
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurol., 12 January 2024
Sec. Pediatric Neurology
Volume 14 - 2023 | https://doi.org/10.3389/fneur.2023.1287559
 Shelei Pan1
Shelei Pan1 Andrew T. Hale2Mackenzie E. Lemieux1
Andrew T. Hale2Mackenzie E. Lemieux1 Dhvanii K. Raval1Thomas P. Garton3Brooke Sadler4,5Kelly B. Mahaney6
Dhvanii K. Raval1Thomas P. Garton3Brooke Sadler4,5Kelly B. Mahaney6 Jennifer M. Strahle1,4,7*
Jennifer M. Strahle1,4,7*Iron physiology is regulated by a complex interplay of extracellular transport systems, coordinated transcriptional responses, and iron efflux mechanisms. Dysregulation of iron metabolism can result in defects in myelination, neurotransmitter synthesis, and neuronal maturation. In neonates, germinal matrix-intraventricular hemorrhage (GMH-IVH) causes iron overload as a result of blood breakdown in the ventricles and brain parenchyma which can lead to post-hemorrhagic hydrocephalus (PHH). However, the precise mechanisms by which GMH-IVH results in PHH remain elusive. Understanding the molecular determinants of iron homeostasis in the developing brain may lead to improved therapies. This manuscript reviews the various roles iron has in brain development, characterizes our understanding of iron transport in the developing brain, and describes potential mechanisms by which iron overload may cause PHH and brain injury. We also review novel preclinical treatments for IVH that specifically target iron. Understanding iron handling within the brain and central nervous system may provide a basis for preventative, targeted treatments for iron-mediated pathogenesis of GMH-IVH and PHH.
Iron homeostasis is critical to a variety of neurodevelopmental processes. Iron deficiency has been linked to impaired myelination (1, 2), altered monoamine neurotransmitter synthesis (2), and reduced hippocampal neuronal metabolism (3) in neonatal rats. Conversely, brain iron overload can also be deleterious (4–10). Therefore, understanding the homeostatic mechanisms that maintain the delicate brain iron balance is important to better understand how we can preserve the neuronal developmental environment after peri- and neonatal iron-related pathology in the central nervous system.
In preterm infants, brain iron homeostasis can be dramatically disrupted by germinal matrix hemorrhage-intraventricular hemorrhage (GMH-IVH) when bleeding from the immature, fragile vascular network of the germinal matrix releases red blood cells (RBCs) which subsequently lyse and release the blood breakdown products iron, hemoglobin, and bilirubin into the germinal matrix (Grade I), ventricles of the brain (Grades II, III), and brain parenchyma (Grade IV). Prognosis and mortality after GMH-IVH are related to the extent of hemorrhage, with higher grades of GMH-IVH associated with the worst neurodevelopmental outcomes (11, 12). In addition, 30% of infants with high grade (Grades III and IV) GMH-IVH develop post-hemorrhagic hydrocephalus (PHH) (13), a progressive dilation of the cerebral ventricles that results in secondary brain injury and for which definitive surgical management is difficult in preterm neonates (14).
While the mechanisms of PHH and other neurological sequelae after GMH-IVH are not clear, the cytotoxic effects of free iron for inducing DNA damage and disrupting oxidative processes are well documented (15), and clinical studies have shown a higher proportion of infants with PHH to have CSF free iron after GMH-IVH compared to control subjects (16). Unbound iron can participate in the Fenton reaction, in which Fe2+ can be oxidized by hydrogen peroxide to form hydroxyl free radicals and Fe3+ (17, 18). These free radicals can subsequently oxidize numerous cellular targets, causing significant damage and cell death. However, how free iron is directly linked to the pathogenesis of PHH and other devastating neurological sequelae after high grade (Grades III-IV) IVH has not been fully elucidated.
Complicating efforts to understand and target iron overload-mediated brain injury following GMH-IVH, iron processing in the developing brain is not as well-understood as it is in the adult brain. Until recently, the primary cellular brain iron transporters reported in the neonatal brain were divalent metal transporter 1 (DMT1), transferrin receptor (TFR), and ferritin, iron transporters that are also involved in iron handling in other epithelial tissues like the intestine. Recent clinical studies have underscored the role of extracellular iron pathway proteins such as transferrin, ceruloplasmin, haptoglobin, and hemopexin that may have a role in CSF iron clearance after IVH (19, 20). Nevertheless, the number of cellular iron transporters characterized in the neonatal brain is small by comparison to the number of iron handling proteins described in adult brain and neonatal peripheral tissues. This qualitative review summarizes our current understanding of iron transport and homeostasis in the brain, as well as the developmental time course of iron pathway protein expression. We also review the role these proteins may have in mediating iron and blood breakdown product clearance after neonatal GMH-IVH.
Iron exists in several different stable states within the human body. The availability of iron in different oxidative states makes it a prime player in intracellular metabolic processes essential to life. In the plasma, circulating iron is primarily bound to the iron binding protein transferrin in the form of Fe3+ (ferric) iron (21). Low (<1 μM) concentrations of non-transferrin bound iron (NTBI) can also be present in the plasma in either Fe2+ (ferrous) or ferric states bound to small organic molecules like citrate (22, 23). Within hemoglobin or myoglobin molecules, iron is much less accessible. In these heme-bound states, ferrous iron is contained within the center of protoporphyrin IX scaffolds (24). Once inside a cell, ferrous iron represents the labile and active pool of iron that is readily used in biological processes, whereas ferric iron is usually stored complexed to the iron-storage protein ferritin (25). Mitochondrial ferritin (FtMt) is one of three ferritins that are encoded separately by the human body (the other two are the cytosolic L and H subunits) and is primarily found in the mitochondria of metabolically active organs like the brain and testis (26, 27). Mitochondria require iron to support the biogenesis of iron–sulfur clusters and heme synthesis (28–30), however close regulation of mitochondrial iron levels is needed to protect the mitochondria from iron-mediated oxidative damage. FtMt is believed to play a role in maintaining this mitochondrial iron homeostasis (31). In addition to ferritin, cellular iron can also be stored in complex with hemosiderin, but the iron in hemosiderin is not as readily available for use. Excess hemosiderin deposits form after hemorrhage and are thought to result from hemoglobin phagocytosis and subsequent heme breakdown into iron and biliverdin (12).
It is important to note that the majority of our understanding of brain iron metabolism is derived from experiments conducted in adult animals. As many of the following cellular iron transport proteins and mechanisms remain largely under characterized in the neonatal brain, and the developmental timelines of the expression of major iron handling proteins in the brain are still not known, it will be crucial to verify these models of brain iron transport and homeostasis in fetal and neonatal animals to advance our understanding of iron-related pathology in the neonatal time period.
The total amount of iron in the body is primarily determined by dietary intake and uptake from the gut (32, 33). Enterocytes, epithelial cells that line the lumen of the intestines, mediate dietary iron absorption in the duodenum and proximal jejunum of the small intestine (34). Non-heme dietary iron exists predominantly in its ferric form and must be reduced to its ferrous form by the ferrireductase duodenal cytochrome B on the apical brush border of enterocytes before it can be absorbed (35). Other ferrireductases may also play a role in converting ferric to ferrous iron (36). Ferrous iron enters the enterocyte via DMT1 expressed on the apical membrane of enterocytes and leaves the enterocyte via ferroportin 1 (FPN1) expressed on the basolateral surface (37, 38). Exported iron is oxidized to its ferric form via a ferroxidase and complexes with a protein (ie. transferrin) or iron-binding small molecule (ie. citrate) to enter the plasma (35, 39).
When the body’s iron stores are sufficient, the liver peptide hormone hepcidin can inhibit iron export from the enterocyte by binding to and causing FPN1 internalization (25). This prevents additional iron entry into the plasma and drives intracellular storage of iron as ferritin (25). Ferritin within the enterocyte that is not used is excreted when enterocytes are sloughed off the intestinal mucosa at the end of their approximately 3-day lifespan (25).
Under physiological conditions, iron is transported into the brain from the circulation through a series of highly regulated and coordinated steps. Early models of iron transport into the brain proposed that iron-bound transferrin can bind to luminal TfR on microvascular endothelial cells of the capillaries and choroid plexus (ChP) that comprise the blood brain barrier (BBB) before being endocytosed and released into the brain extracellular space (i.e., transcytosis) (40–43). In more recent models of receptor mediated endocytosis brought on in part by the identification of DMT1 on brain capillary endothelial cells (BCECs) that form the BBB, endothelial cell iron release is thought to be more nuanced, with endocytosed transferrin dissociating into ferric iron and apotransferrin in the acidified endosome (44). Ferric iron is reduced to ferrous (Fe2+) iron, which is then transported into the cytoplasm of the endothelial cell via DMT1, where it can either be stored intracellularly with ferritin or exported into the interstitial fluid via FPN1 when ferritin is saturated. The ferroxidase ceruloplasmin oxidizes Fe2+ back to Fe3+ (45), which recombines with apotransferrin to re-form transferrin in the interstitium where it can be taken up by glia and neurons. Transferrin-independent mechanisms of iron import into the brain may also exist, including but not limited to putative NTBI import via ferritin receptors (46, 47), however these mechanisms are not well-characterized.
Iron is present in neurons, astrocytes, microglia, and oligodendrocytes where it plays essential roles in cell respiration, neurotransmitter synthesis, myelination, DNA synthesis, and other cellular processes (1, 48–56). Studies in adult mice and rats have revealed several mechanisms by which these cells may take up iron from the interstitial fluid. Adult neurons, which express Tfr, can obtain iron from transferrin (57). In vitro studies have shown neurons also express ferrous iron transporters zinc regulated transporter and iron regulated transporter like protein 8 (Zip8) and Dmt1 at the cell surface to mediate NTBI uptake (57). Similarly, oligodendrocytes in the adult mouse brain express Tfr, and in vitro express the ferritin receptor T-cell immunoglobulin mucin domain 2 (Tim-2) to facilitate NTBI iron uptake via ferritin (48, 49). In vitro, microglia and astrocytes express Tfr and Dmt1 at the cell surface to facilitate iron uptake via transferrin and NTBI, respectively (58–60). There is comparatively less evidence for Tfr and Dmt1 expression on glia in vivo (61). Fpn1 has been detected in neurons and glia, but its expression varies by age and region (62–66).
In non-inflammatory conditions, hepcidin reduces brain iron load by inducing endothelial cell Fpn1 internalization and degradation when interstitial Fe2+ levels rise (67–70). Hepcidin upregulation in inflammation can cause deleterious effects due to its role in inducing Fpn1 internalization in neurons and glia, which in turn increases intracellular iron levels (71). Therefore, regulation of hepcidin is an important and potentially targetable axis of iron homeostasis. It is known that systemic hepcidin produced from the liver can cross the BBB to enter the brain (72), and that increases in brain and systemic hepcidin after hemorrhagic or ischemic parenchymal brain injury lead to increased iron in the brain (72, 73). However, studies in adult rodents have shown that brain iron metabolism is not drastically altered in mouse models in which liver hepcidin production is knocked out (72), suggesting there are additional brain-specific hepcidin regulation pathways.
In vivo studies in adult rats and mice and in vitro studies in rat and human cells have shown that the expression of hepcidin in the brain is controlled through several mechanisms including the interleukin-6/janus kinase 2/signal transducer and activator of transcription 3 (IL-6/JAK2/STAT3), bone morphogenic protein/s-mothers against decapentaplegic (BMP/SMAD), and CCAT enhancer binding (C/EBP) homologous protein (CHOP) pathways (70, 74–80). As a potential mechanism for the neurotoxic effects of hepcidin overexpression in inflammation, it is known that lipopolysaccharide (LPS) released during inflammation stimulates Toll-like receptor 4 (TLR4), a signaling pathway which (1) has previously been shown to underlie ChP-CSF interface inflammation in both post-infectious hydrocephalus and PHH (81), and (2) induces the production and release of critical cytokines like interleukin-6 (IL-6) (82, 83). IL-6 can upregulate hepcidin expression in the brain via the JAK2/STAT3 pathway (76, 77). The BMP/SMAD pathway has similar effects that are specific to hepcidin upregulation in microglia (76), and the CHOP pathway has been shown to play a role in pathology after subarachnoid hemorrhage (SAH) in adult rodents via its effect of upregulating hepcidin expression in neurons (84), thereby preventing iron export out of neurons, increasing neuronal iron content, and inducing apoptosis. Acute increases in brain iron load can also induce hepcidin upregulation (70).
In addition to the bidirectional effects of hepcidin regulation on iron load in the brain, the iron-responsive element (IRE) signaling pathway and cytoplasmic iron regulatory proteins (IRP-1 and IRP-2) play a major role in maintaining brain iron homeostasis at the post-transcriptional level. IRP-1 and IRP-2 are iron and mRNA-binding proteins which can bind to IREs, relatively short and conserved hairpin-loops in the 3′ or 5′ untranslated region (UTR) of IRP target mRNA molecules (85–89). IRP-1 has additional functionality as a cytoplasmic aconitase in iron-rich conditions. Depending on where in the target mRNA the IRE is localized (90, 91), IRP binding can (1) block ribosome binding, translation, and synthesis of key iron pathway proteins including ferritin and Fpn1 (86, 92–97), or (2) stabilize the mRNA to increase the synthesis of iron pathway proteins like Tfr and Dmt1 (87, 98). Iron binding to IRPs decreases their affinity for IREs and induces their dissociation from mRNA molecules (87, 88, 99), offering a mechanism to control iron pathway protein synthesis that is responsive to iron levels in the brain.
Hemorrhage in neonatal IVH most commonly originates from the immature blood vessels of the germinal matrix and is associated with significant morbidity and mortality in preterm infants (100). RBC lysis in the CSF after IVH releases blood breakdown products including bilirubin, hemoglobin, and unbound ferrous iron, the latter of which can be oxidized in the Fenton reaction to form cytotoxic hydroxyl free radicals and ferric iron (12). While iron homeostasis mechanisms keep free iron levels under control in physiologic conditions, IVH may release amounts of hemoglobin and free iron that overwhelm iron handling and clearance systems in the brain.
Free iron in the CSF may be particularly deleterious due to its free access to the ependymal cells that line the surfaces of the ventricles and the CSF-producing cells of the ChP (81), in addition to its proximity to the cells that make up neurodevelopmentally-important periventricular structures including the subventricular zone, white matter, and hippocampus (Figure 1). This may make these cells particularly susceptible to iron uptake and overload. In fact, previous research from Strahle et al. reported subventricular zone iron overload after IVH (101), and Garton et al. demonstrated iron-mediated cell death in hippocampal neurons in a rodent IVH model (102). This was consistent with studies conducted in humans showing perinatal brain injury is associated with smaller hippocampal size in preterm infants (103).
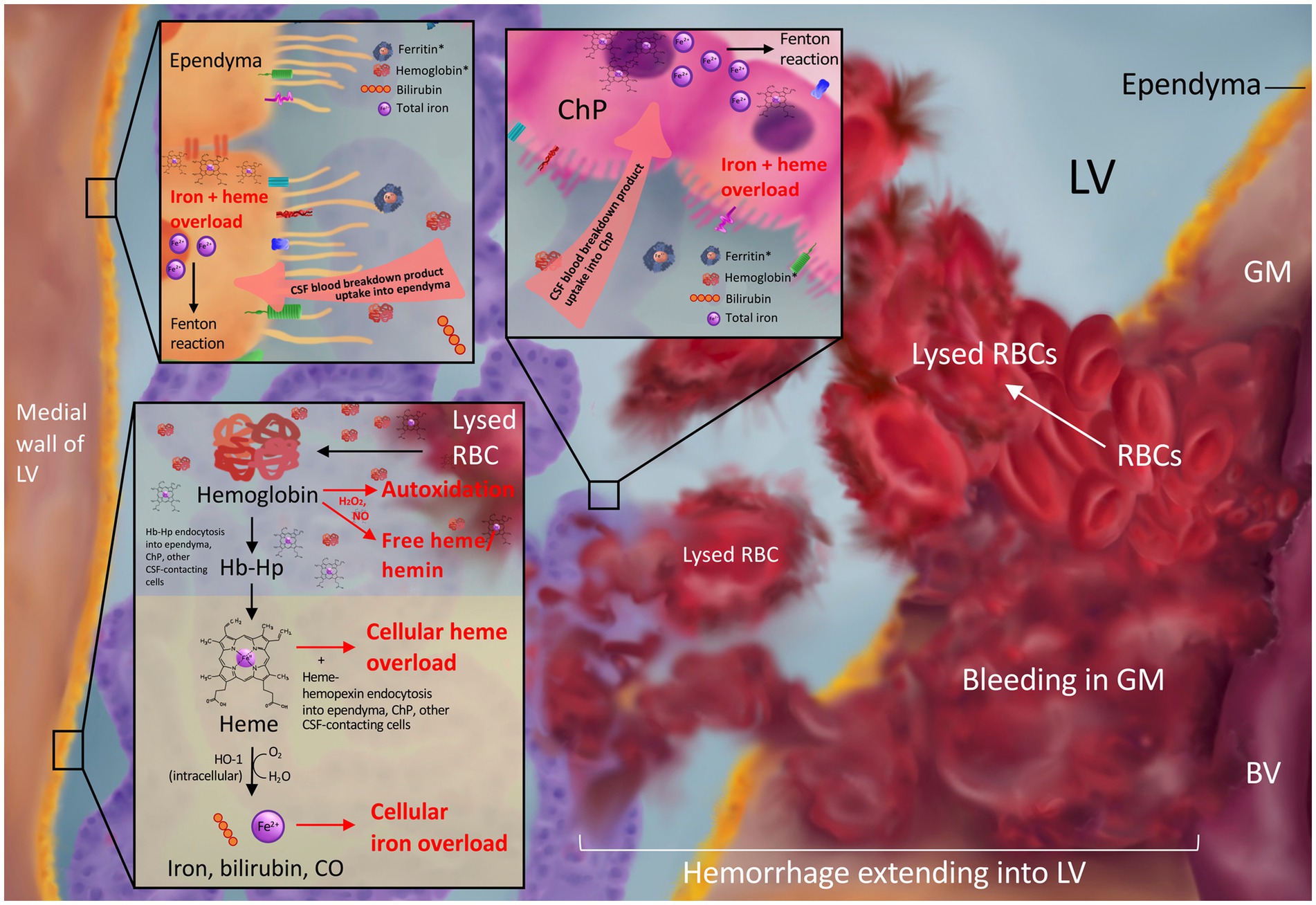
Figure 1. Blood breakdown product release into the CSF after intraventricular extension of germinal matrix hemorrhage. Bleeding from the ruptured immature blood vessels (BV) of the germinal matrix (GM) results in red blood cell (RBC) release into the GM and lateral ventricle (LV) and subsequent lysis to release blood breakdown products including hemoglobin, ferritin, bilirubin, and iron into the CSF. CSF hemoglobin and ferritin (asterisks in top panels) are elevated in the setting of post-hemorrhagic hydrocephalus. Subsequent ependymal and choroid plexus uptake of blood breakdown products from the CSF may lead to toxic iron and heme overload (top panel). RBC lysis in the LVs can release free hemoglobin into the CSF, which may undergo cytotoxic autoxidation in the presence of H2O2 and/or NO and release free heme/hemin into the CSF (bottom panel). Hemoglobin may also be bound and stabilized by the scavenger haptoglobin for cellular uptake and subsequent degradation into heme and Fe2+ ions leading to overload in the setting of GMH-IVH (bottom panel).
Other studies have shown intracellular iron accumulation within perihematomal neurons and glia after GMH in rodents (104); ependymal and subependymal hemosiderin deposition, ferritin expression, and iron accumulation after IVH (105, 106); as well as hemosiderin deposition, ependymal cell death, and subependymal damage in human neonates with IVH-PHH (107). It is also possible that free iron and hemoglobin released into the ventricles after IVH may be transported to distant intraparenchymal regions via the CSF (108), as intraventricular radioactive iron has been shown to distribute to distant anatomic areas of the brain parenchyma in neonatal rats (109).
Recent studies have highlighted the role that the iron handling proteins/scavengers haptoglobin, hemopexin, and ceruloplasmin play in blood and blood breakdown product clearance in the neonatal brain after IVH (19, 20). In this section, we review the developmental time course of iron handling protein expression in the fetal and neonatal brain parenchyma and CSF (Supplementary Table 1). We start with extracellular iron transporters and scavengers in the serum including transferrin, haptoglobin, hemopexin, and ceruloplasmin. We then discuss the expression and localization of the cellular iron transporters ferritin, TFR, DMT1, FPN1, low-density lipoprotein receptor-related protein 1 (LRP1), CD163, and the heme oxygenases. We also discuss proteins that play other key roles in neonatal brain homeostasis, including hepcidin, the IRPs, and amyloid precursor protein (APP). Understanding the molecular mechanisms that mediate iron transport and metabolism in the neonatal brain is necessary to advance our understanding of pathologic iron overload after IVH that leads to inflammation, PHH, and other forms of immediate and delayed injury to the brain.
Transferrin is a glycoprotein that binds to and transports ferric iron through the circulation prior to intracellular delivery. Transferrin is primarily produced by liver hepatocytes, and transferrin-mediated delivery of iron accounts for the majority of iron transport into the brain from the circulation (110, 111). However, the observed rate of iron import into the brain is significantly higher than the rate of transferrin import across the developing BBB (112–114), suggesting there are additional brain endogenous transferrin production mechanisms.
Specifically, transferrin found in the interstitial fluid is produced by oligodendrocytes and the ChP of the lateral and third ventricles (115–118). Endogenous transferrin functions to rapidly bind imported ferric iron ions to mediate delivery to neurons and other cell types. Transferrin has also been shown to play role in oligodendrocyte maturation and enhancing myelinogenesis (119, 120). β-2 transferrin is a desialylated isoform of transferrin synthesized in the brain that is found in the CSF and perilymph only (121).
Brain transferrin levels peak at birth before declining over the first 2–3 postnatal weeks, stabilizing at postnatal day 24 (122–129), and remaining constant throughout the rest of life (113). This decline in mouse and rat brain transferrin levels is region-specific, with a faster rate of decline in the cortex and hindbrain compared to the midbrain (113). In contrast to other cell types, BCEC transferrin expression remains high throughout development (127).
In addition to parenchymal brain transferrin, there is transferrin in fetal rat CSF (130), with a three-fold increase in CSF transferrin from 12 days gestation to 22 days (birth) followed by a significant decrease by postnatal day 10 (130). CSF transferrin has also been detected in human fetuses (131). Transferrin is transported via the CSF to periventricular structures including the medial habenular nucleus, mamillary bodies, interpeduncular nucleus, and brainstem after intraventricular injection into the lateral ventricles of neonatal postnatal day 7 rats (109). The exact role of CSF transferrin in the neonatal brain is not well understood, but it may have a role in transporting iron throughout the developing brain via the CSF.
Mahaney et al. previously demonstrated in humans that there are no significant differences in CSF transferrin levels after low- and high-grade IVH compared to neonates without IVH (19). However, when Strahle et al. followed CSF iron pathway protein levels over time in a separate study, there was an association between longitudinal increases in ventricular CSF transferrin levels after neonatal IVH-PHH and improved cognitive outcomes at 2 years of age (20). Because CSF transferrin is typically fully saturated with ferric iron in physiologic conditions, the blood breakdown products and iron released into the CSF after IVH may overwhelm the iron-binding capabilities of endogenous transferrin. This may lead to high levels of free iron within the ventricular system and subsequent damage to periventricular structures like the hippocampus and white matter (16, 102, 103). Increases in CSF transferrin may thus represent an adaptive physiological mechanism that protects against further injury, however this warrants further investigation.
The hemoglobin-binding scavenger haptoglobin plays an important role in mediating iron recycling and clearance, preventing oxidative damage by sequestering hemoglobin, and facilitating other anti-inflammatory activities in both physiologic and pathologic conditions. Haptoglobin is primarily produced by liver hepatocytes as an approximately 85 kDa multimeric protein with two H chains which can each bind an alpha beta dimer of free extracellular hemoglobin to prevent autoxidation and oxidative tissue damage (132). The haptoglobin-hemoglobin complex is irreversibly stable and is cleared by CD163 receptor-mediated endocytosis followed by intracellular degradation in a variety of cells including macrophages, monocytes, and microglia (133–139). Recent studies in rodents have reported CD163 is upregulated on hippocampal neurons after intracranial hemorrhage (ICH) (140, 141). CSF haptoglobin has also been previously characterized in human neonates (142).
Haptoglobin is present in low quantities in human serum at birth before increasing to adult levels over the first year of life (143, 144). Low levels of haptoglobin synthesis have been identified in the human brain at various stages of fetal development, with the highest haptoglobin expression identified in neurons at 6–8 weeks’ gestation (145). These levels decrease to variable levels of expression at 9–22 weeks’ gestation before rising again at 25–36 weeks’ gestation (145). Compared to neurons, there is less haptoglobin expression within endothelial cells, with variable levels of haptoglobin from 6 to 10 weeks’ gestation that decrease to no expression after 14 weeks’ gestation (145). No haptoglobin was identified in glia at any fetal timepoints evaluated in this study (145).
In a separate study of human fetal brains from 10 to 18 weeks’ gestation, haptoglobin mRNA was not present in brain tissue until 14–18 weeks’ gestation (146). Qualitatively, the authors reported the highest immunopositivity for haptoglobin mRNA at the last timepoint studied (18 weeks) (146). Haptoglobin mRNA levels at postnatal timepoints were lower than those observed during fetal development with regional differences across the basal ganglia, hypothalamus, and cortex (146).
The role of haptoglobin in the context of hemoglobin scavenging after neonatal IVH is still being elucidated. In vivo studies in rabbits have demonstrated that intraventricular injection of haptoglobin attenuates hemoglobin-induced inflammation, cytotoxicity, and structural damage (147). In vitro experiments incubating ChP cells from human neonates with IVH in haptoglobin recapitulate these results (147).
Mahaney et al. previously reported that there are no significant differences in CSF haptoglobin levels between human neonates with and without IVH (all grades) or PHH (19). In conjunction with a study showing haptoglobin expression in the cord blood of premature neonates in response to inflammation is associated with decreased risk of IVH and cerebral palsy (143), these results may suggest that haptoglobin-mediated hemoglobin scavenging mechanisms are exhausted after IVH and that upregulating brain haptoglobin may be a potential target to explore to prevent hemoglobin-mediated neurotoxicity (143). Alternatively, elevated haptoglobin may allow for increased haptoglobin-hemoglobin complex formation and subsequent internalization and degradation into heme and iron in CD163-expressing brain cells, leading to iron overload.
Hemopexin is primarily produced by the liver and then released into the plasma. Analogous to haptoglobin binding of hemoglobin, hemopexin scavenges and binds to heme, a molecule composed of a ferrous iron ion coordinated to a porphyrin ring. Also known as Fe2+ protoporphyrin IX, heme is a precursor to hemoglobin and comprises the non-protein component of hemoglobin. Hemopexin-bound heme is an important co-factor involved in a variety of physiological processes in the brain including neuronal differentiation, growth, and survival (148–152).
