
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Nutr., 31 May 2021
Sec. Clinical Nutrition
Volume 8 - 2021 | https://doi.org/10.3389/fnut.2021.682243
This article is part of the Research TopicComplementary and Alternative Medicine Use in Cancer PatientsView all 8 articles
 Thomas N. Seyfried1*
Thomas N. Seyfried1* Aditya G. Shivane2
Aditya G. Shivane2 Miriam Kalamian3
Miriam Kalamian3 Joseph C. Maroon4
Joseph C. Maroon4 Purna Mukherjee1
Purna Mukherjee1 Giulio Zuccoli5*
Giulio Zuccoli5*Background: Successful treatment of glioblastoma (GBM) remains futile despite decades of intense research. GBM is similar to most other malignant cancers in requiring glucose and glutamine for growth, regardless of histological or genetic heterogeneity. Ketogenic metabolic therapy (KMT) is a non-toxic nutritional intervention for cancer management. We report the case of a 32-year-old man who presented in 2014 with seizures and a right frontal lobe tumor on MRI. The tumor cells were immunoreactive with antibodies to the IDH1 (R132H) mutation, P53 (patchy), MIB-1 index (4–6%), and absent ATRX protein expression. DNA analysis showed no evidence of methylation of the MGMT gene promoter. The presence of prominent microvascular proliferation and areas of necrosis were consistent with an IDH-mutant glioblastoma (WHO Grade 4).
Methods: The patient refused standard of care (SOC) and steroid medication after initial diagnosis, but was knowledgeable and self-motivated enough to consume a low-carbohydrate ketogenic diet consisting mostly of saturated fats, minimal vegetables, and a variety of meats. The patient used the glucose ketone index calculator to maintain his Glucose Ketone Index (GKI) near 2.0 without body weight loss.
Results: The tumor continued to grow slowly without expected vasogenic edema until 2017, when the patient opted for surgical debulking. The enhancing area, centered in the inferior frontal gyrus, was surgically excised. The pathology specimen confirmed IDH1-mutant GBM. Following surgery, the patient continued with a self-administered ketogenic diet to maintain low GKI values, indicative of therapeutic ketosis. At the time of this report (May 2021), the patient remains alive with a good quality of life, except for occasional seizures. MRI continues to show slow interval progression of the tumor.
Conclusion: This is the first report of confirmed IDH1-mutant GBM treated with KMT and surgical debulking without chemo- or radiotherapy. The long-term survival of this patient, now at 80 months, could be due in part to a therapeutic metabolic synergy between KMT and the IDH1 mutation that simultaneously target the glycolysis and glutaminolysis pathways that are essential for GBM growth. Further studies are needed to determine if this non-toxic therapeutic strategy could be effective in providing long-term management for other GBM patients with or without IDH mutations.
Glioblastoma (GBM) has among the highest mortality rates for primary brain tumors and remains poorly manageable. Despite the hype surrounding recent therapies (1–5), median life expectancy following diagnosis remains poor for most GBM patients (6–9). Survival is slightly better, however, for younger GBM patients compared to older GBM patients and for patients with GBM tumors that express IDH1 mutations (10–14). Most GBM patients receive the current standard of care (SOC) involving surgical debulking, radiotherapy, and temozolomide (TMZ) chemotherapy (15, 16). Many GBM patients can also receive corticosteroid medication (dexamethasone) and bevacizumab for managing edema and angiogenesis, respectively. Use of steroids is now under serious reevaluation, as steroids can elevate blood glucose, which is associated with more rapid tumor growth and shortened overall survival (17, 18). The current SOC for GBM has only marginally improved overall survival compared to “best supportive care,” which is ambiguous at best (19, 20). Equally distressing to management failure is evidence that the incidence of GBM is increasing in the United Kingdom (21).
We recently reviewed studies describing the adverse effects that can be associated with the current SOC for GBM management (17). GBM, like most malignant cancers, is driven by glucose and glutamine fermentation through the glycolysis and glutaminolysis pathways, respectively (22–27). The dependency on glucose and glutamine fermentation arises from inefficient oxidative phosphorylation (OxPhos) that is linked to abnormalities in the number, structure, and function of mitochondria in GBM tissue (17, 26–33). Surgical debulking followed by radiotherapy inadvertently increases the availability of glucose and glutamine in the tumor microenvironment (17, 34, 35). TMZ chemotherapy can further damage mitochondria while, at the same time, increasing systemic inflammation and tumor driver mutations (36, 37). Bevacizumab is even more likely than TMZ to cause mitochondrial dysfunction in human brain tumors, and is remarkable in its ability to facilitate distal tumor cell invasion through the neural parenchyma and the perivascular network (38–40). Use of dexamethasone can further accelerate GBM growth by increasing blood glucose levels and glutamine metabolism (25, 41–44). In light of this information, the poor progression free and overall survival experienced by most GBM patients receiving the SOC should not be surprising.
Winter and colleagues coined the term “Ketogenic Metabolic Therapy (KMT)” to describe an anti-neoplastic nutritional strategy, using ketogenic or low-glycemic diets, for the management of malignant gliomas (45). KMT is gaining recognition as a complementary therapeutic strategy for the management of a broad range of cancers in addition to malignant gliomas (17, 19, 45–60). Low carbohydrate, high fat ketogenic diets (KD) reduce the glucose needed to drive aerobic fermentation (Warburg effect) while also elevating ketone bodies, which cannot be effectively metabolized for energy in tumor cells due to defects in mitochondrial OxPhos (17, 26, 45, 56, 61–67). Moreover, ketone body metabolism enhances the ΔG′ATP hydrolysis in normal cells from −56kJ/mole to −59kJ/mole, thus providing normal cells with an energetic advantage over tumor cells (27, 68, 69). Calorie restriction and restricted KD are also anti-angiogenic, anti-inflammatory, anti-invasive, and can kill tumor cells directly through pro-apoptotic mechanisms (17, 62, 70–73). Evidence also shows that therapeutic ketosis can act synergistically with several drugs and procedures to enhance cancer management thus improving both progression free and overall survival (26, 74–76). Hence, KMT targets the multiple drivers of rapid GBM growth while enhancing the metabolic efficiency of normal brain cells (56).
A 26-year-old male from South Devon presented on August 16, 2014 at University Hospitals Plymouth NHS Trust, PL6 8DH, UK, with two episodes of left-sided facial numbness and bilateral tonic-clonic seizures originating from the right temporal lobe. There was no history of malignancy or chronic disorders. The patient's blood pressure was within normal limits (110/70). Laboratory investigation revealed unremarkable blood chemistry, with liver and renal functions within normal limits. Fasting blood glucose and C-Reactive protein were within normal ranges. Prior to therapeutic intervention, the patient's weight, height, and body mass index (BMI) were 63 kg, 180 centimeters, and 19.4 kg/m2, respectively. The contrast enhanced brain MRI (August 22, 2014) showed an intra-axial lesion, centered in the right inferior frontal lobe. The lesion, which was mainly non-enhancing and solid, disclosed the presence of an eccentric contrast enhancing nodule (Figure 1, Panel 1, D).
