 Arlene C. P. Kiyohara1
Arlene C. P. Kiyohara1 Daniel J. Torres1,2
Daniel J. Torres1,2 Ayaka Hagiwara1Jenna Pak1Rachel H. L. H. Rueli1
Ayaka Hagiwara1Jenna Pak1Rachel H. L. H. Rueli1 C. William R. Shuttleworth3
C. William R. Shuttleworth3 Frederick P. Bellinger1*
Frederick P. Bellinger1*- 1Department of Cell and Molecular Biology, John A. Burns School of Medicine, University of Hawaii at Manoa, Honolulu, HI, United States
- 2Pacific Biosciences Research Center, School of Ocean and Earth Science and Technology, University of Hawaii at Manoa, Honolulu, HI, United States
- 3Department of Neurosciences, University of New Mexico, Albuquerque, NM, United States
Selenoprotein P (SELENOP1) is a selenium-rich antioxidant protein involved in extracellular transport of selenium (Se). SELENOP1 also has metal binding properties. The trace element Zinc (Zn2+) is a neuromodulator that can be released from synaptic terminals in the brain, primarily from a subset of glutamatergic terminals. Both Zn2+ and Se are necessary for normal brain function. Although these ions can bind together with high affinity, the biological significance of an interaction of SELENOP1 with Zn2+ has not been investigated. We examined changes in brain Zn2+ in SELENOP1 knockout (KO) animals. Timm-Danscher and N-(6-methoxy-8-quinolyl)-p-toluenesulphonamide (TSQ) staining revealed increased levels of intracellular Zn2+ in the SELENOP1−/− hippocampus compared to wildtype (WT) mice. Mass spectrometry analysis of frozen whole brain samples demonstrated that total Zn2+ was not increased in the SELENOP1−/− mice, suggesting only local changes in Zn2+ distribution. Unexpectedly, live Zn2+ imaging of hippocampal slices with a selective extracellular fluorescent Zn2+ indicator (FluoZin-3) showed that SELENOP1−/− mice have impaired Zn2+ release in response to KCl-induced neuron depolarization. The zinc/metal storage protein metallothionein 3 (MT-3) was increased in SELENOP1−/− hippocampus relative to wildtype, possibly in response to an elevated Zn2+ content. We found that depriving cultured cells of selenium resulted in increased intracellular Zn2+, as did inhibition of selenoprotein GPX4 but not GPX1, suggesting the increased Zn2+ in SELENOP1−/− mice is due to a downregulation of antioxidant selenoproteins and subsequent release of Zn2+ from intracellular stores. Surprisingly, we found increased tau phosphorylation in the hippocampus of SELENOP1−/− mice, possibly resulting from intracellular zinc changes. Our findings reveal important roles for SELENOP1 in the maintenance of synaptic Zn2+ physiology and preventing tau hyperphosphorylation.
Introduction
Within the body, selenium (Se) functions primarily in the form of selenocysteine (Sec), the 21st amino acid, which is incorporated into members of the selenoprotein family (1–3). Selenoprotein P (SELENOP1) is a selenium-rich protein with 10 Sec residues that transports Se in serum from liver to the brain and other organs (4). SELENOP1 is present in the cerebral spinal fluid (CSF) and in the choroid plexus, which releases CSF (5, 6), and in glial cells (7). SELENOP1 has also been described in brain neurons (8, 9), which may be the targets of Se transport. SELENOP1 KO mice have reduced brain selenium and reduced levels of antioxidant selenoproteins such as glutathione peroxidases 1 and 4 (GPX1 and GPX4) (10). Mice with the SELENOP1 gene deletion have deficient hippocampal synaptic function and deficits in spatial learning and long-term potentiation (LTP), a model for learning and memory (11). SELENOP1 is increased in the brain and CSF in Alzheimer's disease (5, 8, 12) and associated with both Alzheimer's and Parkinson's pathology (8, 13).
Se and Zn2+ are both essential trace elements required for proper brain function. Selenium deficiency correlates with impaired cognitive and motor function (14, 15), while Zn2+ deficiency correlates with decreased nerve conduction and impaired cognitive performance (16). Alzheimer's disease is associated with increased brain Zn2+ levels (17). Zn2+ can increase tau phosphorylation (18, 19), which contributes to the formation of neurofibrillary tangles, a hallmark of Alzheimer's disease (20). However, studies have yet to address the biological relevance of the interaction between these elements despite their high affinity for each other and their importance in brain function.
Figure 1A shows SELENOP1's two functional, glycosylated domains: (1) a Se-rich C-terminal domain with 9 S residues, (2) an N-terminal domain with 1 S (U) in U-x-x-C redox motif, 2 histidine-rich metal binding sites (located at residue 204–217 and residue 244–250) and a heparin binding site (4, 21). SELENOP1 also has an N-terminal signal peptide for extracellular secretion, which is cleaved in the Golgi (22).
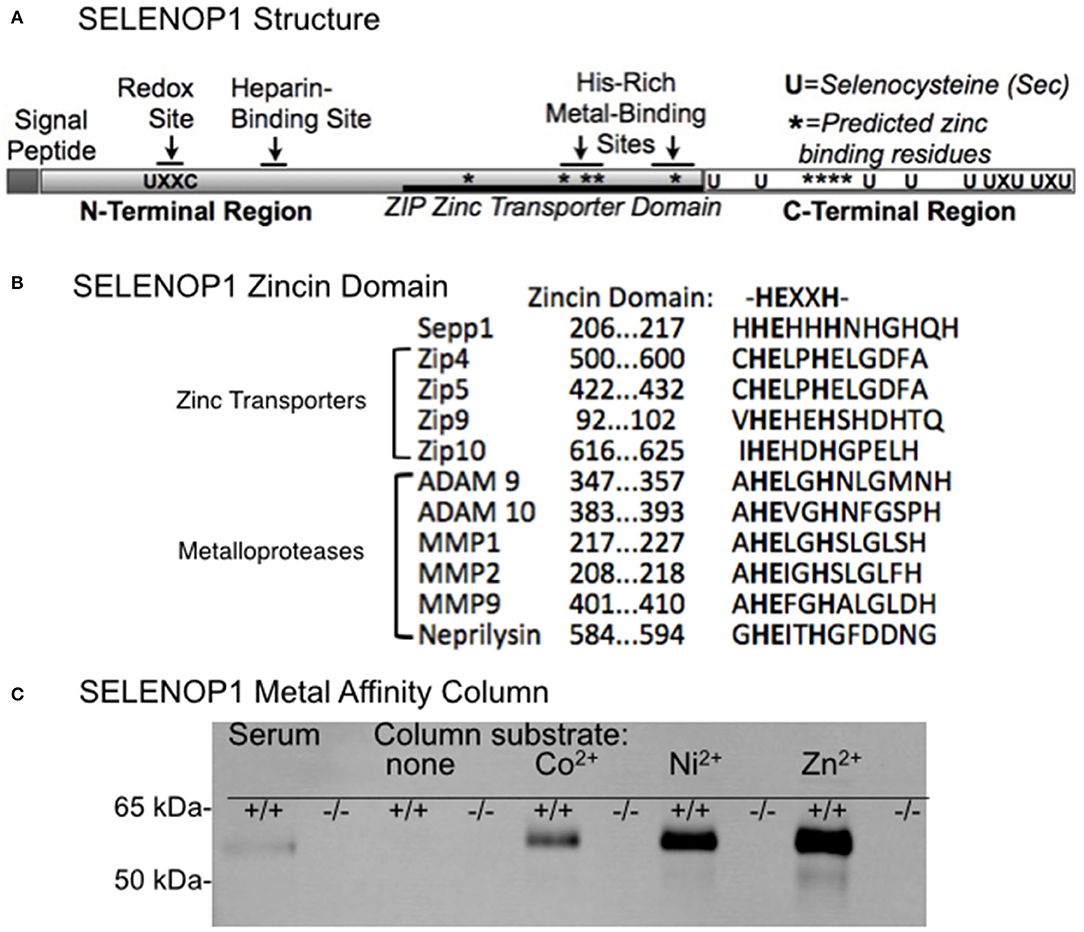
Figure 1. Zinc-binding properties of SELENOP1. (A) Schematic of SELENOP1 structure. The N-terminal region has 1 S (U) in a redox domain, a heparin-binding site and a zinc transporter domain with two metal-binding sites. The C-terminal region has 9 S residues for Se transport. Sites of residues predicted to bind zinc using Predzinc are indicated by *. (B) Alignment of human SELENOP1 metal binding region with other proteins containing the zincin motif. (C) Western blot of metal column eluates after applying mouse serum from WT (+/+) or SELENOP1 KO (−/−) mice to mini columns with agarose only or agarose bound to Co2+, Ni2+, and Zn2+. Untreated SELENOP1−/− and WT serum were also added to the blot as a positive control (left lanes). SELENOP1 protein detected with anti-SELENOP1 antibody (1:1000) had a molecular weight of approximately 55 kDa in the wildtype serum, which was not seen in SELENOP1−/− serum. SELENOP1 was detected in column eluates from wild-type serum applied to all metal columns, but not from columns with agarose only.
