 Guode Wu
Guode Wu Zhenhua Li1
Zhenhua Li1 Jing Li
Jing Li Xin Li
Xin Li Guangyao Liu
Guangyao Liu Pengfei Zhang
Pengfei Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 07 September 2022
Sec. Neurodegeneration
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.998275
This article is part of the Research Topic Molecular Biomarkers in the Prediction, Diagnosis, and Prognosis of Neurodegenerative Diseases View all 22 articles
Globoid cell leukodystrophy (GLD), or Krabbe disease (KD) is a rare neurodegenerative disease, and adult-onset GLD is more even neglected by clinicians. This review provides detailed discussions of the serum enzymes, genes, clinical manifestations, neuroimaging features, and therapies of GLD, with particular emphasis on the characteristics of adult-onset GLD, in an attempt to provide clinicians with in-depth insights into this disease.
Globoid cell leukodystrophy (GLD), or Krabbe disease (KD), is a rare neurodegenerative disorder of the nervous system caused by autosomal-recessive lysosomal dysfunction. Specifically, the mutation in the gene encoding galactosylceramidase (GALC) leads to the deficiency of β-galactosidase activity, which induces the accumulation of neurotoxic galactosylsphingosine [psychosine (PSY)] in oligodendrocytes and Schwann cells, thereby eventually causing progressive demyelination and secondary axonal degeneration in the central (CNS) and peripheral nervous systems (PNS) (Graziano and Cardile, 2015). According to the age of onset, GLD can be clinically classified into early infantile onset, late infantile onset, late childhood onset, adolescent onset, and adult onset, with early infantile phenotype accounting for about 90% of GLD (Duffner et al., 2009a,2012), while late adult onset GLD is rarely reported. Thus, clinicians are less diagnostically sensitive to adult phenotype, especially the late adult onset, which causes frequent misdiagnoses and mistreatment.
The first milestone includes the early 19th century, when the Danish neurologist Knud Haraldsen Krabbe first described the pathological features of five infant patients with injured white matter and cerebellum as “diffuse sclerosis of the brain”. In 1924, Collier (Wenger et al., 2016) provided a full description of the globular swelling of multinucleated bodies (multinucleated macrophages) and astrocytic hyperplasia in infant patients, which may be the histopathological features of this disease.
Second, in the early 1970s, Malone and Suzuki demonstrated an extremely low or lost activity for a lysosomal enzyme used for degrading galactosylceramide (GalCer) in GLD, the kind of lysosomal enzyme known as galactocerebrosidase or galactosylceramide β-galactosidase (GALC). In the settings of the lack of GALC, PSY, with a neurotoxic effect, would accumulate in macrophages and nerve cells, especially oligodendrocytes and Schwann cells, eventually causing hypomyelination, known as the “Psychosine hypothesis” (Suzuki and Suzuki, 1970). Therefore, enzymatic determination of GALC activity is a fundamental method for the early diagnosis and screening of GLD in newborns worldwide, especially in Europe and the United States (Macarov et al., 2011; Schrier Vergano et al., 2022).
Third, in the 1990s, Chen (Chen et al., 1993) first cloned human GALC complementary DNA (cDNA) from the urine of a GLD patient. Specifically, the entire length of GALC cDNA consisted of 3,795 bp, the coding region was 2,007 bp in size, the 5’ untranslated sequence was 47 bp in size, and the 3’ untranslated sequence was 1,741 bp in size. Thus, the researcher mapped the gene to human chromosome 14 by an in situ hybridization with a cDNA probe, specifically chromosome 14q13 region (Cannizzaro et al., 1994).
Undoubtedly, significant efforts have been made throughout the research history of GLD because it is challenging to obtain protein sequencing with sufficient purity from human tissues, including the liver, brain, and even placenta. In the past two decades, a small amount of pure GALC has been purified from urine (Chen and Wenger, 1993). After the amino acid sequence has been obtained, human GALC cDNA has been successfully revealed (Luzi et al., 1995), consisting of 17 exons and 16 introns. Additionally, compared with lesser exons, the introns range in size from 247 bp for intron 2 to about 12 kb for intron 10. Based on the above-mentioned studies, Cannizzaro (Cannizzaro et al., 1994) confirmed that GALC gene is located on human chromosome 14 by linkage analysis, and mapped it to chromosome 14q13 region by an in situ hybridization with a cDNA probe. The GALC gene may encode the precursor protein of GALC, which was processed into a mature form by being cut into 50-kDa N-terminal fragments and 30-kDa C-terminal fragments by proteases in lysosomes (Nagano et al., 1998). Deane (Deane et al., 2011) has discovered the crystal structures of GALC enzyme and revealed three distinctive domain architectures contributing to substrate binding. Besides, some researchers cloned GALC cDNA from other species, including dogs (Victoria et al., 1996), mice (Sakai et al., 1996), and rhesus monkeys (Luzi et al., 1997), which laid a foundation for establishing animal models of GLD for the future.
Early studies preliminarily revealed that the deletion of 30 kb subunits encoding non-functional proteins is the most common pathogenic mutation of GLD, accounting for about 35%–50% of cases (Rafi et al., 1995). This deletion occurs in the C > T transition polymorphism at position 502 of cDNA, inducing the elimination of the coding regions of seven exons, including all 30-kDa subunits and about 15% of 50-kDa subunits. An increasing number of pathogenic gene mutations has been reported in the past three decades (Wenger et al., 2021). Until now, a total of 296 GALC gene mutations related to GLD have been cataloged in the Human Gene Mutation Database (HGMD), including missense mutations, non-sense mutations, deletion, and insertion (Figure 1), and the sites and types of gene mutations have regional and racial variation (Rafi et al., 1996; Kleijer et al., 1997; Xu et al., 2006; Zayed, 2015). Unlike infantile GLD with mutations in the central domain of the coding region, adult GLD has GALC mutations at the N or C terminals, and most mutations are located in GALC gene region encoding 50-kDa subunits (Furuya et al., 1997; Wenger et al., 1997). There is a certain correlation between the clinical manifestations and genotypes of GLD in terms of theory: the type and location of gene mutations would affect GALC protein and its activity; hence, it is ideal to be able to classify a disease-causing variant as “severe” or “mild” to predict as the possible type and severity of GLD a patient will have. For instance, c.857G > A (809G > A) mutation and 30-kb deletion are more common in adult onset (De Gasperi et al., 1996), and p.G286D is common in patients with late childhood GLD (De Gasperi et al., 1999; Tappino et al., 2010). c.169G > A (121G > A) mutation is associated with the late-infantile KD form, especially in Japan (Hossain et al., 2014). and Furthermore, the activity of residual β-galactosidase can be detected in these patients. The exploration into the correlation between the clinical manifestations and genotypes of GLD contributes to predicting patients with GLD during screening, but high variability of polymorphic changes on allelic genes complicates the relationship between genotype and clinical phenotype.
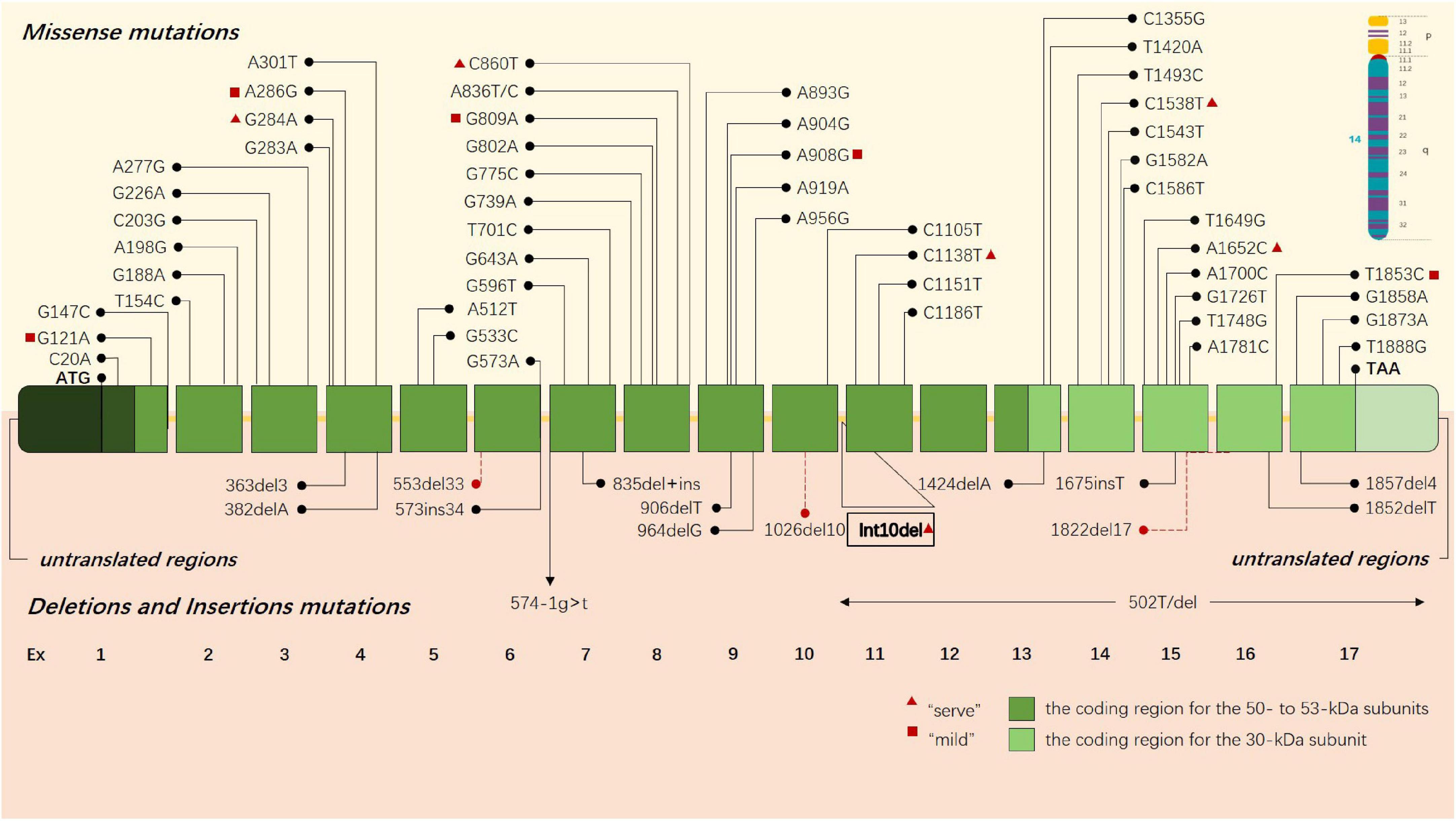
Figure 1. GLD-causing common sites and types of gene mutations are presented in the schematic drawing of GALC gene. Missense mutations are above the gene, and deletions and insertions are shown below the gene. Each box represents an exon, the dark green boxes contain the coding region for the 50- to 53-kDa subunits, and the light green boxes contain the coding region for the 30-kDa subunit. The red triangle indicates that this gene may cause a severe clinical phenotype, and the red square indicates that the gene may lead to a mild clinical phenotype.
We summarized some pathogenic heterozygous mutations associated with adult-onset GLD by gathering and analyzing previously published literature (Table 1).

Table 1. The pathogenic genes associated with adult onset GLD.
Normally, PSY is a cytotoxic lysolipid that can be generated either by the galactosylation of sphingosine by UDP-galactose ceramide galactosyl transferase (CGT) (UDP-galactose-CGT), or by the deacylation of GalCer by N-deacylase (Lgisu, 1989; Kanazawa et al., 2000). Having a critical role in the normal physiological mechanism, UDP-galactose-CGT in oligodendrocytes and Schwann cells participates in the synthesis of myelin lipids and myelin formation (Schulte and Stoffel, 1993; Marcus et al., 2006) and plays a vital role in maintaining the function and structure of membranes of other cells (Dupree et al., 1999). The expression of CGT is high in oligodendrocytes and Schwann cells, but lower in neurons and astrocytes (Schaeren-Wiemers et al., 1995; Pernber et al., 2002). Under In GALC-deficient conditions, PSY would accumulate to high levels in tissues, especially in the brain (Suzuki, 1998). Notably, the primary substrate of GALC is hydrolyzed by GM1 ganglioside β-galactosidase in this situation. Therefore, the level of GalCer in tissues would not be high. Additionally, the number of cells expressing UDP-galactose-CGT may be reduced due to the susceptibility to the toxic effect of PSY, which would further increase the accumulation of PSY (Nagara et al., 1986; Ida et al., 1990; Figure 2).
