
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr. , 03 January 2022
Sec. Nutrition and Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fnut.2021.815456
This article is part of the Research Topic Appetite Control in Obesity View all 6 articles
 Sayani Mukherjee1Silje Skrede2,3
Sayani Mukherjee1Silje Skrede2,3 Edward Milbank4,5,6Ramaroson Andriantsitohaina6
Edward Milbank4,5,6Ramaroson Andriantsitohaina6 Miguel López4,5
Miguel López4,5 Johan Fernø1*
Johan Fernø1*Antipsychotic drugs (APDs) represent a cornerstone in the treatment of schizophrenia and other psychoses. The effectiveness of the first generation (typical) APDs are hampered by so-called extrapyramidal side effects, and they have gradually been replaced by second (atypical) and third-generation APDs, with less extrapyramidal side effects and, in some cases, improved efficacy. However, the use of many of the current APDs has been limited due to their propensity to stimulate appetite, weight gain, and increased risk for developing type 2 diabetes and cardiovascular disease in this patient group. The mechanisms behind the appetite-stimulating effects of the various APDs are not fully elucidated, partly because their diverse receptor binding profiles may affect different downstream pathways. It is critical to identify the molecular mechanisms underlying drug-induced hyperphagia, both because this may lead to the development of new APDs, with lower appetite-stimulating effects but also because such insight may provide new knowledge about appetite regulation in general. Hence, in this review, we discuss the receptor binding profile of various APDs in relation to the potential mechanisms by which they affect appetite.
Schizophrenia, a chronic psychotic illness with a lifetime prevalence ~0.7%, is associated with a long-term reduced quality of life for affected individuals and constitutes a major socio-economic challenge worldwide (1). For patients suffering from schizophrenia, the life expectancy declines by 15–20 years in comparison to unaffected individuals (2). Somatic conditions such as cardiovascular disorders (CVD) have been estimated to account for as much as 60% of the mortality gap (3). In fact, while CVD accounts for 33% of deaths in the general population, the corresponding figure for persons diagnosed with schizophrenia is 75% (4).
Pharmacological treatment with antipsychotic agents is available for the so-called positive (psychotic) symptoms of schizophrenia, such as hallucinations and delusions. The core property of all antipsychotic agents is dopamine 2 (D2) receptor antagonism (or partial agonism) in the central nervous system (CNS) (5). First-generation antipsychotics (FGA), introduced in the early 1950's, can cause serious extrapyramidal adverse effects such as dystonias, parkinsonism, and tardive dyskinesias, the prevalence of which is correlated with the degree of dopamine receptor blockade (6). The so-called second-generation antipsychotics (SGA), introduced from 1993 onwards, are associated with a lower risk of motor dysfunction (7). However, some of these drugs, in particular clozapine and olanzapine, have increased propensity to induce adverse metabolic effects such as pronounced weight gain, dyslipidemia, and diabetes (8, 9). Current evidence indicates that the observed weight gain is largely due to hyperphagic effects of antipsychotics, with a lack of satiety observed in patients and rodent models (10–14). Metabolic adverse effects likely contribute to excess mortality from cardiovascular disease (CVD) in patients, constituting a pressing patient safety issue in psychiatry (4). Interestingly, despite the association of olanzapine and clozapine with a more pronounced risk of weight gain and metabolic adverse effects, they remain widely used because many patients respond well to these agents with regard to symptomatic relief and tolerability (6, 8).
Most antipsychotics have an affinity for a broad range of neurotransmitter receptors in the CNS in addition to the dopamine receptor, some of which have been associated with metabolic side effects. In particular, the risk of weight gain and glucose intolerance has been linked to an affinity to CNS histaminergic (H1), serotonergic (5HT1, 5HT2a, and 5HT2c), and muscarinic M1 and M3 receptors (5, 13, 15). A growing body of publications attempts to unveil the molecular effects of antipsychotics on appetite regulation. The focus of this review is to present an overview of how the receptor binding profiles of various APDs may explain their orexigenic effects and to elucidate the intracellular pathways involved in the regulation of appetite.
In order to understand the relationship between APD receptor binding profile and their propensity to modulate appetite-regulating mechanisms, it is important to understand the CNS anatomy where these interactions take place. An individual's body weight and food intake are primarily controlled by the hypothalamus (Figure 1) (16, 17). The hypothalamus consists of anatomically distinct groups of neurons known as nuclei, including the arcuate nucleus (ARC), ventromedial hypothalamus (VMH), paraventricular nucleus (PVH), and lateral hypothalamus area (LHA) (18). Integrating a wide array of afferent signals, the hypothalamus regulates energy intake and expenditure through signaling mediated by neuromodulators. These include (i) the anorexigenic neuropeptides proopiomelanocortin (POMC), a polypeptide precursor to the endogenous melanocortin receptor agonists (α- and β- melanocyte-stimulating hormones) (19) and cocaine and amphetamine-regulated transcript (CART) (16, 20–23) and (ii) the orexigenic neuropeptide Y (NPY) and agouti-related peptide (AgRP) that acts as an endogenous antagonist and inverse agonist of melanocortin receptors (MC4R) (24). The ARC is considered one of the most important hypothalamic regulators of feeding and contains both anorexigenic and orexigenic neurons. ARC lies adjacent to the third ventricle and median eminence that plays a significant role in metabolic sensing as it contains partially permeable blood-brain barriers, and neurons in this region connect the periphery with the central nervous system.
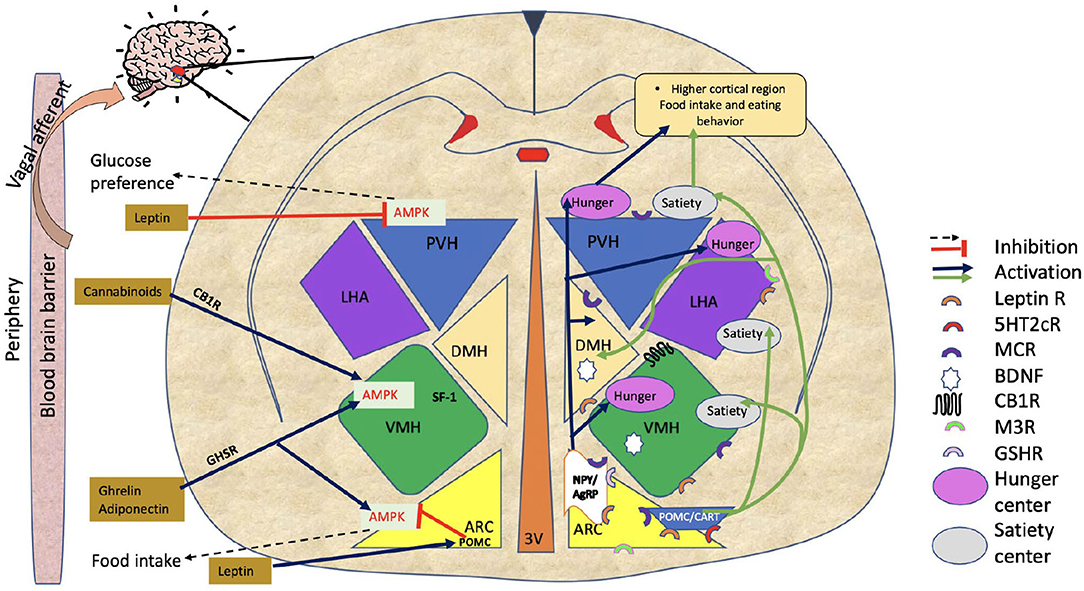
Figure 1. Hypothalamic regulation of food intake. The figure shows hypothalamic regulation of food intake involving various hypothalamus regions (ARC, VMH, LHA, PVH, and DMH). The NPY/AgRP pathway increases food intake, while the POMC/CART pathway triggers satiety. Adiponectin/ ghrelin, and cannabinoids all demonstrate food intake regulation by AMPK activation in the ARC or in VMH. Leptin reduces food intake by directly inhibiting AMPK in PVH or indirectly via POMC neurons in ARC. Black dotted arrows and red lines with blunted ends are showing inhibitory signals. Stimulatory signals are represented by blue and green arrows. On the figure, half circles in different colors show the locations of various receptors through which APDs work. ARC, Arcuate nucleus; VMH, Ventromedial hypothalamus; LHA, Lateral hypothalamus area; PVH, Paraventricular nucleus of hypothalamus; DMH, Dorsomedial hypothalamus; NPY/AgRP, Neuropeptide Y/ agouti-related peptide; POMC, Proopiomelanocortin; CART, Cocaine and amphetamine-regulated transcript; AMPK, AMP-activated protein kinase; M3R, Muscarinic 3 receptor; BDNF, Brain-derived neurotrophic factor; GSH-R, Ghrelin receptor; MCR, Melanocortin receptor; CB1R, Cannabinoid receptor; 5HT2cR, 5-hydroxytryptamine 2c receptor (serotonin receptor); Leptin R, Leptin receptor; APDs Antipsychotics.
The PVH is shown to be involved in neuroendocrine activity, as it produces neuropeptides such as oxytocin (OXT), thyrotropin-releasing hormone (TRH), and corticotropin-releasing hormone. These hormones have traditionally been considered secondary to those controlling food intake and energy expenditure circuitry. Neurons in these circuits respond to energy shortages in two ways. First, they modify the flow of neuroendocrine hormones. Second, they alter the role of preganglionic neurons in the brainstem and spinal cord. Their connections allow them to control energy balance by affecting peripheral tissues, such as the pancreas, liver, spleen, white adipose tissue (WAT), and brown adipose tissue (BAT) (25). Additionally, like ARC, PVH also regulates energy balance by POMC and NPY/AgRP neurons. In the PVH, AgRP neurons stimulate feeding by inhibiting single-minded 1 (SIM1)-expressing target neurons, and activation of POMC exhibits inhibition in food intake (26). It has been reported that SIM1 is also essential for the developmental growth of neurons in the PVH, thereby playing a role in energy balance (27).
The VMH is also significant in regulating glucose metabolism and appetite control (28). VMH neurons express leptin receptors (LEPRs) and estrogen receptors (ERs) (29). According to reports (30), leptin causes the steroidogenic factor-1 (SF1)-positive neurons in the VMH to depolarize and fire more rapidly. Using mice lacking LEPRs on SF1-positive neurons, it has been shown that leptin acts at this site to reduce body weight. These findings emphasize the role of VMH neurons in controlling food intake and diet-induced obesity.
Feeding behavior is also influenced by the mesolimbic pathway (Figure 2). The ventral tegmental area (VTA), positioned in the midbrain, and the nucleus accumbens (NAc), placed in the forebrain, are the two most important structures of this system. The VTA contains dopamine neurons that connect to the NAc. It has been reported that glutamatergic and cholinergic input stimulates dopaminergic neurons, while γ-aminobutyric acid GABAergic input inhibits them (31). Dopamine release at the pre-synaptic level is likely to be responsible for “rewarding” (32). In addition to dopaminergic transmission, cholinergic interneurons deeply penetrate NAc neuronal cells and inhibit dopamine-induced food intake (33).
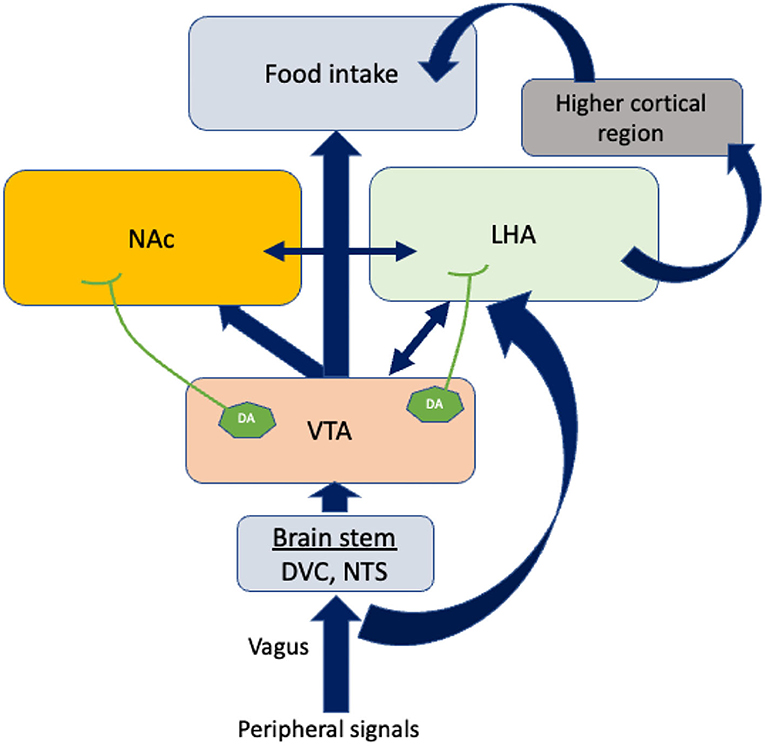
Figure 2. The regulation of food intake by other brain areas. Vagal afferents bring peripheral signals into the VTA by crossing the brain stem. Dopamine neurons (DA) in the VTA project axons to LHA and NAc. In response to the dopamine neurons projected by VTA, LHA connects higher cortical regions to control food intake. The blue arrows indicate the connections between the different brain areas. The double-sided arrows indicate bidirectional connections. VTA, Ventral tegmental area; NAc, Nucleus accumbens; LHA, Lateral hypothalamus area; DA, Dopamine neurons; DVC, Dorso vagal complex.
