
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Mol. Neurosci., 14 December 2022
Sec. Neuroplasticity and Development
Volume 15 - 2022 | https://doi.org/10.3389/fnmol.2022.1043018
This article is part of the Research TopicNew Insights into the Molecular Mechanisms of Autistic Spectrum DisordersView all 9 articles
 Na Wang1†
Na Wang1† Longbao Lv2†Xiaoyi Huang1†Mingqin Shi1Youwu Dai1
Longbao Lv2†Xiaoyi Huang1†Mingqin Shi1Youwu Dai1 Yuanyuan Wei1
Yuanyuan Wei1 Bonan Xu1Chenyang Fu1
Bonan Xu1Chenyang Fu1 Haoyu Huang3Hongling Shi4
Haoyu Huang3Hongling Shi4 Yun Liu3*Xintian Hu2*
Yun Liu3*Xintian Hu2* Dongdong Qin1*
Dongdong Qin1*Autism spectrum disorder (ASD) is a lifelong neurodevelopmental disease, and its diagnosis is dependent on behavioral manifestation, such as impaired reciprocal social interactions, stereotyped repetitive behaviors, as well as restricted interests. However, ASD etiology has eluded researchers to date. In the past decades, based on strong genetic evidence including mutations in a single gene, gene editing technology has become an essential tool for exploring the pathogenetic mechanisms of ASD via constructing genetically modified animal models which validates the casual relationship between genetic risk factors and the development of ASD, thus contributing to developing ideal candidates for gene therapies. The present review discusses the progress in gene editing techniques and genetic research, animal models established by gene editing, as well as gene therapies in ASD. Future research should focus on improving the validity of animal models, and reliable DNA diagnostics and accurate prediction of the functional effects of the mutation will likely be equally crucial for the safe application of gene therapies.
Autism spectrum disorder (ASD) is a lifelong neurodevelopmental disorder characterized by deficits in social communication with restricted and repetitive behaviors (Lord et al., 2018). Approximately 1.5% of the world population is affected by ASD (Schaaf et al., 2020), and about 50%–60% of ASD cases have a genetic etiology (Taylor et al., 2020). Despite the obvious challenges associated with the identification of ASD causes, intensive genetic studies have confirmed that ASD has a strong genetic basis and many susceptibility genes have been identified by genetic analysis (Havdahl et al., 2021). For ASD, the development is centered around the monogenic forms (Weuring et al., 2021). In recent decades, many efforts have been put into developing new ASD animal models established by gene-editing techniques which target mutations introduced into the genome, and then the phenotype is characterized to explore gene expression profiles specific to ASD (Fan et al., 2020). Gene-editing techniques including zinc finger nuclease (ZFN; Hamilton et al., 2014), transcription activator-like effector nuclease (TALEN), clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) systems (Zhang et al., 2019), and Cre-loxp systems (Mclellan et al., 2017) can utilize various modifications to achieve rapid and targeted gene editing, such as site-directed mutagenesis, insertion, knockdown, and combinatorial editing. At the same time, novel gene therapies have been developed on the aforementioned basis which may contribute to identifying ideal therapeutic candidates for ASD. Here, we review the progress in gene editing techniques and genetic research, animal models established by gene editing, as well as gene therapies in ASD, highlight key issues in ASD-associated model studies, and attempt to identify potential avenues for the treatment of ASD. This will provide valuable insights into the application of gene editing techniques for exploring the pathogenetic mechanisms and therapies for ASD.
Gene editing is a genetic engineering technology that can modify, delete, or insert a small piece of DNA at a specific point in the genome of cells and organisms, which holds great promises in the fields of gene detection, gene function regulation, drug research and disease treatment (Cao et al., 2020). Cre-loxp system makes it possible to achieve the precise spatiotemporal control of gene expression by introducing point mutations or insertions/deletions into the host genome. First- and second-generation methods, ZFN and TALEN, respectively, employ nucleases that have both DNA recognition binding domains and DNA cleavage domains (Zhang et al., 2018). The DNA-binding domain in these systems is specially designed to attach to the specific DNA sequence that the DNA nuclease domain cleaves. However, due to their high cost, high off-target likelihood, and complex structure, these first- and second-generation gene editing technologies’ wide range of applications suffer from a low target recognition rate (Huang S. et al., 2021), and now they are replaced by CRISPR/Cas9 which relies on Cas9 and a single guide RNA (sgRNA) to achieve precise cutting to offer ease of design, development and increased efficacy (Kantor et al., 2020). Prior to the introduction of gene editing technologies, natural, physical, or chemical mutagenesis and random insertion of transgenic DNA were the principal approaches used to