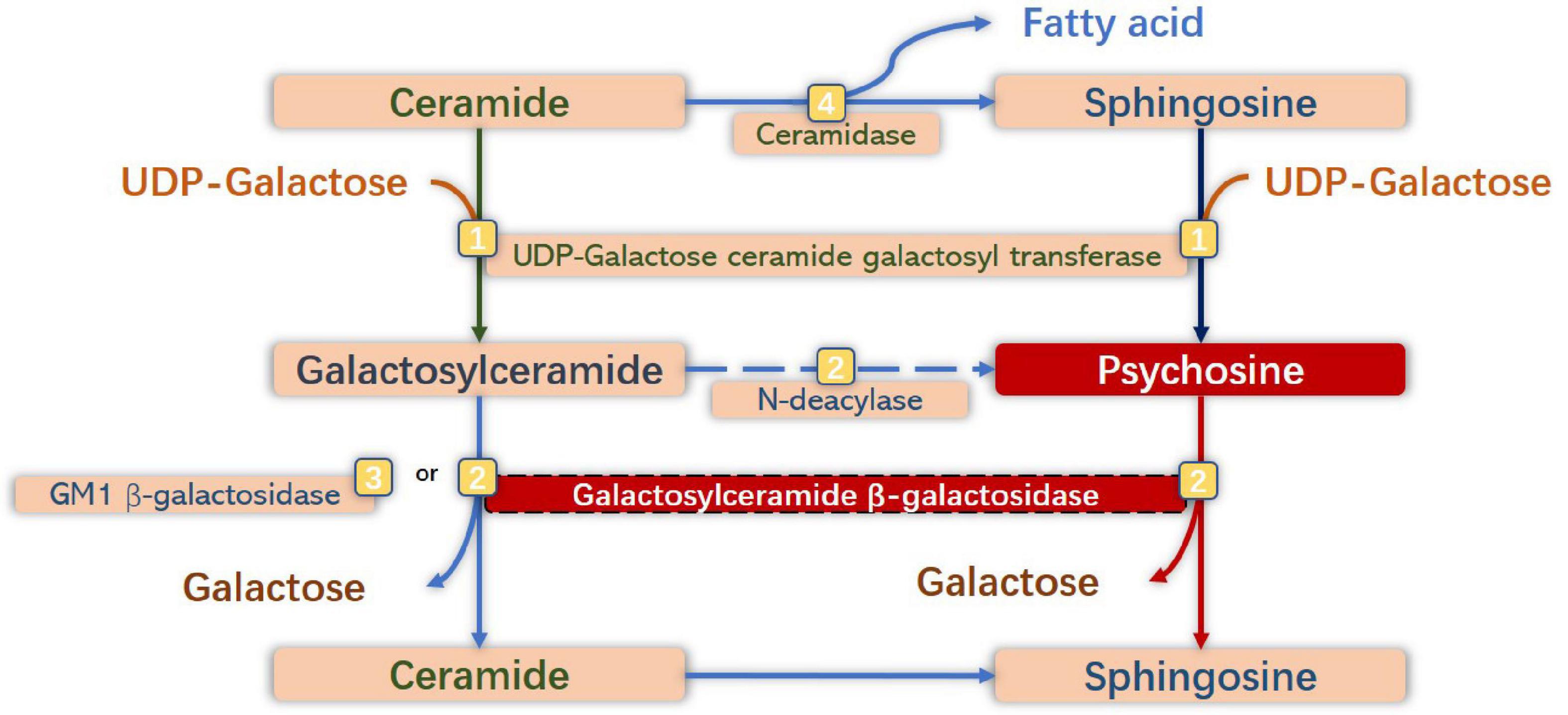
Figure 2. Anabolic and catabolic pathways of galactosylsphingosine [psychosine (PSY)] and galactosylceramide. Galactosylceramide is one of the significant glycosphingolipids of oligodendrocytes (OLs) and Schwann cells, which is synthesized by galactosylation of ceramide by UDP-galactose ceramide galactosyl transferase (UDP-galactose CGT) (Graziano and Cardile, 2015), and is degraded to ceramide by degalactosylation by both galactosylceramide β-galactosidase (GALC) (Duffner et al., 2009a) and GM1 β-galactosidase (Duffner et al., 2012). Sphingosine is synthesized by hydrolysis of ceramide by ceramidase (Wenger et al., 2016). PSY is synthesized by galactosylation of sphingosine by UDP-galactose CGT, and from galactosylceramide by N-deacylase (Suzuki and Suzuki, 1970), and degraded to sphingosine by degalactosylation by GALC. Under GALC-deficient conditions, neurotoxic PSY accumulates to high levels in macrophages (globoid cells) and neural cells, especially in OLs and Schwann cells.
The correlation between the myelination stage and the onset of diseases demonstrates that GLD-induced myelin sheath injuries often occur at the most active myelination stage. The spinal cord is one of the first CNS regions to obtain myelin, and myelination occurs from the gestational age of 20 weeks to 2 years in the spinal cord (Tanaka et al., 1995). Myelination in the pyramidal tract occurs from the gestational age of 25 weeks to 2–3 years (Kinney et al., 1988). Myelination in the posterior corpus callosum occurs from 3 to 8 months after birth to adolescence and adulthood (Barkovich et al., 1988; Courchesne et al., 2000). Myelination in the parieto-occipital white matter begins from at 4–6 months after delivery and reaches 50% at 11–14 months after birth (Bartzokis et al., 2001; Deoni et al., 2011). The most significant changes in myelination occur between midgestation and the end of the second postnatal year. The temporal and spatial pattern of myelination seems to explain the temporal features of myelin sheath abnormalities in GLD. According to studies based on magnetic resonance imaging (MRI), spinal cord involvement precedes intracranial abnormalities in infantile GLD (Vasconcellos and Smith, 1998). In infantile GLD (within 2 years of birth), MR signal abnormalities appear at the active myelination stage, including corticospinal projection fibers (pyramidal tract), posterior corpus callosum, and parietal white matter. Besides, it can also be used to explain that the abnormalities of the cerebellar white matter are only observed in early GLD (including early infantile and late infantile types) but not in adult GLD. We suggest that the myelination of cerebellar white matter begins during pregnancy and reaches the adult level at the age of 3–18 months. The myelination of the cerebellar white matter reaches the adult level before the onset of adult GLD. Hence, adult GLD will not be accompanied by cerebellar lesions or related clinical signs, especially in patients with late adult GLD.
Psychosine is a metabolic by-product of the precursor of GALC causing membrane structure changes due to its detergent-like properties, inducing oligodendrocyte death and myelin sheath loss. However, it has been proved that PSY could induce abnormal membrane structure by changing the structure and function of lipid rafts in animal experiments (White et al., 2011; Hawkins-Salsbury et al., 2013). As has been revealed in many studies, the mechanisms of PSY toxicity in the CNS and PNS also include PSY-induced stress and proapoptosis signaling pathways, PSY-induced immune and inflammatory responses, PSY-induced mitochondrial energy disorder, peripheral nerve injury, and cerebral microvascular dysfunction (neurovascular unit damage) (Won et al., 2016). Multiple signaling pathways are involved in oxidative stress (OS) and apoptosis of oligodendrocytes induced by PSY (Jatana et al., 2002; Hamanoue et al., 2004; Pannuzzo et al., 2010). PSY plays a vital role in the apoptosis of oligodendrocytes by activating stress-activated protein kinases (SAPKs) (Jurewicz et al., 2006). In addition, it can induce the activity and expression of cell death signals and simultaneously inhibit cell survival signals, such as phosphoinositide 3-kinase (PI3K) (Arai and Lo, 2010; Won et al., 2013; Graziano et al., 2016). Multinucleated activated microglia or macrophages can be observed in the brain of patients with GLD and animal models from previous studies, especially the activation and proliferation of astrocytes and the proliferation of glial fibrillary acidic protein (GFAP) in the demyelination area of the white matter, implying that immune and inflammatory reactions mediated by microglia and peripheral immune cells are involved in GLD pathogenesis (Kobayashi et al., 1986; Ransohoff and Brown, 2012). Some researchers have thought that the immune and inflammatory responses of GLD are secondary performance. However, it has been confirmed that PSY-induced infiltration of immune cells and overexpression of cytokines, such as tumor necrosis factor-α, interleukins, and chemokine, can further activate transcription factors, making the up-regulation of inflammatory response appear at an early stage of this disease (Wu et al., 2001; Borda et al., 2008; Ripoll et al., 2011; Hawkins-Salsbury et al., 2012; SSnook et al., 2014). The accumulation of PSY would induce mitochondrial dysfunction and adenosine triphosphate (ATP) consumption (Haq et al., 2003; Giri et al., 2006; Voccoli et al., 2014). Currently, the increased activity of secretory phospholipase A2 (sPLA2) induced by PSY is considered the critical pathological mechanism, and its products can inhibit mitochondrial enzyme activity and destroy mitochondrial integrity (Kalous et al., 1992; Hollie et al., 2014; Wu et al., 2021). AMP-activated protein kinases (AMPKs) can be activated during energy deficiency to produce and preserve ATP (Hardie, 2004; Carling, 2005). The accumulation of PSY down-regulates or even inactivates the activity of AMPKs, accelerating the consumption of ATP in oligodendrocytes, causes mitochondrial dysfunction in oligodendrocytes and their inactivation, which has been confirmed to be irreversible (Giri et al., 2008). According to previous studies, the excessive accumulation of PSY can also inhibit the function of peroxisomes, which would disturb metabolism and energy homeostasis (Khan et al., 2005; Haq et al., 2006). Lower motor neuron disease induced by peripheral nerve demyelination is the most common non-central nervous system disorder in GLD, such as peripheral neuropathy and muscle diseases. As has been revealed in current studies, the toxic effects of PSY might directly mediate injuries in neuron, axon, and neuromuscular junctions (Castelvetri et al., 2011). PSY induces the production of α-synuclein, a neuronal inclusion, leading to abnormalities of neurofilaments by relieving the regulation of PP1 and PP2A phosphatases (Cantuti-Castelvetri et al., 2012) and inducing an axon transport imbalance by activating glycogen synthase kinase 3β (GSK3β) (Cantuti Castelvetri et al., 2013). This results in axon damage and neuron death in the PNS. PSY can reduce peripheral nerve conduction by activating caspase and inhibiting Akt activities, aggravating the damage to neuromuscular junctions.
In this article, we propose that GLD can involve neurovascular units, which may be the ultimate pathological mechanism contributing to the commonalities of CNS degeneration. Abnormalities of microvasculature and disruption of the blood-brain barrier (BBB) in patients with GLD, mainly including perivascular space enlargement, cerebrovascular permeability increases, endothelial morphology changes, macrophage infiltration, and microvascular structure changes, would develop (Giacomini et al., 2015; Belleri and Presta, 2016). It has been confirmed in some studies that the decreased proliferation and migration of endothelial cells induced by PSY and the declined response to angiogenic factors may hinder angiogenesis. The destroyed actin cytoskeleton and the down-regulated expression of tight junction proteins induced by PSY might lead to changes in microvascular ultrastructure and permeability (Belleri et al., 2013). The damage of PSY to oligodendrocytes would cause secondary damage to astrocytes and neurons, accompanied by microvascular injury (Cantuti Castelvetri et al., 2015).
According to the age of onset, GLD can be clinically classified into five types: early infantile type (onset at 0–6 months), late infantile type (onset at 7–12 months), late childhood type (onset at 13 months-10 years), adolescent type (onset at 11–20 years), and adult type (onset at > 21 years) (Hagberg, 1984). There is a close correlation between clinical symptoms and the age of onset in GLD patients. Patients with the early infantile type account for about 85–95% of cases, and the early infantile type has the worst prognosis, and the general median survival time is 8–36 months (Duffner et al., 2011). The clinical course is usually divided into three clinical stages: non-specific symptom stage, symptom progression stage, and end burnt-out stage. At the non-specific symptom stage, infants often present with crying, irritability, poor feeding, vomiting, and myoclonic seizures (Duffner et al., 2009b), accompanied by peripheral neuropathy (Siddiqi et al., 2006). At the symptom progression stage, infants often present with optic atrophy, aggravated epileptic seizures, and the rapid progression of psychomotor degeneration. At the end burnt-out stage, infants completely lose autonomous movement, require nasal feeding, and eventually die of infection and respiratory failure (Chabali et al., 1997). The late infantile type is characterized by progressive deterioration of motor function, spastic paraplegia, cerebellar ataxia, hemiplegia (Fiumara et al., 2011). About 12.5% of patients with late infantile GLD have a visual impairment or even blindness (Lyon et al., 1991). Compared with the early-infantile onset, the late-infantile onset type has a significantly extended lifetime of 2∼14 years (Barone et al., 1996). The late childhood type is a transitional clinical phenotype from late infantile type to adolescent type. The symptoms related to motor functions of the adolescent type are not as significant as those in the early infantile or late infantile types. However, spastic paralysis or cerebellar ataxia can still occur in the adolescent type, and visual dysfunction is the most common clinical symptom in the adolescent type (Phelps et al., 1991). With aging, symptoms would become alleviated in these patients (Arvidsson et al., 1995).
