purified solution was dried with nitrogen at 40°C. Then, the residual was reconstructed with 1.0 mL acetonitrile and filtered by a 0.22 μm polytetrafluoroethene (PTFE) syringe filter (Waters, Beverly, MA, USA).
The mobile phase consisted of 0.5 mmol/L ammonium acetate solution containing 0.1% formic acid (A) and methanol (B). The gradient elution procedure is presented in Table 1. The injection volume was 5 μL. The flow rate was 0.3 mL/min.
The Q Exactive Focus mass spectrometry system was equipped with a HESI ion source using positive ion mode, spray voltage 3.5 kV, and capillary and spray temperatures of 320°C and 250°C, respectively. The sheath and auxiliary gas pressures were set at 45 arb and 8 arb, respectively, and the S-lens RF voltage was 50 V. Both the spray gas and collision gas were nitrogen. Correction solutions (a solution of 2 μg/mL caffeine, 1 μg/mL MRFA, 0.001% Ultramark 1621 and 0.0005% n-butylamine; a solution of 2.9 μg/mL sodium dodecyl sulfate, 5.4 μg/mL sodium taurocholate and 0.001% Ultramark 1621) were used to correct the mass axis once every 7 days. The scanning mode was full MS/dd-MS2 mode. The full MS first-level full scanning range was m/z 100–650, resolution was 70000, automatic gain control AGC and automatic injection time IT were set to 1.0 e6 and 100 ms, respectively; the data-dependent AGC of dd-MS2 was set to 1.0 e5, the resolution was set to 17,500, the maximum IT was set to 60 ms, the separation window was set to 2.0 m/z, the normalized collision energy (NCE) of each compound was set to 20, 40, and 60%, and the dynamic exclusion was set to 8 s.
Compared with triple quadrupole mass spectrometry, Orbitrap high-resolution mass spectrometry is simpler to operate and optimize mass spectrometry conditions (20). First, 12 target standards (concentration 100 μg/L) were scanned by full MS with a resolution of 70,000, and qualitative screening and quantitative detection were carried out according to the accurate mass number of primary parent ions of the target compounds. The quasi-molecular ion peaks of [M + H] +, [M + NH4] +, and [M + Na] + may be produced in the positive ion mode, and the quasi-molecular ion peaks of [M-H]− are mainly produced in the negative ion mode. By comparing the response values of each quasi-molecular ion peak in the two modes, it was found that aflatoxin could produce molecular ions in both positive and negative ion modes, but the response value of [M-H]− was far lower than that of [M + H] +; HT-2 toxin belongs to trichothecenes, and its parent ion can form [M + H] +, [M + Na] + and [M + NH4] +, but the conjugate of [M + Na] + has the highest response value and sensitivity, so HT-2 toxin chooses [M + Na] + as the parent ion. Diacetoxysciroenol can form [M + Na] + and [M + NH4] +, but the response value and sensitivity of [M + Na] + are higher; the other six toxins all have [M + H] + ions in positive ion mode, so positive ion scanning mode was used for detection in this experiment. Then, full MS/dd-MS2 mode was adopted, in which dd-MS2 was used as the confirmation mode. When the parent ion strength reached the set threshold (1 × 106), it automatically triggered secondary mass spectrometry scanning, and the information of secondary fragment ions could be further confirmed by combining the accurate mass number and retention time of primary parent ions. The precise mass number, mass accuracy error and retention time of the 12 mycotoxins are shown in Table 2.
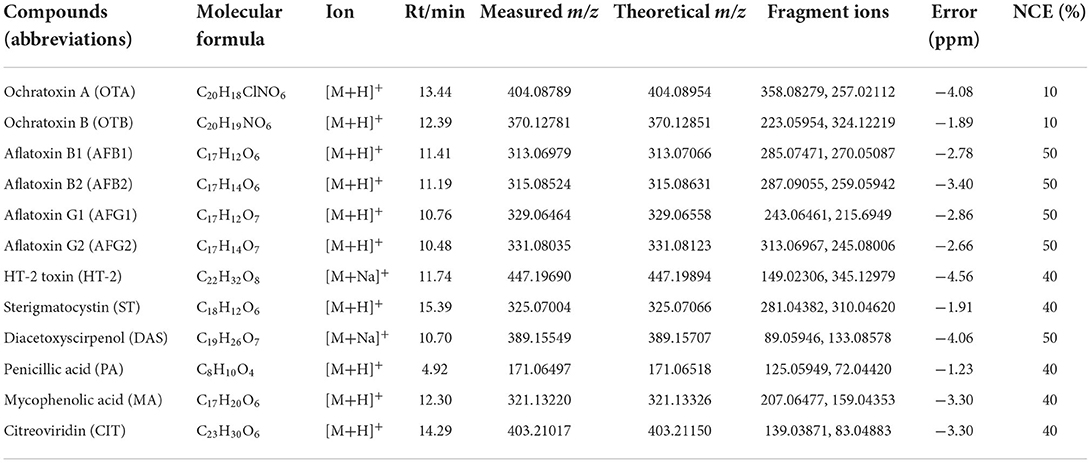
Table 2. HPLC and Orbitrap HRMS parameters of 12 mycotoxins.
The type and proportion of the mobile phase not only affect the retention time and peak shape of the target compound but also affect the ionization efficiency of the target compounds, thus affecting the sensitivity (23–25). In this study, four different mobile phases, A: 0.1% formic acid water-methanol, B: 1 mmol/L ammonium acetate-0.1% formic acid water-methanol, A: 0.1% formic acid water-acetonitrile, and D: 1 mmol/L ammonium acetate-0.1% formic acid water-acetonitrile, were compared on the mass spectral responses of 12 mycotoxins. The results showed that when A was the mobile phase, all toxins had a mass spectrometry response. However, when ammonium acetate is present in the mobile phase, the ionic response of each target substance is obviously enhanced. Therefore, the effects of different concentrations of ammonium acetate (0.1, 0.5, 1, 2, and 5 mmol/L) on the response intensity of mass spectrometry were further optimized. The results showed that when the concentration of ammonium acetate increased to 0.5 mmol/L, the ionic responses of most of the target compounds were enhanced, but the concentration of ammonium acetate continued to increase, but the response values of the other eight target compounds decreased slightly except for four aflatoxins. Therefore, 0.5 mmol/L ammonium acetate-0.1% formic acid aqueous solution was finally selected as the mobile phase. The main organic phases were acetonitrile and methanol. It was found that the peak shape of some target compounds was poor when acetonitrile was used as the mobile phase, so methanol was selected as the organic phase in this study. Therefore, 0.5 mmol/L ammonium acetate-0.1% formic acid aqueous solution-methanol was used as the mobile phase. The chromatographic and mass spectra of 12 mycotoxins after optimization are shown in Figures 1A,B.