generate mutations within targeted cells. However, by their nature, none of these approaches could achieve gene editing at specific desired loci, and they were also disadvantaged by random mutagenic events, low efficiency, as well as time-consuming, laborious, and costly (Yang et al., 2017). Due to the development of improved genetic editing systems, the study of the function of selected genes and their relationship with disease-related phenotypes was simplified. Gene editing techniques are powerful tools to model gene–base disorders, as it allows researchers to precisely study the association between genes or genetic variants and the development of an altered phenotype, which helps to explore the pathogenetic mechanisms of ASD. A schematic diagram of the main gene editing techniques is shown in Figure 1A. The detailed pros and cons of the main gene editing techniques are shown in Table 1.
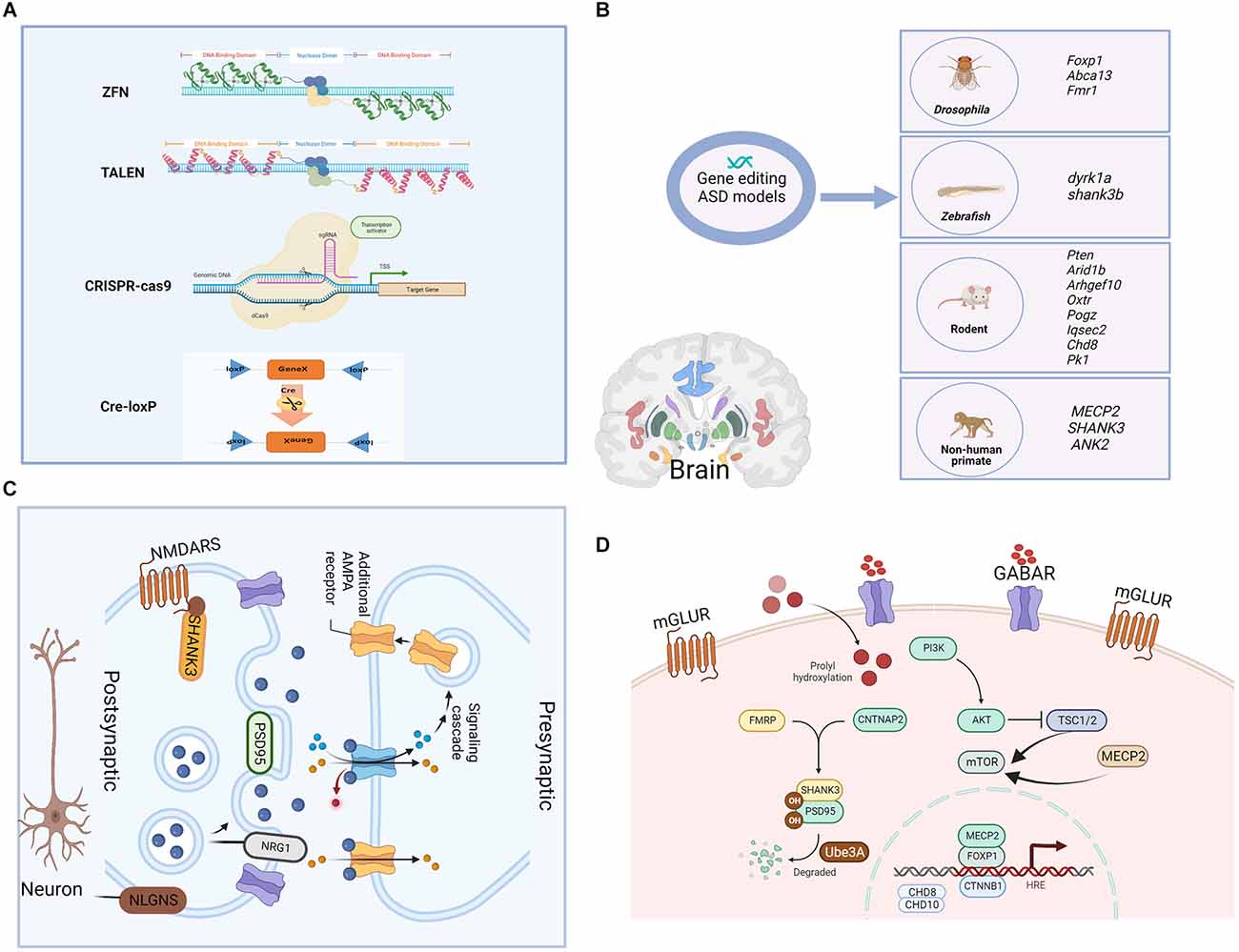
Figure 1. Gene-editing technologies and single gene-edited animal models of ASD as well as the synaptic and cellular functions of modified genes. (A) The main genomic editing technologies available at the moment. (a) Zinc Finger Nucleases—ZFNs; (b) Transcription Activator-Like Effector Nucleases—TALENs; (c) CRISPR/Cas9; and (d) Cre-loxP recombinase system. (B) Single gene-edited animal models of ASD. Functions of the shown genes: Foxp1. forkhead box protein 1, encodes a transcription factor important for the early development of many organ systems, including the brain. Abca13: ATP-binding cassette protein A13, is predicted to have the typical structure of full-size ABC proteins, consisting of two transmembrane domains (TMDs). Fmr1: fragile X mental retardation 1, is primarily associated with neuro/psychiatric risks. dyrk1a: dual specificity tyrosine phosphorylation regulated kinase 1A, is a murine homolog of the Drosophila minibrain gene and has been found to be involved in many biological processes during development and adulthood. shank3b: SH3 and multiple ankyrin repeat domains 3, are predicted to enable ionotropic glutamate receptor binding activity and synaptic receptor adaptor activity. Pten: phosphatase and tensin homolog, is an essential gene for proper control of cell movement and migration. Arid1b: ATT-rich interaction domain 1B, encodes a chromatin remodeling factor, and its haploinsufficiency can cause abnormal gene expression in the brain and induce ASD-like behaviors in mice. Arhgef10: Rho guanine nucleotide exchange factor 10, is a known guanine nucleotide exchange factor (GEF) for RhoA with proposed roles in various diseases. Oxtr: oxytocin receptor, shows close association with prosocial behavior, and produced numerous pro-social effects through intranasal applications of oxytocin. Pogz: pogo transposable element derived with ZNF domain, encodes a multidomain nuclear protein involved in transcriptional regulation and its defective function has been recently associated with a syndromic neurodevelopmental disorder. Iqsec2: isoleucine-glutamine motif and Sec7 domain-containing protein 2, is an X-linked gene that is associated with an autism spectrum disorder. Chd8: chromodomain helicase DNA binding protein 8, functions in several processes that include transcriptional regulation, epigenetic remodeling, promotion of cell proliferation, and regulation of RNA synthesis. Pk1: prokineticin 1, encodes a nuclear receptor that may be a negative regulator of the Wnt/beta-catenin signaling pathway. MECP2: methyl-CpG binding protein 2, is an X chromosome-linked protein coding gene encoding the MECP2 protein, which is important for the function of nerve cells and plays a role in maintaining connections (synapses) between neurons. SHANK3: SH3 and multiple ankyrin repeat domains 3, play a role in the functioning of synapses, and act as a scaffold that supports the connections between neurons, ensuring that the signals sent by one neuron are received by another. ANK2: giant ankyrin 2, plays a role in endocytosis and intracellular protein transport, and is required for synapse stability. (C) Synaptic function of modified genes in the ASD model. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; PSD95, postsynaptic density 95; NRG1, neuregulin; NMDARS, N-methyl D-aspartate (NMDA) receptors; NLGNS, Neuroligins. (D) Cellular function of modified genes in the ASD model. mGLUR, metabotropic glutamate receptors; GABAR, gamma-aminobutyric acid receptor; PI3K, phosphoinositide 3-kinase; TSC1/2, tuberous sclerosis 1/2; mTOR, mammalian target of rapamycin; FMRP: CNTNAP2, contactin associated protein 2; CTNNB1, catenin beta 1.
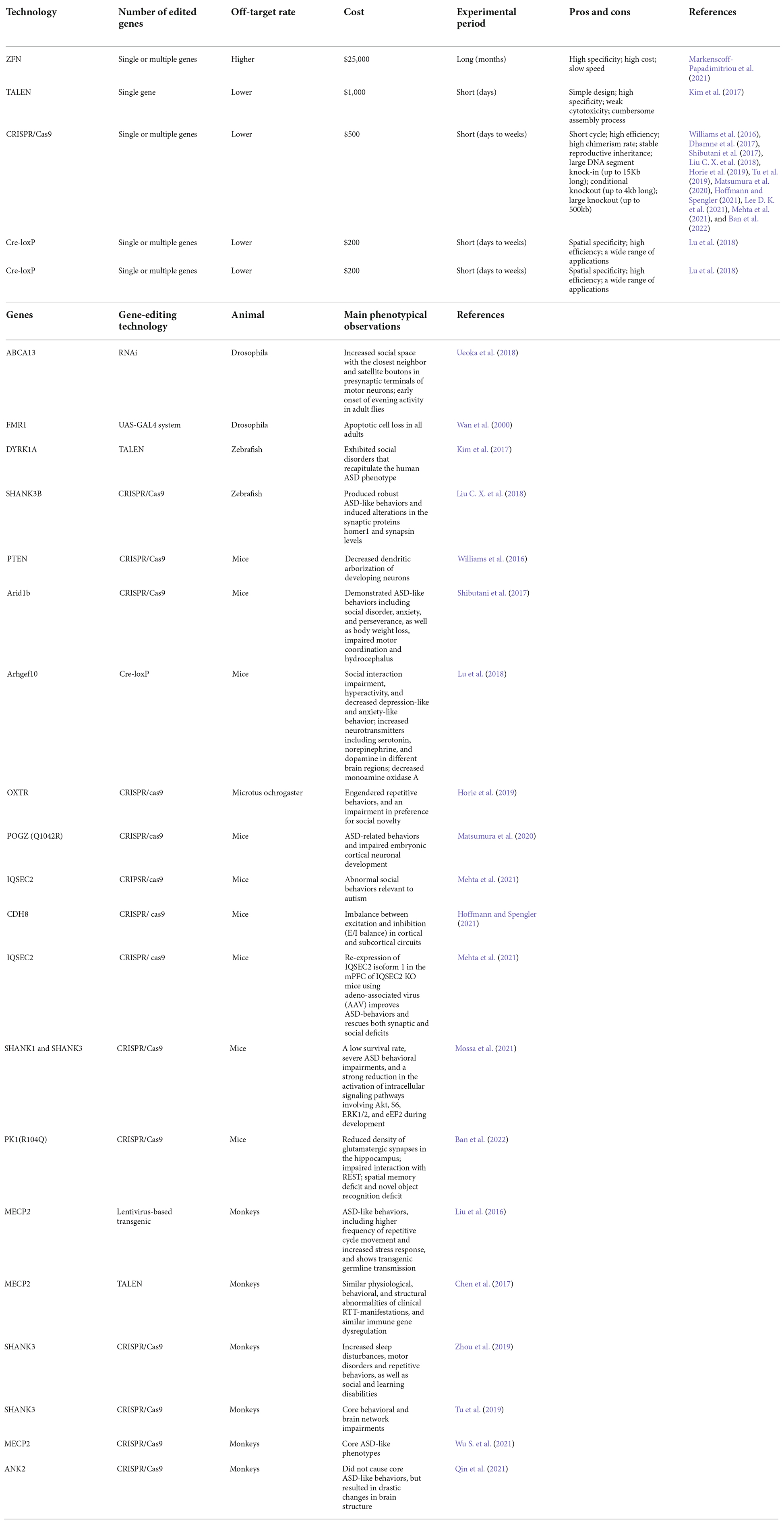
Table 1. Pros and cons of different gene editing techniques, and monogenic ASD animal models established by gene editing.
With the continuous development of high-throughput sequencing, such as genome, transcriptome, and proteome, effective methods have been developed for analysis, identification, verification, and application of biomarkers for ASD. When combined with animal models, they will also finally provide insights into the pathogenesis, accurate diagnosis, and development of novel drugs for ASD.