The caudal brain stem has been shown to play a predominant role in eating processes since it contains all the motoneuron cells involved in different patterns of eating behaviors such as swallowing, suckling, chewing, and licking. Dorsal vagal complex (DVC) premotor neurons are responsible for generating these ingestive patterns. There are three areas in the DVC: the nucleus tractus solitarii (NTS), the area postema, and the dorsal motor nucleus of the vagus nerve. NTS subnuclei are of particular interest because they are the sites where peripheral signals are integrated with motor neurons (Figure 2). Therefore, neurons in the NTS receive dense mechanoreceptor input from the upper gastrointestinal tract (34). Thus, the caudal brain stem comprises some of the normal machineries of hunger and satiation previously believed to be confined to the hypothalamus and the forebrain (35). Further, recent research into neuropeptides, specifically their role in eating behavior, suggests that the caudal brain stem possesses leptin and insulin receptors, glucose-sensing pathways, and neuropeptide mediators that contribute to energy metabolism (1, 36, 37).
Serotonin is a neurotransmitter that regulates food intake and energy expenditure. The first link between serotonin and appetite was described more than 35 years ago (38). As part of the body's homeostatic circuitry, serotonin regulates metabolic signals that communicate energy status and suppress appetite when the body meets its energy requirements. POMC and NPY neurons have been reported to contain serotonin receptors that regulate food intake (39, 40). It has been shown that 5HT2c receptors and serotonin work together to stimulate POMC cleavage, and the inhibition of NPY and AgRP by serotonin at the 5HT1b receptor hinders GABAergic transmission, thereby inhibiting α-MSH and resulting in satiety and thermogenesis.
There are several lines of evidence pointing toward a role for 5HT2c and possibly 5HT1/2a receptors in APD-induced hyperphagia and weight gain (5, 41–43) (Table 1, Figure 3). It is well-known that olanzapine and clozapine show antagonism and have a high affinity for the serotonin 5HT2a and 2c receptors. Additionally, it has been shown that clozapine affects glucose homeostasis by blocking 5HT2a receptors (44). Loss of 5HT2c receptors has been shown to alter feeding behavior and to trigger obesity in mice, and to alter feeding behavior (45). The importance of the 5HT2c receptors in relation to APDs was recently demonstrated in a rodent model, where olanzapine-induced hyperphagia and weight gain are diminished in mice lacking this receptor. Also, treatment with the 5HT2c receptor-specific agonist lorcaserin suppressed hyperphagia and weight gain induced by olanzapine, suggesting pharmacological treatment with this drug as a valid strategy to counteract APD-induced weight gain (46).
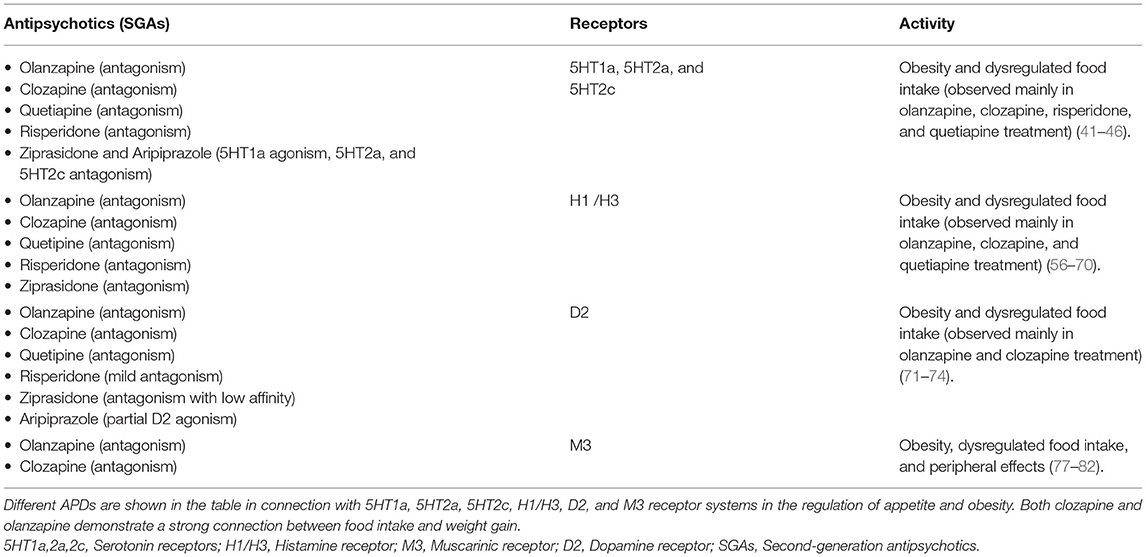
Table 1. APDs and relationship with appetite dysregulation and obesity.
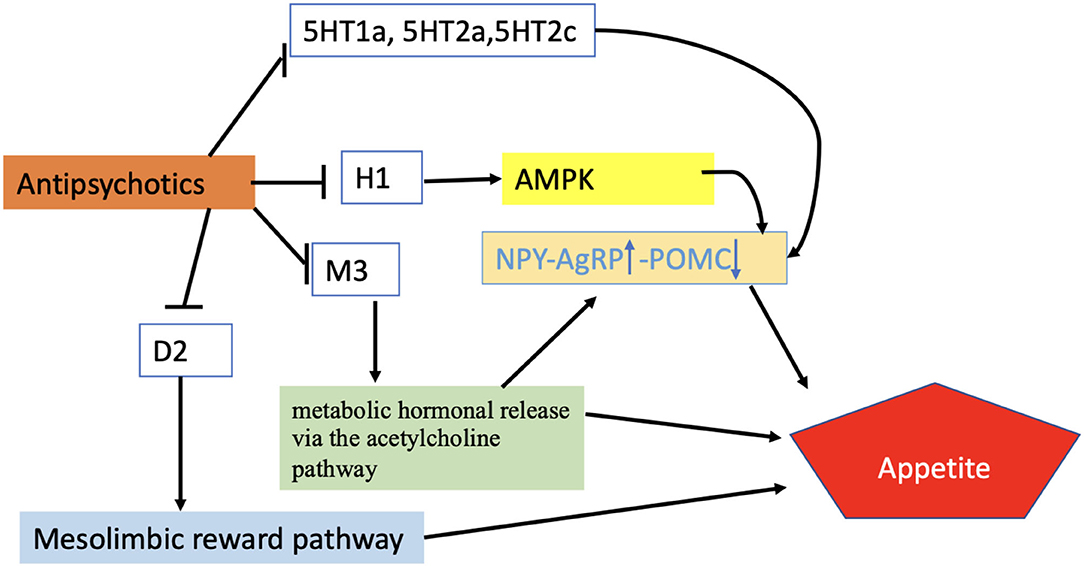
Figure 3. Regulation of appetite by APDs. According to the proposed scheme, APDs enhance appetite through different receptor systems activating AMPK-NPY system, metabolic hormonal release via Ach pathway, or mesolimbic pathway. Blunt-ended black lines indicate inhibitions. Black arrows indicate stimulations. The upward arrow represents the upregulation of the NPY/AgRP signals while the downward arrow indicates the downward regulation of POMC. H1, Histamine receptor; M3, Muscarinic receptor; D2, Dopamine receptor; 5HT1a,2a,2c, Serotonin receptors; AMPK, AMP-activated protein kinase; NPY/AgRP, Neuropeptide Y/ agouti-related peptide; POMC, Proopiomelanocortin.
Quetiapine is another antipsychotic drug with 5HT receptor antagonistic properties that is linked to a significant risk of obesity and hyperphagia (47). The receptor affinity of quetiapine is high for serotonergic 5HT2a-receptors, moderate for dopamine D2-receptors, and low for 5HT2c compared to D2-receptors (48). In a recent case study, it was reported that this receptor binding profile leads to disturbances in sleep, and that quetiapine may cause sleep-related eating disorders. However, the precise mechanism of how quetiapine facilitated sleep-related food intake remains unclear (49).
Risperidone also possesses antagonism of 5HT2 receptors as well as milder dopamine D2 receptor antagonism (50). Kursungoz et al. (51) have found that risperidone affects the expression and plasma concentrations of appetite-regulating hypothalamic peptides by inhibiting 5HT2c receptors. This conclusion is supported by a recent study by Wan and colleagues (52), demonstrating that the 5HT2c receptor-NPY pathways are involved in the stimulatory effects of risperidone on appetite and weight gain in rodents.
Ziprasidone and aripiprazole are two SGAs that are associated with little or no weight gain (53). Ziprasidone acts as an agonist at 5HT1a receptors and as an antagonist at 5HT2a, 5HT2c, and 5HT1b/1d receptors, while aripiprazole has partial agonism at 5HT1a receptors and antagonism at 5HT2 receptors. It has been reported that the coadministration of olanzapine with ziprasidone or aripiprazole does not induce food intake (54), possibly due to the partial agonism effect on 5HT1a receptors that they have in common, but other mechanisms may also be involved.
Some of the orexigenic effects of atypical APDs are believed to be mediated via their effects on histamine H1- and H3-receptors (5, 41, 55, 56) (Table 1, Figure 3). Central histaminergic activity is known to repress food intake, and it has been reported that the administration of specific H1-antagonists caused hyperphagia in both humans and rodents. Accordingly, mice lacking H1 receptors display increased food intake, altered feeding patterns, and obesity (57). In contrast, H3-receptor antagonists have the opposite effect and cause hypophagia (58).
It has been reported that olanzapine-treated rats show a reduction in H1 receptor expression levels in ARC and VMH with an increase in body weight and food intake relative to rats treated with haloperidol or aripiprazole (59). Furthermore, both olanzapine and clozapine have been reported to reduce H1 receptors levels in the VMH, with concomitant stimulation of AMP-activated protein kinase (AMPK), leading to an increase in food intake and weight gain (60). In another study, it has been reported that, by suppressing postsynaptic H1 receptors, olanzapine activates pre-synaptic H3 autoreceptors, reducing histamine synthesis and secretion, aggravating hyperphagia (61). For instance, clozapine acts on H3 auto-receptors (with moderate affinity) to block acetylcholine (ACh) and noradrenaline (NA) release, resulting in dysregulation of appetite (62). Other reports support the involvement of H1 and H3 receptors in antipsychotic-induced weight gain and food intake (63, 64). For instance, betahistine (a potent H1 and H3 agonist) has been combined with APDs (olanzapine/clozapine) and has been found to reduce APD-induced food intake and obesity both in rodents and humans through the H1R-NPY and the H1R-pAMPKα pathways (65–67).
A positron emission tomography study (PET) by Sato et al. (68) revealed high H1-receptor occupancy values in the human brain at low clinical doses of olanzapine and quetiapine, further supporting the role of the histaminergic system in controlling the appetite and obesity in humans.
In summary, direct antagonism of hypothalamic H1 receptors by SGAs can stimulate appetite involving hypothalamus-brainstem circuitry, or weight gain may be influenced by APDs partly by H3 receptors. The decrease in H1 receptor activity induced by APDs may lead to a reduction in histamine release via H3 receptors, thereby increasing the food intake by blocking 5HT, NA, and ACh release.
Dopamine antagonism (or, in the case of e.g., aripiprazole, partial agonism) is the core property of all APDs (Table 1, Figure 3). Two major dopaminergic pathways originating from the VTA (mesolimbic and mesocortical pathways) are particularly involved in hedonic feeding (69). Upon ingestion of palatable foods, dopamine is released into the ventral tegmental area of the brain, resulting in activation of the neural mechanisms connecting the VTA to the NAc by the midbrain.
Researchers have recently shown that dopamine D1 and D2 receptors (D1R and D2R) are found in POMC-positive neurons in the ARCs of rats and mice (70). Hence, these findings support that dysregulation in feeding behavior mainly relies on the dysregulation of dopamine levels as well as receptor activity that can be controlled by APDs.
It has been shown that the APD sulpiride, another D2R antagonist, increases calorie intake, whereas bromocriptine, a specific D2R agonist, counteracts this effect (71). A study by Kaur and Kulkarni (72) shows that SKF 38393 (a D1R agonist) and quinpirole (a D2R agonist) significantly reverse clozapine-induced hyperphagia. Similarly, Cho et al. (73) have shown that the availability of striatal dopamine (DA) D2/3R and body mass index are positively correlated.