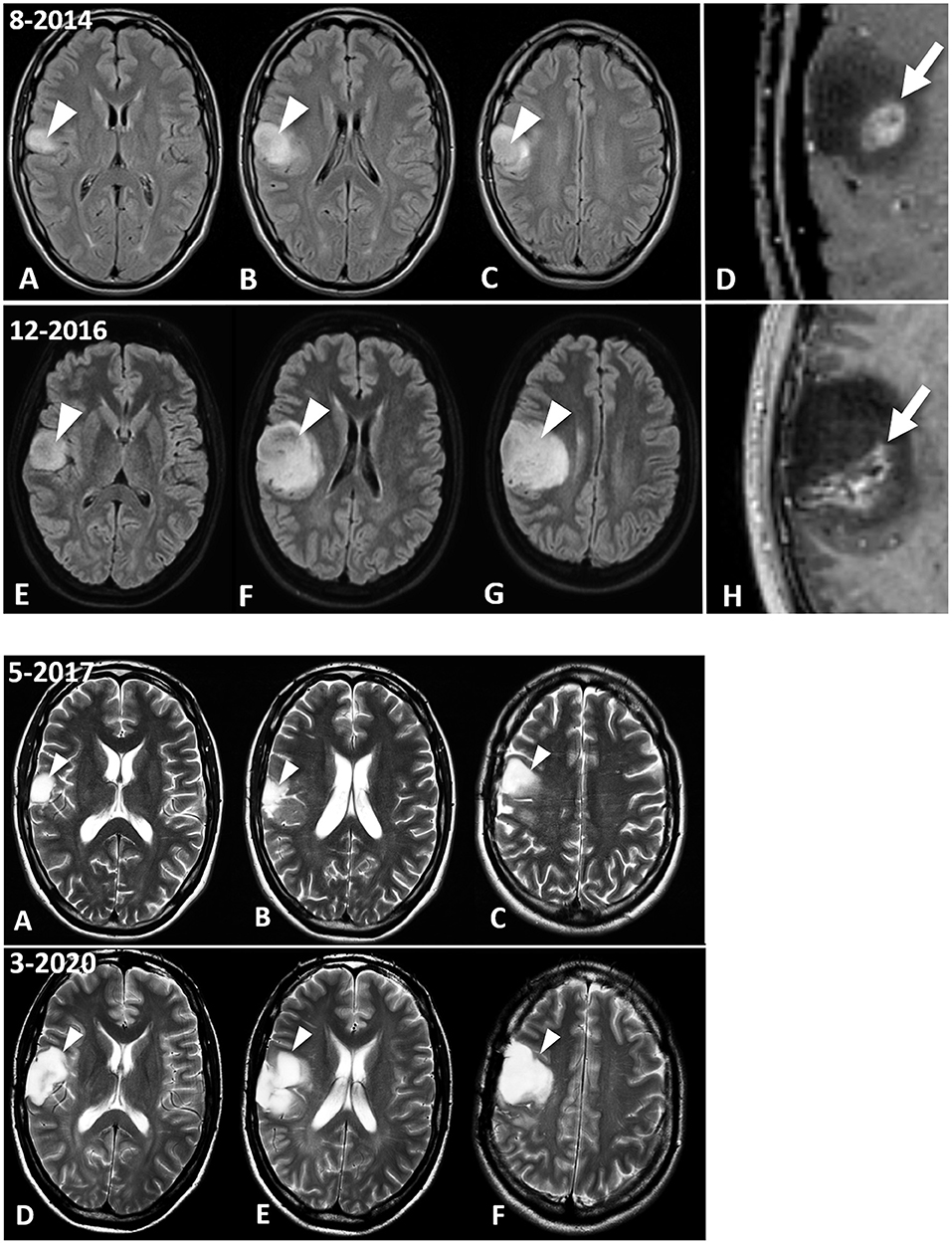
Figure 1. MRI images of the patient's brain tumour. Panel 1. Fluid Attenuated Inversion Recovery (FLAIR) images show the IDH1-mutant GBM at diagnosis in August 2014 (A–D) and ~8 months later, at the last follow-up MRI examination (December 2016) prior to surgical excision of the enhancing nodule (E–H). The lesion discovered on August 2018 is centered in the right inferior frontal lobe (A,B, arrowheads), and abuts the premotor cortex (C, arrowhead). There is an enhancing nodule within the lesion, as seen in the magnified post-contrast image (D) (arrow). This enhancing lesion measured 1.25 mL, which is calculated using the V = ABC/2 formula. Follow-up MRI (E–H) demonstrates interval progression of the non-enhancing tumor (arrowheads). Interval increase in size of the enhancing lesion, measuring 5.97 mL, was also observed (H, arrow). Panel 2 shows the evolution of the lesion between the May 2017 surgical excision (A–C), and in the most recent MRI evaluation, dated March 2020 (D–F). T2-weighted images indicate the residual GBM (arrowheads). (B) shows the T2-hyperintense GBM filling the surgical cavity. The lesion in the premotor cortex is clearly seen in (C). Please note that the surgical debulking involved only the enhancing tumor, while the largest non-enhancing part of the GBM was not excised by the neurosurgeon. The most recent brain MRI (D–F) shows interval increase in size of the GBM (arrowheads), which remains circumscribed to the right frontal region without infiltrating the white matter tracts.
Enhanced MRI of the brain (August 22, 2014) showed a lobar T2 signal abnormality without restricted diffusion and with central ring enhancement centered on the opercula portion of the inferior frontal gyrus. Signal change also extended into the precentral gyrus (Figure 1, Panel 1, C). The preliminary impression was a transforming low-grade glioma. Histopathological analysis (September 16, 2014) of seven cream and white cores of brain biopsy tissue revealed a diffuse cortical infiltration by a paucicellular glial neoplasm composed of predominantly fibrillary and occasional gemistocytic astrocytes. The tumor cells were moderately pleomorphic with infrequent/rare mitosis. There was focal micro-calcification and focal microvascular proliferation (Figure 2A), but no necrosis was about present. The MIB-1 (Ki67) proliferation index was 4–6% (Figure 2B). The tumor cells were immune-reactive with antibodies to mutant IDH1 (R132H) and to patchy P53 expression. DNA analysis showed no evidence of the MGMT gene promoter methylation. The presence of rare mitoses, focal microvascular proliferation and MIB-1 proliferation index was compatible with an IDH-mutant glioblastoma (WHO Grade 4).
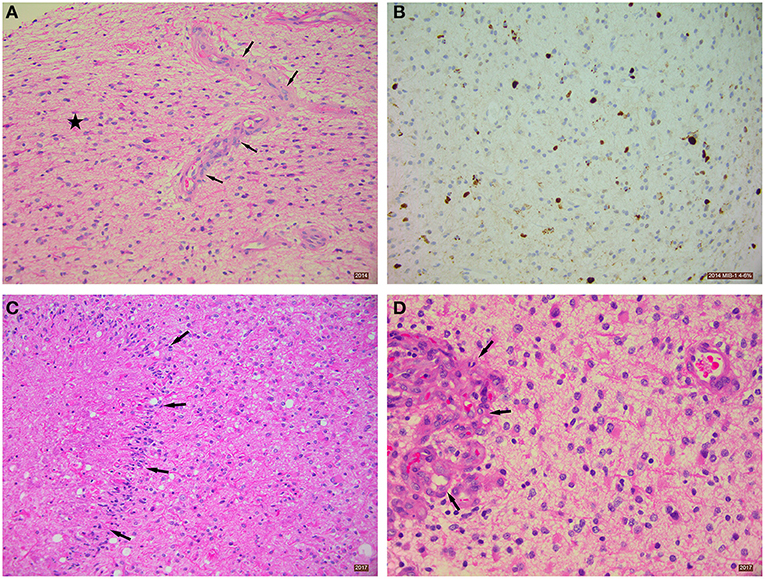
Figure 2. Histopathological analysis of excised brain tumor tissue. (A) Diffusely-infiltrating astrocytic tumor (star) with focal microvascular proliferation (arrows). Tissue taken from 2014 biopsy, H&E stain 200x. (B) Immunohistochemistry using MIB-1 antibody showing a proliferation index of ~4–6%. Tissue taken from 2014 biopsy, Immunostain 200x. (C) Section showing necrosis and palisading of astrocytic tumor cells around the necrotic area (arrows). Tissue taken from 2017 biopsy, H&E stain 200x. (D) Section showing glomeruloid vascular proliferation (arrows). Tissue taken from 2017 biopsy, H&E stain 400x.
Due to the patient's cultural beliefs regarding toxic therapies, he refused the recommended SOC. Instead, he opted for self-administering ketogenic metabolic therapy (KMT) that was initiated 2 weeks after the histopathological diagnosis of GBM. The patient was motivated to educate himself on proper implementation of the diet, replacing the recommended SOC with KMT despite pressure from his healthcare providers to use SOC. The energy composition of his daily diet consisted of fat (1,696 kcal), protein (264 kcal), and carbohydrates (48 kcal) with the addition of MCT oil (medium chain triglycerides). He was prescribed levetiracetam (750 mg, 2x/day) for seizure management, and MCT oil (3 tsp daily with food). He strictly followed the ketogenic diet guidelines found on Patricia Daly's website (https://patriciadaly.com/the-ketogenic-diet-for-cancer), and used the Precision Xtra glucose/ketone meter (Abbott Labs) and the glucose/ketone index calculator to monitor his blood glucose (mmol) and blood β-hydroxybutyrate (β-OHB) (mmol) values (77). It took the patient 2 weeks to enter the predicted zone of therapeutic ketosis, i.e., glucose/ketone index (GKI) values near 2.0 or below, as previously described (77). A second MRI of his tumor conducted on January 24, 2015 revealed no noticeable progression.
Serial MRIs preformed on April 14, 2015; July 17, 2015; November 16, 2015; February 20, 2016; July 9, 2016; and October 29, 2016 revealed evidence of interval slow contrast-enhanced tumor progression over that seen on the original 2014 MRI. MRI evidence of contrast-enhancing disease progression was more concerning on the follow-up MRI from December 15, 2016 (Figure 1, Panel 1, H). In response to these observed changes, the patient opted for an awake debulking craniotomy in April 2017. Excision was uneventful, resulting in gross complete removal of the mixed solid-necrotic contrast enhancing component of the GBM. However, the largest T2 hyperintense part of the GBM remained untouched (Figure 1, Panel 2, A–C). Histological analysis of the tumor tissue showed a diffusely invasive astrocytic tumor with infrequent/rare mitosis, prominent microvascular proliferation (Figure 2C) and areas of necrosis (Figure 2D). The tumor cells expressed mutant IDH1 (R132H), P53 (patchy), and showed loss of nuclear ATRX expression. The overall histological features were in keeping with the diagnosis of IDH1-mutant glioblastoma (WHO Grade 4).
The patient continued with a strict ketogenic diet regimen following tumor debulking and maintained his GKI values at or near 2.0 and below. The following medications were taken for 1 week only after the craniotomy and included Epilim (200 mg), dexamethasone (2 mg), omeprazole (20 mg), and paracetamol (1 g) for post-surgical pain management. Tonic-clonic seizure activity, which increased after the craniotomy, gradually subsided over time. Various supplements were added to the diet that included vitamins, minerals, turmeric, resveratrol, omega-3, and boswellia serrata. No further tumor growth was seen on the MRI preformed on August 17, 2017. As the patient believed that his GBM was under control, he relaxed his adherence to low-carbohydrate foods. This resulted in modest body weight gain (89 kg) and elevated his GKI values to the 5–10 range indicative of increased blood glucose and reduced ketone levels.