Like most iron-containing compounds, heme can also have potentially deleterious effects on surrounding tissues when not bound to hemopexin or other hemoproteins. Free heme can be released from unbound, oxidized hemoglobin in times of haptoglobin saturation and subsequently participate in Fenton reactions. Hemopexin plays a role in blocking heme’s pro-oxidative activity and facilitating cellular heme uptake via CD91/LRP1 (153).
There have been several reports of hemopexin synthesis in neurons and ependyma in the adult human and mouse brain (154–157), however the developmental time-course of hemopexin expression patterns in the parenchyma of the neonatal and postnatal brain is not well-defined. In the human fetal brain, hemopexin protein is expressed in neurons from 6 to 36 weeks’ gestation (145). Hemopexin mRNA has not been identified in the human fetal brain from 10 to 18 weeks’ gestation (146).
Hemopexin has been identified in the neonatal and adult human CSF in physiologic and pathologic states including Alzheimer’s disease (158), diffuse large B cell lymphoma (159), degenerative disk disease (160), SAH, and IVH (19, 161). Mahaney et al. previously reported CSF hemopexin is not elevated after IVH-PHH in human neonates, and CSF hemopexin levels are positively correlated with ceruloplasmin and transferrin levels after IVH-PHH (19). In a separate study in human neonates, Strahle et al. also reported CSF hemopexin is the only iron scavenger that increased between temporary and permanent CSF diversion, and that ventricle size after IVH was inversely correlated with CSF hemopexin levels (20). In the context of previous studies showing the induction of hemopexin expression improves outcomes after ICH in mice (162), and elevated CSF hemopexin is predictive of poor neurological outcomes after SAH in adult humans (163), understanding the role of hemopexin in heme scavenging after IVH represents a pertinent potential therapeutic and diagnostic direction.
Ceruloplasmin is a ferroxidase that oxidizes ferrous iron ions to their ferric form, which is then bound by transferrin. Within the brain, ceruloplasmin is thought to play a critical role in facilitating both cellular iron export (via FPN1) and import to maintain iron homeostasis (164–170). Because ceruloplasmin produced in the liver cannot cross the BBB in significant quantities (171), cells in the adult and neonatal brain, ChP, and retina produce an alternatively spliced form of glycosylphosphatidylinositol (GPI)-anchored ceruloplasmin which comprises the majority of brain endogenous ceruloplasmin (172–174).
In 1988, Møllgård et al. reported ceruloplasmin mRNA expression in the human fetal brain from 14 to 36 weeks’ gestation (145). The protein was identified in neurons starting at 14–18 weeks’ gestation all the way through 36 weeks’ gestation (145). Weak ceruloplasmin expression was also reported in glia from 14 to 22 weeks’ gestation (145).
A more recent study of ceruloplasmin expression in the fetal mouse brain reported GPI-anchored ceruloplasmin protein appeared at embryonic day 12.5 (175), while diffusible ceruloplasmin (defined as ceruloplasmin secreted from the liver and/or released by the GPI anchor) was not detected until embryonic day 17.5 (175). GPI-anchored and diffusible ceruloplasmin levels increased from the time they were initially detected until postnatal day 1, before plateauing at postnatal day 7 and then subsequently decreasing (175). GPI-anchored ceruloplasmin expression on the surface of astrocytes has separately been reported in postnatal day 3 rats (174), however the timeline of postnatal cell-specific and regional expression patterns is not otherwise well-defined.
While low in concentration relative to serum levels, ceruloplasmin has also been reported in fetal and neonatal CSF in humans (6.57 ± 3.53 μg/mL in CSF vs. 20–130 μg/mL in serum) (19, 171, 176). No significant differences in CSF ceruloplasmin concentration over the course of gestation were identified, however sex differences in CSF ceruloplasmin were identified with male fetuses having higher CSF ceruloplasmin concentration than females (171).
Mahaney et al. previously reported no significant differences in CSF ceruloplasmin in humans after neonatal IVH-PHH when compared to control neonates (19), however there were correlations in CSF ceruloplasmin with CSF transferrin and hemopexin. Strahle et al. also reported no significant changes in CSF ceruloplasmin between temporary and permanent CSF diversion in human infants with PHH (20). The role of ceruloplasmin and other ferroxidases in brain iron metabolism throughout development merits further investigation.
Expressed on the apical cell surface, the dimeric transmembrane glycoprotein TFR mediates cellular uptake of ferric iron by binding to its ligand, transferrin, in a pH-dependent and reversible manner (177). The transferrin-TFR complex is internalized by receptor-mediated endocytosis, and subsequent acidification of the endosome causes transferrin to release its ferric iron for transport into the cytosol of the cell (61). Apotransferrin has a high affinity for TFR at low pHs, and they are together recycled back to the plasma membrane via exocytosis.
To facilitate peripheral transferrin-mediated brain iron import, developing BCEC in mice express Tfr from the time they differentiate (178), with the peak in Tfr expression on rat BCECs occurring around postnatal week 2 between postnatal day 10 and postnatal day 21 (179). This coincides with the increase in iron import into the rat brain during the first two postnatal weeks in addition to rapid brain development and growth (180). Tfr expression in BCEC and neurons is also increased in iron deficient conditions in rats (41, 180).
The timeline of Tfr expression on other cells within the brain is distinct from that of BCEC Tfr because while variable low levels of Tfr expression in the neonatal rat ependyma, glia, and cerebral cortex and striatal neurons have been demonstrated as early as postnatal day 5 (181), and Tfr expression has been reported in the rat medial habenula at birth (179), robust and substantive Tfr expression is not seen in until at least postnatal day 15 in rats (181). The peak expression of neuronal Tfr does not occur until postnatal weeks 3–4 after reaching a plateau around postnatal day 21 (179). ChP epithelial cells and hippocampal neurons in rats display virtually no Tfr expression at postnatal days 5 and 10, however become strongly positive by postnatal day 15 (181).
The delayed peak in Tfr expression on rat neurons and glia until at least the third postnatal week has previously been hypothesized to be physiologically linked to the decrease in iron import into the brain from the circulation around the same time and the onset and increase of oligodendrocyte transferrin synthesis during postnatal days 10–25 (118, 179, 180). In conjunction with data showing that iron deficiency enhances brain Tfr expression during all ages (179), it is possible that Tfr expression serves as a compensatory mechanism for neurons and other brain cell types to maintain iron uptake necessary for development and function as overall brain iron levels decrease.
Tfr expression after neonatal GMH-IVH has not been extensively explored and merits further investigation.
DMT1 is a proton-coupled divalent metal ion symporter found in the plasma membrane and endosomes of various cell types in the body (61). In the brain, DMT1 is best known for its role in cellular iron uptake by transporting non-heme ferrous iron from acidified endosomes into the cytosol of neurons after receptor-mediated endocytosis of transferrin from the brain interstitium (182). While there have been several conflicting reports of DMT1 expression on non-neuronal cell types in the brain in vivo (37, 61, 181, 183, 184), DMT1 is now generally accepted to be expressed in low levels in BCEC endosomes to facilitate brain iron uptake across the BBB via receptor-mediated endocytosis of transferrin (182). Dmt1 has also been identified in developing rat ChP epithelial cells and glia including astrocytes and oligodendrocytes (181), however it is likely that there are other non-Dmt1-dependent mechanisms for iron uptake in these cells (61). Dmt1 has also been implicated as a mediator of ferroptosis after brain hemorrhage in rats (185).
First identified in mice in 1995 as the natural resistance-associated macrophage protein 2 (Nramp2) before its functional characterization as the proton-coupled metal-ion transporter divalent cation transporter-1 (Dct-1) in 1997 (37, 186), DMT1 was later understood to have 4 isoforms encoded by the Solute Carrier Family 11-member 2 (SLC11A2) gene that are now known to be differentially expressed across organs and within organelles. Two isoforms have alternative transcripts differing in the 3’ UTR, where one contains an iron response element (Type 1, +IRE) and the other (Type 2, -IRE) does not (187). The third (1A) and fourth (1B) isoforms differ in mRNA processing in the 5′ end, where the 1A transcript starts in Exon 1A, which is upstream of Exon 1B where the 1B isoform transcript starts, and skips over Exon 1B to be spliced directly to Exon 2 (188).
Dmt1 mRNA expression in the neonatal rat brain has been reported as early as postnatal day 3 (189), with the highest regional expression localized to the corpus callosum and on Purkinje cells of cerebellum (190). A separate study identified +IRE mRNA expression and low-level -IRE Dmt1 mRNA expression in the cortex, hippocampus, striatum, and substantia nigra in postnatal week 1 rats (191), which increased in all regions by postnatal week 3 (191). +IRE Dmt1 mRNA subsequently decreased from postnatal weeks 3–28 in the cortex, hippocampus, and striatum, but increased in the substantia nigra (191). Over the same developmental time period, -IRE Dmt1 mRNA increased or stayed relatively constant in the rat hippocampus, striatum, and substantia nigra, but decreased in the cortex (191).
In a study evaluating cellular Dmt1 expression at postnatal days 5, 10, and 15, high levels of Dmt1 expression (isoform unspecified) were identified in blood vessels and ependymal cells at all three timepoints (181). Variable Dmt1 expression was identified in rat ChP epithelial cells from postnatal day 5 to 10 before turning into more robust expression by postnatal day 15 (181, 183). Dmt1 expression in glia followed a similar developmental time course (181, 183). Similarly, neuronal Dmt1 expression was variable at postnatal days 5 and 10 before increasing by postnatal day 15, with slight variations by brain region in the earlier timepoints (183).
Together, these studies indicate low-level Dmt1 expression in the neonatal rat brain that substantially increases over the first 2–3 weeks of rat postnatal development. This timeline mirrors developmental changes in brain iron and transferrin uptake from the circulation, which increase over the first 2–3 postnatal weeks (180), in addition to the peak in TFR expression on brain ECs between postnatal days 10–21 (179).
Ferroportin 1, also known as solute carrier family 40 member 1 (SLC40A1), metal transporter protein 1 (MTP1), and iron-regulated transporter 1 (IREG1), is a transmembrane ferric iron transporter protein and the only known iron transporter responsible for cellular iron export (192). Initially characterized as an iron export protein on the basolateral surface of duodenal enterocytes (193), the basal surface of placental syncytiotrophoblasts (38), and the cytoplasmic compartment of reticuloendothelial cells (96), Fpn1 has more recently been implicated in transporting iron across the abluminal membrane of endothelial cells that comprise the BBB, depositing excess iron into the brain interstitial fluid when ferritin stores are replete (66).
Although the majority of studies of brain Fpn1 localization and function have been performed in adult rodents (63, 194), several studies have reported Fpn1 expression in various cell types across several regions of the developing brain. A 2001 paper by Burdo et al. mentions Fpn1 expression in the embryonic rat central nervous system (195), and a study by Yang et al. describes its expression in BCEC at postnatal day 0 with decreasing expression through postnatal week 8 (196). Fpn1 expression has also been identified in the soma, dendrites, and axons of cortex, striatum, hippocampus, midbrain, brainstem, and cerebellar neurons from postnatal day 7 to 70 (65), with the highest regional expression in the hippocampus (64). Fpn1 levels in the brain generally increase with age, with the lowest expression seen at postnatal week 1 followed by a progressive increase to postnatal week 9 with subsequent declines out to postnatal week 28 (64). Fpn1 has also been identified in postnatal oligodendrocytes and Schwann cells (65, 197).
FPN1 expression has not been studied in the context of neonatal IVH-PHH, however previous investigations have reported decreased Fpn1 expression in the cortex and hippocampus after SAH (198). Injection of hepcidin, a hormone that regulates Fpn1 expression by inducing Fpn1 internalization, further reduced Fpn1 levels after SAH leading to cytotoxic cellular iron overload (198). A separate study demonstrated Fpn1 upregulation in perihematomal brain tissue after intracerebral hemorrhage (199). Experimentally knocking out Fpn1 expression in the striatum with stereotaxic AAV-Cre injection in a Fpn-floxed mouse model significantly worsened iron-related pathology and neurologic outcomes after ICH induction (199). The role of FPN1 in attenuating cellular iron overload after neonatal IVH should explored as a potential therapeutic target.
LRP1, also known as CD91, is a transmembrane heme, hemopexin, and heme-hemopexin complex receptor protein (200). Beyond its roles in mediating heme clearance, LRP1 is also involved in tissue-specific functions including intracellular signaling (201), cell migration (202–204), lipid homeostasis (205–208), and protein scavenging (209). Of LRP1’s CNS-specific functions, two of the most widely studied are BBB regulation and amyloid-beta trafficking clearance (209–211).
Lrp1 expression has previously been reported in mature neurons in the hippocampus, cortex, and cerebellum of the adult rat brain (212, 213), as well as radial glia in the embryonic mouse brain (214). More recently, Lrp1 expression in the CNS was reported to vary by age and cell type (215). In a 2016 study of Lrp1 expression in the embryonic day 13.5 to postnatal day 60 mouse brain, total Lrp1 was reported to peak during postnatal development with stable expression in radial glia, neuroblast, microglia, astrocytes, and neurons through development and adulthood (215). Specifically, Lrp1 was highly expressed in radial glia at embryonic days 13.5–18, astrocytes at postnatal day 5, microglia at embryonic day 13 through postnatal day 60, neuroblasts at embryonic days 13.5–18, and neurons at postnatal day 5-adulthood (215). The proportion of oligodendrocyte precursor cells expressing Lrp1 increases dramatically across embryonic and postnatal brain development, with approximately 69% of oligodendrocyte precursor cells expressing Lrp1 at embryonic day 15.5 and ubiquitous expression in adulthood (215).
While LRP1 expression in the brain is not well-characterized in the context of IVH, Lrp1 upregulation has been reported after ICH (200). Lrp1 has previously been shown to clear heme-hemopexin complexes after ICH (216), and prophylactic intraventricular administration of recombinant human LRP1 protein 20 min before ICH induction in adult mice led to a reduction in hematoma volume, BBB permeability, and other brain injury (216). Lrp1 has also separately been shown to attenuate white matter injury after SAH in rats (217). These results suggest LRP1 has neuroprotective effects, potentially by preventing heme overload and heme-mediated cytotoxicity and deserves further investigation as a therapeutic target after neonatal IVH.
CD163 is a high-affinity scavenger receptor that mediates endocytosis and internalization of the haptoglobin-hemoglobin complex from extracellular spaces including the interstitial fluid (138). While CD163 expression was initially thought to be restricted to macrophages and monocytes, CD163 is expressed in neurons and upregulated after exposure to hemoglobin (102, 140, 218). Soluble CD163 (sCD163) sheds from CD163-positive cells to circulate in the serum and CSF. Subsequent studies have reported that sCD163 scavenges intrathecal hemoglobin-haptoglobin complexes after SAH in adult humans (219).
In addition to its role in scavenging hemoglobin by way of binding and taking up the hemoglobin-haptoglobin complex, CD163 is also involved in anti-inflammatory signaling in macrophages (220). CD163 expression can induce interleukin-10 (IL-10) secretion after binding haptoglobin-hemoglobin complexes containing specific haptoglobin genotypes, which in turn promotes heme oxygenase-1 (HMOX-1) synthesis (221). IL-10 has also been shown to upregulate CD163 expression on monocytes and macrophages (222, 223), while proinflammatory markers like interferon-ɣ are known to decrease expression (223).
Whereas CD163 likely serves an anti-inflammatory role in macrophages, neuronal CD163 may play a deleterious role after ICH and IVH (140, 218, 224). Because hemoglobin is cytotoxic to neurons, which lack key iron sequestering machinery present in macrophages including widespread HMOX-1 induction in response to hemorrhage (106, 225), increased haptoglobin-hemoglobin complex uptake by way of CD163 upregulation post-hemorrhage may lead to iron overload and neuronal cell death.
Consistent with this hypothesis, rat hippocampal neurons expressing CD163 co-localize with phosphorylated-Jun N-terminal kinase (p-JNK) after neonatal IVH (102). As p-JNK is a kinase that plays a key role in the apoptosis cascade, this suggests a mechanism by which CD163 facilitates excessive hemoglobin influx into neurons after IVH to result in cellular ferrous iron overload, oxidative damage, and ultimately cell death (218).
The six-transmembrane epithelial antigen of the prostate (STEAP) family of proteins is comprised of metalloreductases, among which several are ferrireductases that reduce ferric iron to its ferrous state. Of the four identified members of the Steap family (Steap1, Steap2, Steap3, Steap4), Steap2 is arguably best characterized in the brain, where it has been reported in hippocampal neurons and the embryonic mouse ChP (57, 226). At postnatal day 7, Steap2 expression is present in the cerebellar Purkinje cells, and within the superior colliculus, olfactory bulb, and various other anatomic regions in the mouse brain (227). Steap3 was first identified in erythroid precursors where it localizes to endosomes to facilitate transferrin-bound iron uptake (228), however it has also been reported in the lumbar dorsal root ganglion (229, 230). Steap1 and 4 are expressed more ubiquitously, with additional Steap1 upregulation in the prostate and Steap4 enrichment in the bone marrow, placenta, fetal liver, and adipose tissue (226). Within cells, all four Steap proteins partially co-localize with transferrin and TFR, with Steap2 showing the highest degree of co-localization (226).
It is not clear what role the STEAP family of proteins may have in iron homeostasis after intracerebral or intraventricular hemorrhages. STEAP expression and localization in relation to brain iron levels and the specific role of STEAP proteins in the setting of brain iron overload has not yet been evaluated and merits further investigation.
The TIM family of proteins are receptors for H ferritin that mediate ferritin uptake into cells. Tim-2, the rodent ortholog of the human TIM-1 receptor, has been identified on oligodendrocytes as the primary mechanism of oligodendrocyte iron uptake by way of ferritin endocytosis (48, 49, 231, 232). In addition to oligodendrocytes, Tim-2 expression has been identified in neurons, astrocytes, and microvasculature in the mouse brain and is seen at postnatal day 7, 14 and 22 (233).
Ferritin is a major iron storage protein that can be found both intracellularly, where it functions as a ferroxidase to sequester ferrous iron, and extracellularly in the cerebrospinal fluid and serum. Once ferrous iron enters a cell, ferritin binds and uses its di-iron ferroxidase centers to convert it to the ferric form for storage in the ferritin mineral central cavity (234–240). Iron can be subsequently released from ferritin in response to metabolic demand (241).
Ferritin can hold up to 4,500 iron atoms. Early buffer extraction studies showed that about 1/3 of non-heme iron in the brain is stored as ferritin (242). Subsequent reports have estimated that this figure could be as high as 90% (242–244). Therefore, ferritin is a key player in maintaining iron homeostasis by regulating the labile iron pool within the brain.
A developmental study of iron handling proteins in rats showed ferritin levels in the brain peak on postnatal day 2 before decreasing over the course of the first 2 postnatal weeks (113). This downward trend is reversed by postnatal day 17 when brain ferritin levels begin to rise again before stabilizing at high levels similar to those seen at postnatal day 2 by postnatal week 11 (113). 200–500% increases in ferritin levels occur between postnatal day 17 and 2 years, with the most dramatic increases seen in the cortical, pontine, and cerebellar regions (113).
On a cellular level, while one study reported that ferritin mRNA is exclusively found in neurons in the postnatal rat brain, the ferritin protein is in multiple cell types throughout the postnatal brain with differential localization of two functionally distinct ferritin subunits (H and L) across cell types (245). H ferritin, which has redox potential, was identified in oligodendrocytes in postnatal day 21 rats while L ferritin was found in oligodendrocytes, microglia, and astrocytes (245). Neurons express both H and L ferritin (245).
Other studies in adult non-human primates have shown that neurons predominantly express H ferritin (246), and that astrocyte L ferritin expression is primarily localized to the corpus striatum (247). In human fetuses, ferritin has been reported in glia from 6 to 36 weeks’ gestation, with an increase at 19–22 weeks’ gestation (145). More recently, FtMt has been identified in humans as a ferritin that exists only in the mitochondria and whose transcription is not dependent on iron unlike cytosolic ferritins (248). In the context of neurodevelopment, studies in rats have characterized the majority of ferritin-containing cells at postnatal day 5 as microglia, while the majority at postnatal day 30 are oligodendrocytes (247).
In additional to intracellular ferritin within cells in the parenchyma, extracellular CSF ferritin is a valuable indirect measure of brain iron load and has previously been considered as a potential marker for pathology related to Alzheimer’s Disease (249–252), SAH (253), glioblastoma (254), meningitis (255, 256), amyotrophic lateral sclerosis (257), and other disease processes (258).
Specific to IVH-PHH, Strahle et al. recently reported in neonatal humans that longitudinal decreases in CSF ferritin levels between temporary and permanent CSF diversion after PHH are associated with improved scores on cognitive and motor aspects of the Bayley III examination at 2 years of age (20). Larger ventricle size at the time of permanent CSF diversion was also associated with higher levels of CSF ferritin (20). Mahaney et al. also previously showed that elevated CSF ferritin levels were associated with early and severe ventriculomegaly after IVH-PHH (259). These data, in addition to rodent studies showing increases in ferritin positive cells in the hippocampus and periventricular area after IVH, may represent a compensatory increase in ferritin that is reflective of ferritin’s function in preventing iron toxicity in times of overload (260). Specifically, ferritin levels may increase to prevent ferrous iron released into the ventricles after IVH from reacting with H2O2 and producing cytotoxic hydroxyl radicals in the Fenton reaction. Alternatively, elevated CSF ferritin may be secondary to inflammation.
HMOX-1 and HMOX-2 are two of three isoforms of heme oxygenase, an antioxidant enzyme that catalyzes the rate-limiting step of heme degradation. In contrast to the constitutively expressed HMOX-2 that is found in high levels in the brain, HMOX-1 expression in the adult CNS is low at baseline and must be induced by its substrate, heme, and/or a variety of other oxidative stimuli that cause cellular stress (261, 262). Once induced, HMOX-1 degrades heme using NADPH, O2, and cytochrome p450 reductase in a series of three monooxygenation cycles to create the molecule biliverdin in addition to the byproducts carbon monoxide and ferrous iron. Biliverdin can subsequently be converted to bilirubin, a potent antioxidant with anti-inflammatory properties. Ferrous iron can be stored within ferritin, and carbon monoxide can participate in downstream cytoprotective signaling cascades. HMOX-2 degrades heme by the same mechanism.
While robust Hmox-2 expression has been reported in neurons, glia, and blood vessels, Hmox-1 expression in adulthood is limited to hippocampal and olfactory neurons in relatively low levels (263). Using a transgenic Hmox-1-luciferase reporter mouse, Hmox-1 transcription in postnatal day 1 cerebral cortex was shown to be higher than in adult mice (264). Hmox-1 transcription decreases through postnatal days 2–14 before reaching levels similar to those seen in adult mice (264). Similarly, Hmox-1 mRNA and protein levels are highest at postnatal days 1 and 3 before steadily declining over the perinatal time period (264). While Hmox-2 transcription was not been measured in vivo, Hmox-2 mRNA and protein levels in the developing mouse cortex remain relatively constant from embryonic day 14 to postnatal week 6 (264).
As the HMOX-catalyzed heme degradation pathway is one of the only mechanisms for cytotoxic heme removal from cells, HMOX-1 and HMOX-2 likely play essential roles in mediating iron clearance in a variety of iron overload-related pathologies including ICH, IVH in adults after hemorrhagic stroke, and neonatal GMH-IVH. In the context of these diseases, there has been a focus on HMOX-1 as it can be induced by its substrate heme, which is released from hemorrhage (265). Specifically, Hmox-1 is highly expressed in vasculature, microglia, and macrophages adjacent to the area of hemorrhage following ICH induction via collagenase VII-S injection into the caudate putamen in adult mice (266). Iron accumulation in a rat model of adult IVH (identified as T2* MRI hyperintensities) has also been associated with Hmox-1 upregulation (267). Hmox-1 expression in periventricular areas and the hippocampus and cortex is also increased 24 and 72 h after neonatal GMH-IVH in mice (101). In vitro evidence in cells derived from humans, mice, and rats, and in vivo experiments in rodents show that HMOX-1 has regional neuroprotective effects when experimentally upregulated via pharmacologic induction in neurons (268–277), which have physiologically high and ubiquitous Hmox-2 expression but low Hmox-1 expression. Together with HMOX-1’s role in catalyzing heme degradation, these findings may suggest HMOX-1 has a specific function to attenuate heme-related damage after brain hemorrhage and protect against subsequent cellular heme/iron overload.
Hepcidin is a peptide hormone that is primarily produced in the liver. Through binding to and inactivating (via inducing internalization) the cellular iron exporter Fpn1 (278), hepcidin regulates intracellular iron load. While hepcidin synthesis in the brain remains somewhat controversial, studies in adult animals have reported hepcidin protein and/or mRNA expression across the olfactory bulbs, cortex, ChP, corpus callosum, subventricular zone, and hippocampus of rat, mouse, and human brains (279–282).
The role of hepcidin in the neonatal brain is even less well characterized, where developmental changes in hepcidin expression are poorly understood beyond a general increase in hepcidin mRNA from postnatal week 1 to adulthood in the murine cortex, striatum, and hippocampus (281). Strahle et al. previously reported hepcidin in the CSF after neonatal IVH in humans (20), with no changes in CSF hepcidin levels between temporary and permanent CSF diversion for PHH.
IRP-1 and IRP-2 carry out post-transcriptional regulation of cellular iron uptake, storage, and efflux by binding to IREs within the UTRs of mRNAs that code proteins which play crucial roles in brain iron homeostasis. The IREs within the mRNA of the iron exporter FPN1 and the iron storage proteins ferritin H and ferritin L are located in the 5’-UTR, and IRP binding during iron-deficient conditions functions as a post-transcriptional control by inhibiting translation (38, 96, 193, 283–285). TFR and DMT1 mRNA contain IREs within the 3’-UTR (37, 88, 98, 286–288), and IRP binding helps protect the mRNA against endonuclease degradation to promote translation of the proteins to facilitate cellular iron uptake (289, 290). In physiological iron replete conditions, IRPs dissociate from IREs (291, 292), allowing ferritin and Fpn1 translation and TFR and DMT1 mRNA degradation.
Over the course of neurodevelopment, variable Irp-1 and Irp-2 expression has been identified in the neonatal rat brain across ChP epithelia, ependyma, blood vessels, glia, and neurons as early as postnatal day 5 (181), with robust expression of both proteins by postnatal day 15 (181). The percentage of cells with IRP expression varies by anatomic brain region across different ages (181). IRP2 mRNA has been identified in human fetal brain tissue, where it is present in significantly greater quantities than IRP1 mRNA (293).
In the context of neonatal GMH-IVH, Irp2 expression has been reported to decrease 1–5 days after neonatal GMH-IVH induced in postnatal day 7 rodents, an effect that was attenuated by intraperitoneal administration of the iron chelator deferoxamine (294). Irp1 levels did not change in response to GMH-IVH (294).
