
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Nutr. , 19 December 2019
Sec. Nutritional Immunology
Volume 6 - 2019 | https://doi.org/10.3389/fnut.2019.00187
This article is part of the Research Topic The Molecular Aspects of Gluten-Related Disorders View all 7 articles
 Amaia Jauregi-Miguel1
Amaia Jauregi-Miguel1 Izortze Santin2,3
Izortze Santin2,3 Koldo Garcia-Etxebarria1Ane Olazagoitia-Garmendia1Irati Romero-Garmendia1Maialen Sebastian-delaCruz1Iñaki Irastorza4 Spanish Consortium for the Genetics of Celiac Disease
Koldo Garcia-Etxebarria1Ane Olazagoitia-Garmendia1Irati Romero-Garmendia1Maialen Sebastian-delaCruz1Iñaki Irastorza4 Spanish Consortium for the Genetics of Celiac Disease Ainara Castellanos-Rubio1,5*
Ainara Castellanos-Rubio1,5* Jose Ramón Bilbao1,3*
Jose Ramón Bilbao1,3*Celiac disease (CD) patients present a loss of intestinal barrier function due to structural alterations in the tight junction (TJ) network, the most apical unions between epithelial cells. The association of TJ-related gene variants points to an implication of this network in disease susceptibility. This work aims to characterize the functional implication of TJ-related, disease-associated loci in CD pathogenesis. We performed an association study of 8 TJ-related gene variants in a cohort of 270 CD and 91 non-CD controls. The expression level of transcripts located in the associated SNP region was analyzed by RT-PCR in several human tissues and in duodenal biopsies of celiac patients and non-CD controls. (si)RNA-driven silencing combined with gliadin in the Caco2 intestinal cell line was used to analyze the implication of transcripts from the associated region in the regulation of TJ genes. We replicated the association of rs6962966*A variant [p = 0.0029; OR = 1.88 (95%1.24–2.87)], located in an intron of TJ-related MAGI2 coding gene and upstream of RP4-587D13.2 transcript, bioinformatically classified as a long non-coding RNA (lncRNA). The expression of both genes is correlated and constitutively downregulated in CD intestine. Silencing of lncRNA decreases the levels of MAGI2 protein. At the same time, silencing of MAGI2 affects the expression of several TJ-related genes. The associated region is functionally altered in disease, probably affecting CD-related TJ genes.
Celiac disease (CD) is a chronic enteropathy, characterized by villous atrophy and inflammation of the intestinal mucosa, that develops in genetically predisposed individuals. The disease is caused by an inappropriate immune response to ingested gluten, a protein consisting of soluble gliadin and insoluble glutenin components that is found mostly in wheat, but also in barley, rye and oat. There is evidence supporting that various environmental factors, such as repeated gastrointestinal infections (1, 2) or early infant feeding (3, 4) may also contribute to triggering the disease. A lifelong gluten-free diet (GFD) is the only effective treatment for CD that achieves complete remission of the symptoms.
CD is a complex genetic disorder, and multiple genes contribute to disease risk. The most important genetic determinants, Human Leukocyte Antigen (HLA) molecules HLA-DQ2 and HLA-DQ8, are necessary but not sufficient to explain the predisposition to CD. Genome wide association (GWA) and follow-up genetic studies have identified several additional genomic regions associated to the disease, which probably contain CD susceptibility genes, but our understanding of the disease pathogenesis is still limited (5).
In fact, the majority of single-nucleotide polymorphisms (SNPs) associated with increased disease risk are located in non-coding regions of the genome (6–8). Some of them map to long non-coding RNAs (lncRNAs) transcripts longer than 200 bases in length, with no protein-coding potential, that have been described to participate in genomic regulation (9). The analysis of the expression profiles of lncRNAs on autoimmune disease-associated regions has shown that they are enriched in autoimmune-disease loci, suggesting that they may be crucial to explain GWAS findings (10, 11). Although their mechanisms of action are diverse and still not very well-characterized, lncRNAs regulate gene expression at transcriptional and post-transcriptional levels and are involved in a range of developmental processes, adding complexity to our understanding of how gene expression is regulated (12). On the other hand, expression quantitative trait loci (eQTL) analyses have demonstrated that certain SNPs associated with complex diseases can affect the expression of lncRNAs, and thus lncRNA genes can be considered candidate genes for disease-susceptibility (13). In the case of CD, it has recently been shown that a lncRNA in the IL18RAP locus that harbors a disease-associated SNP is a key regulator of genes involved in the NFκB pathway, which is known to be constitutively activated in the small intestine of patients with CD (14, 15).
Apart from GWAS studies, pathway analyses contribute to the reconstruction of altered biological networks that are potentially pathological and could help identify functional candidates that participate to the genetic susceptibility. In the case of CD, although an aberrant immune response is the main force driving the disease, whole genome expression analyses have identified important dysfunctions of other complex biological processes that could be involved in CD development, including alterations in the expression of genes related to intestinal permeability (16–20). Impairment of the epithelial barrier and increased permeability have been shown to be implicated in the development of CD (20) and other gastrointestinal inflammatory diseases like Crohn's Disease (21) and Ulcerative Colitis (22).
The permeability of the intestinal epithelium is dependent on the regulation of intercellular tight junctions (TJ), a continuous, circumferential, belt-like structure at the luminal end of the intercellular space that functions as a barrier. Changes in the expression, distribution, and phosphorylation of TJ proteins have been observed in CD and malfunction of this pathway could have an important role in the augmented intestinal permeability observed in the disease (23, 24). These alterations persist in asymptomatic CD patients who are on GFD (25) and ultrastructural and functional abnormalities in TJs appear also in antibody-negative, asymptomatic first-degree relatives (26), supporting a genetic origin of the pathway alterations and a possible role in the initial stages of the disease. Different studies in European populations have found polymorphisms in several TJ genes, including adapter proteins MAGI2 and PARD3, together with MYO9B, a cytoskeleton protein involved in TJ assembly, that are associated with CD (27–29).
The MAGI proteins (Membrane Associated Guanylate-kinase proteins with Inverted domain arrangement) are evolutionarily conserved scaffolding proteins with several protein–protein interaction domains to coordinate signaling complexes (30). MAGI-2 protein (also known as AIP1/S-SCAM/ARIP1) is mainly expressed in the brain at synaptic junctions but has also been identified in the TJ plaque, where it controls epithelial integrity. Interestingly, apart from the association with CD, linkage analyses have shown that MAGI2 is located in a genomic region harboring susceptibility genes for IBD (31).
With this on mind, we hypothesized that TJ genes could be relevant candidates for gut disorders and specifically for CD. Thus, the aim of the study was to replicate previous association results and to perform gene expression and functional analyses of the potential candidate genes in the associated regions.
CD was diagnosed at the Pediatric Gastroenterology Clinic (Cruces University Hospital), according to the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition criteria in force at the time of recruitment. The study was approved by the ethics committee of Cruces University Hospital (ref. CEIC-E08/59, CEIC-E13/20, and CEIC-E16/46) and informed consent was obtained from patients or their parents. A total of 48 duodenal biopsy samples were taken from 32 patients divided in 3 categories: 16 biopsies from CD patients at the time of diagnosis (symptomatic and on a gluten-containing diet; 10 girls/6 boys; mean age at diagnosis 2.9 years, range 1.3–9.3 years), 16 biopsies from the same patients after at least 2 years on strict GFD (asymptomatic, antibody negative, and with a recovered intestinal epithelium) and 16 samples from non-celiac individuals (5 girls/11 boys; mean age 7.7 years, range 1.1–13.0 years) who had been subjected to endoscopy but had no signs of gut inflammation. After collection, samples were immediately frozen and stored in liquid nitrogen until use.
