 Amira Kohil
Amira Kohil Maha Al-Asmakh
Maha Al-Asmakh Mashael Al-Shafai
Mashael Al-Shafai Annalisa Terranegra
Annalisa Terranegra- 1Department of Biomedical Sciences, College of Health Sciences, QU Health, Qatar University, Doha, Qatar
- 2Biomedical Research Center, Qatar University, Doha, Qatar
- 3Biomedical and Pharmaceutical Research Unit, QU Health, Qatar University, Doha, Qatar
- 4Research Branch, Sidra Medicine, Doha, Qatar
The autoimmune disease, Type 1 Diabetes Mellitus (T1DM), results in the destruction of pancreatic β-cells, and the International Diabetes Federation reports that its incidence is increasing worldwide. T1DM is a complex disease due to the interaction between genetic and environmental factors. Certain dietary patterns and nutrients are known to cause epigenetic modifications in physiological conditions and diseases. However, the interplay between diet and epigenetics is not yet well-understood in the context of T1DM. Several studies have described epigenetic mechanisms involved in the autoimmune reactions that destroy the β-cells, but few explored diet components as potential triggers for epigenetic modifications. Clarifying the link between diet and epigenome can provide new insights into the pathogenesis of T1DM, potentially leading to new diagnostic and therapeutic approaches. In this mini review, we shed light on the influence of the diet-epigenome axis on the pathophysiology of T1DM.
Introduction
T1DM is a metabolic disease resulting from chronic autoimmune destruction of the insulin-producing pancreatic β-cells (1). It primarily affects children and adolescents and can lead to complications including ocular damage, stroke, diabetic ketoacidosis, coma, and kidney failure (2). The disease's incidence is increasing worldwide, with approximately one million cases annually [International Diabetes Federation (IDF) Diabetes Atlas 2014]. This may be due to recent advances in early diagnosis and monitoring of T1DM (1). However, the sedentary lifestyle predominant worldwide and especially in westernized countries has a strong impact toward developing autoimmune disorders, including T1DM (3). This lifestyle, characterized by a high-fat/low-fiber diet and lack of physical activity, is known to strongly modulate the immune system and can lead to T1DM primarily through its impact on T-cells. For example, in the United states, T1DM incidence increased by 21% among young adults (<20 years old) from 2001 to 2009 (4). Moreover, T1DM is highly prevalent among those <19 years of age in countries with a crescent economy and a shift toward a western-like lifestyle, as in the Middle East region, including Kuwait (44.5%), Saudi Arabia (33.5%), and Qatar (12.2%) (5). Similarly, in India the prevalence of T1DM is 31.9 per 100,000, with higher prevalence seen in urban areas compared to rural areas (6). In Korea between 2007 and 2013, the annual incidence of T1DM increased from 2.73 to 5.02 per 100,000 (7). These numbers illustrate the increasing worldwide prevalence of T1DM and indicate a noticeable impact in countries that have recently adopted a westernized lifestyle.
T1DM involves a complex interaction between genetic and environmental factors, and several genetic loci have been linked to the disease (8, 9). This includes strongly-associated genetic variants at the human leukocyte antigens DR and DQ (HLA-DR and -DQ) that confer a high risk for the development of T1DM (10). Variants in other autoantigens, such as those targeting pre-proinsulin, β-cell specific zinc transporter 8, insulinoma-associated antigen 2, the insulin gene, the cytotoxic T-lymphocyte–associated protein-4, and the interleukin-2 receptor are also known to contribute to the susceptibility of T1DM (9, 11, 12). However, genetic factors cannot explain all T1DM cases and sub-phenotypes. Many environmental factors have also been associated with greater risk of T1DM, including infections, dietary factors, old maternal age, psychological stress, use of antibiotics, mode of delivery, and steroids intake (13, 14). Diet and nutrients, among these factors, are known to exert a direct effect on the epigenome (15).
Epigenetic modifications are influenced by the interaction between genetics and the environmental stimuli, resulting in a T1DM specific epigenetic status (16, 17). Moreover, it is proposed that diet can lead to epigenetic changes and immune dysregulation in T1DM pathogenesis with mechanisms similar to what is seen in other inflammatory disorders (18). Since T1DM is a complex disorder a multivariate pathogenesis, a greater understanding of the interplay between diet and epigenetics could provide new avenues for early diagnosis, treatment, and a personalized therapy. This review aims to summarize the role of both diet and epigenome in T1DM development, as well as to clarify the interplay of diet-epigenome in the pathophysiology of T1DM.
The Role of Diet in The Pathogenesis of T1DM
Diet is considered an environmental trigger involved in the development of various metabolic disorders, such as obesity, type 2 diabetes mellitus (T2DM), and T1DM (19–21). Some dietary factors such as fat, protein, and fibers are known to affect the glycemic control in T1DM patients, but the molecular mechanisms by which diet and nutrients impact the development of T1DM are still unknown. Below, we summarize the recent evidences on the role of nutrition in T1DM, focusing mainly on food habits and nutrients that can exert epigenetic changes.
Early Nutrition
Since T1DM and its pre-clinical autoimmunity appear at an early age, several studies highlighted the role of early nutrition in the pathogeneses of T1DM and is also considered a risk factor of the disease. Dietary habits after birth are investigated as potential determinants of individual risk for developing T1DM. Breastfed children, for instance, have shown a lower risk of developing T1DM (22) due to the presence of secretory immunoglobin A, lysosomes, and lactoferrin in breast milk (23). In contrast, increased intake of cow's milk or formula milk, from 6 to 9 months of age, was suggested to increase the risk of developing T1DM (23, 24). This finding is likely, due to the presence of specific fatty acids (e.g., penta-decanoic, myristic, and isomers of monounsaturated palmitoleic acid), which may promote islet autoimmunity (25). Moreover, the intake of hydrolyzed formula (lacking of complex dietary proteins) has no effect on reducing the incidence of T1DM in infants at high risk (26). In addition, the early introduction of cereals that do or do not contain gluten into the diet may promote the development of T1DM, which can be attributed to the immature immune system and undeveloped gut microbiota present in young children, whereas late introduction (after 6 months) of gluten has no effect (27). However, contrasting data showed that delayed introduction of cereals containing gluten is associated with increased risk of developing islet autoimmunity and progression to T1DM (28). Furthermore, one study showed that serum fatty acids, biomarkers of milk, and ruminant meat consumption, are significantly associated with advanced β-cell autoimmunity in children with conferred susceptibility to T1DM compared to seronegative controls (19).
Diet in Adult T1DM Patients
In adults with T1DM, a low-carbohydrate/high-fat diet has been associated with improved glycemic control, shown via low hemoglobin A1c (HbA1c) and less glycemic variability. However, this diet was also linked to an increased risk of hypoglycemia and dyslipidemia (29). In the case of fiber intake, one study showed that low intake of dietary fibers is associated with elevated levels of HbA1c in T1DM patients, thus leading to a poor glycemic control (30). On the other hand, another study showed that a high fiber diet is associated with decreased protein synthesis and degradation (post-absorptive protein turnover) in T1DM male patients (31). Nutritional intake was also found to influence the development of T1DM-related complications. A study conducted by Beretta et al. showed that a diet containing high fibers exhibited a significant reduction in BMI, systolic/diastolic blood pressure, and reduction in energy intake compared to T1DM patients with a low fiber diet (32) development of T1DM-related complications has also been linked to nutritional intake. The intake of complex carbohydrates was linked to the presence of diabetic retinopathy in T1DM adult patients, whereas the intake of unsaturated fat, particularly monounsaturated fatty acids (MUFA), was associated with the absence of diabetes retinopathy (33). In another study, a positive correlation was observed between high intake of saturated fats and proteins and the risk of developing coronary heart disease (CHD) in T1DM patients, while high carbohydrate intake was negatively correlated with CHD (34). Vitamin D also plays important role in regulating both the immune system and metabolic pathways, and several studies have demonstrated that vitamin D supplementation lowers the risk of developing T1DM (35, 36). This effect could be explained by the observation that vitamin D down-regulates the response of T helper-1 lymphocytes (37). However, other investigations did not confirm such effect of vitamin D supplementation (38, 39). These discrepancies could be attributed to the differences in the type of supplement (i.e., cholecalciferol, alpha-calcidol, or calcitriol), the vitamin dosage, the age group of the study participants, and/or the duration of diabetes.