APP is an integral membrane protein found in various tissues and organs including the brain and spinal cord (295). While it does not directly handle iron, APP helps regulate iron homeostasis in neurons, BCECs, and other brain cells by stabilizing cell surface presentation of the iron exporter FPN1 (296–298). APP and Amyloid-Precursor Like Proteins have additional physiological functions in neurodevelopment, where they are thought to contribute to neurogenesis (299–304), neurite growth (305–314), and synaptogenesis (315–318).
Downstream proteolytic processing of APP where APP is cleaved by α-secretase can produce soluble amyloid precursor protein α (sAPPα) (319, 320). APP can also be cleaved by β-secretase and γ-secretase to produce the Aβ1-42 peptide and sAPPβ (321–325), which is a precursor to soluble amyloid-beta. In humans, CSF sAPP is a potential diagnostic biomarker for diseases like Alzheimer’s Disease and Multiple Sclerosis (316, 320, 326), and as a potential predictor of neurodevelopmental outcomes after neonatal GMH-IVH (327, 328).
APP is expressed in the fetal and perinatal brain at various stages of neurodevelopment. In a study investigating App expression in embryonic day 8.5–13.5 mice, APP was identified in hindbrain and spinal cord motor neurons and cranial ganglia neurons as early as embryonic day 9.5 (299). APP expression increased to embryonic day 10.5, with continued increases in intensity and spread to the last time point studied (299). App mRNA was also identified in these brain regions at embryonic day 13.5, suggesting CNS-endogenous App production. Other studies have shown that App mRNA is expressed in the mouse brain at embryonic day 12, with subsequent increases up to 15-fold before it plateaus at postnatal day 10 (299). App has also been identified in radial glial cells in the fetal and neonatal mouse brain (300).
Specific to neonatal IVH, CSF levels of sAPPα are significantly elevated after IVH-PHH in humans compared to control infants (328, 329), and furthermore CSF APP levels are associated with ventricular size after neonatal IVH (327). As APP is released by axonal injury (330–332), this finding may be related to axonal stretch in periventricular regions due to ventricular distention (327). Alternatively, as it is known that APP is upregulated in the presence of free iron (333), and that iron plays a key role in the pathogenesis of PHH after IVH (101), it is possible that APP reflects high brain iron levels after IVH in neonates that develop the most severe ventriculomegaly.
Beyond DMT1, TFR, and FPN1, the localization and developmental expression of few if any cellular iron handling proteins known to be important in iron handling in the adult brain and other tissues outside of the CNS have been reported in the neonatal brain. This section discusses the role of several additional iron handling proteins in peripheral tissues in the neonate, reviews their function in the adult brain, and proposes potential roles they may play in the neonatal brain. It is important to consider additional iron transporters in the neonatal brain as they may play key, therapeutically targetable roles in iron handling after GMH-IVH (Figure 2).
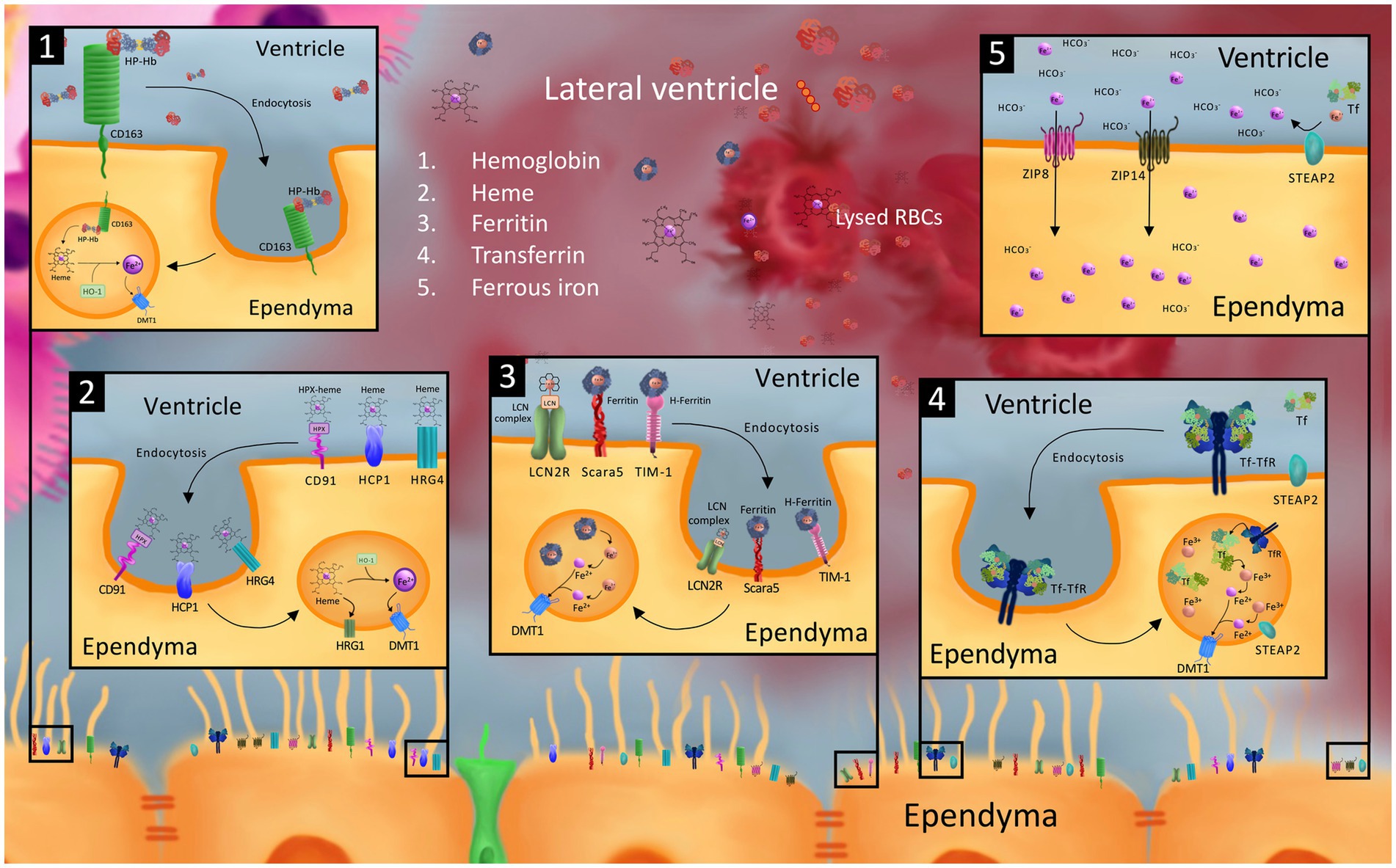
Figure 2. Model of possible cellular iron transporters that may be involved in iron uptake into the ependyma and choroid plexus (ChP) from the CSF after germinal matrix hemorrhage-intraventricular hemorrhage (GMH-IVH). While the exact mechanism of ependymal and choroid plexus uptake of CSF blood and blood breakdown products after GMH-IVH is unknown, it is possible that cellular iron transporters involved in blood breakdown product and iron uptake in other cells of the neonatal and adult brain, neonatal peripheral tissue, and other organs may be involved. Specifically, we model ependymal uptake of hemoglobin-haptoglobin (Hb-Hp) from the CSF via the CD163 receptor (1); heme-hemopexin (hpx) and heme via CD91, heme carrier protein 1 (HCP1), and heme responsive genes 4 and 1 (2); ferritin and Fe3 + −containing lipocalin 2 (LCN) complex via scavenger receptor class A member 5 (Scara5), T cell immunoglobulin and mucin domain containing protein 1 (TIM-1), and LCN2 receptor (LCN2R) (3); transferrin (Tf) via transferrin receptor (TfR) (4); and ferrous iron ions (Fe2+) via the zinc regulated transporter and iron regulated transporter like proteins 8 and 14 (ZIP8 and ZIP14) (5). Intracellular processing in the endosomal compartment to convert receptor-mediated endocytosed blood breakdown product compounds into Fe2+ ions for export into the cytosol via divalent metal transporter 1 (DMT1) and six-transmembrane epithelial antigen of the prostate protein 2 (STEAP2) converting Fe3+ ions into Fe2+ ions are also shown. To our knowledge, the majority of these transporters (including but not limited to ZIP8, ZIP14, Scara5, TIM-1, LCN2R, CD163, CD91, HCP1, HRG4) have not been identified on the ependyma, and are only modeled in this figure as playing a hypothetical role in transporting their corresponding blood breakdown/iron product to guide future investigations.
Heme Responsive Gene 1 (HRG1), also known as solute carrier family 48 member 1 (SLC48A1), is a transmembrane receptor involved in mediating cellular heme homeostasis. Initially identified as an Hrg4 paralog in the C. elegans genome (334), subsequent experiments in zebrafish demonstrated Hrg1 mRNA expression throughout the embryonic CNS (334). Functional experiments revealed that Hrg1 has a role in neurodevelopment and erythropoiesis in the zebrafish embryo (334). Notably, Hrg1 knockdown in zebrafish embryos using antisense morpholinos induced hydrocephalus and yolk tube malformations (334), however the mechanism by which this happens is not clear. Unlike Hrg4 which transports heme from the extracellular environment into the cell, Hrg1 specifically functions to transport endocytosed heme from endosomal compartments into the cytosol (334, 335). In vitro experiments in mouse macrophages undergoing erythrophagocytosis show that Hrg1 transports heme from the phagolysosome out to the cytosol (336, 337).
In adult mammals, HRG1 expression has been identified in brain, kidney, heart, skeletal muscle tissue, liver, lung, placenta, and small intestine in lower quantities (334). Hrg1 protein expression has also been reported on the apical surface of murine renal cortex epithelial cells following neonatal hemolysis (335), and the apical membrane of porcine duodenal enterocytes in piglets fed hemoglobin-enriched diets (338).
Neither the role of HRG1 in the adult brain nor the specific patterns of HRG1 expression in the neonatal brain are well understood. The potential roles of HRG1 in heme transport in the neonatal brain, particularly in connection with cellular heme and iron overload after IVH, merits further investigation alongside other proteins in the HRG family including HRG-3 (339).
Like HRG1, heme carrier protein 1 (HCP1) is a transmembrane protein that transports heme in addition to folate and heme-hemopexin complexes. Also known as solute carrier family 46 member 1 (SLC46A1) and proton-coupled folate transporter (PCFT), Hcp1 was initially characterized on the brush-border membrane of murine duodenal enterocytes where it mediates heme uptake and intestinal heme transport (340). Subsequent studies have reported HCP1 protein and/or mRNA expression on human macrophages and cultured astrocytes (341, 342), and within the human retina and retinal pigment epithelium, and mouse duodenum, liver, and kidney (340, 343).
Within the CNS, variable levels of Hcp1 expression have been identified in the cortex and hippocampus of adult rats (344), and in vitro experiments demonstrated neuronal heme uptake via Hcp1 (344). HCP1 is also expressed on the basolateral surface of the neonatal and adult ChP as a proton symporter that mediates folate transport into the CSF (345). The role of ChP HCP1 in heme transport is not as well-explored.
Lipocalin 2 (LCN2) is an inducible protein secreted by a variety of cell types in the brain, liver, and uterus that mediates intercellular communication, innate immune responses to bacterial infections, and iron homeostasis. Specific to iron, LCN2 mediates transferrin-independent iron delivery and removal from cells by way of sequestering sidephores (346, 347), iron chelators that scavenge ferrous iron with high affinity and specificity. While LCN2 is not well-characterized in the neonatal brain, LCN2 secretion from neurons and glia has previously been reported to be inducible in vitro by exposure to amyloid-beta (348), and in vivo by hemoglobin (349), kainic acid (350), and other compounds. LCN2 is also known as neutrophil gelatinase-associated lipocalin (NGAL), 24p3, and p25, and more archaically siderocalin and uterocalin.
Lcn2 signaling in the adult brain can be upregulated by CNS pathology including spinal cord injury, autoimmune encephalomyelitis, and ICH, and is generally considered to have a role in mediating downstream neurotoxic effects (351–357). Lcn2 is expressed in astrocytes and ECs in the adult mouse brain after middle cerebral artery occlusion in a murine stroke model (358), while the Lcn2 receptor is expressed in neurons, astrocytes, and endothelial cells (358). Similarly, Lcn2 is involved in ischemic stroke reperfusion injury (358, 359) and increases in brain Lcn2 levels after stroke play a role in mediating subsequent brain injury by activating astrocytes (360). LCN2 is also upregulated after traumatic brain injury and has been shown to be a chemokine inducer in the murine adult CNS both in vitro and in vivo (361, 362).
In a mouse model of adult IVH, intraventricular hemoglobin induced Lcn2 upregulation and ventriculomegaly (349) where Lcn2-deficient mice were protected against hemoglobin-induced ventricular dilation, glial activation, and mortality (349). This finding mirrors a report of reduced white matter damage and decreased BBB disruption after SAH in Lcn2-deficient mice compared to wild-type controls (363). Lcn2 causes morphological changes in neuronal dendritic spines to decrease spine density and promote spine elimination in stress conditions (364), supporting its role in responding to adverse and/or noxious stimuli to result in inflammatory or neurologic injury.
LCN2 has been studied in peripheral tissues in developing and neonatal organisms. In vitro Lcn2 expression in embryonic day 13 ureteric bud cells derived from the developing rat kidney was reported to induce mesenchymal cell differentiation into epithelial cells (365). Lcn2 also functions as an iron delivery protein to transport and deliver iron to kidney epithelial progenitors and stroma cells during embryonic organogenesis in vitro (365). In humans, serum, urine, stool, and umbilical cord levels of LRP2 have been used as diagnostic and predictive markers for renal impairment/injury (366–372), necrotizing enterocolitis (373), sepsis (374), and other conditions in preterm neonates with acute and chronic pathologies. While LCN2 has not been reported in the neonatal brain, the Lcn2 receptor megalin is expressed in the neural tube and developing rat and mouse brain where it plays a role in neurodevelopment (375, 376).
Scavenger receptor class A member 5 (SCARA5) belongs to a class of membrane receptors and can recognize and bind a variety of substrates including serum ferritin to mediate ferritin-bound non-transferrin iron uptake and delivery (377, 378). SCARA5 recognizes both H- and L-ferritin in a Ca2+-dependent manner and is expressed in the adult human and mouse retina (379, 380), the developing mouse kidney (377), and epithelial cells in the murine testis, bladder, trachea, adrenal glands, skin, lung, brain, and ovary (378). In vitro experiments in cells derived from human spleen report SCARA5 directly binds to and mediates the intracellular internalization of Von Willebrand Factor, a large multimer that plays an essential role in clotting and hemostasis (381).
While there have been few reports investigating cellular SCARA5 localization in the central nervous system, experiments from an in vitro model of the BBB show SCARA5 in brain endothelial cells mediates substrate uptake into brain endothelial cells to cross the BBB (382). Similar results have been reported in vivo in mice with Scara5-mediated ferritin uptake across the blood-retina barrier (379). Low-level Scara5 expression has been reported on cultured astrocytes derived from neonatal mouse brains (383). In adult humans, SCARA5 is highly enriched in the ChP, with additional expression in the cerebral cortex, basal ganglia, cerebellum, and spinal cord (384).
In a study using Mendelian randomization to identify biomarkers for stroke, SCARA5 levels were associated with a decreased risk of SAH, potentially implicating SCARA5 in baseline risk for hemorrhage (385). SCARA5 also had consistent but non-significant effects on ICH risk (385). It is not clear whether this effect is mediated by SCARA5’s interactions with ferritin, or alternatively its role in binding VWF. SCARA5 expression in the brain has not been investigated after adult or neonatal IVH.
Hypoxia-inducible factor (HIF) is a heterodimeric transcription factor which mediates the adaptive homeostatic response to hypoxia (386, 387). HIF has been studied as a link between iron homeostasis and erythropoiesis (388, 389). HIF binds to hypoxia-responsive elements (HREs) in the regulatory regions of target genes including genes crucial to iron homeostasis like Tfr (390), transferrin (391), ceruloplasmin (392), and Hmox-1 (393).
While the role of HIF in pathologic conditions in the neonatal brain like hypoxic–ischemic injury are well-studied, its role in neonatal brain iron homeostasis in physiologic conditions is not well-explored. HIF subunits and their transcriptional pathways are essential regulators of iron homeostasis in the intestine (389, 394–396). In vivo studies in mice have reported intestinal Dmt1 and Fpn1 are direct transcriptional targets of HIF-2α (394–397), and disrupting Hif-2α signaling in the intestine leads to impaired iron absorption (394). As Hif-1α, Hif-2α, and Hif-1β are expressed in the developing brain and are crucial for healthy brain development (398, 399), HIF may be a targetable aspect of brain iron homeostasis to prevent iron overload after neonatal IVH.
Zinc regulated and Iron regulated transporter-like proteins (ZIP) are a family of metal ion transporters encoded by the SLC39 gene that imports divalent metal ions including Zn2+, Fe2+, and Mn2+ into the cytoplasmic compartment of cells. ZIP8 and ZIP14 are closely related and are the most-studied ZIP proteins in the context of iron trafficking. Both proteins have been identified in vitro on human-derived BCECs that constitute the BBB and hippocampal neurons (57, 400), and Zip8 and Zip14 have separately been shown to mediate cellular NTBI uptake (57, 401, 402). Zip14 may also have additional roles in iron acquisition from endocytosed transferrin in vitro (403). Additional in vitro studies report Zip8 is localized at the cell surface of the neuronal soma and dendrites, where it co-localizes with Steap2 and Tfr, while Zip14 concentrates within the cytosol and nucleus (57). Of note, Zip8 expression has been identified in mouse neural progenitor cells both in vitro and in vivo at E14, with expression significantly decreasing after differentiation (404).
While ZIP-mediated transport of zinc and several other divalent metal ions is known to play a crucial role in healthy development and growth (405, 406), ZIP8 and ZIP14’s roles in iron transport in the neonatal brain are relatively underexplored. Likewise, while a ZIP8 missense variant in humans has been associated with cerebrovascular disease and ICH (407), ZIP8 and ZIP14 expression and localization in the CNS after ICH, SAH, and/or IVH is not well-understood. As Zip8 expression has been shown to be upregulated in cell iron overload in the retina, and knock down of Zip14 is able to prevent iron overload in hepatocytes in a mouse model of hereditary hemochromatosis (408), elucidating ZIP8 and ZIP14 expression patterns (1) in the developing brain, and (2) after hemorrhage represent potential next steps toward understanding ZIP8 and ZIP14 as potential therapeutic targets to prevent brain iron overload after neonatal IVH.
When physiologic mechanisms of iron transport and regulation fail to keep up with accumulating iron levels in the brain after IVH, ferrous iron may react with hydrogen peroxide to generate free radical oxygen species via the Fenton reaction. Hydrogen peroxide is widely available as a small-molecule messenger in the brain, with the mitochondria serving as the major intracellular site of hydrogen peroxide production (409, 410). An imbalance in ferrous iron levels can thus result in rapid and fulminant production of reactive oxygen species that overwhelm the brain’s antioxidant capabilities, leading to oxidative stress.
There are several mechanisms by which oxidative stress is hypothesized to lead to cellular toxicity. Reactive oxygen species can react with a variety of biological macromolecules including lipids, proteins, nucleic acids, and carbohydrates (411). The polyunsaturated fatty acyl side chains in polyunsaturated fatty acids which make up cellular membranes are particularly susceptible to damage via lipid peroxidation (412, 413). This process can disrupt cell and organelle integrity directly via peroxidation of the inner mitochondrial membrane phospholipid cardiolipin (412, 414–416), and indirectly by producing signaling molecules (i.e. those in the NF- κB, mitogen-activated protein kinase, and protein kinase C signaling pathways) capable of inducing both intrinsic and extrinsic apoptotic pathways (415, 417, 418). Lipid peroxidation can also drive ferroptosis, a recently identified mechanism of iron-dependent oxidative stress which leads to non-apoptotic programmed cell death (419), however the precise mechanism by which lipid peroxidation and ferroptosis are connected is not well understood (420). Ferroptosis induction in the ChP has recently been identified as a potential mechanism of cell death after PHH (421, 422), and iron chelators like deferoxamine (DFX) are thought to inhibit lipid peroxidation (415, 423).
In addition to oxidative stress, IVH also induces neuroinflammation and innate neuroimmune activation (424, 425). A recent study using a rat model of IVH induced via intraventricular hemoglobin injection demonstrated acute increases in brain-wide cytokine production and microglia reactivity followed by localized oxidative stress in the white matter (424). Acute ChP and lateral ventricle ependyma inflammation via NF- κB activation has also been reported in rats in response to intraventricular autologous blood (426), as well as inflammation mediated by the TLR4-dependent cytokine TNF-α (147, 427–429). Autologous blood injected into the ventricles of adult mice has also been shown to increase cytokine secretion at the ChP-CSF interface mediated by activation of ChP-associated macrophages (81). Unlike reactive oxygen species generation via the Fenton reaction and oxidative stress, the mechanisms of neuroinflammation after IVH have not specifically been linked to iron overload. While hemoglobin and iron released into the CSF after IVH can increase macrophage and resident microglia activation to facilitate HMOX-1-mediated heme degradation, thrombin, periredoxin 2, methemoglobin, and other blood and blood breakdown product components represent additional candidates (427, 428, 430–433).
Approximately 30% of infants with high grade (grade III or IV) IVH develop PHH (14), an imbalance in the production and efflux of CSF resulting in symptomatic ventriculomegaly. While it is known that blood within the ventricles and brain is the primary risk factor for PHH after IVH, the specific etiology of PHH after IVH is unclear with various potential mechanisms (iron-mediated toxicity, impaired CSF dynamics, inflammation, CSF hypersecretion, ependymal denudation etc.) (12, 81, 424, 434–436).
Strahle et al. previously reported that intraventricular injection of hemoglobin and iron, but not the iron-deficient heme precursor Protoporphyrin IX, results in ventriculomegaly 24 h post-injection in rats (101). Clinical studies have reported elevated CSF non-protein-bound iron in preterm infants with posthemorrhagic ventricular dilation compared to control infants (16). Iron chelation with both peripheral and intraventricular deferoxamine has been shown to reduce ventriculomegaly after IVH in rats (101, 437–440). Together, these results suggest iron is linked to PHH pathogenesis, however the mechanism by which the two are connected is not clear.
One mechanism by which free iron in the CSF and brain parenchyma may lead to PHH is via its cytotoxic effects on specific structures that play key roles in maintaining homeostasis between CSF production and drainage. In one theory of PHH pathogenesis, iron-mediated ependymal and cilia dysfunction is hypothesized to hinder normal CSF circulation and lead to CSF accumulation. E1 ependymal cells, which are one of three subtypes of ependymal cells (E1–E3), line the ventricles and have motile cilia on their apical surface that are damaged and sloughed off by blood breakdown product release into the CSF (441–444). Primary cilium on E2 and E3 ependymal cells, which play a sensory role in detecting chemical and mechanical changes in the CSF (441, 445–447), may also be damaged by iron. The resultant cilia loss and ependymal denudation may (1) expose underlying cells to blood and CSF leading to edema and gliosis of subependymal tissue, (2) impair ventricle wall ependyma-ChP communication and feedback, and (3) alter the velocity, turbulence, and/or vorticity of CSF flow patterns (448). In combination, these effects may contribute to the onset of hydrocephalus by disrupting CSF homeostasis by way of altering CSF flow, however this hypothesis has recently been questioned in light of data suggesting cilia are not the primary drivers of CSF movement in the ventricular system in humans (434, 449–452).
Aberrant CSF production in response to iron-mediated ChP damage and ChP inflammation has been posited as an alternative mechanism of PHH. In a rodent model of neonatal IVH, intraventricular blood was reported to induce early ChP epithelial cell activation and transient increases in ChP Na+/K+/2Cl− co transporter (NKCC1) expression and phosphorylation (436), both which have been linked to subsequent hydrocephalus development and/or increases in PHH severity (436, 453). A separate study also reported ChP transporter activation after IVH and the resultant CSF hypersecretion contribute to PHH onset (454), however it is not clear if this is a direct effect of iron cytotoxicity or if these effects are secondary to inflammation (454–456). Recently, a mechanism by which autologous blood may induce a TLR4-mediated inflammatory response at the ChP has been proposed (81), serving as a potential link between blood breakdown product release into the CSF in IVH, ChP inflammation, CSF hypersecretion, and the onset of PHH.
Other proposed mechanisms of PHH development implicate the complement cascade (457, 458); aquaporins 1, 4, and 5 upregulation (437, 459, 460); CSF pathway obstruction by blood breakdown products, blood clots, debris, or fibrosis secondary to inflammation (12, 461–463); and dysfunction of the blood–brain and blood-CSF barriers (464). Altogether, these proposed mechanisms fall under two major categories – CSF underdrainage and CSF overproduction (465). While it is not entirely clear how iron may be directly linked to either under- or overdrainage, it is clear that intraventricular iron is necessary, but not sufficient, to induce hydrocephalus after IVH. Other genetic factors may underlie development of hydrocephalus in response to IVH (466). Therefore, it will be important to consider how iron may contribute to both CSF under resorption and hypersecretion in tandem to best uncover the mechanism(s) by which iron may be linked to PHH onset.
Iron and blood-breakdown products have been implicated in the pathogenesis of not only PHH, but also direct injury to the brain. Intraventricular injection of hemoglobin in neonatal rats has previously been linked to neuronal degeneration in the hippocampus via the c-Jun N-terminal kinase (JNK) pathway (102). Exposure to intraventricular hemoglobin resulted in significant decreases in hippocampal volume, a finding which was reversable with iron chelation with deferoxamine (102). A separate study conducted in human neonates reported that very preterm infants with high-grade IVH and PHH had the smallest hippocampal volumes and the worst neurodevelopmental outcomes at 2 years of age compared to full-term infants and very preterm infants without brain injury (103). In conjunction with previous studies which demonstrated associations between smaller hippocampal size and worse cognitive and motor outcomes in humans (467, 468), these results together suggest that iron overload and toxicity after IVH may lead to hippocampal neuronal degeneration, which in turn leads to impaired neuromotor developmental outcomes. Furthermore, to directly relate intraventricular blood breakdown product levels with behavioral outcomes, intraventricular hemoglobin injection in rodents has been directly associated with an impaired acoustic startle response (437), a behavioral impairment which was prevented when rodents were treated with intraventricular deferoxamine at the time of IVH (437). Future studies should investigate the precise mechanisms by which iron and blood breakdown product exposure may lead to hippocampal neuron toxicity (ie. oxidative stress, ferroptosis) to identify targetable mechanisms for prophylactic treatment and prevention of IVH-induced brain injury and resultant neurodevelopmental effects.
Despite the devastating long-term neurodevelopmental consequences and mortality risk of PHH, there are currently no curative or preventative solutions available. Up to 34% of very low birth weight infants with persistent ventricular dilation after IVH require surgical intervention for CSF diversion with a shunt (469). While CSF diversion remains the gold standard treatment for hydrocephalus, it is associated with short and long-term complications including shunt failure rates of 33% with a mean time to failure of 344 days (470). Endoscopic third ventriculostomy (ETV) with or withhout ChP cauterization (CPC) is an alternative treatment option however it does not have high success rates for the treatment of PHH (471–474).