Like many other neuropsychiatric disorders, ASD is a high-risk disorder for genetic alterations that affect the expression or function of proteins involved in the formation and maintenance of synapses, or in chromatin remodeling. Genetic abnormalities in ASD can be divided into three categories: single gene mutations (e.g., SH3 and multiple ankyrin repeat domains 3 (Shank3), chromodomains helicase DNA-binding (Chd), methyl-CpG binding protein 2 (MeCP2), contactin associated protein (Cntnap), fragile X messenger ribonucleoprotein (Fmr), ubiquitin protein ligase E3A (Ube3a), TSC complex subunit 1 (Tsc1), TSC complex subunit 2 (Tsc2) and oxytocin receptor (Oxtr); polygenic mutations; and copy number variants including chromosomal duplication, large deletions, inversions, and translocations (Mashayekhi et al., 2021). In this review, we will focus on ASD models based on single gene mutations, which are summarized in Table 1. Most genetic findings implicated in the pathogenesis of ASD to date are inconsistent. However, there are some examples of positive replications. Here, we will review most of the genes that have been consistently related to ASD.
SHANK proteins are multidomain scaffold proteins of the postsynaptic density, which connect neurotransmitter receptors, ion channels, and other membrane proteins to the actin cytoskeleton and G-protein-coupled signaling pathways. They are fundamental to synapse formation and dendritic spine maturation. Gene variants of the Shank family are significantly associated with the development of ASD (Ma et al., 2021). When mutations occur in the Shank gene, it leads to ASD characterized by impairments in social interaction and communication, and restricted behavioral patterns and interests. Moreover, Shank gene family members play important roles in postsynaptic organization by forming complexes containing postsynaptic receptors, ion channels, neurexins (NRXNs), and neuroligins (NLGNs; Medina et al., 2018). SHANK3 and SHANK2, involved in dendritic spine morphogenesis during neuron development, are essential scaffolding proteins found specifically in the postsynaptic density (PSD) of excitatory neurons. Mutations in these two genes can lead to impaired motor coordination, repetitive behavior, and altered social interactions (Woike et al., 2022). In Shank2-deficient mice, synapses, chromatin, ribosomes, mitochondria, GABA (γ-aminobutyric acid) neurons, and oligodendrocytes may show alterations and abnormal functions (Lee D. K. et al., 2021). Mutations in the pre-and post-synaptic adhesion proteins NRXNs and NLGNs have been associated with ASD (Wang et al., 2018). In addition, NLGNs have been shown to interact with postsynaptic scaffolding protein, whereas SHANK3 is required for glutamatergic synaptic maturation. NRXNs act as ligands for NLGNs, ranging from transsynaptic interactions between NRXNs and NLGNs to postsynaptic density complex, including SHANK3 scaffold proteins (Yamagata et al., 2018). Eukaryotic translation initiation factor 4E binding protein 2 (EIF4E-BP2) acts as a regulator of synapse activity and neuronal stem cell renewal by its ability to repress translation initiation, which is involved in synaptic plasticity, learning, and memory formation. The absence of the EIF4E-BP2 leads to the overexpression of NLGN-class cell adhesion molecules. Contactins are axon-associated cell adhesion molecules, play an important role in neuronal network formation and plasticity. Moreover, they are glycosylphosphatidylinositol-anchored neuronal membrane proteins that participate in axon connections in the developing nervous system. Especially, CNTN4 (contactin 4) is an immunoglobulin superfamily domain cell adhesion molecules (IgCAMs) protein with important functions associated with fear memory and is related to altered neuronal morphology and synaptic plasticity of hippocampal neurons (Oguro-Ando et al., 2021). Extensive studies have found that the contactin-associated protein-like (CNTNAP) family played key roles in synaptic development and social behavior. For example, CNTNAP3 has been found to interact with synaptic adhesion proteins (neuroglia 1 and 2), as well as scaffold proteins (PSD95 and gephyrin; Tong et al., 2019). Mice with Cntnap2 mutations showed a reduced frequency of prefrontal projection neural clusters in the cingulate cortex, and this finding revealed a possible selective modulation of Cntnap2 by integrating prefrontal regional connectivity (Liska et al., 2018). MeCP2 is an X chromosome-linked gene that encodes the MECP2 protein, which is widely expressed in the developing and mature brain, acts as a key epigenetic regulator of gene expression, and is essential for neural function (Lu et al., 2020). MECP2 was first found to function as a transcriptional repressor. It binds to the CpG island of methylated DNA and recruits co-repressors, such as the histone deacetylase (HDAC) complex, for transcriptional repression. A number of genetic studies have found that MeCP2 mutation affects synaptic development and neural circuit connectivity in Rett syndrome (RTT). RTT shares some common features with ASD, including motor function deficits, cognitive impairment, and other symptoms associated with mental retardation. Interestingly, MeCP2 has been reported to bind and repress long genes involved in neuronal differentiation and modulation of neuronal function.
Several members of the CHD protein family are involved in synaptic adhesion, axon growth, and guidance which are critical for brain development. Chd8, Chd9, and Chd11 are three of the most widely studied candidate genes in the field of ASD genetics (Wang et al., 2019). In particular, Chd8 encodes a neurite outgrowth-regulating membrane protein cadherin-8, and it is one of the most frequently mutated genes in ASD patients. Its mutation causes an imbalance between excitatory and inhibitory neurotransmitters and has been linked to ASD and learning disability (Suetterlin et al., 2018). The oxytocin receptor (Oxtr) gene showed a close association with prosocial behavior and produced numerous pro-social effects through intranasal applications of oxytocin (Striepens et al., 2011). During brain development, changes of gene methylation occurred in response to environmental changes in the ASD. For example, the Oxtr gene encoding the oxytocin receptor is significantly hypermethylated in the peripheral blood cells and in the temporal cortex of ASD patients, highlighting the reduced oxytocin signaling in ASD. Disconnected (disco)-interacting protein 2 homolog A (DIP2A) protein participates in the synthesis of acetylation-enzyme A (Ac-CoA), which is mainly expressed in brain regions enriched with pyramidal neurons (Ma et al., 2019). The ATT-rich interaction domain 1B (ARID1B) protein encodes a chromatin remodeling factor, and its haploinsufficiency can cause abnormal gene expression in the brain and induce ASD-like behaviors in mice (Shibutani et al., 2017). Engrailed-2 (En2) is a transcription factor that promotes neuronal differentiation of GABAergic neurons in the basal ganglia via TrkB (tropomyosin receptor kinase B)-dependent BDNF (brain-derived neurotrophic factor) signaling (Carratala-Marco et al., 2018).
T-box brain transcription factor 1 (TBR1) is a homodimer involved in axonal projection development and neuronal activation in the amygdala, which plays a critical role in the regulation of social interaction and auditory communication. TBR1 interacts with the forkhead box protein P2 (FOXP2) to cause speech and language disorders (Medvedeva et al., 2019). The Fmr1 gene encodes fragile X mental retardation protein (FMRP), a polyribosome-associated protein playing an important role in protein translation. FMRP targets chromatin modifier genes essential for encoding postsynaptic density proteins (Goin-Kochel et al., 2017). It has been reported that both loss of function and over-expression of UBE3A reduce dendritic branching, suggesting that an appropriate expression level of Ube3a is necessary for normal dendritic pattern (Dindot et al., 2008). ASD-associated proteins such as FMR1 (Jansen et al., 2017), pogo transposable element derived with ZNF domain (POGZ; Zhao et al., 2019), TSC complex subunit 1 (TSC1)/TSC complex subunit 2 (TSC2; Martin et al., 2020), and phosphatase and tensin homolog (PTEN; Grencewicz et al., 2021), are all involved in the regulation of protein synthesis. Additional risk genes for ASD include dual specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1a) (Earl et al., 2017), kinesin family member 1 (Kif1) (Huang Y. et al., 2021, and trafficking protein particle complex subunit 9 (Trappc9) (Cogne et al., 2019). Since the etiology for a substantial portion of ASD remains unknown, these genes may only represent the tip of the “heritable ASD” iceberg.
Current genetic ASD animal models reported in previous studies include transgenic, knock-in, and gene mutations in Drosophila, zebrafish, rodents, and nonhuman primates (Figure 1B), which are based on single gene mutations mainly in the following genes: synaptogenesis and synaptic plasticity-related genes (Shank3, Nrnx1, Nlgns, Nlgn4, Dat, Mglur, Fmrp), chromatin remodeling-related genes (MeCP2, Foxp1, Ctnnb1, Pogz, Chd8, Chd10), protein synthesis-related genes (Tsc1/2, mTor), and protein degradation-related gene (Ube3a). The functions of ASD-related genes in synapses and cells are shown in Figures 1C,D. However, due to inconsistencies of results in most genetic findings implicated in the pathogenesis of ASD, as well as edited genes’ differences between species, we summarized the examples of ASD models of single gene mutations from Drosophila, zebrafish, rodents, and non-human primates.