The Role of Epigenetics in T1DM Pathogenesis
Epigenetics is the activation and inhibition of gene expression by factors that does not affect the DNA sequence itself (40). Such epigenetic changes are affected by genetic variations as well as environmental factors and influencing DNA accessibility by transcription factors leading to the regulation of gene expression (16, 17). It has been shown that epigenetic modifications, mainly DNA methylation, microRNAs, and histone modifications, play an important role in developing various autoimmune disorders, including T1DM (41, 42). Several features of T1DM support the involvement of epigenetics in its pathogenesis. These include high discordance rates among monozygotic (MZ) twins, offspring of an affected father rather than affected mother having a higher risk of developing the disease, and the increased T1DM incidence in genetically stable populations (41, 42) and countries with a westernized lifestyle (3).
DNA Methylation Modifications in T1DM
DNA methylation is defined as the formation of 5-methylcytosine (5-mC) by the attachment of a methyl group on the fifth carbon of cytosine, and it usually occurs in the CpG dinucleotides (43). Three methyltransferases - DNMT1, DNMT3a, and DNMT3b - are primarily responsible for regulating DNA methylation process, which are DNMT1, DNMT3a, and DNMT3b (44). The main function of DNMT1 is to maintain the methylation status in the genome, restoring the methylation pattern in the daughter strands during replication. DNMT3a and DNMT3b, conversely, are responsible for establishing de novo DNA methylation patterns (44). DNA demethylation on the contrary, is a sequential oxidation of 5-mC and the removal of the modified group by thymine DNA glycosylase (TDG) produce cytosine in replacement of 5-mC. The demethylation process also involves Ten-eleven translocation (TET) family dioxygenase enzymes, which are TET1, TET2, and TET3 (45, 46). DNA methylation has been implicated in a number of autoimmune disorders, including T1DM (47). Alterations in DNA methylation may cause changes in the expression of genes responsible for β-cell survival, insulin secretion, and autoimmunity, thereby influencing the development of T1DM (42). This hypothesis is supported by research from Rakyan and colleagues, who generated a genome-wide methylation profile from T1DM-discordant MZ twins. They identified 132 T1DM-associated methylation sites in the promoter regions of genes associated with apoptosis, inflammation, and immune regulation. Additional evidence comes from Stefan and colleagues, who detected 88 differentially-methylated CpG sites in T1DM-discordant MZ twin pairs (48). Although, this is an observational study, the enrollment of MZ twins as study subjects reinforces the value of the findings, providing a strong evidence that DNA methylation plays an integral part in T1DM development and thus not totally explained by genetics.
The INS gene locus is closely involved with T1DM, and its A/T single nucleotide polymorphism (SNP) rs689 in particular has been associated with T1DM development (8). A number of studies found DNA methylation of INS gene promoter in pancreatic β-cells and thymic epithelial cells is significantly implicated in T1DM development (49, 50). One study showed that patients with T1DM have a higher level of methylation at CpG−180 and lower level at methylation of CpG−19,−135, and−234 in INS gene when compared to healthy controls (49). They also found a strong correlation between high methylation levels of CpG −69, −102, −180, −206, and T1DM. However, it is unknown whether these epigenetic changes in T1DM precedes or follows the development of the disease (49). In addition, a study conducted by Rui et al. found that INS gene expression is regulated by the methylation of Ins1 exon-2 and Ins2 exon-1. The study showed, both in NOD mice (in-vivo) and in human β-cells (in-vitro), that pro-inflammatory cytokines could activate methyltransferases, leading to methylation of Ins1 exon-2 and Ins2 exon-1 in the INS genes (50).
Interleukin 2 receptor α-chain gene (IL2RA) encodes the IL-2 receptor, which is highly expressed in regulatory T-cells Tregs and plays a vital role in suppressing autoreactive T-cells. Like the INS gene, epigenetic modulation of the IL2RA gene was implicated in the development of T1DM. One study showed that T1DM patients have a high methylation level at IL2RA CpGs −373 and −456 compared to healthy controls (51). They found that the DNA methylation at IL2RA CpGs −373 was associated with 16 known SNPs to be involved in T1DM.
DNA methylation was also implicated in the complications associated with T1DM, such as diabetic nephropathy (52). Methylation of 19 CpG sites was correlated with the development and onset of diabetic nephropathy. Furthermore, one of the methylated CpG sites was located near the transcription start site of UNC13B (rs13293564), which is known to be involved the development of diabetic nephropathy in T1DM patients (52).
Finally, it is worth noting that epigenetic mechanisms are also implicated in the early development and function of the insulin producing pancreatic β-cells. Different types of epigenetic modifications were found to be involved in the development of pancreatic cells from the endocrine progenitor cells (53). Particularly, a study conducted by Neiman et al. found that promoters of β-cell specific genes, such as Insulin 2 (INS2) and Glucagon genes, were significantly hypermethylated in non-endocrine tissues when compared to β-cells (54). The same study also found different methylation levels of the CpG sites amongst the endocrine cells' subtypes (α-cells, β-cells, and δ-cells), that may implicate a specific control of the gene expression in these cells (54).
The Micro-RNA Modifications in T1DM
The micro-RNA (miRNA) are non-coding RNA molecules ranging from 18 to 22 nucleotides that act as post-transcriptional silencers (55), and are involved in several biological processes, such as cell division, proliferation, and apoptosis (56). In the nucleus, the miRNA is transcribed into primary miRNA by RNA polymerase (RNase) II and III and then processed to precursor miRNA by the complex Drosha/DGCR8. The precursor miRNA is finally processed into the mature miRNA in the cytoplasm by RNase III Dicer complex (57). miRNAs exert their post-transcriptional effect through binding to the 3′ untranslated region of the targeted mRNA, leading to its cleavage, degradation and translation inhibition (58). The effect of miRNAs on gene expression was implicated in different autoimmune disorders, such as autoimmune thyroid diseases (59) and rheumatoid arthritis (60). In the case of T1DM, alterations in miRNA levels may contribute to the disease pathogenesis by affecting multiple pathways, such as insulin secretion, the programmed cell death, the immune system, and the mitogen-activated protein kinase (MAPK) signaling pathway (61). A group of miRNAs (miR-210-3p, miR-155-5p, miR-103a-3p, and miR-146a-5p) was identified as differentially regulated in T1DM, and it was mainly associated with immune system functions (61). Several studies identified the mechanism of single small groups of miRNAs. To mention some, Krishnan et al. found that upregulation of miR-155-5p in human islet derived exosomes targets the mRNA of the transcriptional and immune response regulator gene (TCIM) (62). Another study by Garcia-Diaz et al., found that the upregulation of miR-155-5p has a role in the inflammatory process in T1DM through binding to the toll-like receptors and activating the NF-κB pathway (63). A more recent study identified a different set of miRNAs (miR-142-5p, miR-146a-5p, and miR-223-3p) positively correlated with B lymphocyte, CD4+CD45RO+, and CD4+CD25+ immune cells in T1DM patients (64). Low expression levels of miR-146a-5p were consistently observed in T1DM patients, and it was associated with overproduction of IL-6, which is an important pro-inflammatory cytokine. Therefore, it could be suggested that miR-146a-5p has a negative feedback effect on the NF-κB pathway in regulating the inflammation status in T1DM (63, 65). In addition, the overexpression of miR-23b, miR-98, and miR-590-5p in CD8+ T-cells from T1DM patients was suggested to target apoptotic genes (TRAIL, TRAIL-R2, FAS, and FASLG), resulting in the excessive proliferation of autoreactive T-cell and T1DM development (66). Furthermore, hyperglycemia was found to influence the levels of certain miRNAs, such as miR-125b-5p and miR-365a-3p that were positively correlated with HbA1c levels (67). Circulatory miRNAs can also be used as a biomarker for early detection of T1DM as they are stable and easily detected (61). This was supported by a variety of studies; for example, miR-125-5p and miR-320c were identified in early onset-T1DM patients and were elevated before progression of diabetes in NOD mice (68). Furthermore, urinary miR-377 was found to be positively correlated with HbA1c and urinary albumin creatinine ratio in T1DM patients, which makes it a possible biomarker for diabetic nephropathy (69). Identifying early miRNA biomarkers may help in early diagnosis, treatment and prevention of diabetic complication.