In line with the hypothesis that iron plays a role in PHH pathogenesis, recent pre-clinical studies have investigated iron chelators as a possible treatment to prevent PHH after adult and neonatal IVH (101, 104, 437–440, 475). DFX is an iron chelator that sequesters free iron and has been used in the clinical setting to treat hemochromatosis (476, 477), thalassemia (478–480), and other iron-overload syndromes. Previous research in an adult rat model of IVH-PHH reported that consecutive intraperitoneal deferoxamine administered in 12-h intervals over 7 days starting 3 h post-IVH decreased CSF iron and brain ferritin levels and reduced the incidence of ventriculomegaly 7 and 28 days after the start of DFX treatment (438). Gao et al. also demonstrated intraventricular co-injection of deferoxamine with lysed RBCs markedly reduced ventricular enlargement compared to rats injected with lysed blood cells in a rat model of adult IVH (475). In neonatal rodents, intraventricular administration of deferoxamine at the time of hemorrhage in a neonatal rat model of hemoglobin-induced IVH delays the onset of PHH for 11 days and prevents long-term behavioral deficits (437). In addition, intraperitoneal administration of deferoxamine 2 h after IVH followed by bis in die intraperitoneal administration over 24 h reduced hemoglobin-induced ventricular enlargement (101). These findings have been replicated where deferoxamine reduced ventriculomegaly, improved motor and cognitive function, and prevented other neurological sequelae in a rodent model of neonatal GMH-IVH (439).
In addition to deferoxamine, the tetracycline antibiotic minocycline has also been shown to inhibit ferritin upregulation, reduce edema, and prevent hydrocephalus after hemorrhage induction in a rodent collagenase model of GMH by way of chelating iron to reduce brain iron overload (104). Hemoglobin sequestration compounds including haptoglobin and α1-microglobulin have also been explored as neuroprotective treatment options to attenuate the neurotoxic and inflammatory consequences of excess hemoglobin released in IVH (481, 482). Intraventricular administration of erythropoietin and melatonin, hormones with roles in the survival and maturation of developing neural cells and in reducing neuroinflammation and oxidative stress, has also been shown to preserve neonatal neurodevelopment and prevent ventriculomegaly in a rat model of neonatal chorioamnionitis-IVH (442). The efficacy of melatonin and erythropoietin in combination to prevent PHH after IVH is also currently being evaluated in a clinical trial.
Beyond ongoing research into the efficacy of these and other therapeutic strategies for preventing PHH after IVH, other potential treatment options that address additional aspects of IVH-PHH pathophysiology and warrant further investigation include experimentally up/down regulating iron transporters on ependyma and other brain cells, adapting drugs used clinically for other disorders causing iron overload, and combining these strategies with techniques that improve the delivery of drugs to the brain.
Specifically, one of the cellular iron transporters we discussed in this manuscript, FPN1, may represent a particularly intriguing candidate for experimental up- or downregulation after IVH as the only known non-heme iron exporter that has been identified in mammals. In adult Lewis and LEWzizi rats, Fpn1 is localized to the apical surface of ChP epithelial cells and the basolateral surface of ependymal cells (483). FPN1 expression patterns along the ventricular system of neonates are not as clear and should be characterized for use as a potential target against iron overload in the ependyma, ChP, and/or brain parenchyma.
In addition to targeting the expression of iron transporters to prevent CSF iron from accumulating in cells and resulting in cellular iron overload, facilitating iron removal from the CSF with drugs used clinically for systemic iron overload (ie. previously studied examples of DFX and minocycline) represents an alternative therapeutic avenue. Beyond DFX and minocycline, adapting drugs which have been shown to reverse hemochromatosis and brain siderosis may present alternative options for iron chelation after IVH. For example, Hudson et al and Hale et al previously showed in mice that whole animal and intestine-specific genetic knockout of the lithium-sensitive enzyme bisphosphate 3′-nucleotidase (Bpnt1) enzyme leads to iron deficiency anemia and can rescue hemochromatosis (484, 485). Lithium may thus have potential to reduce iron levels after IVH and can be further investigated as a therapeutic agent; however, lithium may have off target effects.
Finally, the BBB presents a major challenge to brain iron chelation that is not present in treatment of systemic disorders of iron overload. However the BBB, in conjunction with the blood-CSF barrier, can be targeted with techniques that improve the delivery of drugs to the brain. Combining drug candidates with nanoparticles to circumvent the BBB (486–488), using focused ultrasound to open localized regions of the BBB (489–495), and other evolving drug-delivery strategies all represent solutions that may improve the efficacy, efficiency, and breadth of future investigations into the potential therapeutic avenues we describe in this manuscript and should be taken advantage of going forward.
Maintaining the delicate balance in brain iron levels is vital for healthy brain development and is facilitated by a variety of iron handling proteins with specialized roles in brain and cellular iron import and export. GMH-IVH results in major disruptions to brain iron homeostasis during the neonatal time period through release of blood and blood breakdown products into the ventricular system. IVH is thought to be linked to PHH via iron overload-mediated pathogenesis, however the specific mechanisms by which this occurs are not clear. Determining the iron-mediated mechanisms by which IVH results in PHH is complicated due to a relative paucity of understanding for the function of baseline iron transport proteins in the developing brain. Identifying additional iron transport proteins in the neonatal brain and investigating their role in iron handling after neonatal IVH may lead to additional treatment strategies to address and prevent iron-mediated neurological sequelae of GMH-IVH including PHH.
SP: Writing – original draft, Writing – review & editing. AH: Writing – review & editing. ML: Writing – original draft. DR: Writing – original draft. TG: Writing – original draft. BS: Writing – original draft. KM: Writing – original draft. JS: Conceptualization, Writing – original draft, Writing – review & editing.
This work was funded by the National Institutes of Health (R01 NS110793 to JS), Rudy Schulte Research Institute (JS), K12 Neurosurgeon Research Career Development Program (JS), and the Hydrocephalus Association (JS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JW declared a shared parent affiliation with the author TG to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1287559/full#supplementary-material
1. Yu, GSM, Steinkirchner, TM, Rao, GA, and Larkin, EC. Effect of prenatal iron deficiency on myelination in rat pups. Am J Pathol. (1986) 125:620–4.
2. Larkin, EC, and Rao, GA. Importance of fetal and neonatal Iron: adequacy for Normal development of central nervous system. Brain Behav Iron Infant Diet. (1990):43–57.
3. DeUngria, M, Rao, R, Wobken, JD, Luciana, M, Nelson, CA, and Georgieff, MK. Perinatal iron deficiency decreases cytochrome c oxidase (CytOx) activity in selected regions of neonatal rat brain. Pediatr Res. (2000) 48:169–76. doi: 10.1203/00006450-200008000-00009
4. Lavich, IC, de Freitas, BS, Kist, LW, Falavigna, L, Dargél, VA, Köbe, LM, et al. Sulforaphane rescues memory dysfunction and synaptic and mitochondrial alterations induced by brain iron accumulation. Neuroscience. (2015) 301:542–52. doi: 10.1016/j.neuroscience.2015.06.025
5. Li, K, and Reichmann, H. Role of iron in neurodegenerative diseases. J Neural Transm. (2016) 123:389–99. doi: 10.1007/s00702-016-1508-7
6. Rosas, HD, Chen, YI, Doros, G, Salat, DH, Chen, N, Kwong, KK, et al. Alterations in brain transition metals in Huntington disease. Arch Neurol. (2012) 69:887–93. doi: 10.1001/archneurol.2011.2945
7. He, N, Ling, H, Ding, B, Huang, J, Zhang, Y, Zhang, Z, et al. Region-specific disturbed iron distribution in early idiopathic Parkinson’s disease measured by quantitative susceptibility mapping. Hum Brain Mapp. (2015) 36:4407–20. doi: 10.1002/hbm.22928
8. Raven, EP, Lu, PH, Tishler, TA, Heydari, P, and Bartzokis, G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis. (2013) 37:127–36. doi: 10.3233/JAD-130209
9. Budni, P, De Lima, MNM, Polydoro, M, Moreira, JCF, Schroder, N, and Dal-Pizzol, F. Antioxidant effects of selegiline in oxidative stress induced by iron neonatal treatment in rats. Neurochem Res. (2007) 32:965–72. doi: 10.1007/s11064-006-9249-x
10. Nielsen, JE, Jensen, LN, and Krabbe, K. Hereditary haemochromatosis: a case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J Neurol Neurosurg Psychiatry. (1995) 59:318–21. doi: 10.1136/jnnp.59.3.318
11. Merhar, SL, Tabangin, ME, Meinzen-Derr, J, and Schibler, KR. Grade and laterality of intraventricular haemorrhage to predict 18-22 month neurodevelopmental outcomes in extremely low birthweight infants. Acta Paediatr. (2012) 101:414–8. doi: 10.1111/j.1651-2227.2011.02584.x
12. Strahle, J, Garton, HJL, Maher, CO, Muraszko, KM, Keep, RF, and Xi, G. Mechanisms of hydrocephalus after neonatal and adult intraventricular hemorrhage. Transl Stroke Res. (2012) 3:25–38. doi: 10.1007/s12975-012-0182-9
13. Christian, EA, Melamed, EF, Peck, E, Krieger, MD, and McComb, JG. Surgical management of hydrocephalus secondary to intraventricular hemorrhage in the preterm infant. J Neurosurg Pediatr. (2016) 17:278–84. doi: 10.3171/2015.6.PEDS15132
14. Christian, EA, Jin, DL, Attenello, F, Wen, T, Cen, S, Mack, WJ, et al. Trends in hospitalization of preterm infants with intraventricular hemorrhage and hydrocephalus in the United States, 2000-2010. J Neurosurg Pediatr. (2016) 17:260–9. doi: 10.3171/2015.7.PEDS15140
15. Abbaspour, N, Hurrell, R, and Kelishadi, R. Review on iron and its importance for human health. J Res Med Sci. (2014) 19:164–74.
16. Savman, K, Nilsson, UA, Blennow, M, Kjellmer, I, and Whitelaw, A. Non-protein-bound iron is elevated in cerebrospinal fluid from preterm infants with posthemorrhagic ventricular dilatation. Pediatr Res. (2001) 49:208–12. doi: 10.1203/00006450-200102000-00013
17. Duck, KA, and Connor, JR. Iron uptake and transport across physiological barriers. Biometals. (2016) 29:573–91. doi: 10.1007/s10534-016-9952-2
18. Knörle, R . Neuromelanin in Parkinson’s disease: from Fenton reaction to calcium signaling. Neurotox Res. (2018) 33:515–22. doi: 10.1007/s12640-017-9804-z
19. Mahaney, KB, Buddhala, C, Paturu, M, Morales, D, Limbrick, DD, and Strahle, JM. Intraventricular hemorrhage clearance in human neonatal cerebrospinal fluid: associations with hydrocephalus. Stroke. (2020) 51:1712–9. doi: 10.1161/STROKEAHA.119.028744
20. Strahle, JM, Mahaney, KB, Morales, DM, Buddhala, C, Shannon, CN, Wellons, JC, et al. Longitudinal CSF Iron pathway proteins in posthemorrhagic hydrocephalus: associations with ventricle size and neurodevelopmental outcomes. Ann Neurol. (2021) 90:217–26. doi: 10.1002/ana.26133
21. Gosriwatana, I, Loreal, O, Lu, S, Brissot, P, Porter, J, and Hider, RC. Quantification of non-transferrin-bound iron in the presence of unsaturated transferrin. Anal Biochem. (1999) 273:212–20.
22. Patel, M, and Ramavataram, DVSS. Non transferrin bound iron: nature, manifestations and analytical approaches for estimation. Indian J Clin Biochem. (2012) 27:322–32. doi: 10.1007/s12291-012-0250-7
23. Anderson, GJ . Non-transferrin-bound iron and cellular toxicity: editorial. J Gastroenterol Hepatol. (1999) 14:105–8. doi: 10.1046/j.1440-1746.1999.01828.x
24. Ogun, AS, Joy, NV, and Valentine, M. Biochemistry, Heme Synthesis. Treasure Island, FL: StatPearls (2023).
25. Ems, T, Lucia, KS, and Huecker, MR. Biochemistry, Iron Absorption. Treasure Island, FL: StatPearls (2021).
26. Levi, S, Corsi, B, Bosisio, M, Invernizzi, R, Volz, A, Sanford, D, et al. A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem. (2001) 276:24437–40. doi: 10.1074/jbc.C100141200
27. Santambrogio, P, Biasiotto, G, Sanvito, F, Olivieri, S, Arosio, P, and Levi, S. Mitochondrial ferritin expression in adult mouse tissues. J Histochem Cytochem. (2007) 55:1129–37. doi: 10.1369/jhc.7A7273.2007
28. Ponka, P . Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood. (1997) 89:1–25. doi: 10.1182/blood.V89.1.1
29. Lill, R, and Kispal, G. Maturation of cellular Fe-S proteins: an essential function of mitochondria. Trends Biochem Sci. (2000) 25:352–6. doi: 10.1016/S0968-0004(00)01589-9
30. Chitambar, CR . Cellular iron metabolism: mitochondria in the spotlight. Blood. (2005) 105:1844–5. doi: 10.1182/blood-2004-12-4747
31. Levi, S, Ripamonti, M, Dardi, M, Cozzi, A, and Santambrogio, P. Mitochondrial ferritin: its role in physiological and pathological conditions. Cells. (2021) 10:1969. doi: 10.3390/cells10081969
32. Andrews, NC . Forging a field: the golden age of iron biology. Blood. (2008) 112:219–30. doi: 10.1182/blood-2007-12-077388
33. De Domenico, I, McVey Ward, D, and Kaplan, J. Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat Rev Mol Cell Biol. (2008) 9:72–81. doi: 10.1038/nrm2295
34. Fuqua, BK, Vulpe, CD, and Anderson, GJ. Intestinal iron absorption. J Trace Elem Med Biol. (2012) 26:115–9. doi: 10.1016/j.jtemb.2012.03.015
35. McKie, AT, Barrow, D, Latunde-Dada, GO, Rolfs, A, Sager, G, Mudaly, E, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. (1979) 291:1755–9. doi: 10.1126/science.1057206
36. McKie, AT . The role of Dcytb in iron metabolism: an update. Biochem Soc Trans. (2008) 36:1239–41. doi: 10.1042/BST0361239
37. Gunshin, H, Mackenzie, B, Berger, UV, Gunshin, Y, Romero, MF, Boron, WF, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. (1997) 388:482–8. doi: 10.1038/41343
38. Donovan, A, Brownlie, A, Zhou, Y, Shepard, J, Pratt, SJ, Moynihan, J, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. (2000) 403:776–81. doi: 10.1038/35001596
39. Vulpe, CD, Kuo, YM, Murphy, TL, Cowley, L, Askwith, C, Libina, N, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. (1999) 21:195–9. doi: 10.1038/5979
40. Burdo, JR, and Connor, JR. Brain iron uptake and homeostatic mechanisms: An overview. Biometals. (2003) 16:63–75. doi: 10.1023/A:1020718718550
41. Moos, T, and Morgan, EH. Transferrin and transferrin receptor function in brain barrier systems. Cell Mol Neurobiol. (2000) 20:77–95. doi: 10.1023/A:1006948027674
42. Thorstensen, K, and Romslo, I. The role of transferrin in the mechanism of cellular iron uptake. Biochem J. (1990) 271:1–9. doi: 10.1042/bj2710001
43. Richardson, DR, and Ponka, P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta. (1997) 1331:1–40. doi: 10.1016/S0304-4157(96)00014-7
44. Simpson, IA, Ponnuru, P, Klinger, ME, Myers, RL, Devraj, K, Coe, CL, et al. A novel model for brain iron uptake: introducing the concept of regulation. J Cereb Blood Flow Metab. (2015) 35:48–57. doi: 10.1038/jcbfm.2014.168
45. McCarthy, RC, and Kosman, DJ. Ferroportin and exocytoplasmic ferroxidase activity are required for brain microvascular endothelial cell Iron efflux. J Biol Chem. (2013) 288:17932–40. doi: 10.1074/jbc.M113.455428
46. Mills, E, Dong, XP, Wang, F, and Xu, H. Mechanisms of brain iron transport: insight into neurodegeneration and CNS disorders. Fut Med Chem. (2010) 2:51–64. doi: 10.4155/fmc.09.140
47. Fisher, J, Devraj, K, Ingram, J, Slagle-Webb, B, Madhankumar, AB, Liu, X, et al. Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am J Physiol Cell Physiol. (2007) 293:C641–9. doi: 10.1152/ajpcell.00599.2006
48. Todorich, B, Zhang, X, and Connor, JR. H-ferritin is the major source of iron for oligodendrocytes. Glia. (2011) 59:927–35. doi: 10.1002/glia.21164
49. Todorich, B, Zhang, X, Slagle-Webb, B, Seaman, WE, and Connor, JR. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J Neurochem. (2008) 107:1495–505. doi: 10.1111/j.1471-4159.2008.05678.x
50. Ortiz, E, Pasquini, JM, Thompson, K, Felt, B, Butkus, G, Beard, J, et al. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J Neurosci Res. (2004) 77:681–9. doi: 10.1002/jnr.20207
51. Ashkenazi, R, Ben-Shachar, D, and Youdim, MBH. Nutritional iron and dopamine binding sites in the rat brain. Pharmacol Biochem Behav. (1982) 17:43–7. doi: 10.1016/0091-3057(82)90509-3
52. Li, D . Effects of iron deficiency on iron distribution and gamma-aminobutyric acid (GABA) metabolism in young rat brain tissues. Hokkaido Igaky Zasshi. (1998) 73:215–25.
53. Taneja, V, Mishra, K, and Agarwal, KN. Effect of early Iron deficiency in rat on the γ-aminobutyric acid shunt in brain. J Neurochem. (1986) 46:1670–4. doi: 10.1111/j.1471-4159.1986.tb08483.x
54. Kwik-Uribe, CL, Gietzen, D, German, JB, Golub, MS, and Keen, CL. Chronic marginal iron intakes during early development in mice result in persistent changes in dopamine metabolism and myelin composition. J Nutr. (2000) 130:2821–30. doi: 10.1093/jn/130.11.2821
55. Adhami, VM, Husain, R, Husain, R, and Seth, PK. Influence of iron deficiency and lead treatment on behavior and cerebellar and hippocampal polyamine levels in neonatal rats. Neurochem Res. (1996) 21:915–22. doi: 10.1007/BF02532341
56. Beard, JL, and Connor, JR. Iron status and neural functioning. Annu Rev Nutr. (2003) 23:41–58. doi: 10.1146/annurev.nutr.23.020102.075739
57. Ji, C, and Kosman, DJ. Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons. J Neurochem. (2015) 133:668–83. doi: 10.1111/jnc.13040
58. Zhong, MQ, To, Y, Pak, LT, and You, MF. Transferrin receptors on the plasma membrane of cultured rat astrocytes. Exp Brain Res. (1999) 129:0473–6. doi: 10.1007/s002210050916
59. Lis, A, Barone, TA, Paradkar, PN, Plunkett, RJ, and Roth, JA. Expression and localization of different forms of DMT1 in normal and tumor astroglial cells. Mol Brain Res. (2004) 122:62–70. doi: 10.1016/j.molbrainres.2003.11.023
60. Pelizzoni, I, Zacchetti, D, Smith, CP, Grohovaz, F, and Codazzi, F. Expression of divalent metal transporter 1 in primary hippocampal neurons: reconsidering its role in non-transferrin-bound iron influx. J Neurochem. (2012) 120:269–78. doi: 10.1111/j.1471-4159.2011.07578.x
61. Skjørringe, T, Burkhart, A, Johnsen, KB, and Moos, T. Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front Mol Neurosci. (2015) 8:141190. doi: 10.3389/fnmol.2015.00019
62. Mezzanotte, M, Ammirata, G, Boido, M, Stanga, S, and Roetto, A. Activation of the Hepcidin-Ferroportin1 pathway in the brain and astrocytic–neuronal crosstalk to counteract iron dyshomeostasis during aging. Sci Rep. (2022) 12:12. doi: 10.1038/s41598-022-15812-4
63. Boserup, MW, Lichota, J, Haile, D, and Moos, T. Heterogenous distribution of ferroportin-containing neurons in mouse brain. Biometals. (2011) 24:357–75. doi: 10.1007/s10534-010-9405-2
64. Jiang, DH, Ke, Y, Cheng, YZ, Ho, KP, and Qian, ZM. Distribution of ferroportin1 protein in different regions of developing rat brain. Dev Neurosci. (2002) 24:94–8. doi: 10.1159/000065687
65. Moos, T, and Rosengren, NT. Ferroportin in the postnatal rat brain: implications for axonal transport and neuronal export of iron. Semin Pediatr Neurol. (2006) 13:149–57. doi: 10.1016/j.spen.2006.08.003
66. Wu, LJC, Leenders, AGM, Cooperman, S, Meyron-Holtz, E, Smith, S, Land, W, et al. Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res. (2004) 1001:108–17. doi: 10.1016/j.brainres.2003.10.066
67. Du, F, Qian, ZM, Luo, Q, Yung, WH, and Ke, Y. Hepcidin suppresses brain Iron accumulation by downregulating Iron transport proteins in iron-overloaded rats. Mol Neurobiol. (2015) 52:101–14. doi: 10.1007/s12035-014-8847-x
68. Gong, J, Du, F, Qian, ZM, Luo, QQ, Sheng, Y, Yung, WH, et al. Pre-treatment of rats with ad-hepcidin prevents iron-induced oxidative stress in the brain. Free Radic Biol Med. (2016) 90:126–32. doi: 10.1016/j.freeradbiomed.2015.11.016
69. Zhou, YF, Zhang, C, Yang, G, Qian, ZM, Zhang, MW, Ma, J, et al. Hepcidin protects neuron from hemin-mediated injury by reducing Iron. Front Physiol. (2017) 8:332. doi: 10.3389/fphys.2017.00332
70. Vela, D . The dual role of hepcidin in brain iron load and inflammation. Front Neurosci. (2018) 12:740. doi: 10.3389/fnins.2018.00740
71. Urrutia, P, Aguirre, P, Esparza, A, Tapia, V, Mena, NP, Arredondo, M, et al. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem. (2013) 126:541–9. doi: 10.1111/jnc.12244
72. Xiong, XY, Liu, L, Wang, FX, Yang, YR, Hao, JW, Wang, PF, et al. Toll-like receptor 4/MyD88-mediated signaling of hepcidin expression causing brain Iron accumulation, oxidative injury, and cognitive impairment after intracerebral hemorrhage. Circulation. (2016) 134:1025–38. doi: 10.1161/CIRCULATIONAHA.116.021881
73. Ding, H, Yan, CZ, Shi, H, Zhao, YS, Chang, SY, Yu, P, et al. Hepcidin is involved in iron regulation in the ischemic brain. PLoS One. (2011) 6:e25324. doi: 10.1371/journal.pone.0025324
74. Oliveira, SJ, Pinto, JP, Picarote, G, Costa, VM, Carvalho, F, Rangel, M, et al. ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPalpha activity. PLoS One. (2009) 4:e6618. doi: 10.1371/journal.pone.0006618
75. Steinbicker, AU, Bartnikas, TB, Lohmeyer, LK, Leyton, P, Mayeur, C, Kao, SM, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. (2011) 118:4224–30. doi: 10.1182/blood-2011-03-339952
76. Qian, ZM, He, X, Liang, T, Wu, KC, Yan, YC, Lu, LN, et al. Lipopolysaccharides upregulate hepcidin in neuron via microglia and the IL-6/STAT3 signaling pathway. Mol Neurobiol. (2014) 50:811–20. doi: 10.1007/s12035-014-8671-3
77. You, LH, Yan, CZ, Zheng, BJ, Ci, YZ, Chang, SY, Yu, P, et al. Astrocyte hepcidin is a key factor in lps-induced neuronal apoptosis. Cell Death Dis. (2017) 8:8. doi: 10.1038/cddis.2017.93
78. Wang, Q, Du, F, Qian, ZM, Xiao, HG, Zhu, L, Wing, HY, et al. Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology. (2008) 149:3920–5. doi: 10.1210/en.2007-1626
79. Marques, F, Falcao, AM, Sousa, JC, Coppola, G, Geschwind, D, Sousa, N, et al. Altered iron metabolism is part of the choroid plexus response to peripheral inflammation. Endocrinology. (2009) 150:2822–8. doi: 10.1210/en.2008-1610
80. Silvestri, L, Nai, A, Dulja, A, and Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitam Horm. (2019). doi: 10.1016/bs.vh.2019.01.004
81. Robert, SM, Reeves, BC, Kiziltug, E, Duy, PQ, Karimy, JK, Mansuri, MS, et al. The choroid plexus links innate immunity to CSF dysregulation in hydrocephalus. Cells. (2023) 186:764–785.e21. doi: 10.1016/j.cell.2023.01.017
82. Medzhitov, R, and Horng, T. Transcriptional control of the inflammatory response. Nat Rev Immunol. (2009) 9:692–703. doi: 10.1038/nri2634
83. Kawai, T, and Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. (2011) 34:637–50. doi: 10.1016/j.immuni.2011.05.006