Drosophila is used as an important model for ASD research because 75% of human disease-causing genes have functional fly homologs and flies have similar neural properties of the visual and motor systems as humans. Drosophila has a wide range of practical and genetic advantages, such as a short breeding time and a large number of offspring for rapid, large-scale analysis. These features allow efficient high-throughput genetic manipulation and greatly facilitate the discovery of single-gene function and high-level behavior.
A Drosophila fragile X syndrome (FXS) model was developed by using loss-of-function mutants or overexpression of the Fmr1 homolog. In these models, poor synaptic structure was associated with perturbation of neurotransmission and synaptic type-specific alterations in both central and peripheral synapses (Zhang et al., 2001). The results showed that dFmr1 (Drosophila fragile X mental retardation 1) mutant flies displayed considerable defects in the Drosophila stress odorant (dSO) response. Cyclic adenosine monophosphate (cAMP) signaling via PKA was activated after exposure to dSO and several drugs that regulated both cAMP and cyclic guanosine monophosphate (cGMP) levels significantly improved defects in dSO processing in dFmr1 mutant flies (Androschuk et al., 2018).
Na+/H+ exchanger-3 (NHE3) controls the exchange of sodium and hydrogen ions in the cell membrane, which directly affects neural signaling. Reduced Nhe3 expression affects the rate of sodium ion and proton exchange at the cell membrane. The separation between the first and second-order electro-physiological visual responses to homeostatic stimuli in Nhe3 adult mutant flies is strikingly similar to response patterns in human adults with ASD (Vilidaite et al., 2018). Drosophila models phenotypically replicate FXS and Fmr1 knockout mice, and pharmacological tests have been used to develop potential therapies. Altogether, these data reconfirm the suitability of Drosophila as a biomedical research model and its relevance to our understanding of genes and the physiopathology behind ASD.
Zebrafish is a good animal model for neural tube development, which has the advantages of in vitro development, embryonic transparency, and rapid development. It also has the main neural cell types including neurons, astrocytes, oligodendrocytes, and microglia. In zebrafish, common methods to create loss-of-function models include genomic knockout through mutagenesis and morpholino knockdown. Morpholino oligomers can be injected into early zebrafish embryos and transiently knock down the functional targeted genes by binding to the complementary target mRNA and blocking its translation in a manner similar to small interfering RNAs.
CRISPR/Cas9-induced shank3 mutations, which caused ASD-like behaviors in zebrafish, resulted in reduced locomotor activity, and decreased levels of synaptic proteins. Additionally, reduced shank3 expression in the zebrafish model system is responsible for gastrointestinal dysmotility, which resembles gastrointestinal symptoms commonly seen in patients with ASD (Liu C. X. et al., 2018; James et al., 2019). Zebrafish with Cntnap2 mutations were overactive at night, and showed a GABAergic deficit, especially in the forebrain, and sensitivity to drug-induced seizures (Hoffman et al., 2016). A Dyrk1aa knock-out (KO) zebrafish was generated using TALEN-mediated genome editing. KO zebrafish showed social impairments consistent with human phenotypes of ASD (Kim et al., 2017).
Knockdown of pten (phosphatase and tensin homolog) by using antisense morpholino oligonucleotides (MOs) in zebrafish embryos inhibited convergent extension by affecting cell motility and protrusion during gastrulation. pten is an essential gene for proper control of cell movement and migration during zebrafish gastrulation, and regulates actin polymerization by antagonizing PI3 kinase and its downstream effectors AKT1 (AKT serine/threonine kinase 1) and CDC42 (cell division cycle 42; Yeh et al., 2011).
Based on the observation that fmrp knockdown zebrafish embryos exhibited increased anxiety, irritability, and cognitive impairment at 7 dpf (days post-fertilization), researchers concluded that this was an effective DNAzyme-based model for fragile X syndrome, which was associated with impaired craniofacial development and ASD-like behaviors (Medishetti et al., 2020). Although the CRISPR/Cas9 system is arguably the most commonly used gene-editing tool, other mutagenesis methods such as TALEN and ENU (N-ethyl-N-nitrosourea) are often used to create loss-of-function alleles in zebrafish studies. Henceforth, the study in the future should focus on the available genetic strategies applicable to zebrafish to develop reliable models to functionally validate ASD-candidate genes.
Rodent models can recapitulate the molecular genetic defects associated with ASD patients and can serve as effective tools for dissecting the underlying molecular and cellular mechanisms of ASD. Rodent genetic models are developed based on well-studied monogenic or syndromic genes associated with ASD, such as Shank3, MeCP2, Chd8, Pcdh10, Arid1b, Chd11, Chd9, Cntn4, Dip2a, and Casp3. Reduced sociability and social agency presented in adult Shank3 mutant mice are restored through gene re-expression. Lee’s results show that Shank3 disruption can cause a decrease in neurons encoding the experience of other mice, while inducing an increase in neurons encoding their own experience. Recovery of Shank3 expression reversed this coding imbalance and increased sociability within 5–8 weeks (Lee S. et al., 2021). This important finding points to gene therapy as a possible treatment for ASD.
It has been found that survival and phenotypic severity of male MeCP2 null mice improved after neonatal intracranial delivery of single-stranded (ss) AAV9/chicken beta-actin (CBA)-MECP2 vector. Treated mice showed significant improvement in phenotypic severity of motor function and exploratory activity, and normalization of neuronal nuclear volume in transfected cells (Gadalla et al., 2013). In MeCP2-deficient mice, pyramidal neurons of the primary somatosensory cortex exhibited decreased spontaneous firing, decreased spontaneous excitatory synaptic input, and increased inhibitory drive, as well as decreased excitatory synaptic connectivity (Yu et al., 2020). The reversibility of the RTT-like phenotype in mice suggested that MeCP2 gene substitution is a potential therapeutic option for patients. Chd8+/– mice showed increased sensitivity to chemical injury and exhibited delayed dyskinesia, marked low activity, and abnormal ASD-like behavior to social stimuli, and increased synchronous activity in the cortico-hippocampus and auditory-parietal networks (Suetterlin et al., 2018). Female mice carrying a heterozygous mutation of Chd8+/N2373K exhibited suppression of neuronal excitability, inhibitory synaptic transmission, and enhanced neuronal firing, as well as increased gene expression associated with extracellular vesicles and matrix (Jung et al., 2018). Pcdh10+/– male mice showed increased dendritic spine density in immature morphology, while NMDAR expression was decreased, and displayed specific alterations in related behavior and mood (Ferri et al., 2021). Heterozygous Arid1b knockout (hKO) mice exhibit ASD-like traits associated with social behavior, anxiety, and persistence, as well as weight loss, impaired motor coordination, and hydrocephalus (Shibutani et al., 2017). Chd11-null mice, but not Chd9-null mice, have multiple autism-like behavioral changes (Wu N. et al., 2021). The small or missing regions of the anterior commissure of Tbr1+/– mice lead to defective neuronal differentiation in the amygdala, which may serve as a suitable model for determining how autism genes control neuronal circuits, neural activity, and behaviors (Huang et al., 2019). The Cntn4 KO mice model showed increased stress responses, decreased synaptic potentiation, and abnormal dendritic arborization and spines of hippocampal CA1 neurons (Oguro-Ando et al., 2021). Dip2a deficiency results in defective spine morphogenesis, PSD, and reduced synaptic transmission of pyramidal neurons, highlighting the contribution of synaptic protein acetylation to synaptic processing (Ma et al., 2019). Selective loss of the Casp3 gene in the striatum by Cre-loxp-mediated recombination, significantly reduced potassium-induced dopamine release in mice. In addition, loss of Casp3 can lead to impaired social interactions, restricted interests, and repetitive stereotypes in mice (Garcia-Dominguez et al., 2021). Although mice models are valuable for studying the pathogenesis of ASD, species-specific features in behavioral manifestation, as well as brain structure and functions pose a significant challenge to the use of small animals to simulate ASD in humans and translate experimental treatments to the clinic. Specifically, there are considerable differences between rodents and humans in many aspects, such as brain anatomy, cognitive capacity, and behavioral repertoire. Compared with rodents, non-human primates share great similarities with humans in key brain regions implicated in social behaviors.