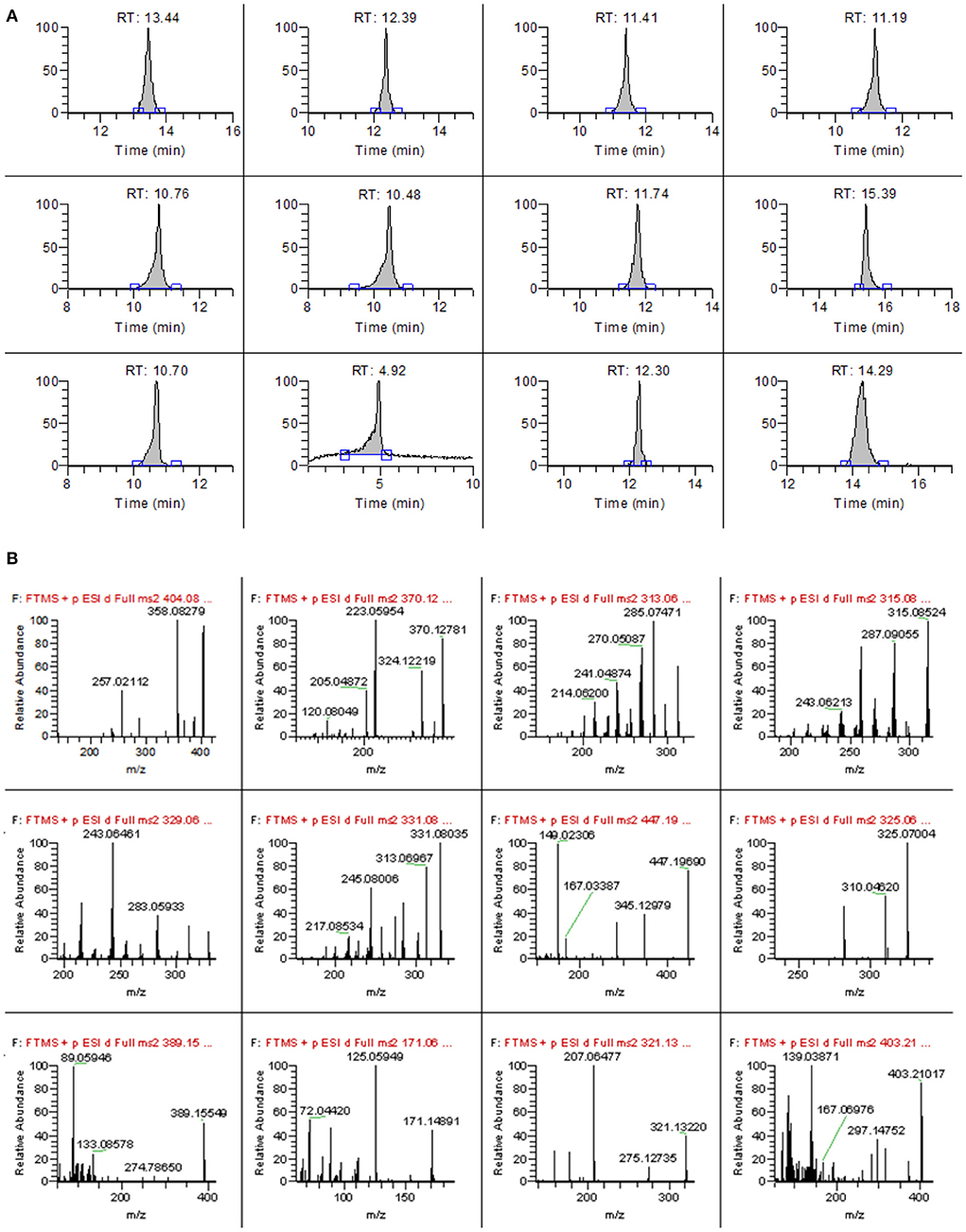
Figure 1. The chromatograms (A) and high-resolution mass spectrum (B) of 12 mycotoxins.
Mycotoxins were mainly extracted by methanol, acetonitrile or a mixture of these two solvents and water in different proportions (26, 27). Therefore, this study compared the extraction efficiency of 12 mycotoxins from soy sauce by six insoluble solvent systems: methanol, acetonitrile, 80% methanol-water, 80% acetonitrile-water, 80% methanol-water-0.1% FA and 80% acetonitrile-water-0.1% FA. The results are shown in Figure 2A. The extraction effect of acetonitrile on 12 mycotoxins was significantly better than that of methanol. The addition of water can significantly improve the extraction rate. Eighty percent acetonitrile-water and 80% acetonitrile-hydr-0.1% FA had little effect on the mycotoxin extraction efficiency, except for HT-2 and penicillin. However, 80% acetonitrile-water was better than 80% acetonitrile-hydr-0.1% FA in the extraction efficiency of HT-2 and penicillic acid. Hence, 80% acetonitrile-water was finally selected as the extraction solvent.