The adult type has the lowest incidence, with chronic progressive pyramidal tract injuries as the main manifestation. Additionally, patients with this type present with chronic progressive spastic paralysis and gait disorders, with or without peripheral nerve injuries, mental disorders, cognitive disorders, bulbar paralysis, and tremor (Henderson et al., 2003; Tokushige et al., 2013). Almost all patients with adult GLD present with chronic progressive spastic paraplegia or walking difficulties, and sometimes the symptoms may be mild (Paiva et al., 2022), but a patient can also present with acute hemiparesis (Mamada et al., 2016). Most patients also develop peripheral nerve injuries (Sabatelli et al., 2002), and isolated peripheral nerve injury occur in some patients (Marks et al., 1997; Adachi et al., 2016). In these patients, asymmetric peripheral nerve conduction abnormalities have been revealed in some electrophysiological studies. Specifically, prolonged F wave incubation period, reduced motor nerve conduction velocity homogeneity, and prolonged distal incubation period are the most common abnormal manifestations, which are consistent with peripheral nerve demyelination (Matsumoto et al., 1996). When the patient is complicated by pes cavus, differential diagnosis with respect to complicated forms of Charcot-Marie-Tooth disease or hereditary spastic paraplegia is necessary (Malandrini et al., 2013). However, a study conducted by Adachi has demonstrates demonstrated that there are many obvious thin thin-myelinated nerve fibers in sural nerves of patients with GLD, and the density of myelinated nerve fibers decreases (Madsen et al., 2019). However, there is no characteristic “onion ball” structure of peronial peroneal myoatrophy, which may be one of the key points for the differential diagnosis between demyelination and dysmyelination. Mild cognitive impairment (MCI) and psychiatric symptoms often occur in patients with adult GLD (Xia et al., 2020). Compared with adolescent GLD, optic atrophy is less likely to occur in adult GLD (Madsen et al., 2019). Notably, cerebellar ataxia would not occur in patients with adult GLD, and this temporal and spatial pattern correlates with the pathological mechanism of this disease. The majority of patients have slow or insidious progress of dyskinesia, and sometimes cognitive impairment, but such patients have survived up to 10–15 years after the initial onset of symptoms, according to the literature (Liao et al., 2014).
It is difficult to evaluate the clinical manifestations of adult GLD in real-world clinical practice, it is impossible to assess whether these patients are completely normal before the age of 20 years or have some mild clinical symptoms that are difficult to observe, hindering the further exploration into adult GLD.
Bilateral symmetrical white matter lesions are a mark of most hereditary white matter lesions and a few secondary white matter lesions. Patchy white matter lesions are common in inflammatory demyelination of the CNS, such as multiple sclerosis, and scattered multiple punctate lesions are common in ischemic white matter lesions (Schiffmann and van der Knaap, 2009). MRI features of the brain have been correlated with the age of onset of GLD patients (Abdelhalim et al., 2014; Muthusamy et al., 2019; Andriescu et al., 2020). For example, white matter hyperintensities (WMHs) mainly involve the cerebellar white matter and the dentate nucleus at the early and late stages of infantile GLD. The cerebellum of adolescent GLD patients is basically normal, but WMHs involve the parietal white matter.
Bilateral WMHs usually occur in adult GLD patients and are limited to the parietal white matter (temporo-parietal junction), corona radiata, optic radiation, proximal corticospinal tract, posterior lateral periventricular white matter, and corpus callosum (Wang et al., 2007; Tokushige et al., 2013), with or without brain and spinal cord atrophy (Bajaj et al., 2002). Noteworthy, cerebellar white matter lesions seldom occur in adult GLD patients. Due to the undefined correlation between gene mutation types and clinical phenotypes, the focus of recent studies has been placed on the classification and scoring of intracranial white matter abnormalities, to identify the correlation of the site and severity of white matter lesions with clinical phenotypes (Cousyn et al., 2019). Based on the analysis and summary of the MRI features of adult GLD patients in our clinical center, we simplified the imaging scoring criteria for GLD to better define the pattern of leukodystrophy and provide treatment opportunities earlier (Figure 3). In the revised scoring criteria, we still highlighted the significance of “continuity of lesions” in the site and severity of white matter lesions, which correlated with the scoring of the lesion site. Discontinuous supratentorial white matter lesions are mainly involved in four parts in the transverse section of brain MRI, including pre- and postcentral gyrus, corona radiate, posterior lateral periventricular white matter, and optic radiations. The degree of white matter lesions of the corpus callosum should be highlighted in the median sagittal section. In theory, the higher the score, the more serious the clinical symptoms. The sensitivity and specificity of the grading scale should still be verified by numerous clinical data.
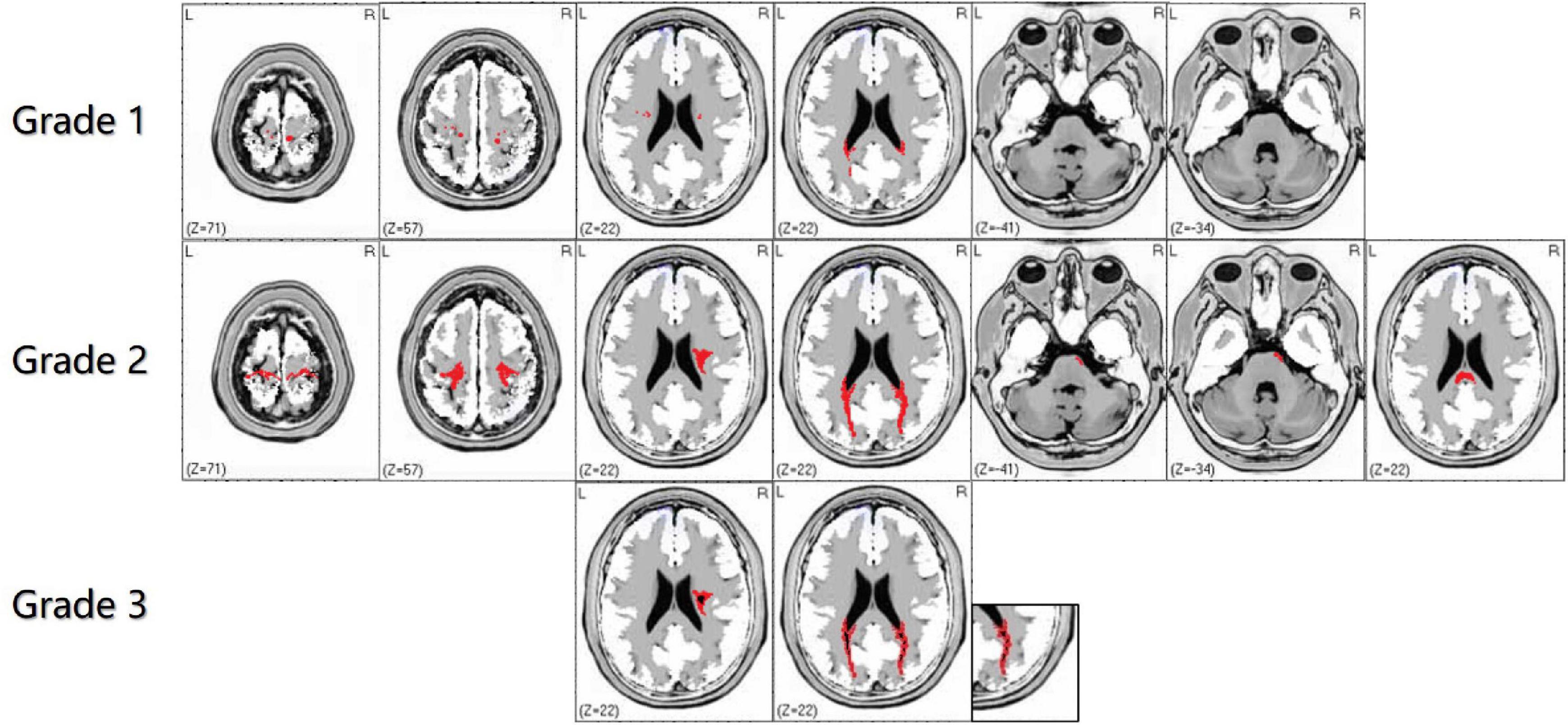
Figure 3. The characteristics of white matter lesions should be graded by imaging, including grade 1: sporadic punctate white matter lesions; grade 2: patchy or continuous linear white matter lesions; grade 3: obvious cavity-like changes in the lesion site. In the supratentorial and infratentorial structures, the damage degree of the corticospinal tract and medial lemniscus should shall be highlighted in the transverse and coronal sections of brain magnetic resonance imaging (MRI). Grade 1 is defined as discontinuous white matter lesions, and grade 2 is defined as continuous white matter lesions. The red area refers to WMHs, and the black part of the red area refers to necrosis or cavity formation in WMHs.
Adult GLD patients usually have a relatively slow progression of white matter lesions. Therefore, the clinical process from an acute attack to rapid aggravation may be misdiagnosed as acute cerebral infarction or inflammatory demyelination of the CNS, such as multiple sclerosis (Debs et al., 2013; Mamada et al., 2016). Therefore, dynamic and longitudinal image follow-up shall be regularly performed to systematically evaluate the accuracy of diagnosis.
Other imaging features include unilateral white matter involvement, advanced brain atrophy with different degrees (Lemmens et al., 2011). Optic neuropathy, optic chiasma lesions, and enhanced oculomotor nerves and trigeminal nerves are common findings in infantile patients (Beslow et al., 2008; Patel et al., 2008; Ganesan et al., 2010; Garcia et al., 2010; Quintas-Neves et al., 2020), which have rarely been reported in adult-onset GLD. Compared with often reported thickened cauda equina nerves in early-infantile and late-infantile Krabbe disease patients (Hwang et al., 2016), brachial plexus enlargement is a more common MRI finding in adult-onset Krabbe disease (Hiyama et al., 2016).
As the study of white matter lesions at an early stage by magnetic resonance spectroscopy (MRS) confirmed, the pattern of metabolic alterations as detected by MRS in infantile-onset, juvenile-onset, and adult-onset has been related to age (Brockmann et al., 2003; Romano et al., 2009), and the concentration of white matter metabolites in adult GLD patients is close to the normal level. Furthermore, the concentration of N-acetylaspartate and N-acetylaspartylglutamate decreases; while, that of choline-containing compounds, creatine, and phosphocreatine increases. There is a slight change in the concentration of myoinositol. The spectra of the gray matter show a slight decrease in the concentration of all metabolites (Udow et al., 2012). Diffusion tensor imaging (DTI) is the most valuable neuroimaging method for CNS demyelination, assess the severity of white matter damage in infantile GLD (Provenzale et al., 2005; Escolar et al., 2009; Wang et al., 2011; Poretti et al., 2014). It may detect early white matter injury in asymptomatic neonates, predict motor functions and prognosis after stem cell transplantation (Poretti et al., 2016).
Enzyme replacement therapy (ERT) has been emphasized in the preliminary studies on GLD treatment. This therapy has been adopted from the treatment of lysosomal storage diseases (Desnick and Schuchman, 2012). However, it is difficult to achieve efficacy owing to the blocking effect of the BBB, especially clinical symptoms exist at diagnosis (Escolar et al., 2005). Currently, hematopoietic stem cell transplantation (HSCT) is the optimal viable therapy for GLD treatment, and these cells are mainly derived from the bone marrow (BM) or umbilical cord blood (UCB) (Wright et al., 2017). Moreover, virus-mediated gene therapy, anti-inflammatory and antioxidant therapy, oligodendrocyte transplantation, and substrate reduction therapy have been subjected to drug tests based on animal models (Mikulka and Sands, 2016). However, a single therapy cannot achieve satisfactory efficacy. GalCer deficiency in GLD can trigger various pathogenic mechanisms, suggesting that there may be multiple targets during the treatment of this disease. Therefore, the focus of recent studies has been placed on different combined therapies based on these therapeutic regimens. Additionally, treatment strategies emphasize symptomatic and supportive treatment from multidisciplinary collaboration for patients (Escolar et al., 2016).