An MRI preformed on October 9, 2018 showed interval progression of the lesion. The patient quickly realized that the regrowth of his tumor might have been linked in part to the relaxation of his dietary rigor. Along with optimization and intensification of the dietary regime, the patient adopted lifestyle interventions including moderate physical training, breathing exercises, and physiological stress management. As of November 2018, the patient has adhered to a two-meal/day schedule with a rigorous time-restricted eating regimen (20 h/day fasting). The diet consisted of eggs, bacon fried in ghee/butter (11:00 h), and steak, lamb chops, beef patties, and liver, all fried in ghee/butter/lard (16:00 h). The patient did not continue with MCT oil after he started the carnivore diet. The patient did not keep a specific food diary. When he was on a restricted calorie ketogenic diet, he would start out by weighing his food and keeping under 2,000 calories/day, but he ended up learning to judge food intake by how hungry he was and ate until he was satiated. Carbohydrates were strictly eliminated from the patient's diet. The patient recognized that a well-formulated animal-based Paleo-carnivore diet would provide most bio-available micronutrients (78). This carnivore nutritional fasting schedule returned the patient's GKI values to 2.0 or below. The patient's BMI normalized to 22.2 (72 kg) at the time of this report. In addition, the patient participated in a breathing program involving breath-holding that increased the weekly average from 15 to 60 s. and lowered the morning average resting heart rate from 80 to 60 bpm. The patient was weaned off all medications except for Zebanex (eslicarbazepine acetate, 1,200 mg) needed for seizure control, which is taken once at 16:00 h. each day. The patient's blood glucose and β-OHB values are shown in Figure 3A over an almost 5-year period, and his computed GKI values over this period are shown in Figure 3B. The raw numbers for these values are presented in the Supplementary Tables.
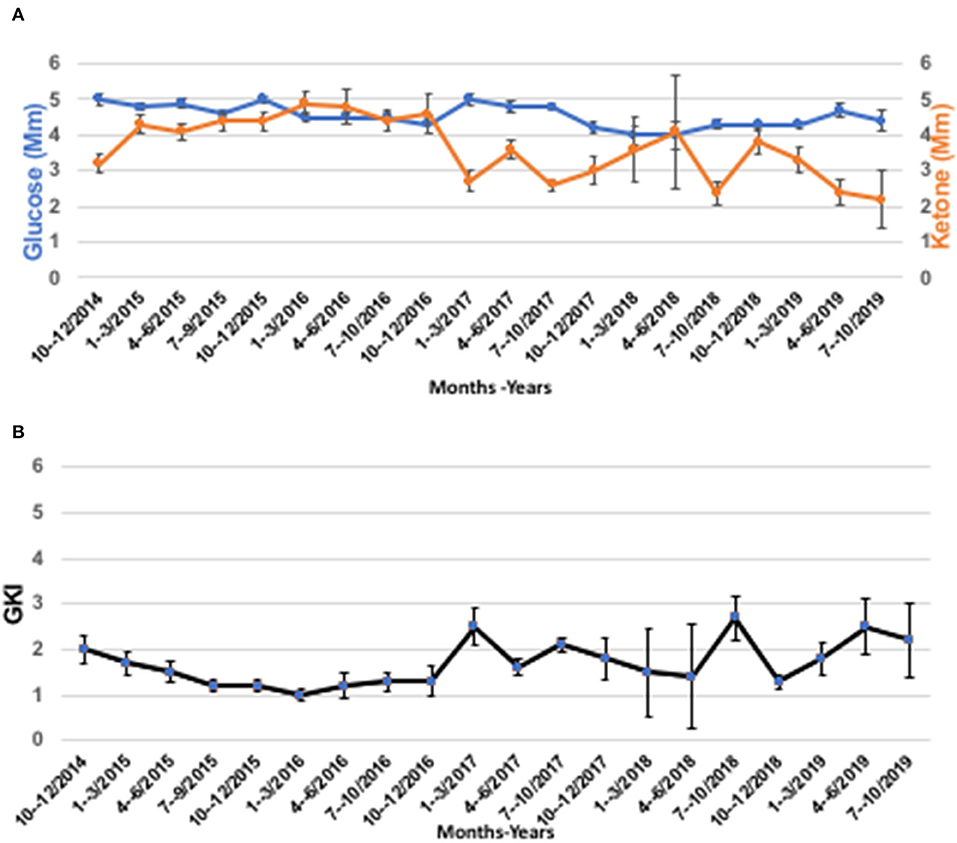
Figure 3. The patient's blood glucose, ketone, and GKI values from 2014 to 2019. (A) Blood glucose, and ketone (β-OHB) values determined using Precision Xtra blood glucose & ketone meter as described in text. (B) GKI values were determined from the individual glucose and ketone values in A using the glucose ketone index calculator, as previously described (77). Individual values were pooled over 3-month time intervals and are expressed as means ± 95% confidence intervals (CI). The number of readings for each data point in A and B ranged from a high of n = 151 (July-September, 2017), to a low of n = 7 (April-June 2018), and are given in the Supplementary Tables 1A–F arranged by year from 2014 to 2019.
An additional seven MRI evaluations, spanning from December 28, 2017 to March 10, 2020, showed continued slow interval progression of disease, without formation of noticeable vasogenic edema. At the time of this report (April 2021), the patient is active with a good quality of life, except for occasional tonic-clonic seizures and no signs of increased intracranial pressure. The patient was a speaker at the September 2018 Childhood and TYA Cancer Conference (http://www.childhoodcancer2018.org.uk/programme.asp; Children with Cancer; London, UK). He maintains a Facebook page that provides updates on his health status (http://www.childhoodcancer2018.org.uk/speakers/pablo-kelly.asp). Figure 4 presents a schematic diagram showing the clinical time course of MRI analysis and dietary treatment.
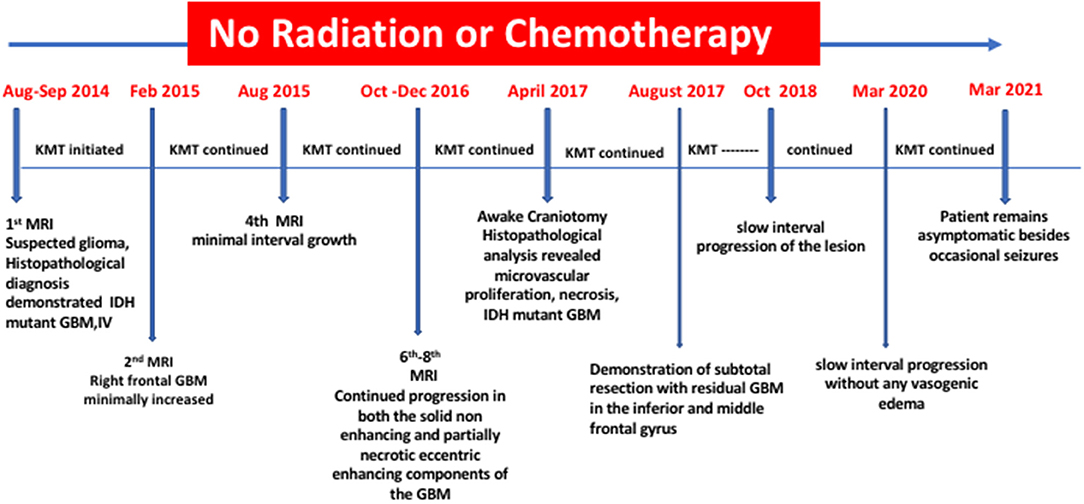
Figure 4. Timeline of clinical course with dates of dietary treatments and MRI.
Although long-term survival is rare in patients with GBM, about 5–13% of GBM patients can survive > 5 years with SOC for reasons that are unclear (79). This case study describes long-term survival and therapeutic management with KMT in a 32-year-old man diagnosed with a histopathologic and radiographically verified IDH1-mutant GBM. Several factors could contribute to the long-term and continued survival of this patient (now at 80 months). First, the patient refused SOC and steroid medication. Due to his preference for non-toxic therapies and the recognized potential of KMT for GBM management, the patient opted for a self-administered KD with various supplements. This strategy, in association with the surgical debulking, could have contributed in part to the slow growth and more effective resection of his GBM. It is well-documented that survival is longer in younger GBM patients (<50 years) than in older GBM patients (> 50 years), and that patients receiving a more complete tumor resection generally survive longer than patients receiving a subtotal resection (10, 80–82). Total or subtotal neurosurgical resection, however, is generally obtained early after diagnosis to achieve a longer survival. In contrast, this patient opted for a watch and wait strategy due to his refusal of the SOC. Consequently, tumor debulking was performed almost 3 years after diagnosis. It is also known that median survival is longer in GBM patients that express the IDH1 (R132H) mutation (31 months) than in patients that express the wild type allele (15 months) (13, 83). While we recognize that the therapeutic response seen in this patient might not be seen in other similarly-treated GBM patients, there are decades of compelling science supporting the mechanisms by which this metabolic therapy could reduce progression of GBM (17).
