 Domenico Sergi
Domenico Sergi Natalie Luscombe-Marsh3
Natalie Luscombe-Marsh3 Nenad Naumovski
Nenad Naumovski Mahinda Abeywardena
Mahinda Abeywardena Nathan O'Callaghan
Nathan O'Callaghan- 1Nutrition and Health Program, Health and Biosecurity, Commonwealth Scientific and Industrial Research Organisation, Adelaide, SA, Australia
- 2Adelaide Medical School, The University of Adelaide, Adelaide, SA, Australia
- 3Diabetes SA, Adelaide, SA, Australia
- 4Faculty of Health, University of Canberra, Canberra, ACT, Australia
- 5Functional Foods and Nutrition Research Laboratory, University of Canberra, Bruce, ACT, Australia
The chain length of saturated fatty acids may dictate their impact on inflammation and mitochondrial dysfunction, two pivotal players in the pathogenesis of insulin resistance. However, these paradigms have only been investigated in animal models and cell lines so far. Thus, the aim of this study was to compare the effect of palmitic (PA) (16:0) and lauric (LA) (12:0) acid on human primary myotubes mitochondrial health and metabolic inflammation. Human primary myotubes were challenged with either PA or LA (500 μM). After 24 h, the expression of interleukin 6 (IL-6) was assessed by quantitative polymerase chain reaction (PCR), whereas Western blot was used to quantify the abundance of the inhibitor of nuclear factor κB (IκBα), electron transport chain complex proteins and mitofusin-2 (MFN-2). Mitochondrial membrane potential and dynamics were evaluated using tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and immunocytochemistry, respectively. PA, contrarily to LA, triggered an inflammatory response marked by the upregulation of IL-6 mRNA (11-fold; P < 0.01) and a decrease in IκBα (32%; P < 0.05). Furthermore, whereas PA and LA did not differently modulate the levels of mitochondrial electron transport chain complex proteins, PA induced mitochondrial fragmentation (37%; P < 0.001), decreased MFN-2 (38%; P < 0.05), and caused a drop in mitochondrial membrane potential (11%; P < 0.01) compared to control, with this effect being absent in LA-treated cells. Thus, LA, as opposed to PA, did not trigger pathogenetic mechanisms proposed to be linked with insulin resistance and therefore represents a healthier saturated fatty acid choice to potentially preserve skeletal muscle metabolic health.
Introduction
There is an exponential increase in the incidence of obesity and type 2 diabetes mellitus (T2DM) in the developed and developing countries, (1). Individuals affected by T2DM are at higher risk of developing severe health consequences including cardiovascular disease, nephropathy, and neuropathy (2). Furthermore, this metabolic disorder is also emerging as a risk factor for the development of neurodegenerative diseases including Parkinson and Alzheimer disease (3, 4). T2DM is characterised by chronic hyperglycaemia, which is a direct consequence of insulin resistance and pancreatic β-cell dysfunction. Particularly, insulin resistance is the hallmark of T2DM and develops in the skeletal muscle decades before pancreatic β-cells become dysfunctional and overt hyperglycaemia develops (5). Therefore, skeletal muscle insulin resistance represents a primary defect in the pathogenesis of T2DM. In terms of the mechanisms underpinning the onset of skeletal muscle insulin resistance, mitochondrial dysfunction and metabolic inflammation have been reported to play a pivotal role (6–8). Mitochondrial dysfunction in the face of increased fatty acid availability promotes the buildup of lipotoxic lipid species, such as ceramides, which disrupt insulin signalling pathway by activating PKC isoform protein kinase Cζ and protein phosphatase 2A, which in turn directly hamper AKT (protein kinase B) activation (9). With regard to metabolic inflammation, the activation of proinflammatory nuclear factor κ light-chain enhancer of activated B cells (NFκB) signalling and c-Jun N-terminal kinases (JNKs) have been reported to impede insulin signal transduction pathway, underpinned by the serine phosphorylation of insulin receptor substrate (10, 11).
Mitochondria bioenergetics and inflammation, apart from being involved in skeletal muscle insulin resistance, are also interrelated processes, with metabolic inflammation being able to impair mitochondrial function and mitochondria themselves, contributing to the activation of intracellular proinflammatory responses (12, 13). Particularly, the activation of the proinflammatory NFκB signalling in response to nutrient overload has been reported to promote mitochondrial dysfunction with a shift in mitochondrial dynamics towards fission (12). Nonetheless, mitochondrial dysfunction may also trigger the activation of proinflammatory pathways by promoting the accumulation of lipotoxic lipid intermediates (14, 15) or via the release of damage-associated molecular patterns such as mitochondrial DNA or increase production of reactive oxygen species (13).
Circulating nutrient excess and particularly the overconsumption of long-chain saturated fatty acids have been reported to trigger metabolic inflammation in a variety of tissues (15–19), as well as mitochondrial dysfunction marked by mitochondrial fission, decreased oxidative capacity, and increased production of reactive species (8, 20–22). These effects have been well-described for palmitic acid (PA), with this long-chain saturated fatty acid being reported to induce mitochondrial dysfunction and metabolic inflammation in a variety of tissues both in the central nervous system and the periphery (12, 23–27). The induction of these pathogenetic mechanisms culminates with the onset of insulin resistance and impaired metabolic health (24, 28). However, not all the saturated fatty acids have been reported to be metabolically detrimental. Indeed, medium-chain saturated fatty acids, such as lauric acid (LA), have been shown not to promote insulin resistance, to be less obesogenic, and to decrease the ratio of total cholesterol to high-density lipoprotein cholesterol compared to longer chain saturated fatty acids (29–32). LA has been reported to be less metabolically detrimental and proinflammatory compared to PA (33). Additionally, LA, as opposed to PA, is β-oxidised more effectively, which, in turn, may prevent the deleterious effects linked with intramyocellular lipid accumulation and lipotoxicity (34). Despite accumulating evidence on the metabolic effect of medium-chain saturated fatty acids on metabolic health, their effect on human skeletal muscle and particularly their ability to modulate inflammatory responses and mitochondrial health remain to be elucidated. Indeed, the studies conducted to date, to our knowledge, reported on the effect of medium-chain fatty acids on mitochondrial function in rodents and cell lines (35, 36); there is therefore a lack of data on human tissues. Furthermore, the activation of proinflammatory NFκB signalling has been shown to trigger mitochondrial fragmentation (12); therefore, fatty acids differing for their ability to trigger the activation of this signalling pathway should also differently affect mitochondrial health.
