 Jaya Kumar
Jaya Kumar Srijit Das
Srijit Das Seong Lin Teoh
Seong Lin Teoh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Nutr., 28 February 2018
Sec. Clinical Nutrition
Volume 5 - 2018 | https://doi.org/10.3389/fnut.2018.00014
Acrylamide (AA) is a water soluble white crystalline solid commonly used in industries. It was listed as an industrial chemical with potential carcinogenic properties. However to date, AA was used to produce polyacrylamide polymer, which was widely used as a coagulant in water treatment; additives during papermaking; grouting material for dams, tunnels, and other underground building constructions. AA in food could be formed during high-temperature cooking via several mechanisms, i.e., formation via acrylic acid which may be derived from the degradation of lipid, carbohydrates, or free amino acids; formation via the dehydration/decarboxylation of organic acids (malic acid, lactic acid, and citric acid); and direct formation from amino acids. The big debate is whether this compound is toxic to human beings or not. In the present review, we discuss the formation of AA in food products, its consumption, and possible link to the development of any cancers. We discuss the body enzymatic influence on AA and mechanism of action of AA on hormone, calcium signaling pathways, and cytoskeletal filaments. We also highlight the deleterious effects of AA on nervous system, reproductive system, immune system, and the liver. The present and future mitigation strategies are also discussed. The present review on AA may be beneficial for researchers, food industry, and also medical personnel.
Acrylamide or 2-propenamide (AA, C3H5NO) is a water soluble white crystalline solid with a relative molecular mass of 71.08 kDa, commonly used in industry (1). In the year 1994, the International Agency for Research on Cancer listed AA as industrial chemicals with potential carcinogenic risk to humans (1). However to date, AA was used to produce polyacrylamide polymer, which remained widely used as a coagulant in water treatment; additives during papermaking; grouting material for dams, tunnels, and other underground building constructions; and as electrophoresis gels (2–5).
Later in the year 2002, AA was detected in heated foods where its formation was temperature dependent (6). Using liquid chromatography–mass spectrometry, moderate levels of AA (5–50 µg/kg) were detected in heated protein-rich foods, but higher contents of AA (150–4,000 µg/kg) were detected in carbohydrate-rich foods (6). Importantly, AA could not be detected in unheated or boiled foods (6). AA in food could be formed during high-temperature cooking via several mechanisms, i.e., formation via acrylic acid which may be derived from the degradation of lipid, carbohydrates, or free amino acids; formation via the dehydration/decarboxylation of organic acids (malic acid, lactic acid, and citric acid); and direct formation from amino acids (7).
Studies have shown that AA is formed mainly from free amino acid asparagine and reducing sugars such as glucose and fructose, during high-temperature cooking through Maillard reactions, a series of non-enzymatic reactions between free amino acids and reducing sugars which is responsible for the flavor and color generated during baking (8, 9). This would explain the formation of AA in cooked food rich in asparagine, e.g., in cereals and potatoes (8). In fact, the concentration of reducing sugars in food is the primary determinant of AA formation, compared to asparagine content (10).
The mean AA contents in potato crisps prepared from different UK-grown potatoes ranged from 131 to 5,360 µg/kg (11). Rosti, a popular Swiss dish, made of grated and fried potatoes, contains an average of 702 µg/kg AA (12). The AA concentration in different commonly consumed breads ranges from <limit of quantification (LOQ) to 695 µg/kg, where the highest AA concentration was detected in wheat bran and whole wheat breads (13). Similarly, AA was also detected in different bakery products: biscuits (LOQ to 2,405.0 µg/kg), sandwich biscuits with cream (112.6–570.4 µg/kg), gingerbread (349.5–955.5 µg/kg), and crackers (347.8–366.1 µg/kg) (14). In addition to potato- and cereal-based food, coffee beverages acquired from coffee vending machines also contain AA concentration which varies from 7.7 to 40.0 µg/L (15).
Estimated AA intakes were measured using food frequency questionnaire, total diet study, hemoglobin adducts of AA, and glycidamide (GA, primary metabolite of AA) (16, 17). For adults, the estimated average AA intakes range from 0.3 to 0.6 µg/kg of body weight per day, where the intake is higher in children (18).
Infant cereal-based foods are important nutrient source for children around the world, and the AA concentration in these cereal-based baby food (ready-to-eat and instant baby foods, candy bars, and cakes) varied between 10 and 60 µg/kg (19). In another study, the mean AA level in cereal-based baby foods ranged from 36 to 604 µg/kg, and it was estimated the mean AA exposures of toddlers from the cereal-based baby food was 1.43 µg/kg of body weight per day (20).
High-income countries have the highest incidence rates for lung, colorectal, breast, and prostate cancers, while low- and middle-income countries have the highest rates of stomach, liver, and esophageal and cervical cancer (21). Global Burden of Disease Cancer Consortium et al. (22) has reported that there were a total of 17.5 million cancer cases globally, 8.7 million deaths, and 208.3 million disability-adjusted life-years, in the year 2015. Between the year 2005 and 2015, incident cancer cases were found to be increased by 33% (22).
Since the discovery of AA in foods, numerous studies have set to explore the carcinogenic potential in humans. Although AA was shown to be carcinogenic in both male and female rodent models, numerous studies reported no statistically significant association between dietary AA intake and various cancers in humans, e.g., pancreatic, prostate, breast, ovarian, and endometrial cancer (23–30). Obon-Santacana et al. (26) analyzed the AA and GA hemoglobin adduct levels in quintiles based on control distributions and showed no effect on overall endometrial cancer risk (HRHbAA;Q5vsQ1: 0.84, 95% CI: 0.49–1.48; HRHbGA;Q5vsQ1: 0.94, 95% CI: 0.54–1.63). Similarly, no clear association were shown between AA intakes and the risk of epithelial ovarian cancer, except some positive associations were observed between second (59.81–78.70) and third (78.80–106.00) quintiles of total AA and GA hemoglobin adduct levels (ORQ2vsQ1, 1.81; 95% CI: 1.06–3.10) and (ORQ3vsQ1, 2.00; 95% CI: 1.16–3.45) (27). Interestingly, Hogervorst et al. (31, 32) showed significant interaction between AA intake and single-nucleotide polymorphisms (SNPs) in AA-metabolizing enzymes cytochrome P450, family 2, subfamily e, polypeptide 1 (CYP2E1) with endometrial cancer risk, and SNPs in the HSD3B1/B2 gene cluster (through effects on progesterone or androgens) with ovarian cancer risk.
Few studies reported possible association between dietary AA intake and cancer risk, e.g., dietary AA may increase the risk for cutaneous malignant melanoma in men (HR: 1.13, 95% CI: 1.01–1.26) per 10 µg increment (33). Similarly, dietary AA may also increase the risk of lymphatic malignancies (i.e., multiple myeloma and follicular lymphoma) in men (34). The adjusted risk of all esophageal tumors combined among Sweden esophageal cancer patients was in the highest quartile of AA intake compared to the lowest, particularly among overweight or obese individuals (35). However, in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort, an association between dietary AA intake and an increased risk of developing esophageal cancer was observed in the middle quartiles, not in the highest quartile (36). A recent study revealed dietary AA intake was associated with increased overall cancer mortality in elderly Chinese (37). When compared to the lowest (<9.9 μg/day) and the highest (>17.1 μg/day) quartile of AA intake, the multivariable hazard ratios were 1.9, 1.9, and 2.0 for the overall, digestive, and respiratory system cancer mortality, respectively (37).