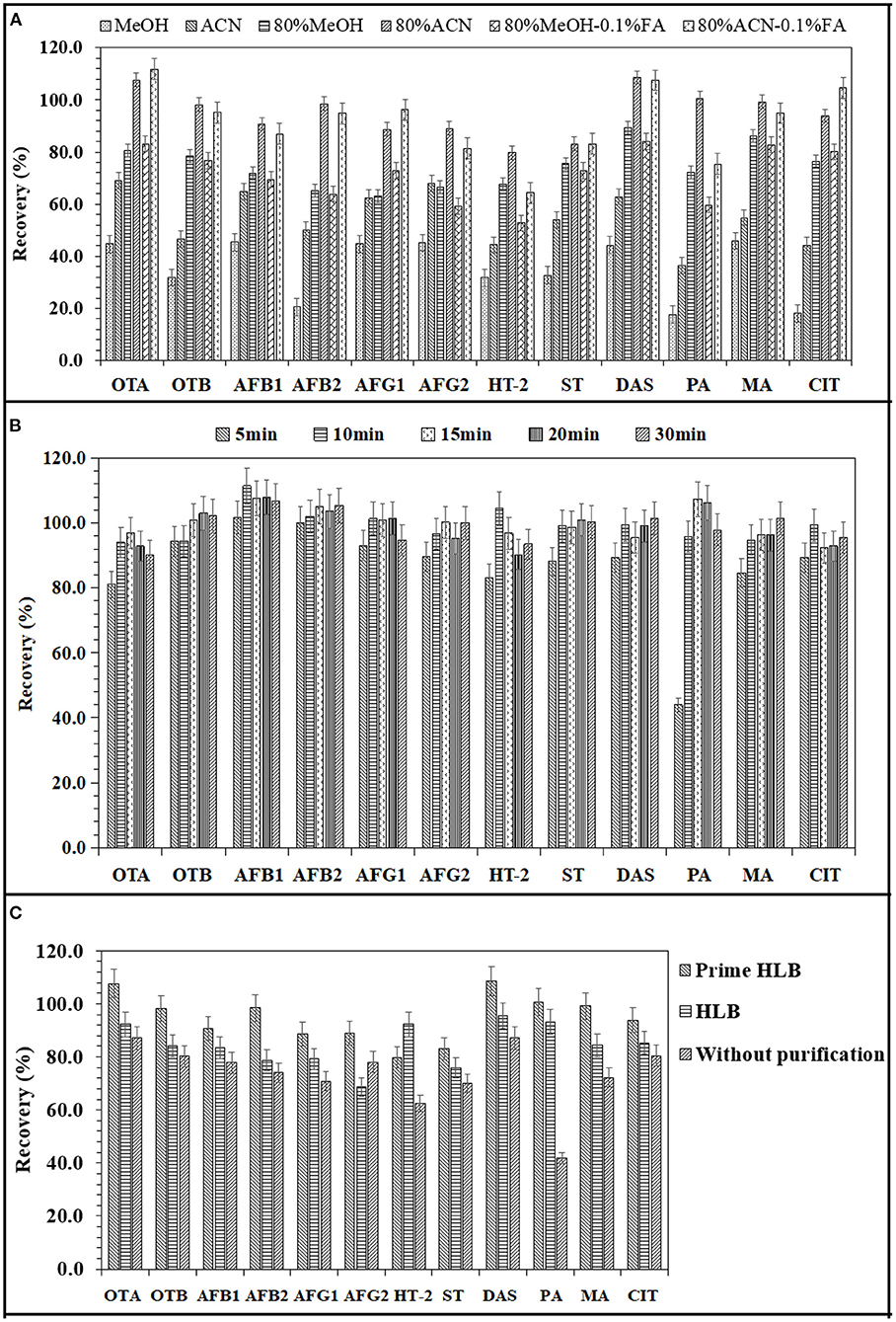
Figure 2. Effects of extraction solvent (A), extraction time (B), and various solid phase extraction columns (C) on the extraction recoveries of 12 mycotoxins.
In this study, 80% acetonitrile-water was used as the extraction solvent, and the effects of different shaking extraction times (5, 10, 15, 20, and 30 min) on the extraction of 12 mycotoxins from condiments were compared. The results are shown in Figure 2B. With increasing extraction time, the recovery rate gradually increased, and after increasing to 10 min, the recovery rate remained basically unchanged. Therefore, a 10 min shaking time was finally selected as the extraction time.
The effects of two different solid phase extraction columns (HLB column and PRiME HLB column) on the recovery of mycotoxins were investigated. The HLB column was a reversed-phase solid phase extraction column, and the impurities were removed by leaching after adsorbing the target substance. Compared with other SPE products, it can remove 95% of common matrix interfering substances (such as phospholipids, fats, salts and proteins) (28). As shown in Figure 2C, the recoveries of 12 mycotoxins were improved by two purification columns. After PRiME HLB purification, the recoveries of mycotoxins ranged from 79.8 to 108.6%, and those obtained by HLB column purification ranged from 68.8 to 93.1%. Without purification, the recoveries of 12 mycotoxins ranged from 41.9 to 87.2%. Compared with the two kinds of solid phase extraction columns, the recovery rate of the impurity adsorption solid phase extraction column was better than that of the target substance adsorption solid phase extraction column except for HT-2 toxin, mainly because the HLB column did not have specific adsorption for a specific toxin, so it is easy to lose the recovery rate in the process of target substance adsorption and impurity elution. Therefore, a PRIME HLB column was selected as the purification column in this experiment.
The matrix is a coextraction interfering substance other than the measured substance in the sample that often competes with the target compound for ionization, has significant interference with the analysis of the measured substance, and affects the accuracy of the determination results (29). These interferences and influences are called the matrix effect. The matrix effect is ion inhibition or ion enhancement. The existence of the matrix effect will affect the accuracy of the determination results (30, 31). The standard solution (analyte concentration is 10 μg/L) was prepared from the matrix extracts of edible oil and condiment blank samples without 12 mycotoxins. The matrix effect (ME) was evaluated by the ratio of the two with reference to the standard solution of the same concentration prepared by pure solvent, that is, the formula ME = B/A, where A and B represent the peak areas of analytes in pure solvent and blank sample matrix solution, respectively. If ME < 0.8, it indicates that the matrix has a significant inhibition on the response of analytes; if ME > 1.2, it indicates that the matrix will significantly enhance the response of analytes; if 0.8 ≤ ME ≤ 1.2, it indicates that the matrix effect is not significant (23, 32). The experimental results are shown in Figure 3. After purification by PRiME HLB column, the matrix effect of 12 mycotoxins in the two matrices is between 0.80 and 1.13 (see Table 2), and when it is not purified, the matrix effect is between 0.64 and 1.08. Some compounds have obvious matrix inhibition, which shows that purification by PRiME HLB column can obviously reduce the matrix effect, which is beneficial to reduce matrix interference and improve accuracy.
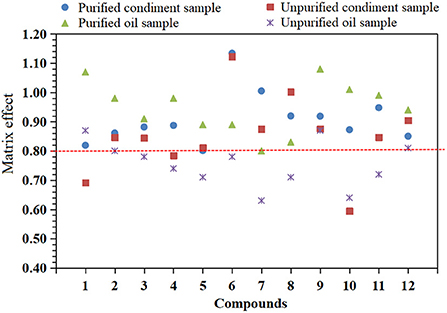
Figure 3. Matrix effects of 12 mycotoxins in purified and unpurified condiments and oil samples.