We hypothesized that SELENOP1, as a metal-binding protein, could have a role in brain Zn2+ homeostasis. In this study, we investigated whether SELENOP1 influences brain Zn2+ by evaluating changes in hippocampal Zn2+ in SELENOP1 KO animals. Here we report that deletion of SELENOP1 alters levels of chelatable Zn2+ and prevents the release of synaptic Zn2+ in mouse hippocampus. These findings indicate an important additional role for SELENOP1 in the regulation of zinc in the brain. They could also have important implications for the treatment of disorders where Zn2+ physiology is impaired.
Materials and Methods
Animals
Mice were group housed on a 12-h light cycle and provided food and water ad libitum. All animals in this study were maintained on diets containing adequate selenium (~0.25 ppm) and zinc (~80 ppm). All mice used were 3–6 months of age and included both male and female mice as indicated. All animal procedures were approved by the University of Hawaii Institutional Animal Care and Use Committee.
SELENOP1−/− mice were obtained from the laboratory of Dr. Raymond Burk at Vanderbilt University. The mutant mice were backcrossed to C57BL/6J for at least ten generations with C57BL/6J mice from Jackson Laboratories to ensure congenic strains. Breeding of SELENOP1+/− mice generated littermates of SELENOP1+/+ and SELENOP1−/− pups, which were used in this study in addition to SELENOP1+/− mice. Genomic DNA extracted from mice tails was used for genotyping PCR using specific primers (forward-ACCTCAGCAATGTGGAGAAGCC, reverse-TGCCCTCTGAGTTTAGCATTG for wild-type, and reverse-GATGATCTGGACGAAGAGCATCA for SELENOP1−/−). Products were run on a 1.5% DNA agarose gel with a SYBR Safe DNA gel stain (Invitrogen) and genotypes were confirmed under UV light.
Timm-Danscher Zn2+ Labeling
The Danscher modification of Timm's zinc stain (“neo-Timm's”) was used to label intracellular chelatable zinc (i.e., not tightly bound to proteins or other molecules) (23). Deeply anesthetized mice received intraperitoneal (IP) injections of 20 mg/kg sodium selenite (2 mg/ml in normal saline). After 2 h, mice were perfused with saline followed by 4% paraformaldehyde (PFA) in saline. Brains were postfixed overnight in 4% PFA, dehydrated serially in 10% and 30% sucrose, and mounted in optimal cutting temperature compound (OCT) for cryostat sectioning. After thoroughly washing in PBS, sections were developed with the IntenSE M silver Enhancement kit (Amersham International) according to the manufacturer's protocol and as previously described (24).
6-Methoxy-8-P-Toluenesulfonamido-Quinoline (TSQ) Stain
TSQ is a Zn2+ fluorophore that binds intracellular chelatable Zn2+ in a 2:1 ligand-to-metal ratio that results in increased fluorescence emission at 490 nm in response to excitation at 360 nm (25). Serial sagittal cryosections (10 μm) of brain hemispheres were mounted on positively charged microscope slides. The slides were immersed with 4.5 μM TSQ (Enzo Lifesciences, UltraPure) in 140 mM sodium barbital and 140 mM sodium acetate buffer (pH 10) for 90 s, as previously described (26) and washed in 0.1% NaCl. TSQ-stained sections were imaged using DAPI filter settings (200 ms, monochrome with a 5x objective). The mean fluorescence intensity of the hippocampal CA1 stratum oriens and stratum radiatum, CA3 mossy fibers, and hilar region were measured with ImageJ software (NIH). The background was measured in unstained areas within lateral ventricles and subtracted from mean TSQ signals.
Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES)
The metal concentrations within frozen right brain hemispheres and liver samples were processed at the Agricultural Diagnostic Service Center (ADSC) run by the College of Tropical Agriculture and Human Resources (CTAHR), University of Hawaii. Dry ash sample preparations were subjected to acid digest before ICP-OES (0.01 ppm detection limit) to measure total brain and liver metal content (Zn2+, Cu2+, Fe2+). Water blanks and solution standards were included with each run to calibrate results.
Protein Extraction and Western Blot
Proteins were extracted from frozen hippocampal tissue using CelLytic MT buffer (Sigma) per the manufacturer's instructions, denatured by heating in Laemmli sample buffer, resolved by SDS-PAGE on a 10–20% gradient Tris-HCl Criterion Precast gel (Bio-Rad Laboratories), and electrically transferred to polyvinylidene difluoride (PVDF) membranes. For detection of Zn2+ regulating proteins, membranes were incubated in anti-metallothionein-3 (1:500, rabbit polyclonal, Biorbyt), and anti-ZnT1 and anti-ZnT3 (1:1000; 1:5000, rabbit polyclonal, Synaptic Systems). For detection of the SELENOP1 protein, membranes were incubated in anti-SELENOP1 (1:1000, rabbit monoclonal, Proteintech). For measurement of tau, antibodies recognizing tau phosphorylated at T231, S214 or S396 (1:1000, Invitrogen) or tau 5 (1:1000, Millipore). Membranes washed and then treated with corresponding secondary antibodies conjugated with infrared fluorophores (1:10,000; Licor). Blots were subsequently treated with anti α-tubulin (1:5000; Novus Biologicals) to control for loading. Membranes were imaged with the Odyssey infrared fluorescence system (LiCor), and densitometry analysis was performed on the Imagestudio software (LiCor).
Metal Agarose Column Purification
To observe SELENOP1 binding to biometals, we used mini spin-columns containing high-density agarose beads conjugated with Zn2+, Ni2+, and Co2+ (Agarose Bead Technologies) to isolate metal-binding proteins from wildtype and SELENOP1−/− mouse serum. A metal-free agarose column served as a negative control. Serum samples were diluted 1:100 in PBS and added to the column, gently shaken for 60 min at 4°C, then spun for 60 s at 800x g to collect flow-through. Columns were washed with increasing concentrations of imidazole (0, 10, and 20 mM) diluted in PBS, and then bound proteins were eluted with 250 mM imidazole diluted in PBS. Eluted proteins were determined with western blot using an anti-SELENOP1 antibody (27).
Live Hippocampal Slice Imaging
To measure stimulus-induced extracellular Zn2+accumulation, hippocampal slices were prepared from 3 to 6 month old SELENOP1 KO and wild-type littermate mice as previously described (28). Following slice preparation, slices were acclimated to room temperature and superfused with oxygenated (95 O2 and 5% CO2 gas mix) artificial cerebral spinal fluid (ACSF, composition in mM: NaCl 130; KCl 3.5; glucose 10; NaHCO3 24; NaH2PO4 1.25; MgSO4 1.5; CaCl2 2.0) for at least 60 min. The CA1 stratum radiatum region of the slices was imaged with a Zeiss laser-scanning microscope using a 10X objective with the pinhole fully opened at 1 frame/s at 640 × 480 resolution. Cell-impermeant FluoZin-3 at 1.5 μM (Molecular Probes) was added to the ACSF to detect extracellular Zn2+ accumulation from hippocampal slices in response to the administration of a depolarizing (35 mM) KCl concentration for 60 s. In some experiments, a slow onset Zn2+ chelator (Ca2+-EDTA) was added to remove contaminating Zn2+ in media and to reduce background fluorescence (29, 30). Fluorescence intensities of hippocampal slices in the CA1 stratum radiatum region upon addition of KCl for each slice were expressed as the fluorescence intensity over the fluorescence during baseline (F/F0). The mean area under the curve during application of high K+ for signals of SELENOP1−/− slices were compared to WT to determine if changes in Zn2+ release were altered.