The C2BBe1 human colon carcinoma cell line (clone of Caco-2) was selected as a well-established in vitro model to study the tight junctions in CD (32, 33). Cells were maintained in DMEM supplemented with 10% heat inactivated FBS, 1% non-essential amino acids and 1% penicillin-streptomycin) in tissue culture flask.
DNA samples from 270 celiac patients and 91 unselected blood donors from the CEGEC (Spanish Consortium for Genetics of Celiac Disease) collection were used for the association study. DNA samples were extracted from whole blood using conventional methods and stored at −80°C until use. The CEGEC study was approved by the Ethics Committees or Institutional Review Board of all participating institutions. Samples were genotyped in the BioMark HD™ platform at the Sequencing and Genotyping Unit of the University of the Basque Country (UPV/EHU). Fluidigm SNP assays were designed according to the protocols recommended by the manufacturer (Fluidigm Corporation, San Francisco, CA, USA) (Table S1). We selected 3 SNPs in the MAGI2 locus, 2 in PARD3, and 3 in MYO9B, based on previously published association results (29, 34, 35). The SNP Genotyping Analysis software by Fluidigm was used for genotype calling. Clusters were evaluated manually, and doubtful calls were recalled or rejected. To ensure population homogeneity, an identity-by-state analysis and multidimensional scaling were performed before standard χ2 association analysis using Plink 1.07 (36).
The protein-coding capacity of RP4-587D13.2 was evaluated in silico. All possible open reading frames (ORFs) were identified using ORF Finder (NCBI) (37) and the coding potential of the transcript was assayed with Phylogenetic Codon Substitution Frequencies (PhyloCSF) based on phylogenetic conservation (38), as well as with the coding potential assessment tool (CPAT) that uses a model built with open reading frame size and coverage together with codon and hexamer usage bias (39).
To analyze the distribution of RP4-587D13.2 RNA in the nuclear and cytoplasmic cell compartments of C2BBe1 cells, nuclei were isolated using hypotonic lysis buffer (10 mM Hepes, 10 mM KCl, 0.1 mM EDTA and %0.8 Triton-X) as previously described. For relative quantification, the amounts of nuclear RP4-587D13.2, Lnc13 (nuclear control) and RPLP0 (cytoplasmic control) RNA were measured by RT-QPCR and compared to the amount of each RNA in whole cells.
For in vitro cell transfection and gliadin stimulation, 5 × 104 cells/well were seeded in 24-well plates with 500 μl of supplemented medium without antibiotics. Reverse transfection was performed with 30 nM of DsiRNA against human MAGI2 (IDT corp. Cat. No. hs.Ri.MAGI2.13.1), RP4-587D13.2 (5′ -GCAGUUUAAUAUCAUGUAUUGAAAA-3′) or negative control DsiRNA (IDT corp. Cat. No. 51-01-14-01) using Lipofectamine RNAimax reagent (Thermo Fisher Scientific, Waltham, MA, USA) and following the manufacturer's instructions. Samples were incubated overnight in 5% CO2 at 37°C before adding fresh medium with antibiotic, then incubated again for a total of 48 h from the start of transfection. Then a well-established protocol was used to mimic gliadin-induced inflammatory response in vitro (40–42). Cells were exposed to 1 mg/ml of pepsin-trypsin digested gliadin (PTG) or BSA (PT-BSA) for 4 h and harvested for RNA or protein extraction. Pepsin-trypsin digest of gliadin (PTG) (Merck, Darmstadt, Germany 1.07185; 9002-07-7, and G3375-25, respectively) was prepared as described previously (43). An enzymatic digest of BSA (PT-BSA) was prepared in the same way and used as a negative control of stimulation.
Whole cell extracts were directly lysed into 2x Laemmli Sample buffer containing 5% β- mercaptoethanol and stored at −20°C until use. Total protein extracts were heat-treated at 95°C for 5 min, fractionated by SDS-PAGE and transferred to nitrocellulose membranes using the Trans-Blot Turbo Transfer apparatus (BioRad, Hercules, California, USA). After incubation with 5% non-fat milk in Tris-buffered saline with 0.05% Tween (TBS-T) for 1 h, the membrane was incubated at 4°C overnight with polyclonal antibodies against MAGI2 (1:500) (ABCAM, Cat. No. ab97343) and β-Tubulin (1:1,000) (ABCAM, Cat. No. ab18207). Membranes were washed three times for 10 min and incubated with horseradish peroxidase-conjugated anti-mouse (1:2,000) or anti-rabbit (1:10,000) antibodies for 1 h at room temperature in 5% BSA in TBS-T (Jackson ImmunoResearch Laboratories, Inc.; Cat No. 111-035-003 and 115-035-003, respectively). Blots were washed with TBS-T three times and visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher, Cat No. 34094) on the ChemiDoc MP system (BioRad, Hercules, California, USA). The quantification of band intensities was done using the Image Lab 4.1 software and results were normalized to β-tubulin.
Total RNA and DNA were prepared with the NucleoSpin RNA kit and NucleoSpin Blood kit, respectively (Macherey-Nagel, Düren, Germany) according to the protocols provided. Frozen biopsy samples were disrupted with disposable plastic pellet pestles prior to nucleic acid extraction. Cells were lysed and homogenized using the buffers supplied with the kits. The concentration and purity of the RNA and DNA were determined by UV absorbance at 260 nm on a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and samples were stored at −80°C until use. To determine the expression of MAGI2 and RP4-587D13.2 across different human tissues, a commercial Human Total RNA Master Panel II (Clontech Laboratories, Inc., Cat. No. 636643) was used.
The expression of MAGI2 and RP4-587D13.2 was analyzed in biopsy samples and caco2 cells. Total RNA was reverse transcribed with the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Inc. Berkeley, CA, USA). Expression of MAGI2 mRNA was determined by real-time (RT)-PCR using specific TaqMan Gene Expression Assays (thermoFisher Scientific) in a Eco Real-Time PCR System (Illumina, Inc., San Diego, CA, USA). The expression of RP4-587D13.2 lncRNA was measured using custom-designed primers (F: 5′-GGTGCTGGAAATTCATCAGTG-3′; R: 5′-TGACCACATGACTGACACCA-3′) and Quantitect SYBR Green master mix (Qiagen, Hilden, Germany). The specificity of the primers was tested by in-silico PCR on the University of California Santa Cruz web server (http://genome.ucsc.edu/cgi-bin/hgPcr) and melting curve analysis were performed in all qPCR reactions.