Moreover, risperidone has been shown to reduce energy expenditure by antagonizing dopamine D2 receptors; thus, risperidone can also lead to weight gain by blocking D2 (74) receptors. Thus, these reports support that the dopaminergic pathway contributes to APD-induced weight gain.
Anorexia nervosa (AN) is a serious eating disorder identified with intense loss of appetite, extreme weight loss, and tremendous concern about gaining weight (75). A model for aspects of AN can be explained in activity-based anorexia (ABA), which is characterized by weight loss, hypophagia, and unusual restlessness in rodents with access to restricted food and running wheels.
It has been found that the antipsychotic amisulpride, which is also a D2/3R antagonist, prevents hypophagia and weight loss during ABA in a similar way to another D2/3R antagonist, eticlopride.
Further, amisulpride has been shown to improve weight loss tendency and hypophagia compared to olanzapine (76). Hence, selective antagonism of D2 and/or D3 receptors have been shown to reduce ABA strongly. According to a recent study on ABA by Fraga et al. (75), it has been indicated that ABA affects brown adipose tissue (BAT) and white adipose tissue (WAT) thermogenesis in significant ways. Moreover, ABA does not alter primary regulators of adipose tissue activity, such as hypothalamic AMPK or endoplasmic reticulum stress signaling, raising the question of whether dopaminergic signaling somehow affects thermogenesis that may play a role in the feeding behavior. There is still a need to determine whether this principle also applies to APD-induced weight gain.
Muscarinic acetylcholine receptors are involved in appetite regulation, but few studies have looked specifically at muscarinic signaling in hypothalamic appetite regulation (77). According to Nakajima et al. (78), the M3 receptor is especially critical to the control of appetite and metabolism in the hypothalamus. M3 receptor activity involves a complex interplay between peripheral and central nervous systems, with M3 receptors controlling metabolic hormones via the vagus nerve linked to the hyperphagic, diabetogenic, and weight gain risks associated with APDs (Table 1, Figure 3). Correlation between muscarinic receptor affinity and weight gain risk (55), as well as correlation with the diabetogenic potential of antipsychotic medicines, has been demonstrated (79). Olanzapine and clozapine are M3 antagonists (77, 80), and both of them have been reported to increase M3R binding density in the ARC, VMH, and DVC, which results in increased weight and food intake (77). The weight gain effect of APDs through the antagonism of the M3 receptor is also supported by findings that cevimeline (an M3 agonist) reduces body weight gain caused by olanzapine treatment (81, 82).
Furthermore, direct M3 receptor antagonism in the hypothalamus and DVC by APDs (olanzapine, clozapine) has been reported to generate peripheral metabolic effects (83). Overall, these references suggest APDs' blockade of M3R in the brain may cause dysregulation of metabolic, hormonal release via the acetylcholine pathway, which may involve hypothalamic neurons that regulate appetite.
Several recent studies have examined the intracellular mechanisms activated by the receptor-binding properties of APDs, as described above. There has been a great deal of attention paid to the AMPK pathway, which is pivotal in the hypothalamic energy system (Figure 1). AMPK, a cellular sensor of energy homeostasis, controls energy balance by modulating hypothalamic fatty acid metabolism (17, 23, 84–86). Increased AMPK activity is a potent orexigenic driver, whereas inhibition of AMPK, particularly in the VMH, increases brown fat thermogenesis (17, 85, 87). The physiological relevance of hypothalamic AMPK on feeding control has been elucidated by the work of Claret and colleagues that demonstrate that genetic ablation of AMPKα2 in POMC and AgRP neurons promote hyperphagic (obese) and hypophagic (lean) phenotypes (88). According to early evidence, pharmacologically or adenovirus-mediated stimulation of medial hypothalamic AMPK enhances appetite by increasing the expression of NPY and AgRP in the brain (89). In addition, hypothalamic AMPK has been shown to regulate dietary selection, first- and second-phase insulin release, lipid metabolism, and gluconeogenesis in the liver, all of which are crucial for energy balance (90–93). AMPK phosphorylation, which activates the enzyme in the hypothalamus, has been explored directly as a means of addressing central mechanisms that influence appetite and weight gain (16, 94).
A variety of receptors may regulate AMPK activity, and the role of AMPK in APD-induced hyperphagia has been examined in several studies. In a study by Kim et al. (60), it has been demonstrated that clozapine and olanzapine stimulate the hypothalamic AMPK through H1 receptor antagonistic activity. Similarly, He et al. (95) show that olanzapine activates AMPK by blocking the H1Rs in the acute stage and induces hyperphagia in female rats. To investigate the specific effect of olanzapine on hypothalamic AMPK, Skrede et al. (96) have used an adenovirus expressing a dominant-negative AMPK to inhibit AMPK-signaling in the ARC or VMH of female rats, followed by olanzapine depot injection. Interestingly, olanzapine's weight-inducing effect is attenuated by inhibition of AMPK in the ARC but not in the VMH, indicating that it is the ARC-specific AMPK activation that drives the orexigenic potential of the drug. It has been reported that olanzapine-induced AMPK-NPY orexigenic signaling could be ameliorated by betahistine co-treatment, suggesting that the H1 receptor-AMPK-NPY pathway plays a role in olanzapine-induced obesity and increase in food intake (65, 66). Furthermore, a study by Chen et al. (97) demonstrated that olanzapine increases AMPK-NPY orexigenic signaling by interfering with the interaction of H1R and ghrelin receptors (GHSR1a) in the hypothalamus of mice.
Intracerebroventricular administration of olanzapine has shown to induce hyperglycemia and activation of AMPK in the hypothalamus of mice (98) via α-1 adrenoreceptor and D2 receptor-AMPK axis.
Polypeptide signaling molecules are also being studied as they are crucial for hypothalamic neuron communication, food intake, and the AMPK pathway (Figure 1). Fernø et al. (99) show that sub-chronic olanzapine treatment increases NPY and AgRP in the ARC and decreases POMC, which is in agreement with the dysregulation of food intake. Palasz et al. (100) observe that the rat amygdala and hippocampal expression of NPY and POMC mRNA differ qualitatively following prolonged administration of olanzapine. Surprisingly, the POMC level in the two tested regions is significantly increased by olanzapine; however, the NPY expression does not change from control to olanzapine. As discussed above, the expression of POMC produces anorexigenic effects. Hence, this paradoxical result can only be explained by the structural and functional differences between the amygdala, hippocampus, and hypothalamus. These changes in neuropeptide expression suggest that olanzapine may also exert a pharmacological effect differently in amygdala pathways. It may be possible that antipsychotics trigger metabolic side effects through this mechanism as well.
Sezlev-Bilecen et al. (101) report that olanzapine administration in male rats decreases mRNA levels of NPY, AgRP, and POMC in the hypothalamus while CART levels are unaffected. Additionally, NPY, AgRP, and α-MSH plasma levels are decreased significantly while CART levels are also decreased. It is possible, therefore, that the inhibitory role of olanzapine on ARC neurons could be a precursor to an imbalance in neurohormone release, contributing to weight gain.
A study conducted in juvenile female rats demonstrated that risperidone (an atypical APD) failed to alter CART levels, although it increased appetite and body mass by increasing mRNA expression of NPY/AgRP in the hypothalamus (102).
We, therefore, conclude that CART, along with other ARC neuropeptides, plays a crucial role in regulating feeding, body weight, and energy metabolism. Nonetheless, the inconsistency in the CART level suggests that further investigation will be required to elucidate the exact mechanism underlying CART action by APDs.
Throughout the last decade, research on endocannabinoids (EC) and the endocannabinoid system (ECS) has contributed to greater insight into their role in food intake (103) (Figure 1). ECS signals are mediated by CB1/CB2 receptors, as well as enzymes responsible for synthesizing and degrading the endogenous ligands attached to these receptors (104). CB1R has been shown to be highly expressed in hypothalamo-cortical and reward-related brain regions, which strongly suggests a direct role in controlling feeding (105, 106).
It has been reported that the functions of endocannabinoids are mediated by AMPK, which regulates energy balance by increasing the adenosine triphosphate/ adenosine monophosphate (ATP/AMP) ratio (107). Specifically, endocannabinoids have been shown to activate AMPK in the hypothalamus and are thus related to appetite (108). On the other hand, endocannabinoids have been shown to reduce the activity of AMPK in peripheral tissues, including adipose tissue, liver, and skeletal muscle (108, 109), leading to impaired metabolism of carbohydrates and lipids. Moreover, the reduction of AMPK can also activate lipogenesis, thereby promoting body mass and reducing energy expenditure. Consequently, CB1Rs have received the most attention because they play a crucial role in energy homeostasis and can treat obesity via rimonabant, a CB1R antagonist (110).
Administration of CB1R antagonists yields a reduction in food intake equally in mice with or without NPY knockout, suggesting that NPY and endocannabinoids promoted food intake independently (111–114). In other studies, it has been shown that sugar and alcohol consumption is increased in rats after administration of CB1R agonists (115, 116). On the contrary, Radziszewska et al. (117) have reported that activation of CB1R with its agonist (WIN 55, 212-2) produces anorexigenic effects. The results of these studies suggest that mostly the activation of CB receptor signaling can increase food consumption and lead to weight gain.
It has been reported that APDs alter cannabinoid receptor-binding density in the DVC of the brainstem and regulate appetite signaling and food intake. Weston-Green et al. (118) have reported that olanzapine significantly inhibits CB receptor binding in the DVC, whereas aripiprazole and haloperidol do not influence the receptor. According to these studies, olanzapine can lead to weight gain by modulating CB receptors in the DVC. According to Lazzari et al. (119), the metabolic side effects of olanzapine treatment can be neutralized by co-administration of CB1R antagonist compounds. In an investigation by Weston-Green et al. (120), it is suggested that a reduction in CB1R density during olanzapine treatment decreases cannabinoid-mediated suppression of GABA and thereby enhances GABAergic transmission to POMC neurons, reducing POMC activity and promoting weight gain.
Brain-derived neurotrophic factor (BDNF) and its receptor, Tyrosine Kinase Receptor B (TyrkB) (121), are involved in the regulation of hypothalamic neural circuitry and feeding behavior and expressed in most hypothalamic nuclei, including VMH, DMH, LH, ARC, and PVH. BDNF neurons in the PVH have been shown to reduce food intake (122), although BDNF has been found to be present in the highest levels in VMH (121, 123, 124). Preclinical studies have shown that reduced BDNF levels, as well as abnormal TrkB signaling, are associated with altered food intake, excessive body weight gain, and metabolic alterations (121, 123–127). These data suggest that reduction in BDNF levels or inhibition of these neurons can lead to hyperphagia and obesity.
APDs have been shown to regulate BDNF levels, suggesting a role for this pathway also in APD-induced weight gain. In a study by Zhang et al. (128), olanzapine-treated patients with pronounced metabolic syndrome have lower serum levels of BDNF than patients with a healthier metabolic profile. In line with this, a preclinical study with haloperidol and risperidone was shown to decrease BDNF levels as well as the levels of TrkB in the brain (129). However, other findings do not support reduced BDNF associated with APD-induced orexigenic effects. In one preclinical study, both clozapine and olanzapine were shown to increase the production of BDNF in the rat brain (130), and in another study, olanzapine diminished the BDNF-lowering effect of the less orexigenic haloperidol (131). In a meta-analysis, Lin (132) show that peripheral BDNF levels are inconsistently affected by antipsychotics in patients with schizophrenia, supporting that the role of BDNF in APD-induced weight gain is complex.
Some of the inconsistent findings may be explained by gender-specific effects. E.g., in one study, BDNF levels were shown to be negatively correlated with body mass index (BMI) gain in female patients but not in male patients (133). This was supported by another study, showing that decreased BDNF serum levels were associated with weight gain in female schizophrenia patients receiving long-term antipsychotic treatment (134). In this study, in 332 patients suffering from chronic schizophrenia treated with APDs, homeostatic model assessment for insulin resistance (HOMA-IR) and BDNF are associated in a differential manner according to. Hence, it is important to consider gender when measuring BDNF levels in patients with schizophrenia and metabolic indicators (134).
As a dynamic endocrine organ, the adipose tissue secretes hormones, peptides, and adipokines that regulate energy, lipid, and glucose homeostasis (135). Leptin was the first adipokine identified to facilitate a link between the adipose tissues and the hypothalamus. Increased fat mass leads to elevated secretion of leptin (136), with subsequent reduction in food intake and increase in energy expenditure (137, 138). Several areas of the hypothalamus are controlled by leptin, including the ARC, VMH, and PVH. By directly activating POMC neurons in the ARC, leptin increases the production of α-MSH. By projecting to MC4R neurons, melanocortin peptide reduces food intake and controls metabolism via energy storage, insulin secretion, and gastrointestinal motility. Moreover, leptin has been shown to inhibit food intake by interacting with dopaminergic neurons in the VTA (139). On the other hand, hedonic feeding suppresses satiety signals. It has been reported that mice lacking D2R were more sensitive to leptin (140–142).