Thus, in consideration of the lack of evidence in experimental models closely mimicking human physiology and considering the putatively less harmful metabolic effect of LA compared to PA, the aim of the present study was to compare the effect of PA and LA on key mechanisms governing in metabolic health. Particularly, we aimed at investigating the effects of these long- and medium-chain saturated fatty acids on mitochondrial dynamics, mitochondrial electron transport chain complex proteins, mitochondrial membrane potential, and metabolic inflammation in human primary myotubes.
Materials and Methods
Human Primary Myoblasts and Reagents
Human primary myoblasts from four different donors were obtained from Cook Myosite (USA). Dulbecco modified eagle low glucose medium (DMEM) containing 5.5 mM glucose, horse serum (HS), heat-inactivated foetal bovine serum (FBS), penicillin–streptomycin solution (10,000 U/ml), Pierce™ BCA protein assay kit, TaqMan assays, TaqMan fast advanced master mix, and rabbit anti–mouse immunoglobulin G (IgG) secondary antibody (Alexa Fluor 488) were from Life Technologies Australia Pty. Ltd. (Mulgrave, Victoria, Australia). The primary antibodies, anti-inhibitor of nuclear factor κB (IκBα), anti–MFN-2, anti–β-actin, and the secondary antibodies, anti–rabbit IgG horseradish peroxidase (HRP)–linked antibody and anti–mouse IgG HRP-linked antibody, were obtained from Cell Signalling Technology. JC-1 mitochondrial membrane potential assay kit, total OXPHOS rodent antibody cocktail, and anti-TOMM20 were from Abcam Australia Pty. Ltd. (Melbourne, Victoria, Australia). PA and LA, as well as low-endotoxin fatty acid–free bovine serum albumin (BSA), were purchased from Sigma–Aldrich (Castle Hill, New South Wales, Australia). Finally, Trans-Blot Turbo Midi 0.2 μm polyvinylidene fluoride (PVDF) Transfer Packs and 4 to 15% Criterion TGX Precast Midi Protein Gel were obtained from Bio-Rad (Gladesville, New South Wales, Australia).
Cell Cultures and Fatty Acid Treatments
Human primary myoblasts derived from the abdominal rectus muscles of four non-type 2 diabetic, non-obese male individuals 31.00 ± 5.67 years of age, body mass index 24.75 ± 1.31 kg/m2 (Human skMDC; Cook Myosite) were maintained in DMEM (low glucose) containing 20% FBS and 100 U/ml penicillin–streptomycin at 37°C under a 5% CO2 atmosphere as previously reported (37). Cells were grown in 25-cm2 flasks, 6-, 24-, or 96-well plates for the assessment gene expression, protein expression by Western blot, immunocytochemistry, or assessment of mitochondrial membrane potential, respectively. Myoblast differentiation was initiated when cells reached 75 to 80% confluence by incubating the cells for 7 days in DMEM containing 2% HS and 100 U/ml penicillin–streptomycin. After 7 days, cells were treated with either low-endotoxin fatty acid–free BSA, 500 μM PA, or 500 μm LA for 24 h in differentiation media, and then samples collected for gene or protein expression or directly assayed for changes in mitochondrial membrane potential and morphology. The concentration of PA used in the present study falls within the circulating physiological range, as previously reported (38). Hence, to compare the effect of PA and LA, these fatty acids were used at the same concentrations to allow for an equimolar comparison.
Fatty Acid Conjugation to BSA
Fatty acids are fat-soluble molecules and therefore insoluble in the cell culture media in their native form; furthermore, free fatty acids travel in the bloodstream bound to albumin. Thus, to allow for fatty acid solubilisation in the cell culture media and mimic fatty acid physiological circulating conditions, both PA and LA were conjugated to BSA as described previously (15). Low-endotoxin BSA was used to prevent potential lipopolysaccharide contamination as reported previously (39). Fatty acids were dissolved in 0.1 M NaOH in a water bath at 70°C to yield a final concentration of 20 mM. BSA was solubilised in serum and penicillin-streptomycin–free DMEM at 55°C and fatty acid mixed with the fatty acid solutions to obtain a 1:4 molar ratio (fatty acids 2 mM: BSA 0.5 mM). The fatty acid–BSA mix was vortexed and then incubated for 10 min at 55°C and cooled to room temperature before being filter sterilised. Conjugated fatty acids were stored at −20°C before use.
RNA Extraction, cDNA Synthesis, and Assessment of Gene Expression by Real-Time PCR–PCR
After the fatty acid treatments, cells were directly lysed in the 25-cm2 flasks using RLT lysis buffer (Qiagen, Chadstone, Victoria, Australia), cell lysates were collected, and total RNA extracted using RNeasy Mini Kit (Qiagen) according to the manufacturer instructions. cDNA was synthesised starting from 0.5 μg of total RNA as a template and using SuperScript II (Life Technologies Australia Pty. Ltd.) as described previously (40). Real-time PCR to assess the expression of IL-6 and ribosomal protein S18 was carried out using CFX Connect 96 real-time PCR detection system (Bio-Rad) employing a two-step cycling program of 95°C for 20 s, and then 40 cycles at 95°C for 3 s and 60°C for 30 s. Each reaction contained 1 μl of cDNA template, 5 μl of TaqMan fast advanced master mix, 3.5 μl of diethylpyrocarbonate-treated water, and 0.5 μl of either of the following Taqman assays: ribosomal protein S18 Hs01375212_g1 and IL-6: Hs00174131_m1. Ct values were normalised to the reference gene ribosomal protein S18 and data analysed using the comparative ΔCt method (41).
Western Blot
Following 24-h incubation with BSA or fatty acids, cells were washed with phosphate-buffered saline (PBS) and directly lysed with RIPA buffer (Sigma–Aldrich) containing Halt Protease and Phosphatase Inhibitor Cocktail (Life Technologies Australia Pty. Ltd.). Cell lysate proteins were quantified using Pierce™ BCA protein assay kit and 6 μg of protein loaded and resolved on a 4 to 15% precast polyacrylamide gel. Proteins were then transferred onto PVDF membranes using a Trans-Blot Turbo Transfer System (Bio-Rad). Non-specific binding sites were blocked by incubating the membranes for 1 h at room temperature with a 5% BSA solution made up in PBS. Membranes were then probed overnight with total OXPHOS rodent antibody cocktail, anti-IκBα, anti–MFN-2, and anti–β-actin followed by three 5-min washes with PBS containing 0.1% Tween-20 (PBST). After the washes, membranes were incubated with HRP-linked secondary antibodies as appropriate. Membranes were finally washed three times for 5 min in PBST before being revealed using Western lightning ECL Pro (PerkinElmer). Western blot bands intensities were quantified by densitometric analysis using NIH ImageJ software (NIH, USA).