Histone Modifications in T1DM
Histones undergo different modifications, such as methylation, acetylation, phosphorylation, and other mechanisms (sumoylation and ubiquitination) in specific amino acid residues located in the N-terminal part (70). The histone methylation involves the addition of methyl groups to lysine or arginine residues, resulting in activation or inhibition of transcription based on the level of modification and the affected region (71), while histone demethylation involve the binding of a lysine specific demethylase-1 (LSD1) to the lysine methylation site on the histone tail, removing the methyl group (72). The histone acetylation involves the addition or removal of acetyl group on lysine residues by the function of histone-acetyltransferases (HATs) and histone-deacetylases (HDACs), respectively (17). Histone acetylation promotes an opened chromatin structure that is more accessible for gene transcription by reducing the electrostatic affinity between protein and DNA (73). Histone ubiquitination, is defined as the addition of ubiquitin molecules to the conserved lysine residues through the function of ubiquitin ligases (74). Furthermore, sumoylation is defined by the attachment of ubiquitin like modifier proteins covalently to histones through the action of ubiquitin analog enzymes (75). Histone modification has been associated with different pathological conditions, including T1DM. One study found a significant increase of H3 lysine-9 di-methylation (H3K9me2) in T1DM patients' lymphocytes compared to healthy controls (76). They also found a strong association between increased H3K9me2 promoter activity in CLTA4 gene (T1DM susceptibility gene) and genes involved in autoimmune and inflammatory pathways, such as TLR, NF-κB, and p38-MAPK (76). Furthermore, T1DM patients were found to exhibit marked variations in H3 lysine-9 acetylation (H3K9Ac) levels at the upstream regions of HLA-DRB1 and HLA-DQB1, which are susceptibility loci strongly associated with T1DM (77). The same study further demonstrated that THP-1 monocytes treated with interferon-γ and TNF-α showed increased expression of HLA-DRB1 and HLA-DQB1 combined with changes in H3K9Ac, similarly to what was observed in T1DM patients (77). Moreover, another study showed an increased level of H4 acetylation in T1DM patients, however it was restricted to T1DM patients with no cardiovascular complications, indicating that histone acetylation may have a protective role against the development of T1DM complications (78). More studies are needed to clarify the mechanism of histone modifications affecting T1DM susceptibility loci and their impact on the development of T1DM.
Diet and Epigenetic Interplay in T1DM Pathogenesis
The interplay between diet and epigenetics and how this link contributes to the pathogenesis of T1DM is yet to be identified. In this review, we suggest that dietary habits as well as specific nutrients may result in certain epigenetic signatures contributing to T1DM development.
In general, three possibilities have been postulated on how nutritional factors modulate DNA methylation; first, by directly providing the substrates needed in the methylation process (methyl-donor nutrients, e.g., methionine, choline, and folate); second, by providing co-factors needed for the function of methyltransferases (e.g., vitamins B2, B6, and B12); third, by altering the activity of the enzymes involved in the methylation process [e.g., polyphenols; (79)].
Most of the dietary factors known to be involved in the pathogenesis of T1DM, such as breastmilk, fibers, MUFA, vitamin D, etc., are also known to have an epigenetic function (80–83), but their mechanism of action is not fully understood in the context of T1DM. We reviewed the major studies identifying a mechanism or a potential link of diet with DNA-methylation, histones modifications and miRNA in T1DM (Table 1).
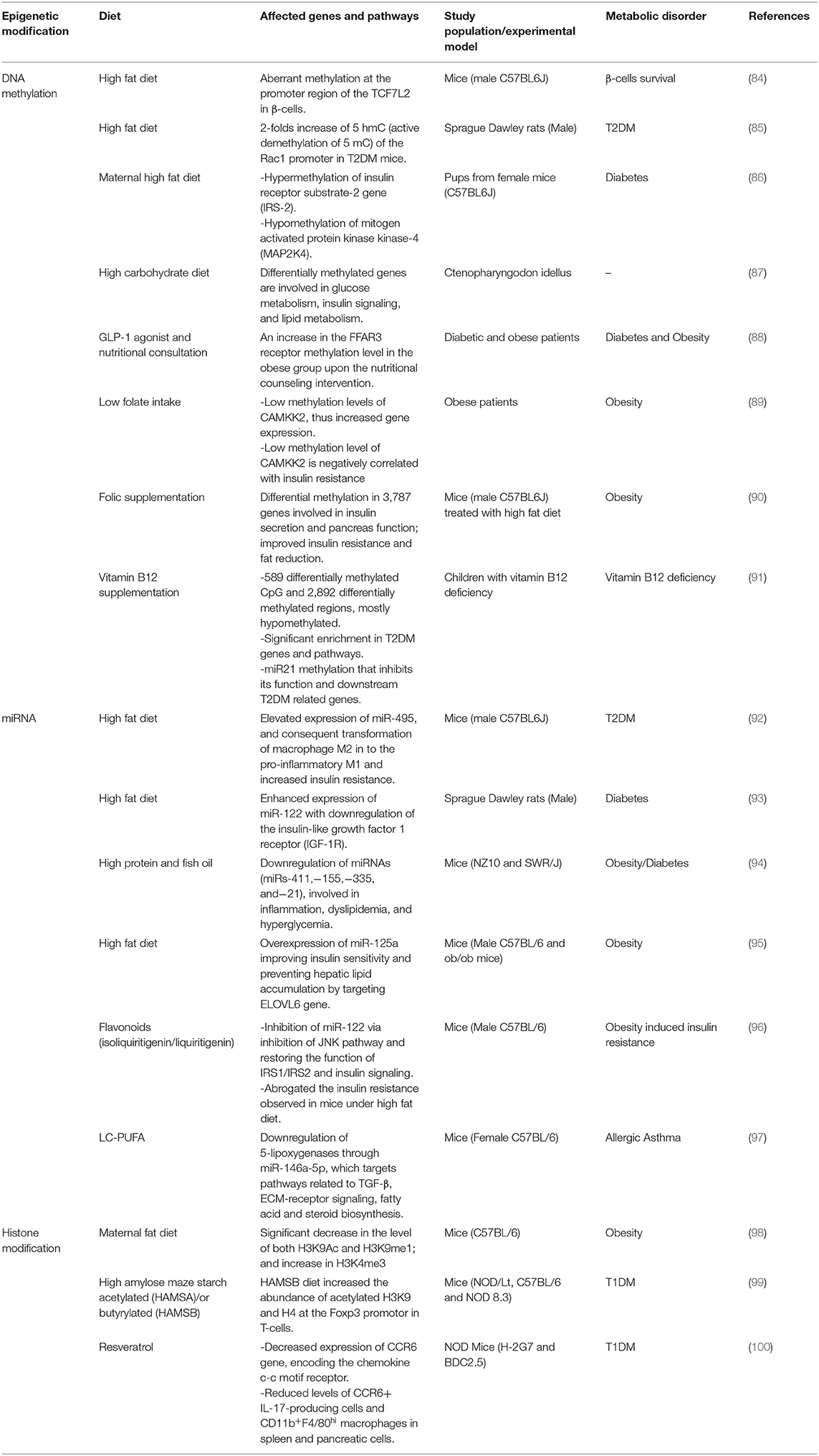
Table 1. The interplay between diet and epigenetic modifications in different metabolic conditions.
Cow's milk allergenicity is considered one of the dietary risk factors for T1DM, as shown in a study where A1 beta-casein from cow milk was associated with an increased T1DM incidence and sub-clinical insulitis across generations (101). The authors suggested that beta-casomorphin peptide (BCM-7) released from A1 beta-casein may cause epigenetic alterations that lead to T1DM development. In gastrointestinal diseases, BCM-7 was found to act as a epigenetic modulator and to differentially methylate genes involved in these diseases (102). It is therefore speculate that the beta-casein effect observed in T1DM can cause differential DNA methylation of similar or other genes.
High intake of fat was reported to be associated with risk of islet autoimmunity, poor glycemic control, and development of T1DM-related complications during both infancy and adulthood in T1DM patients (34, 103). High fat diet was found to epigenetically impact TCF7L2, a transcription factor required for pancreatic β-cell survival (84). An experimental study demonstrated that mice treated with high fat diet showed an aberrant methylation at the promoter region of the TCF7L2 in β-cells compared to mice fed with a normal diet (84). Although defects in TCF7L2 were commonly detected in T2DM patients, a subset of non-obese T1DM patients were also found to have the same defect (104). Another animal study using a high fat induced hyperglycemic rats showed epigenetic involvement in T2DM animals (85). The study showed a significant 2-folds increase of 5 hmC (active demethylation of 5 mC) of the Rac1 promoter in high fat induced T2DM compared to healthy animals and compared to T1DM animals fed on normal diet. Moreover, high intake of saturated fatty acid was associated with low levels of adiponectin in T1DM patients (105) and several studies showed that dietary fatty acids regulate adipocyte function through epigenetic modifications, mainly polyunsaturated fatty acids (PUFA) and saturated fatty acids (86, 106, 107). In a similar human study, the high intake of saturated fatty acids and PUFA was associated with increased DNA methylation level in the adipose tissue, which was correlated with increase in body weight (106). A total of 1,797 genes were deferentially methylated under high PUFA, whereas 125 genes were deferentially methylated in the case of saturated fatty acid intake. An animal-based study found that maternal high fat diet leads to hypermethylation of insulin receptor substrate-2 gene (IRS-2) and hypomethylation of mitogen-activated protein kinase kinase-4 gene (MAP2K4) in mice offspring; thus, decreasing and increasing the gene expression, respectively, and elevating pup's risk of developing diabetes in the future (86). Maternal high fat diet has negative effects on the pancreatic β-cells of male mice offspring, in which proliferation defects and insulin degranulation were detected (107). However, this phenotype was reversed and offspring were protected from developing insulin resistance through early transition from high fat diet to normal fat diet (107).