FluoZin-3 Measurements in Cell Culture
SH-SY5Y cells were plated in 96-well plates and differentiated by exposure to Neurobasal media (Invitrogen) supplemented with B27 (Invitrogen) for 48 hrs. The media was then changed to Roth-Schweizer media (31) but with of 0, 10 or 100 nM. Alternatively, cultures in 10 nM Se were treated with 100 μM mercaptosuccinate (MCS), 0.1 μM RSL-3, 0.1 μM RSL-3 + 100 μM α-tocopherol, or 0.01 μM DMSO as a control for RSL-3. Cells were treated for either 5 days (DMSO, RSL-3, RSL-3 with α-tocopherol) or 7 days (Se concentrations and remaining conditions). Cells were rinsed twice in HEPES buffered saline [HBSS: (in mM) NaCl 146; KCl 3.5; glucose 10; 1.25; MgSO4 1.5; CaCl2 2.0, HEPES 10, NaOH 10]. Cells were loaded with 1 μM FluoZin-3 AM (Thermo Fisher Scientific) with 0.02% pluoronic F-127 for 30 min at 37°C in the dark, then rinsed twice with HBSS, and the fluorescence measured in HBSS. For additional controls, either 100 μM 2, 2′-dithiodipyridine (DTDP), 100 μM tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN), or 100 μM H2O2 were added to the HBSS 10 min before measuring fluorescence. Plates were scanned in a SpectraMax M3 fluorescent plate reader (Molecular Devices) with 494 nm excitation, 516 nm emission. After scanning, cells were fixed in 100% methanol at −20°C overnight, stained with 100 μg propidium iodide (PI), rinsed twice with HBSS, than scanned in HBSS at 536 nm excitation, 617 nm emission. FluoZin-3 fluorescence was normalized to PI fluorescence to correct for any differences in cell density.
Statistical Analysis
All statistical analyses were carried out with Graphpad Prism Software with measurements given as means ± SE. Comparisons between treatments, genotypes and sex were performed by student's unpaired t-test and one-way or two-way analysis of variance (ANOVA), with p < 0.05 considered significant. In general, we did not find sex differences in Zn2+ distribution or release unless shown; otherwise, males and females were averaged together by genotype.
Results
Zn2+ Binding Properties of SELENOP1
Previous studies described the affinity of SELENOP1 for several metals, including Zn2+. However, the biological interaction of SELENOP1 with Zn2+ is unclear. We investigated potential Zn2+-binding domains of SELENOP1. Using the web-based software Predzinc (https://predzinc.bioshu.se/pred/), a web server that predicts zinc-binding proteins and zinc-binding sites from given sequences, we analyzed the SELENOP1 coding sequences for potential zinc-binding sites. We found that a His-residue within the SELENOP1 His-rich metal binding domain as well as two other His residues in the C-terminal region are predicted to be Zn2+ binding motifs, shown in Figure 1A by asterisks (*).
A domain search of the Kyoto Encyclopedia of Genes and Genomes (KEGG) database indicates that the human SELENOP1 protein structure has a zinc-binding domain overlapping one of the His-rich regions, which has homology to the ZIP zinc transporter domain. Additionally, within this sequence is a zincin Zn2+-binding motif, generally found in the metzincin family of metalloproteases (Figure 1B) (32).
Because of the predicted Zn2+-binding region of SELENOP1, we tested SELENOP1's potential to bind Zn2+ ions by passing mouse serum through agarose columns bound to Co2+, Ni2+, or Zn2+, or metal-free as a negative control. Western blot of sample elution from each column purification shows that SELENOP1 binds to Co2+, Ni2+, and Zn2+, but not to the metal-free agarose column (Figure 1C). When serum from SELENOP1−/− mice was used, SELENOP1 immunoreactivity was not detected in eluent from any of the columns. This demonstrates that SELENOP1 is capable of binding different biometals, with a greater amount of binding to Zn2+ compared with Co2+ and Ni2+.
Elevated Levels of Intracellular Zn2+ in SELENOP1–/– Hippocampus
To explore the effects of SELENOP1 on brain Zn2+ homeostasis, we first evaluated intracellular Zn2+ levels in SELENOP1−/− mice using histological methods. Our studies focused on the hippocampus as a zinc-rich region important for learning and memory (33). The Timm-Danscher method (34) revealed a pronounced increase in histologically-detectable Zn2+ in the hippocampus of SELENOP1−/− compared with SELENOP1+/+ hippocampus (Figure 2A). The hilar and CA3 mossy fiber regions had the most intense Zn2+ labeling in both wild-type and SELENOP1−/− animals. However, all layers positive for Zn2+ were increased in KOs, including the stratum radiatum and stratum oriens of the CA1 and CA3 layers.
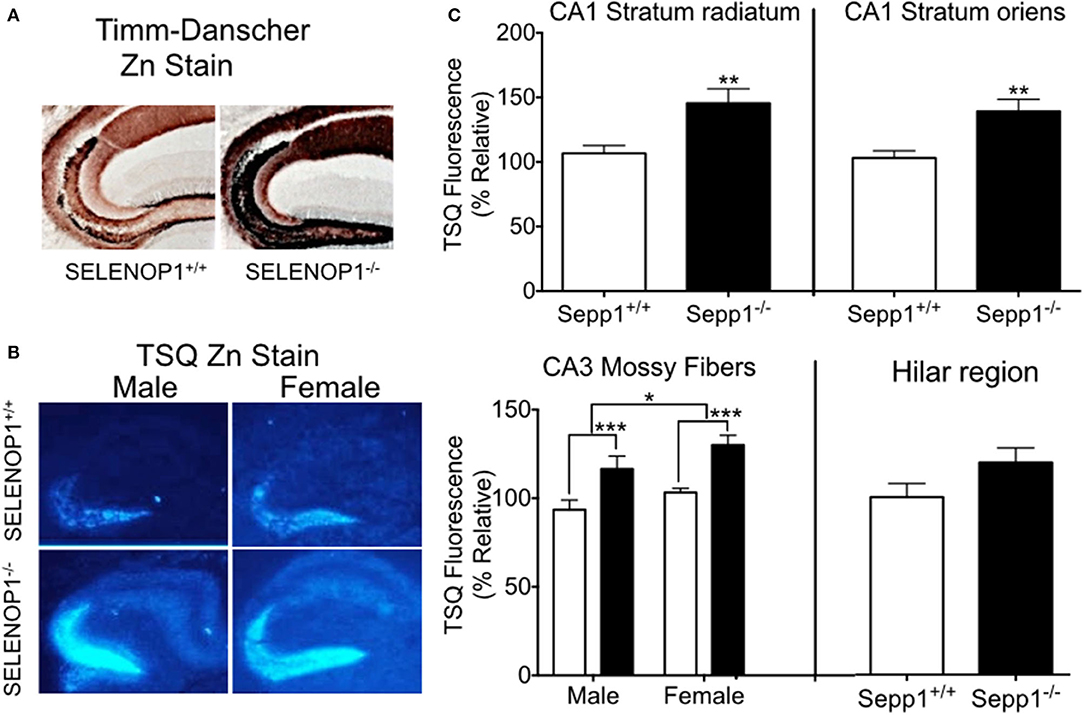
Figure 2. Increased hippocampal zinc in SELENOP1 KO mice. (A) Neo-Timm's (Danscher) stain. Free Zn2+ in wildtype and SELENOP1−/− hippocampal neurons from mice injected with sodium selenite is revealed as a reddish-brown to dark brown color relative to the amount of total Zn2+. (B) Representative images from male and female WT and SELENOP1−/− mouse hippocampus, labeled with the fluorescent Zn2+ indicator, TSQ. (C) Quantitation of TSQ fluorescent label indicates higher Zn2+ levels in SELENOP1−/− animals in all observed regions except for hilar region. The average values for each region were normalized to wild type males. N = 6 animals/group, 5–10 slices/animal. Statistical analysis is performed by Student's unpaired t- test or two-way ANOVA with Bonferroni's post-hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
We further compared bioreactive Zn2+ levels in SELENOP1−/− and wildtype mouse hippocampi using TSQ labeling. Quantitation of TSQ fluorescence also revealed significantly higher levels of intracellular Zn2+ in the CA1 stratum oriens, stratum radiatum, and CA3 mossy fibers of the SELENOP1−/− animals compared to their control (Figures 2B,C). We also observed a sex difference in CA3, with increased Zn2+ in mossy fibers in female compared to male hippocampus regardless of genotype. Unstained hippocampal brain sections showed no fluorescence at TSQ wavelengths, indicating that differences were not due to autofluorescence signals. Hematoxylin staining of sections previously used for TSQ labeling showed no morphological differences between SELENOP1 wildtype and KO hippocampi. Based on our findings, SELENOP1 may be regulating Zn2+ levels directly or through one or more Zn2+ interacting proteins.