The remarkably slow growth of the patient's tumor stands in contrast to previous studies on the MRI growth dynamics of untreated glioblastomas (82). Analysis of 106 untreated GBMs showed a median volume of 17.7 mL at the diagnostic MRI scan, and 27.5 mL at the preoperative scan with an estimated volume doubling time of 49.6 days. Moreover, volume doubling time was significantly faster for smaller tumors at diagnosis (<3.88 mL) than for larger tumors (> 39.88 mL). Previous studies also showed that surgical resection did not significantly increase survival in patients with small tumors no matter what percentage of the tumor could be debulked (84). GBM survival time was estimated at 292 days following immediate surgical resection and 492 days if the first surgical resection debulked 80% of the tumor (84). We used the ABC/2 formula to measure the volume change in the patient's tumor over time (85). The patient's tumor measured 1.25 mL at diagnosis (August, 2014) and grew to 5.97 mL at the time of the preoperative scan (April 2017), a 32-month time interval (Figure 5). The estimated volume doubling time for the patient's tumor was 432 days and his survival time, after resection, is now over 1,400 days. Clearly, the enhancing tumor growth rate and overall survival of this patient is markedly better than those of most previously reported cases. The patient's tumor is consistent with IDH1 mutant GBM with a mass-like morphology > 33% of non-contrast enhancing tumor (nCET), as previously described (86). It is not likely that loss of the ATRX protein or absent MGMT methylation could have contributed to the patient's survival in light of previous information linking these markers to poor survival (87). Could the patient's chosen KMT and the chance acquisition of the IDH1 mutation have contributed to his long-term survival with GBM?
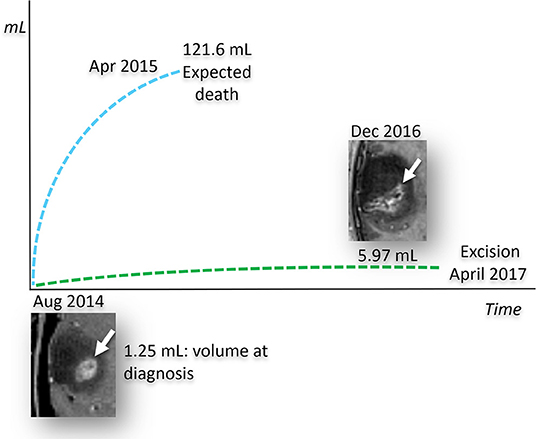
Figure 5. Predicted and observed tumor volume for untreated IDH1-mutant GBM. Measured growth of the patient's tumor compared to its estimated growth, based on the stochastic model of untreated human glioblastomas (84). The size of the tumor, measured at diagnosis in August 2014, is calculated by measuring the post contrast images only, since the current literature does not provide growth models based on the total volume of the tumor represented by the enhancing and non-enhancing tumor. Based on this stochastic model, the predicted growth rate (blue dashed line), shows that the enhancing tumor would have reached a volume incompatible with life around April 2015. In striking contrast to what was expected, the patient's IDH1 mutant GBM, treated with the KMT alone, demonstrated a much lower growth rate (green dashed line). It should be noted that over 70% of the patient's GBM did not enhance as would expected for IDH mutation. The patient's tumor volume measurements were determined from MRI, as described in text.
It is well-known that GBM survival and tumor growth is linked directly to blood glucose levels, i.e., high blood glucose is associated with faster disease progression and shorter survival times (18, 41, 88–97). Glucose is the fuel for aerobic fermentation (Warburg-effect), which is a driver of most malignant cancers including GBM (26, 98). Although the patient did not keep a food diary, he was able to maintain low GKI values with intermittent fasting and his chosen low-carbohydrate food choices. The patient's ability to maintain his GKI values consistently near 2.0 and below would target the Warburg-effect thus inhibiting growth of his tumor and improving his overall survival (99). Reduced blood glucose levels will not only starve the tumor of growth metabolites through glycolysis and one-carbon metabolism, but will also down-regulate the PI3K/Akt/Hif1-1α/mTOR signaling cascades that would further inhibit dysregulated tumor cell growth (17, 58, 100–103). The low GKI values were also in the direction of predicted therapeutic success for reducing lactic acid production (17, 56, 58, 77). Reduced glucose-driven lactic acid would reduce NF-kappa-β-induced inflammation and edema in the tumor microenvironment, thus reducing tumor cell angiogenesis and invasion (17, 58, 70–72, 77, 102, 104). Reduced inflammation in the tumor microenvironment could account in part for the absence of robust vasogenic edema and the slower growth of the GBM seen in our patient, as we observed previously in KMT-treated unirradiated preclinical GBM (75). It is also important to mention that the survival of our patient was much longer than that of most other GBM patients receiving KMT following SOC (45, 54, 59, 105–108). Few of the adult patients treated with KMT in these studies, however, were able to reach or maintain the GKI values predicted to have the greatest therapeutic benefit for managing GBM (77). The avoidance of radiotherapy would also prevent liquefactive necrosis, vascular hyalinization, and rapid tumor progression, as occurred in our previous KMT-treated GBM patient that was of similar age at diagnosis (17, 56). The patient's decision to use KMT as an alternative to SOC and his ability to maintain low GKI values could have contributed in part to his long-term survival and accompanying good quality of life.
The potential mechanism by which the IDH1 mutation might reduce GBM growth and increase survival is discussed below and in Figure 6. We recently described how chemical energy by itself is the central issue for neoplastic cell viability. Tumors cannot grow without ATP, regardless of their cellular or genetic heterogeneity (26). In addition to glucose, glutamine is the other major fermentable fuel that can drive ATP synthesis in most cancers including GBM (24, 26, 114). Glutamine is the only amino acid that can generate ATP synthesis through mSLP in the glutaminolysis pathway (25). While KMT might not be as effective in targeting glutaminolysis as it is in targeting glycolysis, elevation of the patient's blood ketone bodies (β-hydroxybutyrate and acetoacetate), evident from his low GKI values, could indirectly target the glutamine-driven glutaminolysis pathway; also known as the Q-effect (25, 26). ATP synthesis through mitochondrial substrate-level phosphorylation (mSLP) at the succinate CoA ligase reaction (SUCL) in the glutaminolysis pathway can compensate for diminished ATP synthesis through both glycolysis and OxPhos (Figure 6). The synthesis of acetoacetyl-CoA from acetoacetate and β-hydroxybutyrate would siphon off some of the CoA needed for the synthesis of succinyl-CoA thus reducing substrate for ATP synthesis through mSLP (26). Additionally, a reduction in α-ketoglutarate levels through action of the IDH1-induced increase in 2-hydroxyglutarate could further reduce substrate for ATP synthesis through mSLP (25, 26, 112, 115). Recent studies also show that IDH1-derived 2-hydroxyglutarate can facilitate degradation of Hif1-α and thus reduce the Warburg-effect through down-regulation of multiple genes in the glycolytic pathway (113). Further evidence of an inhibitory effect of the IDH1 mutation on glucose consumption and glycolysis was obtained recently from PET analysis (116). The long-term survival of the patient could therefore result in part from a synergistic interaction between his self-directed KMT and the anti-cancer effects of the IDH1-R132H mutation.