Lysosomal storage diseases, such as Fabry disease, are usually treated with ERT (Griffiths, 1989). The core of treatment lies in providing active GALC for dysfunctional cells and tissues in the CNS and PNS. In 2005, Lee (Lee et al., 2005) prolonged the life span of twitcher mice to 47 days by an intraperitoneal injection of recombinant GALC. However, this ERT based on intraperitoneal injection could not provide a long-term effect due to the existence of the BBB. In 2007, Lee (Lee et al., 2007) improved the activity of GALC in the CNS of twitcher mice by injecting recombinant GALC into the ventricle. However, there are two defects in this therapy. First, repeated invasive injections are required to maintain high GALC concentration. Second, multiple injections may be required because GLD widely influences the CNS and PNS. These defects hinder the application of ERTs in clinical practice.
Currently, HSCT is the standard therapy for GLD treatment. GALC-positive donor cells from BM or UCB are transplanted to the CNS to continuously provide GALC and reduce the degree of neuroinflammation (Reddy et al., 2011). Bone marrow transplantation (BMT) is widely used in the early treatment of this disease. At present, umbilical cord blood transplantation can be used for the treatment of patients with early infantile GLD. However, early screening should be performed on newborns (Kwon et al., 2018). The process of hematopoietic stem cells performing a therapeutic role is called cross-correction. Functional GALC secreted by hematopoietic stem cells is ingested and combined with mannose receptors on the surface of defective cells in the CNS through mannose-6-phosphate (M6P), and then transported to lysosomes through endocytosis (Neufeld and Fratantoni, 1970). Both the animal experiment results and the evidence of GLD treatment demonstrate the effectiveness of HSCT, especially for infantile GLD patients before the appearance of symptoms (Krivit et al., 1998). Further, HSCT has been reported to exert immune regulation by reducing the degree of neuroinflammation (Caniglia et al., 2002). Notably, patients receiving HSCT are at risk of severe graft-versus-host disease (GVHD), and this therapy can improve clinical symptoms only to a certain extent.
It has been confirmed in previous animal experiments that virus-mediated gene therapies mediated by adeno-associated virus (AAV) orienting the CNS can improve enzyme activity and enhance behavior and cognitive function in mouse models with lysosomal storage diseases (Passini et al., 2005, 2006). Adenovirus capsid proteins (AAV1, 5, 9, rh10) are currently widely used in GLD treatment (Asokan et al., 2012), and several types of capsid proteins have presented favorable outcomes. After GALC cDNA is delivered by adenovirus to twitcher mice, GALC activity increases several times in the CNS, and the survival time is significantly prolonged compared with untreated mice, by 40–50% (Rafi et al., 2005). Recently, Rafi et al. treated twitcher mice by intravenously injecting AAV2/rh10-mGALC into the tail vein. The results showed that the survival time of treated mice was significantly longer than that of untreated mice, and the GALC activity in the brain, spinal cord, and sciatic nerve reached or exceeded the recombinant wild-type (WT) level (Rafi et al., 2012, 2015a). This implies that multiple injections are not required in virus-mediated gene therapy, which is different from ERT. The efficacy of virus-mediated gene therapy correlates with the improvement of adenovirus vectors, and this therapy is one of the most promising therapies for treating GLD (Rafi, 2016; Nasir et al., 2021; Hordeaux et al., 2022).
PSY-related hyperoxidative stress and inflammatory reactions are also the targets for treating GLD. It has been confirmed through animal experiments that ibudilast, a phosphodiesterase inhibitor, can reduce the degree of tremor and demyelination in twitcher mice, but cannot prolong their life span (Kagitani-Shimono et al., 2005). Luzi (Luzi et al., 2009) used indomethacin, ibuprofen, and minocycline to prolong the life of twi-trs mice (an animal model of GLD). The result has shown that indomethacin could significantly reduce pro-inflammatory cytokines, including interleukin (IL)-6 and tumor necrosis factor (TNF)-α. Paintlia (Paintlia et al., 2015) significantly prolonged the average life span of twitcher mice via a diet therapy with a high level of vitamin D, leading to a decreased accumulation velocity of PSY in the body, and a mitigated degree of demyelination.
Oligodendrocytes in the CNS are one of the targets in treating GLD. Hence, the ultimate goal of treatment is to stabilize the normal function of oligodendrocytes and other myelin sheath cells. Kuai (Kuai et al., 2015) injected oligodendrocyte precursor cells differentiated from normal mouse embryonic stem cells into twitcher mice. However, the researchers obtained unsatisfactory results. Both the motor function and life span could not be improved through this therapy.
Substrate reduction therapy has shown some prospects in the treatment of lysosomal storage diseases. Although this therapy can only alleviate clinical severity and prolong the life span, GLD can still not be eliminated (Parenti et al., 2015). Galactose ceramide galactosyl transferase (CGT) is the enzyme directly responsible for synthesizing PSY in GLD. However, there is no effective inhibitor for this enzyme. Thus, it is necessary to identify the target enzyme upstream of PSY synthesis for achieving effective treatment (LeVine et al., 2000). However, many downstream metabolites related to these upstream enzymes may be affected.
Currently, there are many synergistic regimens based on HSCT, including BMT + endothelial growth factors (Young et al., 2004), BMT + substrate reduction therapy (LeVine et al., 2000), BMT + enzyme replacement therapy (Qin et al., 2012), BMT + virus-mediated gene therapy (Lin et al., 2007; Galbiati et al., 2009; Rafi et al., 2015b), and BMT + gene therapy therapies + substrate reduction therapy (Hawkins-Salsbury et al., 2015). These synergistic regimens outperform single-drug regimens in multiple aspects, including alleviating pathological demyelination, prolonging the life span of twitcher mice, and improving motor function (Bradbury et al., 2021). However, these synergistic regimens are still in the stage of animal experiments, and a long-term research may be required for the real-world application of these regimens for GLD patients.
Among the treatments for globoid cell leukodystrophy, hematopoietic stem cells are used clinically, especially in infants, except for other treatment options, which are basically in the animal experimental stage. There are currently few reports on the treatment of GLD in adults due to the low incidence in this population and the limitations of clinical therapies. According to some case reports, HSCT may be effective in treating adult-onset GLD patients (Sharp et al., 2013). Moreover, the intravenous injection of gamma globulin has been effective in treating adult-onset GLD patients (Fukazawa et al., 2021). Symptomatic and supportive therapies are still the main clinical regimens for treating adult-onset GLD patients.
Despite a systematic review of the pathogenesis, clinical manifestations, and neuroimaging features of globoid cell leukodystrophy, the actual clinical diagnosis of this type of CNS degeneration is mains a challenge. Based on our experience, we tried to provide feasible diagnostic ideas and criteria (Figure 4).

Figure 4. Feasible diagnostic ideas and criteria. If the total score is > 4, it is a confirmed diagnosis; If the total score equals 4 points (without enzyme or gene testing), it is strongly recommended to perform GALC enzyme or genetic testing. If the total score is < 4, differential diagnosis should be focused on, and GALC enzyme or genetic testing should be performed when necessary.
Step 1 is to: assess the patient whether they meet the core clinical symptom of the vast majority of adult-onset GLD, which is “chronic progressive symmetrical spasmodic paralysis”, If the conditions are met, one point is added. Further, an electrophysiological examination should be performed to identify whether it is peripheral neuropathy or evaluate whether it is accompanied by peripheral nerve damage; if the conditions are met, one point is added. If the electrophysiological examination results are abnormal, further brachial plexus root imaging is required (it adds the evidence of diagnosis, but is not used as the diagnostic criteria); if the conditions are met, one point is added.
Step 2 is to: assess patient whether the patient meets the imaging core symptoms of most adult-onset globoid cell leukodystrophy are “mild white matter demyelination of anterior or posterior central gyrus, coronal radiography, posterior horn of the lateral ventricle, and visual radiation” (it adds the evidence of diagnosis, but is not used as the diagnostic criteria); if the conditions are met, one point is added.
Step 3: is to determine the affected GLCA enzyme, or perform genetic testing; if the conditions are met, four points are added.
Through this diagnostic process and strategy, a 67-year-old patient was diagnosed with late adult onset GLD in our clinical center. This patient presented with chronic progressive spastic gait, mild cognitive impairment, and anxiety, and there was no clinical symptom of peripheral nerve involvement. However, the electrophysiological examination results suggested decreased nerve conduction velocity and demyelination. The clinical symptoms of this patient were consistent with the typical clinical manifestations of late adult onset GLD, and this patient has been the oldest case reported in China (Figure 5).
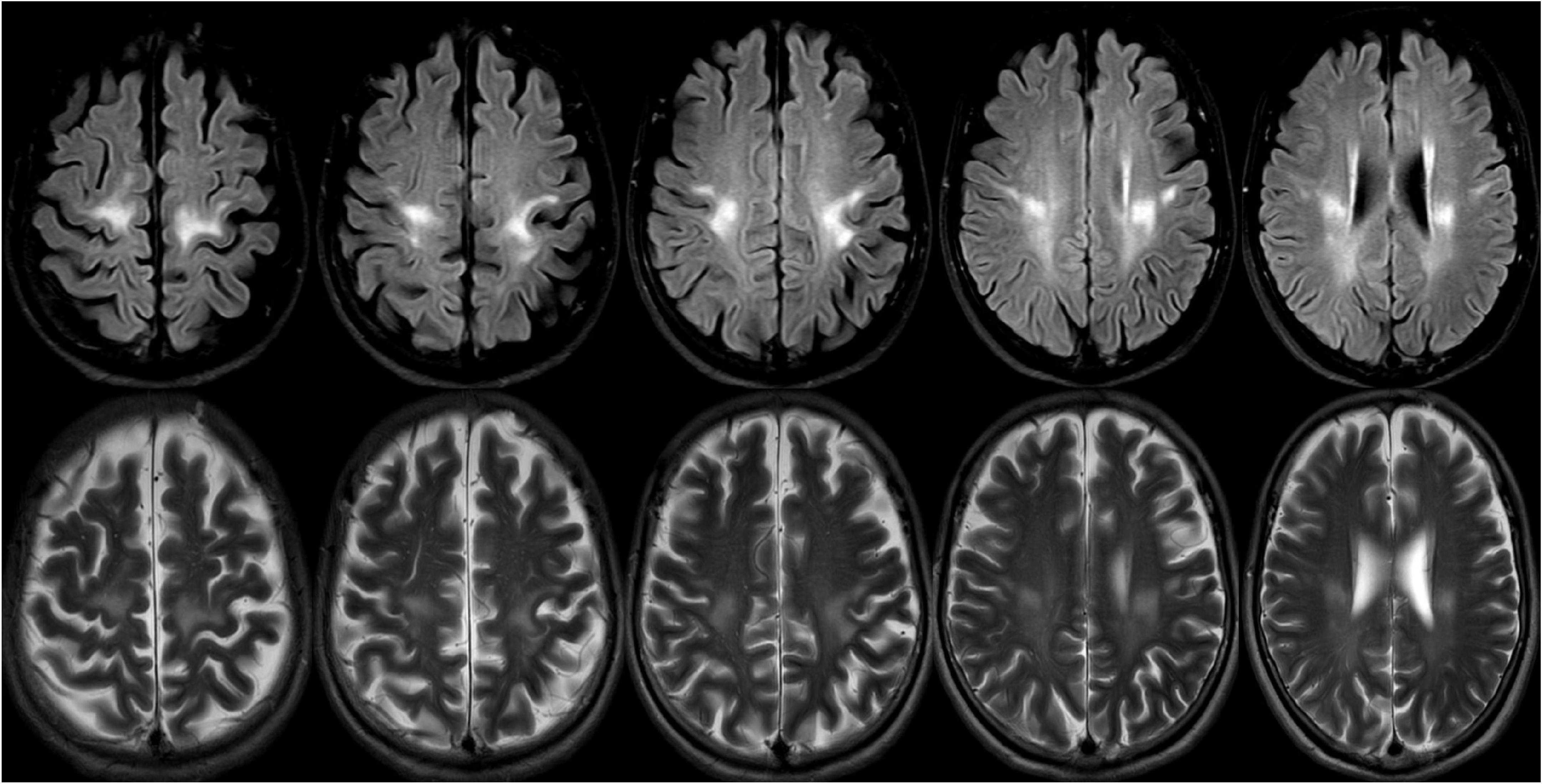
Figure 5. A 69-years-old female presented with a 2-year history of progressive weakness of the lower limbs. A brain magnetic resonance imaging (MRI) revealed abnormal T2 and flair hyperintensities in the periventricular and deep cerebral white matter, and the bilateral subcortical areas of parietal white matter (the lesions were continuous and symmetrically distributed). Reduced galactocerebrosidase activity and GALC gene confirmed the diagnosis of adult late-onset GLD.
Adult-onset GLD is a rare degenerative disease of the CNS caused by abnormal lysosomal storage. This disease is characterized by chronic progressive spastic paralysis and gait disorders, with or without peripheral nerve injury, cognitive impairment, and mental disorders. Although there is no effective therapy for adult-onset GLD, this disease is expected to be eliminated by pharmacological chaperones based on animal models to improve the activity GALC enzyme, viral vectors for gene therapy, and neural progenitor cells injected into the CNS. To treat GLD, clinicians should accurately differentiate it from other acquired white matter demyelination diseases of the CNS based on the clinical and MRI features of this disease, to avoid misdiagnoses.