Despite the anorexigenic effect of leptin, obesity is often associated with elevated leptin levels, indicating leptin resistance. When leptin resistance occurs, energy expenditure becomes unequal to intake, thereby increasing appetite and body weight. Leptin resistance may be caused by a defect in the nutritional regulation of leptin receptor gene expression, which may involve an impairment in signal transducers and activators of transcription (STAT3) signaling (143). However, even with an intact Janus kinase/signal transducers and activators of transcription (JAK2–STAT3) pathway of leptin signaling, hypothalamic neurons may exhibit leptin resistance. AMPK and acyl CoA carboxylase (ACC) are thought to be involved in leptin receptor signaling. The anorexigenic effects of leptin are shown to be dependent on inhibition of hypothalamic AMPK (Figure 1), and leptin resistance may thus result from defects in AMPK-leptin signaling pathways as well as defects in the JAK2-STAT3 pathway. According to Gorobets (144), leptin resistance plays a role in body weight accumulation, and leptin resistance induced by APDs has been suggested to cause body weight gain in psychotic patients (145). For example, Piao et al. (146) demonstrate that risperidone activates extracellular-signal-regulated kinase (ERK) and blocks leptin-induced STAT3 phosphorylation over the course of treatment, enhancing both SOCS3 and SOCS6 mRNA expression. Interestingly, risperidone's impact on leptin and insulin pathways could not be explained by body fat accumulation (147). Other researchers have also examined leptin levels in patients receiving various APDs (mainly clozapine and olanzapine) (148). Several reports suggest that leptin levels in patients with psychosis might be higher than in healthy controls and that they may remain high during APDs therapy (149–151). Potvin et al. reported that olanzapine, clozapine, and quetiapine significantly increase leptin levels and show a correlation between leptin and BMI (149).
Margulska et al. (152) have demonstrated that changes in fasting serum levels of appetite-regulating peptides are not directly associated with the levels of clozapine in patients with schizophrenia. Furthermore, fasting serum levels of other peptides (CART, peptide YY, AgRP, deacylated ghrelin, and obestatin) do not exhibit meaningful correlations.
In summary, APDs have the potential to influence leptin levels directly or indirectly through the effect of obesity and subsequent leptin resistance. Thus, despite of the many studies focusing on this issue, the relationship between leptin levels and antipsychotics remains uncertain.
The “hunger hormone” ghrelin is mainly produced by the stomach and stimulates appetite and appetite-related activities (153) (Figure 1). However, ghrelin has also been found to be distributed in other organs such as the pituitary, the lungs, the pancreas, the gall bladder, the esophagus, the colon, the liver, the spleen, the thyroid, the heart, and in the hypothalamic nuclei (ARC) (154–156). Ghrelin has two different forms, an acylated serine 3 form (ghrelin) and a des-acylated form (active form). The acylation of ghrelin is essential for binding to GHSR1a. Despite the appetite-promoting effect of ghrelin, it's mechanisms of action are not fully understood. An interesting study by Toshinai et al. demonstrates that the des-acylated form of ghrelin induces food intake independently of growth hormone secretagogue receptors (157). Other studies have shown that deficiencies of ghrelin have been linked to binge-eating and obesity (158, 159). Considering the molecular mechanism of appetite regulation, it has been reported that in the ARC and the hindbrain, ghrelin enhances food intake via activation of NPY and AgRP production (160, 161). However, both orexigenic (NPY/AgRP) and anorexigenic (POMC) neurons are shown to be modulated by ghrelin-expressing neurons (162). Moreover, ghrelin has been shown to activate AMPK in the hypothalamus to regulate food intake (163).
During treatment with some APDs, ghrelin has been reported to play a significant role in modulating appetite and energy homeostasis. Researchers have found that serum ghrelin levels in subjects treated with atypical APDs for at least 1 year are significantly higher than controls. An investigation shows a significant increase in total plasma ghrelin levels as well as active ghrelin after olanzapine administration after 6 months (164). Another study by the same group has shown that the level of active and total ghrelin in the risperidone group is significantly greater than that of healthy controls (165). In a preclinical study, it has been shown that olanzapine treatment elevates hypothalamic ghrelin receptor expression in rats (166, 167), supporting that ghrelin signaling plays a role in APD-induced obesity.
On the contrary, in a study on patients with schizophrenia, it was found that circulating ghrelin levels did not increase, but rather decreased, during treatment with risperidone or olanzapine, and there was also no significant difference between the effect of the two drugs (168). Similarly, in a meta-analysis by Goetz and Miller (169), it has been reported that patients with schizophrenia receiving olanzapine exhibit a significant drop in ghrelin levels. The reduction in ghrelin levels following olanzapine therapy may be due to obesity and excess energy intake. However, the drop in ghrelin levels may also be due to stress (170), which is common among psychotic patients. It is therefore questionable whether the drop in ghrelin level is the result of APDs treatment or stress.
Tagami et al. (171) used an electrical impedance-based receptor biosensor assay system (CellKeyTM) to study the effect of olanzapine on ghrelin-mediated GHSR signaling, reporting that olanzapine increased ghrelin-induced GHSR activity. Huang et al. reported that the 5HT2c receptor is dimerized with GHSR1a to inhibit orexigenic signaling, while 5HT2cR antagonists reduce dimerization and increase GHSR1a-induced food consumption (172). However, it is still necessary to establish the relationship between the 5HT2c receptor and GHSR, as well as and other receptor subtypes on appetite and obesity.
Adiponectin is an adipokine secreted by WAT and widely distributed in circulation. Adiponectin is known to promote fatty acid oxidation and insulin sensitivity in peripheral tissues by activating AMPK. A previous report (173) has shown that adiponectin increases AMPK activity in the ARC through its receptor AdipoR1, causing an increase in food intake. Moreover, mice deficient in adiponectin show reduced phosphorylation of AMPK in the ARC, resulting in a decrease in appetite, as well as higher energy expenditure.
Studies have shown that antipsychotic drugs can alter adiponectin levels. In a meta-analysis by Bartoli et al. (174), it has been found that people taking clozapine and olanzapine display lower adiponectin levels than those that are treated with risperidone. The lower adiponectin levels indicate reduced insulin signaling and are normally associated with metabolic disturbances. However, this meta-analysis does not correlate adiponectin levels with weight gain or other metabolic abnormalities. In a study by Lu et al. (175), APDs have been shown to impact adipokines levels and energy homeostasis, and olanzapine decreases the levels of adiponectin more effectively than clozapine. Other studies have found that adiponectin levels are also decreased by antipsychotics independent of adiposity and BMI (176). Thus, both APDs might influence adiponectin levels, but the mechanisms by which they do so have not been identified.
In conclusion, the dysregulated appetite and weight gain associated with atypical APDs seem to be associated with altered serum ghrelin and adiponectin levels or their signaling. Further research is needed to clarify the causality in this link and to explain the inconsistencies observed between several studies.
The possible role of the gut-brain axis in APD appetite regulation has been subject to attention during recent years (Figure 4). The brain is connected to the gut in several ways, including the vagus nerve, the hypothalamic-pituitary-adrenal axis, hormones, and metabolites, as well as through the immune system (177). Additionally, gut microbes have been shown to metabolize ingested nutrients to produce signaling molecules involved in the regulation of the stress axis, energy homeostasis, and obesity (178–180). Also, gut microbes regulate bile acid metabolism to produce a range of metabolites, including short-chain fatty acids (SCFAs), neurotransmitters, tiny protein molecules, and toxins (181). These metabolites may act directly as endocrine factors in addition to transmitting information to the hypothalamic circuitry through the vagal nerve. Both the VMH and ARC act together to sense peripheral metabolic and nutrient signals, thereby controlling appetite and glucose homeostasis (182).
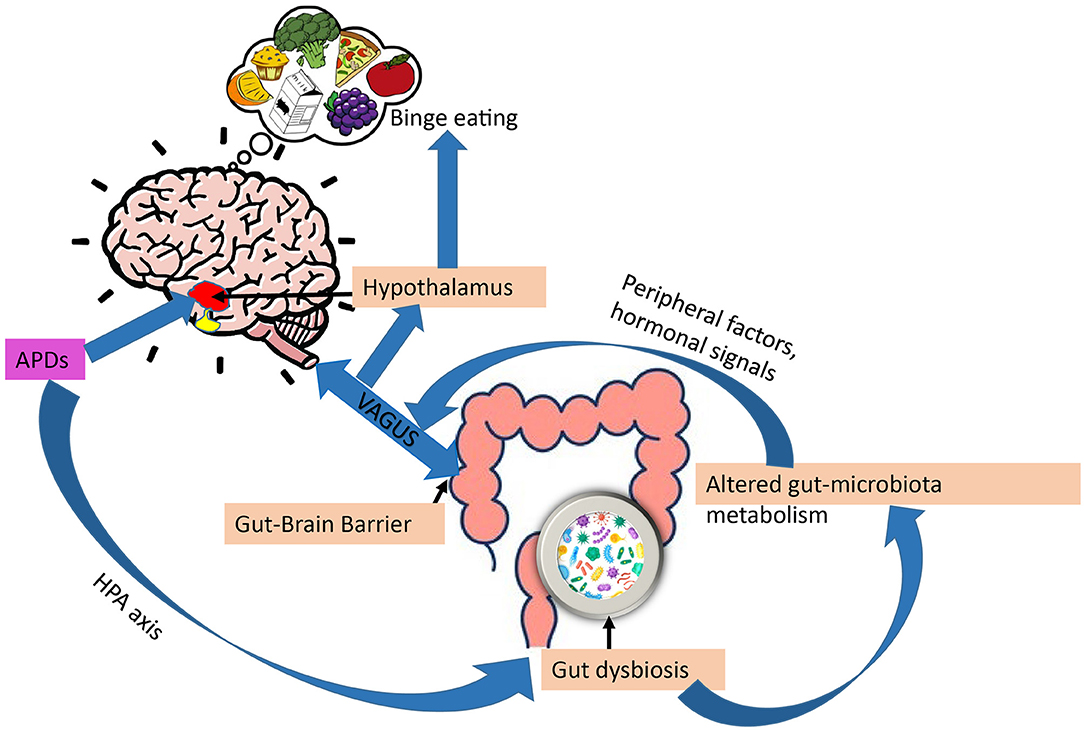
Figure 4. Gut-brain axis and regulation of appetite by APDs. Through impaired stress axis, antipsychotics (APDs) can influence gut microbiota composition, in turn, gut dysbiosis can alter gut-microbiota metabolism, changing the metabolites, leading to increased overeating and weight gain via peripheral (leptin, ghrelin) and vagal afferent signaling.
APDs have been shown to alter gut microbiota compositions. For example, mice treated with olanzapine have altered gut microbiota compositions associated with obesity (183). According to Maier et al. (184), APDs can significantly reduce the level of Akkermansia mucini (human intestinal mucin degradation bacteria) in patients. An inverse relationship between the number of Akkermansia mucini and insulin resistance, as well as inflammation, has been demonstrated in mice. This supports the possibility that APDs may alter glucose metabolism directly via gut dysbiosis, which in turn can contribute to APD-induced obesity (34). Additionally, a study by Bahr et al. (185) has shown that risperidone increases body mass in children, reduces energy expenditure, and alters microbial composition. In another study by Yuan et al. (186) establishes that risperidone treatment increases body weight in first-episode schizophrenia patients and alters their gut microbiota.
We mentioned previously that gut microbiota metabolize food to generate signaling molecules involved in energy homeostasis. By altering the metabolites produced by dysbiosis of the gut, APDs have been shown to disrupt energy homeostasis. SCFAs, the main gut metabolites, have been shown to increase the ratio of AMP/ATP by activating AMPK (187–189). It has been observed that SCFA levels in feces are 20% higher in obese individuals than in lean individuals (190, 191). Therefore, increasing SCFA is related to obesity. A study has shown that olanzapine increases plasma SCFA levels in rats (192, 193). Moreover, antipsychotics have also been reported to change gut- microbiota metabolism through vitamin B6 (194). Thus, confirming APDs can alter gut-microbiota metabolism, thereby contributing to eating behavior and weight gain mediated by the gut-brain axis (195, 196).
Antipsychotics have been shown to impair the hypothalamic-pituitary axis (HPA) axis in psychotic disorders (178). On the other hand, several reports suggest that mental disorders and stress are also associated with gastrointestinal symptoms, and altered gut microbiota may thus be caused by the disease per se. and not by the pharmacological treatment (186, 197, 198). There are many studies demonstrating that the microbiome composition is less healthy in many treatment-naive patients with schizophrenia, bipolar disorder, and major depressive disorder (MDD) with psychotic features in comparison with healthy individuals (186, 199). Thus, this raises the question of whether gut-dysbiosis is mainly caused by APDs or the underlying psychotic disorder.