The diet-epigenetic axis is also implicated in pathways related to glucose metabolism and insulin signaling, as demonstrated in animal models. Whole genome DNA methylation analysis of the grass carp, Ctenopharyngodon idellus, revealed no significant changes in methylation level of metabolic genes between different nutritional conditions (high carbohydrate diet/ normal diet) (87). However, the deferentially methylated genes in Ctenopharyngodon idellus with a high carbohydrate diet were enriched in pathways involved in glucose metabolism, insulin signaling, and lipid metabolism. Most of these obesity and T2DM- involved methylation changes were also observed in mammals (87). Moreover, folic acid (synthetic form of folate) is mainly involved in the methylation processes acting as a methyl donor for the synthetic process of S-adenosylmethionine (108). Low folate intake was associated with the methylation of Calcium/Calmodulin Dependent Protein Kinase Kinase 2 (CAMKK2), which is an important gene involved in glucose homeostasis and adiposity. Obese patients with low folate intake, showed lower methylation levels of CAMKK2, a condition correlated with increased insulin resistance (89). A study conducted by Li and his colleagues found that folate supplementation in mice treated with high fat diet reduced fat mass and improved insulin resistance (90). They also found that deferentially methylated regions associated with folic supplementation mainly affected adipose genes and were involved in pathways related to insulin secretion and pancreatic function. Vitamin B12 also found to be involved in the synthesis of S-adenosylmethionine through acting as a coenzyme that catalyzes the methylation of homocysteine to methionine, supporting its essential role in the methylation process (109). A recent study detected 589 differentially methylated CpG upon vitamin B12 supplementation, in which 73.3% of the genes were hypomethylated (91). In the same study, pathway analysis revealed significant enrichment in pathways related to T2DM, such as glycogen synthesis and adipogenesis pathways. Among the identified genes that were differentially methylated, the authors focused on the differential methylation of miR21, which is known to target different genes involved in T2DM. Their analysis found that miR21 was repressed upon vitamin B12 supplementation leading to inhibiting of its targeted genes (FTO, TCF7L2, CREBBP/CBP, and SIRT1) (91).
There is presently a lack of studies that demonstrate an interaction between diet and miRNA levels in T1DM. However, there are a number of studies that show this correlation in other metabolic disorders, such as in T2DM and obesity [Table 1; (92–95)]. An animal-based study found that mice given high fat diet showed elevated expression of miR-495, which caused the transformation of macrophage M2 into the pro-inflammatory M1 and increased insulin resistance (92). This study has showed the possible mechanism of miR-495 that acts as a negative regulator to the FTO gene and induces adipose tissue inflammation in T2DM mice given high fat intake. Another study showed enhanced expression of miR-122 accompanied with downregulation of insulin-like growth factor 1 receptor (IGF-1R) in the liver of diabetic rats given high fat diet (93). Furthermore, in mice susceptible of developing obesity and diabetes, a diet rich with protein and fish oil has been found to override this genetic susceptibility (94). It has been shown that certain miRNAs (miRs-411, 155, 335, and 21) involved in inflammation, dyslipidemia, and hyperglycemia were downregulated in mice fed with high protein and fish oil diet (94). A study conducted by Liu and colleagues showed that miR-125a level was downregulated in both genetic and dietary (high fat diet) mouse model of obesity (95). In obese mice treated with high fat diet, overexpression of miR-125a improved insulin sensitivity and prevented hepatic lipid accumulation by targeting Elongation of very long chain fatty acids protein 6 (ELOVL6) gene, which is a microsomal enzyme that catalysis the elongation of both saturated fatty acid and MUFA (95). In addition, long chain PUFA (LC-PUFA) was found to modulate miRNA expression in a murine allergic asthma model, where it plays a protective role in the inflammatory process associated with asthma (97). After LC-PUFA supplementation, 21 out of 62 dysregulated miRNAs in asthmatic mice were restored, in which some of the restored miRNAs (mainly miR-146a-5p) are implicated in the function of TGF-β, ECM-receptor signaling, fatty acid and steroid biosynthesis. Mechanistically, the study found that LC-PUFA downregulates 5-lipoxygenases through modulating the expression of miR-146a-5p; thus, acting as an epigenetic regulator. However, it was also found that LC-PUFA downregulates 5-lipoxygenases independent of miR-146a-5p (97). Furthermore, in obese mice fed a high fat diet, flavonoids were shown to inhibit miR-122 dysregulation through the inhibition of JNK, thereby restoring the function of IRS1/IRS2 tyrosine phosphorylation and insulin signaling (96).
The correlation between diet and histone modification in T1DM development is yet to be identified, but several studies were conducted in other metabolic disorders (Table 1). In T1DM, NOD mice treated with resveratrol, a polyphenol known to enhance the activity of sirtuin 1 (NAD dependent histone deacetylase), showed a reduced expression of CCR6 gene, responsible for encoding the chemokine c-c motif receptor (100). It also reduced the level of CCR6+ IL-17-producing cells and CD11b+F4/80hi macrophages in spleen and pancreatic cells (100). One study has identified 21 core histone marks with at least 1.5-folds change in a prediabetic animal model of high-fat diet induced obese mice (110). Diet and histone modification interplay was also implicated in maternal obesity and diabetes, where appropriate fatty acid intake during gestation in mice were found to help offspring to cope with obesogenic conditions (98). In obesity resistant mouse model, a significant decrease was observed in the level of both H3K9Ac and H3K9me1 compared to control mice. However, no significant change was observed of histone markers between obesity resistant mice and obesity prone mice (high fat diet/obesogenic diet), a finding potentially due to the effects of their different diets. In addition, level of H3K4me3 was elevated in both obesity mice models in comparison to healthy controls (98).
The interaction between diet and epigenetics in T1DM pathogenesis could involve the gut microbiota. Specific dietary factors that potentially influence T1DM, including breastfeeding, high fiber intake, and low-fat diet, affect the composition of the gut microbiota and their metabolites (111, 112). Dietary fibers also affect the gut microbial composition as they are digested solely by these organisms, primarily the lower gastrointestinal tract (113). Fermentation of fibers by the gut microbiota produces short-chain fatty acids (SCFA), contributing to gut microbiota diversity (114). These SCFA, such as butyrate, acetate, and propionate, will lead to the specific activation of free fatty acid receptors 2/3 (FFAR2 and FFAR3). This activation inhibits histone deacetylase, leading to inhibition of the inflammatory cascade and the activation of Tregs (18). Administration of dietary fibers to non-obese diabetic (NOD) mice elevated their levels of SCFA, reduced the levels of inflammatory mediators, enhanced the integrity of the gut barrier, and activated Tregs, thereby reducing the incidence of T1DM (99). In patients with T2DM, SCFAs are involved in DNA methylation and histone modifications (88). More specifically, butyrate inhibits histone deacetylases, resulting in inhibition of NF-κB (115) and activation of the MAPK and ERK pathways (116) in intestinal Tregs, thereby down-regulating the pro-inflammatory cascade (117). In addition, in T2DM patients, binding of SCFA to the promoter region of FFAR3 reduces methylation of the CpG islands (88). Since some of the genetic and environmental factors associated with T1DM and T2DM are similar, studies should be designed to identify potentially similar underlying epigenetic mechanisms.
Although epigenetic factors can affect the immune system directly in a manner that leads to T1DM (118), the molecular mechanism(s) underlying these epigenetic changes remain unclear. Our hypothesis is that, as in the case of T2DM and other metabolic conditions, different dietary factors contribute to the pathogenesis of T1DM by inducing epigenetic modifications directly or through the involvement of the gut microbiome [Table 1 and Figure 1; (18, 119)].