As we observed increased levels of intracellular Zn2+ within the hippocampus of SELENOP1−/− mice, we then investigated if the total brain Zn2+ levels are altered by subjecting whole brain hemispheres via ICP-OES to measure total metal content for Zn2+, Cu2+, and Fe2+. There was no significant increase in total brain Zn2+ levels in SELENOP1−/− mice (Table 1). This may indicate that deletion of SELENOP1 results in changes to the distribution of Zn2+ within the brain rather than an increase in total brain Zn2+.
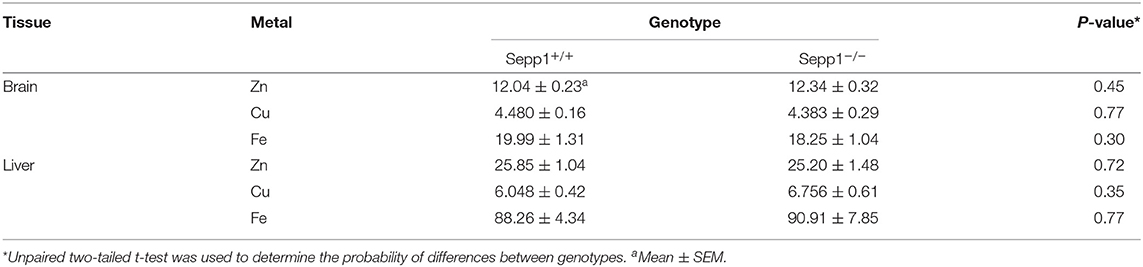
Table 1. Metal content in brain and liver of Sepp1 wildtype (Sepp1+/+) and knockout (Sepp1−/−) mice at the age of 3 months (mg/kg of dry tissue, n = 20 each) measured by ICP-OES.
Expression Levels of Zn2+-Interacting Proteins
We investigated whether changes in Zn2+ regulating proteins could explain differences in Zn2+. We investigated hippocampal expression of the Zn2+ transporters ZnT1 and the vesicle-associated ZnT3, as well as the metal storage protein MT3. Western blot indicated that ZnT1 and ZnT3 proteins were unchanged in SELENOP1−/− animals (Figures 3A,B). However, MT3 protein expression was increased (Figure 3C), suggesting that deletion of the SELENOP1 gene does not affect zinc transport through these major pathways, but rather upregulates expression of the Zn2+ storage protein. The enhanced expression of MT3 may be a result of a feedback mechanism in response to increased Zn2+ levels in the SELENOP1−/− hippocampus.
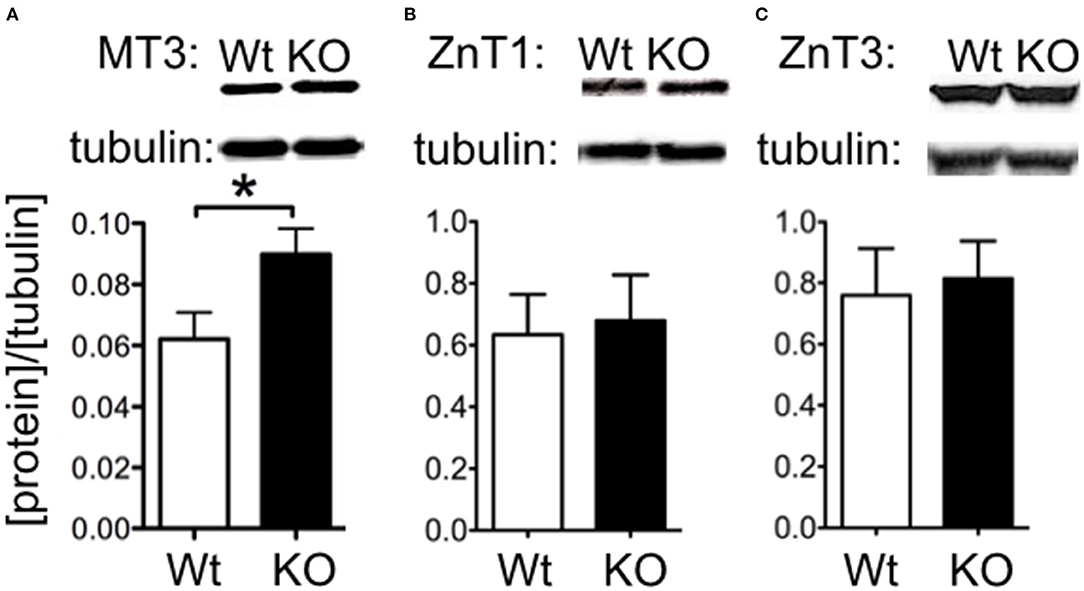
Figure 3. Increased MT3 but not ZnT1 or ZnT3 protein levels in SELENOP1 KO hippocampus. Western blot showing expression levels of Zn2+ transporter proteins, ZnT1 and ZnT3, and Zn2+ storage pool, MT3. Protein densities were normalized to α-tubulin and averaged to male WT values. (A) ZnT1 protein levels were unchanged. (B) ZnT3 protein was also unchanged. (C) Increased MT3 expression. Representative bands are shown above each corresponding bar graph and were found to run at the predicted sizes: ZnT1 (45kDa), ZnT3 (42kDa), and MT3 (40kDa). Statistical analysis is performed by Student's unpaired t-test, *p < 0.05.
Zn2+ Release Is Impaired in SELENOP1–/– Hippocampus in Response to Neuron Depolarization
Most histologically-detectable Zn2+ in hippocampal neurons is vesicular (25). We investigated if the increased Zn2+ levels visualized by TSQ or Timm's staining in SELENOP1−/− mice results in increased extracellular accumulation following depolarizing stimuli designed to promote vesicular release. We imaged extracellular Zn2+ accumulation in hippocampal slices with a selective cell-impermeant fluorescent Zn2+ indicator, Fluozin-3, in the extracellular media. By depolarizing hippocampal cells with the addition of 35 mM KCl, we noticed an increase in fluorescence in slices from wild type mice, indicating release of Zn2+ into the extracellular space (Figures 4A,B). In contrast, we observed a minimal increase in fluorescence in hippocampal slices from SELENOP1−/− mice, indicating negligible Zn2+ release in the SELENOP1−/− hippocampus.