In human and animals, AA is primarily metabolized to an epoxide metabolite, GA, by an enzyme known as CYP2E1 (38, 39). GA may subsequently conjugated with reduced glutathione (GSH) to GSH conjugates by GSH transferases, or GA also can be further metabolized to glyceramide by epoxide hydrolase.
Using CYP2E1-null and wild-type mice, Sumner et al. (40) showed that the absence of CYP2E1 enzyme in the mutant mice led to non-excretion of GA or its metabolites in the urine of 13C-AA-treated mice. Subsequent studies by Ghanayem et al. (39) showed that CYP2E1-mediated oxidation of AA to GA is required for the induction of male germ cell mutagenicity in mice exposed to AA. In their subsequent study, the researchers went further to show that GA through interaction with DNA in the testes/sperm or spermatid protamine formed DNA adducts that leads to male germ cell mutagenicity. In a recent study, Ehlers et al. (41) investigated the effect of low and high doses of GA (1–0.0001 mM) on the expression of genes involved in cancer development using human ovarian and endometrial cancer cell lines and human primary hepatocytes. The authors reported high dose of GA (1 mM) to upregulate the genes involved in oncogenesis such as TF3 (ovarian and endometrial cell lines), dual specific phosphatases DUSP1, DUSP4, and DUSP5 (ovarian cell lines), zinc finger protein gene ZNF746, cYMC, and Bystin-like protein gene (BYSL) (primary human hepatocytes) (41).
GSH is an important antioxidant that prevents cellular damage caused by reactive oxygen species such as peroxides, free radicals, heavy metals, and lipid peroxides (42). GST is a group of enzymes that mainly involved in detoxifying xenobiotics, cell death, and cell proliferation (43). GSH is employed by GST in numerous cellular reactions. Thus, the enzymatic activities of GST depend upon the availability of GSH. Srivastava et al. (44) investigated the effect of single and repeated administration of AA on GSH content and GST activity in rat brain. The researchers reported single administration of AA to deplete GSH content without affecting GST activity. However, repeated administration of AA (50 mg/kg × 10 days) depleted GSH content as well GST activity in the rat brain. Altered GST activity may affect the redox status of the cell. This may subsequently affect various redox-dependent gene expression, cell transformation or proliferation, or even apoptosis may occur (45–48).
Exposure of male F344 rats to AA (50 mg/kg) in drinking water for 14 days resulted in a significant decrease in serum T4 level (49). In contrary, exposure to lower doses of AA (2.5 or 15 mg/kg) had no significant effect on serum T4 or thyroid-stimulating hormone (TSH) level in male F344 rats (50). This led to the suggestion that hormonal mechanism is likely to attribute to thyroid tumorigenesis, at least at high doses of AA (51). Hormonal mechanism is thought to be influenced by decreased circulating levels of T3 or T4 levels, which would subsequently stimulate the release of TSH by the pituitary gland via hypothalamus–pituitary–thyroid axis. Continuous stimulation of the thyroid gland by TSH results in increased proliferation of cells and so the formation of tumor (52). A schematic diagram showing the mechanism of action of AA in thyroid tumor was highlighted in Figure 1.
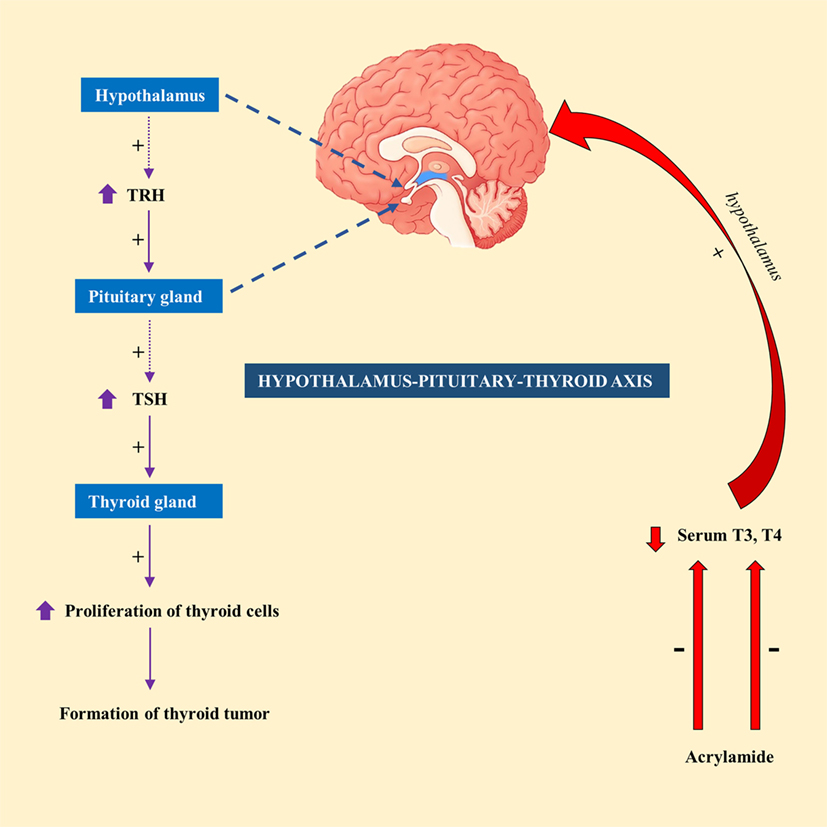
Figure 1. A schematic representation showing the mechanism of action of acrylamide (AA) in thyroid tumor in rats. Exposure to AA reduces serum T3 and T4, which stimulate hypothalamus of hypothalamus-pituitary-thyroid axis to secrete thyrotropin-releasing hormone (TRH). TRH stimulates pituitary gland to release thyroid-stimulating hormone (TSH), which subsequently stimulate thyroid gland to initiate proliferation of cells and eventually development of thyroid tumor.
A recent study correlated calcium signaling and cytoskeletal filaments to genotoxic effects of AA in testes using F344 rats (53). Similar to this finding, existing literature implicates calcium signaling with several types of cancers (54). Binding of AA toward redox-sensitive Cys residues on key regulatory proteins initiate disruption of calcium signaling genes which could lead to reduced expression of calcium signaling genes and disruption of calcium homeostasis. Reduced expression of calcium signaling genes may destabilize microtubules and microfilaments which carry out endo- and excytotic functions (Figure 2). It is hypothesized that disruption in calcium signaling also could lead to cancer development via hormonal actions. However, lack of empirical proof warrants his hypothesis to be further investigated (55).