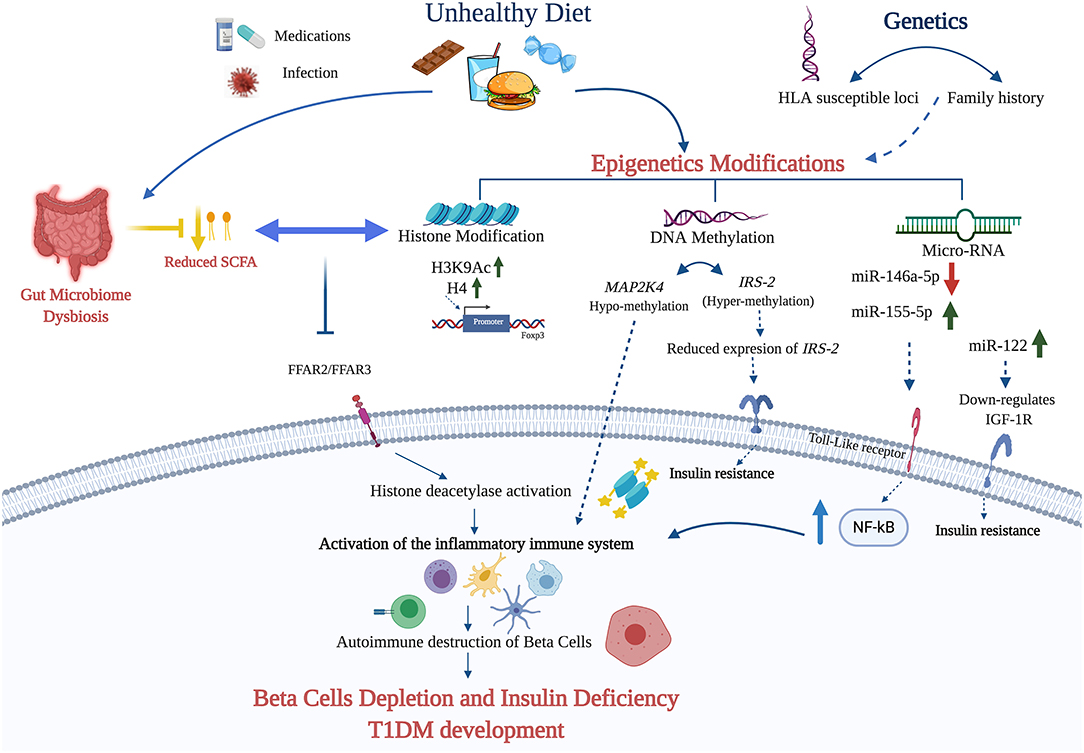
Figure 1. The figure summarizes the potential diet—epigenetic axis in T1DM development. The intake of unhealthy diet may lead to T1DM development through epigenetic modifications (DNA methylation, miRNA, and histone modifications). Diet nutrients can contribute to the hypermethylation and subsequent downregulation of IRS-2 leading to insulin deficiency. In addition, diet can contribute to the up- and down-regulation of certain miRNAs activates NF-kB pathway and the activation of the inflammatory response. The role of histone modification in T1DM is explicated mainly through elevating the expression of H3K9Ac and H4 in the promoter region of Foxp3. Finally, unhealthy diet affects the gut microbiota and its metabolites, such as SCFA. The failure of SCFA binding to the free fatty acid receptors 2/3 (FFAR2 and FFAR3) due to its absence or reduction, leads to the activation of histone deacetylase (HDAC1, HDAC3) and the inflammatory cascade that contribute to β-cell depletion and T1DM development (Created using Biorender.com).
Conclusions
T1DM is a complex disease caused by the interaction of many factors, both genetic and environmental, including epigenetics and dietary factors. Specific dietary patterns and nutrients can exert a direct impact on the immunopathogenesis of T1DM through epigenetic modifications. A deeper understanding of the interplay between the diet and epigenetics will improve our knowledge concerning the pathogenesis of T1DM and it will help identifying new therapeutic targets. We propose here a mechanism by which nutrients can trigger epigenetic modifications leading to β-cells depletion and T1DM development (Figure 1). A better characterization of the specific dietary patterns and nutrients that can exert such effects may help prevent and/or ameliorate T1DM and applying personalized nutrition approaches.
Author Contributions
AT and AK developed the content of the manuscript. AK drafted the manuscript. AT, MA-A, and MA-S discussed the content and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by student grant from Qatar University (QUST-1-CHS-2020-5) and by Sidra Medicine internal funds (SDR200010/SDR400131).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Daniel Ackerman (Insight Editing London) for editing the manuscript during submission.
References
1. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
2. Bjornstad P, Snell-Bergeon JK, Nadeau KJ, Maahs DM. Insulin sensitivity and complications in type 1 diabetes: new insights. World J Diabetes. (2015) 6:8–16. doi: 10.4239/wjd.v6.i1.8
3. Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M. Role of “Western diet” in inflammatory autoimmune diseases. Curr Allergy Asthma Rep. (2014) 14:404. doi: 10.1007/s11882-013-0404-6
4. Xu G, Liu B, Sun Y, Du Y, Snetselaar LG, Hu FB, et al. Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: population based study. BMJ. (2018) 362:k1497. doi: 10.1136/bmj.k1497
5. Saraswathi S, Al-Khawaga S, Elkum N, Hussain K. A systematic review of childhood diabetes research in the Middle East region. Front Endocrinol. (2019) 10:805. doi: 10.3389/fendo.2019.00805
6. Kalra S, Kalra B, Sharma A. Prevalence of type 1 diabetes mellitus in Karnal district, Haryana state, India. Diabetol Metab Syndr. (2010) 2:14–. doi: 10.1186/1758-5996-2-14
7. Lee YB, Han K, Kim B, Jin SM, Lee SE, Jun JE, et al. High proportion of adult cases and prevalence of metabolic syndrome in type 1 diabetes mellitus population in Korea: a nationwide study. Diabetes Metab J. (2019) 43:76–89. doi: 10.4093/dmj.2018.0048
8. Redondo MJ, Steck AK, Pugliese A. Genetics of type 1 diabetes. Pediatr Diabetes. (2018) 19:346–53. doi: 10.1111/pedi.12597
9. Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of type 1 diabetes: what's next? Diabetes. (2010) 59:1561–71. doi: 10.2337/db10-0076
10. Ilonen J, Kiviniemi M, Lempainen J, Simell O, Toppari J, Veijola R, et al. Genetic susceptibility to type 1 diabetes in childhood–estimation of HLA class II associated disease risk and class II effect in various phases of islet autoimmunity. Pediatr Diabetes. (2016) 17:8–16. doi: 10.1111/pedi.12327
11. Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. (2008) 20:111–8. doi: 10.1016/j.coi.2007.11.005
12. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
13. Islam ST, Srinivasan S, Craig ME. Environmental determinants of type 1 diabetes: a role for overweight and insulin resistance. J Paediatrics Child Health. (2014) 50:874–9. doi: 10.1111/jpc.12616
14. Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet. (2016) 387:2340–8. doi: 10.1016/S0140-6736(16)30507-4
15. Ramos-Lopez O, Milagro FI, Allayee H, Chmurzynska A, Choi MS, Curi R, et al. Guide for current nutrigenetic, nutrigenomic, and nutriepigenetic approaches for precision nutrition involving the prevention and management of chronic diseases associated with obesity. Lifestyle Genomics. (2017) 10:43–62. doi: 10.1159/000477729
16. Cerna M. Epigenetic regulation in etiology of type 1 diabetes mellitus. Int J Mol Sci. (2019) 21:36. doi: 10.3390/ijms21010036
17. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. (2016) 17:487. doi: 10.1038/nrg.2016.59
18. Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell. (2016) 64:982–92. doi: 10.1016/j.molcel.2016.10.025
19. Virtanen SM, Niinistö S, Nevalainen J, Salminen I, Takkinen HM, Kääriä S, et al. Serum fatty acids and risk of advanced β-cell autoimmunity: a nested case–control study among children with HLA-conferred susceptibility to type I diabetes. Eur J Clin Nutr. (2010) 64:792–9. doi: 10.1038/ejcn.2010.75
20. Myette-Côté É, Durrer C, Neudorf H, Bammert TD, Botezelli JD, Johnson JD, et al. The effect of a short-term low-carbohydrate, high-fat diet with or without postmeal walks on glycemic control and inflammation in type 2 diabetes: a randomized trial. Am J Physiol Regul Integr Comp Physiol. (2018) 315:R1210–9. doi: 10.1152/ajpregu.00240.2018
21. Foster-Schubert KE, Alfano CM, Duggan CR, Xiao L, Campbell KL, Kong A, et al. Effect of diet and exercise, alone or combined, on weight and body composition in overweight-to-obese postmenopausal women. Obesity. (2012) 20:1628–38. doi: 10.1038/oby.2011.76
22. Frederiksen B, Kroehl M, Lamb MM, Seifert J, Barriga K, Eisenbarth GS, et al. Infant exposures and development of type 1 diabetes mellitus: the Diabetes Autoimmunity Study in the Young (DAISY). JAMA Pediatrics. (2013) 167:808–15. doi: 10.1001/jamapediatrics.2013.317
23. Patelarou E, Girvalaki C, Brokalaki H, Patelarou A, Androulaki Z, Vardavas CJNr. Current evidence on the associations of breastfeeding, infant formula, and cow's milk introduction with type 1 diabetes mellitus: a systematic review. Nutr Rev. (2012) 70:509–19. doi: 10.1111/j.1753-4887.2012.00513.x
24. Hall K, Frederiksen B, Rewers M, Norris JM. Daycare attendance, breastfeeding, and the development of type 1 diabetes: the diabetes autoimmunity study in the young. BioMed Res Int. (2015) 2015:203947. doi: 10.1155/2015/203947