84. Zhao, J, Xiang, X, Zhang, H, Jiang, D, Liang, Y, Qing, W, et al. CHOP induces apoptosis by affecting brain iron metabolism in rats with subarachnoid hemorrhage. Exp Neurol. (2018) 302:22–33. doi: 10.1016/j.expneurol.2017.12.015
85. Leibold, EA, and Munro, HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5′ untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci U S A. (1988) 85:2171–5. doi: 10.1073/pnas.85.7.2171
86. Rouault, TA, Hentze, MW, Caughman, SW, Harford, JB, and Klausner, RD. Binding of a cytosolic protein to the iron-responsive element of human ferritin messenger RNA. Science. (1979) 241:1207–10. doi: 10.1126/science.3413484
87. Müllner, EW, Neupert, B, and Kühn, LC. A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cells. (1989) 58:373–82. doi: 10.1016/0092-8674(89)90851-9
88. Casey, JL, Hentze, MW, Koeller, DM, Wright Caughman, S, Rouault, TA, Klausner, RD, et al. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science. (1979) 240:924–8. doi: 10.1126/science.2452485
89. Menotti, E, Henderson, BR, and Kühn, LC. Translational regulation of mRNAs with distinct IRE sequences by iron regulatory proteins 1 and 2. J Biol Chem. (1998) 273:1821–4. doi: 10.1074/jbc.273.3.1821
90. Goossen, B, and Hentze, MW. Position is the critical determinant for function of iron-responsive elements as translational regulators. Mol Cell Biol. (1992) 12:1959–66. doi: 10.1128/MCB.12.5.1959
91. Butt, J, Kim, HY, Basilion, JP, Cohen, S, Iwai, K, Philpott, CC, et al. Differences in the RNA binding sites of iron regulatory proteins and potential target diversity. Proc Natl Acad Sci U S A. (1996) 93:4345–9. doi: 10.1073/pnas.93.9.4345
92. Gray, NK, and Hentze, MW. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. (1994) 13:3882–91. doi: 10.1002/j.1460-2075.1994.tb06699.x
93. Nk, G, Quick, S, Goossen, B, Constable, A, Hirling, H, Lc, K, et al. Recombinant iron-regulatory factor functions as an iron-responsive-element-binding protein, a translational repressor and an aconitase: a functional assay for translational repression and direct demonstration of the iron switch. Eur J Biochem. (1993) 218:657–67. doi: 10.1111/j.1432-1033.1993.tb18420.x
94. Thomson, AM, Rogers, JT, and Leedman, PJ. Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int J Biochem Cell Biol. (1999) 31:1139–52. doi: 10.1016/S1357-2725(99)00080-1
95. Zahringer, J, Baliga, BS, and Munro, HN. Novel mechanism for translational control in regulation of ferritin synthesis by iron. Proc Natl Acad Sci U S A. (1976) 73:857–61. doi: 10.1073/pnas.73.3.857
96. Abboud, S, and Haile, DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. (2000) 275:19906–12. doi: 10.1074/jbc.M000713200
97. Muckenthaler, M, Gray, NK, and Hentze, MW. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex elF4F. Mol Cell. (1998) 2:383–8. doi: 10.1016/S1097-2765(00)80282-8
98. Gunshin, H, Allerson, CR, Polycarpou-Schwarz, M, Rofts, A, Rogers, JT, Kishi, F, et al. Iron-dependent regulation of the divalent metal ion transporter. FEBS Lett. (2001) 509:309–16. doi: 10.1016/S0014-5793(01)03189-1
99. Guo, B, Yu, Y, and Leibold, EA. Iron regulates cytoplasmic levels of a novel iron-responsive element- binding protein without aconitase activity. J Biol Chem. (1994) 269:24252–60. doi: 10.1016/S0021-9258(19)51075-4
100. Han, RH, McKinnon, A, CreveCoeur, TS, Baksh, BS, Mathur, AM, Smyser, CD, et al. Predictors of mortality for preterm infants with intraventricular hemorrhage: a population-based study. Child’s nervous. System. (2018) 34:2203–13. doi: 10.1007/s00381-018-3897-4
101. Strahle, JM, Garton, T, Bazzi, AA, Kilaru, H, Garton, HJL, Maher, CO, et al. Role of hemoglobin and Iron in hydrocephalus after neonatal intraventricular hemorrhage. Neurosurgery. (2014) 75:696–706. doi: 10.1227/NEU.0000000000000524
102. Garton, TP, He, Y, Garton, HJL, Keep, RF, Xi, G, and Strahle, JM. Hemoglobin-induced neuronal degeneration in the hippocampus after neonatal intraventricular hemorrhage. Brain Res. (2016) 1635:86–94. doi: 10.1016/j.brainres.2015.12.060
103. Strahle, JM, Triplett, RL, Alexopoulos, D, Smyser, TA, Rogers, CE, Limbrick, DD, et al. Impaired hippocampal development and outcomes in very preterm infants with perinatal brain injury. Neuroimage Clin. (2019) 22:101787. doi: 10.1016/j.nicl.2019.101787
104. Guo, J, Chen, Q, Tang, J, Zhang, J, Tao, Y, Li, L, et al. Minocycline-induced attenuation of iron overload and brain injury after experimental germinal matrix hemorrhage. Brain Res. (2015) 1594:115–24. doi: 10.1016/j.brainres.2014.10.046
105. Rypens, F, Avni, EF, Dussaussois, L, David, P, Vermeylen, P, Van Bogaert, P, et al. Hyperechoic thickened ependyma: sonographic demonstration and significance in neonates. Pediatr Radiol. (1994) 24:550–3. doi: 10.1007/BF02012729
106. Garton, T, Keep, RF, Hua, Y, and Xi, G. Brain iron overload following intracranial haemorrhage. Stroke Vasc Neurol. (2016) 1:172–84. doi: 10.1136/svn-2016-000042
107. Fukumizu, M, Takashima, S, and Becker, LE. Glial reaction in periventricular areas of the brainstem in fetal and neonatal posthemorrhagic hydrocephalus and congenital hydrocephalus. Brain and Development. (1996) 18:40–5. doi: 10.1016/0387-7604(95)00103-4
108. Pan, S, Yang, PH, DeFreitas, D, Ramagiri, S, Bayguinov, PO, Hacker, CD, et al. Gold nanoparticle-enhanced X-ray microtomography of the rodent reveals region-specific cerebrospinal fluid circulation in the brain. Nature. Communications. (2023):14, 1–6. doi: 10.1038/s41467-023-36083-1
109. Moos, T, and Morgan, EH. Kinetics and distribution of [59Fe-125I]transferrin injected into the ventricular system of the rat. Brain Res. (1998) 790:115–28. doi: 10.1016/S0006-8993(98)00055-9
110. Kawabata, H, Yang, R, Hirama, T, Vuong, PT, Kawano, S, Gombart, AF, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. (1999) 274:20826–32. doi: 10.1074/jbc.274.30.20826
111. Jefferies, WA, Brandon, MR, Hunt, SV, Williams, AF, Gatter, KC, and Mason, DY. Transferrin receptor on endothelium of brain capillaries. Nature. (1984) 312:162–3. doi: 10.1038/312162a0
112. Dziegielewska, KM, Evans, CA, Malinowska, DH, Møllgård, K, Reynolds, JM, Reynolds, ML, et al. Studies of the development of brain barrier systems to lipid insoluble molecules in fetal sheep. J Physiol. (1979) 292:207–31. doi: 10.1113/jphysiol.1979.sp012847
113. Jane, A, Roskams, I, and Connor, JR. Iron, transferrin, and ferritin in the rat brain during development and aging. J Neurochem. (1994) 63:709–16. doi: 10.1046/j.1471-4159.1994.63020709.x
114. Dwork, AJ, Schon, EA, and Herbert, J. Nonidentical distribution of transferrin and ferric iron in human brain. Neuroscience. (1988) 27:333–45. doi: 10.1016/0306-4522(88)90242-4
115. Aldred, AR, Dickson, PW, Marley, PD, and Schreiber, G. Distribution of transferrin synthesis in brain and other tissues in the rat. J Biol Chem. (1987) 262:5293–7. doi: 10.1016/S0021-9258(18)61187-1
116. Bloch, B, Popovici, T, Levin, MJ, Tuil, D, and Kahn, A. Transferrin gene expression visualized in oligodendrocytes of the rat brain by using in situ hybridization and immunohistochemistry. Proc Natl Acad Sci U S A. (1985) 82:6706–10. doi: 10.1073/pnas.82.19.6706
117. Connor, JR, and Fine, RE. The distribution of transferrin immunoreactivity in the rat central nervous system. Brain Res. (1986) 368:319–28. doi: 10.1016/0006-8993(86)90576-7
118. Connor, JR, and Fine, RE. Development of transferrin-positive oligodendrocytes in the rat central nervous system. J Neurosci Res. (1987) 17:51–9. doi: 10.1002/jnr.490170108
119. Espinosa-Jeffrey, A, Kumar, S, Zhao, PM, Awosika, O, Agbo, C, Huang, A, et al. Transferrin regulates transcription of the MBP gene and its action synergizes with IGF-1 to enhance myelinogenesis in the md rat. Dev Neurosci. (2002) 24:227–41. doi: 10.1159/000065698
120. Espinosa De Los Monteros, A, Kumar, S, Zhao, P, Huang, CJ, Nazarian, R, Pan, T, et al. Transferrin is an essential factor for myelination. Neurochem Res. (1999) 24:235–48. doi: 10.1007/s11064-004-1826-2
121. Nandapalan, V, Watson, ID, and Swift, AC. Beta-2-transferrin and cerebrospinal fluid rhinorrhoea. Clin Otolaryngol Allied Sci. (1996) 21:259–64. doi: 10.1111/j.1365-2273.1996.tb01737.x
122. Markelonis, GJ, Oh, TH, Dion, TL, Bregman, BS, Pugh, MA, Royal, GM, et al. Localization of transferrin within the developing vertebrate nervous system. Rev Neurol (Paris). (1988)
123. Oh, TH, Markelonis, GJ, Royal, GM, and Bregman, BS. Immunocytochemical distribution of transferrin and its receptor in the developing chicken nervous system. Dev Brain Res. (1986) 30:207–20. doi: 10.1016/0165-3806(86)90111-2
124. Reynolds, ML, and Møllgård, K. The distribution of plasma proteins in the neocortex and early allocortex of the developing sheep brain. Anat Embryol (Berl). (1985) 171:41–60. doi: 10.1007/BF00319053
125. Toran-allerand, CD . Coexistence of α-fetoprotein, albumin and transferrin immunoreactivity in neurones of the developing mouse brain. Nature. (1980) 286:733–5. doi: 10.1038/286733a0
126. Møllgård, K, Jacobsen, M, Jacobsen, GK, Clausen, PP, and Saunders, NR. Immunohistochemical evidence for an intracellular localization of plasma proteins in human foetal choroid plexus and brain. Neurosci Lett. (1979) 14:85–90. doi: 10.1016/0304-3940(79)95349-7
127. Dion, TL, Markelonis, GJ, Oh, TH, Bregman, BS, Pugh, MA, Hobbs, SL, et al. Immunocytochemical localization of transferrin and mitochondrial malate dehydrogenase in the developing nervous system of the rat. Dev Neurosci. (1988) 10:152–64. doi: 10.1159/000111965
128. Møllgård, K, Reynolds, ML, Jacobsen, M, Dziegielewska, KM, and Saunders, NR. Differential immunocytochemical staining for fetuin and transferrin in the developing cortical plate. J Neurocytol. (1984) 13:497–502. doi: 10.1007/BF01148077
129. Mllgård, K, Stagaard, M, and Saunders, NR. Cellular distribution of transferrin immunoreactivity in the developing rat brain. Neurosci Lett. (1987) 78:35–40. doi: 10.1016/0304-3940(87)90557-X
130. New, H, Dziegielewska, KM, and Saunders, NR. Transferrin in fetal rat brain and cerebrospinal fluid. Int J Dev Neurosci. (1983) 1:369–73. doi: 10.1016/0736-5748(83)90018-7
131. Adinolfi, M, and Haddad, SA. Levels of plasma proteins in human and rat fetal CSF and the development of the blood CSF barrier. Neuropadiatrie. (1977) 8:345–53. doi: 10.1055/s-0028-1091530
132. Wejman, JC, Hovsepian, D, Wall, JS, Hainfeld, JF, and Greer, J. Structure of haptoglobin and the haptoglobin-hemoglobin complex by electron microscopy. J Mol Biol. (1984) 174:343–68. doi: 10.1016/0022-2836(84)90341-3
133. Roberts, ES, Masliah, E, and Fox, HS. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE). J Neuropathol Exp Neurol. (2004) 63:1255–64. doi: 10.1093/jnen/63.12.1255
134. Fabriek, BO, Dijkstra, CD, and van den Berg, TK. The macrophage scavenger receptor CD163. Immunobiology. (2005) 210:153–60. doi: 10.1016/j.imbio.2005.05.010
135. Lau, SK, Chu, PG, and Weiss, LM. CD163: a specific marker of macrophages in paraffin-embedded tissue samples. Am J Clin Pathol. (2004) 122:794–801. doi: 10.1309/QHD6YFN81KQXUUH6
136. Kim, WK, Alvarez, X, Fisher, J, Bronfin, B, Westmoreland, S, McLaurin, J, et al. CD163 identifies perivascular macrophages in normal and viral encephalitic brains and potential precursors to perivascular macrophages in blood. Am J Pathol. (2006) 168:822–34. doi: 10.2353/ajpath.2006.050215
137. Schaer, CA, Vallelian, F, Imhof, A, Schoedon, G, and Schaer, DJ. CD163-expressing monocytes constitute an endotoxin-sensitive Hb clearance compartment within the vascular system. J Leukoc Biol. (2007) 82:106–10. doi: 10.1189/jlb.0706453
138. Kristiansen, M, Graversen, JH, Jacobsen, C, Sonne, O, Hoffman, HJ, Law, SKA, et al. Identification of the haemoglobin scavenger receptor. Nature. (2001) 409:198–201. doi: 10.1038/35051594
139. Moestrup, SK, and Moller, HJ. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med. (2004) 36:347–54. doi: 10.1080/07853890410033171
140. Liu, R, Cao, S, Hua, Y, Keep, RF, Huang, Y, and Xi, G. CD163 expression in neurons after experimental intracerebral hemorrhage. Stroke. (2017) 48:1369–75. doi: 10.1161/STROKEAHA.117.016850
141. Schaer, CA, Deuel, JW, Bittermann, AG, Rubio, IG, Schoedon, G, Spahn, DR, et al. Mechanisms of haptoglobin protection against hemoglobin peroxidation triggered endothelial damage. Cell Death Differ. (2013) 20:1569–79. doi: 10.1038/cdd.2013.113
142. Häfeli, WE, Rieder, HP, and Kaeser, HE. Perinatal relation of the blood-cerebrospinal fluid barrier and its effect on protein composition of the cerebrospinal fluid. Monatsschr Kinderheilkd. (1985) 133:749–53.
143. Buhimschi, CS, Jablonski, KA, Rouse, DJ, Varner, MW, Reddy, UM, Mercer, BM, et al. Cord blood Haptoglobin, cerebral palsy and death in infants of women at risk for preterm birth: a secondary analysis of a randomised controlled trial. EClinicalMedicine. (2019) 9:11–8. doi: 10.1016/j.eclinm.2019.03.009
144. Kanakoudi, F, Drossou, V, Tzimouli, V, Diamanti, E, Konstantinidis, T, Germenis, A, et al. Serum concentrations of 10 acute-phase proteins in healthy term and preterm infants from birth to age 6 months. Clin Chem. (1995) 41:605–8. doi: 10.1093/clinchem/41.4.605
145. Møllgård, K, Dziegielewska, KM, Saunders, NR, Zakut, H, and Soreq, H. Synthesis and localization of plasma proteins in the developing human brain. Integrity of the fetal blood-brain barrier to endogenous proteins of hepatic origin. Dev Biol. (1988) 128:207–21. doi: 10.1016/0012-1606(88)90283-7
146. Dziegielewska, KM, Saunders, NR, Schejter, EJ, Zakut, H, Zevin-Sonkin, D, Zisling, R, et al. Synthesis of plasma proteins in fetal, adult, and neoplastic human brain tissue. Dev Biol. (1986) 115:93–104. doi: 10.1016/0012-1606(86)90231-9
147. Gram, M, Sveinsdottir, S, Cinthio, M, Sveinsdottir, K, Åkerström, B, Hansson, S, et al. Extracellular haemoglobin – mediator of structural damage, cell death, oxidative stress and inflammation in the choroid plexus following preterm intraventricular haemorrhage. Free Radic Biol Med. (2014) 76:S23–4. doi: 10.1016/j.freeradbiomed.2014.10.475
148. Zhu, Y, Hon, T, Ye, W, and Zhang, L. Heme deficiency interferes with the Ras-mitogen-activated protein kinase signaling pathway and expression of a subset of neuronal genes. Cell Growth Differ. (2002) 13:431–9.
149. Meyer, UA, Schuurmans, MM, and Lindberg, RLP. Acute porphyrias: pathogenesis of neurological manifestations. Semin Liver Dis. (1998) 18:43–52. doi: 10.1055/s-2007-1007139
150. Chernova, T, Nicotera, P, and Smith, AG. Heme deficiency is associated with senescence and causes suppression of N-methyl-D-aspartate receptor subunits expression in primary cortical neurons. Mol Pharmacol. (2006) 69:697–705. doi: 10.1124/mol.105.016675
151. Chernova, T, Steinert, JR, Guerin, CJ, Nicotera, P, Forsythe, ID, and Smith, AG. Neurite degeneration induced by heme deficiency mediated via inhibition of NMDA receptor-dependent extracellular signal-regulated kinase 1/2 activation. J Neurosci. (2007) 27:8475–85. doi: 10.1523/JNEUROSCI.0792-07.2007
152. Sengupta, A, Hon, T, and Zhang, L. Heme deficiency suppresses the expression of key neuronal genes and causes neuronal cell death. Mol Brain Res. (2005) 137:23–30. doi: 10.1016/j.molbrainres.2005.02.007
153. Hvidberg, V, Maniecki, MB, Jacobsen, C, Højrup, P, Møller, HJ, and Moestrup, SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. (2005) 106:2572–9. doi: 10.1182/blood-2005-03-1185
154. Morris, CM, Candy, JM, Edwardson, JA, Bloxham, CA, and Smith, A. Evidence for the localization of haemopexin immunoreactivity in neurones in the human brain. Neurosci Lett. (1993) 149:141–4. doi: 10.1016/0304-3940(93)90756-B
155. Tolosano, E, Cutufia, MA, Hirsch, E, Silengo, L, and Altruda, F. Specific expression in brain and liver driven by the hemopexin promoter in transgenic mice. Biochem Biophys Res Commun. (1996) 218:694–703. doi: 10.1006/bbrc.1996.0124
156. Liu, HM, Atack, JR, and Rapoport, SI. Immunohistochemical localization of intracellular plasma proteins in the human central nervous system. Acta Neuropathol. (1989) 78:16–21. doi: 10.1007/BF00687397
157. Morello, N, Tonoli, E, Logrand, F, Fiorito, V, Fagoonee, S, Turco, E, et al. Haemopexin affects iron distribution and ferritin expression in mouse brain. J Cell Mol Med. (2009) 13:4192–204. doi: 10.1111/j.1582-4934.2008.00611.x
158. Castaño, EM, Roher, AE, Esh, CL, Kokjohn, TA, and Beach, T. Comparative proteomics of cerebrospinal fluid in neuropathologically- confirmed Alzheimer’s disease and non-demented elderly subjects. Neurol Res. (2006) 28:155–63. doi: 10.1179/016164106X98035
159. Zheng, W, Song, YQ, Xie, Y, Lin, NJ, Tu, MF, Liu, WP, et al. Cerebrospinal fluid proteins identification facilitates the differential diagnosis of central nervous system diffuse large B cell lymphoma. J Cancer. (2017) 8:3631–40. doi: 10.7150/jca.20267
160. Lim, TKY, Anderson, KM, Hari, P, Di Falco, M, Reihsen, TE, Wilcox, GL, et al. Evidence for a role of nerve injury in painful intervertebral disc degeneration: a cross-sectional proteomic analysis of human cerebrospinal fluid. J. Pain. (2017) 18:1253–69. doi: 10.1016/j.jpain.2017.06.002
161. Davidsson, P, Folkesson, S, Christiansson, M, Lindbjer, M, Dellheden, B, Blennow, K, et al. Identification of proteins in human cerebrospinal fluid using liquid-phase isoelectric focusing as a prefractionation step followed by two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionisation mass spectrometry. Rapid Commun Mass Spectrom. (2002) 16:2083–8. doi: 10.1002/rcm.834
162. Leclerc, JL, Santiago-Moreno, J, Dang, A, Lampert, AS, Cruz, PE, Rosario, AM, et al. Increased brain hemopexin levels improve outcomes after intracerebral hemorrhage. J Cereb Blood Flow Metab. (2018) 38:1032–46. doi: 10.1177/0271678X16679170
163. Garland, P, Durnford, AJ, Okemefuna, AI, Dunbar, J, Nicoll, JAR, Galea, J, et al. Heme-Hemopexin scavenging is active in the brain and associates with outcome after subarachnoid hemorrhage. Stroke. (2016) 47:872–6. doi: 10.1161/STROKEAHA.115.011956
164. Harris, ZL, Durley, AP, Man, TK, and Gitlin, JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. (1999) 96:10812–7. doi: 10.1073/pnas.96.19.10812
165. Osaki, S, Johnson, DA, and Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J Biol Chem. (1966) 241:2746–51. doi: 10.1016/S0021-9258(18)96527-0
166. Qian, ZM, and Ke, Y. Rethinking the role of ceruloplasmin in brain iron metabolism. Brain Res Rev. (2001) 35:287–94. doi: 10.1016/S0165-0173(01)00056-X
167. Klomp, LWJ, and Gitlin, JD. Expression of the ceruloplasmin gene in the human retina and brain: implications for a pathogenic model in aceruloplasminemia. Hum Mol Genet. (1996) 5:1989–96. doi: 10.1093/hmg/5.12.1989
168. Attieh, ZK, Mukhopadhyay, CK, Seshadrit, V, Tripoulas, NA, and Fox, PL. Ceruloplasmin ferroxidase activity stimulates cellular iron uptake by a trivalent cation-specific transport mechanism. J Biol Chem. (1999) 274:1116–23. doi: 10.1074/jbc.274.2.1116
169. Qian, ZM, Tsoi, YK, Ke, Y, and Wong, MS. Ceruloplasmin promotes iron uptake rather than release in BT325 cells. Exp Brain Res. (2001) 140:369–74. doi: 10.1007/s002210100831
170. Jeong, SY, and David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem. (2003) 278:27144–8. doi: 10.1074/jbc.M301988200
171. Je, B, Aa, F, Collins, M, Marshall, T, Pw, J, Strange, R, et al. Developmental profile of plasma proteins in human fetal cerebrospinal fluid and blood. Neuropathol Appl Neurobiol. (1991) 17:441–56. doi: 10.1111/j.1365-2990.1991.tb00748.x
172. Klomp, LWJ, Farhangrazi, ZS, Dugan, LL, and Gitlin, JD. Ceruloplasmin gene expression in the murine central nervous system. J Clin Investig. (1996) 98:207–15. doi: 10.1172/JCI118768
173. Patel, BN, and David, S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem. (1997) 272:20185–90. doi: 10.1074/jbc.272.32.20185
174. Patel, BN, Dunn, RJ, and David, S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem. (2000) 275:4305–10. doi: 10.1074/jbc.275.6.4305
175. Ducharme, P, Zarruk, JG, David, S, and Paquin, J. The ferroxidase ceruloplasmin influences reelin processing, cofilin phosphorylation and neuronal organization in the developing brain. Mol Cell Neurosci. (2018) 92:104–13. doi: 10.1016/j.mcn.2018.07.005
176. Hultin, S . Mosby's manual of diagnostic and laboratory tests (4th edn). Int J Lab Med. (2012) 49:415. doi: 10.1258/acb.2012.201207
177. Luck, AN, and Mason, AB. Transferrin-mediated cellular Iron delivery. Curr Top Membr. (2012) 69:3–35. doi: 10.1016/B978-0-12-394390-3.00001-X
178. Copp, AJ, Estibeiro, JP, Brook, FA, and Downs, KM. Exogenous transferrin is taken up and localized by the neurulation-stage mouse embryo in vitro. Dev Biol. (1992) 153:312–23. doi: 10.1016/0012-1606(92)90116-X
179. Moos, T, Oates, PS, and Morgan, EH. Expression of the neuronal transferrin receptor is age dependent and susceptible to iron deficiency. J Comp Neurol. (1998) 398:420–30. doi: 10.1002/(SICI)1096-9861(19980831)398:3<420::AID-CNE8>3.0.CO;2-1
180. Taylor, EM, and Morgan, EH. Developmental changes in transferrin and iron uptake by the brain in the rat. Dev Brain Res. (1990) 55:35–42. doi: 10.1016/0165-3806(90)90103-6
181. Siddappa, AJM, Rao, RB, Wobken, JD, Leibold, EA, Connor, JR, and Georgieff, MK. Developmental changes in the expression of iron regulatory proteins and iron transport proteins in the perinatal rat brain. J Neurosci Res. (2002) 68:761–75. doi: 10.1002/jnr.10246
182. Duck, KA, Simpson, IA, and Connor, JR. Regulatory mechanisms for iron transport across the blood-brain barrier. Biochem Biophys Res Commun. (2017) 494:70–5. doi: 10.1016/j.bbrc.2017.10.083
183. Moos, T, Skjoerringe, T, Gosk, S, and Morgan, EH. Brain capillary endothelial cells mediate iron transport into the brain by segregating iron from transferrin without the involvement of divalent metal transporter 1. J Neurochem. (2006) 98:1946–58. doi: 10.1111/j.1471-4159.2006.04023.x
184. Enerson, BE, and Drewes, LR. The rat blood-brain barrier transcriptome. J Cereb Blood Flow Metab. (2006) 26:959–73. doi: 10.1038/sj.jcbfm.9600249
185. Zhang, H, Ostrowski, R, Jiang, D, Zhao, Q, Liang, Y, Che, X, et al. Hepcidin promoted ferroptosis through iron metabolism which is associated with DMT1 signaling activation in early brain injury following subarachnoid hemorrhage. Oxidative Med Cell Longev. (2021) 2021:1–19. doi: 10.1155/2021/9800794
186. Gruenheid, S, Cellier, M, Vidal, S, and Gros, P. Identification and characterization of a second mouse Nramp gene. Genomics. (1995) 25:514–25. doi: 10.1016/0888-7543(95)80053-O
187. Tchernitchko, D, Bourgeois, M, Martin, M-E, and Beaumont, C. Expression of the two mRNA isoforms of the iron transporter Nrmap2/DMTI in mice and function of the iron responsive element. Biochem J. (2002) 363:449–55. doi: 10.1042/bj3630449
188. Hubert, N, and Hentze, MW. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci U S A. (2002) 99:12345–50. doi: 10.1073/pnas.192423399
189. Williams, K, Wilson, MA, and Bressler, J. Regulation and developmental expression of the divalent metal-ion transporter in the rat brain. Cell Mol Biol (Noisy-le-Grand). (2000):46.
