 You-Ri Kang1†
You-Ri Kang1† Tai-Seung Nam1,2*†
Tai-Seung Nam1,2*† Jae-Myung Kim1†
Jae-Myung Kim1† Kyung Wook Kang1,2†
Kyung Wook Kang1,2† Seong-Min Choi1,2†
Seong-Min Choi1,2† Seung-Han Lee1,2†
Seung-Han Lee1,2† Byeong C. Kim1,2†Myeong-Kyu Kim1,2†
Byeong C. Kim1,2†Myeong-Kyu Kim1,2†- 1Department of Neurology, Chonnam National University Hospital, Gwangju, Republic of Korea
- 2Department of Neurology, Chonnam National University Medical School, Gwangju, Republic of Korea
Background: To analyze the clinical phenotype of hereditary spastic paraplegia (HSP) caused by SPG11 mutations (SPG11-HSP).
Methods: Among the 17 patients with sporadic HSP who performed whole exome sequencing analysis, six were diagnosed with SPG11-HSP. The clinical and radiologic findings and the results of the electrodiagnostic and neuropsychologic tests were reviewed retrospectively.
Results: The median age at onset was 16.5 years (range, 13–38 years). Progressive spastic paraparesis was a core feature, and the median spastic paraplegia rating scale score was 24/52 (range, 16–31 points). Additional major symptoms were pseudobulbar dysarthria, intellectual disability, bladder dysfunction, and being overweight. Minor symptoms included upper limbs rigidity and sensory axonopathy. The median body mass index was 26.2 kg/m2 (range, 25.2–32.3 kg/m2). The thin corpus callosum (TCC) was predominant at the rostral body or anterior midbody, and the ears of the lynx sign was seen in all. The follow-up MRI showed the worsening of periventricular white matter (PVWM) signal abnormalities with ventricular widening or the extension of the TCC. Motor evoked potentials (MEP) to the lower limbs showed an absent central motor conduction time (CMCT) in all subjects. The upper limb CMCT was initially absent in three subjects, although it became abnormal in all at the follow-up. The mini-mental state examination median score was 27/30 (range, 26–28) with selective impairment of the attention/calculation domain. The median score of the full-scale intelligence quotient was 48 (range, 42–72) on the Wechsler Adult Intelligence Scale test.
Conclusion: Attention/calculation deficits and being overweight as well as pseudobulbar dysarthria were common additional symptoms in patients with SPG11-HSP. The rostral body and anterior midbody of the corpus callosum were preferentially thinned, especially in the early stage of the disease. The TCC, PVWM signal changes, and MEP abnormality worsened as the disease progressed.
Introduction
Hereditary spastic paraplegia (HSP) is a group of inherited neurological disorders characterized by progressive spastic weakness of the lower limbs due to degeneration of the corticospinal tracts (1). HSP is clinically divided into two subtypes: (a) pure or uncomplicated HSP, which is characterized by difficulty walking due to progressive spastic paraplegia of insidious onset; (b) complicated HSP, which is associated with additional neurologic symptoms or medical conditions, including intellectual disability, dementia, ataxia, extrapyramidal disorders, optic atrophy, peripheral neuropathy, amyotrophy, or epilepsy (1–3). The biallelic mutations in the SPG11 gene encoding the spatacsin protein can lead to complicated HSP (SPG11-HSP). SPG11-HSP is a rare disease with an incidence of 0.35 per 100,000 people and is known to account for 19%–31% of autosomal recessive HSP (4–6). SPG11-HSP may clinically manifest as various neurological symptoms or signs associated with the central and peripheral nervous involvement (5, 7). The thin corpus callosum (TCC) is known as a radiologic hallmark of SPG11-HSP (5, 8). However, TCC cannot be pathognomonic, considering that it is also observed in HSP with other genotypes (8, 9), and several cases of SPG11-HSP without TCC have been reported (10). Thus, a more comprehensive study is needed to understand this rare disease with phenotypic diversity. This study aimed to comprehensively analyze the phenotype of SPG11-HSP, including clinical, radiological, electrodiagnostic, and neuropsychologic features.