Immunocytochemistry and Assessment of Mitochondrial Morphology
Cells were grown and differentiated onto sterile glass coverslips placed in 24-well plates. After differentiation, myotubes were challenged with either BSA, PA, or LA followed by fixation with 4% formaldehyde for 20 min at room temperature. Cells were then washed with PBS and permeabilised using a 0.5% triton X PBS solution for 15 min followed by incubation with 2% BSA dissolved in PBS containing 0.25% Triton X (blocking solution) in order to block non-specific bindings. After permeabilisation and blocking, cells were incubated for 2 h at room temperature with anti-TOMM20 diluted 1:1,000 in permeabilisation solution and then washed with PBS before being incubated with rabbit anti–mouse IgG secondary antibody (Alexa Fluor 488) for 1 h at room temperature in the dark. Incubation with secondary antibody was followed by three washes with PBS. Finally, coverslips were mounted on slides using fluoroshield with DAPI to counterstain the nuclei. Cells were imaged using the EVOS imaging system (Thermo Fisher Scientific, Bothell, WA, USA). Mitochondria morphology was evaluated using the ImageJ macro developed by Dagda et al. (42), as described previously (43). The circularity score was used to define mitochondria morphology, with an increase in the score indicating a higher degree of fragmentation.
Measurement of Mitochondrial Membrane Potential
JC-1 mitochondrial membrane potential assay kit (Abcam Australia Pty. Ltd.) was used to measure mitochondrial membrane potential. JC-1 is a cationic fluorescent dye that accumulates within the mitochondria in a membrane potential-dependent manner. After 24-h exposure to either BSA, PA, or LA, JC-1 was directly administered directly to cell culture media at a final concentration of 10 μM and cells incubated for 10 min at 37°C in the dark. Cells were then washed three times with PBS and JC-1 red fluorescence recorded at 550/615 (excitation/emission) and green fluorescence at 489/535 (excitation/emission) using Victor3 V 1420 Multilabel Counter (PerkinElmer). Finally, the ratio of red to green fluorescence was calculated to quantify mitochondrial membrane potential. The green fluorescence quantified at 489/535 (excitation/emission) is directly proportional to mitochondrial membrane potential, whereas red fluorescence, detected at 550/615 (excitation/emission), represents JC-1 aggregates that accumulate within the mitochondria when membrane potential exceeds −240 mV (44). In light of this, calculating the ratio between red vs. green fluorescence allows the quantification of mitochondrial membrane potential independently of the number of mitochondria assessed. After fluorescence quantification, cells were imaged using the EVOS imaging system to obtain representative images (Thermo Fisher Scientific).
Statistical Analysis
Data are expressed as mean ± SEM and represent the experiments conducted on cells derived from four independent individuals (n = 4) and analysed at least in duplicate as reported in figure legends. Difference between treatments was assessed using repeated-measures one-way analysis of variance followed by Tukey post hoc test using GraphPad Prism 8 for Windows. p < 0.05 was considered statistically significant.
Results
PA, but Not LA, Triggers an Inflammatory Response in Human Primary Myotubes
Metabolic inflammation and particularly the activation of the NFκB signalling pathway was investigated in order to assess the difference between PA and LA in their ability to trigger an inflammatory response. PA decreased the abundance of IκBα (p < 0.05) (Figures 1A,B), a protein that forms a stable complex with NFκB and inhibits its transcriptional activity. However, upon activation of the proinflammatory NFκB signalling, IκBα is phosphorylated by IκB kinase (IKK) and channelled towards proteasomal degradation, thus allowing NFκB to migrate to the nucleus and induce the expression of its gene targets (45). In light of this, PA-induced decrease in IκBα marks the activation of the NFκB signalling pathway, which was further confirmed by the upregulation of IL-6, a NFκB target gene (46), compared to BSA and LA-treated cells (p < 0.01) (Figure 1C). However, despite LA also being a saturated fatty acid, it did not decrease IκBα relative to BSA (p > 0.05) while tended to induce its abundance compared to PA (p = 0.094). Furthermore, LA did not increase IL-6 expression compared to BSA-treated cells (Figure 1C).
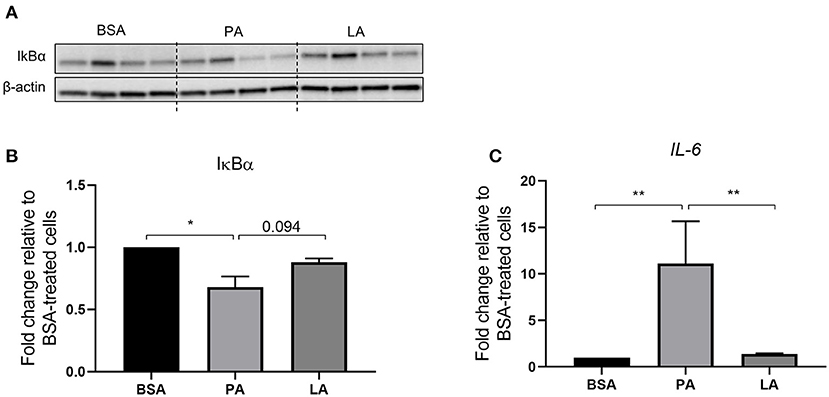
Figure 1. Activation of the IKKβ-NFκB signalling pathway in human primary myotubes in response to saturated fatty acids. (A) Representative Western blot of cellular IκBα abundance, (B) densitometric analysis of IκBα normalised to β-actin, and (C) gene expression analysis of IL-6 by quantitative PCR. Data are reported as means ± SEM, n = 4 cultures from independent donors (means of two replicates). *P < 0.05, **P < 0.01.
Both PA and LA Modulate the Abundance of Mitochondrial Electron Transport Chain Proteins
Mitochondria are key organelles for the catabolism of fatty acids; therefore, the relative abundance of mitochondrial electron transport chain complex proteins was assessed to evaluate whether PA and LA differently modulate their expression. Both PA and LA tended to induce CV alpha subunit (complex V) (p = 0.052 and p = 0.058, respectively) (Figures 2A,B) and upregulated CIII-Core protein 2 (complex III) (p < 0.05) (Figures 2A,C). Furthermore, while PA tended to upregulate CIV subunit I (complex IV) (p = 0.078) compared to BSA (Figures 2A,D), LA did not affect the abundance of this protein (Figures 2A,D). Finally, neither PA nor LA induced the abundance of CII-30 kDa (complex 2) (Figures 2A,E), nor CI subunit NDUFB8 (complex I) (Figures 2A,F).