Overall, the alteration of gut microbiota composition by APDs may disrupt the gut-brain axis, which represents a potential mechanism of action in how these drugs induce appetite and disrupt energy homeostasis.
Several antipsychotic drugs trigger weight gain, which is primarily due to an increase in food consumption. As discussed in this review, this effect is, at least in part, caused by the receptor binding profiles of these drugs and how they affect pathways in brain areas involved in appetite regulation. A high affinity for dopamine D2, serotonin 5HT2c, and histamine H1 receptors seems to be of great importance. Among antipsychotics with a high affinity for the 5HT2c receptor, clozapine and olanzapine cause the greatest weight gain and hyperphagia. Further, antagonistic effects on muscarinic and histaminergic receptors likely contribute to increased food consumption and metabolic dysfunction.
Considering the intercellular mechanisms involved, it has been shown that blocking H1 and 5HT2c receptors stimulate AMPK activity in the hypothalamus, which affects feeding through modulation of neuropeptide expression in ARC.
Other pathways that could be involved in the regulation of APD-induced dysregulated food intake include the endocannabinoid system and BDNF-mediated signaling. APDs seemingly do not have a direct effect on the cannabinoid system in regulating food intake. However, multiple neurotransmitter systems which might indirectly control CB1 receptors are shown to be affected by APDs. For example, olanzapine induces a reduction in CB1R density in both the hypothalamus and brain stem that stimulate the NPY and enhance inhibitory GABAergic input, thereby inhibiting the POMC and causing increased food intake. BDNF is a protein that may affect eating behavior, hormone release and activity, and energy homeostasis. The use of APDs has been shown to alter BDNF levels in the brain, cerebrospinal fluid (CSF), and serum in patients with schizophrenia, as reflected in animal studies. Nevertheless, to what degree BDNF activity may contribute to the obesity-related side effects of APDs is uncertain.
Finally, the enteric nervous system has been shown to have a complex relationship with gut microbiota and the central nervous system that can regulate food intake. Specifically, APDs have been found to affect the gut microbiota composition in such a way that it encourages overeating and weight gain via hormonal (leptin and ghrelin) and nutrient signaling via vagal afferents.
Unfortunately, there are currently no superior strategies available to prevent antipsychotic-induced hyperphagia or obesity. Consequently, a better understanding of the food intake controlling mechanisms that contribute to specific health impairments during APDs may assist in the creation of specific treatment strategies.
SM drafted the article which was critically reviewed by all authors during the development of the article. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was funded by EuroNanoMed III (Grant No. EURONANOMED2019050/ENAMEP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. (2008) 30:67–76. doi: 10.1093/epirev/mxn001
2. Nordentoft M, Wahlbeck K, Hallgren J, Westman J, Osby U, Alinaghizadeh H, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. (2013) 8:e55176. doi: 10.1371/journal.pone.0055176
3. Deh M, Correll CU, Bobes J, Cetkovich-Bakmas M, Cohen D, Asai I, et al. Physical illness in patients with severe mental disorders. I. Prevalence, impact of medications and disparities in health care. World Psychiatry. (2011) 10:52–77. doi: 10.1002/j.2051-5545.2011.tb00014.x
4. Liu NH, Daumit GL, Dua T, Aquila R, Charlson F, Cuijpers P, et al. Excess mortality in persons with severe mental disorders: a multilevel intervention framework and priorities for clinical practice, policy and research agendas. World Psychiatry. (2017) 16:30–40. doi: 10.1002/wps.20384
5. Kaar SJ, Natesan S, McCutcheon R, Howes OD. Antipsychotics: mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology. (2020) 172:107704. doi: 10.1016/j.neuropharm.2019.107704
6. Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. (2013) 382:951–62. doi: 10.1016/S0140-6736(13)60733-3
7. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. (2002) 16:23–45. doi: 10.2165/00023210-200216010-00003
8. Pillinger T, McCutcheon RA, Vano L, Mizuno Y, Arumuham A, Hindley G, et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: a systematic review and network meta-analysis. Lancet Psychiatry. (2020) 7:64–77. doi: 10.1016/S2215-0366(19)30416-X
9. Correll CU, Detraux J, De Lepeleire J, De Hert M. Effects of antipsychotics, antidepressants and mood stabilizers on risk for physical diseases in people with schizophrenia, depression and bipolar disorder. World Psychiatry. (2015) 14:119–36. doi: 10.1002/wps.20204
10. Blouin M, Tremblay A, Jalbert ME, Venables H, Bouchard RH, Roy MA, et al. Adiposity and eating behaviors in patients under second generation antipsychotics. Obesity. (2008) 16:1780–7. doi: 10.1038/oby.2008.277
11. Cooper GD, Pickavance LC, Wilding JP, Halford JC, Goudie AJ. A parametric analysis of olanzapine-induced weight gain in female rats. Psychopharmacology. (2005) 181:80–9. doi: 10.1007/s00213-005-2224-4
12. Hartfield AW, Moore NA, Clifton PG. Effects of clozapine, olanzapine and haloperidol on the microstructure of ingestive behaviour in the rat. Psychopharmacology. (2003) 167:115–22. doi: 10.1007/s00213-002-1368-8
13. Benarroch L, Kowalchuk C, Wilson V, Teo C, Guenette M, Chintoh A, et al. Atypical antipsychotics and effects on feeding: from mice to men. Psychopharmacology. (2016) 233:2629–53. doi: 10.1007/s00213-016-4324-8
14. Mayfield K, Siskind D, Winckel K, Russell AW, Kisely S, Smith G, et al. Glucagon-like peptide-1 agonists combating clozapine-associated obesity and diabetes. J Psychopharmacol. (2016) 30:227–36. doi: 10.1177/0269881115625496
15. Reynolds GP, Kirk SL. Metabolic side effects of antipsychotic drug treatment–pharmacological mechanisms. Pharmacol Ther. (2010) 125:169–79. doi: 10.1016/j.pharmthera.2009.10.010
16. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. (2006) 443:289–95. doi: 10.1038/nature05026
17. Lopez M, Lelliott CJ, Tovar S, Kimber W, Gallego R, Virtue S, et al. Tamoxifen-induced anorexia is associated with fatty acid synthase inhibition in the ventromedial nucleus of the hypothalamus and accumulation of malonyl-CoA. Diabetes. (2006) 55:1327–36. doi: 10.2337/db05-1356
18. Waterson MJ, Horvath TL. Neuronal regulation of energy homeostasis: beyond the hypothalamus and feeding. Cell Metab. (2015) 22:962–70. doi: 10.1016/j.cmet.2015.09.026
19. Baldini G, Phelan KD. The melanocortin pathway and control of appetite-progress and therapeutic implications. J Endocrinol. (2019) 241:R1–R33. doi: 10.1530/JOE-18-0596
20. Hu Z, Cha SH, Chohnan S, Lane MD. Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc Natl Acad Sci USA. (2003) 100:12624–9. doi: 10.1073/pnas.1834402100
21. Carling D. The AMP-activated protein kinase cascade–a unifying system for energy control. Trends Biochem Sci. (2004) 29:18–24. doi: 10.1016/j.tibs.2003.11.005
22. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. (2005) 1:15–25. doi: 10.1016/j.cmet.2004.12.003
23. Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. (2008) 14:539–49. doi: 10.1016/j.molmed.2008.09.007
24. Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. (2010) 30:2472–9. doi: 10.1523/JNEUROSCI.3118-09.2010
25. Hill JW. PVN pathways controlling energy homeostasis. Indian J Endocrinol Metab. (2012) 16:S627–36. doi: 10.4103/2230-8210.105581
26. Kublaoui BM, Holder JL Jr, Gemelli T, Zinn AR. Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Mol Endocrinol. (2006) 20:2483–92. doi: 10.1210/me.2005-0483
27. Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Levy E, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. (2001) 10:1465–73. doi: 10.1093/hmg/10.14.1465
28. Zhang J, Chen D, Sweeney P, Yang Y. An excitatory ventromedial hypothalamus to paraventricular thalamus circuit that suppresses food intake. Nat Commun. (2020) 11:6326. doi: 10.1038/s41467-020-20093-4
29. Lopez M, Tena-Sempere M. Estrogens and the control of energy homeostasis: a brain perspective. Trends Endocrinol Metab. (2015) 26:411–21. doi: 10.1016/j.tem.2015.06.003
30. Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. (2006) 49:191–203. doi: 10.1016/j.neuron.2005.12.021
31. van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. (2012) 73:1184–94. doi: 10.1016/j.neuron.2012.02.016
32. Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. (2012) 2:33–41. doi: 10.1016/j.celrep.2012.05.011
33. Avena NM, Rada PV. Cholinergic modulation of food and drug satiety and withdrawal. Physiol Behav. (2012) 106:332–6. doi: 10.1016/j.physbeh.2012.03.020
34. Schneeberger M, Gomis R, Claret M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J Endocrinol. (2014) 220:T25–46. doi: 10.1530/JOE-13-0398
35. Grill HJ, Kaplan JM. The neuroanatomical axis for control of energy balance. Front Neuroendocrinol. (2002) 23:2–40. doi: 10.1006/frne.2001.0224
36. Niu SN, Huang ZB, Wang H, Rao XR, Kong H, Xu J, et al. Brainstem Hap1-Ahi1 is involved in insulin-mediated feeding control. FEBS Lett. (2011) 585:85–91. doi: 10.1016/j.febslet.2010.11.059
37. Zsombok A, Smith BN. Plasticity of central autonomic neural circuits in diabetes. Biochim Biophys Acta. (2009) 1792:423–31. doi: 10.1016/j.bbadis.2008.12.001
38. Blundell JE. Serotonin and appetite. Neuropharmacology. (1984) 23:1537–51. doi: 10.1016/0028-3908(84)90098-4
39. Bonn M, Schmitt A, Lesch KP, Van Bockstaele EJ, Asan E. Serotonergic innervation and serotonin receptor expression of NPY-producing neurons in the rat lateral and basolateral amygdaloid nuclei. Brain Struct Funct. (2013) 218:421–35. doi: 10.1007/s00429-012-0406-5
40. Sohn JW, Xu Y, Jones JE, Wickman K, Williams KW, Elmquist JK. Serotonin 2C receptor activates a distinct population of arcuate pro-opiomelanocortin neurons via TRPC channels. Neuron. (2011) 71:488–97. doi: 10.1016/j.neuron.2011.06.012
41. Kroeze WK, Hufeisen SJ, Popadak BA, Renock SM, Steinberg S, Ernsberger P, et al. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. (2003) 28:519–26. doi: 10.1038/sj.npp.1300027
42. Davies MA, Setola V, Strachan RT, Sheffler DJ, Salay E, Hufeisen SJ, et al. Pharmacologic analysis of non-synonymous coding h5-HT2A SNPs reveals alterations in atypical antipsychotic and agonist efficacies. Pharmacogenomics J. (2006) 6:42–51. doi: 10.1038/sj.tpj.6500342
43. Nasrallah HA. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol Psychiatry. (2008) 13:27–35. doi: 10.1038/sj.mp.4002066
44. Joshi RS, Singh SP, Panicker MM. 5-HT2A deletion protects against Clozapine-induced hyperglycemia. J Pharmacol Sci. (2019) 139:133–5. doi: 10.1016/j.jphs.2018.11.015
45. Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. (1995) 374:542–6. doi: 10.1038/374542a0
46. Lord CC, Wyler SC, Wan R, Castorena CM, Ahmed N, Mathew D, et al. The atypical antipsychotic olanzapine causes weight gain by targeting serotonin receptor 2C. J Clin Invest. (2017) 127:3402–6. doi: 10.1172/JCI93362