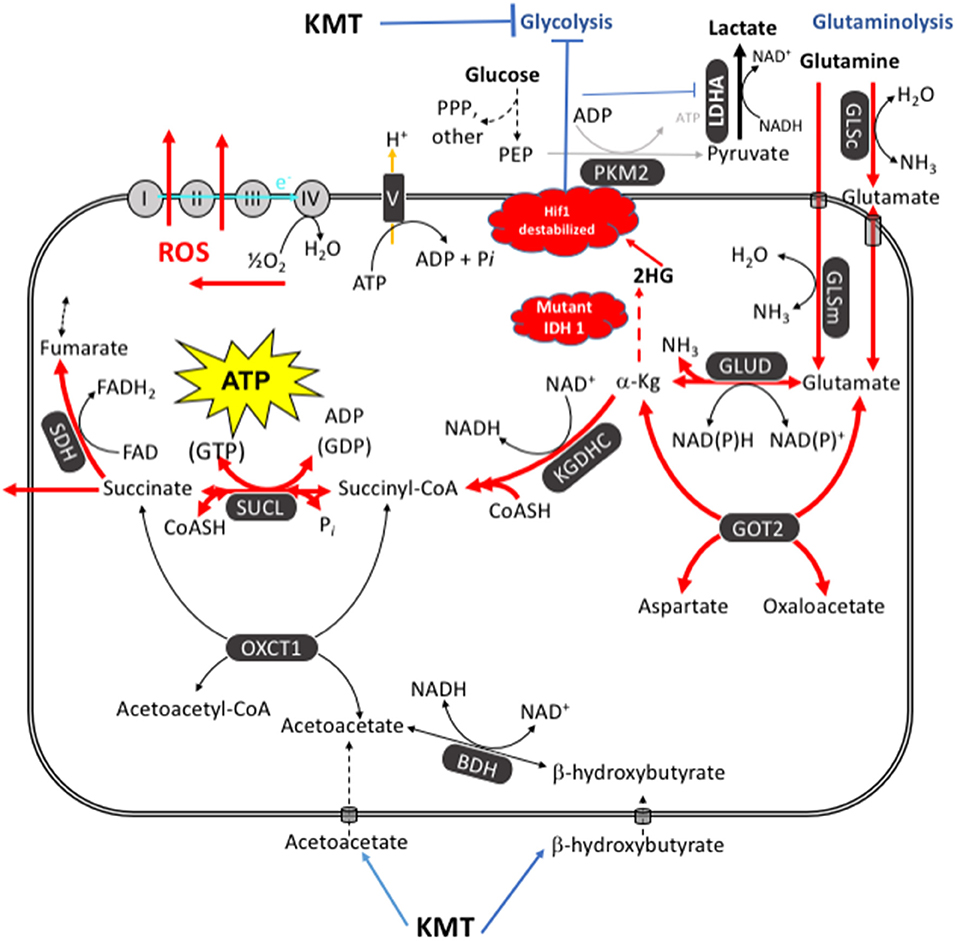
Figure 6. KMT/IDH1 synergistic interaction for targeting GBM energy metabolism. KMT can reduce glucose availability for glycolysis while also interfering with the glutaminolysis pathway. Glutamine-driven mitochondrial substrate-level phosphorylation (mSLP), in the glutaminolysis pathway, is a major source of ATP synthesis for GBM cells (25, 26). The glutaminolysis pathway (red) becomes dominant in tumor cells with inefficient OxPhos and that express the dimeric PKM2 isoform. PKM2 is expressed in GBM and produces less ATP through glycolysis than does the PKM1 isoform (109–111). The elevation of ketone bodies (β-hydroxybutyrate and acetoacetate) through KMT could indirectly reduce ATP synthesis through the succinate CoA ligase (SUCL) reaction by diverting CoA from succinate to acetoacetate. The IDH1 mutation could further reduce ATP synthesis through mSLP by increasing synthesis of 2-hydroxyglutarate from α-ketoglutarate and thus reducing the succinyl CoA substrate for the SUCL reaction (26, 112). Besides its potential effect in reducing glutaminolysis, 2-hydroxyglutarate can also target multiple HIF1α-responsive genes and enzymes in the glycolysis pathway thus limiting synthesis of metabolites and one-carbon metabolism needed for rapid tumor growth (25, 26, 103, 113). The down-regulation of Hif1-α-regulated lactate dehydrogenase A (LDHA), through the action of both KMT and the IDH1 mutation, would reduce extracellular lactate levels thus reducing microenvironment inflammation and tumor cell invasion. Hence, the simultaneous inhibition of glycolysis and glutaminolysis through the synergistic effects of KMT and the IDH1 mutation will stress the majority of signaling pathways necessary for rapid GBM growth. BDH, β-hydroxybutyrate dehydrogenase; FAD, flavin adenine dinucleotide; GLSc, glutaminase, cytosolic; GLSm, glutaminase; mitochondrial; GLUD, glutamate dehydrogenase; GOT2, aspartate aminotransferase; KGDHC, α-ketoglutarate dehydrogenase complex; LDHA, lactate dehydrogenase A; NME, nucleoside diphosphate kinase; OXCT1, succinyl-CoA:3-ketoacid coenzyme A transferase 1; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PEP, phosphoenolpyruvate; PKM2, pyruvate kinase M2; SDH, succinate dehydrogenase; SUCL, succinate-CoA ligase. Reprinted with modifications from Seyfried et al. (26).
If a capability is truly important for the biology of tumors, then its inhibition should be considered as a therapeutic strategy for effective management (99, 117). The capability in this case is the fermentation metabolism needed for the synthesis of growth metabolites and ATP through the glycolytic and glutaminolysis pathways (26). As GBM “stem cells” are more dependent on glucose than on glutamine for growth, whereas GBM “mesenchymal cells” are more dependent on glutamine than on glucose, it becomes essential to inhibit both the glycolysis and the glutaminolysis pathways simultaneously to achieve maximal GBM management (24, 75). The glutamine-addicted GBM mesenchymal cells arise from neoplastic microglia/macrophages (118, 119). A recent study showed that expression of the macrophage/microglial marker, CD163, was lower in an IDH1-mutant GBM than in IDH1 wild-type GBM (120). CD163 is a biomarker for glutamine-dependent neoplastic macrophages in tumor tissue (119, 121). Hence, a synergistic interaction between the effects of the IDH1 mutation and KMT could simultaneously down-regulate both the Warburg-effect and the Q-effect in the GBM neoplastic cell populations thus providing a novel mechanism contributing to the long-term survival of our patient.
Although N-of-one single subject studies have been considered the ultimate strategy for individualizing medicine (122), we cannot predict if the therapeutic response to KMT seen in our GBM patient will also be seen in other similarly treated GBM patients, especially those with tumors that are wild-type at the IDH1 locus. For those GBM patients not fortunate enough to have acquired the spontaneous IDH1 mutation in their tumor, glutamine targeting drugs used with KMT will be necessary to reduce tumor growth. Our recent bench-to-bedside translational studies show that simultaneous targeting of glucose and glutamine availability, using KMT and the pan-glutaminase inhibitor, 6-diazo-5-oxo-L-norleucine (DON), can significantly prolong survival in preclinical syngeneic glioblastomas (75). It is also important to note that ketogenic diets can facilitate delivery of small-molecule therapeutic drugs through the blood brain barrier without toxicity (75, 123, 124). As GBM, like most malignant cancers, is dependent on fermentation for ATP synthesis and survival, the simultaneous restriction of fermentable fuels, i.e., glucose and glutamine, while elevating non-fermentable ketone bodies, offers a non-toxic therapeutic strategy for managing GBM. Further studies will be needed to test this hypothesis in other patients diagnosed with GBM.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
TS: wrote the manuscript and assisted in data presentation and analysis. AS: conducted the histological report. MK: provided information on nutritional status and helped write the paper. JM: re-evaluate the patient's data and edit the paper. PM: evaluated data and assisted in manuscript preparation. GZ: analysis of MRI images and helped in writing the paper. All authors contributed to the article and approved the submitted version.
MK was employed by Dietary Therapies LLC.
The authors declare that this study received funding from the Foundation for Metabolic Cancer Therapies, CrossFit Inc., The Nelson and Claudia Peltz Family Foundation, Lewis Topper, The John and Kathy Garcia Foundation, Edward Miller, the patient himself, the George Yu Foundation, Kenneth Rainin Foundation, Children with Cancer UK, and the Boston College Research Expense Fund. The funders were not involved in the study design, collection, analysis, interpretation of data, and the writing of this article or the decision to submit it for publication.
We thank the Foundation for Metabolic Cancer Therapies, CrossFit Inc., The Nelson and Claudia Peltz Family Foundation, Lewis Topper, The John and Kathy Garcia Foundation, Edward Miller, the patient himself, the George Yu Foundation, Kenneth Rainin Foundation, Children with Cancer UK, and the Boston College Research Expense Fund for their support. We also thank Misha Sakharoff for assisting the patient with dietary recommendations, breathing exercises, and mindfulness techniques to reduce stress. We also thank Kevin Cardenas for technical assistance. Finally, we thank the two reviewers for their valuable recommendations to improve the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2021.682243/full#supplementary-material
1. Polivka J, Polivka J, Holubec L, Kubikova T, Priban V, Hes O, et al. Advances in experimental targeted therapy and immunotherapy for patients with glioblastoma multiforme. Anticancer Res. (2017) 37:21–33. doi: 10.21873/anticanres.11285
2. Fabbro-Peray P, Zouaoui S, Darlix A, Fabbro M, Pallud J, Rigau V, et al. Association of patterns of care, prognostic factors, and use of radiotherapy-temozolomide therapy with survival in patients with newly diagnosed glioblastoma: a French national population-based study. J Neurooncol. (2018) 142:91–101. doi: 10.1007/s11060-018-03065-z
3. Geraldo LHM, Garcia C, da Fonseca ACC, Dubois LGF, de Sampaio ESTCL, Spohr TCL, et al. Glioblastoma therapy in the age of molecular medicine. Trends Cancer. (2019) 5:46–65. doi: 10.1016/j.trecan.2018.11.002
4. Wegman-Ostrosky T, Reynoso-Noveron N, Mejia-Perez SI, Sanchez-Correa TE, Alvarez-Gomez RM, Vidal-Millan S, et al. Clinical prognostic factors in adults with astrocytoma: historic cohort. Clin Neurol Neurosurg. (2016) 146:116–22. doi: 10.1016/j.clineuro.2016.05.002
5. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs. maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. (2017) 318:2306–16. doi: 10.1001/jama.2017.18718
6. Gutiontov SI. The median isn't the message: revisited. J Clin Oncol. (2020) 38:1112–4. doi: 10.1200/JCO.19.03078
7. Fatehi M, Hunt C, Ma R, Toyota BD. Persistent disparities in survival for patients with glioblastoma. World Neurosurg. (2018) 120:e511–6. doi: 10.1016/j.wneu.2018.08.114