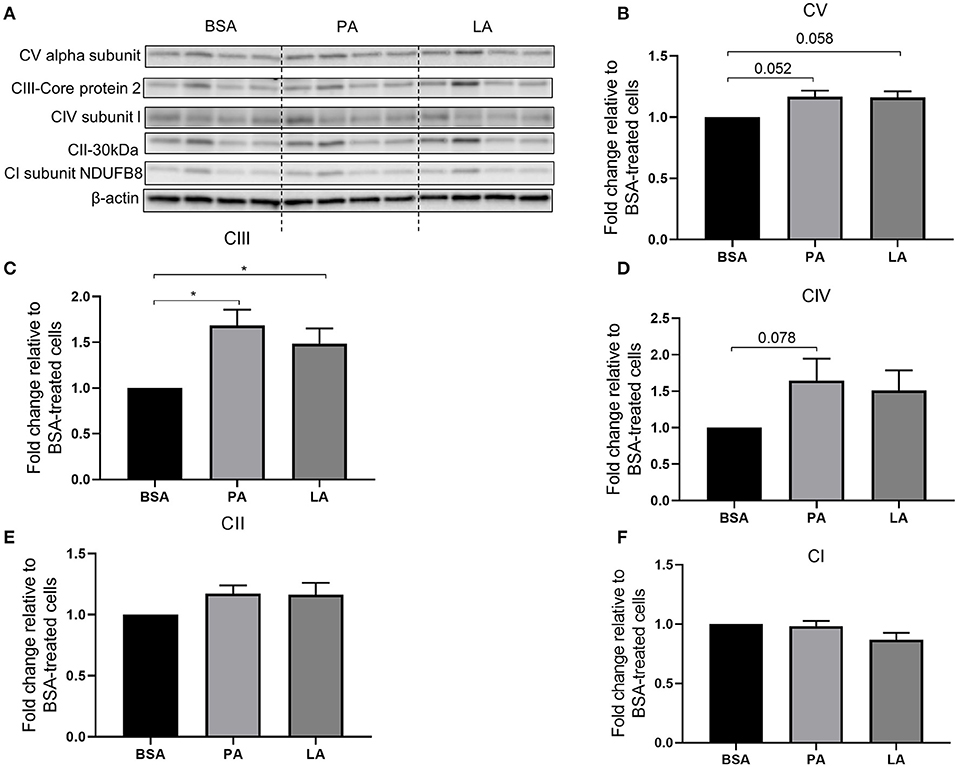
Figure 2. Relative abundance of electron transport chain complex proteins in response to palmitic or lauric acid. (A) Representative Western blot of CV alpha subunit (complex V), CIII-Core protein 2 (complex III), CIV subunit I (complex IV), CII-30kDa (complex II), CI subunit NDUFB8 (complex I). Densitometric analysis normalised to β-actin of CV alpha subunit (complex V) (B); CIII-Core protein 2 (complex III) (C); CIV subunit I (complex IV) (D); CII-30kDa (complex II) (E); CI subunit NDUFB8 (complex I) (F). Data are reported as means ± SEM, n = 4 cultures from independent donors (means of two replicates). *P < 0.05.
PA Promotes Mitochondrial Fragmentation
Mitochondria go through cycles of fusion and fission, termed mitochondrial dynamics. Mitochondrial dynamics represents a key modulator of mitochondrial oxidative capacity, allowing the cells to adapt to short-term changes in fuel availability (47). PA induced an increase in mitochondrial fragmentation as indicated by a drop in the circularity score compared to BSA-treated cells (p < 0.001) (Figures 3A,B,D). However, LA, despite being a saturated fatty acid, did not induce mitochondria fragmentation (Figures 3C,D), with LA-treated myotubes maintaining a tubular mitochondrial network comparable to cells challenged with PA (Figure 3C). MFN-2 is a key protein responsible for promoting mitochondrial fusion (48) with its overexpression ameliorating PA-induced insulin resistance (49). Therefore, to further confirm the effect of PA on mitochondria dynamics, its expression was assessed in response to fatty acid challenges. While PA downregulated MFN-2 compared to myotubes treated with BSA (p < 0.05) (Figures 3E,F), the same effect was not elicited by LA (Figures 3E,F), confirming that PA and LA differently regulate mitochondrial dynamics, with PA promoting a shift towards fission.
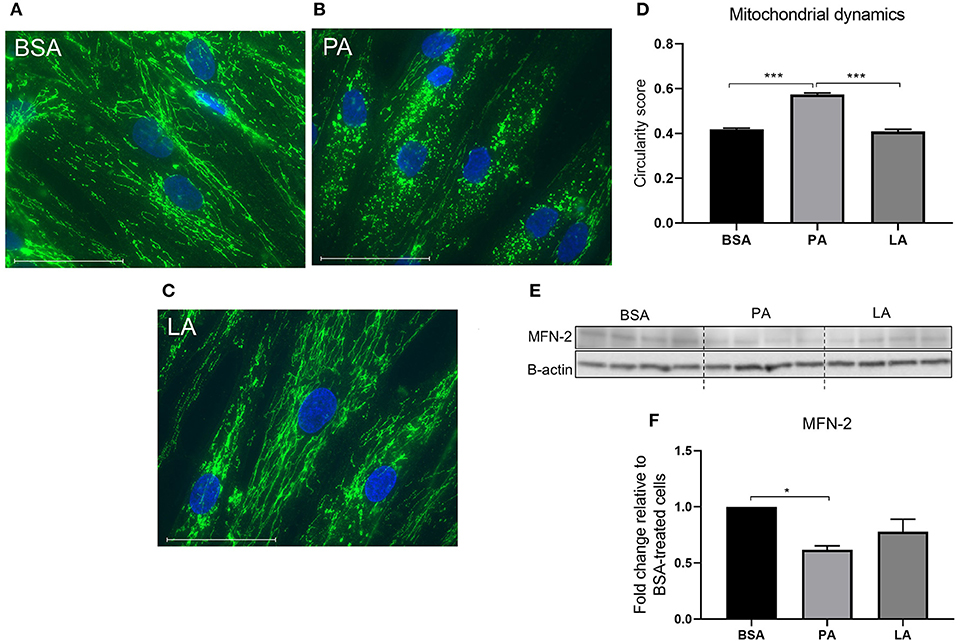
Figure 3. The effect of palmitic and lauric acid on mitochondrial dynamics. Myotubes were immunostained with anti-TOMM20 and fluorescence detected using the EVOS imaging system following 24 incubation with BSA (A), PA (B), and LA (C). Nuclei were counterstained with DAPI (blue). Scale bar = 10 μm. (B) PA induced mitochondrial fragmentation. (D) Image analysis of fluorescence-labelled mitochondria, values represent mitochondrial circularity score, with a higher score indicating increased mitochondrial circularity and fragmentation. (E) Representative Western blot reflecting the abundance of MFN-2. (F) Densitometric analysis of MFN-2 normalised to β-actin. Data are reported as means ± SEM, n = 4 cultures from independent donors. Image analysis was performed on 10 different fields per treatment on each independent cell line. Western blot data are the means of three replicates. *P < 0.05, ***P < 0.001.