Materials and methods
Subjects
We searched for patients undergoing follow-ups at the outpatient neurology clinic after being diagnosed with sporadic HSP. Whole exome sequencing (WES) was performed to identify the causative gene in 17 patients, of which 5 were classified into pure HSP and 12 were into complicated HSP. Six patients with complicated HSP were considered as having SPG11-HSP. In this study, the diagnosis of SPG11-HSP is defined as the presence of slowly progressive spastic paraparesis, TCC on MRI scan, identification of pathogenic mutations in the SPG11 gene, but no significant variants identified in more than 80 other HSP-related genes. We retrospectively reviewed the medical records of six subjects, including two (subject number (SN)-5 and SN-6) identical twins. Five known pathogenic and two novel variants in the SPG11 gene were identified (Table 1). The twins carried an additional pathogenic heterozygous c.950G > A/p.Arg317Gln variant in the CLCN1 gene (11, 12). A detailed description of the WES analysis, the flowchart of patient selection, and information on all genetic variants found in the SPG11 gene in this study are provided in the Supplementary materials. This project was approved by the institutional review board of the Chonnam National University Hospital (CNUH-2020-018).
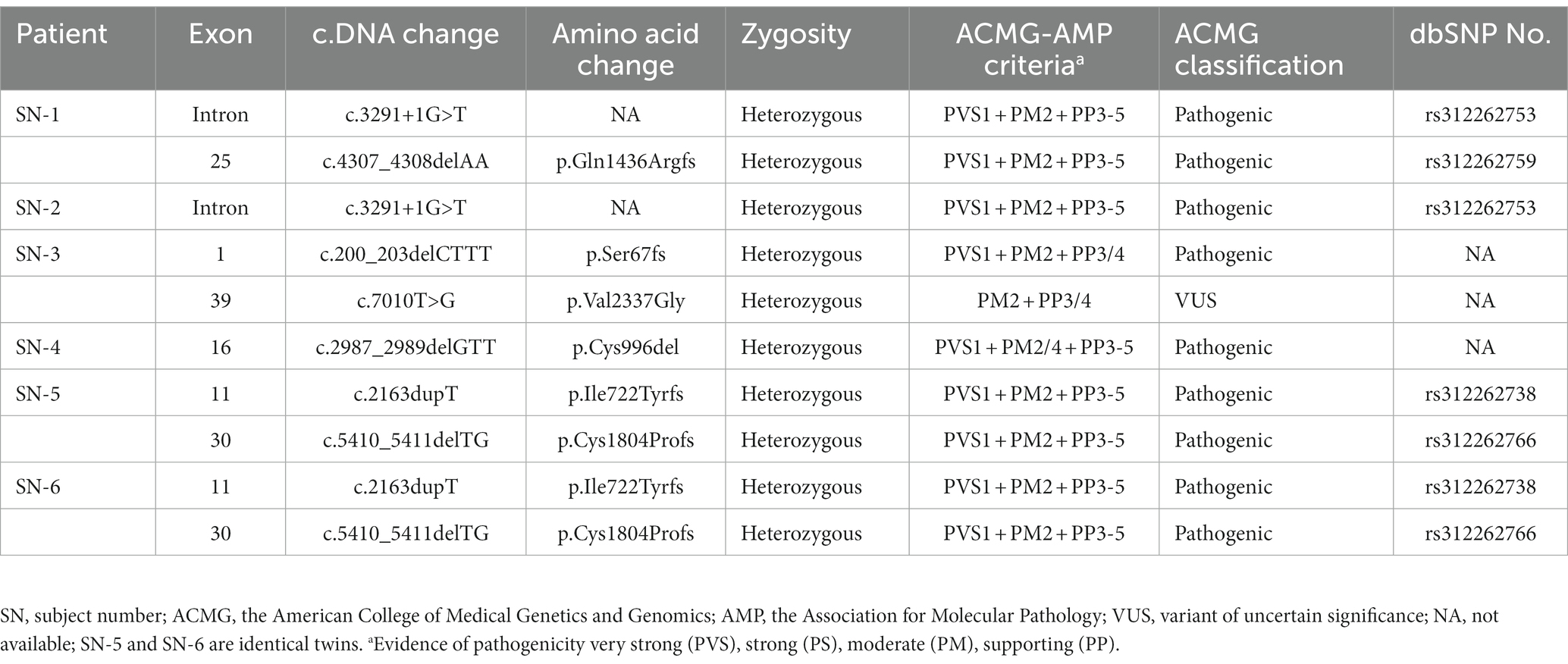
Table 1. Variants identified in the SPG11 gene.