Rodent models have been widely used to model human genetic disorders, while the evolutionary differences between these two species have also been the focus of discussion regarding translational potentials. Non-human primates (NHPs) are considered to be better models for ASD than rodents because of their strong similarity to humans in terms of brain anatomy. Therefore, the use of NHPs to model human disease is of great importance, especially for neurodevelopmental disorders. Lentivirus-based transgenic cynomolgus monkeys overexpressing MECP2 in the brain exhibited ASD-like behaviors, including a higher frequency of repetitive circular locomotion, increased stress responses, and fewer interactive behaviors. Interestingly, the cognitive functions of transgenic monkeys were essentially normal, even though some showed stereotypic cognitive behaviors (Liu et al., 2016). TALEN-edited MeCP2 mutant cynomolgus monkeys showed similar phenotypes to ASD patients, including disruption of social interaction, social withdrawal, sleep disturbances, and reduced response to pain (Chen et al., 2017). Adeno-associated viruses (AAV)-delivered CRISPR/Cas9 gene editing can efficiently induce deletion of MECP2 in the hippocampus of adolescent rhesus monkeys, and elicit core ASD-like phenotypes, including defects in social communication and interaction, hyposensitivity to sensory inputs, hypo-activity during the daytime, abnormal sleep patterns and hand motions (Wu S. et al., 2021). After CRISPR/Cas9-mediated disruption of SHANK3, mutant monkeys displayed core behavioral abnormalities, including impaired social interaction and repetitive behavior, apparent stereotypic movements, and reduced brain network activity detected by positron emission computed tomography (PET). Impaired social interaction and stereotypic behavior were significantly improved after fluoxetine treatment (Tu et al., 2019). Another study reported germline-transmissible mutations of SHANK3 in cynomolgus macaques and their F1 offspring. The founder mutants displayed sleep disturbances, motor deficits, and increased repetitive behaviors, as well as social and learning impairment characteristic of ASD (Liu et al., 2019). Giant ankyrin 2 (ANK2), a strong candidate gene involved in nonsyndromic ASD, was depleted in cynomolgus and rhesus monkeys using CRISPR/Cas9 (Qin et al., 2021). Surprisingly, the depletion of ANK2 in monkeys did not cause the core symptoms of ASD that had been observed in a mouse model (Yang et al., 2019). Rather, mutant monkeys showed typical abnormalities in brain development, including significant enlargement of lateral ventricles and loss of gray matter. This suggests that the functions of ANK2 are evolutionarily divergent between rodents and primates. This finding provides further evidence that non-human primates are much better suited to recapitulate ASD-like behavior, which has great potential for the development of effective ASD treatments. The animal models of ASD are shown in Table 1.
Rodent ASD models are faced with the challenges of relating the rodent brain to the human brain and rodent behaviors to human behaviors. Rodents are separated from humans by more than 70 million years of evolution (Gibbs et al., 2004). While, NHPs diverge from human evolution closer to 25 million years ago (Qin et al., 2019). In comparison to rodents, NHPs exhibit greater similarity to humans in genetic variation, brain development, brain size, functionally specialized brain structures and, ultimately, ASD-like behaviors. The brain regions implicated in social processing, such as the frontal, temporal, and anterior cingulate cortices as well as the amygdala, are either not well-developed or nonexistent in rodents, but are better developed in NHPs. Like humans, NHPs live in a complex social structure and have evolved a sophisticated social communication and interaction system, such as communicating with a variety of facial expressions, vocalizations, and body postures (Bauman and Schumann, 2018). However, reciprocal social behavior is limited in rodents, who rely heavily on olfactory communication. This is because not all brain regions implicated in ASD are well-developed in rodents, and genetic differences may also contribute to it. Undoubtedly, laboratory rats and mice provide ideal animal models for ASD research and comparative medicine studies due to their small size, ease of maintenance, short life cycle, and abundant genetic resources (Bryda, 2013). NHPs are ongoingly contributing to identify neural circuits and patterns related to behaviors affected by ASD. In the future, we should strengthen collaborative efforts between rodents and NHPs models to give full play to their advantages, and ultimately translate results from animal models into preventative strategies and/or novel therapies for human ASD.
Rodents have been widely used to study the pathogenesis of ASD, but it is not yet known whether we can accurately simulate and identify symptoms and mechanisms of ASD using rodent models. Non-human primates are not only related to humans in terms of genetic evolution, brain development, brain function, and social organization, but also exhibit very similar behavioral phenotypes. Cloning of macaque monkeys by somatic cell nucleus transfer (SCNT) can allow the generation of monkeys with uniform genetic backgrounds. This is very useful to expand ASD models with uniform genetic backgrounds and allows researchers to increase the significance level of the results (Liu Z. et al., 2018, Liu et al., 2019).
The monogenic aspect and severity of syndromic ASD make it an ideal candidate for gene therapy. The development of an antisense oligonucleotide (ASO) targeting toxic CGG repeats in FMR1 mRNA, reduced the number of pathogenic transcripts of FMR1 mRNA both in vitro and in vivo (Derbis et al., 2021). An ASO targeted to the native antisense transcript was able to un-silence the paternal allele and restore normal UBE3A expression in vitro and in vivo (Meng et al., 2015). The ASO targeting UBE3A-ATS was developed by two pharmaceutical companies and is currently in phase 1 or 2 clinical trials (Markati et al., 2021). Human hemopoietic stem cells were first transduced with an LV containing the UBE3A gene, and subsequently engrafted in the mouse. Eight weeks after birth, both newborn and adult mice showed normalized UBE3A levels, as well as phenotypic rescue (Adhikari et al., 2021). CRISPR/Cas9 has also been considered for the treatment of Angelman syndrome (AS) by knocking out the UBE3A antisense transcript, which can silence transcription. This strategy has been shown to be effective in AS mouse models and has ameliorated the disease phenotype (Wolter et al., 2020). Gene editing permanently changes the genome and can be a new therapy for ASD. Gene therapies with transient effects, such as ASOs, non-coding RNA, and RNA editing, which leave the genome unedited and require repeated administration, may have the advantage of being more controllable and reversible. These differences highlight important issues related to gene therapy, including dosing, administration, and safety (Weuring et al., 2021).
ZFN enables targeted homologous recombination in model organisms and human cells, which can edit single or multiple genes with high specificity. However, its application in gene therapy is significantly limited due to high off-target rate, slow speed, and high cost. TALEN is a highly specific single gene editing technology with a low off-target rate, short period, and weak cytotoxicity, but the cumbersome assembly process limits its application in ASD therapy. CRIPSR/Cas9 is not only used for genome editing but also adopted for transcriptional perturbation and epigenetic modulation. Whereas, the application of CRIPSR/Cas9 in ASD therapy is still a challenge due to low specificity. Although Cre/loxP provides cell- and time-specificity of gene editing, there is currently a limited application in ASD therapy.
Gene editing has the advantage of assessing gene interactions and the relationships between genes and phenotypes. ASD is a neurodevelopmental disease with considerable heterogeneity. Previous studies do not support the causality between the brain regions and ASD behaviors because of the apparent technical limitations in human studies. A variety of animal models designed to reproduce genetic or idiopathic ASD have highlighted potential therapeutic approaches targeting the core behaviors and underlying mechanisms of ASD. Gene re-expression with AAV vectors can reverse behavioral phenotypes or recover animal models from ASD symptoms, and these studies create a platform for clinical translation of various single gene forms of ASD. However, major obstacles remain pending issues, especially in most ASD disorders which are characterized by variable penetrance, epistasis, and a phenotype determined by the interaction between genetic and environmental factors. Furthermore, whether, or in what case, epigenetic factors can preclude reversibility, is still unclear. Future research should focus on improving the validity of animal models, also reliable DNA diagnostics and accurate prediction of the functional effects of the mutation will likely be equally crucial for the safe application of gene treatments.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the National Natural Science Foundation of China (31960178, 82160923); Applied Basic Research Programs of Science and Technology Commission Foundation of Yunnan Province (2019FA007); Key Laboratory of Traditional Chinese Medicine for Prevention and Treatment of Neuropsychiatric Diseases, Yunnan Provincial Department of Education; Scientific Research Projects for High-level Talents of Yunnan University of Chinese Medicine (2019YZG01); Young Top-Notch Talent in 10,000 Talent Program of Yunnan Province (YNWR-QNBJ-2019-235); National Science and Technology Innovation 2030 Major Program (2021ZD0200900).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adhikari, A., Copping, N. A., Beegle, J., Cameron, D. L., Deng, P., O’Geen, H., et al. (2021). Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum. Mol. Genet. 30, 1067–1083. doi: 10.1093/hmg/ddab104
Androschuk, A., He, R. X., Weber, S., Rosenfelt, C., and Bolduc, F. V. (2018). Stress odorant sensory response dysfunction in Drosophila fragile X syndrome mutants. Front. Mol. Neurosci. 11:242. doi: 10.3389/fnmol.2018.00242
Ban, Y., Yu, T., Wang, J., Wang, X., Liu, C., Baker, C., et al. (2022). Mutation of the murine Prickle1 (R104Q) causes phenotypes analogous to human symptoms of epilepsy and autism. Exp. Neurol. 347:113880. doi: 10.1016/j.expneurol.2021.113880
Bauman, M. D., and Schumann, C. M. (2018). Advances in nonhuman primate models of autism: integrating neuroscience and behavior. Exp. Neurol. 299, 252–265. doi: 10.1016/j.expneurol.2017.07.021
Bryda, E. C. (2013). The mighty mouse: the impact of rodents on advances in biomedical research. Mo. Med. 110, 207–211.