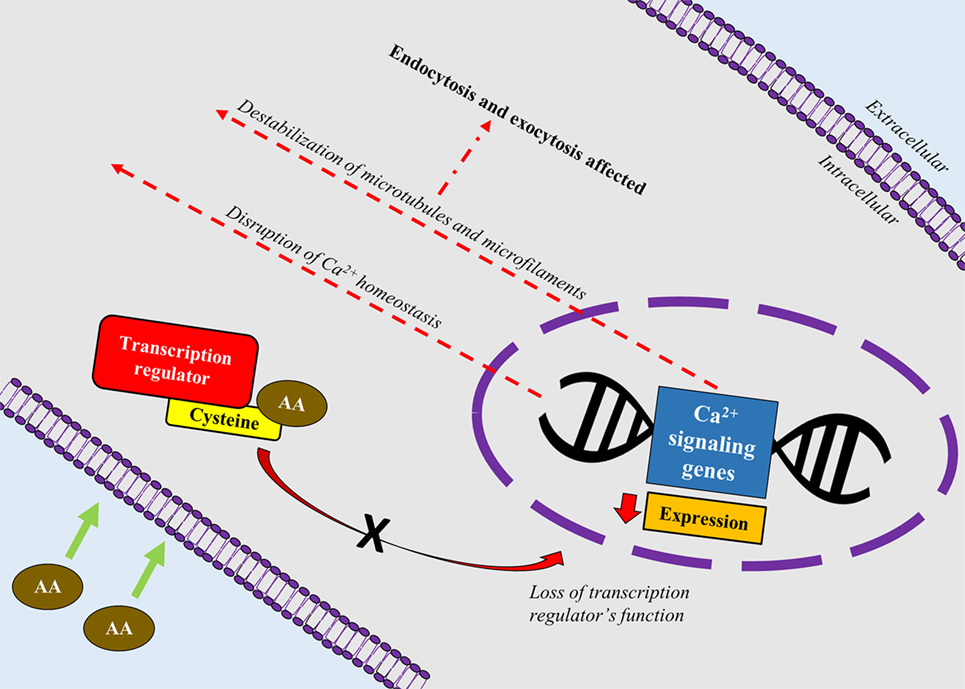
Figure 2. A schematic depiction correlating calcium signaling and cytoskeletal filaments to genotoxic effects of acrylamide (AA) in rats’ testes. AA binds toward redox-sensitive Cys residues on key regulatory proteins such as transcription factors, which disrupts the protein’s function. Loss of the transcription regulator’s function causes reduced expression of calcium signaling genes and disruption of calcium homeostasis. Reduced expression of calcium signaling genes also destabilizes microtubules and microfilaments which carry out endo- and excytotic functions.
The idea of cytoskeletal proteins mediating the genotoxic effects of AA were implicitly corroborated using various study models (55, 56). Cytoskeletal proteins are tasked with multitude of functions that integral for cell survival such as movement of vesicles and cell compartments, regulation of cell division (57), and cell mechanics (58). Few studies reported AA to bind directly to cytoskeletal proteins (59–61). Given the direct functional relation between the cytoskeletal proteins and cell divisions (mitosis/meiosis), AA’s disruptive effects pose various adverse effects, including delays in cell cycle (62), heritable translocation of chromosomes (63), chromosomal aberrations (64), blocks in mitotic and meiotic process (59, 65), and aneuploidy (66).
The primary targets of AA are kinesin motors which are proteins critically involved in cell divisions (67). Kinesins are motor proteins that move along microtubules, fueled by hydrolysis of ATP. Movement of kinesins aids various cellular functions including the transport of cellular cargo from central toward cell periphery. As tailored to their functions, the most common feature of kinesin is presence of motor domain for microtubule binding and ATPase activities. Kinesins are categorized as N (N-terminal), C (C-terminal), and M or I-type (middle or also known as non-motor) based on the location of motor domain within the macromolecule (68). AA was shown to inhibit kinesin in neurons (60) and testes (61). In 2007, Sickles and colleagues investigated the effects of AA, glycidamide (AA’s toxic metabolite), and propionamide (non-neurotoxic metabolite) on function of specific kinesins, KIFC5A (member of kinesin-14 subfamily with motor domain located at the carboxy terminus of the polypeptide), and KRP2 (member of kinesin-12 subfamily with motor domain located more toward the center of the polypeptide). Generally, KIFC5A stabilizes spindle pole by crosslinking the adjacent microtubules (69, 70) and KRP2 regulates microtubule dynamics and chromosome segregation during anaphase (71). The researchers reported AA and glycinamide to specifically inhibit both KIFC5A and KRP2 kinesins (61). Their findings were similar to their previous findings where AA and glycinamide inhibited neuronal N-type kinesins (60). Despite their findings which were convincing at in vitro level, the authors concluded that the carcinogenicity of AA and glycinamide were inconclusive and this warrants future studies to further elucidate the conundrum.
The neurotoxic effects of AA is the only toxic effect proven to be harmful on humans via occupational exposure (72, 73). The clinical signs of AA neurotoxicity were peripheral neuropathies such as upper and lower limb numbness and tingling (74). Chronic administration of AA to laboratory animals resulted in skeletal muscle weakness and ataxia (72, 75, 76). Numerous excellent reviews have covered in-depth literature on the neurotoxicity effects of AA (73, 77). The mechanism underlying the neurotoxicity of AA is inconclusive. However, several mechanisms were proposed to mediate the neurotoxic effects of AA. Interaction of AA with kinesin motor protein in neurons impaired fast anterograde transport of nerve growth factors from cell body to periphery, resulting in nerve death (60). AA was also shown to inhibit uptake of neurotransmitter into striatal synaptic vesicle through possible interaction with sulfhydryl groups on specific proteins and thereby disrupting presynaptic release of neurotransmitters (78).
To date, reproductive toxicity of AA in humans is unheard of. In laboratory animals, exposure to high levels of AA proven to cause reproductive toxicity (79, 80). Various concentration range of AA and duration of AA exposure have been experimented on the reproductive toxic effects of AA in laboratory animals which resulted in decreased fertility rates, decreased litter sizes, abnormal sperm, decreased sperm counts, reduced copulatory activity, and decreased weight of testes (81–83). Further corroborating the reproductive toxicity of AA, Yilmaz et al. (84) investigated the effects of AA and GA exposure on mouse Leydig and Sertoli cells. The researchers showed exposure to AA and GA to reduce cell viability and increase oxidative stress and apoptosis in both Leydig and Sertoli cells (84).
Several theories were proposed to delineate the mechanistic effects of AA on reproductive toxicity. One of the theories relate the neurotoxic effects of AA to mating behaviors. AA-induced peripheral neuropathies such as reduced hind-limb function could impair copulatory behavior, mounting responses, and intromission (83), subsequently affecting proper deposition of sperm in the vagina and uterus and also the resulting hormonal changes (85). AA-induced inhibition of kinesin motor proteins, which are also found in flagella of sperm in addition to nervous system, could affect sperm motility and fertilization events (80, 86, 87). Regarding hormonal mechanism of action, AA reduced serum testosterone and prolactin levels (88) which could lead to testicular atrophy, affects sperm development and motility (81, 89).
The literature on the immunotoxic potential of AA is limited. In rats, exposure to AA significantly decreased the weight of spleen, thymus, and mesenteric lymph nodes (90). Orally administered AA reduced alpha-naphthyl acetate esterase-positive lymphocytes counts and also caused lesions in ileal Peyer’s patches in a dose-dependent manner (91). In other words, AA was detrimental toward gut-associated lymphoid tissue and peripheral blood lymphocytes in rats (91). Fang et al. (92) reported decreased terminal body weight, thymus and spleen weights, and lymphocyte counts following exposure to AA in female BALB/c mice. The researchers also reported significant reduction in the percentage of natural killer cells, interleukin-6, and concanavalin A-induced splenocyte proliferation (92). Kim et al. (93) demonstrated AA-induced senescence in C57BL/6 male mice macrophages via ATF3-mediated pathway. The researchers also associated the AA-induced senescence to ROS production, activation of p38 and JNK signaling pathways, and also increased expression of p53 (Table 1) (93).