ZL and JL contributed to the study design and drafted the manuscript. GW and XL edited the manuscript. JZ, PZ, and GL contributed to MRI of the data in the literature. MW supervised the writing of the manuscript and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Natural Science Foundation of Gansu Province, China (grant no. 21JR1RA121), the Natural Science Foundation of Gansu Province, China (grant no. 21JR1RA132), and Scientific research topics of Traditional Chinese Medicine and Health Commission of Gansu Province, China (grant no. GZKZ-2021-9).
We thank all the subjects who participated in our study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelhalim, A. N., Alberico, R. A., Barczykowski, A. L., and Duffner, P. K. (2014). Patterns of magnetic resonance imaging abnormalities in symptomatic patients with Krabbe disease correspond to phenotype. Pediatr. Neurol. 50, 127–134. doi: 10.1016/j.pediatrneurol.2013.10.001
Adachi, H., Ishihara, K., Tachibana, H., Oka, N., Higuchi, Y., Takashima, H., et al. (2016). Adult-onset Krabbe disease presenting with an isolated form of peripheral neuropathy. Muscle Nerve 54, 152–157. doi: 10.1002/mus.25067
Andriescu, E. C., Russo, S. N., and Pérez, C. A. (2020). Teaching neuroImages: Infantile-onset Krabbe disease with tigroid appearance of the white matter. Neurology 94, e1964–e1965. doi: 10.1212/WNL.0000000000009380
Arai, K., and Lo, E. H. (2010). Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J. Neurosci. Res. 88, 758–763. doi: 10.1002/jnr.22256
Arvidsson, J., Hagberg, B., Månsson, J. E., and Svennerholm, L. (1995). Late onset globoid cell leukodystrophy (Krabbe’s disease)–Swedish case with 15 years of follow-up. Acta Paediatr. 84, 218–221. doi: 10.1111/j.1651-2227.1995.tb13616.x
Asokan, A., Schaffer, D. V., and Samulski, R. J. (2012). The AAV vector toolkit: Poised at the clinical crossroads. Mol. Ther. 20, 699–708. doi: 10.1038/mt.2011.287
Bajaj, N. P., Waldman, A., Orrell, R., Wood, N. W., and Bhatia, K. P. (2002). Familial adult onset of Krabbe’s disease resembling hereditary spastic paraplegia with normal neuroimaging. J. Neurol. Neurosurg. Psychiatry 72, 635–638. doi: 10.1136/jnnp.72.5.635
Barkovich, A. J., Kjos, B. O., Jackson, D. E. Jr., and Norman, D. (1988). Normal maturation of the neonatal and infant brain: MR imaging at 1.5 T. Radiology 166(1 Pt 1), 173–180. doi: 10.1148/radiology.166.1.3336675
Barone, R., Brühl, K., Stoeter, P., Fiumara, A., Pavone, L., and Beck, M. (1996). Clinical and neuroradiological findings in classic infantile and late-onset globoid-cell leukodystrophy (Krabbe disease). Am. J. Med. Genet. 63, 209–217. doi: 10.1002/(SICI)1096-8628(19960503)63:1<209::AID-AJMG37>3.0.CO;2-Q
Bartzokis, G., Beckson, M., Lu, P. H., Nuechterlein, K. H., Edwards, N., and Mintz, J. (2001). Age-related changes in frontal and temporal lobe volumes in men: A magnetic resonance imaging study [published correction appears in Arch Gen Psychiatry 2001 Aug;58(8):774]. Arch. Gen. Psychiatry 58, 461–465. doi: 10.1001/archpsyc.58.5.461
Belleri, M., and Presta, M. (2016). Endothelial cell dysfunction in globoid cell leukodystrophy. J. Neurosci. Res. 94, 1359–1367. doi: 10.1002/jnr.23744
Belleri, M., Ronca, R., Coltrini, D., Nico, B., Ribatti, D., Poliani, P. L., et al. (2013). Inhibition of angiogenesis by β-galactosylceramidase deficiency in globoid cell leukodystrophy. Brain 136(Pt 9), 2859–2875. doi: 10.1093/brain/awt215
Beslow, L. A., Schwartz, E. S., and Bönnemann, C. G. (2008). Thickening and enhancement of multiple cranial nerves in conjunction with cystic white matter lesions in early infantile Krabbe disease. Pediatr. Radiol. 38, 694–696. doi: 10.1007/s00247-008-0763-7
Borda, J. T., Alvarez, X., Mohan, M., Ratterree, M. S., Phillippi-Falkenstein, K., Lackner, A. A., et al. (2008). Clinical and immunopathologic alterations in rhesus macaques affected with globoid cell leukodystrophy. Am. J. Pathol. 172, 98–111. doi: 10.2353/ajpath.2008.070404
Bradbury, A. M., Bongarzone, E. R., and Sands, M. S. (2021). Krabbe disease: New hope for an old disease. Neurosci. Lett. 752:135841. doi: 10.1016/j.neulet.2021.135841
Brockmann, K., Dechent, P., Wilken, B., Rusch, O., Frahm, J., and Hanefeld, F. (2003). Proton MRS profile of cerebral metabolic abnormalities in Krabbe disease. Neurology 60, 819–825. doi: 10.1212/01.wnl.0000049469.29011.e9
Caniglia, M., Rana, I., Pinto, R. M., Fariello, G., Caruso, R., Angioni, A., et al. (2002). Allogeneic bone marrow transplantation for infantile globoid-cell leukodystrophy (Krabbe’s disease). Pediatr. Transplant. 6, 427–431. doi: 10.1034/j.1399-3046.2002.02026.x
Cannizzaro, L. A., Chen, Y. Q., Rafi, M. A., and Wenger, D. A. (1994). Regional mapping of the human galactocerebrosidase gene (GALC) to 14q31 by in situ hybridization. Cytogenet. Cell Genet. 66, 244–245. doi: 10.1159/000133703
Cantuti Castelvetri, L., Givogri, M. I., Hebert, A., Smith, B., Song, Y., Kaminska, A., et al. (2013). The sphingolipid psychosine inhibits fast axonal transport in Krabbe disease by activation of GSK3β and deregulation of molecular motors. J. Neurosci. 33, 10048–10056. doi: 10.1523/JNEUROSCI.0217-13.2013
Cantuti Castelvetri, L., Maravilla, E., Marshall, M., Tamayo, T., D’auria, L., Monge, J., et al. (2015). Mechanism of neuromuscular dysfunction in Krabbe disease. J. Neurosci. 35, 1606–1616. doi: 10.1523/JNEUROSCI.2431-14.2015
Cantuti-Castelvetri, L., Zhu, H., Givogri, M. I., Chidavaenzi, R. L., Lopez-Rosas, A., and Bongarzone, E. R. (2012). Psychosine induces the dephosphorylation of neurofilaments by deregulation of PP1 and PP2A phosphatases. Neurobiol. Dis. 46, 325–335. doi: 10.1016/j.nbd.2012.01.013
Carling, D. (2005). AMP-activated protein kinase: Balancing the scales. Biochimie 87, 87–91. doi: 10.1016/j.biochi.2004.10.017
Castelvetri, L. C., Givogri, M. I., Zhu, H., Smith, B., Lopez-Rosas, A., Qiu, X., et al. (2011). Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta Neuropathol. 122, 35–48. doi: 10.1007/s00401-011-0814-2
Chabali, R., Matre, W. M., and Greene, M. K. (1997). Infant with irritability, feeding problems, and progressive developmental abnormalities presenting repeatedly to a pediatric emergency department. Pediatr. Emerg. Care 13, 123–126. doi: 10.1097/00006565-199704000-00011
Chen, Y. Q., Rafi, M. A., de Gala, G., and Wenger, D. A. (1993). Cloning and expression of cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum. Mol. Genet. 2, 1841–1845. doi: 10.1093/hmg/2.11.1841
Chen, Y. Q., and Wenger, D. A. (1993). Galactocerebrosidase from human urine: Purification and partial characterization. Biochim. Biophys. Acta 1170, 53–61. doi: 10.1016/0005-2760(93)90175-9
Courchesne, E., Chisum, H. J., Townsend, J., Cowles, A., Covington, J., Egaas, B., et al. (2000). Normal brain development and aging: Quantitative analysis at in vivo MR imaging in healthy volunteers. Radiology 216, 672–682. doi: 10.1148/radiology.216.3.r00au37672
Cousyn, L., Law-Ye, B., Pyatigorskaya, N., Debs, R., Froissart, R., Piraud, M., et al. (2019). Brain MRI features and scoring of leukodystrophy in adult-onset Krabbe disease. Neurology 93, e647–e652. doi: 10.1212/WNL.0000000000007943
De Gasperi, R., Gama Sosa, M. A., Sartorato, E., Battistini, S., Raghavan, S., and Kolodny, E. H. (1999). Molecular basis of late-life globoid cell leukodystrophy. Hum. Mutat. 14, 256–262. doi: 10.1002/(SICI)1098-1004(1999)14:3<256::AID-HUMU9>3.0.CO;2-6
De Gasperi, R., Gama Sosa, M. A., Sartorato, E. L., Battistini, S., MacFarlane, H., Gusella, J. F., et al. (1996). Molecular heterogeneity of late-onset forms of globoid-cell leukodystrophy [published correction appears in Am J Hum Genet 1997 May;60(5):1264]. Am. J. Hum. Genet. 59, 1233–1242.