8. Johnson DR, Ma DJ, Buckner JC, Hammack JE. Conditional probability of long-term survival in glioblastoma: a population-based analysis. Cancer. (2012) 118:5608–13. doi: 10.1002/cncr.27590
9. Alvarez-Torres MDM, Fuster-Garcia E, Reynes G, Juan-Albarracin J, Chelebian E, Oleaga L, et al. Differential effect of vascularity between long- and short-term survivors with IDH1/2 wild-type glioblastoma. NMR Biomed. (2021) 34:e4462. doi: 10.1002/nbm.4462
10. Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, et al. Long-term survival with glioblastoma multiforme. Brain. (2007) 130:2596–606. doi: 10.1093/brain/awm204
11. Shinojima N, Kochi M, Hamada JI, Hideo N, Yano S, Makino K, et al. The influence of sex and the presence of giant cells on postoperative long-term survival in adult patients with supratentorial glioblastoma multiforme. J Neurosurgery. (2004) 95:249–57. doi: 10.3171/jns.2004.101.2.0219
12. Perry JR, Laperriere N, O'Callaghan CJ, Brandes AA, Menten J, Phillips C, et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med. (2017) 376:1027–37. doi: 10.1056/NEJMoa1611977
13. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 DD. and IDH2 mutations in gliomas. N Engl J Med. (2009) 360:765–73. doi: 10.1056/NEJMoa0808710
14. Salford LG, Brun A, Nirfalk S. Ten-year survival among patients with supratentorial astrocytomas grade III and IV. J Neurosurg. (1988) 69:506–9. doi: 10.3171/jns.1988.69.4.0506
15. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
16. Brandes AA, Franceschi E, Ermani M, Tosoni A, Albani F, Depenni R, et al. Pattern of care and effectiveness of treatment for glioblastoma patients in the real world: Results from a prospective population-based registry. Could survival differ in a high-volume center? Neurooncol Pract. (2014) 1:166–71. doi: 10.1093/nop/npu021
17. Seyfried TN, Shelton L, Arismendi-Morillo G, Kalamian M, Elsakka A, Maroon J, et al. Provocative question: should ketogenic metabolic therapy become the standard of care for glioblastoma? Neurochem Res. (2019) 44:2392–404. doi: 10.1007/s11064-019-02795-4
18. Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, et al. Corticosteroids compromise survival in glioblastoma. Brain. (2016) 139:1458–71. doi: 10.1093/brain/aww046
19. Seyfried TN, Flores R, Poff AM, D'Agostino DP, Mukherjee P. Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Lett. (2015) 356:289–300. doi: 10.1016/j.canlet.2014.07.015
20. Zafar SY, Currow D, Abernethy AP. Defining best supportive care. J Clin Oncol. (2008) 26:5139–40. doi: 10.1200/JCO.2008.19.7491
21. Philips A, Henshaw DL, Lamburn G, O'Carroll MJ. Brain tumours: rise in glioblastoma multiforme incidence in England 1995-2015 suggests an adverse environmental or lifestyle factor. J Environ Public Health. (2018) 2018:7910754. doi: 10.1155/2018/2170208
22. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) 16:749. doi: 10.1038/nrc.2016.114
23. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
24. Oizel K, Chauvin C, Oliver L, Gratas C, Geraldo F, Jarry U, et al. Efficient mitochondrial glutamine targeting prevails over glioblastoma metabolic plasticity. Clin Cancer Res. (2017) 23:6292–304. doi: 10.1158/1078-0432.CCR-16-3102
25. Chinopoulos C, Seyfried TN. Mitochondrial substrate-level phosphorylation as energy source for glioblastoma: review and hypothesis. ASN Neuro. (2018) 10:1759091418818261. doi: 10.1177/1759091418818261
26. Seyfried TN, Arismendi-Morillo G, Mukherjee P, Chinopoulos C. On the origin of ATP synthesis in cancer. iScience. (2020) 23:101761. doi: 10.1016/j.isci.2020.101761
27. Seyfried TN, Mukherjee P. Targeting energy metabolism in brain cancer: review and hypothesis. Nutr Metab. (2005) 2:30. doi: 10.1186/1743-7075-2-30
28. Deighton RF, Le Bihan T, Martin SF, Barrios-Llerena ME, Gerth AM, Kerr LE, et al. The proteomic response in glioblastoma in young patients. J Neuro-Oncol. (2014) 119:79–89. doi: 10.1007/s11060-014-1474-6
29. Deighton RF, Le Bihan T, Martin SF, Gerth AM, McCulloch M, Edgar JM, et al. Interactions among mitochondrial proteins altered in glioblastoma. J Neuro-Oncol. (2014) 118:247–56. doi: 10.1007/s11060-014-1430-5
30. Oudard S, Boitier E, Miccoli L, Rousset S, Dutrillaux B, Poupon MF. Gliomas are driven by glycolysis: putative roles of hexokinase, oxidative phosphorylation and mitochondrial ultrastructure. Anticancer Res. (1997) 17:1903–11.
31. Feichtinger RG, Weis S, Mayr JA, Zimmermann F, Geilberger R, Sperl W, et al. Alterations of oxidative phosphorylation complexes in astrocytomas. Glia. (2014) 62:514–25. doi: 10.1002/glia.22621
32. Ordys BB, Launay S, Deighton RF, McCulloch J, Whittle IR. The role of mitochondria in glioma pathophysiology. Mol Neurobiol. (2010) 42:64–75. doi: 10.1007/s12035-010-8133-5
33. Arismendi-Morillo GJ, Castellano-Ramirez AV. Ultrastructural mitochondrial pathology in human astrocytic tumors: potentials implications pro-therapeutics strategies. J Electron Microsc. (2008) 57:33–9. doi: 10.1093/jmicro/dfm038
34. Bergsneider M, Hovda DA, Shalmon E, Kelly DF, Vespa PM, Martin NA, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. (1997) 86:241–51. doi: 10.3171/jns.1997.86.2.0241
35. Rohlenova K, Veys K, Miranda-Santos I, De Bock K, Carmeliet P. Endothelial cell metabolism in health and disease. Trends Cell Biol. (2018) 28:224–36. doi: 10.1016/j.tcb.2017.10.010
36. Oliva CR, Nozell SE, Diers A, McClugage SG 3rd, Sarkaria JN, Markert JM, et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem. (2010) 285:39759–67. doi: 10.1074/jbc.M110.147504
37. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. (2014) 343:189–93. doi: 10.1126/science.1239947
38. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. (2009) 15:220–31. doi: 10.1016/j.ccr.2009.01.027
39. Nanegrungsunk D, Apaijai N, Yarana C, Sripetchwandee J, Limpastan K, Watcharasaksilp W, et al. Bevacizumab is superior to Temozolomide in causing mitochondrial dysfunction in human brain tumors. Neurol Res. (2016) 38:285–93. doi: 10.1080/01616412.2015.1114233
40. de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. (2010) 12:233–42. doi: 10.1093/neuonc/nop027
41. Klement RJ, Champ CE. Corticosteroids compromise survival in glioblastoma in part through their elevation of blood glucose levels. Brain. (2017) 140:e16. doi: 10.1093/brain/aww324
42. Arcuri C, Tardy M, Rolland B, Armellini R, Menghini AR, Bocchini V. Glutamine synthetase gene expression in a glioblastoma cell-line of clonal origin: regulation by dexamethasone and dibutyryl cyclic AMP. Neurochem Res. (1995) 20:1133–9. doi: 10.1007/BF00995375
43. Wong ET, Lok E, Gautam S, Swanson KD. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br J Cancer. (2015) 113:232–41. doi: 10.1038/bjc.2015.238
44. Seyfried TN, Shelton LM, Mukherjee P. Does the existing standard of care increase glioblastoma energy metabolism? Lancet Oncol. (2010) 11:811–3. doi: 10.1016/S1470-2045(10)70166-2
45. Winter SF, Loebel F, Dietrich J. Role of ketogenic metabolic therapy in malignant glioma: a systematic review. Crit Rev Oncol Hematol. (2017) 112:41–58. doi: 10.1016/j.critrevonc.2017.02.016
46. Iyikesici MS. Feasibility study of metabolically supported chemotherapy with weekly carboplatin/paclitaxel combined with ketogenic diet, hyperthermia and hyperbaric oxygen therapy in metastatic non-small cell lung cancer. Int J Hyperthermia. (2019) 36:446–55. doi: 10.1080/02656736.2019.1589584
47. Iyikesici MS, Slocum AK, Slocum A, Berkarda FB, Kalamian M, Seyfried TN. Efficacy of metabolically supported chemotherapy combined with ketogenic diet, hyperthermia, and hyperbaric oxygen therapy for stage IV triple-negative breast cancer. Cureus. (2017) 9:e1445. doi: 10.7759/cureus.1445
48. Seyfried TN, Mukherjee P, Iyikesici MS, Slocum A, Kalamian M, Spinosa JP, et al. Consideration of ketogenic metabolic therapy as a complementary or alternative approach for managing breast cancer. Front Nutr. (2020) 7:21. doi: 10.3389/fnut.2020.00021
49. Khodabakhshi A, Akbari ME, Mirzaei HR, Seyfried TN, Kalamian M, Davoodi SH. Effects of Ketogenic metabolic therapy on patients with breast cancer: a randomized controlled clinical trial. Clin Nutr. (2020) 40:751–8. doi: 10.1016/j.clnu.2020.06.028
50. Kalamian M. KETO for CANCER: Ketogenic Metabolic Therapy as a Targeted Nutritional Strategy. White River Junction, VT: Chelsea Green (2017).