Cao, J. X., Wang, Y. L., and Wang, Z. X. (2020). Advances in precise regulation of CRISPR/Cas9 gene editing technology. Yi Chuan 42, 1168–1177. doi: 10.16288/j.yczz.20-069
Carratala-Marco, F., Andreo-Lillo, P., Martinez-Morga, M., Escamez-Martinez, T., Botella-Lopez, A., Bueno, C., et al. (2018). Clinical phenotypes associated to engrailed 2 gene alterations in a series of neuropediatric patients. Front. Neuroanat. 12:61. doi: 10.3389/fnana.2018.00061
Chen, Y., Yu, J., Niu, Y., Qin, D., Liu, H., Li, G., et al. (2017). Modeling rett syndrome using TALEN-edited MECP2 mutant cynomolgus monkeys. Cell 169, 945–955.e10. doi: 10.1016/j.cell.2017.04.035
Cogne, B., Ehresmann, S., Beauregard-Lacroix, E., Rousseau, J., Besnard, T., Garcia, T., et al. (2019). Missense variants in the histone acetyltransferase complex component gene TRRAP cause autism and syndromic intellectual disability. Am. J. Hum. Genet. 104, 530–541. doi: 10.1016/j.ajhg.2019.01.010
Derbis, M., Kul, E., Niewiadomska, D., Sekrecki, M., Piasecka, A., Taylor, K., et al. (2021). Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 12:1265. doi: 10.1038/s41467-021-21021-w
Dhamne, S. C., Silverman, J. L., Super, C. E., Lammers, S., Hameed, M. Q., Modi, M. E., et al. (2017). Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism 8:26. doi: 10.1186/s13229-017-0142-z
Dindot, S. V., Antalffy, B. A., Bhattacharjee, M. B., and Beaudet, A. L. (2008). The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17, 111–118. doi: 10.1093/hmg/ddm288
Earl, R. K., Turner, T. N., Mefford, H. C., Hudac, C. M., Gerdts, J., Eichler, E. E., et al. (2017). Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol. Autism 8:54. doi: 10.1186/s13229-017-0173-5
Fan, C., Gao, Y., Liang, G., Huang, L., Wang, J., Yang, X., et al. (2020). Transcriptomics of Gabra4 knockout mice reveals common NMDAR pathways underlying autism, memory and epilepsy. Mol. Autism 11:13. doi: 10.1186/s13229-020-0318-9
Ferri, S. L., Dow, H. C., Schoch, H., Lee, J. Y., Brodkin, E. S., and Abel, T. (2021). Age- and sex-specific fear conditioning deficits in mice lacking Pcdh10, an autism associated gene. Neurobiol. Learn. Mem. 178:107364. doi: 10.1016/j.nlm.2020.107364
Gadalla, K. K., Bailey, M. E., Spike, R. C., Ross, P. D., Woodard, K. T., Kalburgi, S. N., et al. (2013). Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol. Ther. 21, 18–30. doi: 10.1038/mt.2012.200
Garcia-Dominguez, I., Suarez-Pereira, I., Santiago, M., Perez-Villegas, E. M., Bravo, L., Lopez-Martin, C., et al. (2021). Selective deletion of Caspase-3 gene in the dopaminergic system exhibits autistic-like behaviour. Prog. Neuropsychopharmacol. Biol. Psychiatry 104:110030. doi: 10.1016/j.pnpbp.2020.110030
Gibbs, R. A., Weinstock, G. M., Metzker, M. L., Muzny, D. M., Sodergren, E. J., Scherer, S., et al. (2004). Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521. doi: 10.1038/nature02426
Goin-Kochel, R. P., Trinh, S., Barber, S., and Bernier, R. (2017). Gene disrupting mutations associated with regression in autism spectrum disorder. J. Autism Dev. Disord. 47, 3600–3607. doi: 10.1007/s10803-017-3256-4
Grencewicz, D. J., Romigh, T., Thacker, S., Abbas, A., Jaini, R., Luse, D., et al. (2021). Redefining the PTEN promoter: identification of novel upstream transcription start regions. Hum. Mol. Genet. 30, 2135–2148. doi: 10.1093/hmg/ddab175
Hamilton, S. M., Green, J. R., Veeraragavan, S., Yuva, L., Mccoy, A., Wu, Y., et al. (2014). Fmr1 and Nlgn3 knockout rats: novel tools for investigating autism spectrum disorders. Behav. Neurosci. 128, 103–109. doi: 10.1037/a0035988
Havdahl, A., Niarchou, M., Starnawska, A., Uddin, M., van der Merwe, C., and Warrier, V. (2021). Genetic contributions to autism spectrum disorder. Psychol. Med. 51, 2260–2273. doi: 10.1037/a0035988
Hoffmann, A., and Spengler, D. (2021). Single-cell transcriptomics supports a role of CHD8 in autism. Int. J. Mol. Sci. 22:3261. doi: 10.3390/ijms22063261
Hoffman, E. J., Turner, K. J., Fernandez, J. M., Cifuentes, D., Ghosh, M., Ijaz, S., et al. (2016). Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron 89, 725–733. doi: 10.1016/j.neuron.2015.12.039
Horie, K., Inoue, K., Suzuki, S., Adachi, S., Yada, S., Hirayama, T., et al. (2019). Oxytocin receptor knockout prairie voles generated by CRISPR/Cas9 editing show reduced preference for social novelty and exaggerated repetitive behaviors. Horm. Behav. 111, 60–69. doi: 10.1016/j.yhbeh.2018.10.011
Huang, Y., Jiao, J., Zhang, M., Situ, M., Yuan, D., Lyu, P., et al. (2021). [A study on KIF1A gene missense variant analysis and its protein expression and structure profiles of an autism spectrum disorder family trio]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 38, 620–625. doi: 10.3760/cma.j.cn511374-20210120-00060
Huang, S., Yan, Y., Su, F., Huang, X., Xia, D., Jiang, X., et al. (2021). Research progress in gene editing technology. Front. Biosci. (Landmark Ed) 26, 916–927. doi: 10.52586/4997
Huang, T. N., Yen, T. L., Qiu, L. R., Chuang, H. C., Lerch, J. P., and Hsueh, Y. P. (2019). Haploinsufficiency of autism causative gene Tbr1 impairs olfactory discrimination and neuronal activation of the olfactory system in mice. Mol. Autism 10:5. doi: 10.1186/s13229-019-0257-5
James, D. M., Kozol, R. A., Kajiwara, Y., Wahl, A. L., Storrs, E. C., Buxbaum, J. D., et al. (2019). Intestinal dysmotility in a zebrafish (Danio rerio) shank3a;shank3b mutant model of autism. Mol. Autism 10:3. doi: 10.1186/s13229-018-0250-4
Jansen, A., Dieleman, G. C., Smit, A. B., Verhage, M., Verhulst, F. C., Polderman, T., et al. (2017). Gene-set analysis shows association between FMRP targets and autism spectrum disorder. Eur. J. Hum. Genet. 25, 863–868. doi: 10.1038/ejhg.2017.55
Jung, H., Park, H., Choi, Y., Kang, H., Lee, E., Kweon, H., et al. (2018). Sexually dimorphic behavior, neuronal activity and gene expression in Chd8-mutant mice. Nat. Neurosci. 21, 1218–1228. doi: 10.1038/s41593-018-0208-z
Kantor, A., McClements, M. E., and MacLaren, R. E. (2020). CRISPR-Cas9 DNA base-editing and prime-editing. Int. J. Mol. Sci. 21:6240. doi: 10.3390/ijms21176240
Kim, O. H., Cho, H. J., Han, E., Hong, T. I., Ariyasiri, K., Choi, J. H., et al. (2017). Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 8:50. doi: 10.1186/s13229-017-0168-2
Lee, S., Kang, H., Jung, H., Kim, E., and Lee, E. (2021). Gene dosage- and age-dependent differential transcriptomic changes in the prefrontal cortex of Shank2-mutant mice. Front. Mol. Neurosci. 14:683196. doi: 10.3389/fnmol.2021.683196