The expression levels of TJ-related genes were measured by RT-PCR on a Fluidigm BioMark dynamic array system following the 48.48 Fast Real-Time PCR Workflow (Fluidigm Corporation, San Francisco, CA, USA). Candidate genes with central functions in the TJ and TJ-related Toll Like Receptor (TLR) networks according to the KEGG database (http://www.genome.jp/kegg) were selected (Table S2). The reverse transcribed cDNA from each sample was preamplified using the Fluidigm Gene Expression Specific Target Amplification kit (Fluidigm Corporation, San Francisco, CA, USA) and amplified using TaqMan Universal PCR master mix (Thermo Fisher Scientific, Waltham, MA, USA). Complete results are available at the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with accession number GSE84729. The expression of the housekeeping gene RPLP0 (large ribosomal protein) was quantified in each experiment and used as an endogenous control of input RNA. The relative expression of each gene was calculated using the accurate Ct method as previously described (44).
The Commercial TaqMan assays probes (Thermo Fisher Scientific, Waltham, MA, USA) used for expression analyses are listed in Table S2. Probes that spanned an exon–exon junction were chosen to avoid the detection of genomic DNA.
In order to determine whether the risk attributed to the newly identified variant rs6962966 was related to gene expression in CD patients, the tag SNP rs2691527 (r2 = 1 with associated SNP rs6962966) was genotyped in the duodenal biopsy samples on a Eco Real-Time PCR System (Illumina, Inc., San Diego, CA, USA) using a predesigned TaqMan Genotyping assay (Thermo Fisher Scientific, Assay ID 4351379) and TaqMan genotyping Mastermix (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analyses were carried out using GraphPad Prism version 5.0 software (GraphPad Software Inc, La Jolla, CA, USA) to assess differences among groups. Non-parametric, paired (Wilcoxon matched-pairs test) tests were performed for the comparison of the expression levels between CD patients at diagnosis and after 2 years on GFD, and the Mann–Whitney U test was used to compare each disease group to the controls and unpaired t test to in the silencing experiments. Coexpression was assessed with Spearman's correlation test. Heat-maps were constructed using the FiRe Macro v2.2 for Microsoft Excel from the University of Fribourg available at http://www.unifr.ch/plantbio/FiRe/main.html. Only 2-tailed p-values below 0.05 were considered significant.
We analyzed 8 SNPs located in MAGI2, PARD3, and MYO9B, previously associated with CD. Their frequencies in CD patients and controls are listed on Table 1. We detected an association between SNP rs6962966 (GRCh38/hg38; chr7:77,804,123) in an intronic region of MAGI2 and CD risk. The SNP was in Hardy-Weinberg equilibrium in the control group and the frequency of the minor allele was higher in patients, augmenting the risk to develop CD 1.88-fold (95% CI = 1.24–2.87; p = 0.0029). No other SNPs in MAGI2, PARD3, or MYO9B were associated with CD.
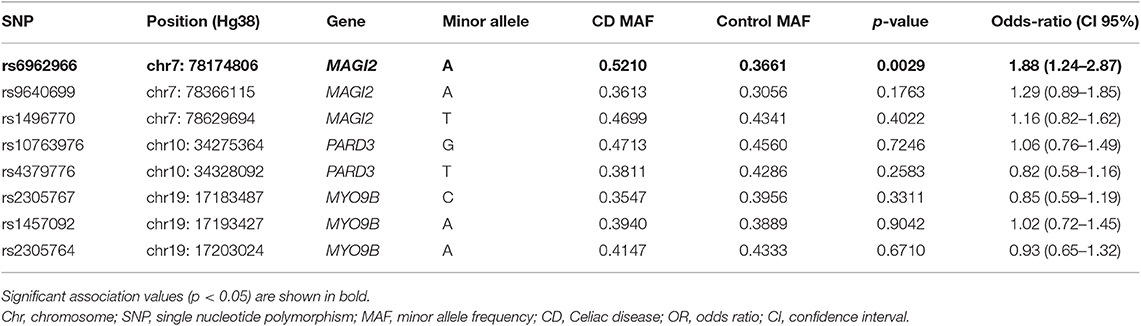
Table 1. Association analysis of single nucleotide polymorphisms in TJ-related candidate genes in CD.
In order to characterize the effects of the associated intronic SNP on disease risk, we analyzed its genomic context in search for putative regulatory elements that could be affected causing alterations in gene regulation. An analysis of current UCSC Genome browser annotations showed that protein-coding gene MAGI2 hosts different types of non-coding genes, including processed transcripts, pseudogenes, and RNA genes. The great majority of them are located in introns and no specific function or effect on their host gene have been described so far. As an exception, the antisense gene MAGI2-AS3 overlaps the first exon of MAGI2 and has been recently linked to breast cancer (45). Regarding the intron where the CD-associated rs6962966 SNP falls, we found that it also harbors a non-characterized transcript, named RP4-587D13.2 (Figure 1).
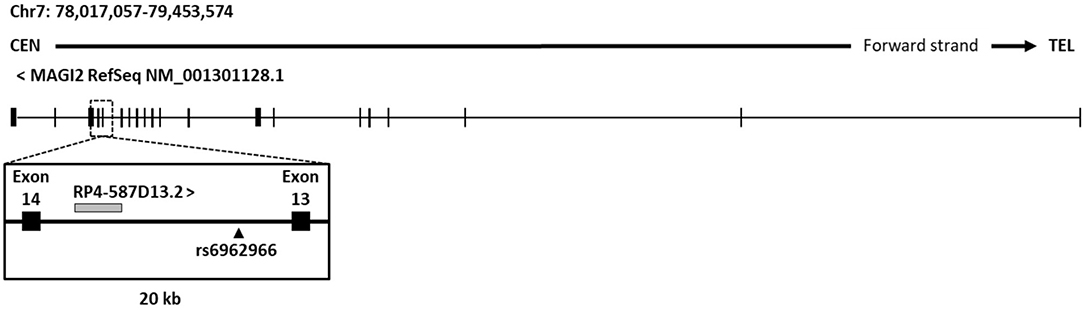
Figure 1. Genomic context of CD associated SNP rs6969266. The reference sequence of MAGI2 (NCBI) and genomic annotations of a 20-kb region around candidate SNP rs6962966 are shown.
In order to characterize the region around the associated SNP, we performed expression analysis of both RP4-587D13.2 and MAGI2 across different human tissues. The results showed that these transcripts are ubiquotiusly expressed in all human tissues analyzed, including the intestine, the CD target tissue, and are predominantly expressed in brain (Figure 2A). It can be observed that both present a similar expression pattern and are positively correlated (p = 0.0007) across tissues. We next evaluated the protein-coding capacity of RP4-587D13.2 in silico using three different approaches. Overall, the lack of functional ORFs as well as the low phylogenetic conservation and coding probability of the region suggests that RP4-587D13.2 is a long non-coding transcript (Table S3).