In this method, pure solvent was used to dilute the standard solution step by step to the lowest concentration that could be detected by the instrument, and the samples were injected repeatedly. According to the standard deviation of the test results, the detection limit (LOD, S/N ≥ 3) was determined by three times the signal-to-noise ratio, and the quantification limit (LOQ, S/N ≥ 10) was determined by 10 times the signal-to-noise ratio (33, 34). Regression analysis was carried out with the peak area (y) of the exact mass-to-charge ratio of the target parent ion as the ordinate and the compound concentration (x, μg/L) as the abscissa, and the linear regression equation, linear range and detection limit of each compound were obtained, as shown in Table 3. The peak area of 12 mycotoxins showed a good linear relationship with their mass concentrations; the determination coefficient R2 was > 0.998, the detection limit ranged from 0.12 to 1.2 μg/L, and the quantitative limit ranged from 0.4 to 4.0 μg/L, which indicated that the method had good sensitivity. The applied regulatory levels and standards of mycotoxins vary in the different regions of the world. The current maximum levels for aflatoxins set by the European Commission (EC) are 2 μg/kg for AFB1 and 4 μg/kg for total aflatoxins in various foods. These are to be extended to cover spices with limits of 5 and 10 μg/kg for AFB1 and total aflatoxins, respectively (35). Apart from aflatoxins, the maximum levels for OTA, DON, ZEN, and FBs (sum of FB1 and FB2) in various foods are also stipulated in Commission Regulation (EC) No 1881/2006 and in its amendments (36). The sensitivity parameters in this developed method are adequate to the legal limit requirements.
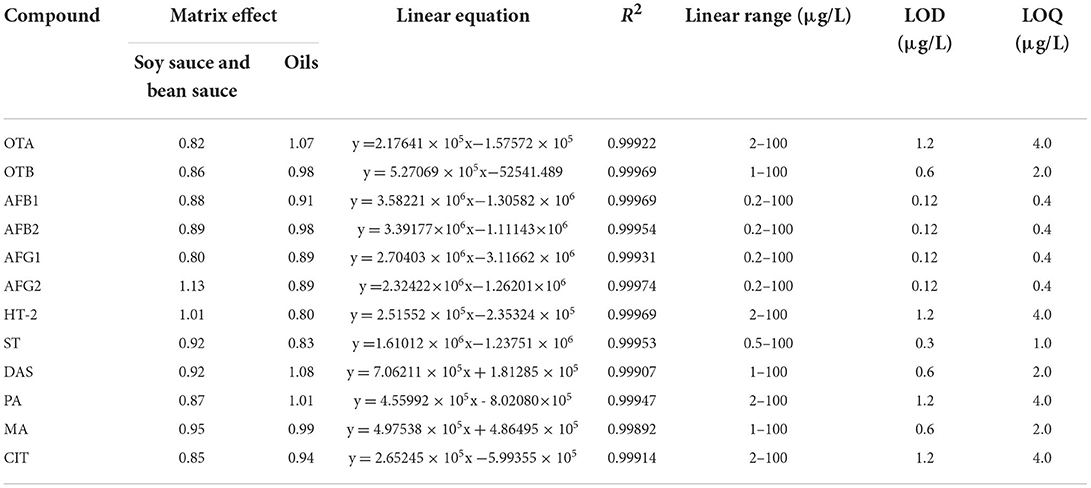
Table 3. Matrix effect, linear ranges, regression equations, determination coefficients, LODs, and LOQs for 12 mycotoxins.
The blank substrate samples of edible oil and condiment without the target substance to be tested were selected for the standard addition recovery experiment. Standard solutions with three concentration levels of LOQ, 2× LOQ, and 10 × LOQ were added, and six parallel samples were made for each standard addition concentration. The recovery rate was calculated, and the results are shown in Table 3. Table 4 shows that the average recovery rate of 12 mycotoxins in soy sauce and bean sauce is between 78.4 and 106.8%, and the precision RSD (n = 6) is between 1.2% and 9.7%. The average recoveries in edible oil ranged from 78.3 to 115.6%, and the precision RSD (n = 6) ranged from 0.9 to 8.6%.
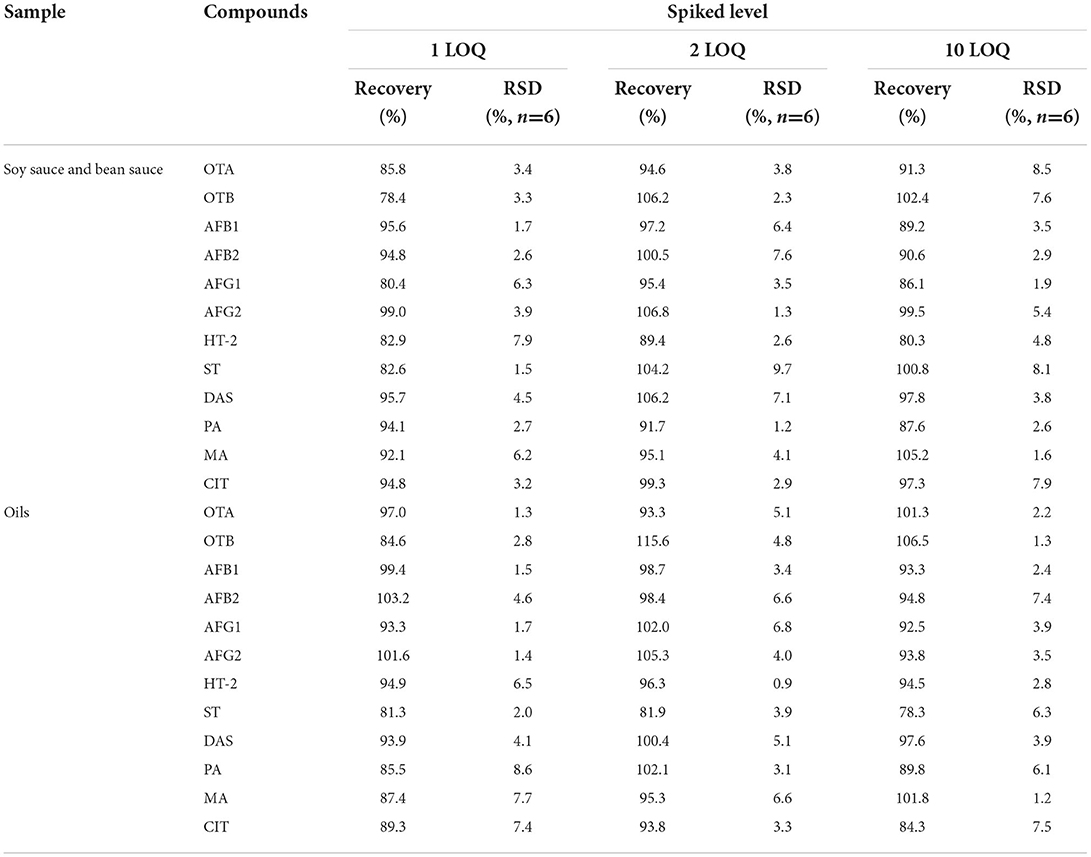
Table 4. Recoveries and RSDs (n = 6) of blank samples fortified with 12 mycotoxins.