De Stefano, N., Dotti, M. T., Mortilla, M., Pappagallo, E., Luzi, P., Rafi, M. A., et al. (2000). Evidence of diffuse brain pathology and unspecific genetic characterization in a patient with an atypical form of adult-onset Krabbe disease. J. Neurol. 247, 226–228. doi: 10.1007/s004150050571
Deane, J. E., Graham, S. C., Kim, N. N., Stein, P. E., McNair, R., Cachón-González, M. B., et al. (2011). Insights into Krabbe disease from structures of galactocerebrosidase. Proc. Natl. Acad. Sci. U.S.A. 108, 15169–15173. doi: 10.1073/pnas.1105639108
Debs, R., Froissart, R., Aubourg, P., Papeix, C., Douillard, C., Degos, B., et al. (2013). Krabbe disease in adults: Phenotypic and genotypic update from a series of 11 cases and a review. J. Inherit. Metab. Dis. 36, 859–868. doi: 10.1007/s10545-012-9560-4
Deoni, S. C., Mercure, E., Blasi, A., Gasston, D., Thomson, A., Johnson, M., et al. (2011). Mapping infant brain myelination with magnetic resonance imaging. J. Neurosci. 31, 784–791. doi: 10.1523/JNEUROSCI.2106-10.2011
Desnick, R. J., and Schuchman, E. H. (2012). Enzyme replacement therapy for lysosomal diseases: Lessons from 20 years of experience and remaining challenges. Annu. Rev. Genomics Hum. Genet. 13, 307–335. doi: 10.1146/annurev-genom-090711-163739
Duffner, P. K., Barczykowski, A., Jalal, K., Yan, L., Kay, D. M., and Carter, R. L. (2011). Early infantile Krabbe disease: Results of the World-Wide Krabbe Registry. Pediatr. Neurol. 45, 141–148. doi: 10.1016/j.pediatrneurol.2011.05.007
Duffner, P. K., Barczykowski, A., Kay, D. M., Jalal, K., Yan, L., Abdelhalim, A., et al. (2012). Later onset phenotypes of Krabbe disease: Results of the world-wide registry. Pediatr. Neurol. 46, 298–306. doi: 10.1016/j.pediatrneurol.2012.02.023
Duffner, P. K., Caggana, M., Orsini, J. J., Wenger, D. A., Patterson, M. C., Crosley, C. J., et al. (2009a). Newborn screening for Krabbe disease: The New York State model. Pediatr. Neurol. 40, 245–255. doi: 10.1016/j.pediatrneurol.2008.11.010
Duffner, P. K., Jalal, K., and Carter, R. L. (2009b). The hunter’s hope Krabbe family database. Pediatr. Neurol. 40, 13–18. doi: 10.1016/j.pediatrneurol.2008.08.011
Dupree, J. L., Girault, J. A., and Popko, B. (1999). Axo-glial interactions regulate the localization of axonal paranodal proteins. J. Cell Biol. 147, 1145–1152. doi: 10.1083/jcb.147.6.1145
Escolar, M. L., Poe, M. D., Provenzale, J. M., Richards, K. C., Allison, J., Wood, S., et al. (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N. Engl. J. Med. 352, 2069–2081. doi: 10.1056/NEJMoa042604
Escolar, M. L., Poe, M. D., Smith, J. K., Gilmore, J. H., Kurtzberg, J., Lin, W., et al. (2009). Diffusion tensor imaging detects abnormalities in the corticospinal tracts of neonates with infantile Krabbe disease. AJNR Am. J. Neuroradiol. 30, 1017–1021. doi: 10.3174/ajnr.A1476
Escolar, M. L., West, T., Dallavecchia, A., Poe, M. D., and LaPoint, K. (2016). Clinical management of Krabbe disease. J. Neurosci. Res. 94, 1118–1125. doi: 10.1002/jnr.23891
Fiumara, A., Barone, R., Arena, A., Filocamo, M., Lissens, W., Pavone, L., et al. (2011). Krabbe leukodystrophy in a selected population with high rate of late onset forms: Longer survival linked to c.121G>A (p.Gly41Ser) mutation. Clin. Genet. 80, 452–458. doi: 10.1111/j.1399-0004.2010.01572.x
Fukazawa, R., Takeuchi, H., Oka, N., Shibuya, T., Sakai, N., and Fujii, A. (2021). Adult Krabbe disease that was successfully treated with intravenous immunoglobulin. Intern. Med. 60, 1283–1286. doi: 10.2169/internalmedicine.6094-20
Furuya, H., Kukita, Y., Nagano, S., Sakai, Y., Yamashita, Y., Fukuyama, H., et al. (1997). Adult onset globoid cell leukodystrophy (Krabbe disease): Analysis of galactosylceramidase cDNA from four Japanese patients. Hum. Genet. 100, 450–456. doi: 10.1007/s004390050532
Galbiati, F., Givogri, M. I., Cantuti, L., Rosas, A. L., Cao, H., van Breemen, R., et al. (2009). Combined hematopoietic and lentiviral gene-transfer therapies in newborn Twitcher mice reveal contemporaneous neurodegeneration and demyelination in Krabbe disease. J. Neurosci. Res. 87, 1748–1759. doi: 10.1002/jnr.22006
Ganesan, K., Desai, S., and Hegde, A. (2010). Multiple cranial nerve enhancement: Uncommon imaging finding in early infantile Krabbe’s disease. J. Neuroimaging 20, 195–197. doi: 10.1111/j.1552-6569.2008.00308.x
Garcia, A. M., Morais, N. M., Ohlweiler, L., Winckler, M. I., Ranzan, J., Artigalás, O. A., et al. (2010). Optic nerve enlargement and leukodystrophy: An unusual finding of the infantile form of Krabbe disease. Arq. Neuropsiquiatr. 68, 816–818. doi: 10.1590/s0004-282x2010000500029
Giacomini, A., Ackermann, M., Belleri, M., Coltrini, D., Nico, B., Ribatti, D., et al. (2015). Brain angioarchitecture and intussusceptive microvascular growth in a murine model of Krabbe disease. Angiogenesis 18, 499–510. doi: 10.1007/s10456-015-9481-6
Giri, S., Khan, M., Nath, N., Singh, I., and Singh, A. K. (2008). The role of AMPK in psychosine mediated effects on oligodendrocytes and astrocytes: Implication for Krabbe disease. J. Neurochem. 105, 1820–1833. doi: 10.1111/j.1471-4159.2008.05279.x
Giri, S., Khan, M., Rattan, R., Singh, I., and Singh, A. K. (2006). Krabbe disease: Psychosine-mediated activation of phospholipase A2 in oligodendrocyte cell death. J. Lipid Res. 47, 1478–1492. doi: 10.1194/jlr.M600084-JLR200
Graziano, A. C., and Cardile, V. (2015). History, genetic, and recent advances on Krabbe disease. Gene 555, 2–13. doi: 10.1016/j.gene.2014.09.046
Graziano, A. C., Parenti, R., Avola, R., and Cardile, V. (2016). Krabbe disease: Involvement of connexin43 in the apoptotic effects of sphingolipid psychosine on mouse oligodendrocyte precursors. Apoptosis 21, 25–35. doi: 10.1007/s10495-015-1183-4
Griffiths, G. (1989). The structure and function of a mannose 6-phosphate receptor-enriched, pre-lysosomal compartment in animal cells. J. Cell Sci. Suppl. 11, 139–147. doi: 10.1242/jcs.1989.supplement_11.11
Hagberg, B. (1984). Krabbe’s disease: Clinical presentation of neurological variants. Neuropediatrics 15(Suppl.), 11–15. doi: 10.1055/s-2008-1052374
Hamanoue, M., Yoshioka, A., Ohashi, T., Eto, Y., and Takamatsu, K. (2004). NF-kappaB prevents TNF-alpha-induced apoptosis in an oligodendrocyte cell line. Neurochem. Res. 29, 1571–1576. doi: 10.1023/b:nere.0000029571.39497.56
Haq, E., Contreras, M. A., Giri, S., Singh, I., and Singh, A. K. (2006). Dysfunction of peroxisomes in twitcher mice brain: A possible mechanism of psychosine-induced disease. Biochem. Biophys. Res. Commun. 343, 229–238. doi: 10.1016/j.bbrc.2006.02.131
Haq, E., Giri, S., Singh, I., and Singh, A. K. (2003). Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J. Neurochem. 86, 1428–1440. doi: 10.1046/j.1471-4159.2003.01941.x
Hardie, D. G. (2004). The AMP-activated protein kinase pathway–new players upstream and downstream. J. Cell Sci. 117(Pt 23), 5479–5487. doi: 10.1242/jcs.01540
Harzer, K., Knoblich, R., Rolfs, A., Bauer, P., and Eggers, J. (2002). Residual galactosylsphingosine (psychosine) beta-galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clin. Chim. Acta 317, 77–84. doi: 10.1016/s0009-8981(01)00791-4
Hawkins-Salsbury, J. A., Parameswar, A. R., Jiang, X., Schlesinger, P. H., Bongarzone, E., Ory, D. S., et al. (2013). Psychosine, the cytotoxic sphingolipid that accumulates in globoid cell leukodystrophy, alters membrane architecture. J. Lipid Res. 54, 3303–3311. doi: 10.1194/jlr.M039610
Hawkins-Salsbury, J. A., Qin, E. Y., Reddy, A. S., Vogler, C. A., and Sands, M. S. (2012). Oxidative stress as a therapeutic target in globoid cell leukodystrophy. Exp. Neurol. 237, 444–452. doi: 10.1016/j.expneurol.2012.07.013
Hawkins-Salsbury, J. A., Shea, L., Jiang, X., Hunter, D. A., Guzman, A. M., Reddy, A. S., et al. (2015). Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J. Neurosci. 35, 6495–6505. doi: 10.1523/JNEUROSCI.4199-14.2015
Henderson, R. D., MacMillan, J. C., and Bradfield, J. M. (2003). Adult onset Krabbe disease may mimic motor neurone disease. J. Clin. Neurosci. 10, 638–639. doi: 10.1016/s0967-5868(02)00302-8
Hiyama, T., Masumoto, T., Hara, T., Kunimatsu, A., Mamada, N., Kiyotaka, N., et al. (2016). Enlargement of the brachial plexus on magnetic resonance imaging: A novel finding in adult-onset Krabbe disease. BJR Case Rep. 2:20150213. doi: 10.1259/bjrcr.20150213
Hollie, N. I., Cash, J. G., Matlib, M. A., Wortman, M., Basford, J. E., Abplanalp, W., et al. (2014). Micromolar changes in lysophosphatidylcholine concentration cause minor effects on mitochondrial permeability but major alterations in function. Biochim. Biophys. Acta 1841, 888–895. doi: 10.1016/j.bbalip.2013.11.013
Hordeaux, J., Jeffrey, B. A., Jian, J., Choudhury, G. R., Michalson, K., Mitchell, T. W., et al. (2022). Efficacy and safety of a Krabbe disease gene therapy. Hum. Gene Ther. 33, 499–517. doi: 10.1089/hum.2021.245
Hossain, M. A., Otomo, T., Saito, S., Ohno, K., Sakuraba, H., Hamada, Y., et al. (2014). Late-onset Krabbe disease is predominant in Japan and its mutant precursor protein undergoes more effective processing than the infantile-onset form. Gene 534, 144–154. doi: 10.1016/j.gene.2013.11.003
Hwang, M., Zuccoli, G., Panigrahy, A., Rodriguez, D., Poe, M. D., and Escolar, M. L. (2016). Thickening of the cauda equina roots: A common finding in Krabbe disease. Eur. Radiol. 26, 3377–3382. doi: 10.1007/s00330-016-4233-6
Iacono, S., Del Giudice, E., Leon, A., La Bella, V., and Spataro, R. (2022). A novel compound heterozygous mutation in GALC associated with adult-onset Krabbe disease: Case report and literature review. Neurogenetics 23, 157–165. doi: 10.1007/s10048-021-00682-1
Ida, H., Eto, Y., and Maekawa, K. (1990). Biochemical pathogenesis of demyelination in globoid cell leukodystrophy (Krabbe’s disease): The effects of psychosine upon oligodendroglial cell culture. Acta Paediatr. Jpn. 32, 20–26. doi: 10.1111/j.1442-200x.1990.tb00779.x
Jatana, M., Giri, S., and Singh, A. K. (2002). Apoptotic positive cells in Krabbe brain and induction of apoptosis in rat C6 glial cells by psychosine. Neurosci. Lett. 330, 183–187. doi: 10.1016/s0304-3940(02)00655-9
Jurewicz, A., Matysiak, M., Andrzejak, S., and Selmaj, K. (2006). TRAIL-induced death of human adult oligodendrocytes is mediated by JNK pathway. Glia 53, 158–166. doi: 10.1002/glia.20249
Kagitani-Shimono, K., Mohri, I., Fujitani, Y., Suzuki, K., Ozono, K., Urade, Y., et al. (2005). Anti-inflammatory therapy by ibudilast, a phosphodiesterase inhibitor, in demyelination of twitcher, a genetic demyelination model. J. Neuroinflammation 2:10. doi: 10.1186/1742-2094-2-10
Kalous, M., Rauchová, H., and Drahota, Z. (1992). The effect of lysophosphatidylcholine on the activity of various mitochondrial enzymes. Biochim. Biophys. Acta 1098, 167–171. doi: 10.1016/s0005-2728(05)80332-9
Kanazawa, T., Nakamura, S., Momoi, M., Yamaji, T., Takematsu, H., Yano, H., et al. (2000). Inhibition of cytokinesis by a lipid metabolite, psychosine. J. Cell Biol. 149, 943–950. doi: 10.1083/jcb.149.4.943
Khan, M., Haq, E., Giri, S., Singh, I., and Singh, A. K. (2005). Peroxisomal participation in psychosine-mediated toxicity: Implications for Krabbe’s disease. J. Neurosci. Res. 80, 845–854. doi: 10.1002/jnr.20529
Kinney, H. C., Brody, B. A., Kloman, A. S., and Gilles, F. H. (1988). Sequence of central nervous system myelination in human infancy. II. Patterns of myelination in autopsied infants. J. Neuropathol. Exp. Neurol. 47, 217–234. doi: 10.1097/00005072-198805000-00003
Kleijer, W. J., Keulemans, J. L., van der Kraan, M., Geilen, G. G., van der Helm, R. M., Rafi, M. A., et al. (1997). Prevalent mutations in the GALC gene of patients with Krabbe disease of Dutch and other European origin. J. Inherit. Metab. Dis. 20, 587–594. doi: 10.1023/a:1005315311165
Kobayashi, S., Chiu, F. C., Katayama, M., Sacchi, R. S., Suzuki, K., and Suzuki, K. (1986). Expression of glial fibrillary acidic protein in the CNS and PNS of murine globoid cell leukodystrophy, the twitcher. Am. J. Pathol. 125, 227–243.