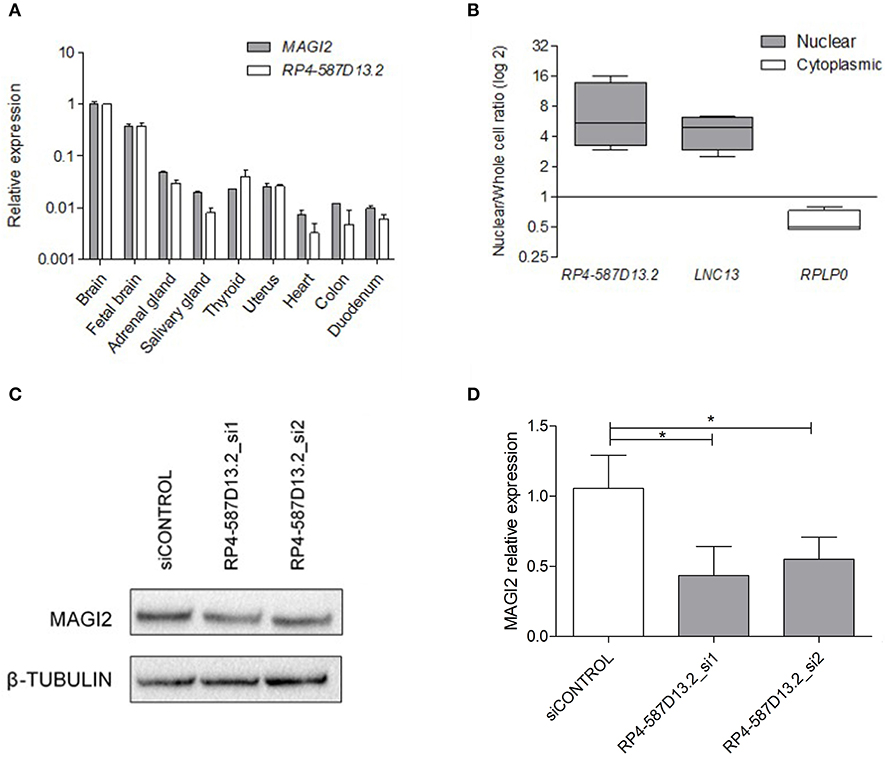
Figure 2. Characterization of RPL4-587D13.2 expression in human tissues and the C2BBe1 cell line. (A) mRNA expression of MAGI2 and RPL4-587D13.2 in RNA from 9 human tissues. Values are represented relative to the tissue with the highest expression levels (brain). (B) Subcellular localization of RPL4-587D13.2 in C2BBe1 cells. The box-plots show the ratio of nuclear to whole cell transcripts, represented as the maximum and minimum values and the mean (n = 3); LNC13 and RPLP0 are used as nuclear and cytoplasmic controls, respectively. (C) Representative western blot and (D) quantification of MAGI2 protein levels in C2BBe1 cells silenced for RPL4-587D13.2 (siRNA_1 and siRNA_2). The mean and standard deviation of MAGI2 expression relative to the control siRNA are shown (n = 3) (*0.01 < p < 0.05).
The cellular localization of the lncRNA was evaluated in C2BBe1 intestinal human cells, and results revealed that RP4-587D13.2 accumulates preferentially in the nucleus (Figure 2B). The nuclear localization of RP4-587D13.2 together with its coexpression with the neighboring MAGI2 suggests that this lncRNA could contribute to the regulation of MAGI2 expression. To test this hypothesis, we performed siRNA silencing of RP4-587D13.2 in C2Bbbe1 intestinal cells using two different siRNAs. We managed to reduce the expression of RP4-587D13.2 by 46% in intestinal cells (Figure S1) which in turn resulted in a 56% decrease in MAGI2 protein (Figures 2C,D), confirming that this lncRNA is somehow affecting the regulation of the coding gene.
In order to determine the functional significance of the MAGI2 region in the CD characteristic dysregulation of the TJ pathway, the expression of a TJ-related gene panel was analyzed in the C2BBe1 intestinal cell line. Due to the difficulty of efficiently silencing the lncRNA, probably because of its nuclear location, the effects in downstream gene expression were assessed by MAGI2 silencing. As expected, the efficiency of MAGI2 silencing was above 59% (Figures 3A,B) and silenced cells showed altered expression of CLDN1, F11R, PARD6A, PPP2R3A, TICAM2, and ZAK (Figure 3C). Interestingly, upon PTG stimulation, there was an additional decrease of MAGI2 and a stronger effect on the TJ-related genes in MAGI2-silenced cells: the changes observed in PARD6A and ZAK were more pronounced, and alterations extended to other genes of the pathway, including GNAI1 and CLDN2.
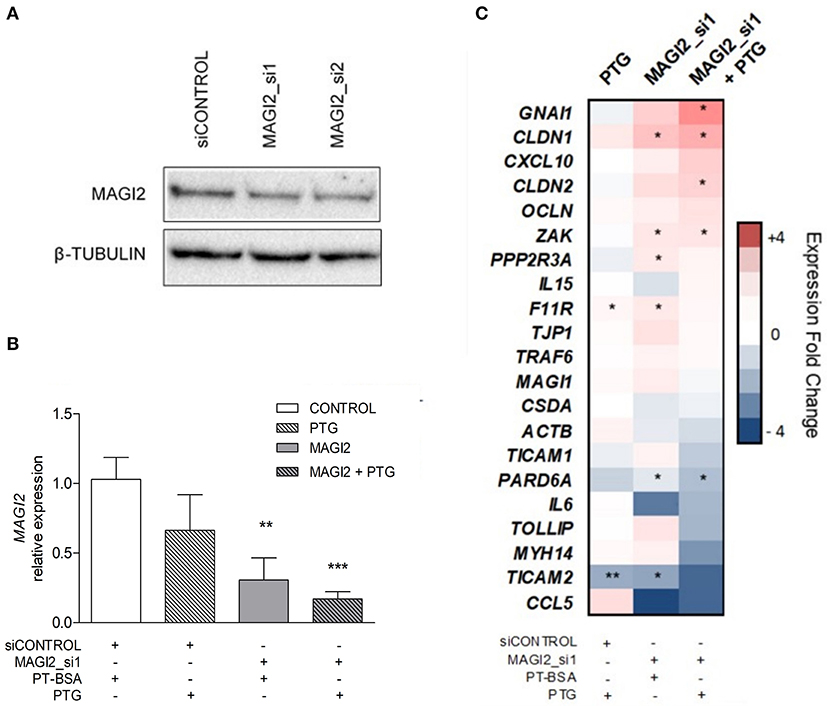
Figure 3. Effects of MAGI2 silencing and gliadin on the expression of a TJ-related genes in C2BBe1 intestinal cells. (A) Western blot of MAGI2 protein upon silencing in C2BBe1 cells. (B) Quantification of MAGI2 protein levels under different cell treatments, relative to control cells (transfected with control siRNA and incubated with PT-BSA). Mean and standard deviation are shown (n = 3). (C) Heat map of candidate gene expression in different conditions; PTG stimulation, siRNA mediated MAGI2 silencing (MAGI2_si1) and PTG stimulation of MAGI2_si1 cells. Expression levels are shown as fold-change relative to the average of control samples (silenced with a control siRNA and incubated with PT-BSA). Columns represent treatment categories and lines candidate genes (*0.01 < p < 0.05; **0.001 < p < 0.01; ***p < 0.001).
Since the region harboring RP4-587D13.2 and MAGI2 is associated to CD and we have observed that both genes can influence the expression of other TJ-related genes, we analyzed their expression in biopsies from CD patients and controls. As shown in Figures 4A,B, both MAGI2 and RP4-587D13.2 show significantly lower expression levels in the duodenum of active CD patients, and this downregulation persisted after GFD treatment. The correlation between MAGI2 and RP4-587D13.2 gene expression previously observed in different human tissues (Figure 2A) was also present in duodenal biopsies (r = 0.4033; p = 0.027) (Figure S2).