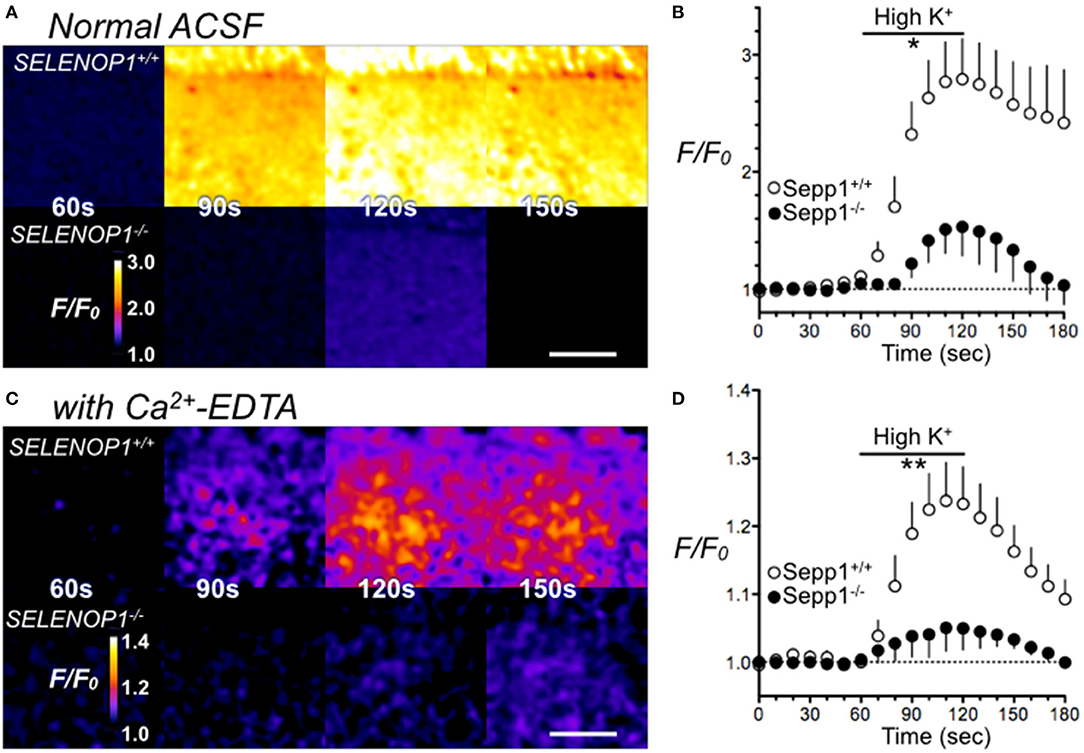
Figure 4. Impaired Zn2+ release in SELENOP1 KO hippocampus. (A) Zn2+ release from hippocampal slices of SELENOP1−/− and WT mice visualized with Fluozin-3 following depolarization induced with ACSF containing high (35 mM) KCl solution. Fluorescence in SELENOP1−/− hippocampal slices was significantly reduced compared to WT animals. (A) Representative Fluozin-3 images from WT and SELENOP1−/− animals at 60s, 90s, 120s and 150s, after adding high K+ from 60 to 120 s. (B) Data plotted as relative fluorescence (F/F0) over time with measurements averaged every 10 s. WT, n = 6 slices, 3 mice; SELENOP1−/−, n = 5 slices, 3 mice. (C) Zn2+ release visualized with Fluozin-3 in ACSF with the addition of Zn2+ chelator, Ca2+-EDTA to reduce background fluorescence, following application of ACSF with high (35 mM) KCl. Representative Fluozin-3 images from WT and SELENOP1−/− animals at 60, 90, 120, and 150 s, after adding high K+ from 60 to 120 s. (D) Data plotted as relative fluorescence (F/F0) over time with measurements averaged every 5 s. WT, n = 3 slices, 3 mice; SELENOP1−/−, n = 4 slices, 4 mice; Fluorescence in SELENOP1−/− hippocampal slices was significantly reduced compared to WT animals. Statistical analysis was performed by Student's unpaired t-test comparing the mean area under the curve for each genotype between 60 and 120 s, *p < 0.05, **p < 0.01.
A high background fluorescence recording from Zn2+ contaminants in the reagents used could possibly mask Zn2+ release. To reduce background fluorescence, we also imaged slices with the Zn2+ chelator, Ca2+-EDTA, added to the ACSF. Ca2+-EDTA enables removal of basal levels of extracellular Zn2+, but its slow kinetics do not prevent the detection of synaptically-evoked Zn2+ accumulation (35). Even in the presence of Ca2+-EDTA, we still observed significantly larger FluoZin-3 increases in wild-type hippocampus slices relative to SELENOP1−/− slices upon cell depolarization (Figures 4C,D). The baseline fluorescence (F0) was not significantly different between SELENOP1+/+ and SELENOP1−/− slices in either normal ACSF for Ca2+-EDTA ACSF.
Selenium Deficiency Releases Intracellular Zn2+
We hypothesized that increased oxidation from Se-deficient conditions could result in reduced intracellular chelation of Zn2+. We tested this by measuring the fluorescence of cell-permeable FluoZin-3 in cultured SH-SY5Y cells. As shown in Figure 5A, release of intracellular zinc by DTDP, a thiol oxidizer which liberates intracellular Zn2+, significantly increased FluoZin-3 fluorescence. However, the intracellular Zn2+ chelator TPEN did not reduce FluoZin-3 fluorescence, indicating that free non-chelated Zn2+ levels were too low to be detectable. We also found that cells grown in 0 Se culture media had significantly increased fluorescence compared with cells grown in our baseline media with 10 nM Se (Figure 5B). Oxidation with H2O2 greatly increased FluoZin-3 fluorescence, demonstrating that oxidation could free Zn2+ from stores. To test if a reduction in activity of the H2O2-reducing selenoprotein GPX1, which depends on Se for synthesis, could lead to increased intracellular Zn2+, we exposed cells to the GPX1 inhibitor mercaptosuccinate (MCS) (Figure 5C). MCS did not alter fluorescence, however, we found that RSL-3, an inhibitor of the phospholipid peroxidase selenoprotein GPX4, increased intracellular Zn2+ (Figure 5D). This increase was prevented by the vitamin E compound α-tocopherol, which reduces lipid peroxidation. These findings suggest that a deficiency in Se can increase free intracellular Zn2+ by causing a reduction in GPX4, which results in increased oxidation of lipids.
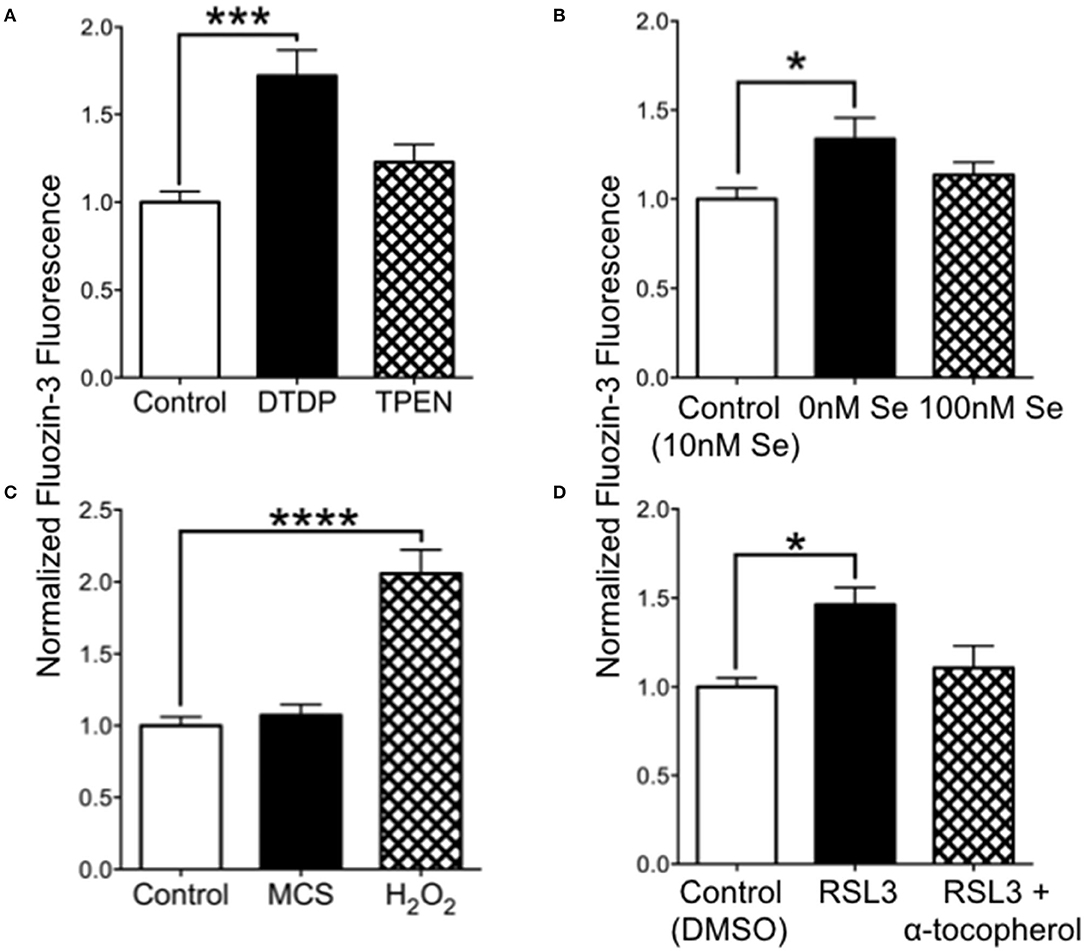
Figure 5. Intracellular Zn2+levels are increased by 0 Se and GPX4 inhibitor RSL-3. (A) Relative FluoZin-3 fluorescence of SH-SY5Y cells. Cells exposed to DTDP to release zinc showed increased fluorescence, while cells exposed to PTEN to reduce intracellular zinc had no change in fluorescence. (B) FluoZin-3 fluorescence was increased in cells grown in media with 0 nM Se relative to control media (10 nM), while 100 nM Se did not alter intracellular zinc. (C) Hydrogen peroxide but not GPX1 inhibitor mercaptosuccinate (MCS) increase intracellular zinc. (D) The GPX4 inhibitor RSL-3 increased intracellular zinc, which was reversed when RSL-3 was added with α-tocopherol. Statistical analysis is performed by one-way ANOVA followed by Dunnett's multiple comparison test. *p < 0.05, ***p < 0.005, ****p < 0.0001.