PA and LA Differently Regulate Mitochondrial Membrane Potential
Considering mitochondrial dynamics being closely related to function (47), mitochondrial membrane potential was assessed to evaluate whether PA-induced mitochondrial fragmentation also resulted in a decrease in mitochondrial function. In agreement with its effect on mitochondrial fragmentation and downregulation of MFN-2, PA also caused a drop in mitochondrial membrane potential relative to myotubes treated with both BSA and LA (p < 0.01) (Figures 4A–D). Contrarily to PA, however, LA did not induce a drop in mitochondrial membrane potential relative to cells exposed to BSA alone (Figures 4A,B,D), indicating that the carbon-chain length, and not only the presence of double bonds, also plays an important role in dictating the impact of fatty acids on mitochondrial function.
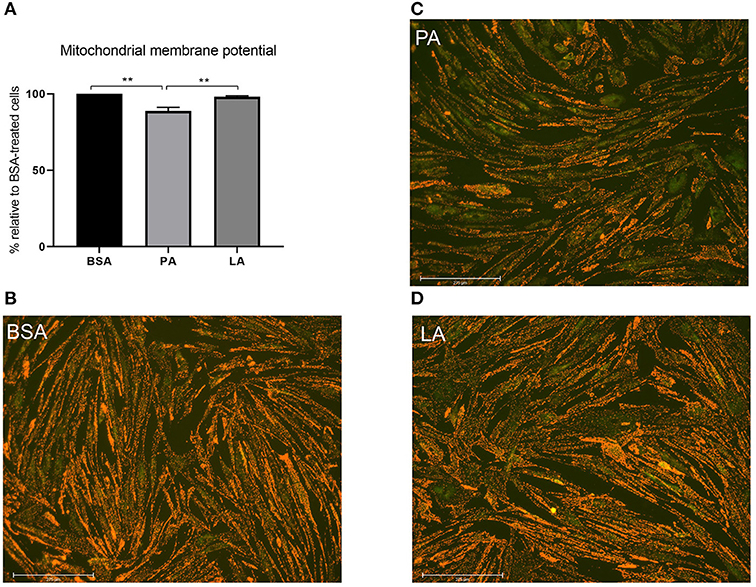
Figure 4. The impact of palmitic and lauric acid on mitochondrial membrane potential. (A) Mitochondrial membrane potential calculated as red to green JC-1 fluorescence ratio. Representative images of JC-1–derived red and green fluorescence of myotubes treated with BSA (B), PA (C), and LA (D). Scale bar = 275 μM. Data are reported as means ± SEM, n = 4 cultures from independent donors (means of three replicates). **P < 0.01.
Discussion
The results of this study highlight the contrasting effects of PA and LA on metabolic inflammation, mitochondrial dynamics, and function in human primary myotubes, providing mechanistic insights on the effects of long- vs. medium-chain saturated fatty acids on metabolic health (31, 32) and particularly on insulin sensitivity (29, 50).
Metabolic inflammation, defined as a sterile inflammatory response (51) and typical of obesity and the metabolic syndrome (52), has been widely described for its ability to induce insulin resistance in metabolically active tissues (6, 51, 53, 54). Thus, the inability of LA to trigger an inflammatory response as opposed to PA may, at least in part, explain the different metabolic effects of medium- compared to long-chain saturated fatty acids (35, 55, 56). Particularly, the metabolic fate of PA and LA may be held responsible for their contrasting effects on inflammation. Indeed, LA compared to PA can directly enter the mitochondria for β-oxidation independently of carnitine palmitoyl transferase-dependent conjugation with carnitine (57), which therefore facilitate its catabolism. Increased fatty acid catabolism prevents the accumulation of lipotoxic lipid species, such as ceramides and diacylglycerols (32), which, in turn, has been shown to be, at least in part, responsible for long-chain saturated fatty acid-induced inflammation (14, 15). Furthermore, this confirms the inability of LA to trigger the activation of NFκB via the Toll-like receptor 4 (39).
Interestingly, PA and LA modulate the abundance of the mitochondrial electron transport chain complex proteins, suggesting that both fatty acids trigger adaptive responses aimed at increasing fatty acid catabolism. This notion, in particular, is supported by the induction of CIII-Core protein 2 (complex III). Indeed, there is a close physical and functional interaction between the mitochondrial electron transport chain supercomplexes and fatty acid oxidation enzymes (58, 59). Particularly, reducing equivalents resulting from the first reaction of the β-oxidation pathway in the mitochondrial matrix, catalysed by acyl-CoA dehydrogenases, are transferred to mitochondrial complex III, which directly interacts with electron flavoprotein dehydrogenase, the enzyme responsible for the oxidation of electron transfer flavoprotein (60). Thus, the induction of mitochondrial complex III in response to PA and LA challenge suggests myotubes mounting a response to cope with increased fatty acid availability. Nonetheless, despite promoting similar changes with regard to mitochondrial electron transport chain complex proteins, PA and LA differently impact upon mitochondrial dynamics with PA, contrarily to LA, downregulating MTF-2 and promoting mitochondria fragmentation. Importantly, a shift in mitochondrial dynamics towards fission is associated with a decrease in mitochondrial oxidative capacity (20) and therefore a compromised ability to effectively catabolise PA in excess. On the contrary, LA did not negatively impact mitochondrial dynamics maintaining a fused mitochondrial network, which, in turn, has been reported to promote fatty acid utilisation (21). This supports the notion that LA may be β-oxidised more effectively compared to PA (34), thereby preventing lipotoxicity and the downstream effects on skeletal muscle metabolic health. However, despite this being supported by previous evidence (34), it remains to be fully investigated by directly quantifying fatty acid β-oxidation (61) or tracking fatty acid metabolic fate by lipidomics (62). Furthermore, whereas LA did not impair mitochondrial membrane potential, exposure of myotubes to PA induced a drop in mitochondrial membrane potential, which further confirms that LA, compared to PA, preserved mitochondrial oxidative capacity as already reported in C2C12 myotubes (35). Nevertheless, despite assessing mitochondrial dynamics and mitochondrial membrane potential, which represent two key discriminants of mitochondrial function (20, 44, 47), a limitation of the present study is related to the fact that we did not directly assess mitochondrial oxidative capacity or oxygen consumption. Thus, the direct impact of PA and LA on mitochondrial oxidative capacity remains to be fully elucidated in human skeletal muscle. However, our data suggest that PA and LA differently regulate mitochondrial oxidative capacity, as demonstrated by mitochondrial fragmentation and the drop in mitochondrial membrane potential induced by PA.