47. Rasmussen H, Ebdrup BH, Oranje B, Pinborg LH, Knudsen GM, Glenthoj B. Neocortical serotonin2A receptor binding predicts quetiapine associated weight gain in antipsychotic-naive first-episode schizophrenia patients. Int J Neuropsychopharmacol. (2014) 17:1729–36. doi: 10.1017/S1461145714000777
48. Tarazi FI, Zhang K, Baldessarini RJ. Long-term effects of olanzapine, risperidone, and quetiapine on serotonin 1A, 2A and 2C receptors in rat forebrain regions. Psychopharmacology. (2002) 161:263–70. doi: 10.1007/s00213-002-1016-3
49. Heathman JC, Neal DW, Thomas CR. Sleep-related eating disorder associated with quetiapine. J Clin Psychopharmacol. (2014) 34:658–60. doi: 10.1097/JCP.0000000000000198
50. Nyberg S, Farde L, Eriksson L, Halldin C, Eriksson B. 5-HT2 and D2 dopamine receptor occupancy in the living human brain. A PET study with risperidone. Psychopharmacology. (1993) 110:265–72. doi: 10.1007/BF02251280
51. Kursungoz C, Ak M, Yanik T. Effects of risperidone treatment on the expression of hypothalamic neuropeptide in appetite regulation in Wistar rats. Brain Res. (2015) 1596:146–55. doi: 10.1016/j.brainres.2014.10.070
52. Wan XQ, Zeng F, Huang XF, Yang HQ, Wang L, Shi YC, et al. Risperidone stimulates food intake and induces body weight gain via the hypothalamic arcuate nucleus 5-HT2c receptor-NPY pathway. CNS Neurosci Ther. (2020) 26:558–66. doi: 10.1111/cns.13281
53. Dayabandara M, Hanwella R, Ratnatunga S, Seneviratne S, Suraweera C, de Silva VA. Antipsychotic-associated weight gain: management strategies and impact on treatment adherence. Neuropsychiatr Dis Treat. (2017) 13:2231–41. doi: 10.2147/NDT.S113099
54. Snigdha S, Thumbi C, Reynolds GP, Neill JC. Ziprasidone and aripiprazole attenuate olanzapine-induced hyperphagia in rats. J Psychopharmacol. (2008) 22:567–71. doi: 10.1177/0269881107081519
55. Matsui-Sakata A, Ohtani H, Sawada Y. Receptor occupancy-based analysis of the contributions of various receptors to antipsychotics-induced weight gain and diabetes mellitus. Drug Metab Pharmacokinet. (2005) 20:368–78. doi: 10.2133/dmpk.20.368
56. Deng C, Weston-Green K, Huang X-F. The role of histaminergic H1 and H3 receptors in food intake: a mechanism for atypical antipsychotic-induced weight gain? Progr Neuro Psychopharmacol Biol Psychiatry. (2010) 34:1–4. doi: 10.1016/j.pnpbp.2009.11.009
57. Masaki T, Chiba S, Yasuda T, Noguchi H, Kakuma T, Watanabe T, et al. Involvement of hypothalamic histamine H1 receptor in the regulation of feeding rhythm and obesity. Diabetes. (2004) 53:2250–60. doi: 10.2337/diabetes.53.9.2250
58. Attoub S, Moizo L, Sobhani I, Laigneau JP, Lewin MJ, Bado A. The H3 receptor is involved in cholecystokinin inhibition of food intake in rats. Life Sci. (2001) 69:469–78. doi: 10.1016/S0024-3205(01)01138-9
59. Han M, Deng C, Burne THJ, Newell KA, Huang X-F. Short- and long-term effects of antipsychotic drug treatment on weight gain and H1 receptor expression. Psychoneuroendocrinology. (2008) 33:569–80. doi: 10.1016/j.psyneuen.2008.01.018
60. Kim SF, Huang AS, Snowman AM, Teuscher C, Snyder SH. From the Cover: Antipsychotic drug-induced weight gain mediated by histamine H1 receptor-linked activation of hypothalamic AMP-kinase. Proc Natl Acad Sci USA. (2007) 104:3456–9. doi: 10.1073/pnas.0611417104
61. Davoodi N, Kalinichev M, Clifton PG. Comparative effects of olanzapine and ziprasidone on hypophagia induced by enhanced histamine neurotransmission in the rat. Behav Pharmacol. (2008) 19:121–8. doi: 10.1097/FBP.0b013e3282f62c66
62. Schlicker E, Marr I. The moderate affinity of clozapine at H3 receptors is not shared by its two major metabolites and by structurally related and unrelated atypical neuroleptics. Naunyn Schmiedebergs Arch Pharmacol. (1996) 353:290–4. doi: 10.1007/BF00168630
63. Lian J, Huang XF, Pai N, Deng C. Ameliorating antipsychotic-induced weight gain by betahistine: mechanisms and clinical implications. Pharmacol Res. (2016) 106:51–63. doi: 10.1016/j.phrs.2016.02.011
64. Smith RC, Maayan L, Wu R, Youssef M, Jing Z, Sershen H, et al. Betahistine effects on weight-related measures in patients treated with antipsychotic medications: a double-blind placebo-controlled study. Psychopharmacology (Berl). (2018) 235:3545–58.
65. Lian J, Huang X-F, Pai N, Deng C. Betahistine ameliorates olanzapine-induced weight gain through modulation of histaminergic, NPY and AMPK pathways. Psychoneuroendocrinology. (2014) 48:77–86. doi: 10.1016/j.psyneuen.2014.06.010
66. Lian J, Huang XF, Pai N, Deng C. Preventing olanzapine-induced weight gain using betahistine: a study in a rat model with chronic olanzapine treatment. PLoS ONE. (2014) 9:e104160. doi: 10.1007/s00213-018-5079-1
67. Barak N, Beck Y, Albeck JH. Betahistine decreases olanzapine-induced weight gain and somnolence in humans. J Psychopharmacol. (2016) 30:237–41. doi: 10.1177/0269881115626349
68. Sato H, Ito C, Hiraoka K, Tashiro M, Shibuya K, Funaki Y, et al. Histamine H1 receptor occupancy by the new-generation antipsychotics olanzapine and quetiapine: a positron emission tomography study in healthy volunteers. Psychopharmacology. (2015) 232:3497–505. doi: 10.1007/s00213-015-4002-2
69. Coccurello R, Maccarrone M. Hedonic eating and the “delicious circle”: from lipid-derived mediators to brain dopamine and back. Front Neurosci. (2018) 12:271. doi: 10.3389/fnins.2018.00271
70. Romanova IV, Derkach KV, Mikhrina AL, Sukhov IB, Mikhailova EV, Shpakov AO. The leptin, dopamine and serotonin receptors in hypothalamic POMC-neurons of normal and obese rodents. Neurochem Res. (2018) 43:821–37. doi: 10.1007/s11064-018-2485-z
71. Baptista T, Parada M, Hernandez L. Long term administration of some antipsychotic drugs increases body weight and feeding in rats. Are D2 dopamine receptors involved? Pharmacol Biochem Behav. (1987) 27:399–405. doi: 10.1016/0091-3057(87)90340-6
72. Kaur G, Kulkarni SK. Studies on modulation of feeding behavior by atypical antipsychotics in female mice. Prog Neuropsychopharmacol Biol Psychiatry. (2002) 26:277–85. doi: 10.1016/S0278-5846(01)00266-4
73. Cho SS, Yoon EJ, Kim SE. Asymmetry of dopamine D2/3 receptor availability in dorsal putamen and body mass index in non-obese healthy males. Exp Neurobiol. (2015) 24:90–4. doi: 10.5607/en.2015.24.1.90
74. Grant S, Fitton A. Risperidone. A review of its pharmacology and therapeutic potential in the treatment of schizophrenia. Drugs. (1994) 48:253–73. doi: 10.2165/00003495-199448020-00009
75. Fraga A, Rial-Pensado E, Nogueiras R, Ferno J, Dieguez C, Gutierrez E, et al. Activity-based anorexia induces browning of adipose tissue independent of hypothalamic AMPK. Front Endocrinol. (2021) 12:669980. doi: 10.3389/fendo.2021.669980
76. Klenotich SJ, Ho EV, McMurray MS, Server CH, Dulawa SC. Dopamine D2/3 receptor antagonism reduces activity-based anorexia. Transl Psychiatry. (2015) 5:e613. doi: 10.1038/tp.2015.109
77. Weston-Green K, Huang XF, Lian J, Deng C. Effects of olanzapine on muscarinic M3 receptor binding density in the brain relates to weight gain, plasma insulin and metabolic hormone levels. Eur Neuropsychopharmacol. (2012) 22:364–73. doi: 10.1016/j.euroneuro.2011.09.003
78. Nakajima K, Jain S, Ruiz de Azua I, McMillin SM, Rossi M, Wess J. Minireview: novel aspects of M3 muscarinic receptor signaling in pancreatic beta-cells. Mol Endocrinol. (2013) 27:1208–16. doi: 10.1210/me.2013-1084
79. Silvestre JS, Prous J. Research on adverse drug events. I. Muscarinic M3 receptor binding affinity could predict the risk of antipsychotics to induce type 2 diabetes. Methods Find Exp Clin Pharmacol. (2005) 27:289–304. doi: 10.1358/mf.2005.27.5.908643
80. Johnson DE, Yamazaki H, Ward KM, Schmidt AW, Lebel WS, Treadway JL, et al. Inhibitory effects of antipsychotics on carbachol-enhanced insulin secretion from perifused rat islets: role of muscarinic antagonism in antipsychotic-induced diabetes and hyperglycemia. Diabetes. (2005) 54:1552–8. doi: 10.2337/diabetes.54.5.1552
81. Lian J, Deng C. The dosage-dependent effects of cevimeline in preventing olanzapine-induced metabolic side-effects in female rats. Pharmacol Biochem Behav. (2020) 191:172878. doi: 10.1016/j.pbb.2020.172878
82. Han M, Lian J, Su Y, Deng C. Cevimeline co-treatment attenuates olanzapine-induced metabolic disorders via modulating hepatic M3 muscarinic receptor: AMPKalpha signalling pathway in female rats. J Psychopharmacol. (2021) 2021:2698811211050549. doi: 10.1177/02698811211050549
83. Liu X, Wu Z, Lian J, Hu CH, Huang XF, Deng C. Time-dependent changes and potential mechanisms of glucose-lipid metabolic disorders associated with chronic clozapine or olanzapine treatment in rats. Sci Rep. (2017) 7:2762. doi: 10.1038/s41598-017-02884-w
84. Lopez M, Lage R, Saha AK, Perez-Tilve D, Vazquez MJ, Varela L, et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. (2008) 7:389–99. doi: 10.1016/j.cmet.2008.03.006
85. Lopez M, Varela L, Vazquez MJ, Rodriguez-Cuenca S, Gonzalez CR, Velagapudi VR, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med. (2010) 16:1001–8. doi: 10.1038/nm.2207
86. Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. (2004) 428:569–74. doi: 10.1038/nature02440
87. Lopez M. AMPK Wars: the VMH strikes back, return of the PVH. Trends Endocrinol Metab. (2018) 29:135–7. doi: 10.1016/j.tem.2018.01.004
88. Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. (2007) 117:2325–36. doi: 10.1172/JCI31516
89. Chen H, Vlahos R, Bozinovski S, Jones J, Anderson GP, Morris MJ. Effect of short-term cigarette smoke exposure on body weight, appetite and brain neuropeptide Y in mice. Neuropsychopharmacology. (2005) 30:713–9. doi: 10.1038/sj.npp.1300597
90. Yang CS, Lam CK, Chari M, Cheung GW, Kokorovic A, Gao S, et al. Hypothalamic AMP-activated protein kinase regulates glucose production. Diabetes. (2010) 59:2435–43. doi: 10.2337/db10-0221
91. Okamoto S, Asgar NF, Yokota S, Saito K, Minokoshi Y. Role of the alpha2 subunit of AMP-activated protein kinase and its nuclear localization in mitochondria and energy metabolism-related gene expressions in C2C12 cells. Metabolism. (2019) 90:52–68. doi: 10.1016/j.metabol.2018.10.003
92. Kume S, Kondo M, Maeda S, Nishio Y, Yanagimachi T, Fujita Y, et al. Hypothalamic AMP-activated protein kinase regulates biphasic insulin secretion from pancreatic beta cells during fasting and in type 2 diabetes. EBioMedicine. (2016) 13:168–80. doi: 10.1016/j.ebiom.2016.10.038
93. Park S, Kim DS, Kang S, Shin BK. Chronic activation of central AMPK attenuates glucose-stimulated insulin secretion and exacerbates hepatic insulin resistance in diabetic rats. Brain Res Bull. (2014) 108:18–26. doi: 10.1016/j.brainresbull.2014.08.002
94. Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. (2007) 47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304
95. He M, Zhang Q, Deng C, Wang H, Lian J, Huang XF. Hypothalamic histamine H1 receptor-AMPK signaling time-dependently mediates olanzapine-induced hyperphagia and weight gain in female rats. Psychoneuroendocrinology. (2014) 42:153–64. doi: 10.1016/j.psyneuen.2014.01.018
96. Skrede S, Martins L, Berge RK, Steen VM, Lopez M, Ferno J. Olanzapine depot formulation in rat: a step forward in modelling antipsychotic-induced metabolic adverse effects. Int J Neuropsychopharmacol. (2014) 17:91–104. doi: 10.1017/S1461145713000862