51. Weber DD, Aminzadeh-Gohari S, Tulipan J, Catalano L, Feichtinger RG, Kofler B. Ketogenic diet in the treatment of cancer - Where do we stand? Mol Metab. (2019) 33:102–21. doi: 10.1016/j.molmet.2019.06.026
52. Klein P, Tyrlikova I, Zuccoli G, Tyrlik A, Maroon JC. Treatment of glioblastoma multiforme with “classic” 4:1 ketogenic diet total meal replacement. Cancer Metab. (2020) 8:24. doi: 10.1186/s40170-020-00230-9
53. Maroon J, Bost J, Amos A, Zuccoli G. Restricted calorie ketogenic diet for the treatment of glioblastoma multiforme. J Child Neurol. (2013) 28:1002–8. doi: 10.1177/0883073813488670
54. Zuccoli G, Marcello N, Pisanello A, Servadei F, Vaccaro S, Mukherjee P, et al. Metabolic management of glioblastoma multiforme using standard therapy together with a restricted ketogenic diet: case report. Nutr Metab. (2010) 7:33. doi: 10.1186/1743-7075-7-33
55. Panhans CM, Gresham G, Amaral JL, Hu J. Exploring the feasibility and effects of a ketogenic diet in patients with CNS malignancies: a retrospective case series. Front Neurosci. (2020) 14:390. doi: 10.3389/fnins.2020.00661
56. Elsakka AMA, Bary MA, Abdelzaher E, Elnaggar M, Kalamian M, Mukherjee P, et al. Management of glioblastoma multiforme in a patient treated with ketogenic metabolic therapy and modified standard of care: a 24-month follow-up. Front Nutr. (2018) 5:20. doi: 10.3389/fnut.2018.00020
57. Schwartz KA, Noel M, Nikolai M, Chang HT. Investigating the Ketogenic Diet As Treatment for Primary Aggressive Brain Cancer: Challenges and Lessons Learned. Front Nutr. (2018) 5:11. doi: 10.3389/fnut.2018.00011
58. Martuscello RT, Vedam-Mai V, McCarthy DJ, Schmoll ME, Jundi MA, Louviere CD, et al. A supplemented high-fat low-carbohydrate diet for the treatment of glioblastoma. Clin Cancer Res. (2016) 22:2482–95. doi: 10.1158/1078-0432.CCR-15-0916
59. Rieger J, Bahr O, Maurer GD, Hattingen E, Franz K, Brucker D, et al. ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. Int J Oncol. (2014) 44:1843–52. doi: 10.3892/ijo.2014.2382
60. Rieger J, Steinbach JP. To diet or not to diet - that is still the question. Neuro Oncol. (2016) 18:1035–6. doi: 10.1093/neuonc/now131
61. Maroon JC, Seyfried TN, Donohue JP, Bost J. The role of metabolic therapy in treating glioblastoma multiforme. Surg Neurol Int. (2015) 6:61. doi: 10.4103/2152-7806.155259
62. Zhou W, Mukherjee P, Kiebish MA, Markis WT, Mantis JG, Seyfried TN. The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr Metab. (2007) 4:5. doi: 10.1186/1743-7075-4-5
63. Kiebish MA, Han X, Cheng H, Chuang JH, Seyfried TN. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. J Lipid Res. (2008) 49:2545–56. doi: 10.1194/jlr.M800319-JLR200
64. Chang HT, Olson LK, Schwartz KA. Ketolytic and glycolytic enzymatic expression profiles in malignant gliomas: implication for ketogenic diet therapy. Nutr Metab. (2013) 10:47. doi: 10.1186/1743-7075-10-47
65. Fredericks M, Ramsey RB. 3-Oxo acid coenzyme A transferase activity in brain and tumors of the nervous system. J Neurochem. (1978) 31:1529–31. doi: 10.1111/j.1471-4159.1978.tb06581.x
66. Maurer GD, Brucker DP, Baehr O, Harter PN, Hattingen E, Walenta S, et al. Differential utilization of ketone bodies by neurons and glioma cell lines: a rationale for ketogenic diet as experimental glioma therapy. BMC Cancer. (2011) 11:315. doi: 10.1186/1471-2407-11-315
67. Gabriel AM, Alan CR, Thomas NS. Ultrastructural characterization of the Mitochondria-associated membranes abnormalities in human astrocytomas: Functional and therapeutics implications. Ultrastruct Pathol. (2017) 41:234–44. doi: 10.1080/01913123.2017.1300618
68. Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. (2004) 70:309–19. doi: 10.1016/j.plefa.2003.09.007
69. Veech RL, Todd King M, Pawlosky R, Kashiwaya Y, Bradshaw PC, Curtis W. The “great” controlling nucleotide coenzymes. IUBMB Life. (2019) 71:565–79. doi: 10.1002/iub.1997
70. Mukherjee P, El-Abbadi MM, Kasperzyk JL, Ranes MK, Seyfried TN. Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br J Cancer. (2002) 86:1615–21. doi: 10.1038/sj.bjc.6600298
71. Mukherjee P, Mulrooney TJ, Marsh J, Blair D, Chiles TC, Seyfried TN. Differential effects of energy stress on AMPK phosphorylation and apoptosis in experimental brain tumor and normal brain. Mol Cancer. (2008) 7:37. doi: 10.1186/1476-4598-7-37
72. Mulrooney TJ, Marsh J, Urits I, Seyfried TN, Mukherjee P. Influence of caloric restriction on constitutive expression of NF-kappaB in an experimental mouse astrocytoma. PLoS ONE. (2011) 6:e18085. doi: 10.1371/journal.pone.0018085
73. Shelton LM, Huysentruyt LC, Mukherjee P, Seyfried TN. Calorie restriction as an anti-invasive therapy for malignant brain cancer in the VM mouse. ASN Neuro. (2010) 2:e00038. doi: 10.1042/AN20100002
74. Poff AM, Ari C, Seyfried TN, D'Agostino DP. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systemic metastatic cancer. PLoS ONE. (2013) 8:e65522. doi: 10.1371/journal.pone.0065522
75. Mukherjee P, Augur ZM, Li M, Hill C, Greenwood B, Domin MA, et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun Biol. (2019) 2:200. doi: 10.1038/s42003-019-0455-x
76. Klement RJ. Beneficial effects of ketogenic diets for cancer patients: a realist review with focus on evidence and confirmation. Med Oncol. (2017) 34:132. doi: 10.1007/s12032-017-0991-5
77. Meidenbauer JJ, Mukherjee P, Seyfried TN. The glucose ketone index calculator: a simple tool to monitor therapeutic efficacy for metabolic management of brain cancer. Nutr Metab. (2015) 12:12. doi: 10.1186/s12986-015-0009-2
78. Toth C, Clemens Z. Halted progression of soft palate cancer in a patient treated with the paleolithic ketogenic diet alone: a 20-months follow-up. Am J Med Case Rep. (2016) 4:288–92. doi: 10.12691/ajmcr-4-8-8
79. Park CK, Bae JM, Park SH. Long-term survivors of glioblastoma are a unique group of patients lacking universal characteristic features. Neurooncol Adv. (2020) 2:vdz056. doi: 10.1093/noajnl/vdz056
80. Ahmadloo N, Kani AA, Mohammadianpanah M, Nasrolahi H, Omidvari S, Mosalaei A, et al. Treatment outcome and prognostic factors of adult glioblastoma multiforme. J Egypt Natl Canc Inst. (2013) 25:21–30. doi: 10.1016/j.jnci.2012.11.001
81. Barakat MK, Belal AM, Fadel SH, Gamal H. Outcome of high grade gliomas in limited resource country (10 years'experience in Alexandria University Oncology Center 2003-2012). J Brain Tumors Neurooncol. (2016) 1:1–9. doi: 10.4172/2475-3203.1000111
82. Stensjoen AL, Solheim O, Kvistad KA, Haberg AK, Salvesen O, Berntsen EM. Growth dynamics of untreated glioblastomas in vivo. Neuro Oncol. (2015) 17:1402–11. doi: 10.1093/neuonc/nov029
83. Lv S, Teugels E, Sadones J, Quartier E, Huylebrouck M, S DUF, et al. Correlation between IDH1 gene mutation status and survival of patients treated for recurrent glioma. Anticancer Res. (2011) 31:4457–63.