Krivit, W., Shapiro, E. G., Peters, C., Wagner, J. E., Cornu, G., Kurtzberg, J., et al. (1998). Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy. N. Engl. J. Med. 338, 1119–1126. doi: 10.1056/NEJM199804163381605
Kuai, X. L., Ni, R. Z., Zhou, G. X., Mao, Z. B., Zhang, J. F., Yi, N., et al. (2015). Transplantation of mouse embryonic stem cell-derived oligodendrocytes in the murine model of globoid cell leukodystrophy. Stem Cell Res. Ther. 6:30. doi: 10.1186/s13287-015-0024-2
Kwon, J. M., Matern, D., Kurtzberg, J., Wrabetz, L., Gelb, M. H., Wenger, D. A., et al. (2018). Consensus guidelines for newborn screening, diagnosis and treatment of infantile Krabbe disease. Orphanet. J. Rare Dis. 13:30. doi: 10.1186/s13023-018-0766-x
Lee, W. C., Courtenay, A., Troendle, F. J., Stallings-Mann, M. L., Dickey, C. A., DeLucia, M. W., et al. (2005). Enzyme replacement therapy results in substantial improvements in early clinical phenotype in a mouse model of globoid cell leukodystrophy. FASEB J. 19, 1549–1551. doi: 10.1096/fj.05-3826fje
Lee, W. C., Tsoi, Y. K., Troendle, F. J., DeLucia, M. W., Ahmed, Z., Dicky, C. A., et al. (2007). Single-dose intracerebroventricular administration of galactocerebrosidase improves survival in a mouse model of globoid cell leukodystrophy. FASEB J. 21, 2520–2527. doi: 10.1096/fj.06-6169com
Lemmens, R., Piessens, F., Demaerel, P., and Robberecht, W. (2011). Unilateral white matter involvement in Krabbe disease. Arch. Neurol. 68, 130–131. doi: 10.1001/archneurol.2010.331
LeVine, S. M., Pedchenko, T. V., Bronshteyn, I. G., and Pinson, D. M. (2000). L-cycloserine slows the clinical and pathological course in mice with globoid cell leukodystrophy (twitcher mice). J. Neurosci. Res. 60, 231–236. doi: 10.1002/(SICI)1097-4547(20000415)60:2<231::AID-JNR12>3.0.CO;2-E
Lgisu, H. (1989). Psychosine: A “toxin” produced in the brain–its mechanism of action. J. UOEH 11, 487–493. doi: 10.7888/juoeh.11.487
Liao, P., Gelinas, J., and Sirrs, S. (2014). Phenotypic variability of Krabbe disease across the lifespan. Can. J. Neurol. Sci. 41, 5–12. doi: 10.1017/s0317167100016188
Lim, S. M., Choi, B. O., Oh, S. I., Choi, W. J., Oh, K. W., Nahm, M., et al. (2016). Patient fibroblasts-derived induced neurons demonstrate autonomous neuronal defects in adult-onset Krabbe disease. Oncotarget 7, 74496–74509. doi: 10.18632/oncotarget.12812
Lin, D., Donsante, A., Macauley, S., Levy, B., Vogler, C., and Sands, M. S. (2007). Central nervous system-directed AAV2/5-mediated gene therapy synergizes with bone marrow transplantation in the murine model of globoid-cell leukodystrophy. Mol. Ther. 15, 44–52. doi: 10.1038/sj.mt.6300026
Luzi, P., Abraham, R. M., Rafi, M. A., Curtis, M., Hooper, D. C., and Wenger, D. A. (2009). Effects of treatments on inflammatory and apoptotic markers in the CNS of mice with globoid cell leukodystrophy. Brain Res. 1300, 146–158. doi: 10.1016/j.brainres.2009.09.017
Luzi, P., Rafi, M. A., Victoria, T., Baskin, G. B., and Wenger, D. A. (1997). Characterization of the rhesus monkey galactocerebrosidase (GALC) cDNA and gene and identification of the mutation causing globoid cell leukodystrophy (Krabbe disease) in this primate. Genomics 42, 319–324. doi: 10.1006/geno.1997.4744
Luzi, P., Rafi, M. A., and Wenger, D. A. (1995). Structure and organization of the human galactocerebrosidase (GALC) gene. Genomics 26, 407–409. doi: 10.1016/0888-7543(95)80230-j
Luzi, P., Rafi, M. A., and Wenger, D. A. (1996). Multiple mutations in the GALC gene in a patient with adult-onset Krabbe disease. Ann. Neurol. 40, 116–119. doi: 10.1002/ana.410400119
Lyon, G., Hagberg, B., Evrard, P., Allaire, C., Pavone, L., and Vanier, M. (1991). Symptomatology of late onset Krabbe’s leukodystrophy: The European experience. Dev. Neurosci. 13, 240–244. doi: 10.1159/000112167
Macarov, M., Zlotogora, J., Meiner, V., Khatib, Z., Sury, V., Mengistu, G., et al. (2011). Genetic screening for Krabbe disease: Learning from the past and looking to the future. Am. J. Med. Genet. Part A 155A, 574–576. doi: 10.1002/ajmg.a.33815
Madsen, A. M. H., Wibrand, F., Lund, A. M., Ek, J., Dunø, M., and Østergaard, E. (2019). Genotype and phenotype classification of 29 patients affected by Krabbe disease. JIMD Rep. 46, 35–45. doi: 10.1002/jmd2.12007
Malandrini, A., D’Eramo, C., Palmeri, S., Gaudiano, C., Gambelli, S., Sicurelli, F., et al. (2013). Peripheral neuropathy in late-onset Krabbe disease: Report of three cases. Neurol. Sci. 34, 79–83. doi: 10.1007/s10072-012-0956-6
Mamada, N., Nakamagoe, K., Shioya, A., Furuta, J., Sakai, N., Ishii, A., et al. (2016). Adult-onset Krabbe disease presenting as acute hemiparesis and progressive demyelination detected by diffusion-weighted imaging. J. Neurol. Sci. 367, 326–328. doi: 10.1016/j.jns.2016.06.026
Marcus, J., Honigbaum, S., Shroff, S., Honke, K., Rosenbluth, J., and Dupree, J. L. (2006). Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia 53, 372–381. doi: 10.1002/glia.20292
Marks, H. G., Scavina, M. T., Kolodny, E. H., Palmieri, M., and Childs, J. (1997). Krabbe’s disease presenting as a peripheral neuropathy. Muscle Nerve 20, 1024–1028.
Matsumoto, R., Oka, N., Nagahama, Y., Akiguchi, I., and Kimura, J. (1996). Peripheral neuropathy in late-onset Krabbe’s disease: Histochemical and ultrastructural findings. Acta Neuropathol. 92, 635–639. doi: 10.1007/s004010050573
Meng, X., Li, Y., Lian, Y., Li, Y., Du, L., Xie, N., et al. (2020). A new compound heterozygous mutation in adult-onset Krabbe disease. Int. J. Neurosci. 130, 1267–1271. doi: 10.1080/00207454.2020.1731504
Mikulka, C. R., and Sands, M. S. (2016). Treatment for Krabbe’s disease: Finding the combination. J. Neurosci. Res. 94, 1126–1137. doi: 10.1002/jnr.23822
Muthusamy, K., Sudhakar, S. V., Thomas, M., Yoganathan, S., Christudass, C. S., Chandran, M., et al. (2019). Revisiting magnetic resonance imaging pattern of Krabbe disease - Lessons from an Indian cohort. J. Clin. Imaging Sci. 9:25. doi: 10.25259/JCIS-18-2019
Nagano, S., Yamada, T., Shinnoh, N., Furuya, H., Taniwaki, T., and Kira, J. (1998). Expression and processing of recombinant human galactosylceramidase. Clin. Chim. Acta 276, 53–61. doi: 10.1016/s0009-8981(98)00095-3
Nagara, H., Ogawa, H., Sato, Y., Kobayashi, T., and Suzuki, K. (1986). The twitcher mouse: Degeneration of oligodendrocytes in vitro. Brain Res. 391, 79–84. doi: 10.1016/0165-3806(86)90009-x
Nasir, G., Chopra, R., Elwood, F., and Ahmed, S. S. (2021). Krabbe disease: Prospects of finding a cure using AAV gene therapy. Front. Med. 8:760236. doi: 10.3389/fmed.2021.760236
Neufeld, E. F., and Fratantoni, J. C. (1970). Inborn errors of mucopolysaccharide metabolism. Science 169, 141–146. doi: 10.1126/science.169.3941.141
Paintlia, M. K., Singh, I., and Singh, A. K. (2015). Effect of vitamin D3 intake on the onset of disease in a murine model of human Krabbe disease. J. Neurosci. Res. 93, 28–42. doi: 10.1002/jnr.23476
Paiva, A. R. B., Fonseca Neto, R. E., Afonso, C. L., Freua, F., Nóbrega, P. R., and Kok, F. (2022). Incidental magnetic resonance imaging findings leading to an unusual diagnosis: Adult onset Krabbe disease. Eur. J. Neurol. 29, 1859–1862. doi: 10.1111/ene.15298
Pannuzzo, G., Cardile, V., Costantino-Ceccarini, E., Alvares, E., Mazzone, D., and Perciavalle, V. (2010). A galactose-free diet enriched in soy isoflavones and antioxidants results in delayed onset of symptoms of Krabbe disease in twitcher mice. Mol. Genet. Metab. 100, 234–240. doi: 10.1016/j.ymgme.2010.03.021
Parenti, G., Andria, G., and Ballabio, A. (2015). Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 66, 471–486. doi: 10.1146/annurev-med-122313-085916
Passini, M. A., Dodge, J. C., Bu, J., Yang, W., Zhao, Q., Sondhi, D., et al. (2006). Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J. Neurosci. 26, 1334–1342. doi: 10.1523/JNEUROSCI.2676-05.2006
Passini, M. A., Macauley, S. L., Huff, M. R., Taksir, T. V., Bu, J., Wu, I. H., et al. (2005). AAV vector-mediated correction of brain pathology in a mouse model of Niemann-Pick A disease. Mol. Ther. 11, 754–762. doi: 10.1016/j.ymthe.2005.01.011
Patel, B., Gimi, B., Vachha, B., Agadi, S., and Koral, K. (2008). Optic nerve and chiasm enlargement in a case of infantile Krabbe disease: Quantitative comparison with 26 age-matched controls. Pediatr. Radiol. 38, 697–699. doi: 10.1007/s00247-008-0849-2
Pernber, Z., Molander-Melin, M., Berthold, C. H., Hansson, E., and Fredman, P. (2002). Expression of the myelin and oligodendrocyte progenitor marker sulfatide in neurons and astrocytes of adult rat brain. J. Neurosci. Res. 69, 86–93. doi: 10.1002/jnr.10264
Phelps, M., Aicardi, J., and Vanier, M. T. (1991). Late onset Krabbe’s leukodystrophy: A report of four cases. J. Neurol. Neurosurg. Psychiatry 54, 293–296. doi: 10.1136/jnnp.54.4.293
Poretti, A., Meoded, A., Bunge, M., Fatemi, A., Barrette, P., Huisman, T. A., et al. (2014). Novel diffusion tensor imaging findings in Krabbe disease. Eur. J. Paediatr. Neurol. 18, 150–156. doi: 10.1016/j.ejpn.2013.09.008
Poretti, A., Meoded, A., and Fatemi, A. (2016). Diffusion tensor imaging: A biomarker of outcome in Krabbe’s disease. J. Neurosci. Res. 94, 1108–1115. doi: 10.1002/jnr.23769
Provenzale, J. M., Escolar, M., and Kurtzberg, J. (2005). Quantitative analysis of diffusion tensor imaging data in serial assessment of Krabbe disease. Ann. N. Y. Acad. Sci. 1064, 220–229. doi: 10.1196/annals.1340.040
Qin, E. Y., Hawkins-Salsbury, J. A., Jiang, X., Reddy, A. S., Farber, N. B., Ory, D. S., et al. (2012). Bone marrow transplantation increases efficacy of central nervous system-directed enzyme replacement therapy in the murine model of globoid cell leukodystrophy. Mol. Genet. Metab. 107, 186–196. doi: 10.1016/j.ymgme.2012.05.021
Quintas-Neves, M., Xavier, S. A., and Soares-Fernandes, J. P. (2020). Sixth cranial nerve involvement in early onset Krabbe disease. Neuropediatrics 51, 307–308. doi: 10.1055/s-0040-1701661
Rafi, M. A. (2016). Gene therapy for CNS diseases - Krabbe disease. Bioimpacts 6, 69–70. doi: 10.15171/bi.2016.09
Rafi, M. A., Luzi, P., Chen, Y. Q., and Wenger, D. A. (1995). A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum. Mol. Genet. 4, 1285–1289. doi: 10.1093/hmg/4.8.1285
Rafi, M. A., Luzi, P., Zlotogora, J., and Wenger, D. A. (1996). Two different mutations are responsible for Krabbe disease in the Druze and Moslem Arab populations in Israel. Hum. Genet. 97, 304–308. doi: 10.1007/BF02185759
Rafi, M. A., Rao, H. Z., Luzi, P., Curtis, M. T., and Wenger, D. A. (2012). Extended normal life after AAVrh10-mediated gene therapy in the mouse model of Krabbe disease. Mol. Ther. 20, 2031–2042. doi: 10.1038/mt.2012.153