The activation of the proinflammatory NFκB signalling in response to nutrient overload, including an excess of PA, has been recently reported to promote mitochondrial dysfunction, also underlaid by an increase in mitochondrial fission and a significant decrease in respiratory capacity (12). The same effect was reported herein, with PA inducing the activation of NFκB, which was paralleled by an increase in mitochondrial fragmentation and a drop in mitochondrial membrane potential, an effect completely absent in myotubes challenged with LA, albeit fatty acids being used at equimolar concentrations. Despite being tempting to blame metabolic inflammation for the effects of PA on mitochondria, the fact that metabolic inflammation may be a direct consequence of mitochondrial dysfunction rather than a cause must not be overlooked. Indeed, mitochondrial dysfunction in the face of increased PA availability favours the accumulation of ceramides, which, as already described, promote inflammation in metabolically active tissues (14). LA instead failed to trigger the activation of NFκB, which on the one hand may be responsible for the inability of this fatty acid to compromise mitochondrial health, but on the other may depend on LA failing to promote mitochondrial dysfunction and lipotoxicity (63, 64). Independently on whether mitochondrial fragmentation and the drop in mitochondrial membrane potential are a consequence or a cause of metabolic inflammation, these are both pathophysiological mechanisms associated with skeletal muscle insulin resistance, which, however, was not directly assessed in the experimental model used as part of this study. Therefore, the ability of PA to trigger these mechanisms, as opposed to LA, further supports the contrasting effects of these fatty acids on metabolic health, previously reported (29–33). Thus, despite PA and LA being both saturated fatty acids, their chain lengths, C12:0 vs. C16:0, respectively, appear to be an important discriminant in dictating the effects of these fatty acids on mitochondrial health as well as inflammation.
In conclusion, this study provides novel insights on the putative mechanisms responsible for the different metabolic outcomes of saturated fatty acids differing for their chain length (29, 30, 55, 65) in a physiologically relevant in vitro model closely mimicking human physiology: human primary myotubes. These findings support the possibility that the prevention of metabolic inflammation and mitochondrial dysfunction may be key mechanisms in preserving skeletal muscle insulin sensitivity in response to diets enriched in medium-chain triglycerides, as already demonstrated in rodents (50). Thus, LA, despite being a saturated fatty acid, did not induce the activation of pathophysiological mechanisms linked with insulin resistance, and therefore, eucaloric substitution of LA for PA may represent a valid strategy to preserve skeletal muscle metabolic health.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon reasonable request.
Author Contributions
DS developed the hypothesis, performed the experiments, analysed results, and wrote the manuscript. DS, NO'C, and MA designed the experiments and coordinated the study. NL-M, NN, MA, and NO'C critically reviewed the manuscript and provided inputs on experimental procedures. All authors read and approved the final version of the manuscript.
Funding
This work was supported by The Commonwealth Scientific and Industrial Research Organisation (CSIRO)'s Precision Health Future Science Platform (FSP). DS was the recipient of a Commonwealth Scientific and Industrial Research Organisation (CSIRO; Australia) Postdoctoral Research Fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cho NH, Shaw JE, Karuranga S, Huang Y, Da Rocha Fernandes JD, Ohlrogge AW, et al. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
2. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. (2005) 54:1615–25. doi: 10.2337/diabetes.54.6.1615
3. Lee HJ, Seo HI, Cha HY, Yang YJ, Kwon SH, Yang SJ. Diabetes and alzheimer's disease: mechanisms and nutritional aspects. Clin Nutr Res. (2018) 7:229–40. doi: 10.7762/cnr.2018.7.4.229
4. Sergi D, Renaud J, Simola N, Martinoli MG. Diabetes, a contemporary risk for parkinson's disease: epidemiological and cellular evidences. Front Aging Neurosci. (2019) 11:302. doi: 10.3389/fnagi.2019.00302
5. Defronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. (2009) 32 (Suppl 2):S157–63. doi: 10.2337/dc09-S302
6. Wei Y, Chen K, Whaley-Connell AT, Stump CS, Ibdah JA, Sowers JR. Skeletal muscle insulin resistance: role of inflammatory cytokines and reactive oxygen species. Am J Physiol Regul Integr Comp Physiol. (2008) 294:R673–80. doi: 10.1152/ajpregu.00561.2007
7. Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connections. (2015) 4:R1–R15. doi: 10.1530/EC-14-0092
8. Sergi D, Naumovski N, Heilbronn LK, Abeywardena M, O'callaghan N, Lionetti L, et al. Mitochondrial (Dys)function and insulin resistance: from pathophysiological molecular mechanisms to the impact of diet. Front Physiol. (2019) 10:532. doi: 10.3389/fphys.2019.00532
9. Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metabolism. (2012) 15:585–94. doi: 10.1016/j.cmet.2012.04.002
10. Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE. (2005) 2005:pe4. doi: 10.1126/stke.2682005pe4
11. Solinas G, Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J. (2010) 24:2596–611. doi: 10.1096/fj.09-151340
12. Nisr RB, Shah DS, Ganley IG, Hundal HS. Proinflammatory NFkB signalling promotes mitochondrial dysfunction in skeletal muscle in response to cellular fuel overloading. Cell Mol Life Sci. (2019) 76:4887–904. doi: 10.1007/s00018-019-03148-8