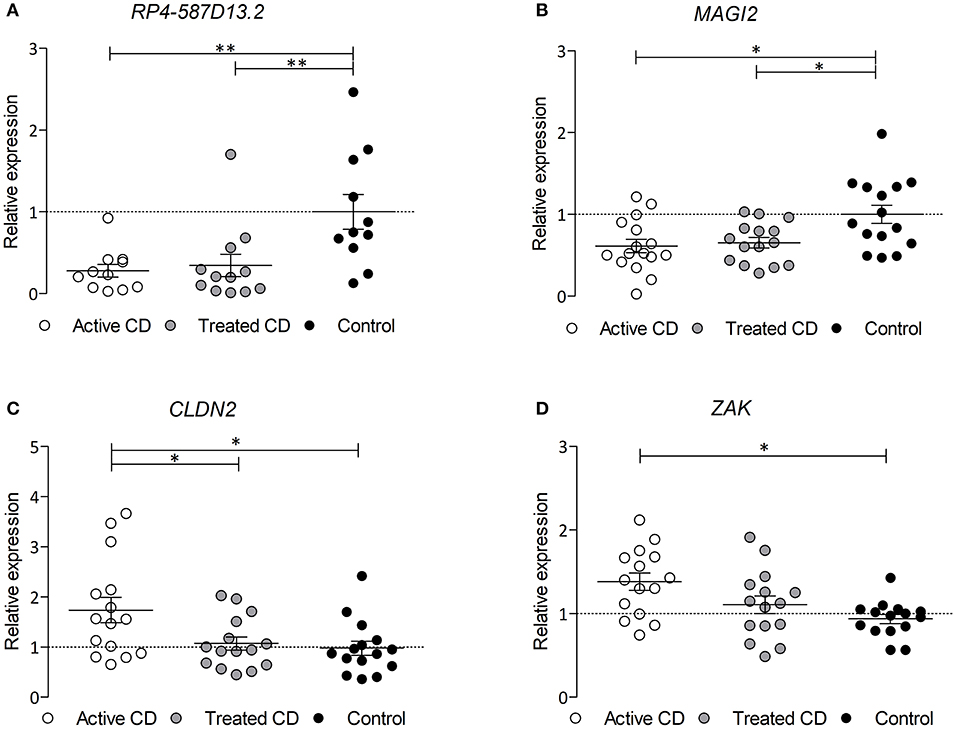
Figure 4. mRNA expression of (A) RPL4-587D13.2, (B) MAGI2, (C) CLDN2, and (D) ZAK in duodenal biopsies from CD patients at diagnosis (white dots), treated CD patients (gray dots) and non-Celiac controls (black dots) represented as mean and SEM, relative to the average of the control samples (*0.01 < p < 0.05; **0.001 < p < 0.01).
In order to assess whether the TJ genes that are altered by the downregulation of the MAGI2 region are also altered in CD, we analyzed their expression in CD and control biopsies. Two of the genes, CLDN2 and ZAK, are overexpressed in the intestine of active CD patients (on a gluten containing diet) compared to treated and control samples (Figures 4C,D), confirming what had been previously observed in vitro (Figure 3C).
Despite the limited number of biological samples representing all three genotypes in our study, we also searched for relationships between the risk genotypes and the expression level of the MAGI2 region genes. As the presence of all three genotypes in every group was not possible, heterozygous, and risk-allele homozygous samples were combined. The results of eQTL analysis showed that the presence of the risk allele was not significantly associated with decreased expression of either transcript in the MAGI2 region (Figures 5A,B). However, when we analyzed the correlation of both transcripts based on the genotype we observed that the samples harboring the risk allele showed a stronger correlation (r = 0.6364; p = 0.0402) than those with no risk allele (r = 0.4286; p = 0.4194) (Figures 5C,D) suggesting that the risk allele strengthens the interaction of both genes thus affecting the regulation of downstream genes.
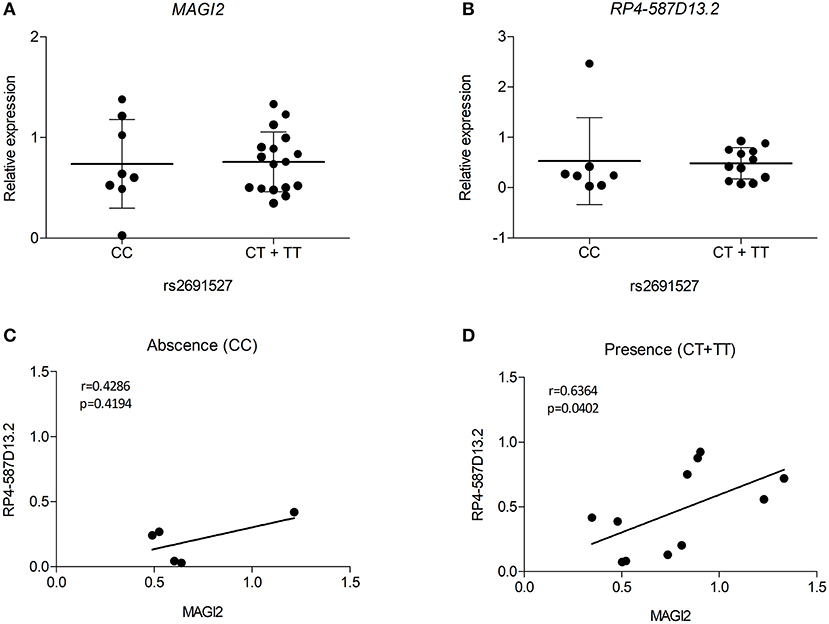
Figure 5. eQTL analyses for (A) MAGI2 and (B) RP4-587D13.2 in duodenal biopsies from patients with CD and non-CD controls. Scatter plots show expression relative to the control mean in patients lacking or harboring the tag SNP rs2691527*T allele. Spearman correlation between MAGI2 and RP4-587D13.2 expression levels according to the (C) absence or (D) presence of tag SNP rs2691527*T allele.
Polymorphisms in TJ-related genes MAGI2, PARD3, and MYO9B that potentially influence the homeostasis of the intestinal barrier have been associated with risk of gastrointestinal disorders like CD and IBD (27, 29, 46). However, attempts to replicate those associations in different cohorts have sometimes failed, highlighting the heterogeneity and complexity of common human disorders (34, 35, 47). In this study, we have been able to replicate the association with rs6962966 (an intronic variant in MAGI2), previously identified in Dutch and British cohorts (29), in a Spanish CD cohort.