Rafi, M. A., Rao, H. Z., Luzi, P., Luddi, A., Curtis, M. T., and Wenger, D. A. (2015a). Intravenous injection of AAVrh10-GALC after the neonatal period in twitcher mice results in significant expression in the central and peripheral nervous systems and improvement of clinical features. Mol. Genet. Metab. 114, 459–466. doi: 10.1016/j.ymgme.2014.12.300
Rafi, M. A., Rao, H. Z., Luzi, P., and Wenger, D. A. (2015b). Long-term improvements in lifespan and pathology in CNS and PNS after BMT plus one intravenous injection of AAVrh10-GALC in twitcher mice. Mol. Ther. 23, 1681–1690. doi: 10.1038/mt.2015.145
Rafi, M. A., Zhi Rao, H., Passini, M. A., Curtis, M., Vanier, M. T., Zaka, M., et al. (2005). AAV-mediated expression of galactocerebrosidase in brain results in attenuated symptoms and extended life span in murine models of globoid cell leukodystrophy. Mol. Ther. 11, 734–744. doi: 10.1016/j.ymthe.2004.12.020
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin. Invest. 122, 1164–1171. doi: 10.1172/JCI58644
Reddy, A. S., Kim, J. H., Hawkins-Salsbury, J. A., Macauley, S. L., Tracy, E. T., Vogler, C. A., et al. (2011). Bone marrow transplantation augments the effect of brain- and spinal cord-directed adeno-associated virus 2/5 gene therapy by altering inflammation in the murine model of globoid-cell leukodystrophy. J. Neurosci. 31, 9945–9957. doi: 10.1523/JNEUROSCI.1802-11.2011
Ripoll, C. B., Flaat, M., Klopf-Eiermann, J., Fisher-Perkins, J. M., Trygg, C. B., Scruggs, B. A., et al. (2011). Mesenchymal lineage stem cells have pronounced anti-inflammatory effects in the twitcher mouse model of Krabbe’s disease. Stem Cells 29, 67–77. doi: 10.1002/stem.555
Romano, A., De Simone, R., Fasoli, F., Ferrante, M., Cipriani, V., Fantozzi, L. M., et al. (2009). Selective white matter involvement in a patient with late onset Krabbe disease: MR, MR spectroscopy, and diffusion tensor study. J. Neuroimaging 19, 191–193. doi: 10.1111/j.1552-6569.2008.00258.x
Sabatelli, M., Quaranta, L., Madia, F., Lippi, G., Conte, A., Lo Monaco, M., et al. (2002). Peripheral neuropathy with hypomyelinating features in adult-onset Krabbe’s disease. Neuromuscul. Disord. 12, 386–391. doi: 10.1016/s0960-8966(01)00285-1
Sakai, N., Inui, K., Tatsumi, N., Fukushima, H., Nishigaki, T., Taniike, M., et al. (1996). Molecular cloning and expression of cDNA for murine galactocerebrosidase and mutation analysis of the twitcher mouse, a model of Krabbe’s disease. J. Neurochem. 66, 1118–1124. doi: 10.1046/j.1471-4159.1996.66031118.x
Schaeren-Wiemers, N., van der Bijl, P., and Schwab, M. E. (1995). The UDP-galactose:ceramide galactosyltransferase: Expression pattern in oligodendrocytes and Schwann cells during myelination and substrate preference for hydroxyceramide. J. Neurochem. 65, 2267–2278. doi: 10.1046/j.1471-4159.1995.65052267.x
Schiffmann, R., and van der Knaap, M. S. (2009). Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology 72, 750–759. doi: 10.1212/01.wnl.0000343049.00540.c8
Schrier Vergano, S. A., Kanungo, S., and Arnold, G. (2022). Making decisions about Krabbe disease newborn screening. Pediatrics 149:e2021053175. doi: 10.1542/peds.2021-053175
Schulte, S., and Stoffel, W. (1993). Ceramide UDPgalactosyltransferase from myelinating rat brain: Purification, cloning, and expression. Proc. Natl. Acad. Sci. U.S.A. 90, 10265–10269. doi: 10.1073/pnas.90.21.10265
Sharp, M. E., Laule, C., Nantel, S., Mädler, B., Aul, R. B., Yip, S., et al. (2013). Stem cell transplantation for adult-onset Krabbe disease: Report of a case. JIMD Rep. 10, 57–59. doi: 10.1007/8904_2012_203
Siddiqi, Z. A., Sanders, D. B., and Massey, J. M. (2006). Peripheral neuropathy in Krabbe disease: Electrodiagnostic findings. Neurology 67, 263–267. doi: 10.1212/01.wnl.0000230153.34613.84
SSnook, E. R., Fisher-Perkins, J. M., Sansing, H. A., Lee, K. M., Alvarez, X., MacLean, A. G., et al. (2014). Innate immune activation in the pathogenesis of a murine model of globoid cell leukodystrophy. Am. J. Pathol. 184, 382–396. doi: 10.1016/j.ajpath.2013.10.011
Suzuki, K. (1998). Twenty five years of the “psychosine hypothesis”: A personal perspective of its history and present status. Neurochem. Res. 23, 251–259. doi: 10.1023/a:1022436928925
Suzuki, K., and Suzuki, Y. (1970). Globoid cell leucodystrophy (Krabbe’s disease): Deficiency of galactocerebroside beta-galactosidase. Proc. Natl. Acad. Sci. U.S.A. 66, 302–309. doi: 10.1073/pnas.66.2.302
Tanaka, S., Mito, T., and Takashima, S. (1995). Progress of myelination in the human fetal spinal nerve roots, spinal cord and brainstem with myelin basic protein immunohistochemistry. Early Hum. Dev. 41, 49–59. doi: 10.1016/0378-3782(94)01608-r
Tappino, B., Biancheri, R., Mort, M., Regis, S., Corsolini, F., Rossi, A., et al. (2010). Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum. Mutat. 31, E1894–E1914. doi: 10.1002/humu.21367
Tokushige, S., Sonoo, T., Maekawa, R., Shirota, Y., Hanajima, R., Terao, Y., et al. (2013). Isolated pyramidal tract impairment in the central nervous system of adult-onset Krabbe disease with novel mutations in the GALC gene. Brain Dev. 35, 579–581. doi: 10.1016/j.braindev.2012.08.004
Udow, S., Bunge, M., Ryner, L., Mhanni, A. A., and Salman, M. S. (2012). Prolonged survival and serial magnetic resonance imaging/magnetic resonance spectroscopy changes in infantile Krabbe disease. Pediatr. Neurol. 47, 299–302. doi: 10.1016/j.pediatrneurol.2012.06.015
Vasconcellos, E., and Smith, M. (1998). MRI nerve root enhancement in Krabbe disease. Pediatr. Neurol. 19, 151–152. doi: 10.1016/s0887-8994(98)00033-2
Victoria, T., Rafi, M. A., and Wenger, D. A. (1996). Cloning of the canine GALC cDNA and identification of the mutation causing globoid cell leukodystrophy in West Highland White and Cairn terriers. Genomics 33, 457–462. doi: 10.1006/geno.1996.0220
Voccoli, V., Tonazzini, I., Signore, G., Caleo, M., and Cecchini, M. (2014). Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis. 5:e1529. doi: 10.1038/cddis.2014.483
Wang, C., Melberg, A., Weis, J., Månsson, J. E., and Raininko, R. (2007). The earliest MR imaging and proton MR spectroscopy abnormalities in adult-onset Krabbe disease. Acta Neurol. Scand. 116, 268–272. doi: 10.1111/j.1600-0404.2007.00867.x
Wang, Y., Gupta, A., Liu, Z., Zhang, H., Escolar, M. L., Gilmore, J. H., et al. (2011). DTI registration in atlas based fiber analysis of infantile Krabbe disease. Neuroimage 55, 1577–1586. doi: 10.1016/j.neuroimage.2011.01.038
Wenger, D. A., Luzi, P., and Rafi, M. A. (2021). Advances in the diagnosis and treatment of Krabbe disease. Int. J. Neonatal Screen. 7:57. doi: 10.3390/ijns7030057
Wenger, D. A., Rafi, M. A., and Luzi, P. (1997). Molecular genetics of Krabbe disease (globoid cell leukodystrophy): Diagnostic and clinical implications. Hum. Mutat. 10, 268–279. doi: 10.1002/(SICI)1098-1004(1997)10:4<268::AID-HUMU2>3.0.CO;2-D
Wenger, D. A., Rafi, M. A., and Luzi, P. (2016). Krabbe disease: One Hundred years from the bedside to the bench to the bedside. J. Neurosci. Res. 94, 982–989. doi: 10.1002/jnr.23743
White, A. B., Galbiati, F., Givogri, M. I., Lopez Rosas, A., Qiu, X., van Breemen, R., et al. (2011). Persistence of psychosine in brain lipid rafts is a limiting factor in the therapeutic recovery of a mouse model for Krabbe disease. J. Neurosci. Res. 89, 352–364. doi: 10.1002/jnr.22564
Won, J. S., Kim, J., Paintlia, M. K., Singh, I., and Singh, A. K. (2013). Role of endogenous psychosine accumulation in oligodendrocyte differentiation and survival: Implication for Krabbe disease. Brain Res. 1508, 44–52. doi: 10.1016/j.brainres.2013.02.024
Won, J. S., Singh, A. K., and Singh, I. (2016). Biochemical, cell biological, pathological, and therapeutic aspects of Krabbe’s disease. J. Neurosci. Res. 94, 990–1006. doi: 10.1002/jnr.23873
Wright, M. D., Poe, M. D., DeRenzo, A., Haldal, S., and Escolar, M. L. (2017). Developmental outcomes of cord blood transplantation for Krabbe disease: A 15-year study. Neurology 89, 1365–1372. doi: 10.1212/WNL.0000000000004418
Wu, L., Liao, X., Yang, S., and Gan, S. (2021). Krabbe disease associated with mitochondrial dysfunction in a Chinese family. Front. Neurol. 12:750095. doi: 10.3389/fneur.2021.750095
Wu, Y. P., McMahon, E. J., Matsuda, J., Suzuki, K., Matsushima, G. K., and Suzuki, K. (2001). Expression of immune-related molecules is downregulated in twitcher mice following bone marrow transplantation. J. Neuropathol. Exp. Neurol. 60, 1062–1074. doi: 10.1093/jnen/60.11.1062
Xia, Z., Wenwen, Y., Xianfeng, Y., Panpan, H., Xiaoqun, Z., and Zhongwu, S. (2020). Adult-onset Krabbe disease due to a homozygous GALC mutation without abnormal signals on an MRI in a consanguineous family: A case report. Mol. Genet. Genomic Med. 8:e1407. doi: 10.1002/mgg3.1407
Xu, C., Sakai, N., Taniike, M., Inui, K., and Ozono, K. (2006). Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correlation. J. Hum. Genet. 51, 548–554. doi: 10.1007/s10038-006-0396-3
Yang, Y., Ren, X., Xu, Q., Wang, C., Liu, H., and He, X. (2013). Four novel GALC gene mutations in two Chinese patients with Krabbe disease. Gene 519, 381–384. doi: 10.1016/j.gene.2013.02.010
Young, P. P., Fantz, C. R., and Sands, M. S. (2004). VEGF disrupts the neonatal blood-brain barrier and increases life span after non-ablative BMT in a murine model of congenital neurodegeneration caused by a lysosomal enzyme deficiency. Exp. Neurol. 188, 104–114. doi: 10.1016/j.expneurol.2004.03.007
Zayed, H. (2015). Krabbe disease in the arab world. J. Pediatr. Genet. 4, 1–8. doi: 10.1055/s-0035-1554981
Zhang, C., Liu, Z., and Dong, H. (2021). Two cases of female Chinese adult-onset krabbe disease with one novel mutation and a review of literature. J. Mol. Neurosci. 71, 1185–1192. doi: 10.1007/s12031-020-01742-1
Zhang, T., Yan, C., Ji, K., Lin, P., Chi, L., Zhao, X., et al. (2018). Adult-onset Krabbe disease in two generations of a Chinese family. Ann. Transl. Med. 6:174. doi: 10.21037/atm.2018.04.30
Keywords: globoid cell leukodystrophy, Krabbe disease, adult-onset GLD, galactosylceramidase, GALC, psychosine
Citation: Wu G, Li Z, Li J, Li X, Wang M, Zhang J, Liu G and Zhang P (2022) A neglected neurodegenerative disease: Adult-onset globoid cell leukodystrophy. Front. Neurosci. 16:998275. doi: 10.3389/fnins.2022.998275
Received: 19 July 2022; Accepted: 18 August 2022;
Published: 07 September 2022.
Edited by:
Yuzhen Xu, The Second Affiliated Hospital of Shandong First Medical University, ChinaReviewed by:
Qihua Chen, Central South University, ChinaCopyright © 2022 Wu, Li, Li, Li, Wang, Zhang, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manxia Wang, d214MzIyQGFsaXl1bi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.