13. Rocha M, Apostolova N, Diaz-Rua R, Muntane J, Victor VM. Mitochondria and T2D: role of autophagy, ER stress, and inflammasome. Trends Endocrinol Metab. (2020) 31:725–41. doi: 10.1016/j.tem.2020.03.004
14. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. (2016) 57:2099–114. doi: 10.1194/jlr.R066514
15. Sergi D, Morris AC, Kahn DE, Mclean FH, Hay EA, Kubitz P, et al. Palmitic acid triggers inflammatory responses in N42 cultured hypothalamic cells partially via ceramide synthesis but not via TLR4. Nutr Neurosci. (2018) 23:1–14. doi: 10.1080/1028415X.2018.1501533
16. Pillon NJ, Arane K, Bilan PJ, Chiu TT, Klip A. Muscle cells challenged with saturated fatty acids mount an autonomous inflammatory response that activates macrophages. Cell Commun Signal. (2012) 10:30. doi: 10.1186/1478-811X-10-30
17. Palomer X, Pizarro-Delgado J, Barroso E, Vazquez-Carrera M. Palmitic and oleic acid: the yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocr Metab. (2018) 29:178–90. doi: 10.1016/j.tem.2017.11.009
18. Sergi D, Campbell FM, Grant C, Morris AC, Bachmair E-M, Koch C, et al. SerpinA3N is a novel hypothalamic gene upregulated by a high-fat diet and leptin in mice. Genes Nutr. (2018) 13:28. doi: 10.1186/s12263-018-0619-1
19. Sergi D, Williams LM. Potential relationship between dietary long-chain saturated fatty acids and hypothalamic dysfunction in obesity. Nutr Rev. (2020) 78:261–77. doi: 10.1093/nutrit/nuz056
20. Jheng H.-F., Tsai P.-J., Guo S.-M.-H., Chang C.-S., Su I.-J., et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. (2012) 32:309–19. doi: 10.1128/MCB.05603-11
21. Lionetti L, Mollica MP, Donizzetti I, Gifuni G, Sica R, Pignalosa A, et al. High-lard and high-fish-oil diets differ in their effects on function and dynamic behaviour of rat hepatic mitochondria. PLoS ONE. (2014) 9:e92753. doi: 10.1371/journal.pone.0092753
22. Putti R, Migliaccio V, Sica R, Lionetti L. Skeletal muscle mitochondrial bioenergetics and morphology in high fat diet induced obesity and insulin resistance: focus on dietary fat source. Front Physiol. (2015) 6:426. doi: 10.3389/fphys.2015.00426
23. Reynoso R, Salgado LM, Calderon V. High levels of palmitic acid lead to insulin resistance due to changes in the level of phosphorylation of the insulin receptor and insulin receptor substrate-1. Mol Cell Biochem. (2003) 246:155–62. doi: 10.1007/978-1-4615-0298-2_22
24. Shinjo S, Jiang S, Nameta M, Suzuki T, Kanai M, Nomura Y, et al. Disruption of the mitochondria-associated ER membrane (MAM) plays a central role in palmitic acid-induced insulin resistance. Exp Cell Res. (2017) 359:86–93. doi: 10.1016/j.yexcr.2017.08.006
25. Alnahdi A, John A, Raza H. Augmentation of glucotoxicity, oxidative stress, apoptosis and mitochondrial dysfunction in HepG2 cells by palmitic acid. Nutrients. (2019) 11:1979. doi: 10.3390/nu11091979
26. Guo A, Li K, Xiao Q. Fibroblast growth factor 19 alleviates palmitic acid-induced mitochondrial dysfunction and oxidative stress via the AMPK/PGC-1alpha pathway in skeletal muscle. Biochem Biophys Res Commun. (2020) 526:1069–76. doi: 10.1016/j.bbrc.2020.04.002
27. Sergi D, Luscombe-Marsh N, Heilbronn LK, Birch-Machin M, Naumovski N, Lionetti L, et al. The inhibition of metabolic inflammation by EPA is associated with enhanced mitochondrial fusion and insulin signaling in human primary myotubes. J Nutr. (2021) 151:810–9. doi: 10.1093/jn/nxaa430
28. Sears B, Perry M. The role of fatty acids in insulin resistance. Lipids Health Dis. (2015) 14:121. doi: 10.1186/s12944-015-0123-1
29. Wein S, Wolffram S, Schrezenmeir J, Gasperikova D, Klimes I, Sebokova E. Medium-chain fatty acids ameliorate insulin resistance caused by high-fat diets in rats. Diabetes Metab Res Rev. (2009) 25:185–94. doi: 10.1002/dmrr.925
30. Nagao K, Yanagita T. Medium-chain fatty acids: functional lipids for the prevention and treatment of the metabolic syndrome. Pharmacol Res. (2010) 61:208–12. doi: 10.1016/j.phrs.2009.11.007
31. Mumme K, Stonehouse W. Effects of medium-chain triglycerides on weight loss and body composition: a meta-analysis of randomized controlled trials. J Acad Nutr Diet. (2015) 115:249–63. doi: 10.1016/j.jand.2014.10.022
32. Mccarty MF, Dinicolantonio JJ. Lauric acid-rich medium-chain triglycerides can substitute for other oils in cooking applications and may have limited pathogenicity. Open Heart. (2016) 3:e000467. doi: 10.1136/openhrt-2016-000467
33. Saraswathi V, Kumar N, Gopal T, Bhatt S, Ai W, Ma C, et al. Lauric acid versus palmitic acid: effects on adipose tissue inflammation, insulin resistance, and non-alcoholic fatty liver disease in obesity. Biology. (2020) 9:346. doi: 10.3390/biology9110346
34. Delany JP, Windhauser MM, Champagne CM, Bray GA. Differential oxidation of individual dietary fatty acids in humans. Am J Clin Nutr. (2000) 72:905–11. doi: 10.1093/ajcn/72.4.905
35. Montgomery MK, Osborne B, Brown SH, Small L, Mitchell TW, Cooney GJ, et al. Contrasting metabolic effects of medium- versus long-chain fatty acids in skeletal muscle. J Lipid Res. (2013) 54:3322–33. doi: 10.1194/jlr.M040451
36. Tham YY, Choo QC, Muhammad TST, Chew CH. Lauric acid alleviates insulin resistance by improving mitochondrial biogenesis in THP-1 macrophages. Mol Biol Rep. (2020) 47:9595–607. doi: 10.1007/s11033-020-06019-9
37. Makinen S, Nguyen YH, Skrobuk P, Koistinen HA. Palmitate and oleate exert differential effects on insulin signalling and glucose uptake in human skeletal muscle cells. Endocr Connect. (2017) 6:331–9. doi: 10.1530/EC-17-0039
38. Abdelmagid SA, Clarke SE, Nielsen DE, Badawi A, El-Sohemy A, Mutch DM, et al. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS ONE. (2015) 10:e0116195. doi: 10.1371/journal.pone.0116195