97. Chen X, Yu Y, Zheng P, Jin T, He M, Zheng M, et al. Olanzapine increases AMPK-NPY orexigenic signaling by disrupting H1R-GHSR1a interaction in the hypothalamic neurons of mice. Psychoneuroendocrinology. (2020) 114:104594. doi: 10.1016/j.psyneuen.2020.104594
98. Ikegami M, Ikeda H, Ohashi T, Kai M, Osada M, Kamei A, et al. Olanzapine-induced hyperglycemia: possible involvement of histaminergic, dopaminergic and adrenergic functions in the central nervous system. Neuroendocrinology. (2013) 98:224–32. doi: 10.1159/000356119
99. Ferno J, Varela L, Skrede S, Vazquez MJ, Nogueiras R, Dieguez C, et al. Olanzapine-induced hyperphagia and weight gain associate with orexigenic hypothalamic neuropeptide signaling without concomitant AMPK phosphorylation. PLoS ONE. (2011) 6:e20571. doi: 10.1371/journal.pone.0020571
100. Palasz A, Bandyszewska M, Rojczyk E, Wiaderkiewicz R. Effect of extended olanzapine administration on POMC and neuropeptide Y mRNA levels in the male rat amygdala and hippocampus. Pharmacol Rep. (2016) 68:292–6. doi: 10.1016/j.pharep.2015.09.012
101. Sezlev-Bilecen D, Ak M, Yanik T. Dysregulation of hypothalamic modulation in olanzapine treated male rats. Prog Neuropsychopharmacol Biol Psychiatry. (2016) 71:103–7. doi: 10.1016/j.pnpbp.2016.06.012
102. Lian J, De Santis M, He M, Deng C. Risperidone-induced weight gain and reduced locomotor activity in juvenile female rats: the role of histaminergic and NPY pathways. Pharmacol Res. (2015) 95–6:20–6. doi: 10.1016/j.phrs.2015.03.004
103. Li C, Jones PM, Persaud SJ. Role of the endocannabinoid system in food intake, energy homeostasis and regulation of the endocrine pancreas. Pharmacol Ther. (2011) 129:307–20. doi: 10.1016/j.pharmthera.2010.10.006
104. Duffy SS, Hayes JP, Fiore NT, Moalem-Taylor G. The cannabinoid system and microglia in health and disease. Neuropharmacology. (2021) 190:108555. doi: 10.1016/j.neuropharm.2021.108555
105. Mastinu A, Pira M, Pani L, Pinna GA, Lazzari P. NESS038C6, a novel selective CB1 antagonist agent with anti-obesity activity and improved molecular profile. Behav Brain Res. (2012) 234:192–204. doi: 10.1016/j.bbr.2012.06.033
106. Yang D, Xu L, Guo F, Sun X, Zhang D, Wang M. Orexin-A and endocannabinoid signaling regulate glucose-responsive arcuate nucleus neurons and feeding behavior in obese rats. Neuropeptides. (2018) 69:26–38. doi: 10.1016/j.npep.2018.04.001
107. Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. (2006) 574:7–15. doi: 10.1113/jphysiol.2006.108944
108. Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem. (2005) 280:25196–201. doi: 10.1074/jbc.C500175200
109. Liu YL, Connoley IP, Wilson CA, Stock MJ. Effects of the cannabinoid CB1 receptor antagonist SR141716 on oxygen consumption and soleus muscle glucose uptake in Lep(ob)/Lep(ob) mice. Int J Obes. (2005) 29:183–7. doi: 10.1038/sj.ijo.0802847
110. Leite CE, Mocelin CA, Petersen GO, Leal MB, Thiesen FV. Rimonabant: an antagonist drug of the endocannabinoid system for the treatment of obesity. Pharmacol Rep. (2009) 61:217–24. doi: 10.1016/S1734-1140(09)70025-8
111. Addy C, Wright H, Van Laere K, Gantz I, Erondu N, Musser BJ, et al. The acyclic CB1R inverse agonist taranabant mediates weight loss by increasing energy expenditure and decreasing caloric intake. Cell Metab. (2008) 7:68–78. doi: 10.1016/j.cmet.2007.11.012
112. Aronne LJ, Finer N, Hollander PA, England RD, Klioze SS, Chew RD, et al. Efficacy and safety of CP-945,598, a selective cannabinoid CB1 receptor antagonist, on weight loss and maintenance. Obesity. (2011) 19:1404–14. doi: 10.1038/oby.2010.352
113. Han JH, Shin H, Rho JG, Kim JE, Son DH, Yoon J, et al. Peripheral cannabinoid 1 receptor blockade mitigates adipose tissue inflammation via NLRP3 inflammasome in mouse models of obesity. Diabetes Obes Metab. (2018) 20:2179–89. doi: 10.1111/dom.13350
114. Khansari MR, Panahi N, Hosseinzadeh S, Zendehdel M. Effect of cannabinoid-serotonin interactions in the regulation of neuropeptide Y1 receptors expression in rats: the role of CB1 and 5-HT2C receptor. Comp Clin Path. (2020) 29:561–71. doi: 10.1007/s00580-019-03081-z
115. Gallate JE, Saharov T, Mallet PE, McGregor IS. Increased motivation for beer in rats following administration of a cannabinoid CB1 receptor agonist. Eur J Pharmacol. (1999) 370:233–40. doi: 10.1016/S0014-2999(99)00170-3
116. Rivera P, Blanco E, Bindila L, Alen F, Vargas A, Rubio L, et al. Pharmacological activation of CB2 receptors counteracts the deleterious effect of ethanol on cell proliferation in the main neurogenic zones of the adult rat brain. Front Cell Neurosci. (2015) 9:379. doi: 10.3389/fncel.2015.00379
117. Radziszewska E, Bojanowska E. Effects of glucagon-like peptide-1 receptor stimulation and blockade on food consumption and body weight in rats treated with a cannabinoid CB1 receptor agonist WIN 55,212-2. Med Sci Monit Basic Res. (2013) 19:6–11. doi: 10.12659/MSMBR.883726
118. Weston-Green K, Huang X-F, Han M, Deng C. The effects of antipsychotics on the density of cannabinoid receptors in the dorsal vagal complex of rats: implications for olanzapine-induced weight gain. Int J Neuropsychopharmacol. (2008) 11:8560. doi: 10.1017/S1461145708008560
119. Lazzari P, Serra V, Marcello S, Pira M, Mastinu A. Metabolic side effects induced by olanzapine treatment are neutralized by CB1 receptor antagonist compounds co-administration in female rats. Eur Neuropsychopharmacol. (2017) 27:667–78. doi: 10.1016/j.euroneuro.2017.03.010
120. Weston-Green K, Huang XF, Deng C. Alterations to melanocortinergic, GABAergic and cannabinoid neurotransmission associated with olanzapine-induced weight gain. PLoS ONE. (2012) 7:e33548. doi: 10.1371/journal.pone.0033548
121. Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. (1997) 378:135–57. doi: 10.1002/(SICI)1096-9861(19970203)378:1<135::AID-CNE8>3.0.CO;2-5
122. Urabe H, Kojima H, Chan L, Terashima T, Ogawa N, Katagi M, et al. Haematopoietic cells produce BDNF and regulate appetite upon migration to the hypothalamus. Nat Commun. (2013) 4:1526. doi: 10.1038/ncomms2536
123. Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci. (2007) 27:14265–74. doi: 10.1523/JNEUROSCI.3308-07.2007
124. Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. (2003) 6:736–42. doi: 10.1038/nn1073
125. Tran PV, Akana SF, Malkovska I, Dallman MF, Parada LF, Ingraham HA. Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J Comp Neurol. (2006) 498:637–48. doi: 10.1002/cne.21070
126. Bariohay B, Lebrun B, Moyse E, Jean A. Brain-derived neurotrophic factor plays a role as an anorexigenic factor in the dorsal vagal complex. Endocrinology. (2005) 146:5612–20. doi: 10.1210/en.2005-0419
127. Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. (2012) 64:238–58. doi: 10.1124/pr.111.005108
128. Zhang C, Fang X, Yao P, Mao Y, Cai J, Zhang Y, et al. Metabolic adverse effects of olanzapine on cognitive dysfunction: a possible relationship between BDNF and TNF-alpha. Psychoneuroendocrinology. (2017) 81:138–43. doi: 10.1016/j.psyneuen.2017.04.014
129. Angelucci F, Mathe AA, Aloe L. Brain-derived neurotrophic factor and tyrosine kinase receptor TrkB in rat brain are significantly altered after haloperidol and risperidone administration. J Neurosci Res. (2000) 60:783–94. doi: 10.1002/1097-4547(20000615)60:6<783::AID-JNR11>3.0.CO;2-M
130. Bai O, Chlan-Fourney J, Bowen R, Keegan D, Li XM. Expression of brain-derived neurotrophic factor mRNA in rat hippocampus after treatment with antipsychotic drugs. J Neurosci Res. (2003) 71:127–31. doi: 10.1002/jnr.10440
131. Shoval G, Weizman A. The possible role of neurotrophins in the pathogenesis and therapy of schizophrenia. Eur Neuropsychopharmacol. (2005) 15:319–29. doi: 10.1016/j.euroneuro.2004.12.005
132. Lin P-Y. Increase in brain-derived neurotrophic factor in patients with schizophrenia treated with olanzapine: a systemic review and meta-analysis. J Exp Clin Med. (2012) 4:119–24. doi: 10.1016/j.jecm.2012.02.002
133. Yang F, Wang K, Du X, Deng H, Wu HE, Yin G, et al. Sex difference in the association of body mass index and BDNF levels in Chinese patients with chronic schizophrenia. Psychopharmacology. (2018) 236:753–62. doi: 10.1007/s00213-018-5107-1
134. Yang Y, Zhang Y, Wang J, Ning X, Zhang Y, Zhao T, et al. Sex differences in the association of HOMA-IR index and BDNF in Han Chinese patients with chronic schizophrenia. Front Psychiatr. (2021) 12:656230. doi: 10.3389/fpsyt.2021.656230
135. Kwok KHM, Lam KSL, Xu A. Heterogeneity of white adipose tissue: molecular basis and clinical implications. Exp Mol Med. (2016) 48:e215-e. doi: 10.1038/emm.2016.5
136. Benbaibeche H, Bounihi A, Koceir EA. Leptin level as a biomarker of uncontrolled eating in obesity and overweight. Irish J Med Sci. (2020) 190:155–61. doi: 10.1007/s11845-020-02316-1
137. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. (1998) 395:763–70. doi: 10.1038/27376
138. Diéguez C, Vazquez MJ, Romero A, López M, Nogueiras R. Hypothalamic control of lipid metabolism: focus on leptin, ghrelin and melanocortins. Neuroendocrinology. (2011) 94:1–11. doi: 10.1159/000328122
139. Hommel JD, Trinko R, Sears RM, Georgescu D, Liu Z-W, Gao X-B, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. (2006) 51:801–10. doi: 10.1016/j.neuron.2006.08.023
140. Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology. (1997) 138:4489–92. doi: 10.1210/endo.138.10.5570
141. Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Morgan PJ, et al. Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J Neuroendocrinol. (1996) 8:733–5. doi: 10.1046/j.1365-2826.1996.05161.x
142. Kim KS, Yoon YR, Lee HJ, Yoon S, Kim S-Y, Shin SW, et al. Enhanced hypothalamic leptin signaling in mice lacking dopamine D2 receptors. J Biol Chem. (2010) 285:8905–17. doi: 10.1074/jbc.M109.079590
143. Sahu A. Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol. (2003) 24:225–53. doi: 10.1016/j.yfrne.2003.10.001
144. Gorobets LN. Contribution of leptin to the formation of neuroleptic obesity in patients with schizophrenia during antipsychotic therapy. Bull Exp Biol Med. (2009) 146:348–50. doi: 10.1007/s10517-008-0294-0
145. Singh R, Bansal Y, Medhi B, Kuhad A. Antipsychotics-induced metabolic alterations: recounting the mechanistic insights, therapeutic targets and pharmacological alternatives. Eur J Pharmacol. (2019) 844:231–40. doi: 10.1016/j.ejphar.2018.12.003
146. Piao L, Park J, Li Y, Shin S, Shin S, Kong G, et al. SOCS3 and SOCS6 are required for the risperidone-mediated inhibition of insulin and leptin signaling in neuroblastoma cells. Int J Mol Med. (2014) 33:1364–70. doi: 10.3892/ijmm.2014.1693
147. Srisawasdi P, Vanwong N, Hongkaew Y, Puangpetch A, Vanavanan S, Intachak B, et al. Impact of risperidone on leptin and insulin in children and adolescents with autistic spectrum disorders. Clin Biochem. (2017) 50:678–85. doi: 10.1016/j.clinbiochem.2017.02.003
148. Ragguett R-M, Hahn M, Messina G, Chieffi S, Monda M, De Luca V. Association between antipsychotic treatment and leptin levels across multiple psychiatric populations: an updated meta-analysis. Hum Psychopharmacol Clin Exp. (2017) 32:2631. doi: 10.1002/hup.2631
149. Potvin S, Zhornitsky S, Stip E. Antipsychotic-induced changes in blood levels of leptin in schizophrenia: a meta-analysis. Can J Psychiatry. (2015) 60(3Suppl.2):S26–34.