84. Ma Z, Niu B, Phan TA, Stensjoen AL, Ene C, Woodiwiss T, et al. Stochastic growth pattern of untreated human glioblastomas predicts the survival time for patients. Sci Rep. (2020) 10:6642. doi: 10.1038/s41598-020-63394-w
85. Sreenivasan SA, Madhugiri VS, Sasidharan GM, Kumar RV. Measuring glioma volumes: a comparison of linear measurement based formulae with the manual image segmentation technique. J Cancer Res Ther. (2016) 12:161–8. doi: 10.4103/0973-1482.153999
86. Lasocki A, Gaillard F, Tacey M, Drummond K, Stuckey S. Morphologic patterns of noncontrast-enhancing tumor in glioblastoma correlate with IDH1 mutation status and patient survival. J Clin Neurosci. (2018) 47:168–73. doi: 10.1016/j.jocn.2017.09.007
87. Rahman M, Kresak J, Yang C, Huang J, Hiser W, Kubilis P, et al. Analysis of immunobiologic markers in primary and recurrent glioblastoma. J Neurooncol. (2018) 137:249–57. doi: 10.1007/s11060-017-2732-1
88. Champ CE, Klement RJ. Commentary on “Strong adverse prognostic impact of hyperglycemic episodes during adjuvant chemoradiotherapy of glioblastoma multiforme”. Strahlenther Onkol. (2015) 191:281–2. doi: 10.1007/s00066-014-0788-9
89. Decker M, Sacks P, Abbatematteo J, De Leo E, Brennan M, Rahman M. The effects of hyperglycemia on outcomes in surgical high-grade glioma patients. Clin Neurol Neurosurg. (2019) 179:9–13. doi: 10.1016/j.clineuro.2019.02.011
90. Strowd RE, Cervenka MC, Henry BJ, Kossoff EH, Hartman AL, Blakeley JO. Glycemic modulation in neuro-oncology: experience and future directions using a modified Atkins diet for high-grade brain tumors. Neurooncol Pract. (2015) 2:127–36. doi: 10.1093/nop/npv010
91. Link TW, Woodworth GF, Chaichana KL, Grossman SA, Mayer RS, Brem H, et al. Hyperglycemia is independently associated with post-operative function loss in patients with primary eloquent glioblastoma. J Clin Neurosci. (2012) 19:996–1000. doi: 10.1016/j.jocn.2011.09.031
92. McGirt MJ, Chaichana KL, Gathinji M, Attenello F, Than K, Ruiz AJ, et al. Persistent outpatient hyperglycemia is independently associated with decreased survival after primary resection of malignant brain astrocytomas. Neurosurgery. (2008) 63:286–91. doi: 10.1227/01.NEU.0000315282.61035.48
93. Seyfried TN, Sanderson TM, El-Abbadi MM, McGowan R, Mukherjee P. Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br J Cancer. (2003) 89:1375–82. doi: 10.1038/sj.bjc.6601269
94. Derr RL, Ye X, Islas MU, Desideri S, Saudek CD, Grossman SA. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J Clin Oncol. (2009) 27:1082–6. doi: 10.1200/JCO.2008.19.1098
95. Mayer A, Vaupel P, Struss HG, Giese A, Stockinger M, Schmidberger H. Strong adverse prognostic impact of hyperglycemic episodes during adjuvant chemoradiotherapy of glioblastoma multiforme. Strahlenther Onkol. (2014) 190:933–8. doi: 10.1007/s00066-014-0696-z
96. Tieu MT, Lovblom LE, McNamara MG, Mason W, Laperriere N, Millar BA, et al. Impact of glycemia on survival of glioblastoma patients treated with radiation and temozolomide. J Neurooncol. (2015) 124:119–26. doi: 10.1007/s11060-015-1815-0
97. Zhao S, Cai J, Li J, Bao G, Li D, Li Y, et al. Bioinformatic profiling identifies a glucose-related risk signature for the malignancy of glioma and the survival of patients. Mol Neurobiol. (2016) 54:8203–10. doi: 10.1007/s12035-016-0314-4
98. Yu M, Chen S, Hong W, Gu Y, Huang B, Lin Y, et al. Prognostic role of glycolysis for cancer outcome: evidence from 86 studies. J Cancer Res Clin Oncol. (2019) 145:967–99. doi: 10.1007/s00432-019-02847-w
99. Seyfried TN, Yu G, Maroon JC, D'Agostino DP. Press-pulse: a novel therapeutic strategy for the metabolic management of cancer. Nutr Metab. (2017) 14:19. doi: 10.1186/s12986-017-0178-2
100. Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. (2016) 7:33440–50. doi: 10.18632/oncotarget.7961
101. Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, et al. M. G. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell. (2013) 155:397–409. doi: 10.1016/j.cell.2013.09.025
102. Marsh J, Mukherjee P, Seyfried TN. Akt-dependent proapoptotic effects of dietary restriction on late-stage management of a phosphatase and tensin homologue/tuberous sclerosis complex 2-deficient mouse astrocytoma. Clin Cancer Res. (2008) 14:7751–62. doi: 10.1158/1078-0432.CCR-08-0213
103. Zhang K, Xu P, Sowers JL, Machuca DF, Mirfattah B, Herring J, et al. Proteome analysis of hypoxic glioblastoma cells reveals sequential metabolic adaptation of one-carbon metabolic pathways. Mol Cell Proteomics. (2017) 16:1906–21. doi: 10.1074/mcp.RA117.000154
104. Jiang YS, Wang FR. Caloric restriction reduces edema and prolongs survival in a mouse glioma model. J Neuro-Oncol. (2013) 114:25–32. doi: 10.1007/s11060-013-1154-y
105. Woolf EC, Syed N, Scheck AC. Tumor metabolism, the ketogenic diet and beta-hydroxybutyrate: novel approaches to adjuvant brain tumor therapy. Front Mol Neurosci. (2016) 9:122. doi: 10.3389/fnmol.2016.00122
106. Artzi M, Liberman G, Vaisman N, Bokstein F, Vitinshtein F, Aizenstein O, et al. D. Changes in cerebral metabolism during ketogenic diet in patients with primary brain tumors: 1H-MRS study. J Neurooncol. (2017) 132:267–75. doi: 10.1007/s11060-016-2364-x
107. Champ CE, Palmer JD, Volek JS, Werner-Wasik M, Andrews DW, Evans JJ, et al. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J Neuro-Oncol. (2014) 117:125–31. doi: 10.1007/s11060-014-1362-0
108. Schwartz K, Chang HT, Nikolai M, Pernicone J, Rhee S, Olson K, et al. Treatment of glioma patients with ketogenic diets: report of two cases treated with an IRB-approved energy-restricted ketogenic diet protocol and review of the literature. Cancer Metab. (2015) 3:3. doi: 10.1186/s40170-015-0129-1
109. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. (2010) 463:364–8. doi: 10.1038/nature08697
110. Mukherjee J, Phillips JJ, Zheng S, Wiencke J, Ronen SM, Pieper RO. Pyruvate kinase M2 expression, but not pyruvate kinase activity, is up-regulated in a grade-specific manner in human glioma. PLoS ONE. (2013) 8:e57610. doi: 10.1371/journal.pone.0057610
111. Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. (2014) 53:75–87. doi: 10.1016/j.molcel.2013.11.001
112. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
113. Chesnelong C, Chaumeil MM, Blough MD, Al-Najjar M, Stechishin OD, Chan JA, et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. (2014) 16:686–95. doi: 10.1093/neuonc/not243
114. Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C, et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med. (2015) 7:274ra217. doi: 10.1126/scitranslmed.aaa1009
115. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. (2009) 462:739–44. doi: 10.1038/nature08617
116. Liu FM, Gao YF, Kong Y, Guan Y, Zhang J, Li SH, et al. The diagnostic value of lower glucose consumption for IDH1 mutated gliomas on FDG-PET. BMC Cancer. (2021) 21:83. doi: 10.1186/s12885-021-07797-6
117. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
118. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Critical Rev Oncogenesis. (2013) 18:43–73. doi: 10.1615/CritRevOncog.v18.i1-2.40
119. Huysentruyt LC, Akgoc Z, Seyfried TN. Hypothesis: are neoplastic macrophages/microglia present in glioblastoma multiforme? ASN Neuro. (2011) 3:e00064. doi: 10.1042/AN20110011
120. Rahimi Koshkaki H, Minasi S, Ugolini A, Trevisi G, Napoletano C, Zizzari IG, et al. Immunohistochemical characterization of immune infiltrate in tumor microenvironment of glioblastoma. J Pers Med. (2020) 10:112. doi: 10.3390/jpm10030112
121. Garvin S, Oda H, Arnesson LG, Lindstrom A, Shabo I. Tumor cell expression of CD163 is associated to postoperative radiotherapy and poor prognosis in patients with breast cancer treated with breast-conserving surgery. J Cancer Res Clin Oncol. (2018) 144:1253–63. doi: 10.1007/s00432-018-2646-0
122. Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Per Med. (2011) 8:161–73. doi: 10.2217/pme.11.7
123. Denny CA, Heinecke KA, Kim YP, Baek RC, Loh KS, Butters TD, et al. Restricted ketogenic diet enhances the therapeutic action of N-butyldeoxynojirimycin towards brain GM2 accumulation in adult Sandhoff disease mice. J Neurochem. (2010) 113:1525–35. doi: 10.1111/j.1471-4159.2010.06733.x
Keywords: standard of care, glycolysis, glutaminolysis, fasting, mitochondrial substrate level phosphorylation (mSLP), 2-hydroxyglutarate, carnivore diet
Citation: Seyfried TN, Shivane AG, Kalamian M, Maroon JC, Mukherjee P and Zuccoli G (2021) Ketogenic Metabolic Therapy, Without Chemo or Radiation, for the Long-Term Management of IDH1-Mutant Glioblastoma: An 80-Month Follow-Up Case Report. Front. Nutr. 8:682243. doi: 10.3389/fnut.2021.682243
Received: 18 March 2021; Accepted: 07 May 2021;
Published: 31 May 2021.
Edited by:
Kathryn Knecht, Loma Linda University, United StatesReviewed by:
Rainer Johannes Klement, Leopoldina Hospital, GermanyCopyright © 2021 Seyfried, Shivane, Kalamian, Maroon, Mukherjee and Zuccoli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas N. Seyfried, VGhvbWFzLnNleWZyaWVkQGJjLmVkdQ==; Giulio Zuccoli, Z2l1bGlvLnp1Y2NvbGlAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.