190. Knutson, M, Menzies, S, Connor, J, and Wessling-Resnick, M. Developmental, regional, and cellular expression of SFT/UbcH5A and DMT1 mRNA in brain. J Neurosci Res. (2004) 76:633–41. doi: 10.1002/jnr.20113
191. Ke, Y, Chang, YZ, Duan, XL, Du, JR, Zhu, L, Wang, K, et al. Age-dependent and iron-independent expression of two mRNA isoforms of divalent metal transporter 1 in rat brain. Neurobiol Aging. (2005) 26:739–48. doi: 10.1016/j.neurobiolaging.2004.06.002
192. Ward, DM, and Kaplan, J. Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta, Mol Cell Res. (2012) 1823:1426–33. doi: 10.1016/j.bbamcr.2012.03.004
193. McKie, AT, Marciani, P, Rolfs, A, Brennan, K, Wehr, K, Barrow, D, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. (2000) 5:299–309. doi: 10.1016/S1097-2765(00)80425-6
194. Burkhart, A, Skjørringe, T, Johnsen, KB, Siupka, P, Thomsen, LB, Nielsen, MS, et al. Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood-brain barrier. Mol Neurobiol. (2016) 53:7237–53. doi: 10.1007/s12035-015-9582-7
195. Burdo, JR, Menzies, SL, Simpson, IA, Garrick, LM, Garrick, MD, Dolan, KG, et al. Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J Neurosci Res. (2001) 66:1198–207. doi: 10.1002/jnr.1256
196. Yang, WM, Jung, KJ, Lee, MO, Lee, YS, Lee, YH, Nakagawa, S, et al. Transient expression of iron transport proteins in the capillary of the developing rat brain. Cell Mol Neurobiol. (2011) 31:93–9. doi: 10.1007/s10571-010-9558-0
197. Mietto, BS, Jhelum, P, Schulz, K, and David, S. Schwann cells provide iron to axonal mitochondria and its role in nerve regeneration. J Neurosci. (2021) 41:7300–13. doi: 10.1523/JNEUROSCI.0900-21.2021
198. Tan, G, Liu, L, He, Z, Sun, J, Xing, W, and Sun, X. Role of hepcidin and its downstream proteins in early brain injury after experimental subarachnoid hemorrhage in rats. Mol Cell Biochem. (2016) 418:31–8. doi: 10.1007/s11010-016-2730-1
199. Bao, WD, Zhou, XT, Zhou, LT, Wang, F, Yin, X, Lu, Y, et al. Targeting miR-124/Ferroportin signaling ameliorated neuronal cell death through inhibiting apoptosis and ferroptosis in aged intracerebral hemorrhage murine model. Aging Cell. (2020) 19:e13235. doi: 10.1111/acel.13235
200. Wang, G, Manaenko, A, Shao, A, Ou, Y, Yang, P, Budbazar, E, et al. Low-density lipoprotein receptor-related protein-I facilitates heme scavenging after intracerebral hemorrhage in mice. J Cereb Blood Flow Metab. (2017) 37:1299–310. doi: 10.1177/0271678X16654494
201. Boucher, P, Liu, P, Gotthardt, M, Hiesberger, T, Anderson, RGW, and Herz, J. Platelet-derived growth factor mediates tyrosine phosphorylation of the cytoplasmic domain of the low density lipoprotein receptor-related protein in caveolae. J Biol Chem. (2002) 277:15507–13. doi: 10.1074/jbc.M200428200
202. Spijkers, PPEM, Da Costa, MP, Westein, E, Gahmberg, CG, Zwaginga, JJ, and Lenting, PJ. LDL-receptor-related protein regulates β2-integrin-mediated leukocyte adhesion. Blood. (2005) 105:170–7. doi: 10.1182/blood-2004-02-0498
203. Orr, AW, Pedraza, CE, Pallero, MA, Elzie, CA, Goicoechea, S, Strickland, DK, et al. Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J Cell Biol. (2003) 161:1179–89. doi: 10.1083/jcb.200302069
204. Cao, C, Lawrence, DA, Li, Y, Von Arnim, CAF, Herz, J, Su, EJ, et al. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. (2006) 25:1860–70. doi: 10.1038/sj.emboj.7601082
205. Nykjaer, A, Bengtsson-Olivecrona, G, Lookene, A, Moestrup, SK, Petersen, CM, Weber, W, et al. The α2-macroglobulin receptor/low density lipoprotein receptor-related protein binds lipoprotein lipase and β-migrating very low density lipoprotein associated with the lipase. J Biol Chem. (1993) 268:15048–55. doi: 10.1016/S0021-9258(18)82436-X
206. Merkel, M, Kako, Y, Radner, H, Cho, IS, Ramasamy, R, Brunzell, JD, et al. Catalytically inactive lipoprotein lipase expression in muscle of transgenic mice increases very low density lipoprotein uptake: direct evidence that lipoprotein lipase bridging occurs in vivo. Proc Natl Acad Sci U S A. (1998) 95:13841–6. doi: 10.1073/pnas.95.23.13841
207. Chappel, DA, Inoue, I, Fry, GL, Pladet, MW, Bowen, SL, et al. Cellular catabolism of normal very low density lipoproteins via the low density lipoprotein receptor-related protein/α2-macroglobulin receptor is induced by the C-terminal domain of lipoprotein lipase. J Biol Chem. (1994):269.
208. Kounnas, MZ, Chappell, DA, Wong, H, Argraves, WS, and Strickland, DK. The cellular internalization and degradation of hepatic lipase is mediated by low density lipoprotein receptor-related protein and requires cell surface proteoglycans. J Biol Chem. (1995) 270:9307–12. doi: 10.1074/jbc.270.16.9307
209. Lillis, AP, Van, DLB, Murphy-ullrich, JE, Dudley, K, and Strickland, DK. The low density lipoprotein receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. NIH-PA Author Manuscript. (2009) 88:887–918. doi: 10.1152/physrev.00033.2007
210. Strickland, DK, Au, DT, Cunfer, P, and Muratoglu, SC. Low-density lipoprotein receptor-related protein-1: role in the regulation of vascular integrity. Arterioscler Thromb Vasc Biol. (2014) 34:487–98. doi: 10.1161/ATVBAHA.113.301924
211. Kanekiyo, T, and Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front Aging Neurosci. (2014) 6:93. doi: 10.3389/fnagi.2014.00093
212. Bu, G, Maksymovitch, EA, Nerbonne, JM, and Schwartz, AL. Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J Biol Chem. (1994) 269:29874–82. doi: 10.1016/S0021-9258(17)32340-2
213. Wolf, BB, Lopes, MBS, VandenBerg, SR, and Gonias, SL. Characterization and immunohistochemical localization of α2- macroglobulin receptor (low-density lipoprotein receptor-related protein) in human brain. Am J Pathol. (1992) 141:37–42.
214. Hennen, E, Safina, D, Haussmann, U, Wörsdörfer, P, Edenhofer, F, Poetsch, A, et al. A LewisX glycoprotein screen identifies the low density lipoprotein receptor-related protein 1 (LRP1) as a modulator of oligodendrogenesis in mice. J Biol Chem. (2013) 288:16538–45. doi: 10.1074/jbc.M112.419812
215. Auderset, L, Cullen, CL, and Young, KM. Low density lipoprotein-receptor related protein 1 is differentially expressed by neuronal and glial populations in the developing and mature mouse central nervous system. PLoS One. (2016) 11:e0155878. doi: 10.1371/journal.pone.0155878
216. Wang, G, Manaenko, A, Shao, A, Ou, Y, Yang, P, Budbazar, E, et al. Low-density lipoprotein receptor-related protein-1 facilitates heme scavenging after intracerebral hemorrhage in mice. J Cereb Blood Flow Metab. (2017) 37:1299–1310.
217. Peng, J, Pang, J, Huang, L, Enkhjargal, B, Zhang, T, Mo, J, et al. LRP1 activation attenuates white matter injury by modulating microglial polarization through Shc1/PI3K/Akt pathway after subarachnoid hemorrhage in rats. Redox Biol. (2019) 21:101121. doi: 10.1016/j.redox.2019.101121
218. Garton, T, Keep, RF, Hua, Y, and Xi, G. CD163, a hemoglobin/Haptoglobin scavenger receptor, after intracerebral hemorrhage: functions in microglia/macrophages versus neurons. Transl Stroke Res. (2017) 8:612–6. doi: 10.1007/s12975-017-0535-5
219. Galea, J, Cruickshank, G, Teeling, JL, Boche, D, Garland, P, Perry, VH, et al. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J Neurochem. (2012) 121:785–92. doi: 10.1111/j.1471-4159.2012.07716.x
220. Kowal, K, Silver, R, Sławińska, E, Bielecki, M, Chyczewski, L, and Kowal-Bielecka, O. CD163 and its role in inflammation. Folia Histochem Cytobiol. (2011) 49:365–74. doi: 10.5603/FHC.2011.0052
221. Landis, RC, Philippidis, P, Domin, J, Boyle, JJ, and Haskard, DO. Haptoglobin genotype-dependent anti-inflammatory signaling in CD163 + macrophages. Int J Inflam. (2013) 2013:1–7. doi: 10.1155/2013/980327
222. Sulahian, TH, Högger, P, Wahner, AE, Wardwell, K, Goulding, NJ, Sorg, C, et al. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. (2000) 12:1312–21. doi: 10.1006/cyto.2000.0720
223. Buechler, C, Ritter, M, Orsó, E, Langmann, T, Klucken, J, and Schmitz, G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol. (2000) 67:97–103. doi: 10.1002/jlb.67.1.97
224. Chen-Roetling, J, and Regan, RF. Haptoglobin increases the vulnerability of CD163-expressing neurons to hemoglobin. J Neurochem. (2016) 139:586–95. doi: 10.1111/jnc.13720
225. Zhang, Z, Liu, Y, Zhang, Z, Luo, B, and Schluesener, HJ. Lesional accumulation of heme oxygenase-1+ microglia/macrophages in rat traumatic brain injury. Neuroreport. (2013) 24:281–6. doi: 10.1097/WNR.0b013e32835f2810
226. Ohgami, RS, Campagna, DR, McDonald, A, and Fleming, MD. The Steap proteins are metalloreductases. Blood. (2006) 108:1388–94. doi: 10.1182/blood-2006-02-003681
227. GENSAT GENSAT project at Rockefeller University, mouse brain atlas, image navigator. Available at:http://gensat.org/imagenavigator.jsp?imageID=70906#batches (Accessed June 25, 2023)
228. Ohgami, RS, Campagna, DR, Greer, EL, Antiochos, B, McDonald, A, Chen, J, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. (2005) 37:1264–9. doi: 10.1038/ng1658
229. UniProt . Steap3 – STEAP family member 3 - Mus musculus (mouse) | UniProtKB | UniProt. (n.d.) Available at:https://www.uniprot.org/uniprotkb/E9QN92/entry (Accessed June 25, 2023)
230. Rocha, SM, Socorro, S, Passarinha, LA, and Maia, CJ. Comprehensive landscape of STEAP family members expression in human cancers: unraveling the potential usefulness in clinical practice using integrated bioinformatics analysis. Data (Basel). (2022) 7:64. doi: 10.3390/data7050064
231. Hulet, SW, Heyliger, SO, Powers, S, and Connor, JR. Oligodendrocyte progenitor cells internalize ferritin via clathrin- dependent receptor mediated endocytosis. J Neurosci Res. (2000) 61:52–60. doi: 10.1002/1097-4547(20000701)61:1<52::AID-JNR6>3.0.CO;2-T
232. Han, J, Seaman, WE, Di, X, Wang, W, Willingham, M, Torti, FM, et al. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS One. (2011) 6:e23800. doi: 10.1371/journal.pone.0023800
233. Chiou, B, Neely, EB, Mcdevitt, DS, Simpson, IA, and Connor, JR. Transferrin and H-ferritin involvement in brain iron acquisition during postnatal development: impact of sex and genotype. J Neurochem. (2020) 152:381. doi: 10.1111/jnc.14834
234. Bou-Abdallah, F . The iron redox and hydrolysis chemistry of the ferritins. Biochim Biophys Acta Gen Subj. (2010) 1800:719–31. doi: 10.1016/j.bbagen.2010.03.021
235. Chandramouli, B, Bernacchioni, C, Di Maio, D, Turano, P, and Brancato, G. Electrostatic and structural bases of Fe2+ translocation through ferritin channels. J Biol Chem. (2016) 291:25617–28. doi: 10.1074/jbc.M116.748046
236. Zhao, G, Bou-Abdallah, F, Arosio, P, Levi, S, Janus-Chandler, C, and Chasteen, ND. Multiple pathways for mineral core formation in mammalian apoferritin. The role of hydrogen peroxide. Biochemistry. (2003) 42:3142–50. doi: 10.1021/bi027357v
237. Liu, X, and Theil, EC. Ferritin reactions: direct identification of the site for the diferric peroxide reaction intermediate. Proc Natl Acad Sci U S A. (2004) 101:8557–62. doi: 10.1073/pnas.0401146101
238. Salgado, JC, Olivera-Nappa, A, Gerdtzen, ZP, Tapia, V, Theil, EC, Conca, C, et al. Mathematical modeling of the dynamic storage of iron in ferritin. BMC Syst Biol. (2010) 4:147. doi: 10.1186/1752-0509-4-147
239. Narayanan, S, Shahbazian-Yassar, R, and Shokuhfar, T. Transmission electron microscopy of the iron oxide core in ferritin proteins: current status and future directions. J Phys D Appl Phys. (2019) 52:453001. doi: 10.1088/1361-6463/ab353b
240. Theil, EC . Ferritin: the protein nanocage and iron biomineral in health and in disease. Inorg Chem. (2013) 52:12223–33. doi: 10.1021/ic400484n
241. Kidane, TZ, Sauble, E, and Linder, MC. Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol. (2006) 291:C445–55. doi: 10.1152/ajpcell.00505.2005
242. Hallgren, B, and Sourander, P. The effect of age on the non-Haemin iron in the human brain. J Neurochem. (1958) 3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x
243. Octave, JN, Schneider, YJ, Trouet, A, and Crichton, RR. Iron uptake and utilization by mammalian cells. I: cellular uptake of transferrin and iron. Trends Biochem Sci. (1983) 8:217–20. doi: 10.1016/0968-0004(83)90217-7
244. Double, KL, Maywald, M, Schmittel, M, Riederer, P, and Gerlach, M. In vitro studies of ferritin iron release and neurotoxicity. J Neurochem. (1998) 70:2492–9. doi: 10.1046/j.1471-4159.1998.70062492.x
245. Han, J, Day, JR, Connor, JR, and Beard, JL. H and L ferritin subunit mRNA expression differs in brains of control and iron-deficient rats. J Nutr. (2002) 132:2769–74. doi: 10.1093/jn/132.9.2769
246. Connor, JR, Boeshore, KL, Benkovic, SA, and Menzies, SL. Isoforms of ferritin have a specific cellular distribution in the brain. J Neurosci Res. (1994) 37:461–5. doi: 10.1002/jnr.490370405
247. Cheepsunthorn, P, Palmer, C, and Connor, JR. Cellular distribution of ferritin subunits in postnatal rat brain. J Comp Neurol. (1998) 400:73–86. doi: 10.1002/(SICI)1096-9861(19981012)400:1<73::AID-CNE5>3.0.CO;2-Q
248. Bou-Abdallah, F, Santambrogio, P, Levi, S, Arosio, P, and Chasteen, ND. Unique iron binding and oxidation properties of human mitochondrial ferritin: a comparative analysis with human H-chain ferritin. J Mol Biol. (2005) 347:543:554. doi: 10.1016/j.jmb.2005.01.007
249. Ayton, S, Janelidze, S, Kalinowski, P, Palmqvist, S, Belaidi, AA, Stomrud, E, et al. CSF ferritin in the clinicopathological progression of Alzheimer’s disease and associations with APOE and inflammation biomarkers. J Neurol Neurosurg Psychiatry. (2023) 94:211–9. doi: 10.1136/jnnp-2022-330052
250. Ayton, S, Faux, NG, and Bush, AI. Association of cerebrospinal fluid ferritin level with preclinical cognitive decline in APOE-ε4 carriers. JAMA Neurol. (2017) 74:122–5. doi: 10.1001/jamaneurol.2016.4406
251. Kuiper, MA, Mulder, C, van Kamp, GJ, Scheltens, P, and Wolters, EC. Cerebrospinal fluid ferritin levels of patients with Parkinson’s disease, Alzheimer’s disease, and multiple system atrophy. J Neural Transm Park Dis Dement Sect. (1994) 7:109–14. doi: 10.1007/BF02260965
252. Ayton, S, Faux, NG, Bush, AI, Weiner, MW, Aisen, P, Petersen, R, et al. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat Commun. (2015) 6:6760. doi: 10.1038/ncomms7760
253. Petzold, A, Worthington, V, Appleby, I, Kerr, ME, Kitchen, N, and Smith, M. Cerebrospinal fluid ferritin level, a sensitive diagnostic test in late-presenting subarachnoid hemorrhage. J Stroke Cerebrovasc Dis. (2011) 20:489–93. doi: 10.1016/j.jstrokecerebrovasdis.2010.02.021
254. Sato, Y, Honda, Y, Asoh, T, Oizumi, K, Ohshima, Y, and Honda, E. Cerebrospinal fluid ferritin in glioblastoma: evidence for tumor synthesis. J Neuro-Oncol. (1998) 40:47–50. doi: 10.1023/A:1006078521790
255. Takahashi, S, Oki, J, Miyamoto, A, Moriyama, T, Asano, A, Inyaku, F, et al. Beta-2-microglobulin and ferritin in cerebrospinal fluid for evaluation of patients with meningitis of different etiologies. Brain Develop. (1999) 21:192–9. doi: 10.1016/S0387-7604(99)00017-0
256. Sindic, CJM, Collet-Cassart, D, Cambiaso, CL, Masson, PL, and Laterre, EC. The clinical relevance of ferritin concentration in the cerebrospinal fluid. J Neurol Neurosurg Psychiatry. (1981) 44:329–33. doi: 10.1136/jnnp.44.4.329
257. Paydarnia, P, Mayeli, M, Shafie, M, Agah, E, Hasani, SA, Jazani, MR, et al. Alterations of the serum and CSF ferritin levels and the diagnosis and prognosis of amyotrophic lateral sclerosis. eNeurologicalSci. (2021) 25:100379. doi: 10.1016/j.ensci.2021.100379
258. Menna-Barreto, M, Bianchini, O, Jardim, C, Steinstrasser, LE, and Bernardi, R. 4-14-02 cerebrospinal fluid ferritin levels in HTLV-II and HTLV-I infected patients. J Neurol Sci. (1997) 150:S231. doi: 10.1016/S0022-510X(97)85998-9
259. Mahaney, KB, Buddhala, C, Paturu, M, Morales, DM, Smyser, CD, Limbrick, DD, et al. Elevated cerebrospinal fluid iron and ferritin associated with early severe ventriculomegaly in preterm posthemorrhagic hydrocephalus. J Neurosurg Pediatr. (2022) 30:169–76. doi: 10.3171/2022.4.PEDS21463
260. Harrison, PM, and Arosio, P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta Bioenerg. (1996) 1275:161–203. doi: 10.1016/0005-2728(96)00022-9
261. Nitti, M, Piras, S, Brondolo, L, Marinari, UM, Pronzato, MA, and Furfaro, AL. Heme oxygenase 1 in the nervous system: does it favor neuronal cell survival or induce neurodegeneration? Int J Mol Sci. (2018) 19:2260. doi: 10.3390/ijms19082260
262. Was, H, Dulak, J, and Jozkowicz, A. Heme oxygenase-1 in tumor biology and therapy. Curr Drug Targets. (2012) 11:1551–70. doi: 10.2174/1389450111009011551
263. Barañano, DE, and Snyder, SH. Neural roles for heme oxygenase: contrasts to nitric oxide synthase. Proc Natl Acad Sci U S A. (2001) 98:10996–1002. doi: 10.1073/pnas.191351298
264. Zhao, H, Wong, RJ, Nguyen, X, Kalish, F, Mizobuchi, M, Vreman, HJ, et al. Expression and regulation of heme oxygenase isozymes in the developing mouse cortex. Pediatr Res. (2006) 60:518–23. doi: 10.1203/01.PDR.0000242374.21415.f5
265. Ferrandiz, M, and Devesa, I. Inducers of Heme Oxygenase-1. Curr Pharm Des. (2008) 14:473–86. doi: 10.2174/138161208783597399
266. Wang, J, and Doré, S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. (2007) 130:1643–52. doi: 10.1093/brain/awm095
267. Chen, Z, Gao, C, Hua, Y, Keep, RF, Muraszko, K, and Xi, G. Role of iron in brain injury after intraventricular hemorrhage. Stroke. (2011) 42:465–70. doi: 10.1161/STROKEAHA.110.602755
268. Zhang, F, Wang, S, Zhang, M, Weng, Z, Li, P, Gan, Y, et al. Pharmacological induction of heme oxygenase-1 by a triterpenoid protects neurons against ischemic injury. Stroke. (2012) 43:1390–7. doi: 10.1161/STROKEAHA.111.647420
269. Hung, SY, Liou, HC, Kang, KH, Wu, RM, Wen, CC, and Fu, WM. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol Pharmacol. (2008) 74:1564–75. doi: 10.1124/mol.108.048611
270. Lin, WP, Xiong, GP, Lin, Q, Chen, XW, Zhang, LQ, Shi, JX, et al. Heme oxygenase-1 promotes neuron survival through down-regulation of neuronal NLRP1 expression after spinal cord injury. J Neuroinflammation. (2016) 13:52. doi: 10.1186/s12974-016-0521-y
271. Dong, F, Wang, S, Wang, Y, Yang, X, Jiang, J, Wu, D, et al. Quercetin ameliorates learning and memory via the Nrf2-ARE signaling pathway in D-galactose-induced neurotoxicity in mice. Biochem Biophys Res Commun. (2017) 491:636–41. doi: 10.1016/j.bbrc.2017.07.151
272. Catino, S, Paciello, F, Miceli, F, Rolesi, R, Troiani, D, Calabrese, V, et al. Ferulic acid regulates the Nrf2/heme oxygenase-1 system and counteracts trimethyltin-induced neuronal damage in the human neuroblastoma cell line SH-SY5Y. Front Pharmacol. (2016) 6:305. doi: 10.3389/fphar.2015.00305
273. Seo, JY, Kim, BR, Oh, J, and Kim, JS. Soybean-derived phytoalexins improve cognitive function through activation of Nrf2/HO-1 signaling pathway. Int J Mol Sci. (2018) 19:268. doi: 10.3390/ijms19010268
274. Le, WD, Xie, WJ, and Appel, SH. Protective role of heme oxygenase-1 in oxidative stress-induced neuronal injury. J Neurosci Res. (1999) 56:652–8. doi: 10.1002/(SICI)1097-4547(19990615)56:6<652::AID-JNR11>3.0.CO;2-5
275. Ye, F, Li, X, Li, L, Yuan, J, and Chen, J. T-BHQ provides protection against Lead neurotoxicity via Nrf2/ pathway. Oxidative Med Cell Longev. (2016) 2016:1–15. doi: 10.1155/2016/2075915
276. Takizawa, S, Hirabayashi, H, Matsushima, K, Tokuoka, K, and Shinohara, Y. Induction of heme oxygenase protein protects neurons in cortex and striatum, but not in hippocampus, against transient forebrain ischemia. J Cereb Blood Flow Metab. (1998) 18:559–69. doi: 10.1097/00004647-199805000-00011
277. Chen, K, Gunter, K, and Maines, MD. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J Neurochem. (2000) 75:304–13. doi: 10.1046/j.1471-4159.2000.0750304.x
278. Nemeth, E, Tuttle, MS, Powelson, J, Vaughn, MD, Donovan, A, Ward, DMV, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. (1979) 2004:306.
279. Raha, AA, Vaishnav, RA, Friedland, RP, Bomford, A, and Raha-Chowdhury, R. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol Commun. (2014) 1:2. doi: 10.1186/2051-5960-1-55
280. Raha-Chowdhury, R, Raha, AA, Forostyak, S, Zhao, JW, Stott, SRW, and Bomford, A. Expression and cellular localization of hepcidin mRNA and protein in normal rat brain. BMC Neurosci. (2015) 16:24. doi: 10.1186/s12868-015-0161-7
281. Wang, SM, Fu, LJ, Duan, XL, Crooks, DR, Yu, P, Qian, ZM, et al. Role of hepcidin in murine brain iron metabolism. Cell Mol Life Sci. (2010) 67:123–33. doi: 10.1007/s00018-009-0167-3
282. Zechel, S, Huber-Wittmer, K, and Von Halbach, OB. Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J Neurosci Res. (2006) 84:790–800. doi: 10.1002/jnr.20991
283. Hentze, MW, Caughman, SW, Rouault, TA, Barriocanal, JG, Dancis, A, Harford, JB, et al. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. (1979) 238:1570–3. doi: 10.1126/science.3685996
284. Wilkinson, N, and Pantopoulos, K. The IRP/IRE system in vivo: insights from mouse models. Front Pharmacol. (2014) 5:176. doi: 10.3389/fphar.2014.00176
285. Theil, EC . Regulation of ferritin and transferrin receptor mRNAs. J Biol Chem. (1990) 265:4771–4. doi: 10.1016/S0021-9258(19)34036-0
286. Garrick, MD, Dolan, KG, Horbinski, C, Ghio, AJ, Higgins, D, Porubcin, M, et al. DMT1: a mammalian transporter for multiple metals. Biometals. (2003) 16:41–54. doi: 10.1023/a:1020702213099
287. Müllner, EW, and Kühn, LC. A stem-loop in the 3′ untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cells. (1988) 53:815–25.
288. Casey, JL, Koeller, DM, Ramin, VC, Klausner, RD, and Harford, JB. Iron regulation of transferrin receptor mRNA levels requires iron-responsive elements and a rapid turnover determinant in the 3′ untranslated region of the mRNA. EMBO J. (1989) 8:3693–9. doi: 10.1002/j.1460-2075.1989.tb08544.x
289. Yoshinaga, M, Nakatsuka, Y, Vandenbon, A, Ori, D, Uehata, T, Tsujimura, T, et al. Regnase-1 maintains iron homeostasis via the degradation of transferrin receptor 1 and prolyl-hydroxylase-domain-containing protein 3 mRNAs. Cell Rep. (2017) 19:1614–30. doi: 10.1016/j.celrep.2017.05.009
290. Corral, VM, Schultz, ER, Eisenstein, RS, and Connell, GJ. Roquin is a major mediator of iron-regulated changes to transferrin receptor-1 mRNA stability. iScience. (2021) 24:102360. doi: 10.1016/j.isci.2021.102360
291. Rouault, TA, and Maio, N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J Biol Chem. (2017) 292:12744–53. doi: 10.1074/jbc.R117.789537
292. Wang, H, Shi, H, Rajan, M, Canarie, ER, Hong, S, Simoneschi, D, et al. FBXL5 regulates IRP2 stability in Iron homeostasis via an oxygen-responsive [2Fe2S] cluster. Mol Cell. (2020) 78:31–41.e5. doi: 10.1016/j.molcel.2020.02.011
293. Samaniego, F, Chin, J, Iwai, K, Rouault, TA, and Klausner, RD. Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. Structure, function, and post- translational regulation. J Biol Chem. (1994) 269:30904–10. doi: 10.1016/S0021-9258(18)47367-X
294. Li, Q, Ding, Y, Krafft, P, Wan, W, Yan, F, Wu, G, et al. Targeting germinal matrix hemorrhage-induced overexpression of sodium-coupled bicarbonate exchanger reduces Posthemorrhagic hydrocephalus formation in neonatal rats. J Am Heart Assoc. (2018) 7:7. doi: 10.1161/JAHA.117.007192
295. Puig, KL, and Combs, CK. Expression and function of APP and its metabolites outside the central nervous system. Exp Gerontol. (2013) 48:608–11. doi: 10.1016/j.exger.2012.07.009
296. McCarthy, RC, Park, Y, and Kosman, DJ. sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep. (2014) 15:809–15. doi: 10.15252/embr.201338064
297. Wong, BX, Tsatsanis, A, Lim, LQ, Adlard, PA, Bush, AI, and Duce, JA. β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One. (2014) 9:e114174. doi: 10.1371/journal.pone.0114174
298. Duce, JA, Tsatsanis, A, Cater, MA, James, SA, Robb, E, Wikhe, K, et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cells. (2010) 142:857–67. doi: 10.1016/j.cell.2010.08.014
299. Salbaum, JM, and Ruddle, FH. Embryonic expression pattern of amyloid protein precursor suggests a role in differentiation of specific subsets of neurons. J Exp Zool. (1994) 269:116–27.