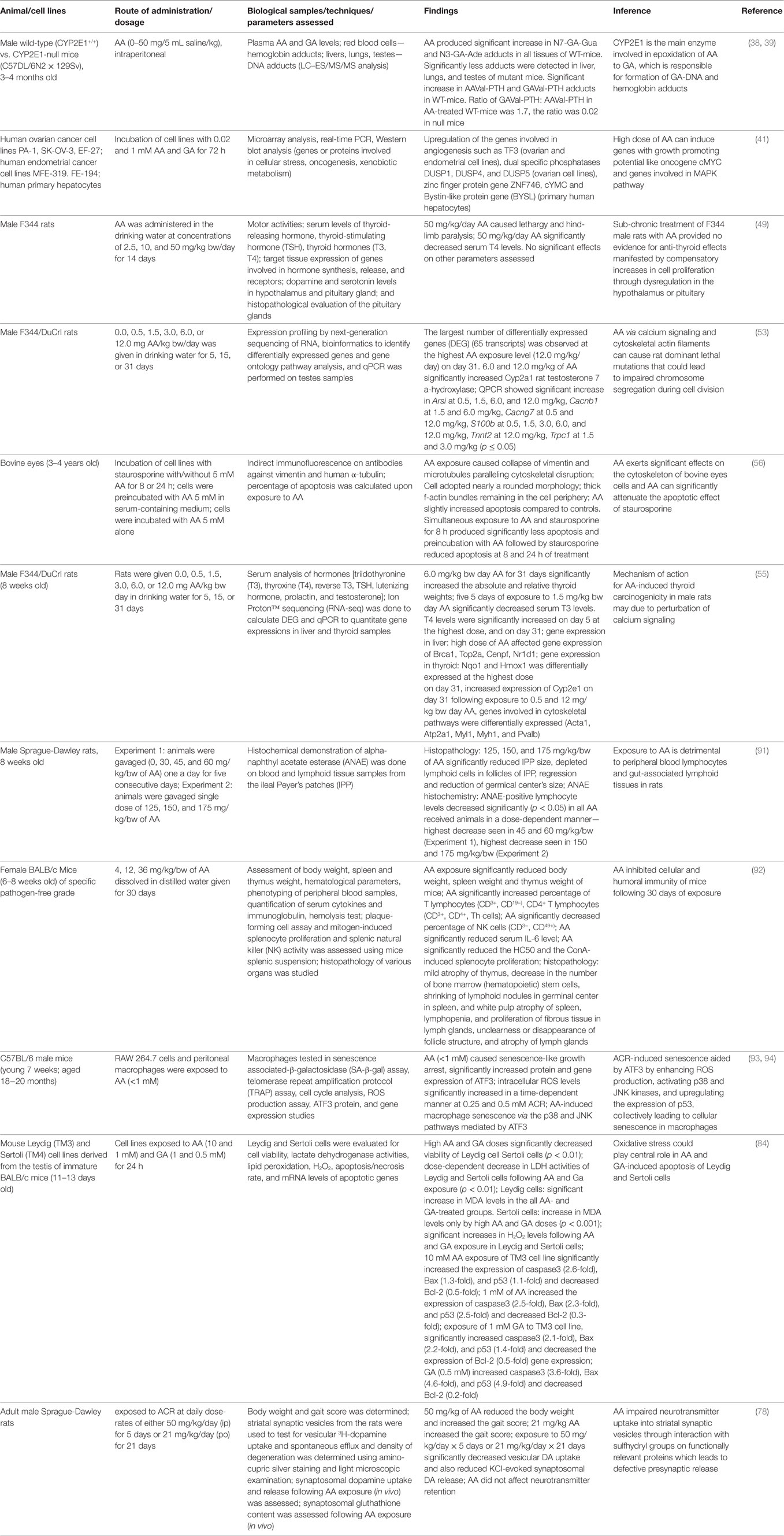
Table 1. Acrylamide (AA) exposure-induced toxicity.
Similar to the reproductive toxicity, there were no reports of AA hepatotoxicity in humans, although AA metabolism takes place in the liver. However, numerous studies have reported the harmful effect of dietary AA in the liver of experimental animals due to oxidative stress (95, 96). A high dose of 25 mg/kg AA administrated for 21 days resulted in significant decrease of liver GSH level and total antioxidant status in experimental adult rats (95). AA administration also lead to decrease of liver enzymes (e.g., aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase), superoxide dismutase, catalase activities, while total oxidant status and malondialdehyde levels increased (95). Moreover, the total cholesterol, triglyceride, and low-density lipoprotein cholesterol levels were increased, while high-density lipoprotein cholesterol was decreased following AA administration (96). Waterborne exposure of zebrafish in water containing AA (300 ppm) resulted in acute death within 26 h along with elevation of blood glucose, triglyceride, hepatic inflammation, and fat accumulation in the liver (94).
In the year 2005, European Food Safety Authority and the Joint Food and Agriculture Organization of the United Nations (FAO)/World Health Organization committee recommended to employ “margin of exposure (MOE)” approach to evaluate the risk assessment of AA (7, 97). The numerical value of MOE represents the ratio between the dose–response curve results in tumors in experimental animals and human dietary intake. Based on the modified risk assessment, the MOE value of AA showed the chemical to impose a severe human health concern compared to other chemicals, even the likes of benzopyrene (98).
Numerous strategies were shown to be able to inhibit or mitigate AA formation, both in model systems and actual food systems. These strategies include pH modification, decreasing the heating temperature and time, choosing raw materials with fewer precursors, adding the different exogenous additives, and process technology interventions (e.g., acids, amino acids, hydrogen carbonates, proteins, or antioxidant) (99, 100).
The AA formation in bread are significantly influenced by the yeast amount, fermentation time, fermentation temperature, and yeast types used in bread baking (101). Treatment with asparaginase produced from Cladosporium sp. (to convert the precursor asparagine to aspartic acid) to the wheat-based dough were able to reduce 97 and 73% of AA formation in the crust and crumb regions of the bread (102). Similarly, the use of asparaginase was able to cause 90% reduction in AA content after using a combination of asparaginase treatment and blanching of potato slices (103). In a separate study, asparaginase was also experimented on chilled French fries which reduced the AA content in the fries up to 90% without affecting final taste of the products (104).
Numerous studies used antioxidants as additives to inhibit AA formation. The inhibition of AA formation was in correlation with antioxidant activity of the additives (105). The addition of flavonoids significantly inhibited AA formation during microwave heat processing, where the inhibition strength were shown to be related to the number of phenolic hydroxyls of flavonoids (105). In addition, phenolic antioxidants also demonstrated an inhibiting effect in AA formation, with the most significant reductions (≈60%) observed for caffeoylquinic acids (100). Alternatively, addition of natural herbal extract also demonstrated inhibition of AA formation. Addition of antioxidants of bamboo leaves (AOB) and acylated AOB were able to reduce AA formation in fried potato crisps ranging from 30.7 to 46.9% (106). Similarly, the addition of green tea extract into burgers and nuggets coating were able to reduce AA formation (107).