Human MAGI2 is a relatively large gene (~1.5 Mbps) that maps to chromosome 7q21 and encodes the membrane-associated, guanylate kinase inverted 2 tight junction protein, involved in epithelial integrity. The CD-associated allele has been proposed to be involved in MAGI2 gene function, but no studies have been performed to determine the possible effects of the CD-associated polymorphism. The candidate gene selection that follows SNP association studies is often aprioristic, and functional studies are the only unbiased approach to characterize the associated region. Therefore, in order to define the role of this intronic variant in CD pathogenesis, we scrutinized the genomic region around rs6962966 and found that, apart from the coding gene MAGI2, the region also harbors RP4-587D13.2, a long RNA transcript represented by a cluster of expressed sequence tags (ESTs) covering 721 bp that maps 3,9 kb upstream (centromeric) of the CD-associated SNP. In silico analysis showed that this transcript is a long non-coding intronic RNA of unknown biological function. Recent studies have evidenced that intronic regions are key sources of regulatory non-coding RNAs that are able to regulate the expression of other genes through diverse mechanisms (48). Interestingly, the antisense lncRNA MAGI2-AS3 overlapping the first exon of MAGI2 has been recently related to tumor suppression in several cancers including breast, lung and hepatocellular carcinoma (45, 49, 50). In the case of lncRNA RP4-587D13.2, located within the intron harboring the CD-associated rs6962966, both its differential tissue distribution and preferential nuclear localization suggest that it is a functional, regulatory transcript. Interestingly, RP4-587D13.2 and MAGI2 show a concordant expression profile across human tissues, including the small intestine, indicating that RP4-587D13.2 could be processed from the same pre-mRNA and could be functionally related to MAGI2. Moreover, MAGI2 shows decreased protein levels upon silencing of this intronic lncRNA, suggesting that RP4-587D13.2 could be involved in the transcriptional (or post-transcriptional) regulation of MAGI2 expression and thus in the coordination of the expression of genes that are relevant to the TJ pathway. However, these regulatory relationships between the intronic lncRNA and the protein transcribed by its host locus MAGI2 must be confirmed and the mechanisms that underlie such regulatory behaviors remain to be fully understood.
We observed that the expression of several TJ-related genes is altered in an in vitro model that mimics the context of CD, where the expression of MAGI2 is repressed and intestinal cells are exposed to digested gliadin (PTG), suggesting that both MAGI2, and the upstream RP4-587D13.2 are necessary for a correct function of the pathway. Moreover, the decrease in expression of both genes, together with the alteration observed in several other TJ genes in the disease tissue suggests that the MAGI2 region could be involved in the regulation of the network. Interestingly, in the in vitro model, gliadin stimulation enhanced the effects of gene-silencing, resembling what is observed for CLDN2 and ZAK in biopsy samples from active and treated CD patients. On the other hand, the expression of MAGI2 and RP4-587D13.2 is downregulated in the duodenum of active CD patients and persists even after more than 2 years on GFD and an apparent recovery from intestinal atrophy, supporting a constitutive alteration that could have a genetic origin. However, several other TJ genes that are altered in the PTG-challenged in vitro model (and in active CD) are normalized in GFD-treated patients, stressing the role of gluten-mediated induction of some of the alterations. The constitutive alteration of gene expression in the MAGI2 region could promote an increased response to ingested gluten, probably leading to a higher disruption of TJ structures and to an enhanced intestinal permeability. Overall, the constitutive alteration of gene expression, together with the presence of an associated genetic variant point to a dysfunction that is present before the clinical onset of the disease, as has been previously suggested for the enhanced intestinal permeability that occurs in CD (25).
Several studies have described that more than half of the disease-associated SNPs correlate with the expression of at least one adjacent gene (51) acting as cis-eQTL. The eQTL analysis in our samples did not find a link between the risk allele and the reduction of MAGI2 or RP4-587D13.2 expression in intestinal biopsy samples. However, there was a stronger correlation between the expression of both genes in the presence of the risk allele, suggesting that the associated SNP could be influencing the regulation of MAGI2 exerted by RP4-587D13.2. The effects of disease-associated SNPs go far beyond the simplistic idea of transcriptional control of an adjacent locus and the complexity of the mechanisms involved in the functional translation of associated genetic variants make it difficult to explain their role in the disease. Further functional studies and large meta-analyses might be able to pinpoint the causal variants within a disease-associated locus and determine their downstream effects in greater detail (52).
In summary, we have demonstrated that the MAGI2 genomic region is associated with CD and presents transcriptional alterations that seem to be relevant for disease pathogenesis. We postulate that RP4-587D13.2 is regulating MAGI2 expression, that this relationship seems to be affected by the associated SNP and that the constitutive reduced expression of both transcripts in patients could affect the TJ pathway, which is crucial for intestinal permeability, and enhance the response to the ingested gluten. Further studies are needed to verify the functionality of this intronic lncRNA and the complex regulatory relationships between RP4-587D13.2, its host loci MAGI2 and disease. All this will help to understand the complexity of the interactions between the environmental trigger, genetic polymorphisms and gene expression.
The datasets generated for this study can be found in the Gene Expression Omnibus GSE84729.
The studies involving human participants were reviewed and approved by Ethics Committee of Cruces University Hospital (ref. CEIC-E08/59, CEIC-E13/20 and CEICE16/46). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
AJ-M designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. IS contributed with the experimental design and procedures and data analysis. KG-E analyzed and interpreted data. AO-G performed experiments and analyzed data. IR-G and MS performed experiments and collected samples. II performed sample collection. AC-R conceived the ideas of experimental design of the study, performed experiments, analyzed and interpreted data, and provided revisions to scientific and grammatical content of the manuscript. JB conceived the ideas of experimental design of the study, interpreted data, provided revisions to scientific, and grammatical content of the manuscript and provided funding.
This work was partially funded by the Basque Department of Education grant IT1281-19 and ISCIII Research Project PI16/00258, cofunded by the European Union ERDF, A way to make Europe to JB. AC-R is supported by an Ikerbasque Fellowship and funded by a research project grant 2017111082 from the Basque Goverment. IS was funded by a research project grant 2015111068 from the Basque Department of Health. AJ-M and AO-G are predoctoral fellows funded by FPI grants from the Basque Department of Education, Universities and Research and IR-G and MS are predoctoral fellows funded by the University of Basque Country.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CEGEC: Participants are members from different Spanish universities and university hospitals that collaborated in collecting the DNA samples to create a DNA collection for research in celiac disease. Montserrat Alsina, María Esteve and Laura Pardo (CATLAB-Hospital Universitari Mutua de Terrassa-Barcelona); Luis Castaño, Pablo Oliver and Juan Carlos Vitoria (Hospital Universitario de Cruces, Barakaldo-Bizkaia); Luis Rodrigo, Carlos López-Larrea and Antonio López-Vázquez (Hospital General de Asturias, Oviedo-Asturias); Idoia Hualde and Zuriñe García (Hospital de Txagorritxu, Vitoria-Gasteiz); Blanca Hernández, María Antonia Ramos-Arroyo and Félix Sánchez-Valverde (Hospital Virgen del Camino, Pamplona-Iruña); Carme Farré, Teresa Marqués and Pere Vilar (Hospital Sant Joan de Deu, Esplugues de Llobregat-Barcelona), Eduardo Arranz and José Antonio Garrote (Universidad de Valladolid). Consortium participants, other than JB, did not participate in the study design, analysis and interpretation of data, writing of the report and in the decision to submit the report for publication. Technical and human support provided by SGIker (UPV/EHU, MICINN, GV/EJ, ERDF, and ESF) is gratefully acknowledged.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2019.00187/full#supplementary-material
BSA, Bovine serum albumin; CD, Celiac disease; CEGEC, Spanish Consortium for the Genetics of Celiac Disease; CPAT, Coding potential assessment tool; DsiRNA, Dicer-substrate short interfering RNAs; EST, expressed sequence tag; eQTL, Expression quantitative trait loci; GFD, Gluten-free diet; GWA, Genome wide association; HLA, Human Leukocyte Antigen; IBD, Inflammatory bowel disease; lncRNA, Long non-coding RNA; OR, Odds ratio; ORF, Open reading frame; PhyloCSF, Phylogenetic Codon Substitution Frequencies; PT-BSA, Pepisn-trypsin digested BSA; PTG, Pepsin-trypsin digested gliadin; RT-qPCR, Quantitative reverse transcription PCR; si-RNA, Small interfering RNA; SNP, Single-nucleotide polymorphism; TBS-T, Tris-buffered saline with Tween; TJ, Tight Junction; TLR, Toll-like receptor; WB, Western blot.