AFB2 was detected in 4 soy sauce and bean sauce samples (S2, S5, S11, and S13) under optimized experimental conditions with contents of 1.8–22.1 μg/kg (Table 5). AFB1 was detected in 6 edible oil samples (S15, S16, S18, S20, S23, S24) with contents ranging from 0.9 to 4.7 μg/kg. Sterigmatocystin was detected in S3 and S15 with contents of 2.9 and 2.1 μg/kg, respectively. Mycophenolic acid was detected in S3 at a concentration of 7.5 μg/kg. It could be inferred from the data that AFB2 is easily produced in soy sauce and bean sauce, and AFB1 is easily produced in edible oil. AFBs represent a global public health issue, as they are responsible for significant adverse health issues affecting consumers worldwide. AFB1, due to its toxic, mutagenic, immunotoxic, teratogenic, and carcinogenic effects on humans and animals, is classified as a group 1 carcinogen in the International Agency for Research on Cancer (IARC) classification of carcinogenic substances (37). To avoid or minimize health concerns, the EU has also established maximum tolerable limits for AFs in chillies as 10 μg/kg for total and 5 μg/kg for AFB1 (38). Therefore, the control of AFBs, especially AFB1 and AFB2, is particularly crucial due to their high contents in soy sauce, bean sauce and edible oil. Moreover, the need for normative updates regarding legal limits for sterigmatocystin and mycophenolic acid should be considered based on their biological toxicity, as they were detected in several samples.
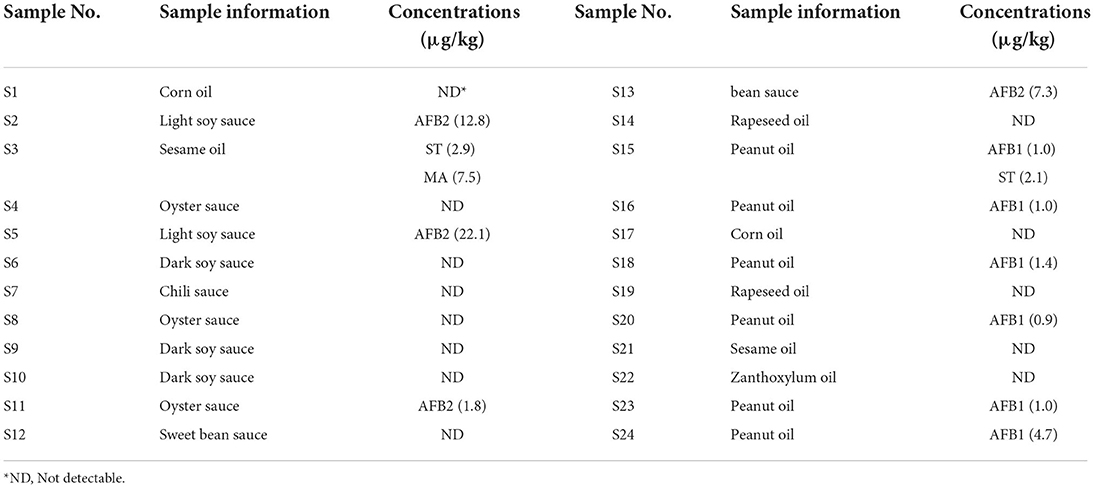
Table 5. Concentrations of mycotoxins detected in the condiments and oils.
The comparison of this established method with other reported methods for the determination of mycotoxins in oil and sauce samples is summarized in Table 6. Table 6 clearly shows that HPLC and liquid chromatography with tandem mass spectrometry (LC–MS/MS) are the most commonly used methods for the determination of mycotoxins in separate matrices (38–41, 43, 44). For the pretreatment method, the QuEChERS procedure (quick, easy, cheap, effective, rugged and safe) seems to be the most commonly used technique (9, 39, 43–46). However, all these methods listed in Table 6 are suitable for the determination of several mycotoxins in either vegetable oil samples or sauce samples. The method established in our work can be used for the analysis of 12 mycotoxins in not only edible oil samples but also soy sauce and bean sauce samples. The PRiME HLB solid phase extraction combined with HPLC-Orbitrap HRMS developed in the present work shows rapid extraction time as well as favorable linearity, LODs, recoveries and RSDs, which are comparable or superior in comparison with other analytical methods. In addition, this is also the first study to develop an accurate method for the determination of sterigmatocystin (ST), diacetoxyscirpenol (DAS), penicillic acid (PA), mycophenolic acid (MA), and citreoviridin (CIT) in foodstuffs.