The research studies conducted on AA described the potential health risk of fried and baked carbohydrate-rich foods. There is clear evidence to show that AA is carcinogenic in rodents. However, there is paucity of facts depicting the health risk in humans as evident from numerous epidermiological and toxicological studies. There are lacunae in the past studies, especially with regard to lack of findings in estimating dietary AA intake through questionnaires and AA content database in foods, lack of repeated exposure estimations, and lack of statistical power to detect small increases in risk. Nevertheless, it is important that efforts should continue to reduce AA levels in food products. Further studies are suggested in human beings to show the detrimental effect of AA.
Conceptual framework and design: SD. Searched references: ST and JK. Drafted manuscript: ST, SD, and JK. Critically revised the manuscript: ST, SD, and JK.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge the Research Secretariat, Universiti Kebangsaan Malaysia Medical Centre.
1. Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Some Industrial Chemicals. Lyon, France: WHO (1994). 560 p.
2. Rong H, Gao B, Zhao Y, Sun S, Yang Z, Wang Y, et al. Advanced lignin-acrylamide water treatment agent by pulp and paper industrial sludge: synthesis, properties and application. J Environ Sci (2013) 25(12):2367–77. doi:10.1016/S1001-0742(12)60326-X
3. Wuethrich A, Haddad PR, Quirino JP. Zero net-flow in capillary electrophoresis using acrylamide based hydrogel. Analyst (2014) 139:3722–6. doi:10.1039/C4AN00557K
4. Wei T, Zhang D, Chen L. The kinetics study and reaction mechanism of acrylate grouting materials. Bulg Chem Commun (2015) 47:89–92.
5. Lenze CJ, Peksa CA, Sun W, Hoeger IC, Salas C, Hubbe MA. Intact and broken cellulose nanocrystals as model nanoparticles to promote dewatering and fine-particle retention during papermaking. Cellulose (2016) 23(6):3951–62. doi:10.1007/s10570-016-1077-9
6. Tareke E, Rydberg P, Karlsson P, Eriksson S, Tornqvist M. Analysis of acrylamide, a carcinogen formed in heated foodstuffs. J Agric Food Chem (2002) 50(17):4998–5006. doi:10.1021/jf020302f
7. WHO and FAO. Health Implications of Acrylamide in Food. Geneva: World Health Organization (2002).
8. Mottram DS, Wedzicha BL, Dodson AT. Food chemistry: acrylamide is formed in the Maillard reaction. Nature (2002) 419:448–9. doi:10.1038/419448a
9. Brathen E, Knutsen SH. Effect of temperature and time on the formation of acrylamide in starch-based and cereal model systems, flat breads and bread. Food Chem (2005) 92(4):693–700. doi:10.1016/j.foodchem.2004.08.030
10. Muttucumaru N, Powers SJ, Elmore JS, Dodson A, Briddon A, Mottram DS, et al. Acrylamide-forming potential of potatoes grown at different locations, and the ratio of free asparagine to reducing sugars at which free asparagine becomes a limiting factor for acrylamide formation. Food Chem (2017) 220:76–86. doi:10.1016/j.foodchem.2016.09.199
11. Elmore JS, Briddon A, Dodson AT, Muttucumaru N, Halford NG, Mottram DS. Acrylamide in potato crisps prepared from 20 UK-grown varieties: effects of variety and tuber storage time. Food Chem (2015) 182:1–8. doi:10.1016/j.foodchem.2015.02.103
12. McCombie G, Biedermann M, Biedermann-Brem S, Suter G, Eicher A, Pfefferle A. Acrylamide in a fried potato dish (rosti) from restaurants in Zurich, Switzerland. Food Addit Contam (2016) 9(1):21–6. doi:10.1080/19393210.2015.1102974
13. Gunduz CPB, Cengiz MF. Acrylamide contents of commonly consumed bread types in Turkey. Int J Food Prop (2015) 18(4):833–41. doi:10.1080/10942912.2013.877028
14. Negoita M, Culetu A. Application of an accurate and validated method for identification and quantification of acrylamide in bread, biscuits and other bakery products using GC-MS/MS system. J Braz Chem Soc (2016) 27(10):1782–91. doi:10.5935/0103-5053.20160059
15. Mesias M, Morales FJ. Acrylamide in coffee: estimation of exposure from vending machines. J Food Compos Anal (2016) 48:8–12. doi:10.1016/j.jfca.2016.02.005
16. Wilson KM, Vesper HW, Tocco P, Sampson L, Rosen J, Hellenas KE, et al. Validation of a food frequency questionnaire measurement of dietary acrylamide intake using hemoglobin adducts of acrylamide and glycidamide. Cancer Causes Control (2009) 20(3):269–78. doi:10.1007/s10552-008-9241-7
17. Kadawathagedara M, Tong ACH, Heude B, Forhan A, Charles MA, Sirot V, et al. Dietary acrylamide intake during pregnancy and anthropometry at birth in the French EDEN mother-child cohort study. Environ Res (2016) 149:189–96. doi:10.1016/j.envres.2016.05.019
18. Mucci LA, Wilson KM. Acrylamide intake through diet and human cancer risk. J Agric Food Chem (2008) 56(15):6013–9. doi:10.1021/jf703747b
19. Michalak J, Gujska E, Kuncewicz A. RP-HPLC-DAD studies on acrylamide in cereal-based baby foods. J Food Compos Anal (2013) 32(1):68–73. doi:10.1016/j.jfca.2013.08.006
20. Cengiz MF, Gunduz CP. Acrylamide exposure among Turkish toddlers from selected cereal-based baby food samples. Food Chem Toxicol (2013) 60:514–9. doi:10.1016/j.fct.2013.08.018
21. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends: an update. Cancer Epidemiol Biomarkers Prev (2016) 25(1):16–27. doi:10.1158/1055-9965.EPI-15-0578
22. Global Burden of Disease Cancer Consortium, Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol (2017) 3(4):524–48. doi:10.1001/jamaoncol.2016.5688
23. Wilson KM, Giovannucci E, Stampfer MJ, Mucci LA. Dietary acrylamide and risk of prostate cancer. Int J Cancer (2012) 131(2):479–87. doi:10.1002/ijc.26383
24. Virk-Baker MK, Nagy TR, Barnes S, Groopman J. Dietary acrylamide and human cancer: a systematic review of literature. Nutr Cancer (2014) 66(5):774–90. doi:10.1080/01635581.2014.916323
25. Pelucchi C, Bosetti C, Galeone C, La Vecchia C. Dietary acrylamide and cancer risk: an updated meta-analysis. Int J Cancer (2015) 136(12):2912–22. doi:10.1002/ijc.29339
26. Obon-Santacana M, Freisling H, Peeters PH, Lujan-Barroso L, Ferrari P, Boutron-Ruault MC, et al. Acrylamide and glycidamide hemoglobin adduct levels and endometrial cancer risk: a nested case-control study in nonsmoking postmenopausal women from the EPIC cohort. Int J Cancer (2016) 138(5):1129–38. doi:10.1002/ijc.29853
27. Obon-Santacana M, Lujan-Barroso L, Travis RC, Freisling H, Ferrari P, Severi G, et al. Acrylamide and glycidamide hemoglobin adducts and epithelial ovarian cancer: a nested case-control study in nonsmoking postmenopausal women from the EPIC cohort. Cancer Epidemiol Biomarkers Prev (2016) 25(1):127–34. doi:10.1158/1055-9965.EPI-15-0822