150. Atmaca M, Kuloglu M, Tezcan E, Ustundag B. Serum leptin and triglyceride levels in patients on treatment with atypical antipsychotics. J Clin Psychiatry. (2003) 64:598–604. doi: 10.4088/JCP.v64n0516
151. Pena R, Marquina D, Serrano A, Elfakih Y, Debaptista E, Carrizo E, et al. Frequency of abnormal correlation between leptin and the body mass index during first and second generation antipsychotic drug treatment. Schizophr Res. (2008) 106:315–9. doi: 10.1016/j.schres.2008.08.031
152. Margulska A, Kozłowska E, Wysokiński A. Effect of clozapine dose and concentration on fasting concentration of appetite regulating peptides. Psychiatry Res. (2018) 260:473–7. doi: 10.1016/j.psychres.2017.12.018
153. Higgins SC, Gueorguiev M, Korbonits M. Ghrelin, the peripheral hunger hormone. Ann Med. (2009) 39:116–36. doi: 10.1080/07853890601149179
154. Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology. (2000) 141:4255–61. doi: 10.1210/endo.141.11.7757
155. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. (1999) 402:656–60. doi: 10.1038/45230
156. Korbonits M, Kojima M, Kangawa K, Grossman AB. Presence of ghrelin in normal and adenomatous human pituitary. Endocrine. (2001) 14:101–4. doi: 10.1385/ENDO:14:1:101
157. Toshinai K, Mondal M, Shimbara T, Yamaguchi H, Date Y, Kangawa K, et al. Ghrelin stimulates growth hormone secretion and food intake in aged rats. Mech Ageing Dev. (2007) 128:182–6. doi: 10.1016/j.mad.2006.10.001
158. Geliebter A, Gluck ME, Hashim SA. Plasma ghrelin concentrations are lower in binge-eating disorder. J Nutr. (2005) 135:1326–30. doi: 10.1093/jn/135.5.1326
159. Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. (2001) 50:707–9. doi: 10.2337/diabetes.50.4.707
160. Varela L, Vazquez MJ, Cordido F, Nogueiras R, Vidal-Puig A, Dieguez C, et al. Ghrelin and lipid metabolism: key partners in energy balance. J Mol Endocrinol. (2010) 46:R43–63. doi: 10.1677/JME-10-0068
161. Lage R, Vázquez MJ, Varela L, Saha AK, Vidal-Puig A, Nogueiras R, et al. Ghrelin effects on neuropeptides in the rat hypothalamus depend on fatty acid metabolism actions on BSX but not on gender. FASEB J. (2010) 24:2670–9. doi: 10.1096/fj.09-150672
162. Schaeffer M, Langlet F, Lafont C, Molino F, Hodson DJ, Roux T, et al. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Nat Acad Sci USA. (2013) 110:1512–7. doi: 10.1073/pnas.1212137110
163. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. (2004) 279:12005–8. doi: 10.1074/jbc.C300557200
164. Murashita M, Kusumi I, Inoue T, Takahashi Y, Hosoda H, Kangawa K, et al. Olanzapine increases plasma ghrelin level in patients with schizophrenia. Psychoneuroendocrinology. (2005) 30:106–10. doi: 10.1016/j.psyneuen.2004.05.008
165. Murashita M, Inoue T, Kusumi I, Nakagawa S, Itoh K, Tanaka T, et al. Glucose and lipid metabolism of long-term risperidone monotherapy in patients with schizophrenia. Psychiatry Clin Neurosci. (2007) 61:54–8. doi: 10.1111/j.1440-1819.2007.01610.x
166. Davey KJ, O'Mahony SM, Schellekens H, O'Sullivan O, Bienenstock J, Cotter PD, et al. Gender-dependent consequences of chronic olanzapine in the rat: effects on body weight, inflammatory, metabolic and microbiota parameters. Psychopharmacology. (2012) 221:155–69. doi: 10.1007/s00213-011-2555-2
167. Zhang Q, Deng C, Huang X-F. The role of ghrelin signalling in second-generation antipsychotic-induced weight gain. Psychoneuroendocrinology. (2013) 38:2423–38. doi: 10.1016/j.psyneuen.2013.07.010
168. Togo T, Hasegawa K, Miura S, Hosojima H, Kojima K, Shoji M, et al. Serum ghrelin concentrations in patients receiving olanzapine or risperidone. Psychopharmacology. (2004) 172:230–2. doi: 10.1007/s00213-003-1642-4
169. Goetz RL, Miller BJ. Meta-analysis of ghrelin alterations in schizophrenia: effects of olanzapine. Schizophr Res. (2019) 206:21–6. doi: 10.1016/j.schres.2018.11.036
170. Spencer SJ, Xu L, Clarke MA, Lemus M, Reichenbach A, Geenen B, et al. Ghrelin regulates the hypothalamic-pituitary-adrenal axis and restricts anxiety after acute stress. Biol Psychiatry. (2012) 72:457–65. doi: 10.1016/j.biopsych.2012.03.010
171. Tagami K, Kashiwase Y, Yokoyama A, Nishimura H, Miyano K, Suzuki M, et al. The atypical antipsychotic, olanzapine, potentiates ghrelin-induced receptor signaling: an in vitro study with cells expressing cloned human growth hormone secretagogue receptor. Neuropeptides. (2016) 58:93–101. doi: 10.1016/j.npep.2015.12.010
172. Huang XF, Weston-Green K, Yu Y. Decreased 5-HT2cR and GHSR1a interaction in antipsychotic drug-induced obesity. Obes Rev. (2018) 19:396–405. doi: 10.1111/obr.12638
173. Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. (2007) 6:55–68. doi: 10.1016/j.cmet.2007.06.003
174. Bartoli F, Crocamo C, Clerici M, Carrà G. Second-generation antipsychotics and adiponectin levels in schizophrenia: a comparative meta-analysis. Eur Neuropsychopharmacol. (2015) 25:1767–74. doi: 10.1016/j.euroneuro.2015.06.011
175. Lu M-L, Wang T-N, Lin T-Y, Shao W-C, Chang S-H, Chou J-Y, et al. Differential effects of olanzapine and clozapine on plasma levels of adipocytokines and total ghrelin. Progr Neuro Psychopharmacol Biol Psychiatry. (2015) 58:47–50. doi: 10.1016/j.pnpbp.2014.12.001
176. Sapra M, Lawson D, Iranmanesh A, Varma A. Adiposity-independent hypoadiponectinemia as a potential marker of insulin resistance and inflammation in schizophrenia patients treated with second generation antipsychotics. Schizophr Res. (2016) 174:132–6. doi: 10.1016/j.schres.2016.04.051
177. Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. (2015) 28:203–9.
178. Misiak B, Łoniewski I, Marlicz W, Frydecka D, Szulc A, Rudzki L, et al. The HPA axis dysregulation in severe mental illness: can we shift the blame to gut microbiota? Progr Neuro Psychopharmacol Biol Psychiatry. (2020) 102:109951. doi: 10.1016/j.pnpbp.2020.109951
179. Donohoe Dallas R, Garge N, Zhang X, Sun W, O'Connell Thomas M, Bunger Maureen K, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metabol. (2011) 13:517–26. doi: 10.1016/j.cmet.2011.02.018
180. Kao AC-C, Chan KW, Anthony DC, Lennox BR, Burnet PWJ. Prebiotic reduction of brain histone deacetylase (HDAC) activity and olanzapine-mediated weight gain in rats, are acetate independent. Neuropharmacology. (2019) 150:184–91. doi: 10.1016/j.neuropharm.2019.02.014
181. Oliphant K, Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome. (2019) 7:91. doi: 10.1186/s40168-019-0704-8
182. Wu Q, Lemus MB, Stark R, Bayliss JA, Reichenbach A, Lockie SH, et al. The temporal pattern of cfos activation in hypothalamic, cortical, and brainstem nuclei in response to fasting and refeeding in male mice. Endocrinology. (2014) 155:840–53. doi: 10.1210/en.2013-1831
183. Heimesaat MM, Morgan AP, Crowley JJ, Nonneman RJ, Quackenbush CR, Miller CN, et al. The antipsychotic olanzapine interacts with the gut microbiome to cause weight gain in mouse. PLoS ONE. (2014) 9:115225. doi: 10.1371/journal.pone.0115225
184. Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. (2018) 555:623–8. doi: 10.1038/nature25979
185. Bahr SM, Tyler BC, Wooldridge N, Butcher BD, Burns TL, Teesch LM, et al. Use of the second-generation antipsychotic, risperidone, and secondary weight gain are associated with an altered gut microbiota in children. Transl Psychiatry. (2015) 5:e652-e. doi: 10.1038/tp.2015.135
186. Yuan X, Zhang P, Wang Y, Liu Y, Li X, Kumar BU, et al. Changes in metabolism and microbiota after 24-week risperidone treatment in drug naïve, normal weight patients with first episode schizophrenia. Schizophr Res. (2018) 201:299–306. doi: 10.1016/j.schres.2018.05.017
187. Sleeth ML, Thompson EL, Ford HE, Zac-Varghese SEK, Frost G. Free fatty acid receptor 2 and nutrient sensing: a proposed role for fibre, fermentable carbohydrates and short-chain fatty acids in appetite regulation. Nutr Res Rev. (2010) 23:135–45. doi: 10.1017/S0954422410000089
188. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. (2013) 54:2325–40. doi: 10.1194/jlr.R036012
189. Peterson J, den Besten G, Gerding A, van Dijk TH, Ciapaite J, Bleeker A, et al. Protection against the metabolic syndrome by guar gum-derived short-chain fatty acids depends on peroxisome proliferator-activated receptor γ and glucagon-like peptide-1. PLoS ONE. (2015) 10:136364. doi: 10.1371/journal.pone.0136364
190. de la Cuesta-Zuluaga J, Mueller N, Álvarez-Quintero R, Velásquez-Mejía E, Sierra J, Corrales-Agudelo V, et al. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients. (2018) 11:10051. doi: 10.3390/nu11010051
191. Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. (2010) 18:190–5. doi: 10.1038/oby.2009.167
192. Kao AC-C, Spitzer S, Anthony DC, Lennox B, Burnet PWJ. Prebiotic attenuation of olanzapine-induced weight gain in rats: analysis of central and peripheral biomarkers and gut microbiota. Transl Psychiatry. (2018) 8:116. doi: 10.1038/s41398-018-0116-8
193. Bretler T, Weisberg H, Koren O, Neuman H. The effects of antipsychotic medications on microbiome and weight gain in children and adolescents. BMC Med. (2019) 17:1. doi: 10.1186/s12916-019-1346-1
194. Xu Y, Shao M, Fang X, Tang W, Zhou C, Hu X, et al. Antipsychotic-induced gastrointestinal hypomotility and the alteration in gut microbiota in patients with schizophrenia. Brain Behav Immun. (2022) 99:119–29. doi: 10.1016/j.bbi.2021.09.014
195. Kelly JR, Borre Y, O' Brien C, Patterson E, El Aidy S, Deane J, et al. Transferring the blues: depression-associated gut microbiota induces neurobehavioural changes in the rat. J Psychiatr Res. (2016) 82:109–18. doi: 10.1016/j.jpsychires.2016.07.019
196. Vich Vila A, Collij V, Sanna S, Sinha T, Imhann F, Bourgonje AR, et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat Commun. (2020) 11:14177. doi: 10.1038/s41467-019-14177-z
197. Sobański JA, Klasa K, Mielimaka M, Rutkowski K, Dembińska E, Müldner-Nieckowski Ł, et al. The crossroads of gastroenterology and psychiatry – what benefits can psychiatry provide for the treatment of patients suffering from gastrointestinal symptoms. Gastroenterol Rev. (2015) 4:222–8. doi: 10.5114/pg.2015.51213
198. Sherwin E, Sandhu KV, Dinan TG, Cryan JF. May the force be with you: the light and dark sides of the microbiota–gut–brain axis in neuropsychiatry. CNS Drugs. (2016) 30:1019–41. doi: 10.1007/s40263-016-0370-3
Keywords: hypothalamus, appetite control, mechanisms, hyperphagia, antipsychotics
Citation: Mukherjee S, Skrede S, Milbank E, Andriantsitohaina R, López M and Fernø J (2022) Understanding the Effects of Antipsychotics on Appetite Control. Front. Nutr. 8:815456. doi: 10.3389/fnut.2021.815456
Received: 15 November 2021; Accepted: 10 December 2021;
Published: 03 January 2022.
Edited by:
Tatiana Zilberter, Independent Researcher, Marseille, FranceReviewed by:
Gavin P. Reynolds, Sheffield Hallam University, United KingdomCopyright © 2022 Mukherjee, Skrede, Milbank, Andriantsitohaina, López and Fernø. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johan Fernø, am9oYW4uZmVybm9AdWliLm5v
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.