25. Lamb MM, Miller M, Seifert JA, Frederiksen B, Kroehl M, Rewers M, et al. The effect of childhood cow's milk intake and HLA-DR genotype on risk of islet autoimmunity and type 1 diabetes: the diabetes autoimmunity study in the young. Pediatr Diabetes. (2015) 16:31–8. doi: 10.1111/pedi.12115
26. Writing Group for the TRIGR Study Group, Knip M, Åkerblom HK, Al Taji E, Becker D, Bruining J, et al. Effect of hydrolyzed infant formula vs. conventional formula on risk of type 1 diabetes: the TRIGR randomized clinical trial. JAMA. (2018) 319:38–48. doi: 10.1001/jama.2017.19826
27. Chmiel R, Beyerlein A, Knopff A, Hummel S, Ziegler A-G, Winkler C. Early infant feeding and risk of developing islet autoimmunity and type 1 diabetes. Acta Diabetol. (2015) 52:621–4. doi: 10.1007/s00592-014-0628-5
28. Uusitalo U, Lee H-S, Andrén Aronsson C, Vehik K, Yang J, Hummel S, et al. Early infant diet and islet autoimmunity in the TEDDY study. Diabetes Care. (2018) 41:522–30. doi: 10.2337/dc17-1983
29. Leow ZZX, Guelfi KJ, Davis EA, Jones TW, Fournier PA. The glycaemic benefits of a very-low-carbohydrate ketogenic diet in adults with type 1 diabetes mellitus may be opposed by increased hypoglycaemia risk and dyslipidaemia. Diabetic Med. (2018) 35:1258–63. doi: 10.1111/dme.13663
30. Balk S, Schoenaker D, Mishra GD, Toeller M, Chaturvedi N, Fuller JH, et al. Association of diet and lifestyle with glycated haemoglobin in type 1 diabetes participants in the EURODIAB prospective complications study. Eur J Clin Nutr. (2016) 70:229–36. doi: 10.1038/ejcn.2015.110
31. Bruttomesso D, Tessari P. A high-fiber diet decreases postabsorptive protein turnover but does not alter insulin sensitivity in men with type 1 diabetes mellitus. J Nutr. (2019) 149:596–604. doi: 10.1093/jn/nxy300
32. Beretta MV, Bernaud FR, Nascimento C, Steemburgo T, Rodrigues TC. Higher fiber intake is associated with lower blood pressure levels in patients with type 1 diabetes. Arch Endocrinol Metab. (2018) 62:47–54. doi: 10.20945/2359-3997000000008
33. Granado-Casas M, Ramírez-Morros A, Martín M, Real J, Alonso N, Valldeperas X, et al. Type 1 diabetic subjects with diabetic retinopathy show an unfavorable pattern of fat intake. Nutrients. (2018) 10:1184. doi: 10.3390/nu10091184
34. Snell-Bergeon JK, Chartier-Logan C, Maahs DM, Ogden LG, Hokanson JE, Kinney GL, et al. Adults with type 1 diabetes eat a high-fat atherogenic diet that is associated with coronary artery calcium. Diabetologia. (2009) 52:801–9. doi: 10.1007/s00125-009-1280-4
35. Aljabri KS, Bokhari SA, Khan MJ. Glycemic changes after vitamin D supplementation in patients with type 1 diabetes mellitus and vitamin D deficiency. Ann Saudi Med. (2010) 30:454–8. doi: 10.4103/0256-4947.72265
36. Habibian N, Amoli MM, Abbasi F, Rabbani A, Alipour A, Sayarifard F, et al. Role of vitamin D and vitamin D receptor gene polymorphisms on residual beta cell function in children with type 1 diabetes mellitus. Pharmacol Rep. (2019) 71:282–8. doi: 10.1016/j.pharep.2018.12.012
37. Khoo A-L, Joosten I, Michels M, Woestenenk R, Preijers F, He X-H, et al. 1, 25-Dihydroxyvitamin D3 inhibits proliferation but not the suppressive function of regulatory T cells in the absence of antigen-presenting cells. Immunology. (2011) 134:459–68. doi: 10.1111/j.1365-2567.2011.03507.x
38. Simpson M, Brady H, Yin X, Seifert J, Barriga K, Hoffman M, et al. No association of vitamin D intake or 25-hydroxyvitamin D levels in childhood with risk of islet autoimmunity and type 1 diabetes: the Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia. (2011) 54:2779. doi: 10.1007/s00125-011-2278-2
39. Bizzarri C, Pitocco D, Napoli N, Di Stasio E, Maggi D, Manfrini S, et al. No protective effect of calcitriol on β-cell function in recent-onset type 1 diabetes: the IMDIAB XIII trial. Diabetes Care. (2010) 33:1962–3. doi: 10.2337/dc10-0814
40. Nicoglou A, Merlin F. Epigenetics: a way to bridge the gap between biological fields. Stud History Philos Biol Biomed Sci. (2017) 66:73–82. doi: 10.1016/j.shpsc.2017.10.002
41. Jerram ST, Dang MN, Leslie RD. The role of epigenetics in type 1 diabetes. Curr Diabetes Rep. (2017) 17:89. doi: 10.1007/s11892-017-0916-x
42. Zullo A, Sommese L, Nicoletti G, Donatelli F, Mancini FP, Napoli CJTR. Epigenetics and type 1 diabetes: mechanisms and translational applications. Trans Res. (2017) 185:85–93. doi: 10.1016/j.trsl.2017.05.002
43. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. (2006) 31:89–97. doi: 10.1016/j.tibs.2005.12.008
44. Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. (2011) 12:647–56. doi: 10.1038/embor.2011.110
45. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. (2009) 324:930–5. doi: 10.1126/science.1170116
46. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. (2013) 502:472–9. doi: 10.1038/nature12750
47. Kindt AS, Fuerst RW, Knoop J, Laimighofer M, Telieps T, Hippich M, et al. Allele-specific methylation of type 1 diabetes susceptibility genes. J Autoimmunity. (2018) 89:63–74. doi: 10.1016/j.jaut.2017.11.008
48. Stefan M, Zhang W, Concepcion E, Yi Z, Tomer Y. DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J Autoimmun. (2014) 50:33–7. doi: 10.1016/j.jaut.2013.10.001
49. Fradin D, Le Fur S, Mille C, Naoui N, Groves C, Zelenika D, et al. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS ONE. (2012) 7:e36278. doi: 10.1371/journal.pone.0036278
50. Rui J, Deng S, Lebastchi J, Clark PL, Usmani-Brown S, Herold KC. Methylation of insulin DNA in response to proinflammatory cytokines during the progression of autoimmune diabetes in NOD mice. Diabetologia. (2016) 59:1021–9. doi: 10.1007/s00125-016-3897-4
51. Belot M-P, Fradin D, Mai N, Le Fur S, Zélénika D, Kerr-Conte J, et al. CpG methylation changes within the IL2RA promoter in type 1 diabetes of childhood onset. PLoS ONE. (2013) 8:e68093. doi: 10.1371/journal.pone.0068093
52. Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics. (2010) 3:33. doi: 10.1186/1755-8794-3-33
53. Golson ML, Kaestner KH. Epigenetics in formation, function, and failure of the endocrine pancreas. Mol Metab. (2017) 6:1066–76. doi: 10.1016/j.molmet.2017.05.015
54. Neiman D, Moss J, Hecht M, Magenheim J, Piyanzin S, Shapiro AMJ, et al. Islet cells share promoter hypomethylation independently of expression, but exhibit cell-type-specific methylation in enhancers. Proc Natl Acad Sci USA. (2017) 114:13525–30. doi: 10.1073/pnas.1713736114
55. Sommese L, Benincasa G, Lanza M, Sorriento A, Schiano C, Lucchese R, et al. Novel epigenetic-sensitive clinical challenges both in type 1 and type 2 diabetes. J Diabetes Comp. (2018) 32:1076–84. doi: 10.1016/j.jdiacomp.2018.08.012
56. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
57. Chuang JC, Jones PA. Epigenetics and MicroRNAs. Pediatric Res. (2007) 61:24–9. doi: 10.1203/pdr.0b013e3180457684
58. Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. (2010) 79:351–79. doi: 10.1146/annurev-biochem-060308-103103