28. Pelucchi C, Galeone C, Negri E, Bosetti C, Serraino D, Montella M, et al. Dietary acrylamide and the risk of endometrial cancer: an Italian case-control. Nutr Cancer (2016) 68(2):187–92. doi:10.1080/01635581.2016.1142585
29. Kotemori A, Ishihara J, Zha L, Liu R, Sawada N, Iwasaki M, et al. Dietary acrylamide intake and risk of breast cancer: the Japan Public Health Center-based Prospective Study. Cancer Sci (2017) 1–11. doi:10.1111/cas.13496
30. Pelucchi C, Rosato V, Bracci PM, Li D, Neale RE, Lucenteforte E, et al. Dietary acrylamide and the risk of pancreatic cancer in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol (2017) 28(2):408–14. doi:10.1093/annonc/mdw618
31. Hogervorst JG, van den Brandt PA, Godschalk RW, van Schooten FJ, Schouten LJ. The influence of single nucleotide polymorphisms on the association between dietary acrylamide intake and endometrial cancer risk. Sci Rep (2016) 6:34902. doi:10.1038/srep34902
32. Hogervorst JGF, van den Brandt PA, Godschalk RWL, van Schooten FJ, Schouten LJ. Interactions between dietary acrylamide intake and genes for ovarian cancer risk. Eur J Epidemiol (2017) 32(5):431–41. doi:10.1007/s10654-017-0244-0
33. Lipunova N, Schouten LJ, van den Brandt PA, Hogervorst JG. A prospective cohort study on dietary acrylamide intake and the risk for cutaneous malignant melanoma. Eur J Cancer Prev (2016) 26(6):528–31. doi:10.1097/CEJ.0000000000000268
34. Bongers ML, Hogervorst JG, Schouten LJ, Goldbohm RA, Schouten HC, van den Brandt PA. Dietary acrylamide intake and the risk of lymphatic malignancies: the Netherlands Cohort Study on diet and cancer. PLoS One (2012) 7(6):e38016. doi:10.1371/journal.pone.0038016
35. Lin Y, Lagergren J, Lu Y. Dietary acrylamide intake and risk of esophageal cancer in a population-based case-control study in Sweden. Int J Cancer (2011) 128(3):676–81. doi:10.1002/ijc.25608
36. Lujan-Barroso L, Gonzalez CA, Slimani N, Obon-Santacana M, Ferrari P, Freisling H, et al. Dietary intake of acrylamide and esophageal cancer risk in the European Prospective Investigation into Cancer and Nutrition cohort. Cancer Causes Control (2014) 25(5):639–46. doi:10.1007/s10552-014-0359-5
37. Liu ZM, Tse LA, Ho SC, Wu S, Chen B, Chan D, et al. Dietary acrylamide exposure was associated with increased cancer mortality in Chinese elderly men and women: a 11-year prospective study of Mr. and Ms. OS Hong Kong. J Cancer Res Clin Oncol (2017) 143(11):2317–26. doi:10.1007/s00432-017-2477-4
38. Ghanayem BI, McDaniel LP, Churchwell MI, Twaddle NC, Snyder R, Fennell TR, et al. Role of CYP2E1 in the epoxidation of acrylamide to glycidamide and formation of DNA and hemoglobin adducts. Toxicol Sci (2005) 88(2):311–8. doi:10.1093/toxsci/kfi307
39. Ghanayem BI, Witt KL, El-Hadri L, Hoffler U, Kissling GE, Shelby MD, et al. Comparison of germ cell mutagenicity in male CYP2E1-null and wild-type mice treated with acrylamide: evidence supporting a glycidamide-mediated effect. Biol Reprod (2005) 72(1):157–63. doi:10.1095/biolreprod.104.033308
40. Sumner SC, Fennell TR, Moore TA, Chanas B, Gonzalez F, Ghanayem BI. Role of cytochrome P4502E1 in the metabolism of acrylamide and acrylonitrile in mice. Chem Res Toxicol (1999) 12(11):1110–6. doi:10.1021/tx990040k
41. Ehlers A, Lenze D, Broll H, Zagon J, Hummel M, Lampen A. Dose dependent molecular effects of acrylamide and glycidamide in human cancer cell lines and human primary hepatocytes. Toxicol Lett (2013) 217(2):111–20. doi:10.1016/j.toxlet.2012.12.017
42. Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol (2003) 66(8):1499–503. doi:10.1016/S0006-2952(03)00504-5
43. Laborde E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ (2010) 17:1373–80. doi:10.1038/cdd.2010.80
44. Srivastava S, Sabri MI, Agrawal AK, Seth PK. Effect of single and repeated doses of acrylamide and bis-acrylamide on glutathione-S-transferase and dopamine receptors in rat brain. Brain Res (1986) 371(2):319–23. doi:10.1016/0006-8993(86)90369-0
45. Banerjee S, Segal A. In vitro transformation of C3H/10T1/2 and NIH/3T3 cells by acrylonitrile and acrylamide. Cancer Lett (1986) 32(3):293–304. doi:10.1016/0304-3835(86)90182-5
46. Abate C, Patel L, Rauscher FJ, Curran T. Redox regulation of fos and jun DNA-binding activity in vitro. Science (1990) 249(4973):1157–61. doi:10.1126/science.2118682
47. Tsuda H, Shimizu CS, Taketomi MK, Hasegawa MM, Hamada A, Kawata KM, et al. Acrylamide; induction of DNA damage, chromosomal aberrations and cell transformation without gene mutations. Mutagenesis (1993) 8(1):23–9. doi:10.1093/mutage/8.1.23
48. Schulze-Osthoff K, Los M, Baeuerle PA. Redox signalling by transcription factors NF-κB and AP-1 in lymphocytes. Biochem Pharmacol (1995) 50(6):735–41. doi:10.1016/0006-2952(95)02011-Z
49. Bowyer JF, Latendresse JR, Delongchamp RR, Muskhelishvili L, Warbritton AR, Thomas M, et al. The effects of subchronic acrylamide exposure on gene expression, neurochemistry, hormones, and histopathology in the hypothalamus-pituitary-thyroid axis of male Fischer 344 rats. Toxicol Appl Pharmacol (2008) 230(2):208–15. doi:10.1016/j.taap.2008.02.028
50. Khan MA, Davis CA, Foley GL, Friedman MA, Hansen LG. Changes in thyroid gland morphology after acute acrylamide exposure. Toxicol Sci (1999) 47(2):151–7. doi:10.1093/toxsci/47.2.151
51. Dourson M, Hertzberg R, Allen B, Haber L, Parker A, Kroner O, et al. Evidence-based dose-response assessment for thyroid tumorigenesis from acrylamide. Regul Toxicol Pharmacol (2008) 52(3):264–89. doi:10.1016/j.yrtph.2008.08.004
52. Meek ME, Bucher JR, Cohen SM, Dellarco V, Hill RN, Lehman-McKeeman LD, et al. A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol (2003) 33(6):591–653. doi:10.1080/713608373
53. Recio L, Friedman M, Marroni D, Maynor T, Chepelev NL. Impact of acrylamide on calcium signaling and cytoskeletal filaments in testes from F344 rat. Int J Toxicol (2017) 36(2):124–32. doi:10.1177/1091581817697696
54. Stewart TA, Yapa KT, Monteith GR. Altered calcium signaling in cancer cells. Biochim Biophys Acta (2015) 1848(10 Pt B):2502–11. doi:10.1016/j.bbamem.2014.08.016
55. Chepelev NL, Gagne R, Maynor T, Kuo B, Hobbs CA, Recio L, et al. Transcriptional profiling of male F344 rats suggests the involvement of calcium signaling in the mode of action of acrylamide-induced thyroid cancer. Food Chem Toxicol (2017) 107(Pt A):186–200. doi:10.1016/j.fct.2017.06.019
56. Arocena M. Effect of acrylamide on the cytoskeleton and apoptosis of bovine lens epithelial cells. Cell Biol Int (2006) 30(12):1007–12. doi:10.1016/j.cellbi.2006.07.008