1. Plot L, Amital H. Infectious associations of Celiac disease. Autoimmun Rev. (2009) 8:316–9. doi: 10.1016/j.autrev.2008.10.001
2. Mårild K, Kahrs CR, Tapia G, Stene LC, Størdal K. Infections and risk of Celiac disease in childhood: a prospective nationwide cohort study. Am J Gastroenterol. (2015) 110:1475–84. doi: 10.1038/ajg.2015.287
3. Andrén Aronsson C, Lee HS, Koletzko S, Uusitalo U, Yang J, Virtanen SM, et al. Effects of gluten intake on risk of Celiac disease: a case-control study on a Swedish birth cohort. Clin Gastroenterol Hepatol. (2016) 14:403–9.e3. doi: 10.1016/j.cgh.2015.09.030
4. Mårild K, Dong F, Lund-Blix NA, Seifert J, Barón AE, Waugh KC, et al. Gluten intake and risk of Celiac disease. Am J Gastroenterol. (2019) 114:1307–14. doi: 10.14309/ajg.0000000000000255
5. Kumar V, Wijmenga C, Withoff S. From genome-wide association studies to disease mechanisms: Celiac disease as a model for autoimmune diseases. Semin Immunopathol. (2012) 34:567–80. doi: 10.1007/s00281-012-0312-1
6. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. (2012) 337:1190–5. doi: 10.1126/science.1222794
7. Ricaño-Ponce I, Wijmenga C. Mapping of immune-mediated disease genes. Annu Rev Genomics Hum Genet. (2013) 14:325–53. doi: 10.1146/annurev-genom-091212-153450
8. Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin. (2015) 8:57. doi: 10.1186/s13072-015-0050-4
9. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. (2012) 22:1775–89. doi: 10.1101/gr.132159.111
10. Hrdlickova B, Kumar V, Kanduri K, Zhernakova DV, Tripathi S, Karjalainen J, et al. Expression profiles of long non-coding RNAs located in autoimmune disease-associated regions reveal immune cell-type specificity. Genome Med. (2014) 6:88. doi: 10.1186/s13073-014-0088-0
11. Plaza-Izurieta L, Fernandez-Jimenez N, Irastorza I, Jauregi-Miguel A, Romero-Garmendia I, Vitoria JC, et al. Expression analysis in intestinal mucosa reveals complex relations among genes under the association peaks in Celiac disease. Eur J Hum Genet. (2015) 23:1100–5. doi: 10.1038/ejhg.2014.244
12. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. (2011) 43:904–14. doi: 10.1016/j.molcel.2011.08.018
13. Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. (2013) 9:e1003201. doi: 10.1371/journal.pgen.1003201
14. Maiuri MC, De Stefano D, Mele G, Fecarotta S, Greco L, Troncone R, et al. Nuclear factor kappaB is activated in small intestinal mucosa of Celiac patients. J Mol Med. (2003) 81:373–9. doi: 10.1007/s00109-003-0440-0
15. Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PHR, et al. A long noncoding RNA associated with susceptibility to Celiac disease. Science. (2016) 352:91–5. doi: 10.1126/science.aad0467
16. Juuti-Uusitalo K, Mäki M, Kaukinen K, Collin P, Visakorpi T, Vihinen M, et al. cDNA microarray analysis of gene expression in coeliac disease jejunal biopsy samples. J Autoimmun. (2004) 22:249–65. doi: 10.1016/j.jaut.2003.12.001
17. Diosdado B, Wapenaar MC, Franke L, Duran KJ, Goerres MJ, Hadithi M, et al. A microarray screen for novel candidate genes in coeliac disease pathogenesis. Gut. (2004) 53:944–51. doi: 10.1136/gut.2003.018374
18. Castellanos-Rubio A, Santin I, Martin-Pagola A, Irastorza I, Castaño L, Vitoria JC, et al. Long-term and acute effects of gliadin on small intestine of patients on potentially pathogenic networks in Celiac disease. Autoimmunity. (2010) 43:131–9. doi: 10.3109/08916930903225229
19. Quinn EM, Coleman C, Molloy B, Dominguez Castro P, Cormican P, Trimble V, et al. Transcriptome analysis of CD4+ T cells in coeliac disease reveals imprint of BACH2 and IFNγ Regulation. PLoS ONE. (2015) 10:e0140049. doi: 10.1371/journal.pone.0140049
20. Schulzke JD, Bentzel CJ, Schulzke I, Riecken EO, Fromm M. Epithelial tight junction structure in the jejunum of children with acute and treated Celiac sprue. Pediatr Res. (1998) 43:435–41. doi: 10.1203/00006450-199804000-00001
21. Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. (1986) 105:883–5. doi: 10.7326/0003-4819-105-6-883
22. Büning C, Geissler N, Prager M, Sturm A, Baumgart DC, Büttner J, et al. Increased small intestinal permeability in ulcerative colitis: rather genetic than environmental and a risk factor for extensive disease? Inflamm Bowel Dis. (2012) 18:1932–9. doi: 10.1002/ibd.22909
23. Ciccocioppo R, Finamore A, Ara C, Di Sabatino A, Mengheri E, Corazza GR. Altered expression, localization, and phosphorylation of epithelial junctional proteins in Celiac disease. Am J Clin Pathol. (2006) 125:502–11. doi: 10.1309/DTYRA91G8R0KTM8M
24. Schumann M, Siegmund B, Schulzke JD, Fromm M. Celiac disease: role of the epithelial barrier. Cell Mol Gastroenterol Hepatol. (2017) 3:150–62. doi: 10.1016/j.jcmgh.2016.12.006
25. Bjarnason I, Peters TJ. In vitro determination of small intestinal permeability: demonstration of a persistent defect in patients with coeliac disease. Gut. (1984) 25:145–50. doi: 10.1136/gut.25.2.145
26. Mishra A, Prakash S, Sreenivas V, Das TK, Ahuja V, Gupta SD, et al. Structural and functional changes in the tight junctions of asymptomatic and serology-negative first-degree relatives of patients with Celiac disease. J Clin Gastroenterol. (2016) 50:551–60. doi: 10.1097/MCG.0000000000000436
27. Monsuur AJ, de Bakker PI, Alizadeh BZ, Zhernakova A, Bevova MR, Strengman E, et al. Myosin IXB variant increases the risk of Celiac disease and points toward a primary intestinal barrier defect. Nat Genet. (2005) 37:1341–4. doi: 10.1038/ng1680
28. Amundsen SS, Vatn M, Wijmenga C, Sollid LM, Lie BA, Lie BA. Association analysis of MYO9B gene polymorphisms and inflammatory bowel disease in a Norwegian cohort. Tissue Antigens. (2006) 68:249–52. doi: 10.1111/j.1399-0039.2006.00665.x