Lee, D. K., Li, S. W., Bounni, F., Friedman, G., Jamali, M., Strahs, L., et al. (2021). Reduced sociability and social agency encoding in adult Shank3-mutant mice are restored through gene re-expression in real time. Nat. Neurosci. 24, 1243–1255. doi: 10.1038/s41593-021-00888-4
Liska, A., Bertero, A., Gomolka, R., Sabbioni, M., Galbusera, A., Barsotti, N., et al. (2018). Homozygous loss of autism-risk gene CNTNAP2 results in reduced local and long-range prefrontal functional connectivity. Cereb. Cortex 28, 1141–1153. doi: 10.1093/cercor/bhx022
Liu, Z., Cai, Y., Liao, Z., Xu, Y., Wang, Y., Wang, Z., et al. (2019). Cloning of a gene-edited macaque monkey by somatic cell nuclear transfer. Natl. Sci. Rev. 6, 101–108. doi: 10.1093/nsr/nwz003
Liu, Z., Cai, Y., Wang, Y., Nie, Y., Zhang, C., Xu, Y., et al. (2018). Cloning of macaque monkeys by somatic cell nuclear transfer. Cell 172, 881–887.e7. doi: 10.1016/j.cell.2018.01.020
Liu, C. X., Li, C. Y., Hu, C. C., Wang, Y., Lin, J., Jiang, Y. H., et al. (2018). CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism 9:23. doi: 10.1186/s13229-018-0204-x
Liu, Z., Li, X., Zhang, J. T., Cai, Y. J., Cheng, T. L., Cheng, C., et al. (2016). Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 530, 98–102. doi: 10.1038/nature16533
Lord, C., Elsabbagh, M., Baird, G., and Veenstra-Vanderweele, J. (2018). Autism spectrum disorder. Lancet 392, 508–520. doi: 10.1016/S0140-6736(18)31129-2
Lu, D. H., Liao, H. M., Chen, C. H., Tu, H. J., Liou, H. C., Gau, S. S., et al. (2018). Impairment of social behaviors in Arhgef10 knockout mice. Mol. Autism 9:11. doi: 10.1186/s13229-018-0197-5
Lu, Z., Liu, Z., Mao, W., Wang, X., Zheng, X., Chen, S., et al. (2020). Locus-specific DNA methylation of Mecp2 promoter leads to autism-like phenotypes in mice. Cell Death Dis. 11:85. doi: 10.1038/s41419-020-2290-x
Ma, S. L., Chen, L. H., Lee, C. C., Lai, K., Hung, S. F., Tang, C. P., et al. (2021). Genetic overlap between attention deficit/hyperactivity disorder and autism spectrum disorder in SHANK2 gene. Front. Neurosci. 15:649588. doi: 10.3389/fnins.2021.649588
Ma, J., Zhang, L. Q., He, Z. X., He, X. X., Wang, Y. J., Jian, Y. L., et al. (2019). Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLoS Biol. 17:e3000461. doi: 10.1371/journal.pbio.3000461
Markati, T., Duis, J., and Servais, L. (2021). Therapies in preclinical and clinical development for Angelman syndrome. Expert Opin. Investig. Drugs 30, 709–720. doi: 10.1080/13543784.2021.1939674
Markenscoff-Papadimitriou, E., Binyameen, F., Whalen, S., Price, J., Lim, K., Ypsilanti, A. R., et al. (2021). Autism risk gene POGZ promotes chromatin accessibility and expression of clustered synaptic genes. Cell Rep. 37:110089. doi: 10.1016/j.celrep.2021.110089
Martin, P., Wagh, V., Reis, S. A., Erdin, S., Beauchamp, R. L., Shaikh, G., et al. (2020). TSC patient-derived isogenic neural progenitor cells reveal altered early neurodevelopmental phenotypes and rapamycin-induced MNK-eIF4E signaling. Mol. Autism 11:2. doi: 10.1186/s13229-019-0311-3
Mashayekhi, F., Mizban, N., Bidabadi, E., and Salehi, Z. (2021). The association of SHANK3 gene polymorphism and autism. Minerva Pediatr. (Torino) 73, 251–255. doi: 10.23736/S2724-5276.16.04539-4
Matsumura, K., Seiriki, K., Okada, S., Nagase, M., Ayabe, S., Yamada, I., et al. (2020). Pathogenic POGZ mutation causes impaired cortical development and reversible autism-like phenotypes. Nat. Commun. 11:859. doi: 10.1038/s41467-020-14697-z
Mclellan, M. A., Rosenthal, N. A., and Pinto, A. R. (2017). Cre-loxP-mediated recombination: general principles and experimental considerations. Curr. Protoc. Mouse Biol. 7, 1–12. doi: 10.1002/cpmo.22
Medina, M. A., Andrade, V. M., Caracci, M. O., Avila, M. E., Verdugo, D. A., Vargas, M. F., et al. (2018). Wnt/β-catenin signaling stimulates the expression and synaptic clustering of the autism-associated Neuroligin 3 gene. Transl. Psychiatry 8:45. doi: 10.1038/s41398-018-0093-y
Medishetti, R., Rani, R., Kavati, S., Mahilkar, A., Akella, V., Saxena, U., et al. (2020). A DNAzyme based knockdown model for Fragile-X syndrome in zebrafish reveals a critical window for therapeutic intervention. J. Pharmacol. Toxicol. Methods 101:106656. doi: 10.1016/j.vascn.2019.106656
Medvedeva, V. P., Rieger, M. A., Vieth, B., Mombereau, C., Ziegenhain, C., Ghosh, T., et al. (2019). Altered social behavior in mice carrying a cortical Foxp2 deletion. Hum. Mol. Genet. 28, 701–717. doi: 10.1093/hmg/ddy372
Mehta, A., Shirai, Y., Kouyama-Suzuki, E., Zhou, M., Yoshizawa, T., Yanagawa, T., et al. (2021). IQSEC2 deficiency results in abnormal social behaviors relevant to autism by affecting functions of neural circuits in the medial prefrontal cortex. Cells 10:2724. doi: 10.3390/cells10102724
Meng, L., Ward, A. J., Chun, S., Bennett, C. F., Beaudet, A. L., and Rigo, F. (2015). Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518, 409–412. doi: 10.1038/nature13975
Mossa, A., Pagano, J., Ponzoni, L., Tozzi, A., Vezzoli, E., Sciaccaluga, M., et al. (2021). Developmental impaired Akt signaling in the Shank1 and Shank3 double knock-out mice. Mol. Psychiatry 26, 1928–1944. doi: 10.1038/s41380-020-00979-x
Oguro-Ando, A., Bamford, R. A., Sital, W., Sprengers, J. J., Zuko, A., Matser, J. M., et al. (2021). Cntn4, a risk gene for neuropsychiatric disorders, modulates hippocampal synaptic plasticity and behavior. Transl. Psychiatry 11:106. doi: 10.1038/s41398-021-01223-y
Qin, D., Wu, S., Chen, Y., and Hu, X. (2019). Behavioral screening tools for identifying autism in macaques: existing and promising tests. Brain Res. Bull. 146, 87–93. doi: 10.1016/j.brainresbull.2018.12.018
Qin, D. D., Zhou, J. K., He, X. C., Shen, X. Y., Li, C., Chen, H. Z., et al. (2021). Depletion of giant ANK2 in monkeys causes drastic brain volume loss. Cell Discov. 7:113. doi: 10.1038/s41421-021-00336-4
Schaaf, C. P., Betancur, C., Yuen, R., Parr, J. R., Skuse, D. H., Gallagher, L., et al. (2020). A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat. Rev. Genet. 21, 367–376. doi: 10.1038/s41576-020-0231-2
Shibutani, M., Horii, T., Shoji, H., Morita, S., Kimura, M., Terawaki, N., et al. (2017). Arid1b haploinsufficiency causes abnormal brain gene expression and autism-related behaviors in mice. Int. J. Mol. Sci. 18:1872. doi: 10.3390/ijms18091872