57. Alberts B, Watson J, Bray D, Lewis J. Molecular Biology of the Cell. 5th ed. New York: Garland Science (2008).
58. Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature (2010) 463(7280):485–92. doi:10.1038/nature08908
59. Sickles DW, Welter DA, Friedman MA. Acrylamide arrests mitosis and prevents chromosome migration in the absence of changes in spindle microtubules. J Toxicol Environ Health (1995) 44(1):73–86. doi:10.1080/15287399509531944
60. Sickles DW, Brady ST, Testino A, Friedman MA, Wrenn RW. Direct effect of the neurotoxicant acrylamide on kinesin-based microtubule motility. J Neurosci Res (1996) 46(1):7–17. doi:10.1002/(SICI)1097-4547(19961001)46:1<7::AID-JNR2>3.0.CO;2-P
61. Sickles DW, Sperry AO, Testino A, Friedman M. Acrylamide effects on kinesin-related proteins of the mitotic/meiotic spindle. Toxicol Appl Pharmacol (2007) 222(1):111–21. doi:10.1016/j.taap.2007.04.006
62. Gassner P, Adler ID. Induction of hypoploidy and cell cycle delay by acrylamide in somatic and germinal cells of male mice. Mutat Res (1996) 367(4):195–202. doi:10.1016/S0165-1218(96)90077-4
63. Adler ID, Gonda H, Hrabe de Angelis M, Jentsch I, Otten IS, Speicher MR. Heritable translocations induced by dermal exposure of male mice to acrylamide. Cytogenet Genome Res (2004) 104(1–4):271–6. doi:10.1159/000077501
64. Marchetti F, Lowe X, Bishop J, Wyrobek AJ. Induction of chromosomal aberrations in mouse zygotes by acrylamide treatment of male germ cells and their correlation with dominant lethality and heritable translocations. Environ Mol Mutagen (1997) 30(4):410–7. doi:10.1002/(SICI)1098-2280(1997)30:4<410:AID-EM6>3.0.CO;2-M
65. Adler ID, Zouh R, Schmid E. Perturbation of cell division by acrylamide in vitro and in vivo. Mutat Res (1993) 301(4):249–54. doi:10.1016/0165-7992(93)90065-4
66. Cao J, Beisker W, Nusse M, Adler ID. Flow cytometric detection of micronuclei induced by chemicals in poly- and normochromatic erythrocytes of mouse peripheral blood. Mutagenesis (1993) 8(6):533–41. doi:10.1093/mutage/8.6.533
67. Heald R. Motor function in the mitotic spindle. Cell (2000) 102(4):399–402. doi:10.1016/S0092-8674(00)00044-1
68. Marx A, Hoenger A, Mandelkow E. Structures of kinesin motor proteins. Cell Motil Cytoskeleton (2009) 66(11):958–66. doi:10.1002/cm.20392
69. Gaglio T, Dionne MA, Compton DA. Mitotic spindle poles are organized by structural and motor proteins in addition to centrosomes. J Cell Biol (1997) 138(5):1055–66. doi:10.1083/jcb.138.5.1055
70. Karabay A, Walker RA. Identification of microtubule binding sites in the Ncd tail domain. Biochemistry (1999) 38(6):1838–49. doi:10.1021/bi981850i
71. Walczak CE, Verma S, Mitchison TJ. XCTK2: a kinesin-related protein that promotes mitotic spindle assembly in Xenopus laevis egg extracts. J Cell Biol (1997) 136(4):859–70. doi:10.1083/jcb.136.4.859
72. Spencer PS, Schaumburg HH. A review of acrylamide neurotoxicity. Part I. Properties, uses and human exposure. Can J Neurol Sci (1974) 1(2):143–50. doi:10.1017/S0317167100019739
73. LoPachin RM. The changing view of acrylamide neurotoxicity. Neurotoxicology (2004) 25(4):617–30. doi:10.1016/j.neuro.2004.01.004
74. Hagmar L, Tornqvist M, Nordander C, Rosen I, Bruze M, Kautiainen A, et al. Health effects of occupational exposure to acrylamide using hemoglobin adducts as biomarkers of internal dose. Scand J Work Environ Health (2001) 27(4):219–26. doi:10.5271/sjweh.608
75. McCollister DD, Oyen F, Rowe VK. Toxicology of acrylamide. Toxicol Appl Pharmacol (1964) 6(2):172–81. doi:10.1016/0041-008X(64)90103-6
76. Miller MS, Spencer PS. The mechanisms of acrylamide axonopathy. Annu Rev Pharmacol Toxicol (1985) 25:643–66. doi:10.1146/annurev.pa.25.040185.003235
77. Erkekoglu P, Baydar T. Acrylamide neurotoxicity. Nutr Neurosci (2014) 17(2):49–57. doi:10.1179/1476830513Y.0000000065
78. LoPachin RM, Barber DS, He D, Das S. Acrylamide inhibits dopamine uptake in rat striatal synaptic vesicles. Toxicol Sci (2006) 89(1):224–34. doi:10.1093/toxsci/kfj005
79. Dearfield KL, Abernathy CO, Ottley MS, Brantner JH, Hayes PF. Acrylamide: its metabolism, developmental and reproductive effects, genotoxicity, and carcinogenicity. Mutat Res (1988) 195(1):45–77. doi:10.1016/0165-1110(88)90015-2
80. Tyl RW, Friedman MA. Effects of acrylamide on rodent reproductive performance. Reprod Toxicol (2003) 17(1):1–13. doi:10.1016/S0890-6238(02)00078-3
81. Hashimoto K, Tanii H. Mutagenicity of acrylamide and its analogues in Salmonella typhimurium. Mutat Res Lett (1985) 158(3):129–33. doi:10.1016/0165-1218(85)90075-8
82. Sakamoto J, Hashimoto K. Reproductive toxicity of acrylamide and related compounds in mice – effects on fertility and sperm morphology. Arch Toxicol (1986) 59(4):201–5. doi:10.1007/BF00290538
83. Zenick H, Hope E, Smith MK. Reproductive toxicity associated with acrylamide treatment in male and female rats. J Toxicol Environ Health (1986) 17(4):457–72. doi:10.1080/15287398609530840
84. Yilmaz BO, Yildizbayrak N, Aydin Y, Erkan M. Evidence of acrylamide- and glycidamide-induced oxidative stress and apoptosis in Leydig and Sertoli cells. Hum Exp Toxicol (2017) 36(12):1225–35. doi:10.1177/0960327116686818
85. Exon JH. A review of the toxicology of acrylamide. J Toxicol Environ Health (2006) 9(5):397–412. doi:10.1080/10937400600681430
86. Tyl RW, Marr MC, Myers CB, Ross WP, Friedman MA. Relationship between acrylamide reproductive and neurotoxicity in male rats. Reprod Toxicol (2000) 14(2):147–57. doi:10.1016/S0890-6238(00)00066-6
87. Tyla RW, Friedman MA, Losco PE, Fisher LC, Johnson KA, Strother DE, et al. Rat two-generation reproduction and dominant lethal study of acrylamide in drinking water. Reprod Toxicol (2000) 14(5):385–401. doi:10.1016/S0890-6238(00)00097-6
88. Ali SF, Hong JS, Wilson WE, Uphouse LL, Bondy SC. Effect of acrylamide on neurotransmitter metabolism and neuropeptide levels in several brain regions and upon circulating hormones. Arch Toxicol (1983) 52(1):35–43. doi:10.1007/BF00317980
89. Burek JD, Albee RR, Beyer JE, Carreon RM, Morden DC, Wade CE, et al. Subchronic toxicity of acrylamide administered to rats in the drinking water followed by up to 144 days of recovery. J Environ Pathol Toxicol (1980) 4:157–82.