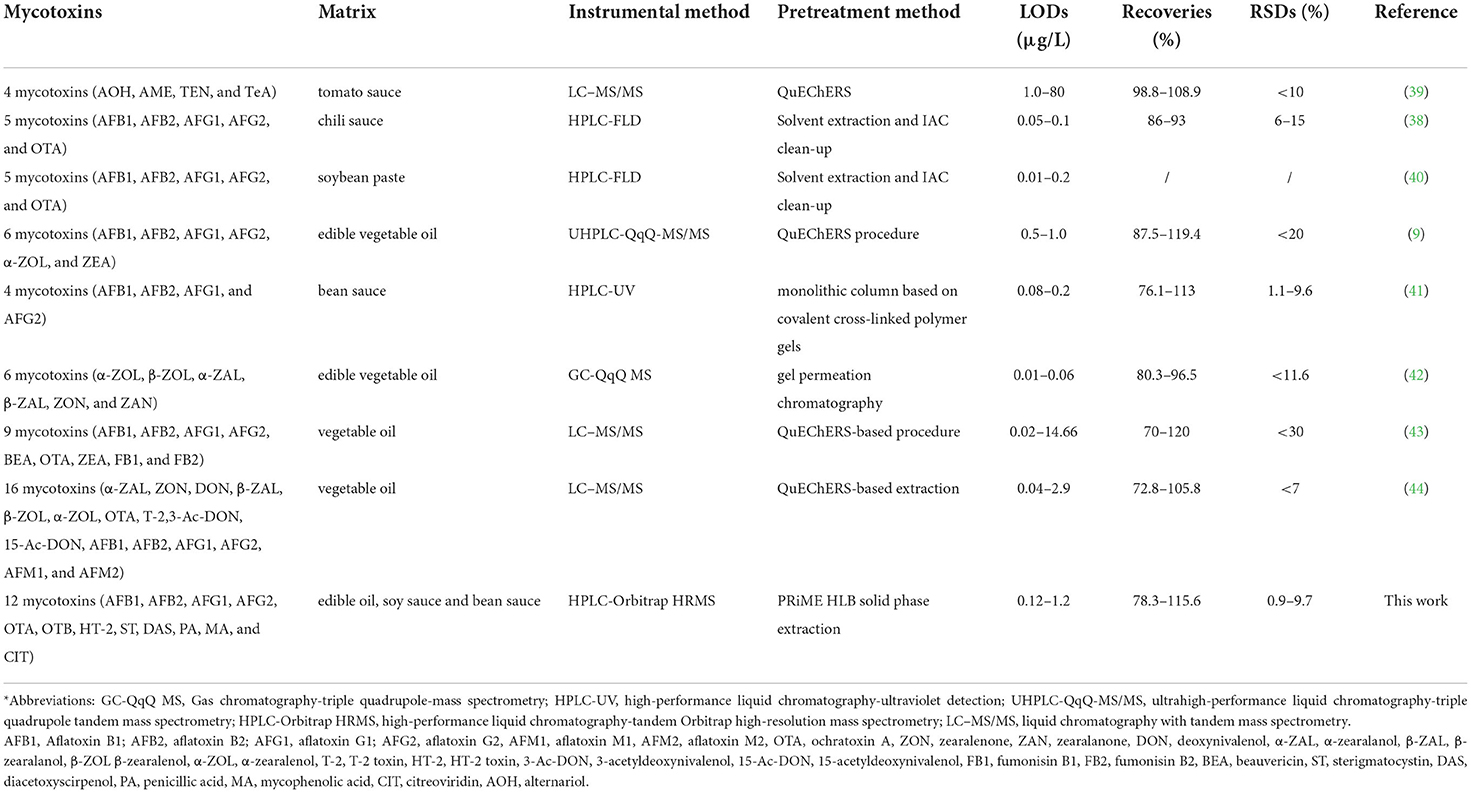
Table 6. Comparison with other methods reported in the literature*.
Aiming at the present situation that edible oil, soy sauce and bean sauce are easily contaminated by many mycotoxins simultaneously, a new impurity adsorption purification technology was adopted, and a simultaneous determination method for rapid screening and quantitative determination of 12 mycotoxins by HPLC-Orbitrap HRMS was established. The pretreatment operation is simple and rapid. The method realizes simultaneous analysis and detection of various mycotoxins, with high stability and sensitivity, strong specificity and good reproducibility. The LODs of the 12 mycotoxins were 0.12–1.2 μg/L. The average recoveries in edible oil, soy sauce and bean sauce were 78.3–115.6% with RSDs of 0.9–9.7%. Therefore, the developed method can be used as a quantitative method for the determination of various mycotoxins in edible oils, soy sauce and bean sauce.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
DL prepared and wrote the manuscript. JG, JC, and ML corrected, revised, and improved the manuscript. CZ and YX, modified the Tables and Figures in the manuscript. HD and XX designed the study, supervised, reviewed, and finalized the manuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported by Guangdong Provincial Key R&D Programme (No. 2021B0707060001), project (No. 2020PT01) Science and Technology Project of Chaozhou Branch of Chemistry and Chemical Engineering Guangdong Laboratory (No. HJL202202B007), and Characteristic Innovation Project of Universities in Guangdong province (2022KTSCX058).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2022.1001671/full#supplementary-material
1. Wan J, Chen B, Rao J. Occurrence and preventive strategies to control mycotoxins in cereal-based food. Comprehens Rev Food Sci Food Saf. (2020) 19:928–53. doi: 10.1111/1541-4337.12546
2. Xu X, Xu X, Han M, Qiu S, Hou X. Development of a modified QuEChERS method based on magnetic multiwalled carbon nanotubes for the simultaneous determination of veterinary drugs, pesticides and mycotoxins in eggs by UPLC-MS/MS. Food Chem. (2019) 276:419–26. doi: 10.1016/j.foodchem.2018.10.051
3. Yang Y, Li G, Wu D, Liu J, Li X, Luo P, et al. Recent advances on toxicity and determination methods of mycotoxins in foodstuffs. Trends Food Sci Technol. (2020) 96:233–252. doi: 10.1016/j.tifs.2019.12.021
4. Abdolmaleki K, Khedri S, Alizadeh L, Javanmardi F, Oliveira CA, Khaneghah AM, et al. The mycotoxins in edible oils: an overview of prevalence, concentration, toxicity, detection and decontamination techniques. Trends Food Sci and Technology. (2021) 115:500–11. doi: 10.1016/j.tifs.2021.06.057
5. Arroyo-Manzanares N, Campillo N, López-García I, Hernández-Córdoba M, Viñas P. High-Resolution mass spectrometry for the determination of mycotoxins in biological samples. A review. Microchem J. (2021) 166:106197. doi: 10.1016/j.microc.2021.106197
6. Khodaei D, Javanmardi F, Khaneghah AM. The global overview of the occurrence of mycotoxins in cereals: a three-year survey. Curr Opin Food Sci. (2021) 39:36–42. doi: 10.1016/j.cofs.2020.12.012
7. Martins C, Vidal A, De Boevre, De Saeger M, Nunes S, Torres C, et al. Exposure assessment of Portuguese population to multiple mycotoxins: the human biomonitoring approach. Int J Hygiene Environ Health. (2019) 222:913–25. doi: 10.1016/j.ijheh.2019.06.010
8. Sarmast E, Fallah AA, Jafari T, Khaneghah AM. Occurrence and fate of mycotoxins in cereals and cereal-based products: a narrative review of systematic reviews and meta-analyses studies. Current Opinion in Food Science. (2021) 39:68–75. doi: 10.1016/j.cofs.2020.12.013
9. Hidalgo-Ruiz JL, Romero-González R, Martínez Vidal JL, Garrido Frenich A. A rapid method for the determination of mycotoxins in edible vegetable oils by ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chemistry. (2019) 288:22–28. doi: 10.1016/j.foodchem.2019.03.003
10. Bessaire T, Mujahid C, Mottier P, Desmarchelier A. Multiple mycotoxins determination in food by LC-MS/MS: an international collaborative study. Toxins. (2019) 11:658. doi: 10.3390/toxins11110658
11. Romera D, Mateo EM, Mateo-Castro R, Gomez JV, Gimeno-Adelantado JV, Jimenez M, et al. Determination of multiple mycotoxins in feedstuffs by combined use of UPLC–MS/MS and UPLC–QTOF–MS. Food Chem. (2018) 267:140–8. doi: 10.1016/j.foodchem.2017.11.040
12. Turner NW, Subrahmanyam S, Piletsky SA. Analytical methods for determination of mycotoxins: a review. Anal Chim Acta. (2009) 632:168–80. doi: 10.1016/j.aca.2008.11.010
13. Anastassiades M, Lehotay SJ, Štajnbaher D, Schenck FJ. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int. (2003) 86:412–31. doi: 10.1093/jaoac/86.2.412