59. Martínez-Hernández R, Serrano-Somavilla A, Ramos-Leví A, Sampedro-Nuñez M, Lens-Pardo A, De Nova JLM, et al. Integrated miRNA and mRNA expression profiling identifies novel targets and pathological mechanisms in autoimmune thyroid diseases. EBioMedicine. (2019) 50:329–42. doi: 10.1016/j.ebiom.2019.10.061
60. Fernández-Ruiz JC, Ramos-Remus C, Sánchez-Corona J, Castillo-Ortiz JD, Castañeda-Sánchez JJ, Bastian Y, et al. Analysis of miRNA expression in patients with rheumatoid arthritis during remission and relapse after a 5-year trial of tofacitinib treatment. Int Immunopharmacol. (2018) 63:35–42. doi: 10.1016/j.intimp.2018.07.028
61. Assmann TS, Recamonde-Mendoza M, Puñales M, Tschiedel B, Canani LH, Crispim DJDr, et al. MicroRNA expression profile in plasma from type 1 diabetic patients: case-control study and bioinformatic analysis. Diabetes Res Clin Pract. (2018) 141:35–46. doi: 10.1016/j.diabres.2018.03.044
62. Krishnan P, Syed F, Jiyun Kang N, Mirmira RG, Evans-Molina C. Profiling of RNAs from human islet-derived exosomes in a model of type 1 diabetes. Int J Mol Sci. (2019) 20:5903. doi: 10.3390/ijms20235903
63. García-Díaz DF, Pizarro C, Camacho-Guillén P, Codner E, Soto N, Pérez-Bravo F. Expression of miR-155, miR-146a, and miR-326 in T1D patients from Chile: relationship with autoimmunity and inflammatory markers. Arch Endocrinol Metab. (2018) 62:34–40. doi: 10.20945/2359-3997000000006
64. Garavelli S, Bruzzaniti S, Tagliabue E, Prattichizzo F, Di Silvestre D, Perna F, et al. Blood co-circulating extracellular microRNAs and immune cell subsets associate with type 1 diabetes severity. Int J Mol Sci. (2020) 21:477. doi: 10.3390/ijms21020477
65. O'neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of toll-like receptor signalling. Nat Rev Immunol. (2011) 11:163–75. doi: 10.1038/nri2957
66. De Jong V, Van der Slik A, Laban S, van‘t Slot R, Koeleman B, Zaldumbide A, et al. Survival of autoreactive T lymphocytes by microRNA-mediated regulation of apoptosis through TRAIL and Fas in type 1 diabetes. Genes Immunity. (2016) 17:342–8. doi: 10.1038/gene.2016.29
67. Satake E, Pezzolesi MG, Md Dom ZI, Smiles AM, Niewczas MA, Krolewski AS. Circulating miRNA profiles associated with hyperglycemia in patients with type 1 diabetes. Diabetes. (2018) 67:1013–23. doi: 10.2337/db17-1207
68. Liu L, Yan J, Xu H, Zhu Y, Liang H, Pan W, et al. Two novel MicroRNA biomarkers related to β-cell damage and their potential values for early diagnosis of type 1 diabetes. J Clin Endocrinol Metab. (2018) 103:1320–9. doi: 10.1210/jc.2017-01417
69. El-Samahy MH, Adly AA, Elhenawy YI, Ismail EA, Pessar SA, Mowafy ME, et al. Urinary miRNA-377 and miRNA-216a as biomarkers of nephropathy and subclinical atherosclerotic risk in pediatric patients with type 1 diabetes. J Diabetes Complications. (2018) 32:185–92. doi: 10.1016/j.jdiacomp.2017.10.014
70. Taudt A, Colomé-Tatché M, Johannes F. Genetic sources of population epigenomic variation. Nat Rev Genet. (2016) 17:319–32. doi: 10.1038/nrg.2016.45
71. Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta. (2014) 1839:1362–72. doi: 10.1016/j.bbagrm.2014.02.007
72. Horton JR, Gale M, Yan Q, Cheng X. The molecular basis of histone demethylation. In: Kaneda A, Tsukada YI, editors. DNA and Histone Methylation as Cancer Targets. Cham: Springer International Publishing. (2017). p. 151–219. doi: 10.1007/978-3-319-59786-7_7
73. Gräff J, Tsai L-H. Histone acetylation: molecular mnemonics on the chromatin. Nat Rev Neurosci. (2013) 14:97–111. doi: 10.1038/nrn3427
74. Cao J, Yan Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front Oncol. (2012) 2:26. doi: 10.3389/fonc.2012.00026
75. Ryu H-Y, Zhao D, Li J, Su D, Hochstrasser M. Histone sumoylation promotes Set3 histone-deacetylase complex-mediated transcriptional regulation. Nucleic Acids Res. (2020) 48:12151–68. doi: 10.1093/nar/gkaa1093
76. Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes. (2008) 57:3189–98. doi: 10.2337/db08-0645
77. Miao F, Chen Z, Zhang L, Liu Z, Wu X, Yuan Y-C, et al. Profiles of epigenetic histone post-translational modifications at type 1 diabetes susceptible genes. J Biol Chem. (2012) 287:16335–45. doi: 10.1074/jbc.M111.330373
78. Chen SS, Jenkins AJ, Majewski H. Elevated plasma prostaglandins and acetylated histone in monocytes in Type 1 diabetes patients. Diabet Med. (2009) 26:182–6. doi: 10.1111/j.1464-5491.2008.02658.x
79. Jiménez-Chillarón JC, Díaz R, Martínez D, Pentinat T, Ramón-Krauel M, Ribó S, et al. The role of nutrition on epigenetic modifications and their implications on health. Biochimie. (2012) 94:2242–63. doi: 10.1016/j.biochi.2012.06.012
80. Sherwood WB, Bion V, Lockett GA, Ziyab AH, Soto-Ramírez N, Mukherjee N, et al. Duration of breastfeeding is associated with leptin (LEP) DNA methylation profiles and BMI in 10-year-old children. Clin Epigenetics. (2019) 11:128. doi: 10.1186/s13148-019-0727-9
81. Matt SM, Allen JM, Lawson MA, Mailing LJ, Woods JA, Johnson RW. Butyrate and dietary soluble fiber improve neuroinflammation associated with aging in mice. Front Immunol. (2018) 9:1832. doi: 10.3389/fimmu.2018.01832
82. González-Becerra K, Ramos-Lopez O, Barrón-Cabrera E, Riezu-Boj JI, Milagro FI, Martínez-López E, et al. Fatty acids, epigenetic mechanisms and chronic diseases: a systematic review. Lipids Health Dis. (2019) 18:178. doi: 10.1186/s12944-019-1120-6
83. Obeid R, Hübner U, Bodis M, Graeber S, Geisel J. Effect of adding B-vitamins to vitamin D and calcium supplementation on CpG methylation of epigenetic aging markers. Nutr Metab Cardiovasc Dis. (2018) 28:411–7. doi: 10.1016/j.numecd.2017.12.006
84. Hu Y, Shi P, He K, Zhu YQ, Yang F, Yang M, et al. Methylation of Tcf712 promoter by high-fat diet impairs β-cell function in mouse pancreatic islets. Diabetes Metab Res Rev. (2018) 34:e2980. doi: 10.1002/dmrr.2980
85. Kowluru RA. Retinopathy in a diet-induced type 2 diabetic rat model and role of epigenetic modifications. Diabetes. (2020) 69:689–98. doi: 10.2337/db19-1009
86. Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, et al. A maternal high-fat diet induces DNA methylation changes that contribute to glucose intolerance in offspring. Front Endocrinol. (2019) 10:871. doi: 10.3389/fendo.2019.00871
87. Cai W-J, Liang X-F, Yuan X-C, Li A-X, He S. Changes of DNA methylation pattern in metabolic pathways induced by high-carbohydrate diet contribute to hyperglycemia and fat deposition in grass carp (Ctenopharyngodon idellus). Front Endocrinol. (2020) 11:398. doi: 10.3389/fendo.2020.00398
88. Remely M, Aumueller E, Merold C, Dworzak S, Hippe B, Zanner J, et al. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene. (2014) 537:85–92. doi: 10.1016/j.gene.2013.11.081
89. Ramos-Lopez O, Samblas M, Milagro FI, Zulet MA, Mansego ML, Riezu-Boj JI, et al. Association of low dietary folate intake with lower CAMKK2 gene methylation, adiposity, and insulin resistance in obese subjects. Nutr Res. (2018) 50:53–62. doi: 10.1016/j.nutres.2017.11.007