90. Zaidi SI, Raisuddin S, Singh KP, Jafri A, Husain R, Husain MM, et al. Acrylamide induced immunosuppression in rats and its modulation by 6-MFA, an interferon inducer. Immunopharmacol Immunotoxicol (1994) 16(2):247–60. doi:10.3109/08923979409007093
91. Yener Y, Sur E, Telatar T, Oznurlu Y. The effect of acrylamide on alpha-naphthyl acetate esterase enzyme in blood circulating lymphocytes and gut associated lymphoid tissues in rats. Exp Toxicol Pathol (2013) 65(1–2):143–6. doi:10.1016/j.etp.2011.07.002
92. Fang J, Liang CL, Jia XD, Li N. Immunotoxicity of acrylamide in female BALB/c mice. Biomed Environ Sci (2014) 27(6):401–9. doi:10.3967/bes2014.069
93. Kim KH, Park B, Rhee DK, Pyo S. Acrylamide induces senescence in macrophages through a process involving ATF3, ROS, p38/JNK, and a telomerase-independent pathway. Chem Res Toxicol (2015) 28(1):71–86. doi:10.1021/tx500341z
94. Kim SM, Baek JM, Lim SM, Kim JY, Kim J, Choi I, et al. Modified lipoproteins by acrylamide showed more atherogenic properties and exposure of acrylamide induces acute hyperlipidemia and fatty liver changes in zebrafish. Cardiovasc Toxicol (2015) 15(4):300–8. doi:10.1007/s12012-014-9294-7
95. Gedik S, Erdemli ME, Gul M, Yigitcan B, Gozukara Bag H, Aksungur Z, et al. Hepatoprotective effects of crocin on biochemical and histopathological alterations following acrylamide-induced liver injury in Wistar rats. Biomed Pharmacother (2017) 95:764–70. doi:10.1016/j.biopha.2017.08.139
96. Ghorbel I, Elwej A, Chaabene M, Boudawara O, Marrakchi R, Jamoussi K, et al. Effects of acrylamide graded doses on metallothioneins I and II induction and DNA fragmentation: biochemical and histomorphological changes in the liver of adult rats. Toxicol Ind Health (2017) 33(8):611–22. doi:10.1177/0748233717696613
97. EFSA. Opinion of the scientific committee on a request from EFSA related to a harmonised approach for risk assessment of substances which are both genotoxic and carcinogenic. EFSA J (2005) 3(10):282. doi:10.2903/j.efsa.2005.282
98. WHO. Evaluation of Certain Contaminants in Food: Seventy-Second Report of the Joint FAO/WHO Expert Committee on Food Additives. Geneva: World Health Organization (2011).
99. Liu Y, Wang P, Chen F, Yuan Y, Zhu Y, Yan H, et al. Role of plant polyphenols in acrylamide formation and elimination. Food Chem (2015) 186:46–53. doi:10.1016/j.foodchem.2015.03.122
100. Constantinou C, Koutsidis G. Investigations on the effect of antioxidant type and concentration and model system matrix on acrylamide formation in model Maillard reaction systems. Food Chem (2016) 197(Pt A):769–75. doi:10.1016/j.foodchem.2015.11.037
101. Katsaiti T, Granby K. Mitigation of the processing contaminant acrylamide in bread by reducing asparagine in the bread dough. Food Add Contam (2016) 33(9):1402–10. doi:10.1080/19440049.2016.1217068
102. Mohan Kumar NS, Shimray CA, Indrani D, Manonmani HK. Reduction of acrylamide formation in sweet bread with l-asparaginase treatment. Food Bioprocess Technol (2014) 7(3):741–8. doi:10.1007/s11947-013-1108-6
103. Pedreschi F, Mariotti S, Granby K, Risum J. Acrylamide reduction in potato chips by using commercial asparaginase in combination with conventional blanching. Food Sci Technol (2011) 44(6):1473–6. doi:10.1016/j.lwt.2011.02.004
104. Medeiros Vinci R, Mestdagh F, Van Poucke C, Kerkaert B, de Muer N, Denon Q, et al. Implementation of acrylamide mitigation strategies on industrial production of French fries: challenges and pitfalls. J Agric Food Chem (2011) 59(3):898–906. doi:10.1021/jf1042486
105. Cheng J, Chen X, Zhao S, Zhang Y. Antioxidant-capacity-based models for the prediction of acrylamide reduction by flavonoids. Food Chem (2015) 168:90–9. doi:10.1016/j.foodchem.2014.07.008
106. Ma X, Wang E, Lu Y, Wang Y, Ou S, Yan R. Acylation of antioxidant of bamboo leaves with fatty acids by lipase and the acylated derivatives’ efficiency in the inhibition of acrylamide formation in fried potato crisps. PLoS One (2015) 10(6):e0130680. doi:10.1371/journal.pone.0130680
Keywords: acrylamide, food, nutrition, cancer, mechanism, human
Citation: Kumar J, Das S and Teoh SL (2018) Dietary Acrylamide and the Risks of Developing Cancer: Facts to Ponder. Front. Nutr. 5:14. doi: 10.3389/fnut.2018.00014
Received: 24 October 2017; Accepted: 12 February 2018;
Published: 28 February 2018
Edited by:
Marcelo Perim Baldo, Unimontes, BrazilReviewed by:
Dora Il’yasova, Georgia State University, United StatesCopyright: © 2018 Kumar, Das and Teoh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seong Lin Teoh, dGVvaHNlb25nbGluQHlhaG9vLmNvbS5teQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.