Striepens, N., Kendrick, K. M., Maier, W., and Hurlemann, R. (2011). Prosocial effects of oxytocin and clinical evidence for its therapeutic potential. Front. Neuroendocrinol. 32, 426–450. doi: 10.1016/j.yfrne.2011.07.001
Suetterlin, P., Hurley, S., Mohan, C., Riegman, K., Pagani, M., Caruso, A., et al. (2018). Altered neocortical gene expression, brain overgrowth and functional over-connectivity in Chd8 haploinsufficient mice. Cereb. Cortex 28, 2192–2206. doi: 10.1093/cercor/bhy058
Taylor, M. J., Rosenqvist, M. A., Larsson, H., Gillberg, C., D’Onofrio, B. M., Lichtenstein, P., et al. (2020). Etiology of autism spectrum disorders and autistic traits over time. JAMA Psychiatry 77, 936–943. doi: 10.1001/jamapsychiatry.2020.0680
Tong, D. L., Chen, R. G., Lu, Y. L., Li, W. K., Zhang, Y. F., Lin, J. K., et al. (2019). The critical role of ASD-related gene CNTNAP3 in regulating synaptic development and social behavior in mice. Neurobiol. Dis. 130:104486. doi: 10.1016/j.nbd.2019.104486
Tu, Z., Zhao, H., Li, B., Yan, S., Wang, L., Tang, Y., et al. (2019). CRISPR/Cas9-mediated disruption of SHANK3 in monkey leads to drug-treatable autism-like symptoms. Hum. Mol. Genet. 28, 561–571. doi: 10.1093/hmg/ddy367
Ueoka, I., Kawashima, H., Konishi, A., Aoki, M., Tanaka, R., Yoshida, H., et al. (2018). Novel Drosophila model for psychiatric disorders including autism spectrum disorder by targeting of ATP-binding cassette protein A. Exp. Neurol. 300, 51–59. doi: 10.1016/j.expneurol.2017.10.027
Vilidaite, G., Norcia, A. M., West, R., Elliott, C., Pei, F., Wade, A. R., et al. (2018). Autism sensory dysfunction in an evolutionarily conserved system. Proc. Biol. Sci. 285:20182255. doi: 10.1098/rspb.2018.2255
Wan, L., Dockendorff, T. C., Jongens, T. A., and Dreyfuss, G. (2000). Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol. 20, 8536–8547. doi: 10.1128/MCB.20.22.8536-8547.2000
Wang, J., Gong, J., Li, L., Chen, Y., Liu, L., Gu, H., et al. (2018). Neurexin gene family variants as risk factors for autism spectrum disorder. Autism Res. 11, 37–43. doi: 10.1002/aur.1881
Wang, C., Pan, Y. H., Wang, Y., Blatt, G., and Yuan, X. B. (2019). Segregated expressions of autism risk genes Cdh11 and Cdh9 in autism-relevant regions of developing cerebellum. Mol. Brain 12:40. doi: 10.1186/s13041-019-0461-4
Weuring, W., Geerligs, J., and Koeleman, B. P. C. (2021). Gene therapies for monogenic autism spectrum disorders. Genes (Basel) 12:1667. doi: 10.3390/genes12111667
Williams, M. R., Fricano-Kugler, C. J., Getz, S. A., Skelton, P. D., Lee, J., Rizzuto, C. P., et al. (2016). A retroviral CRISPR-Cas9 system for cellular autism-associated phenotype discovery in developing neurons. Sci. Rep. 6:25611. doi: 10.1038/srep25611
Woike, D., Wang, E., Tibbe, D., Hassani, N. F., Failla, A. V., Kibaek, M., et al. (2022). Mutations affecting the N-terminal domains of SHANK3 point to different pathomechanisms in neurodevelopmental disorders. Sci. Rep. 12:902. doi: 10.1038/s41598-021-04723-5
Wolter, J. M., Mao, H., Fragola, G., Simon, J. M., Krantz, J. L., Bazick, H. O., et al. (2020). Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 587, 281–284. doi: 10.1038/s41586-020-2835-2
Wu, S., Li, X., Qin, D., Zhang, L., Cheng, T., Chen, Z., et al. (2021). Induction of core symptoms of autism spectrum disorder by in vivo CRISPR/Cas9-based gene editing in the brain of adolescent rhesus monkeys. Sci. Bull. 66, 937–946. doi: 10.1016/j.scib.2020.12.017
Wu, N., Wang, Y., Jia, J. Y., Pan, Y. H., and Yuan, X. B. (2021). Association of CDH11 with autism spectrum disorder revealed by matched-gene co-expression analysis and mouse behavioral studies. Neurosci. Bull. 38, 29–46. doi: 10.1007/s12264-021-00770-0
Yamagata, A., Goto-Ito, S., Sato, Y., Shiroshima, T., Maeda, A., Watanabe, M., et al. (2018). Structural insights into modulation and selectivity of transsynaptic neurexin-LRRTM interaction. Nat. Commun. 9:3964. doi: 10.1038/s41467-018-06333-8
Yang, R., Walder-Christensen, K. K., Kim, N., Wu, D., Lorenzo, D. N., Badea, A., et al. (2019). ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc. Natl. Acad. Sci. U S A 116, 15262–15271. doi: 10.1073/pnas.1904348116
Yang, N., Wang, R., and Zhao, Y. (2017). Revolutionize genetic studies and crop improvement with high-throughput and genome-scale CRISPR/Cas9 gene editing technology. Mol. Plant 10, 1141–1143. doi: 10.1016/j.molp.2017.08.001
Yeh, C. M., Liu, Y. C., Chang, C. J., Lai, S. L., Hsiao, C. D., and Lee, S. J. (2011). Ptenb mediates gastrulation cell movements via Cdc42/AKT1 in zebrafish. PLoS One 6:e18702. doi: 10.1371/journal.pone.0018702
Yu, B., Yuan, B., Dai, J. K., Cheng, T. L., Xia, S. N., He, L. J., et al. (2020). Reversal of social recognition deficit in adult mice with MECP2 duplication via normalization of MeCP2 in the medial prefrontal cortex. Neurosci. Bull. 36, 570–584. doi: 10.1007/s12264-020-00467-w
Zhang, Y. Q., Bailey, A. M., Matthies, H. J., Renden, R. B., Smith, M. A., Speese, S. D., et al. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603. doi: 10.1016/s0092-8674(01)00589-x
Zhang, Y., Massel, K., Godwin, I. D., and Gao, C. (2018). Applications and potential of genome editing in crop improvement. Genome Biol. 19:210. doi: 10.1186/s13059-018-1586-y
Zhang, H. X., Zhang, Y., and Yin, H. (2019). Genome editing with mRNA encoding ZFN, TALEN and Cas9. Mol. Ther. 27, 735–746. doi: 10.1016/j.ymthe.2019.01.014
Zhao, W., Tan, J., Zhu, T., Ou, J., Li, Y., Shen, L., et al. (2019). Rare inherited missense variants of POGZ associate with autism risk and disrupt neuronal development. J. Genet. Genomics 46, 247–257. doi: 10.1016/j.jgg.2019.04.002
Keywords: autism spectrum disorder, gene editing, animal model, pathogenesis, therapy
Citation: Wang N, Lv L, Huang X, Shi M, Dai Y, Wei Y, Xu B, Fu C, Huang H, Shi H, Liu Y, Hu X and Qin D (2022) Gene editing in monogenic autism spectrum disorder: animal models and gene therapies. Front. Mol. Neurosci. 15:1043018. doi: 10.3389/fnmol.2022.1043018
Received: 13 September 2022; Accepted: 22 November 2022;
Published: 14 December 2022.
Edited by:
Katsuhiko Tabuchi, Shinshu University, JapanReviewed by:
J. Peter H. Burbach, University Medical Center Utrecht, NetherlandsCopyright © 2022 Wang, Lv, Huang, Shi, Dai, Wei, Xu, Fu, Huang, Shi, Liu, Hu and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongdong Qin, cWluZG9uZzEwOEAxNjMuY29t; Yun Liu, bGl1eXVuQGV0eXkuY24=; Xintian Hu, eHRodUBtYWlsLmtpei5hYy5jbg==
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.