Elevated Tau Phosphorylation in SELENOP1 Knockout Hippocampus
Zn2+ promotes tau phosphorylation leading to neurofibrillary tangle formation (18). We questioned whether tau phosphorylation could be altered in the SELENOP1−/− mouse. We performed western blot analysis to compare specific pTau sites to total tau protein. We found that phosphorylation at threonine 231 and at serine 396 were significantly increased in SELENOP1−/− mice (Figure 6A). However, phosphorylation at the serine 214 site was unchanged (Figures 6B,C). Deletion of SELENOP1−/− thus results in a site-specific increase in tau phosphorylation.
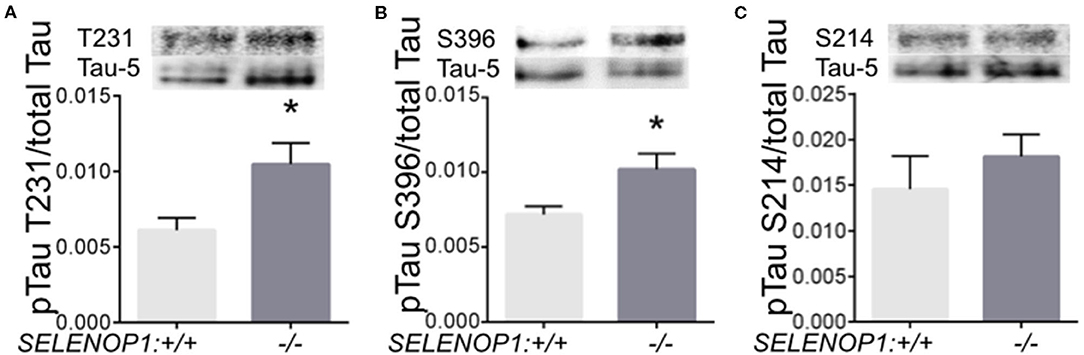
Figure 6. Deletion of SELENOP1 increases tau phosphorylation in mouse hippocampus. Western blots of phospho-specific tau antibodies relative to the Tau-5 antibody, which recognizes total tau. Blot images are superimposed above graphs. (A) SELENOP1 deletion resulted in increased tau phosphorylation at threonine 231. (B) Serine 396 was also significantly reduced in SELENOP1 knockouts. (C) No change in phosphorylation at serine 214. N = 4 animals/group. Statistical analysis is performed by Student's unpaired t-test, *p < 0.05.
Discussion
Our findings show that deletion of SELENOP1 increased free (chelatable) intracellular Zn2+ levels. However, release of synaptic zinc was impaired. Although the Zn2+ storage protein MT3 was elevated without change to the zinc transporters ZnT1 or ZnT3, we showed that selenium deficiency could induce release of Zn2+ from stores, likely through a decrease of the selenoprotein GPX4 and a subsequent increase in lipid peroxidation. Interestingly, GPX4 helps to prevent neurodegeneration though ferroptosis (36). Lastly, we demonstrated an increase in site-specific phosphorylation of tau. These findings suggest that SELENOP1 plays a role in regulating storage of intracellular Zn2+. This role may be important for preventing tau hyperphosphorylation in AD.
Dietary supplementation with selenium in the form of sodium selenate reduces tau phosphorylation to potentially reduce neurofibrillary tangle formation (37, 38). Selenate can act as an agonist for protein phosphatase 2A (PP2A), which targets tau phosphorylation (39). Interestingly, Zn2+ is an inhibitor of PP2A, and may promote neurofibrillary tangle formation (18). Hyper-phosphorylation of tau leads to neurofibrillary tangle formation, and de-phosphorylation by PP2A should reduce tangle formation. However, dietary selenate supplementation also upregulates the expression of selenoproteins (40, 41). We have previously reported that a reduction in selenoprotein S can promote tau phosphorylation (42). In a type 2 early clinical trial for selenate supplementation in Alzheimer's disease, selenate could increase brain selenium in some patients, and the increase in brain selenium correlated with lack of decline in performance on the Mini-Mental Status Examination (MMSE) (43). Thus, brain selenoproteins, including SELENOP1, may be important for preventing Alzheimer's pathology.
Zn2+ metabolism is altered in AD, resulting in abnormally enriched Zn2+ environments within the AD brain (44). Zinc-binding sites on the Aβ peptide result in Zn2+ mediated aggregation of Aβ and amyloid plaque formation. Furthermore, both the neuroprotective role of SELENOP1 against Aβ toxicity and the role of Zn2+ in protecting the cell against oxidative damage, could be working together to reduce the levels of Aβ stress observed in AD pathology, however more studies need to be done to further elucidate their contribution to alleviating oxidative stress.
We did not observe an increase in total brain Zn2+ levels with ICP-OES, suggesting only changes in local Zn2+ distribution. The absence of SELENOP1 could result in increased oxidative stress in the brain and lead to Zn2+ release from MT3 (45), possibly inducing an upregulation of the Zn2+ storage protein. In the absence of SELENOP1, we observed a seemingly paradoxical impairment of Zn2+ release despite an overall increase in intracellular chelatable Zn2+. This was surprising, since most chelatable Zn2+ is thought to be localized to synaptic vesicles (46). Overexpression of MT3 in SELENOP1−/− mice may affect the subcellular distribution of Zn2+ by limiting the amount of free Zn2+ available for loading into the synaptic vesicles. The protein Reelin can increase release of a subset of synaptic vesicles (47). This increase is dependent on Reelin binding to ApoER2. SELENOP1 is another ligand for ApoER2 (48, 49), and thus may also modulate vesicle release, possibly including zincergic vesicles. A decrease in release of Zn2+ vesicles could result in a “back-up” of these vesicles, which could contribute to the observed increase in chelatable Zn2+. The APOE ε4 allele of the major ligand ApoE for ApoER2, increases risk of Alzheimer's disease (50). Interestingly, the APOE ε4 allele is also associated with decreased selenium in the brain (51), again suggesting a possible role for selenium and selenoproteins in preventing Alzheimer's disease.
We found an interesting sex difference in Zn2+ levels in the CA3 region of the hippocampus (Figure 2). Female mice generally had higher Zn2+ levels but with some variability. Previous studies have shown higher Zn2+ levels in female wild-type mice and AD mouse models (52, 53), although these studies did not agree on the age of sex differences. The differences suggest Zn2+ as a possible explanation for the increased risk of AD in women (54). Researchers recently discovered that estrogen increases hippocampal Zn2+, which was cycle-dependent in female mice (55, 55), which may explain the increased Zn2+ in other studies as well as the variability of the current results.