39. Erridge C, Samani NJ. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler Thromb Vasc Biol. (2009) 29:1944–49. doi: 10.1161/ATVBAHA.109.194050
40. Williams LM, Campbell FM, Drew JE, Koch C, Hoggard N, Rees WD, et al. The development of diet-induced obesity and glucose intolerance in C57BL/6 mice on a high-fat diet consists of distinct phases. PLoS ONE. (2014) 9:e106159. doi: 10.1371/journal.pone.0106159
41. Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. (1999) 270:41–9. doi: 10.1006/abio.1999.4085
42. Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress. Autophagy. (2008) 4:770–82. doi: 10.4161/auto.6458
43. Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. (2017) 119:315–26. doi: 10.1016/j.acthis.2017.03.001
44. Canto C, Pich S, Paz JC, Sanches R, Martinez V, Orpinell M, et al. Neuregulins increase mitochondrial oxidative capacity and insulin sensitivity in skeletal muscle cells. Diabetes. (2007) 56:2185–93. doi: 10.2337/db06-1726
45. Mathes E, O'dea EL, Hoffmann A, Ghosh G. NF-kappaB dictates the degradation pathway of IkappaBalpha. EMBO J. (2008) 27:1357–67. doi: 10.1038/emboj.2008.73
46. Wang Y, Mao L, Zhang L, Zhang L, Yang M, Zhang Z, et al. Adoptive regulatory t-cell therapy attenuates subarachnoid hemor-rhage-induced cerebral inflammation by suppressing TLR4/NF-B signaling pathway. Curr Neurovasc Res. (2016) 13:121–6. doi: 10.2174/1567202613666160314151536
47. Ferree A, Shirihai O. Mitochondrial dynamics: the intersection of form and function. Adv Exp Med Biol. (2012) 748:13–40. doi: 10.1007/978-1-4614-3573-0_2
48. Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell. (2016) 61:683–94. doi: 10.1016/j.molcel.2016.02.022
49. Tubbs E, Chanon S, Robert M, Bendridi N, Bidaux G, Chauvin MA, et al. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes. (2018) 67:636–50. doi: 10.2337/db17-0316
50. Turner N, Hariharan K, Tidang J, Frangioudakis G, Beale SM, Wright LE, et al. Enhancement of muscle mitochondrial oxidative capacity and alterations in insulin action are lipid species dependent: potent tissue-specific effects of medium-chain fatty acids. Diabetes. (2009) 58:2547–54. doi: 10.2337/db09-0784
51. Mcnelis JC, Olefsky JM. Macrophages, immunity, metabolic disease. Immunity. (2014) 41:36–48. doi: 10.1016/j.immuni.2014.05.010
52. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Ann Rev Immunol. (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
53. Kennedy A, Martinez K, Chuang CC, Lapoint K, Mcintosh M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. Journal Nutr. (2009) 139:1–4. doi: 10.3945/jn.108.098269
54. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. (2017) 542:177–85. doi: 10.1038/nature21363
55. Wilson DE, Chan IF, Stevenson KB, Horton SC, Schipke C. Eucaloric substitution of medium chain triglycerides for dietary long chain fatty acids in acquired total lipodystrophy: effects on hyperlipoproteinemia and endogenous insulin resistance. J Clin Endocrinol Metab. (1983) 57:517–23. doi: 10.1210/jcem-57-3-517
56. Bach AC, Ingenbleek Y, Frey A. The usefulness of dietary medium-chain triglycerides in body weight control: fact or fancy? J Lipid Res. (1996) 37:708–26. doi: 10.1016/S0022-2275(20)37570-2
57. Hoppel CL. Carnitine and carnitine palmitoyltransferase in fatty acid oxidation and ketosis. Fed Proc. (1982) 41:2853–7.
58. Parker A, Engel PC. Preliminary evidence for the existence of specific functional assemblies between enzymes of the beta-oxidation pathway and the respiratory chain. Biochem J. (2000) 345 (Pt 3):429–35. doi: 10.1042/bj3450429
59. Wang Y, Mohsen AW, Mihalik SJ, Goetzman ES, Vockley J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J Biol Chem. (2010) 285:29834–41. doi: 10.1074/jbc.M110.139493
60. Wang Y, Palmfeldt J, Gregersen N, Makhov AM, Conway JF, Wang M, et al. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J Biol Chem. (2019) 294:12380–91. doi: 10.1074/jbc.RA119.008680
61. Wensaas AJ, Rustan AC, Just M, Berge RK, Drevon CA, Gaster M. Fatty acid incubation of myotubes from humans with type 2 diabetes leads to enhanced release of beta-oxidation products because of impaired fatty acid oxidation: effects of tetradecylthioacetic acid and eicosapentaenoic acid. Diabetes. (2009) 58:527–35. doi: 10.2337/db08-1043
62. Gallego SF, Hermansson M, Liebisch G, Hodson L, Ejsing CS. Total fatty acid analysis of human blood samples in one minute by high-resolution mass spectrometry. Biomolecules. (2018) 9:7. doi: 10.3390/biom9010007
63. Mayorek N, Bar-Tana J. Medium chain fatty acids as specific substrates for diglyceride acyltransferase in cultured hepatocytes. J Biol Chem. (1983) 258:6789–92. doi: 10.1016/S0021-9258(18)32291-9
64. Brandes R, Mayorek N, Berry E, Arad R, Bar-Tana J. The specificity of triacylglycerol synthesis for medium-chain fatty acids in rat and human adipose preparations. Biochim Biophys Acta. (1985) 836:63–6. doi: 10.1016/0005-2760(85)90220-6
Keywords: fatty acids, mitochondria, metabolic inflammation, skeletal muscle, lauric acid, palmitic acid
Citation: Sergi D, Luscombe-Marsh N, Naumovski N, Abeywardena M and O'Callaghan N (2021) Palmitic Acid, but Not Lauric Acid, Induces Metabolic Inflammation, Mitochondrial Fragmentation, and a Drop in Mitochondrial Membrane Potential in Human Primary Myotubes. Front. Nutr. 8:663838. doi: 10.3389/fnut.2021.663838
Received: 03 February 2021; Accepted: 22 April 2021;
Published: 31 May 2021.
Edited by:
Guillermo Lopez Lluch, Universidad Pablo de Olavide, SpainReviewed by:
Sandro Massao Hirabara, Universidade Cruzeiro Do Sul, BrazilDaniel José Moreno Fernández-Ayala, Universidad Pablo de Olavide, Spain
Copyright © 2021 Sergi, Luscombe-Marsh, Naumovski, Abeywardena and O'Callaghan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domenico Sergi, ZC44OEBsaXZlLml0