14. Facorro R, Llompart M, Dagnac T. Combined (d)SPE-QuEChERS extraction of mycotoxins in mixed feed rations and analysis by high performance liquid chromatography-high-resolution mass spectrometry. Toxins. (2020) 12:206. doi: 10.3390/toxins12030206
15. Pereira V, Fernandes J, Cunha S. Mycotoxins in cereals and related foodstuffs: a review on occurrence and recent methods of analysis. Trends Food Sci Technol. (2014) 36:96–136. doi: 10.1016/j.tifs.2014.01.005
16. Wu Z, He D, Cui B, Jin Z, Xu E, Yuan C, et al. Trimer-based aptasensor for simultaneous determination of multiple mycotoxins using SERS and fluorimetry. Microchimica Acta. (2020) 187:1–7. doi: 10.1007/s00604-020-04487-1
17. Dong H, Xian Y, Xiao K, Wu Y, Zhu L, He J, et al. Development and comparison of single-step solid phase extraction and QuEChERS clean-up for the analysis of 7 mycotoxins in fruits and vegetables during storage by UHPLC-MS/MS. Food Chemistry. (2019) 274:471–9. doi: 10.1016/j.foodchem.2018.09.035
18. Soleimany F, Jinap S, Abas F. Determination of mycotoxins in cereals by liquid chromatography tandem mass spectrometry. Food Chem. (2012) 130:1055–60. doi: 10.1016/j.foodchem.2011.07.131
19. Dong H, Xian Y, Li H, Wu Y, Bai W, Zeng X, et al. Analysis of heterocyclic aromatic amine profiles in Chinese traditional bacon and sausage based on ultrahigh-performance liquid chromatography–quadrupole-Orbitrap high-resolution mass spectrometry (UHPLC–Q-Orbitrap-HRMS). Food Chem. (2020) 310:125937. doi: 10.1016/j.foodchem.2019.125937
20. Liang M, Xian Y, Wang B, Hou X, Wang L, Guo X, et al. High throughput analysis of 21 perfluorinated compounds in drinking water, tap water, river water and plant effluent from southern China by supramolecular solvents-based microextraction coupled with HPLC-Orbitrap HRMS. Environ Pollution. (2020) 263:114389. doi: 10.1016/j.envpol.2020.114389
21. Xian Y, Liang M, Wu Y, Wang B, Hou X, Dong H, et al. Fluorine and nitrogen functionalized magnetic graphene as a novel adsorbent for extraction of perfluoroalkyl and polyfluoroalkyl substances from water and functional beverages followed by HPLC-Orbitrap HRMS determination. Sci Total Environ. (2020) 723:138103. doi: 10.1016/j.scitotenv.2020.138103
22. Xu Y, Li H, Liang J, Ma J, Yang J, Zhao X, et al. High-throughput quantification of eighteen heterocyclic aromatic amines in roasted and pan-fried meat on the basis of high performance liquid chromatography-quadrupole-orbitrap high resolution mass spectrometry. Food Chem. (2021) 361:130147. doi: 10.1016/j.foodchem.2021.130147
23. Dong H, Xiao K. Modified QuEChERS combined with ultra high performance liquid chromatography tandem mass spectrometry to determine seven biogenic amines in Chinese traditional condiment soy sauce. Food Chemistry. (2017) 229:502–8. doi: 10.1016/j.foodchem.2017.02.120
24. Dong H, Xiao K, Xian Y, Wu Y, Zhu L. A novel approach for simultaneous analysis of perchlorate (ClO[[sb]]4[[/s]]−) and bromate (BrO[[sb]]3[[/s]]−) in fruits and vegetables using modified QuEChERS combined with ultrahigh performance liquid chromatography-tandem mass spectrometry. Food Chemistry. (2018) 270:196–203. doi: 10.1016/j.foodchem.2018.07.091
25. Dong H, Zeng X, Bai W. Solid phase extraction with high polarity Carb/PSA as composite fillers prior to UPLC-MS/MS to determine six bisphenols and alkylphenols in trace level hotpot seasoning. Food Chemistry. (2018) 258:206–13. doi: 10.1016/j.foodchem.2018.03.074
26. Campone L, Rizzo S, Piccinelli AL, Celano R, Pagano I, Russo M, et al. Determination of mycotoxins in beer by multi heart-cutting two-dimensional liquid chromatography tandem mass spectrometry method. Food Chem. (2020) 318:126496. doi: 10.1016/j.foodchem.2020.126496
27. Silva AS, Brites C, Pouca AV, Barbosa J, Freitas A. UHPLC-ToF-MS method for determination of multi-mycotoxins in maize: development and validation. Curr Res Food Sci. (2019) 1:1–7. doi: 10.1016/j.crfs.2019.07.001
28. Hu J, Liang M, Xian Y, Chen R, Wang L, Hou X, et al. Development and validation of a multianalyte method for quantification of aflatoxins and bongkrekic acid in rice and noodle products using PRiME-UHPLC-MS/MS method. Food Chem. (2022) 395:133598. doi: 10.1016/j.foodchem.2022.133598
29. Arce-López B, Lizarraga E, Flores-Flores M, Irigoyen Á, González-Peñas E. Development and validation of a methodology based on Captiva EMR-lipid clean-up and LC-MS/MS analysis for the simultaneous determination of mycotoxins in human plasma. Talanta. (2020) 206:120193. doi: 10.1016/j.talanta.2019.120193
30. Xian Y, Dong H, Wu Y, Guo X, Hou X, Wang B, et al. QuEChERS-based purification method coupled to ultrahigh performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) to determine six quaternary ammonium compounds (QACs) in dairy products. Food Chem. (2016) 212:96–103. doi: 10.1016/j.foodchem.2016.05.151