90. Li W, Tang R, Ma F, Ouyang S, Liu Z, Wu J. Folic acid supplementation alters the DNA methylation profile and improves insulin resistance in high-fat-diet-fed mice. J Nutr Biochem. (2018) 59:76–83. doi: 10.1016/j.jnutbio.2018.05.010
91. Yadav DK, Shrestha S, Lillycrop KA, Joglekar CV, Pan H, Holbrook JD, et al. Vitamin B12 supplementation influences methylation of genes associated with type 2 diabetes and its intermediate traits. Epigenomics. (2018) 10:71–90. doi: 10.2217/epi-2017-0102
92. Hu F, Tong J, Deng B, Zheng J, Lu C. MiR-495 regulates macrophage M1/M2 polarization and insulin resistance in high-fat diet-fed mice via targeting FTO. Pflügers Archiv Eur J Physiol. (2019) 471:1529–37. doi: 10.1007/s00424-019-02316-w
93. Dong L, Hou X, Liu F, Tao H, Zhang Y, Zhao H, et al. Regulation of insulin resistance by targeting the insulin-like growth factor 1 receptor with microRNA-122-5p in hepatic cells. Cell Biol Int. (2019) 43:553–64. doi: 10.1002/cbin.11129
94. Adi N, Adi J, Lassance-Soares RM, Kurlansky P, Yu H, Webster KA. High protein/fish oil diet prevents hepatic steatosis in NONcNZO10 mice; association with diet/genetics-regulated micro-RNAs. J Diabetes Metab. (2016) 7:676. doi: 10.4172/2155-6156.1000676
95. Liu R, Wang M, Li E, Yang Y, Li J, Chen S, et al. Dysregulation of microRNA-125a contributes to obesity-associated insulin resistance and dysregulates lipid metabolism in mice. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158640. doi: 10.1016/j.bbalip.2020.158640
96. Yang YM, Seo SY, Kim TH, Kim SG. Decrease of microRNA-122 causes hepatic insulin resistance by inducing protein tyrosine phosphatase 1B, which is reversed by licorice flavonoid. Hepatology. (2012) 56:2209–20. doi: 10.1002/hep.25912
97. Fussbroich D, Kohnle C, Schwenger T, Driessler C, Dücker RP, Eickmeier O, et al. A combination of LCPUFAs regulates the expression of miRNA-146a-5p in a murine asthma model and human alveolar cells. Prostaglandins Other Lipid Mediat. (2020) 147:106378. doi: 10.1016/j.prostaglandins.2019.106378
98. Attig L, Vigé A, Gabory A, Karimi M, Beauger A, Gross M-S, et al. Dietary alleviation of maternal obesity and diabetes: increased resistance to diet-induced obesity transcriptional and epigenetic signatures. PLoS ONE. (2013) 8:e66816. doi: 10.1371/journal.pone.0066816
99. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol. (2017) 18:552. doi: 10.1038/ni.3713
100. Lee SM, Yang H, Tartar DM, Gao B, Luo X, Ye SQ, et al. Prevention and treatment of diabetes with resveratrol in a non-obese mouse model of type 1 diabetes. Diabetologia. (2011) 54:1136–46. doi: 10.1007/s00125-011-2064-1
101. Chia JSJ, McRae JL, Enjapoori AK, Lefèvre CM, Kukuljan S, Dwyer KM. Dietary cows' milk protein A1 beta-casein increases the incidence of T1D in NOD Mice. Nutrients. (2018) 10:1291. doi: 10.3390/nu10091291
102. Trivedi MS, Hodgson NW, Walker SJ, Trooskens G, Nair V, Deth RC. Epigenetic effects of casein-derived opioid peptides in SH-SY5Y human neuroblastoma cells. Nutr Metab. (2015) 12:54. doi: 10.1186/s12986-015-0050-1
103. Niinistö S, Takkinen H-M, Erlund I, Ahonen S, Toppari J, Ilonen J, et al. Fatty acid status in infancy is associated with the risk of type 1 diabetes-associated autoimmunity. Diabetologia. (2017) 60:1223–33. doi: 10.1007/s00125-017-4280-9
104. Redondo M, Grant S, Davis A, Greenbaum C, Biobank TDE. Dissecting heterogeneity in paediatric type 1 diabetes: association of TCF7L2 rs7903146 TT and low-risk human leukocyte antigen (HLA) genotypes. Diabetic Med. (2017) 34:286–90. doi: 10.1111/dme.13123
105. Eccel Prates R, Beretta MV, Nascimento FV, Bernaud FR, de Almeira JC, Rodrigues TC. Saturated fatty acid intake decreases serum adiponectin levels in subjects with type 1 diabetes. Diabetes Res Clin Pract. (2016) 116:205–11. doi: 10.1016/j.diabres.2016.03.019
106. Perfilyev A, Dahlman I, Gillberg L, Rosqvist F, Iggman D, Volkov P, et al. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: a randomized controlled trial. Am J Clin Nutr. (2017) 105:991–1000. doi: 10.3945/ajcn.116.143164
107. Liu Z, Ding Z, Lynch EC, McCauley N, Zhou Y, Zhang KK, et al. Pregestational diet transition to normal-fat diet avoids the deterioration of pancreatic β-cell function in male offspring induced by maternal high-fat diet. J Nutr Biochem. (2020) 86:108495. doi: 10.1016/j.jnutbio.2020.108495
108. Salbaum JM, Kappen C. Genetic and epigenomic footprints of folate. Progr Mol Biol Trans Sci. (2012) 108:129–58. doi: 10.1016/B978-0-12-398397-8.00006-X
109. Audhya T. Role of B Vitamins in Biological Methylation. Health Diagnostics and Research Institute. (2012).
110. Nie L, Shuai L, Zhu M, Liu P, Xie Z-F, Jiang S, et al. The landscape of histone modifications in a high-fat Diet-Induced Obese (DIO) mouse model. Mol Cell Proteomics. (2017) 16:1324–34. doi: 10.1074/mcp.M117.067553
111. Lecomte V, Kaakoush NO, Maloney CA, Raipuria M, Huinao KD, Mitchell HM, et al. Changes in gut microbiota in rats fed a high fat diet correlate with obesity-associated metabolic parameters. PLoS ONE. (2015) 10:e0126931. doi: 10.1371/journal.pone.0126931
112. Prieto I, Hidalgo M, Segarra AB, Martínez-Rodríguez AM, Cobo A, Ramirez M, et al. Influence of a diet enriched with virgin olive oil or butter on mouse gut microbiota and its correlation to physiological and biochemical parameters related to metabolic syndrome. PLoS ONE. (2018) 13:e0190368. doi: 10.1371/journal.pone.0190368
113. Eswaran S, Muir J, Chey WD. Fiber and functional gastrointestinal disorders. Am J Gastroenterol. (2013) 108:718. doi: 10.1038/ajg.2013.63
114. Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. (2011) 5:220–30. doi: 10.1038/ismej.2010.118
115. Segain JP, Raingeard de la Blétière D, Bourreille A, Leray V, Gervois N, Rosales C, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn's disease. Gut. (2000) 47:397–403. doi: 10.1136/gut.47.3.397
116. Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. (2013) 145:396–406.e10. doi: 10.1053/j.gastro.2013.04.056
117. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. (2013) 341:569–73. doi: 10.1126/science.1241165
118. Durazzo M, Ferro A, Gruden G. Gastrointestinal microbiota and type 1 diabetes mellitus: the state of art. J Clin Med. (2019) 8:1843. doi: 10.3390/jcm8111843
Keywords: type 1 diabetes, diet, epigenetics, histone modifications, DNA methylation, micro-RNA
Citation: Kohil A, Al-Asmakh M, Al-Shafai M and Terranegra A (2021) The Interplay Between Diet and the Epigenome in the Pathogenesis of Type-1 Diabetes. Front. Nutr. 7:612115. doi: 10.3389/fnut.2020.612115
Received: 30 September 2020; Accepted: 22 December 2020;
Published: 28 January 2021.
Edited by:
Rosita Gabbianelli, University of Camerino, ItalyReviewed by:
Melita Vidakovic, University of Belgrade, SerbiaSharon Ross, National Cancer Institute (NCI), United States
Copyright © 2021 Kohil, Al-Asmakh, Al-Shafai and Terranegra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annalisa Terranegra, YXRlcnJhbmVncmFAc2lkcmEub3Jn