SELENOP1 contains a putative metal-binding domain that can potentially bind Zn2+ with a high affinity. Our results demonstrate that SELENOP1 is capable of binding Zn2+ as well as Co2+ and Ni2+. However, our finding that selenium deficiency and inhibition of GPX4 suggest that the absence of selenium and loss of antioxidant selenoprotein function in the SELENOP1 KO mice is responsible for the increased Zn2+ levels. The reason for the presence of the functional metal-binding domain of SELENOP1 remains unknown. It is possible that Zn2+ binding could alter the affinity of SELENOP1 for the ApoER2 receptor, allowing for regulation of Se by excess Zn2+. Additionally, the homology of the Zn2+ binding domain with metalloproteases such as ADAM10 and neprilysin (Figure 1) opens the possibility of an enzymatic role in proteolysis. ADAM10 has a protective role in Alzheimer's disease as a putative α-secretase that promotes non-amyloidogenic cleavage of amyloid precursor protein, preventing amyloid beta formation (56).
The data presented here indicate that SELENOP1 may play a crucial role in the maintenance of brain Zn2+. The SELENOP1 gene can affect Zn2+ metabolism and synaptic release from neuronal synapses. Zn2+ is increased in Alzheimer's disease and interacts with amyloid beta (17), and can also promote tau phosphorylation (18). Thus, Zn2+-binding properties of SELENOP1 could contribute to the association of SELENOP1 with amyloid beta plaques in Alzheimer's disease (8). The SELENOP1-Zn2+ interaction has potentially important implications in neuronal function and synaptic physiology.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by University of Hawaii IACUC.
Author Contributions
FB, CS, DT, and AK designed the research. DT, AH, JP, and RR performed the research. FB analyzed the data and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants 2P20GM103466, G12MD007601, and F32DK124963, and the Hawaii Community Foundation grant ADVC-49233.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Ann Hashimoto for technical assistance, Marla Berry, Robert Nichols, Suguru Kurokawa and Tessi Sherrin for helpful comments and suggestions, and Robert Huang at the University of Hawaii Agricultural Diagnostic Service Center for performing mass spectroscopy.
References
1. Bellinger FP, Raman AV, Reeves MA, Berry MJ. Regulation and function of selenoproteins in human disease. Biochem J. (2009) 422:11–22. doi: 10.1042/BJ20090219
2. Pillai R, Uyehara-Lock JH, Bellinger FP. Selenium and selenoprotein function in brain disorders. IUBMB Life. (2014) 66:229–39. doi: 10.1002/iub.1262
3. Cardoso BR, Roberts BR, Bush AI, Hare DJ. Selenium, selenoproteins and neurodegenerative diseases. Metallomics. (2015) 7:1213–28. doi: 10.1039/C5MT00075K
4. Burk RF, Hill KE. Selenoprotein P-expression, functions, and roles in mammals. Biochim Biophys Acta. (2009) 1790:1441–7. doi: 10.1016/j.bbagen.2009.03.026
5. Rueli RH, Parubrub AC, Dewing AS, Hashimoto AC, Bellinger MT, Weeber EJ, et al. Increased Selenoprotein P in choroid plexus and cerebrospinal fluid in Alzheimer's disease brain. J Alzheimers Dis. (2015) 44:379–83. doi: 10.3233/JAD-141755
6. Solovyev N, Drobyshev E, Blume B, Michalke B. Selenium at the neural barriers: areview. Front Neurosci. (2021) 15:630016. doi: 10.3389/fnins.2021.630016
7. Steinbrenner H, Sies H. Selenium homeostasis and antioxidant selenoproteins in brain: implications for disorders in the central nervous system. Arch Biochem Biophys. (2013) 536:152–7. doi: 10.1016/j.abb.2013.02.021
8. Bellinger FP, He QP, Bellinger MT, Lin Y, Raman AV, White LR, et al. Association of selenoprotein p with Alzheimer's pathology in human cortex. J Alzheimers Dis. (2008) 15:465–72. doi: 10.3233/JAD-2008-15313
9. Scharpf M, Schweizer U, Arzberger T, Roggendorf W, Schomburg L, Kohrle J. Neuronal and ependymal expression of selenoprotein P in the human brain. J Neural Transm. (2007) 114:877–84. doi: 10.1007/s00702-006-0617-0
10. Hoffmann PR, Höge SC, Li P-A, Hoffmann FW, Hashimoto AC, Berry MJ. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. (2007) 35:3963–73. doi: 10.1093/nar/gkm355
11. Peters MM, Hill KE, Burk RF, Weeber EJ. Altered hippocampus synaptic function in selenoprotein P deficient mice. Mol Neurodegener. (2006) 1:12. doi: 10.1186/1750-1326-1-12
12. Takemoto AS, Berry MJ, Bellinger FP. Role of selenoprotein P in Alzheimer's disease. Ethn Dis. (2010) 20:S1–92–5.
13. Bellinger FP, Raman AV, Rueli RH, Bellinger MT, Dewing AS, Seale LA, et al. Changes in selenoprotein P in substantia nigra and putamen in Parkinson's disease. J Parkinsons Dis. (2012) 2:115–26. doi: 10.3233/JPD-2012-11052
14. Akbaraly TN, Hininger-Favier I, Carriere I, Arnaud J, Gourlet V, Roussel AM, et al. Plasma selenium over time and cognitive decline in the elderly. Epidemiology. (2007) 18:52–8. doi: 10.1097/01.ede.0000248202.83695.4e
15. Gao S, Jin Y, Hall KS, Liang C, Unverzagt FW, Ji R, et al. Selenium level and cognitive function in rural elderly Chinese. Am J Epidemiol. (2007) 165:955–65. doi: 10.1093/aje/kwk073
16. Maret W, Sandstead HH. Zinc requirements and the risks and benefits of zinc supplementation. J Trace Elem Med Biol. (2006) 20:3–18. doi: 10.1016/j.jtemb.2006.01.006
17. Adlard PA, Bush AI. Metals and Alzheimer's disease. J Alzheimers Dis. (2006) 10:145–63. doi: 10.3233/JAD-2006-102-303
18. Sun X-Y, Wei Y-P, Xiong Y, Wang X-C, Xie A-J, Wang X-L, et al. Synaptic released zinc promotes tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J Biol Chem. (2012) 287:11174–82. doi: 10.1074/jbc.M111.309070
19. Xiong Y, Jing X-P, Zhou X-W, Wang X-L, Yang Y, Sun X-Y, et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol Aging. (2013) 34:745–56. doi: 10.1016/j.neurobiolaging.2012.07.003
20. Brion JP, Anderton BH, Authelet M, Dayanandan R, Leroy K, Lovestone S, et al. Neurofibrillary tangles and tau phosphorylation. Biochem Soc Symp. (2001) 67:81–88. doi: 10.1042/bss0670081
21. Burk RF, Hill KE. Regulation of selenium metabolism and transport. Annu Rev Nutr. (2015) 35:109–34. doi: 10.1146/annurev-nutr-071714-034250
22. Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr. (2005) 25:215–35. doi: 10.1146/annurev.nutr.24.012003.132120
23. Danscher G, Stoltenberg M. Zinc-specific autometallographic in vivo selenium methods: tracing of zinc-enriched (ZEN) terminals, ZEN pathways, and pools of zinc ions in a multitude of other ZEN cells. J Histochem Cytochem. (2005) 53:141–53. doi: 10.1369/jhc.4R6460.2005
24. Danscher G, Norgaard JO, Baatrup E. Autometallography: tissue metals demonstrated by a silver enhancement kit. Histochemistry. (1987) 86:465–69. doi: 10.1007/BF00500618
25. Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci USA. (1999) 96:1716–21. doi: 10.1073/pnas.96.4.1716
26. Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. (2005) 6:449–62. doi: 10.1038/nrn1671
27. Seale LA, Hashimoto AC, Kurokawa S, Gilman CL, Seyedali A, Bellinger FP, et al. Disruption of the selenocysteine lyase-mediated selenium recycling pathway leads to metabolic syndrome in mice. Mol Cell Biol. (2012) 32:4141–54. doi: 10.1128/MCB.00293-12
28. Bark C, Bellinger FP, Kaushal A, Mathews JR, Partridge LD, Wilson MC. Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J Neurosci. (2004) 24:8796–805. doi: 10.1523/JNEUROSCI.1940-04.2004
29. Kay AR. Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn. J Neurosci. (2003) 23:6847–55. doi: 10.1523/JNEUROSCI.23-17-06847.2003
30. Kay AR. Detecting and minimizing zinc contamination in physiological solutions. BMC Physiol. (2004) 4:4. doi: 10.1186/1472-6793-4-4
31. Roth S, Zhang S, Chiu J, Wirth EK, Schweizer U. Development of a serum-free supplement for primary neuron culture reveals the interplay of selenium and vitamin E in neuronal survival. J Trace Elem Med Biol. (2010) 24:130–37. doi: 10.1016/j.jtemb.2010.01.007
32. Gomis-Ruth FX. Structural aspects of the metzincin clan of metalloendopeptidases. Mol Biotechnol. (2003) 24:157–202. doi: 10.1385/MB:24:2:157
33. Tamano H, Koike Y, Nakada H, Shakushi Y, Takeda A. Significance of synaptic Zn 2+ signaling in zincergic and non-zincergic synapses in the hippocampus in cognition. J Trace Elem Med Biol. (2016) 38:93–98. doi: 10.1016/j.jtemb.2016.03.003
34. Stoltenberg M, Bruhn M, Sondergaard C, Doering P, West MJ, Larsen A, et al. Immersion autometallographic tracing of zinc ions in Alzheimer beta-amyloid plaques. Histochem Cell Biol. (2005) 123:605–11. doi: 10.1007/s00418-005-0787-0
35. Qian J, Noebels JL. Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol. (2005) 566:747–58. doi: 10.1113/jphysiol.2005.089276
36. Cardoso BR, Hare DJ, Bush AI, Roberts BR. Glutathione peroxidase 4: a new player in neurodegeneration. Mol Psychiatry. (2017) 22:328–35. doi: 10.1038/mp.2016.196
37. Corcoran NM, Martin D, Hutter-Paier B, Windisch M, Nguyen T, Nheu L, et al. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer's disease model. J Clin Neurosci. (2010) 17:1025–33. doi: 10.1016/j.jocn.2010.04.020
38. Eersel J, van Ke YD, Liu X, Delerue F, Kril JJ, Götz J, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer's disease models. Proc Natl Acad Sci US A. (2010) 107:13888–93. doi: 10.1073/pnas.1009038107
39. Chen S, Li B, Grundke-Iqbal I, Iqbal K. I1PP2A affects tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J Biol Chem. (2008) 283:10513–21. doi: 10.1074/jbc.M709852200
40. Bosse AC, Pallauf J, Hommel B, Sturm M, Fischer S, Wolf NM, et al. Impact of selenite and selenate on differentially expressed genes in rat liver examined by microarray analysis. Biosci Rep. (2011) 30:293–306. doi: 10.1042/BSR20090089
41. Hoefig CS, Renko K, Köhrle J, Birringer M, Schomburg L. Comparison of different selenocompounds with respect to nutritional value vs. Toxicity using liver cells in culture. J Nutr Bioch. (2011) 22:945–55. doi: 10.1016/j.jnutbio.2010.08.006
42. Rueli RH, Torres DJ, Dewing AS, Kiyohara AC, Barayuga SM, Bellinger MT, et al. Selenoprotein S reduces endoplasmic reticulum stress-induced phosphorylation of tau: potential role in selenate mitigation of tau pathology. J Alzheimers Dis. (2017) 55:749–62. doi: 10.3233/JAD-151208
43. Cardoso BR, Apolinário D, Silva Bandeira V, Busse AL, Magaldi RM, Jacob-Filho W, et al. Correction to: Effects of Brazil nut consumption on selenium status and cognitive performance in older adults with mild cognitive impairment: a randomized controlled pilot trial. Eur J Nutr. (2021) 60:557. doi: 10.1007/s00394-020-02443-6
44. Farlow MR, Graham SM, Alva G. Memantine for the treatment of Alzheimer's disease: tolerability and safety data from clinical trials. Drug Saf. (2008) 31:577–85. doi: 10.2165/00002018-200831070-00003
45. Maret W. Cellular zinc and redox states converge in the metallothionein/thionein pair. J Nutr. (2003) 133:1460S−2S. doi: 10.1093/jn/133.5.1460S
46. Frederickson CJ, Bush AI. Synaptically released zinc: physiological functions and pathological effects. Biometals. (2001) 14:353–66. doi: 10.1023/A:1012934207456
47. Bal M, Leitz J, Reese AL, Ramirez DM, Durakoglugil M, Herz J, et al. Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron. (2013) 80:934–46. doi: 10.1016/j.neuron.2013.08.024
48. Burk RF, Hill KE, Olson GE, Weeber EJ, Motley AK, Winfrey VP, et al. Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J Neurosci. (2007) 27:6207–11. doi: 10.1523/JNEUROSCI.1153-07.2007
49. Kurokawa S, Bellinger FP, Hill KE, Burk RF, Berry MJ. Isoform-specific binding of selenoprotein P to the beta-propeller domain of apolipoprotein E receptor 2 mediates selenium supply. J Biol Chem. (2014) 289:9195–207. doi: 10.1074/jbc.M114.549014
50. Querfurth HW, LaFerla FM. Alzheimer's Disease. N Engl J Med. (2010) 362:329–44. doi: 10.1056/NEJMra0909142
51. Cardoso BR, Hare DJ, Lind M, McLean CA, Volitakis I, Laws SM, et al. The APOE ε4 Allele Is Associated with Lower Selenium Levels in the Brain: Implications for Alzheimer's Disease. ACS Chem Neurosci. (2017) 8:1459–64. doi: 10.1021/acschemneuro.7b00014
52. Nakashima AS, Oddo S, Laferla FM, Dyck RH. Experience-dependent regulation of vesicular zinc in male and female 3xTg-AD mice. Neurobiol Aging. (2010) 31:605–13. doi: 10.1016/j.neurobiolaging.2008.05.028
53. Lee JY, Cho E, Seo JW, Hwang JJ, Koh JY. Alteration of the cerebral zinc pool in a mouse model of Alzheimer disease. J Neuropathol Exp Neurol. (2012) 71:211–22. doi: 10.1097/NEN.0b013e3182417387
54. Li R, Singh M. Sex differences in cognitive impairment and Alzheimer's disease. Front Neuroendocrinol. (2014) 35:385–403. doi: 10.1016/j.yfrne.2014.01.002
55. Padilla-Gomez E, Beltran-Campos V, Montes S, Diaz-Ruiz A, Quirarte GL, Rios C, et al. Effect of 17ss-estradiol on zinc content of hippocampal mossy fibers in ovariectomized adult rats. Biometals. (2012) 25:1129–39. doi: 10.1007/s10534-012-9575-1
Keywords: Selenoprotein P, zinc, Alzheimer's disease, tau, selenium
Citation: Kiyohara ACP, Torres DJ, Hagiwara A, Pak J, Rueli RHLH, Shuttleworth CWR and Bellinger FP (2021) Selenoprotein P Regulates Synaptic Zinc and Reduces Tau Phosphorylation. Front. Nutr. 8:683154. doi: 10.3389/fnut.2021.683154
Received: 20 March 2021; Accepted: 26 May 2021;
Published: 01 July 2021.
Edited by:
Cristiane Cominetti, Universidade Federal de Goiás, BrazilReviewed by:
Yao Wang, University of California, San Francisco, United StatesPatricia Regina Manzine, Federal University of São Carlos, Brazil
Copyright © 2021 Kiyohara, Torres, Hagiwara, Pak, Rueli, Shuttleworth and Bellinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederick P. Bellinger, ZmJAaGF3YWlpLmVkdQ==