300. Trapp, BD, and Hauer, PE. Amyloid precursor protein is enriched in radial glia: implications for neuronal development. J Neurosci Res. (1994) 37:538–50. doi: 10.1002/jnr.490370413
301. Hayashi, Y, Kashiwagi, K, Ohta, J, Nakajima, M, Kawashima, T, and Yoshikawa, K. Alzheimer amyloid protein precursor enhances proliferation of neural stem cells from fetal rat brain. Biochem Biophys Res Commun. (1994) 205:936–43. doi: 10.1006/bbrc.1994.2755
302. Ohsawa, I, Takamura, C, Morimoto, T, Ishiguro, M, and Kohsaka, S. Amino-terminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. Eur J Neurosci. (1999) 11:1907–13. doi: 10.1046/j.1460-9568.1999.00601.x
303. Young-Pearse, TL, Bai, J, Chang, R, Zheng, JB, Loturco, JJ, and Selkoe, DJ. A critical function for β-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. (2007) 27:14459–69. doi: 10.1523/JNEUROSCI.4701-07.2007
304. Freude, KK, Penjwini, M, Davis, JL, LaFerla, FM, and Blurton-Jones, M. Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J Biol Chem. (2011) 286:24264–74. doi: 10.1074/jbc.M111.227421
305. Milward, EA, Papadopoulos, R, Fuller, SJ, Moir, RD, Small, D, Beyreuther, K, et al. The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. (1992) 9:129–37. doi: 10.1016/0896-6273(92)90228-6
306. Hoareau, C, Borrell, V, Soriano, E, Krebs, MO, Prochiantz, A, and Allinquant, B. Amyloid precursor protein cytoplasmic domain antagonizes reelin neurite outgrowth inhibition of hippocampal neurons. Neurobiol Aging. (2008) 29:542–53. doi: 10.1016/j.neurobiolaging.2006.11.012
307. Chasseigneaux, S, Dinc, L, Rose, C, Chabret, C, Coulpier, F, Topilko, P, et al. Secreted amyloid precursor protein β and secreted amyloid precursor protein α induce axon outgrowth in vitro through egr1 signaling pathway. PLoS One. (2011) 6:e16301. doi: 10.1371/journal.pone.0016301
308. Zhou, F, Gong, K, Song, B, Ma, T, van Laar, T, Gong, Y, et al. The APP intracellular domain (AICD) inhibits Wnt signalling and promotes neurite outgrowth. Biochim Biophys Acta Mol Cell Res. (2012) 1823:1233–41. doi: 10.1016/j.bbamcr.2012.05.011
309. Soldano, A, Okray, Z, Janovska, P, Tmejová, K, Reynaud, E, Claeys, A, et al. The Drosophila homologue of the amyloid precursor protein is a conserved modulator of Wnt PCP signaling. PLoS Biol. (2013) 11:e1001562. doi: 10.1371/journal.pbio.1001562
310. Mora, N, Almudi, I, Alsina, B, Corominas, M, and Serras, F. β amyloid protein precursor-like (Appl) is a Ras1/MAPK-regulated gene required for axonal targeting in Drosophila photoreceptor neurons. J Cell Sci. (2013) 126:53–9. doi: 10.1242/jcs.114785
311. Rama, N, Goldschneider, D, Corset, V, Lambert, J, Pays, L, and Mehlen, P. Amyloid precursor protein regulates netrin-1-mediated commissural axon outgrowth. J Biol Chem. (2012) 287:30014–23. doi: 10.1074/jbc.M111.324780
312. Magara, F, Müller, U, Li, ZW, Lipp, HP, Weissmann, C, Stagljar, M, et al. Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the β-amyloid-precursor protein. Proc Natl Acad Sci U S A. (1999) 96:4656–61. doi: 10.1073/pnas.96.8.4656
313. Young-Pearse, TL, Chen, AC, Chang, R, Marquez, C, and Selkoe, DJ. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. (2008) 3:15. doi: 10.1186/1749-8104-3-15
314. Nicolas, M, and Hassan, BA. Amyloid precursor protein and neural development. Development (Cambridge). (2014) 141:2543–8. doi: 10.1242/dev.108712
315. Wang, P, Yang, G, Mosier, DR, Chang, P, Zaidi, T, Gong, YD, et al. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-like protein 2. J Neurosci. (2005) 25:1219–25. doi: 10.1523/JNEUROSCI.4660-04.2005
316. Schubert, W, Prior, R, Weidemann, A, Dircksen, H, Multhaup, G, Masters, CL, et al. Localization of Alzheimer βA4 amyloid precursor protein at central and peripheral synaptic sites. Brain Res. (1991) 563:184–94. doi: 10.1016/0006-8993(91)91532-6
317. Ashley, J, Packard, M, Ataman, B, and Budnik, V. Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/mint. J Neurosci. (2005) 25:5943–55. doi: 10.1523/JNEUROSCI.1144-05.2005
318. Torroja, L, Packard, M, Gorczyca, M, White, K, and Budnik, V. The Drosophila β-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci. (1999) 19:7793–803. doi: 10.1523/JNEUROSCI.19-18-07793.1999
319. Esch, FS, Keim, PS, Beattie, EC, Blacher, RW, Culwell, AR, Oltersdorf, T, et al. Cleavage of amyloid β peptide during constitutive processing of its precursor. Science. (1979) 248:1122–4. doi: 10.1126/science.2111583
320. Brinkmalm, G, Brinkmalm, A, Bourgeois, P, Persson, R, Hansson, O, Portelius, E, et al. Soluble amyloid precursor protein α and β in CSF in Alzheimer’s disease. Brain Res. (2013) 1513:117–26. doi: 10.1016/j.brainres.2013.03.019
321. Cai, H, Wang, Y, McCarthy, D, Wen, H, Borchelt, DR, Price, DL, et al. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat Neurosci. (2001) 4:233–4. doi: 10.1038/85064
322. Andreasson, U, Portelius, E, Andersson, ME, Blennow, K, and Zetterberg, H. Aspects of β-amyloid as a biomarker for Alzheimer’s disease. Biomark Med. (2007) 1:59–78. doi: 10.2217/17520363.1.1.59
323. Vassar, R, Bennett, BD, Babu-Khan, S, Kahn, S, Mendiaz, EA, Denis, P, et al. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. (1979) 286:735–41. doi: 10.1126/science.286.5440.735
324. Mishra, S, and Caflisch, A. Dynamics in the active site of β-secretase: a network analysis of atomistic simulations. Biochemistry. (2011) 50:9328–39. doi: 10.1021/bi2011948
325. Sinha, S, Anderson, JP, Barbour, R, Basi, GS, Caccaveffo, R, Davis, D, et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature. (1999) 402:537–40. doi: 10.1038/990114
326. Johansson, K, Wasling, P, Novakova, L, Sjödin, S, Brinkmalm, A, Brinkmalm, G, et al. Cerebrospinal fluid amyloid precursor protein as a potential biomarker of fatigue in multiple sclerosis: a pilot study. Mult Scler Relat Disord. (2022) 63:103846. doi: 10.1016/j.msard.2022.103846
327. Morales, DM, Holubkov, R, Inder, TE, Ahn, HC, Mercer, D, Rao, R, et al. Cerebrospinal fluid levels of amyloid precursor protein are associated with ventricular size in post-hemorrhagic hydrocephalus of prematurity. PLoS One. (2015) 10:e0115045. doi: 10.1371/journal.pone.0115045
328. Morales, DM, Silver, SA, Morgan, CD, Mercer, D, Inder, TE, Holtzman, DM, et al. Lumbar cerebrospinal fluid biomarkers of posthemorrhagic hydrocephalus of prematurity: amyloid precursor protein, soluble amyloid precursor protein α, and L1 cell adhesion molecule. Neurosurgery. (2017) 80:82–90. doi: 10.1227/NEU.0000000000001415
329. Morales, DM, Townsend, RR, Malone, JP, Ewersmann, CA, Macy, EM, Inder, TE, et al. Alterations in protein regulators of neurodevelopment in the cerebrospinal fluid of infants with posthemorrhagic hydrocephalus of prematurity. Mol Cell Proteomics. (2012) 11:M111.011973. doi: 10.1074/mcp.M111.011973
330. Gentleman, SM, Nash, MJ, Sweeting, CJ, Graham, DI, and Roberts, GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. (1993) 160:139–44.
331. Sherriff, FE, Bridges, LR, and Sivaloganathan, S. Early detection of axonal injury after human head trauma using immunocytochemistry for β-amyloid precursor protein. Acta Neuropathol (1994) 87,:55:62, doi: 10.1007/BF00386254
332. Gleckman, AM, Bell, MD, Evans, RJ, and Smith, TW. Diffuse axonal injury in infants with nonaccidental craniocerebral trauma: enhanced detection by beta-amyloid precursor protein immunohistochemical staining. Arch Pathol Lab Med. (1999) 123:146–51.
333. Rogers, JT, Randall, JD, Cahill, CM, Eder, PS, Huang, X, Gunshin, H, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem. (2002) 277:45518–28. doi: 10.1074/jbc.M207435200
334. Rajagopal, A, Rao, AU, Amigo, J, Tian, M, Upadhyay, SK, Hall, C, et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature. (2008) 453:1127–31. doi: 10.1038/nature06934
335. Bednarz, A, Lipiński, P, Starzyński, RR, Tomczyk, M, Nowak, W, Mucha, O, et al. Role of the kidneys in the redistribution of heme-derived iron during neonatal hemolysis in mice. Sci Rep. (2019) 9:11102. doi: 10.1038/s41598-019-47414-y
336. Delaby, C, Rondeau, C, Pouzet, C, Willemetz, A, Pilard, N, Desjardins, M, et al. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS One. (2012) 7:e42199. doi: 10.1371/annotation/d301d160-bbe0-4d48-847f-b8a57aa853c0
337. White, C, Yuan, X, Schmidt, PJ, Bresciani, E, Samuel, TK, Campagna, D, et al. HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. (2013) 17:261–70. doi: 10.1016/j.cmet.2013.01.005
338. Staron, R, Lipinski, P, Lenartowicz, M, Bednarz, A, Gajowiak, A, Smuda, E, et al. Dietary hemoglobin rescues young piglets from severe iron deficiency anemia: duodenal expression profile of genes involved in heme iron absorption. PLoS One. (2017) 12:e0181117. doi: 10.1371/journal.pone.0181117
339. Chen, C, Samuel, TK, Sinclair, J, Dailey, HA, and Hamza, I. An intercellular heme-trafficking protein delivers maternal heme to the embryo during development in c. elegans. Cells. (2011) 145:720–31. doi: 10.1016/j.cell.2011.04.025
340. Shayeghi, M, Latunde-Dada, GO, Oakhill, JS, Laftah, AH, Takeuchi, K, Halliday, N, et al. Identification of an intestinal heme transporter. Cells. (2005) 122:789–801. doi: 10.1016/j.cell.2005.06.025
341. Dang, TN, Bishop, GM, Dringen, R, and Robinson, SR. The putative heme transporter HCP1 is expressed in cultured astrocytes and contributes to the uptake of hemin. Glia. (2010) 58:55–65. doi: 10.1002/glia.20901
342. Schaer, CA, Vallelian, F, Imhof, A, Schoedon, G, and Schaer, DJ. Heme carrier protein (HCP-1) spatially interacts with the CD163 hemoglobin uptake pathway and is a target of inflammatory macrophage activation. J Leukoc Biol. (2008) 83:325–33. doi: 10.1189/jlb.0407226
343. Sharma, S, Dimasi, D, Bröer, S, Kumar, R, and Della, NG. Heme carrier protein 1 (HCP1) expression and functional analysis in the retina and retinal pigment epithelium. Exp Cell Res. (2007) 313:1251–9. doi: 10.1016/j.yexcr.2007.01.019
344. Li, H, Zhang, C, Shen, H, Shen, Z, Wu, L, Mo, F, et al. Physiological stress-induced corticosterone increases heme uptake via KLF4-HCP1 signaling pathway in hippocampus neurons. Sci Rep. (2017) 7:5745. doi: 10.1038/s41598-017-06058-6
345. Zhao, R, Aluri, S, and Goldman, ID. The proton-coupled folate transporter (PCFT-SLC46A1) and the syndrome of systemic and cerebral folate deficiency of infancy: hereditary folate malabsorption. Mol Asp Med. (2017) 53:57–72. doi: 10.1016/j.mam.2016.09.002
346. Devireddy, LR, Hart, DO, Goetz, DH, and Green, MR. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cells. (2010) 141:1006–17. doi: 10.1016/j.cell.2010.04.040
347. Bao, G, Clifton, M, Hoette, TM, Mori, K, Deng, SX, Qiu, A, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol. (2010) 6:602–9. doi: 10.1038/nchembio.402
348. Mesquita, SD, Ferreira, AC, Falcao, AM, Sousa, JC, Oliveira, TG, Correia-Neves, M, et al. Lipocalin 2 modulates the cellular response to amyloid beta. Cell Death Differ. (2014) 21:1588–99. doi: 10.1038/cdd.2014.68
349. Shishido, H, Toyota, Y, Hua, Y, Keep, RF, and Xi, G. Role of lipocalin 2 in intraventricular haemoglobin-induced brain injury. Stroke Vasc Neurol. (2016) 1:37–43. doi: 10.1136/svn-2016-000009
350. Shin, HJ, Jeong, EA, Lee, JY, An, HS, Jang, HM, Ahn, YJ, et al. Lipocalin-2 deficiency reduces oxidative stress and neuroinflammation and results in attenuation of kainic acid-induced hippocampal cell death. Antioxidants. (2021) 10:10. doi: 10.3390/antiox10010100
351. Rathore, KI, Berard, JL, Redensek, A, Chierzi, S, Lopez-Vales, R, Santos, M, et al. Lipocalin 2 plays an immunomodulatory role and has detrimental effects after spinal cord injury. J Neurosci. (2011) 31:13412–9. doi: 10.1523/JNEUROSCI.0116-11.2011
352. Bi, F, Huang, C, Tong, J, Qiu, G, Huang, B, Wu, Q, et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A. (2013) 110:4069–74. doi: 10.1073/pnas.1218497110
353. Lee, S, Lee, WH, Lee, MS, Mori, K, and Suk, K. Regulation by lipocalin-2 of neuronal cell death, migration, and morphology. J Neurosci Res. (2012) 90:540–50. doi: 10.1002/jnr.22779
354. Lee, S, Lee, J, Kim, S, Park, J-Y, Lee, W-H, Mori, K, et al. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol. (2007) 179:3231–41. doi: 10.4049/jimmunol.179.5.3231
355. Lee, S, Park, JY, Lee, WH, Kim, H, Park, HC, Mori, K, et al. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci. (2009) 29:234–49. doi: 10.1523/JNEUROSCI.5273-08.2009
356. Berard, JL, Zarruk, JG, Arbour, N, Prat, A, Yong, VW, Jacques, FH, et al. Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia. (2012) 60:1145–59. doi: 10.1002/glia.22342
357. Dong, M, Xi, G, Keep, RF, and Hua, Y. Role of iron in brain lipocalin 2 upregulation after intracerebral hemorrhage in rats. Brain Res. (2013) 1505:86–92. doi: 10.1016/j.brainres.2013.02.008
358. Jin, M, Kim, JH, Jang, E, Lee, YM, Soo Han, H, Woo, DK, et al. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. (2014) 34:1306–14. doi: 10.1038/jcbfm.2014.83
359. Wang, G, Weng, YC, Chiang, IC, Huang, YT, Liao, YC, Chen, YC, et al. Neutralization of lipocalin-2 diminishes stroke-reperfusion injury. Int J Mol Sci. (2020) 21:1–20. doi: 10.3390/ijms21176253
360. Zhao, N, Xu, X, Jiang, Y, Gao, J, Wang, F, Xu, X, et al. Lipocalin-2 may produce damaging effect after cerebral ischemia by inducing astrocytes classical activation. J Neuroinflammation. (2019) 16:168. doi: 10.1186/s12974-019-1556-7
361. Russell, NH, Black, RT, Lee, NN, Doperalski, AE, Reeves, TM, and Phillips, LL. Time-dependent hemeoxygenase-1, lipocalin-2 and ferritin induction after non-contusion traumatic brain injury. Brain Res. (2019) 1725:146466. doi: 10.1016/j.brainres.2019.146466
362. Lee, S, Kim, J-H, Kim, J-H, Seo, J-W, Han, H-S, Lee, W-H, et al. Lipocalin-2 is a chemokine inducer in the central nervous system. J Biol Chem. (2011) 286:43855–70. doi: 10.1074/jbc.M111.299248
363. Toyota, Y, Wei, J, Xi, G, Keep, RF, and Hua, Y. White matter T2 hyperintensities and blood-brain barrier disruption in the hyperacute stage of subarachnoid hemorrhage in male mice: the role of lipocalin-2. CNS Neurosci Ther. (2019) 25:1207–14. doi: 10.1111/cns.13221
364. Mucha, M, Skrzypiec, AE, Schiavon, E, Attwood, BK, Kucerova, E, and Pawlak, R. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc Natl Acad Sci U S A. (2011) 108:18436–41. doi: 10.1073/pnas.1107936108
365. Yang, J, Goetz, D, Li, JY, Wang, W, Mori, K, Setlik, D, et al. An iron delivery pathway mediated by a lipocalin. Mol Cell. (2002) 10:1045–56. doi: 10.1016/S1097-2765(02)00710-4
366. Song, Y, Sun, S, Yu, Y, Li, G, Song, J, Zhang, H, et al. Diagnostic value of neutrophil gelatinase-associated lipocalin for renal injury in asphyxiated preterm infants. Exp Ther Med. (2017) 13:1245–8. doi: 10.3892/etm.2017.4107
367. Sellmer, A, Bech, BH, Bjerre, JV, Schmidt, MR, Hjortdal, VE, Esberg, G, et al. Urinary neutrophil gelatinase-associated Lipocalin in the evaluation of patent ductus arteriosus and AKI in very preterm neonates: a cohort study. BMC Pediatr. (2017) 17:7. doi: 10.1186/s12887-016-0761-0
368. Naunova-Timovska, S, Cekovska, S, Sahpazova, E, and Tasić, V. Neutrophil gelatinase-associated lipocalin as an early biomarker of acute kidney injury in newborns. Acta Clin Croat. (2020) 59:55. doi: 10.20471/acc.2020.59.01.07
369. Mikulić, V, Rogić, D, Mikulić, I, Ljubić, K, Ćuk, A, Tomić, V, et al. Urine neutrophil gelatinase-associated lipocalin concentration in healthy newborns during the first three postnatal days. Biochem Med (Zagreb). (2020) 30:1–5. doi: 10.11613/BM.2020.030706
370. Baumert, M, Surmiak, P, Więcek, A, and Walencka, Z. Serum NGAL and copeptin levels as predictors of acute kidney injury in asphyxiated neonates. Clin Exp Nephrol. (2017) 21:658–64. doi: 10.1007/s10157-016-1320-6
371. Huynh, TK, Bateman, DA, Parravicini, E, Lorenz, JM, Nemerofsky, SL, Sise, ME, et al. Reference values of urinary neutrophil gelatinase-associated lipocalin in very low birth weight infants. Pediatr Res. (2009) 66:528. doi: 10.1203/PDR.0b013e3181baa3dd
372. Surmiak, P, Baumert, M, Fiala, M, Walencka, Z, and Więcek, A. Umbilical neutrophil gelatinase-associated lipocalin level as an early predictor of acute kidney injury in neonates with hypoplastic left heart syndrome. Biomed Res Int. (2015) 2015:360209. doi: 10.1155/2015/360209
373. Thibault, MP, Tremblay, É, Horth, C, Fournier-Morin, A, Grynspan, D, Babakissa, C, et al. Lipocalin-2 and calprotectin as stool biomarkers for predicting necrotizing enterocolitis in premature neonates. Pediatr Res. (2022) 91:129. doi: 10.1038/s41390-021-01680-7
374. Midan, D, El-Gendy, F, Abo Elalla, D, and Kotb, M. Clinical assessment of neutrophil gelatinase-associated lipocalin as a potential diagnostic marker for neonatal sepsis: a prospective cohort study. Ann Med. (2022) 54:1725–31. doi: 10.1080/07853890.2022.2091789
375. Spoelgen, R, Hammes, A, Anzenberger, U, Zechner, D, Andersen, OM, Jerchow, B, et al. LRP2/megalin is required for patterning of the ventral telencephalon. Development. (2005) 132:405–14. doi: 10.1242/dev.01580
376. Fisher, CE, and Howie, SEM. The role of megalin (LRP-2/Gp330) during development. Dev Biol. (2006) 296:279–97. doi: 10.1016/j.ydbio.2006.06.007
377. Li, JY, Paragas, N, Ned, RM, Qiu, A, Viltard, M, Leete, T, et al. Scara5 is a ferritin receptor mediating non-transferrin Iron delivery. Dev Cell. (2009) 16:35. doi: 10.1016/j.devcel.2008.12.002
378. Jiang, Y, Oliver, P, Davies, KE, and Platt, N. Identification and characterization of murine SCARA5, a novel class A scavenger receptor that is expressed by populations of epithelial cells. J Biol Chem. (2006) 281:11834–45. doi: 10.1074/jbc.M507599200
379. Mendes-Jorge, L, Ramos, D, Valenҫa, A, López-Luppo, M, Pires, VMR, Catita, J, et al. L-ferritin binding to Scara5: a New Iron traffic pathway potentially implicated in retinopathy. PLoS One. (2014) 9:e106974. doi: 10.1371/journal.pone.0106974
380. Yu, B, Cheng, C, Wu, Y, Guo, L, Kong, D, Zhang, Z, et al. Interactions of ferritin with scavenger receptor class a members. J Biol Chem. (2020) 295:15727–41. doi: 10.1074/jbc.RA120.014690
381. Swystun, LL, Ogiwara, K, Lai, JD, Ojala, JRM, Rawley, O, Lassalle, F, et al. The scavenger receptor SCARA5 is an endocytic receptor for von Willebrand factor expressed by littoral cells in the human spleen. J Thromb Haemost. (2019) 17:1384. doi: 10.1111/jth.14521
382. Srimanee, A, Regberg, J, Hällbrink, M, Vajragupta, O, and Langel, Ü. Role of scavenger receptors in peptide-based delivery of plasmid DNA across a blood–brain barrier model. Int J Pharm. (2016) 500:128–35. doi: 10.1016/j.ijpharm.2016.01.014
383. Kurronen, A, Pihlaja, R, Pollari, E, Kanninen, K, Storvik, M, Wong, G, et al. Adult and neonatal astrocytes exhibit diverse gene expression profiles in response to beta amyloid ex vivo. World J Neurosci. (2012) 2:57–67. doi: 10.4236/wjns.2012.22009
384. Brain tissue expression of SCARA5 - Summary – the human protein atlas. Available at:https://www.proteinatlas.org/ENSG00000168079-SCARA5/brain
385. Chong, M, Sjaarda, J, Pigeyre, M, Mohammadi-Shemirani, P, Lali, R, Shoamanesh, A, et al. Novel drug targets for ischemic stroke identified through Mendelian randomization analysis of the blood proteome. Circulation. (2019) 140:819–30. doi: 10.1161/CIRCULATIONAHA.119.040180
386. Fandrey, J, and Gassmann, M. Oxygen sensing and the activation of the hypoxia inducible factor 1 (HIF-1). Adv Exp Med Biol. (2009). doi: 10.1007/978-90-481-2259-2_23
387. Mylonis, I, Sembongi, H, Befani, C, Liakos, P, Siniossoglou, S, and Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J Cell Sci. (2012) 125:3485–93. doi: 10.1242/jcs.106682
388. Shah, YM, and Xie, L. Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology. (2014) 146:630–42. doi: 10.1053/j.gastro.2013.12.031
389. Rishi, G, and Subramaniam, VN. The relationship between systemic iron homeostasis and erythropoiesis. Biosci Rep. (2017) 37:BSR20170195. doi: 10.1042/BSR20170195
390. Lok, CN, and Ponka, P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. (1999) 274:24147–52. doi: 10.1074/jbc.274.34.24147
391. Rolfs, A, Kvietikova, I, Gassmann, M, and Wenger, RH. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J Biol Chem. (1997) 272:20055–62. doi: 10.1074/jbc.272.32.20055
392. Mukhopadhyay, CK, Mazumder, B, and Fox, PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem. (2000) 275:21048–54. doi: 10.1074/jbc.M000636200
393. Lee, PJ, Jiang, BH, Chin, BY, Iyer, NV, Alam, J, Semenza, GL, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem. (1997) 272:5375–81. doi: 10.1074/jbc.272.9.5375
394. Shah, YM, Matsubara, T, Ito, S, Yim, SH, and Gonzalez, FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following Iron deficiency. Cell Metab. (2009) 9:152–64. doi: 10.1016/j.cmet.2008.12.012
395. Wang, D, Wang, LH, Zhao, Y, Lu, YP, and Zhu, L. Hypoxia regulates the ferrous iron uptake and reactive oxygen species level via divalent metal transporter 1 (DMT1) exon1B by hypoxia-inducible factor-1. IUBMB Life. (2010) 62:629–36. doi: 10.1002/iub.363
396. Taylor, M, Qu, A, Anderson, ER, Matsubara, T, Martin, A, Gonzalez, FJ, et al. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology. (2011) 140:2044–55. doi: 10.1053/j.gastro.2011.03.007
397. Mastrogiannaki, M, Matak, P, Keith, B, Simon, MC, Vaulont, S, and Peyssonnaux, C. HIF-2α, but not HIF-1α, promotes iron absorption in mice. J Clin Investig. (2009) 119:1159–66. doi: 10.1172/JCI38499
398. Jain, S, Maltepe, E, Lu, MM, Simon, C, and Bradfield, CA. Expression of ARNT, ARNT2, HIF1α, HIF2α and Ah receptor mRNAs in the developing mouse. Mech Dev. (1998) 73:117–23. doi: 10.1016/S0925-4773(98)00038-0
399. Kleszka, K, Leu, T, Quinting, T, Jastrow, H, Pechlivanis, S, Fandrey, J, et al. Hypoxia-inducible factor-2α is crucial for proper brain development. Sci Rep. (2020) 10:10. doi: 10.1038/s41598-020-75838-4
400. Steimle, BL, Smith, FM, and Kosman, DJ. The solute carriers ZIP8 and ZIP14 regulate manganese accumulation in brain microvascular endothelial cells and control brain manganese levels. J Biol Chem. (2019) 294:19197–208. doi: 10.1074/jbc.RA119.009371
401. Liuzzi, JP, Aydemir, F, Nam, H, Knutson, MD, and Cousins, RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A. (2006) 103:13612–7. doi: 10.1073/pnas.0606424103
402. Pinilla-Tenas, JJ, Sparkman, BK, Shawki, A, Illing, AC, Mitchell, CJ, Zhao, N, et al. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am J Physiol Cell Physiol. (2011) 301:C862–71. doi: 10.1152/ajpcell.00479.2010