31. Xian Y, Guo X, Hou X, Wang L, Wu Y, Chen L, et al. A modified quick, easy, cheap, effective, rugged, and safe cleanup method followed by liquid chromatography-tandem mass spectrometry for the rapid analysis of perchlorate, bromate and hypophosphite in flour. J Chromatography A. (2017) 1526:31–8. doi: 10.1016/j.chroma.2017.10.047
32. Zeng X, Bai W, Xian Y, Dong H, Luo D. Application of QuEChERS-based purification coupled with isotope dilution gas chromatography-mass spectrometry method for the determination of N-nitrosamines in soy sauce. Anal Methods. (2016) 8:5248–54. doi: 10.1039/C6AY01169A
33. González-Jartín JM, Rodríguez-Cañás I, Alfonso A, Sainz MJ, Vieytes MR, Gomes A, et al. Multianalyte method for the determination of regulated, emerging and modified mycotoxins in milk: QuEChERS extraction followed by UHPLC–MS/MS analysis. Food Chem. (2021) 356:129647. doi: 10.1016/j.foodchem.2021.129647
34. Rodríguez-Carrasco Y, Castaldo L, Gaspari A, Graziani G, Ritieni A. Development of an UHPLC-Q-Orbitrap HRMS method for simultaneous determination of mycotoxins and isoflavones in soy-based burgers. LWT-Food Sci Technol. (2019) 99:34–42. doi: 10.1016/j.lwt.2018.09.046
35. Gilbert J, Anklam E. Validation of analytical methods for determining mycotoxins in foodstuffs. TrAC Trends Anal Chem. (2002) 21:468–86. doi: 10.1016/S0165-9936(02)00604-0
36. Eskola M, Kos G, Elliott CT, Hajšlová J, Mayar S, Krska R, et al. Worldwide contamination of food-crops with mycotoxins: Validity of the widely cited ‘FAO estimate’ of 25%. Crit Rev Food Sci Nutr. (2020) 60:2773–89. doi: 10.1080/10408398.2019.1658570
37. Jallow A, Xie H, Tang X, Qi Z, Li P. Worldwide aflatoxin contamination of agricultural products and foods: From occurrence to control. Comprehens Rev Food Sci Food Saf. (2021) 20:2332–81. doi: 10.1111/1541-4337.12734
38. Iqbal SZ, Asi MR, Zuber M, Akhtar J, Jawwad Saif M. Natural occurrence of aflatoxins and ochratoxin A in commercial chilli and chilli sauce samples. Food Control. (2012) 30:621–5. doi: 10.1016/j.foodcont.2012.09.003
39. De Berardis, De Paola S, Montevecchi EL, Garbini G, Masino D, Antonelli F, et al. Determination of four Alternaria alternata mycotoxins by QuEChERS approach coupled with liquid chromatography-tandem mass spectrometry in tomato-based and fruit-based products. Food Res Int. (2018) 106:677–85. doi: 10.1016/j.foodres.2018.01.032
40. Jeong SE, Chung SH, Hong SY. Natural occurrence of aflatoxins and ochratoxin A in meju and soybean paste produced in South Korea. App Biol Chem. (2019) 62:65. doi: 10.1186/s13765-019-0472-y
41. Wei T, Chen Z, Li G, Zhang Z. A monolithic column based on covalent cross-linked polymer gels for online extraction and analysis of trace aflatoxins in food sample. J Chromatography A. (2018) 1548:27–36. doi: 10.1016/j.chroma.2018.03.015
42. Qian M, Zhang H, Wu L, Jin N, Wang J, Jiang K, et al. (2014). (2015). Simultaneous determination of zearalenone and its derivatives in edible vegetable oil by gel permeation chromatography and gas chromatography–triple quadrupole mass spectrometry. Food Chemistry, 166, 23–28. doi: 10.1016/j.foodchem.2014.05.133
43. Junsai T, Poapolathep S, Sutjarit S, Giorgi M, Zhang Z, Logrieco A, et al. Determination of multiple mycotoxins and their natural occurrence in edible vegetable oils using liquid chromatography–tandem mass spectrometry. Foods. (2021) 10:2795. doi: 10.3390/foods10112795
44. Zhao H, Chen X, Shen C, Qu B. Determination of 16 mycotoxins in vegetable oils using a QuEChERS method combined with high-performance liquid chromatography-tandem mass spectrometry. Food Addi Contamin: Part A. (2017) 34:255–64. doi: 10.1080/19440049.2016.1266096
45. Payá P, Anastassiades M, Mack D, Sigalova I, Tasdelen B, Oliva J, et al. Analysis of pesticide residues using the quick easy cheap effective rugged and safe (QuEChERS) pesticide multiresidue method in combination with gas and liquid chromatography and tandem mass spectrometric detection. Anal Bioanal Chem. (2007) 389:1697–714. doi: 10.1007/s00216-007-1610-7
Keywords: PRiME HLB, HPLC-Orbitrap HRMS, edible oil, mycotoxin, soy sauce
Citation: Luo D, Guan J, Dong H, Chen J, Liang M, Zhou C, Xian Y and Xu X (2022) Simultaneous determination of twelve mycotoxins in edible oil, soy sauce and bean sauce by PRiME HLB solid phase extraction combined with HPLC-Orbitrap HRMS. Front. Nutr. 9:1001671. doi: 10.3389/fnut.2022.1001671
Received: 23 July 2022; Accepted: 09 September 2022;
Published: 28 September 2022.
Edited by:
A. M. Abd El-Aty, Cairo University, EgyptReviewed by:
Thierry Dagnac, Galician Agency for Food Quality, SpainCopyright © 2022 Luo, Guan, Dong, Chen, Liang, Zhou, Xian and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Dong, ZG9uZ2hhb0B6aGt1LmVkdS5jbg==; Xiaofei Xu, eGZ4dWZlQGdkb3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.