29. Wapenaar MC, Monsuur AJ, van Bodegraven AA, Weersma RK, Bevova MR, Linskens RK, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitisAn unusual case of ascites. Gut. (2007) 57:463–7. doi: 10.1136/gut.2007.133132
30. Dunn HA, Ferguson SSG. PDZ protein regulation of G protein–coupled receptor trafficking and signaling pathways. Mol Pharmacol. (2015) 88:624–39. doi: 10.1124/mol.115.098509
31. Satsangi J, Parkes M, Louis E, Hashimoto L, Kato N, Welsh K, et al. Two stage genome–wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet. (1996) 14:199–202. doi: 10.1038/ng1096-199
32. Olivares M, Laparra M, Sanz Y. Influence of Bifidobacterium longum CECT 7347 and gliadin peptides on intestinal epithelial cell proteome. J Agric Food Chem. (2011) 59:7666–71. doi: 10.1021/jf201212m
33. Rauhavirta T, Oittinen M, Kivistö R, Männistö PT, Garcia-Horsman JA, Wang Z, et al. Are transglutaminase 2 inhibitors able to reduce gliadin-induced toxicity related to Celiac disease? A proof-of-concept study. J Clin Immunol. (2013) 33:134–42. doi: 10.1007/s10875-012-9745-5
34. Amundsen SS, Monsuur AJ, Wapenaar MC, Lie BA, Ek J, Gudjónsdóttir AH, et al. Association analysis of MYO9B gene polymorphisms with Celiac disease in a Swedish/Norwegian cohort. Hum Immunol. (2006) 67:341–5. doi: 10.1016/j.humimm.2006.03.020
35. Chen YQ, Zhang L, Lv XY, Wang HZ. Lack of association between MYO9B gene polymorphisms and susceptibility to coeliac disease in caucasians: evidence from a meta-analysis. Immunol Invest. (2016) 45:396–405. doi: 10.3109/08820139.2016.1156692
36. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. (2007) 81:559–75. doi: 10.1086/519795
37. Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci USA. (2001) 98:6336–41. doi: 10.1073/pnas.101133498
38. Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. (2011) 27:i275–82. doi: 10.1093/bioinformatics/btr209
39. Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. (2013) 41:e74. doi: 10.1093/nar/gkt006
40. Junker Y, Zeissig S, Kim SJ, Barisani D, Wieser H, Leffler DA, et al. Wheat amylase trypsin inhibitors drive intestinal inflammation via activation of toll-like receptor 4. J Exp Med. (2012) 209:2395–408. doi: 10.1084/jem.20102660
41. Gujral N, Suh JW, Sunwoo HH. Effect of anti-gliadin IgY antibody on epithelial intestinal integrity and inflammatory response induced by gliadin. BMC Immunol. (2015) 16:41. doi: 10.1186/s12865-015-0104-1
42. Manai F, Azzalin A, Gabriele F, Martinelli C, Morandi M, Biggiogera M, et al. The in vitro effects of enzymatic digested gliadin on the functionality of the autophagy process. Int J Mol Sci. (2018) 19:635. doi: 10.3390/ijms19020635
43. Bondar C, Plaza-Izurieta L, Fernandez-Jimenez N, Irastorza I, Withoff S, Wijmenga C, et al. THEMIS and PTPRK in Celiac intestinal mucosa: coexpression in disease and after in vitro gliadin challenge. Eur J Hum Genet. (2014) 22:358–62. doi: 10.1038/ejhg.2013.136
44. Martín-Pagola A, Pérez-Nanclares G, Ortiz L, Vitoria JC, Hualde I, Zaballa R, et al. MICA response to gliadin in intestinal mucosa from Celiac patients. Immunogenetics. (2004) 56:549–54. doi: 10.1007/s00251-004-0724-8
45. Yang Y, Yang H, Xu M, Zhang H, Sun M, Mu P, et al. Long non-coding RNA (lncRNA) MAGI2-AS3 inhibits breast cancer cell growth by targeting the Fas/FasL signalling pathway. Hum Cell. (2018) 31:232–41. doi: 10.1007/s13577-018-0206-1
46. Sánchez E, Alizadeh BZ, Valdigem G, Ortego-Centeno N, Jiménez-Alonso J, de Ramón E, et al. MYO9B gene polymorphisms are associated with autoimmune diseases in Spanish population. Hum Immunol. (2007) 68:610–5. doi: 10.1016/j.humimm.2007.03.006
47. Wolters VM, Xu W, Zhao X, Walters TD, Griffiths AM, Silverberg MS, et al. Replication of genetic variation in the MYO9B gene in Crohn's disease. Hum Immunol. (2011) 72:592–7. doi: 10.1016/j.humimm.2011.03.025
48. Louro R, Smirnova AS, Verjovski-Almeida S. Long intronic noncoding RNA transcription: expression noise or expression choice? Genomics. (2009) 93:291–8. doi: 10.1016/j.ygeno.2008.11.009
49. Yin Z, Ma T, Yan J, Shi N, Zhang C, Lu X, et al. LncRNA MAGI2-AS3 inhibits hepatocellular carcinoma cell proliferation and migration by targeting the miR-374b-5p/SMG1 signaling pathway. J Cell Physiol. (2019) 234:18825–36. doi: 10.1002/jcp.28521
50. Hao XZ, Yang K. LncRNA MAGI2-AS3 suppresses the proliferation and invasion of non-small cell lung carcinoma through miRNA-23a-3p/PTEN axis. Eur Rev Med Pharmacol Sci. (2019) 23:7399–407. doi: 10.26355/eurrev_201909_18848
51. Dubois PCA, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for Celiac disease influencing immune gene expression. Nat Genet. (2010) 42:295–302. doi: 10.1038/ng.543
Keywords: celiac disease, tight junction, association analysis, expression analysis, MAGI2, long non-coding RNA
Citation: Jauregi-Miguel A, Santin I, Garcia-Etxebarria K, Olazagoitia-Garmendia A, Romero-Garmendia I, Sebastian-delaCruz M, Irastorza I, Spanish Consortium for the Genetics of Celiac Disease, Castellanos-Rubio A and Bilbao JR (2019) MAGI2 Gene Region and Celiac Disease. Front. Nutr. 6:187. doi: 10.3389/fnut.2019.00187
Received: 29 August 2019; Accepted: 03 December 2019;
Published: 19 December 2019.
Edited by:
Carmen Gianfrani, Institute of Biochemistry and Cell Biology (CNR), ItalyReviewed by:
Yanfei Zhang, Geisinger Health System, United StatesCopyright © 2019 Jauregi-Miguel, Santin, Garcia-Etxebarria, Olazagoitia-Garmendia, Romero-Garmendia, Sebastian-delaCruz, Irastorza, Spanish Consortium for the Genetics of Celiac Disease, Castellanos-Rubio and Bilbao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ainara Castellanos-Rubio, YWluYXJhLmNhc3RlbGxhbm9zQGVodS5ldXM=; Jose Ramón Bilbao, am9zZXJhbW9uLmJpbGJhb0BlaHUuZXVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.