403. Zhao, N, Gao, J, Enns, CA, and Knutson, MD. ZRT/IRT-like protein 14 (ZIP14) promotes the cellular assimilation of iron from transferrin. J Biol Chem. (2010) 285:32141–50. doi: 10.1074/jbc.M110.143248
404. Nishikawa, M, Mori, H, and Hara, M. Analysis of ZIP (Zrt-, Irt-related protein) transporter gene expression in murine neural stem/progenitor cells. Environ Toxicol Pharmacol. (2017) 53:81–8. doi: 10.1016/j.etap.2017.05.008
405. Hojyo, S, Fukada, T, Shimoda, S, Ohashi, W, Bin, BH, Koseki, H, et al. The zinc transporter SLC39A14/ZIP14 controls G-protein coupled receptor-mediated signaling required for systemic growth. PLoS One. (2011) 6:e18059. doi: 10.1371/journal.pone.0018059
406. Gálvez-Peralta, M, He, L, Jorge-Nebert, LF, Wang, B, Miller, ML, Eppert, BL, et al. Zip8 zinc transporter: indispensable role for both multiple-organ organogenesis and hematopoiesis in utero. PLoS One. (2012) 7:e36055. doi: 10.1371/journal.pone.0036055
407. McCoy, TH, Pellegrini, AM, and Perlis, RH. Using phenome-wide association to investigate the function of a schizophrenia risk locus at SLC39A8. Transl Psychiatry. (2019) 9:45. doi: 10.1038/s41398-019-0386-9
408. Jenkitkasemwong, S, Wang, CY, Coffey, R, Zhang, W, Chan, A, Biel, T, et al. SLC39A14 is required for the development of hepatocellular Iron overload in murine models of hereditary hemochromatosis. Cell Metab. (2015) 22:138–50. doi: 10.1016/j.cmet.2015.05.002
409. Boveris, A, and Cadenas, E. Mitochondrial production of hydrogen peroxide regulation by nitric oxide and the role of ubisemiquinone. IUBMB Life. (2000) 50:245–50. doi: 10.1080/713803732
410. Bao, L, Avshalumov, MV, Patel, JC, Lee, CR, Miller, EW, Chang, CJ, et al. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J Neurosci. (2009) 29:9002–10. doi: 10.1523/JNEUROSCI.1706-09.2009
411. Andrés Juan, C, Pérez, M, de la Lastra, J, Plou, FJ, Pérez-Lebeña, E, and Reinbothe, S. Molecular sciences the chemistry of reactive oxygen species (ROS) revisited: outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int J Mol Sci. (2021) 22:22. doi: 10.3390/ijms22094642
412. Kannan, K, and Jain, SK. Oxidative stress and apoptosis. Pathophysiology. (2000) 7:153–63. doi: 10.1016/S0928-4680(00)00053-5
413. Xiao, M, Zhong, H, Xia, L, Tao, Y, and Yin, H. Pathophysiology of mitochondrial lipid oxidation: role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic Biol Med. (2017) 111:316–27. doi: 10.1016/j.freeradbiomed.2017.04.363
414. Kagan, VE, Tyurin, VA, Jiang, J, Tyurina, YY, Ritov, VB, Amoscato, AA, et al. Cytochrome C acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. (2005) 1:223–32. doi: 10.1038/nchembio727
415. Su, LJ, Zhang, JH, Gomez, H, Murugan, R, Hong, X, Xu, D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxidative Med Cell Longev. (2019) 2019:1–13. doi: 10.1155/2019/5080843
416. Zhong, H, Xiao, M, Zarkovic, K, Zhu, M, Sa, R, Lu, J, et al. Mitochondrial control of apoptosis through modulation of cardiolipin oxidation in hepatocellular carcinoma: a novel link between oxidative stress and cancer. Free Radic Biol Med. (2017) 102:67–76. doi: 10.1016/j.freeradbiomed.2016.10.494
417. Chaudhary, P, Sharma, R, Sharma, A, Vatsyayan, R, Yadav, S, Singhal, SS, et al. Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry. (2010) 49:6263–75. doi: 10.1021/bi100517x
418. Elkin, ER, Harris, SM, and Loch-Caruso, R. Trichloroethylene metabolite S-(1,2-dichlorovinyl)-L-cysteine induces lipid peroxidation-associated apoptosis via the intrinsic and extrinsic apoptosis pathways in a first-trimester placental cell line. Toxicol Appl Pharmacol. (2018) 338:30–42. doi: 10.1016/j.taap.2017.11.006
419. Dixon, SJ, Lemberg, KM, Lamprecht, MR, Skouta, R, Zaitsev, EM, Gleason, CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cells. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
420. Feng, H, and Stockwell, BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
421. Meng, Z, Liu, C, Chen, C, Di, F, Zhang, S, Liang, X, et al. The induction of ferroptosis in posthemorrhagic hydrocephalus. Brain Res. (2023) 1798:148133. doi: 10.1016/j.brainres.2022.148133
422. Sevensky, R, Newville, JC, Tang, HL, Robinson, S, and Jantzie, LL. Cumulative damage: cell death in posthemorrhagic hydrocephalus of prematurity. Cells. (2021) 10:1911. doi: 10.3390/cells10081911
423. Doll, S, and Conrad, M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. (2017) 69:423–34. doi: 10.1002/iub.1616
424. Goulding, DS, Caleb Vogel, R, Gensel, JC, Morganti, JM, Stromberg, AJ, and Miller, BA. Acute brain inflammation, white matter oxidative stress, and myelin deficiency in a model of neonatal intraventricular hemorrhage. J Neurosurg Pediatr. (2020) 26:613–23. doi: 10.3171/2020.5.PEDS20124
425. Gram, M, Sveinsdottir, S, Ruscher, K, Hansson, SR, Cinthio, M, Åkerström, B, et al. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J Neuroinflammat. (2013) 10:100. doi: 10.1186/1742-2094-10-100
426. Simard, PF, Tosun, C, Melnichenko, L, Ivanova, S, Gerzanich, V, and Simard, JM. Inflammation of the choroid plexus and ependymal layer of the ventricle following intraventricular hemorrhage. Transl Stroke Res. (2011) 2:227–31. doi: 10.1007/s12975-011-0070-8
427. Karimy, JK, Reeves, BC, Damisah, E, Duy, PQ, Antwi, P, David, W, et al. Inflammation in acquired hydrocephalus: pathogenic mechanisms and therapeutic targets. Nat Rev Neurol. (2020) 16:285–96. doi: 10.1038/s41582-020-0321-y
428. Gram, M, Sveinsdottir, S, Cinthio, M, Sveinsdottir, K, Hansson, SR, Mörgelin, M, et al. Extracellular hemoglobin - mediator of inflammation and cell death in the choroid plexus following preterm intraventricular hemorrhage. J Neuroinflammation. (2014) 11:200. doi: 10.1186/s12974-014-0200-9
429. Miller, BA, Pan, S, Yang, PH, Wang, C, Trout, AL, DeFreitas, D, et al. Modeling neonatal intraventricular hemorrhage through intraventricular injection of hemoglobin. J Vis Exp. (2022). doi: 10.3791/63345-v
430. Holste, KG, Xia, F, Ye, F, Keep, RF, and Xi, G. Mechanisms of neuroinflammation in hydrocephalus after intraventricular hemorrhage: a review. Fluids Barriers CNS. (2022) 19:28. doi: 10.1186/s12987-022-00324-0
431. Peng, K, Koduri, S, Xia, F, Gao, F, Hua, Y, Keep, RF, et al. Impact of sex differences on thrombin-induced hydrocephalus and white matter injury: the role of neutrophils. Fluids Barriers CNS. (2021) 18:38. doi: 10.1186/s12987-021-00273-0
432. Zamorano, M, Olson, SD, Haase, C, Herrera, JJ, Huang, S, Sequeira, DJ, et al. Innate immune activation and white matter injury in a rat model of neonatal intraventricular hemorrhage are dependent on developmental stage. Exp Neurol. (2023) 367:114472. doi: 10.1016/j.expneurol.2023.114472
433. Gram, M, Sveinsdottir, S, Vallius, S, Sveinsdottir, K, Cinthio, M, and Ley, D. PO-0395 scavenging of extracellular haemoglobin modifies the monocyte-macrophage recruitment and differentiation in the intraventricular space following Ivh. Arch Dis Child. (2014) 99:A375.1–A375. doi: 10.1136/archdischild-2014-307384.1041
434. Duy, PQ, Greenberg, ABW, Butler, WE, and Kahle, KT. Rethinking the cilia hypothesis of hydrocephalus. Neurobiol Dis. (2022) 175:105913. doi: 10.1016/j.nbd.2022.105913
435. Karimy, JK, Zhang, J, Kurland, DB, Theriault, BC, Duran, D, Stokum, JA, et al. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med. (2017) 23:997–1003. doi: 10.1038/nm.4361
436. Sadegh, C, Xu, H, Sutin, J, Fatou, B, Gupta, S, Pragana, A, et al. Choroid plexus-targeted NKCC1 overexpression to treat post-hemorrhagic hydrocephalus. Neuron. (2023) 111:1591–1608.e4. doi: 10.1016/j.neuron.2023.02.020
437. Ramagiri, S, Pan, S, DeFreitas, D, Yang, PH, Raval, DK, Wozniak, DF, et al. Deferoxamine prevents neonatal posthemorrhagic hydrocephalus through choroid plexus-mediated Iron clearance. Transl Stroke Res. (2022) 14:704–22. doi: 10.1007/s12975-022-01092-7
438. Meng, H, Li, F, Hu, R, Yuan, Y, Gong, G, Hu, S, et al. Deferoxamine alleviates chronic hydrocephalus after intraventricular hemorrhage through iron chelation and Wnt1/Wnt3a inhibition. Brain Res. (2015) 1602:44–52. doi: 10.1016/j.brainres.2014.08.039
439. Klebe, D, Krafft, PR, Hoffmann, C, Lekic, T, Flores, JJ, Rolland, W, et al. Acute and delayed Deferoxamine treatment attenuates long-term sequelae after germinal matrix hemorrhage in neonatal rats. Stroke. (2014) 45:2475–9. doi: 10.1161/STROKEAHA.114.005079
440. Chen, Q, Tang, J, Tan, L, Guo, J, Tao, Y, Li, L, et al. Intracerebral hematoma contributes to hydrocephalus after intraventricular hemorrhage via aggravating iron accumulation. Stroke. (2015) 46:2902–8. doi: 10.1161/STROKEAHA.115.009713
441. Mirzadeh, Z, Merkle, FT, Soriano-Navarro, M, Garcia-Verdugo, JM, and Alvarez-Buylla, A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. (2008) 3:265–78. doi: 10.1016/j.stem.2008.07.004
442. Robinson, S, Conteh, FS, Oppong, AY, Yellowhair, TR, Newville, JC, El, DN, et al. Extended combined neonatal treatment with erythropoietin plus melatonin prevents posthemorrhagic hydrocephalus of prematurity in rats. Front Cell Neurosci. (2018) 12:322. doi: 10.3389/fncel.2018.00322
443. Liu, C, Cao, Y, Wang, HX, Zhao, L, Chen, YX, Zhong, KH, et al. Necrostatin-1 decreases necroptosis and inflammatory markers after intraventricular hemorrhage in mice. Neural Regen Res. (2022) 17:2710. doi: 10.4103/1673-5374.339488
444. Lummis, NC, Sánchez-Pavón, P, Kennedy, G, Frantz, AJ, Kihara, Y, Blaho, VA, et al. LPA1/3 overactivation induces neonatal posthemorrhagic hydrocephalus through ependymal loss and ciliary dysfunction. Sci Adv. (2019) 5:5. doi: 10.1126/sciadv.aax2011
445. Mirzadeh, Z, Kusne, Y, Duran-Moreno, M, Cabrales, E, Gil-Perotin, S, Ortiz, C, et al. Bi- and uniciliated ependymal cells define continuous floor-plate-derived tanycytic territories. Nat Commun. (2017) 8:13759. doi: 10.1038/ncomms13759
446. Del Brio, MA, Riera, P, Garcia, JM, and Alvarez-Uria, M. Cell types of the third ventricle wall of the rabbit (Oryctolagus cuniculus). J Submicrosc Cytol Pathol. (1991) 23:147–57.
447. Singla, V, and Reiter, JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. (1979) 313:629–33. doi: 10.1126/science.1124534
448. Dawes, W . Secondary brain injury following neonatal intraventricular hemorrhage: the role of the ciliated ependyma. Front Pediatr. (2022) 10:887606. doi: 10.3389/fped.2022.887606
449. Greitz, D . Cerebrospinal fluid circulation and associated intracranial dynamics. A radiologic investigation using MR imaging and radionuclide cisternography. Acta Radiol Suppl. (1993) 386:1–23.
450. Greitz, D, Wirestam, R, Franck, A, Nordell, B, Thomsen, C, and Ståhlberg, F. Pulsatile brain movement and associated hydrodynamics studied by magnetic resonance phase imaging – the Monro-Kellie doctrine revisited. Neuroradiology. (1992) 34:370–80. doi: 10.1007/BF00596493
451. Yamada, S . Cerebrospinal fluid physiology: visualization of cerebrospinal fluid dynamics using the magnetic resonance imaging time-spatial inversion pulse method. Croat Med J. (2014) 55:337–46. doi: 10.3325/cmj.2014.55.337
452. Olstad, EW, Ringers, C, Hansen, JN, Wens, A, Brandt, C, Wachten, D, et al. Ciliary beating compartmentalizes cerebrospinal fluid flow in the brain and regulates ventricular development. Curr Biol. (2019) 29:229–241.e6. doi: 10.1016/j.cub.2018.11.059
453. Xu, H, Fame, RM, Sadegh, C, Sutin, J, Naranjo, C, Syau, D, et al. Choroid plexus NKCC1 mediates cerebrospinal fluid clearance during mouse early postnatal development. Nature. Communications. (2021) 12:1–16. doi: 10.1038/s41467-020-20666-3
454. Lolansen, SD, Rostgaard, N, Barbuskaite, D, Capion, T, Olsen, MH, Norager, NH, et al. Posthemorrhagic hydrocephalus associates with elevated inflammation and CSF hypersecretion via activation of choroidal transporters. Fluids Barriers CNS. (2022) 19:62. doi: 10.1186/s12987-022-00360-w
455. Cherian, S, Thoresen, M, Silver, IA, Whitelaw, A, and Love, S. Transforming growth factor-βs in a rat model of neonatal posthaemorrhagic hydrocephalus. Neuropathol Appl Neurobiol. (2004) 30:585–600. doi: 10.1111/j.1365-2990.2004.00588.x
456. Zhang, Z, Tan, Q, Guo, P, Huang, S, Jia, Z, Liu, X, et al. NLRP3 inflammasome-mediated choroid plexus hypersecretion contributes to hydrocephalus after intraventricular hemorrhage via phosphorylated NKCC1 channels. J Neuroinflammation. (2022) 19:163. doi: 10.1186/s12974-022-02530-x
457. Alshareef, M, Mallah, K, Vasas, T, Alawieh, A, Borucki, D, Couch, C, et al. A role of complement in the pathogenic sequelae of mouse neonatal germinal matrix hemorrhage. Int J Mol Sci. (2022) 23:2943. doi: 10.3390/ijms23062943
458. Hatchell, D, Alshareef, M, Vasas, T, Guglietta, S, Borucki, D, Guo, C, et al. A role for P-selectin and complement in the pathological sequelae of germinal matrix hemorrhage. J Neuroinflammation. (2023) 20:143. doi: 10.1186/s12974-023-02828-4
459. Gu, W, Li, F, Zhang, W, and Jia, P. Expression and significance of aquaporin protein in sprague-dawley rats after experimental intraventricular hemorrhage. Cell Mol Biol. (2016) 62:59–62.
460. Sveinsdottir, S, Gram, M, Cinthio, M, Sveinsdottir, K, Mörgelin, M, and Ley, D. Altered expression of aquaporin 1 and 5 in the choroid plexus following preterm intraventricular hemorrhage. Dev Neurosci. (2014) 36:542–51. doi: 10.1159/000366058
461. Pang, D, Sclabassi, RJ, and Horton, JA. Lysis of intraventricular blood clot with urokinase in a canine model: part 3. Effects of intraventricular urokinase on clot lysis and posthemorrhagic hydrocephalus. Neurosurgery. (1986) 19:553–72. doi: 10.1227/00006123-198610000-00010
462. Hill, A, Shackelford, GD, and Volpe, JJ. A potential mechanism of pathogenesis for early posthemorrhagic hydrocephalus in the premature newborn. Pediatrics. (1984) 73:19–21. doi: 10.1542/peds.73.1.19
463. Larroche, JC . Post-haemorrhagic hydrocephalus in infancy. Anatomical study. Biol Neonate. (1972) 20:287–99.
464. Di, HX, Le, CS, Zhang, HM, Shang, DS, Tong, LS, and Gao, F. Thrombin disrupts vascular endothelial-cadherin and leads to hydrocephalus via protease-activated receptors-1 pathway. CNS Neurosci Ther. (2019) 25:1142–50. doi: 10.1111/cns.13129
465. Ben-Shoshan,, and David, S. CSF hypersecretion versus impaired CSF absorption in posthemorrhagic hydrocephalus: a systematic review. Acta neurochi. (2023) 165:3271–3287. doi: 10.1007/s00701-023-05746-9
466. Hale, AT, Bastarache, L, Morales, DM, Wellons, JC, Limbrick, DD, and Gamazon, ER. Multi-omic analysis elucidates the genetic basis of hydrocephalus. Cell Rep. (2021) 35:109085. doi: 10.1016/j.celrep.2021.109085
467. Martinussen, M, Flanders, DW, Fischl, B, Busa, E, Løhaugen, GC, Psychol, C, et al. Segmental brain volumes and cognitive and perceptual correlates in 15-year-old adolescents with low birth weight. J Pediatr. (2009) 155:848–853.e1. doi: 10.1016/j.jpeds.2009.06.015
468. Thompson, DK, Wood, SJ, Doyle, LW, Warfield, SK, Lodygensky, GA, Anderson, PJ, et al. Neonate hippocampal volumes: prematurity, perinatal predictors, and 2-year outcome. Ann Neurol. (2008) 63:642–51. doi: 10.1002/ana.21367
469. Murphy, BP, Inder, TE, Rooks, V, Taylor, GA, Anderson, NJ, Mogridge, N, et al. Posthaemorrhagic ventricular dilatation in the premature infant: natural history and predictors of outcome. Arch Dis Child Fetal Neonatal Ed. (2002) 87:37F–341F. doi: 10.1136/fn.87.1.F37
470. Riva-Cambrin, J, Kestle, JRW, Holubkov, R, Butler, J, Kulkarni, AV, Drake, J, et al. Risk factors for shunt malfunction in pediatric hydrocephalus: a multicenter prospective cohort study. J Neurosurg Pediatr. (2016) 17:382–90. doi: 10.3171/2015.6.PEDS14670
471. Stone, SSD, and Warf, BC. Combined endoscopic third ventriculostomy and choroid plexus cauterization as primary treatment for infant hydrocephalus: a prospective North American series. J Neurosurg Pediatr. (2014) 14:439–46. doi: 10.3171/2014.7.PEDS14152
472. Kulkarni, AV, Riva-Cambrin, J, Rozzelle, CJ, Naftel, RP, Alvey, JS, Reeder, RW, et al. Endoscopic third ventriculostomy and choroid plexus cauterization in infant hydrocephalus: a prospective study by the hydrocephalus clinical research network. J Neurosurg Pediatr. (2018) 21:214–23. doi: 10.3171/2017.8.PEDS17217
473. Kulkarni, AV, Riva-Cambrin, J, Browd, SR, Drake, JM, Holubkov, R, Kestle, JRW, et al. Endoscopic third ventriculostomy and choroid plexus cauterization in infants with hydrocephalus: a retrospective hydrocephalus clinical research network study. J Neurosurg Pediatr. (2014) 14:224–9. doi: 10.3171/2014.6.PEDS13492
474. Riva-Cambrin, J, Kestle, JRW, Rozzelle, CJ, Naftel, RP, Alvey, JS, Reeder, RW, et al. Predictors of success for combined endoscopic third ventriculostomy and choroid plexus cauterization in a North American setting: a hydrocephalus clinical research network study. J Neurosurg Pediatr. (2019) 24:128–38. doi: 10.3171/2019.3.PEDS18532
475. Gao, C, Du, H, Hua, Y, Keep, RF, Strahle, J, and Xi, G. Role of red blood cell lysis and iron in hydrocephalus after intraventricular hemorrhage. J Cereb Blood Flow Metab. (2014) 34:1070–5. doi: 10.1038/jcbfm.2014.56
476. Young, N . Treatment of primary hemochromatosis with deferoxamine. J Am Med Assoc. (1979) 241:1152. doi: 10.1001/jama.1979.03290370056031
477. Nielsen, P, Fischer, R, Buggisch, P, and Janka-Schaub, G. Effective treatment of hereditary haemochromatosis with desferrioxamine in selected cases. Br J Haematol. (2003) 123:952–3. doi: 10.1046/j.1365-2141.2003.04708.x
478. Borgna-Pignatti, C, Rugolotto, S, De Stefano, P, Zhao, H, Cappellini, MD, Del Vecchio, GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. (2004) 89:1187–93.
479. Gabutti, V, and Piga, A. Results of long-term Iron-chelating therapy. Acta Haematol. (1996) 95:26–36. doi: 10.1159/000203853
480. Brittenham, GM, Cohen, AR, McLaren, CE, Martin, MB, Griffith, PM, Nienhuis, AW, et al. Hepatic iron stores and plasma ferritin concentration in patients with sickle cell anemia and thalassemia major. Am J Hematol. (1993) 42:81–5. doi: 10.1002/ajh.2830420116
481. Romantsik, O, Agyemang, AA, Sveinsdóttir, S, Rutardóttir, S, Holmqvist, B, Cinthio, M, et al. The heme and radical scavenger α1-microglobulin (A1M) confers early protection of the immature brain following preterm intraventricular hemorrhage. J Neuroinflammation. (2019) 16:122. doi: 10.1186/s12974-019-1486-4
482. Agyemang, AA, Sveinsdóttir, K, Vallius, S, Sveinsdóttir, S, Bruschettini, M, Romantsik, O, et al. Cerebellar exposure to cell-free hemoglobin following preterm intraventricular hemorrhage: causal in cerebellar damage? Transl Stroke Res. (2017) 8:461–73. doi: 10.1007/s12975-017-0539-1
483. Wimmer, I, Scharler, C, Kadowaki, T, Hillebrand, S, Scheiber-Mojdehkar, B, Ueda, S, et al. Iron accumulation in the choroid plexus, ependymal cells and CNS parenchyma in a rat strain with low-grade haemolysis of fragile macrocytic red blood cells. Brain Pathol. (2021) 31:333–45. doi: 10.1111/bpa.12920
484. Hale, AT, Brown, RE, Luka, Z, Hudson, BH, Matta, P, Williams, CS, et al. Modulation of sulfur assimilation metabolic toxicity overcomes anemia and hemochromatosis in mice. Adv Biol Regul. (2020) 76:100694. doi: 10.1016/j.jbior.2020.100694
485. Hudson, BH, Hale, AT, Irving, RP, Li, S, and York, JD. Modulation of intestinal sulfur assimilation metabolism regulates iron homeostasis. Proc Natl Acad Sci U S A. (2018) 115:3000–5. doi: 10.1073/pnas.1715302115
486. Guillama Barroso, G, Narayan, M, Alvarado, M, Armendariz, I, Bernal, J, Carabaza, X, et al. Nanocarriers as potential drug delivery candidates for overcoming the blood-brain barrier: challenges and possibilities. ACS Omega. (2020) 5:12583–95. doi: 10.1021/acsomega.0c01592
487. Zhou, Y, Peng, Z, Seven, ES, and Leblanc, RM. Crossing the blood-brain barrier with nanoparticles. J Control Release. (2018) 270:290–303. doi: 10.1016/j.jconrel.2017.12.015
488. Hersh, AM, Alomari, S, and Tyler, BM. Crossing the blood-brain barrier: advances in nanoparticle technology for drug delivery in neuro-oncology. Int J Mol Sci. (2022) 23:4153. doi: 10.3390/ijms23084153
489. Chen, H, Chen, CC, Acosta, C, Wu, SY, Sun, T, and Konofagou, EE. A new brain drug delivery strategy: Focused ultrasound-enhanced intranasal drug delivery. PLoS One. (2014) 9:e108880. doi: 10.1371/journal.pone.0108880
490. Hynynen, K, McDannold, N, Vykhodtseva, N, Raymond, S, Weissleder, R, Jolesz, FA, et al. Focal disruption of the blood-brain barrier due to 260-kHz ultrasound bursts: a method for molecular imaging and targeted drug delivery. J Neurosurg. (2006) 105:445–54. doi: 10.3171/jns.2006.105.3.445
491. Hynynen, K, McDannold, N, Sheikov, NA, Jolesz, FA, and Vykhodtseva, N. Local and reversible blood-brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. NeuroImage. (2005) 24:12–20. doi: 10.1016/j.neuroimage.2004.06.046
492. Mesiwala, AH, Farrell, L, Wenzel, HJ, Silbergeld, DL, Crum, LA, Winn, HR, et al. High-intensity focused ultrasound selectively disrupts the blood-brain barrier in vivo. Ultrasound Med Biol. (2002) 28:389–400. doi: 10.1016/S0301-5629(01)00521-X
493. McDannold, NJ, Vykhodtseva, NI, and Hynynen, K. Microbubble contrast agent with focused ultrasound to create brain lesions at low power levels: MR imaging and histologic study in rabbits. Radiology. (2006) 241:95–106. doi: 10.1148/radiol.2411051170
494. Hynynen, K, McDannold, N, Vykhodtseva, N, and Jolesz, FA. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology. (2001) 220:640–6. doi: 10.1148/radiol.2202001804
Keywords: germinal matrix hemorrhage, intraventricular hemorrhage, posthemorrhagic hydrocephalus, iron overload, choroid plexus, ependyma, ferroportin, iron transporters
Citation: Pan S, Hale AT, Lemieux ME, Raval DK, Garton TP, Sadler B, Mahaney KB and Strahle JM (2024) Iron homeostasis and post-hemorrhagic hydrocephalus: a review. Front. Neurol. 14:1287559. doi: 10.3389/fneur.2023.1287559
Edited by:
Diego Iacono, Neuroscience – Uniformed Services University of the Health Sciences (USU), United StatesReviewed by:
Sirena Soriano, Houston Methodist Research Institute, United StatesCopyright © 2024 Pan, Hale, Lemieux, Raval, Garton, Sadler, Mahaney and Strahle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer M. Strahle, c3RyYWhsZWpAd3VzdGwuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.