Methods
The clinical characteristics were established by obtaining the following: (1) Clinical findings included demographic information, including body mass index (BMI), subjective symptoms, and neurological examinations. The spastic paraplegia rating scale (SPRS) was measured (13). (2) Radiological findings included the TCC and periventricular white matter (PVWM) signal abnormalities. Midsagittal T1-weighted images were selected to assess the affected regions of the corpus callosum, which was divided into seven subdivisions according to Witelson’s guidelines: region 1 (rostrum), region 2 (genu), region 3 (rostral body), region 4 (anterior midbody), region 5 (posterior midbody), region 6 (isthmus), and region 7 (splenium) (14, 15). PVWM signal abnormalities were evaluated on an axial fluid-attenuated inversion recovery (FLAIR) image, and ventricular widening was investigated. (3) Electrodiagnostic findings included the results from the motor evoked potentials (MEP) and somatosensory evoked potentials (SEP), the nerve conduction study (NCS), and electromyography (EMG). Central motor conduction time (CMCT) was measured by subtracting the peripheral motor conduction time (PMCT) from the onset latencies of the MEP. PMCT was calculated by adding F-wave latency and M-wave latency measured in the posterior tibial and ulnar nerves and then subtracting 1 msec, reflecting the turnaround time at the anterior horn cell and dividing by two. Thus, the formula used for calculating the CMCT is as follows: CMCT (msec) = MEP – (F-latency + M-latency – 1)/2 (16). (4) Neuropsychologic findings included the results of the Korean version of the mini-mental state examination (K-MMSE) and the Korean Wechsler Adult Intelligence Scale (K-WAIS) test for the evaluation of the intelligence quotient (IQ).
Results
The subjects were between 17 and 41 years old. All subjects had no family history of neurodegenerative diseases. Three subjects (SN-1, SN-2, and SN-3) had a history of intellectual disability. Five subjects were educated for over 9 years, including the highly educated one (SN-4) who graduated from university. The median BMI was 26.2 kg/m2 (range, 25.2–32.3 kg/m2). All subjects presented with slowly progressive spasticity in the lower limbs. The median age at the onset of gait disturbance was 16.5 years (range, 13–38 years); it developed in late adolescence in five subjects, while the other (SN-4) experienced it in her late thirties. Five of the six subjects could walk independently at the initial examination. All became dependent on ambulatory assistive devices (AAD) with time, and the median duration from onset to use of AAD was 5 years. The median SPRS score was 24/52 (range, 16–31 points). Learning difficulties and urinary or bowel incontinence were common. One subject (SN-1) complained of rigidity or stiffness in the upper limbs, another (SN-4) had paresthesia in the lower limbs, and the twins (SN-5 and SN-6) had experienced painful muscle stiffness in their calves since their early teens. Neurological examinations generally revealed pseudobulbar dysarthria, positive Babinski sign and ankle clonus on both sides, and Achilles tendon contracture. Calf muscle hypertrophy was seen in the twins. The clinical manifestations are summarized in Table 2.
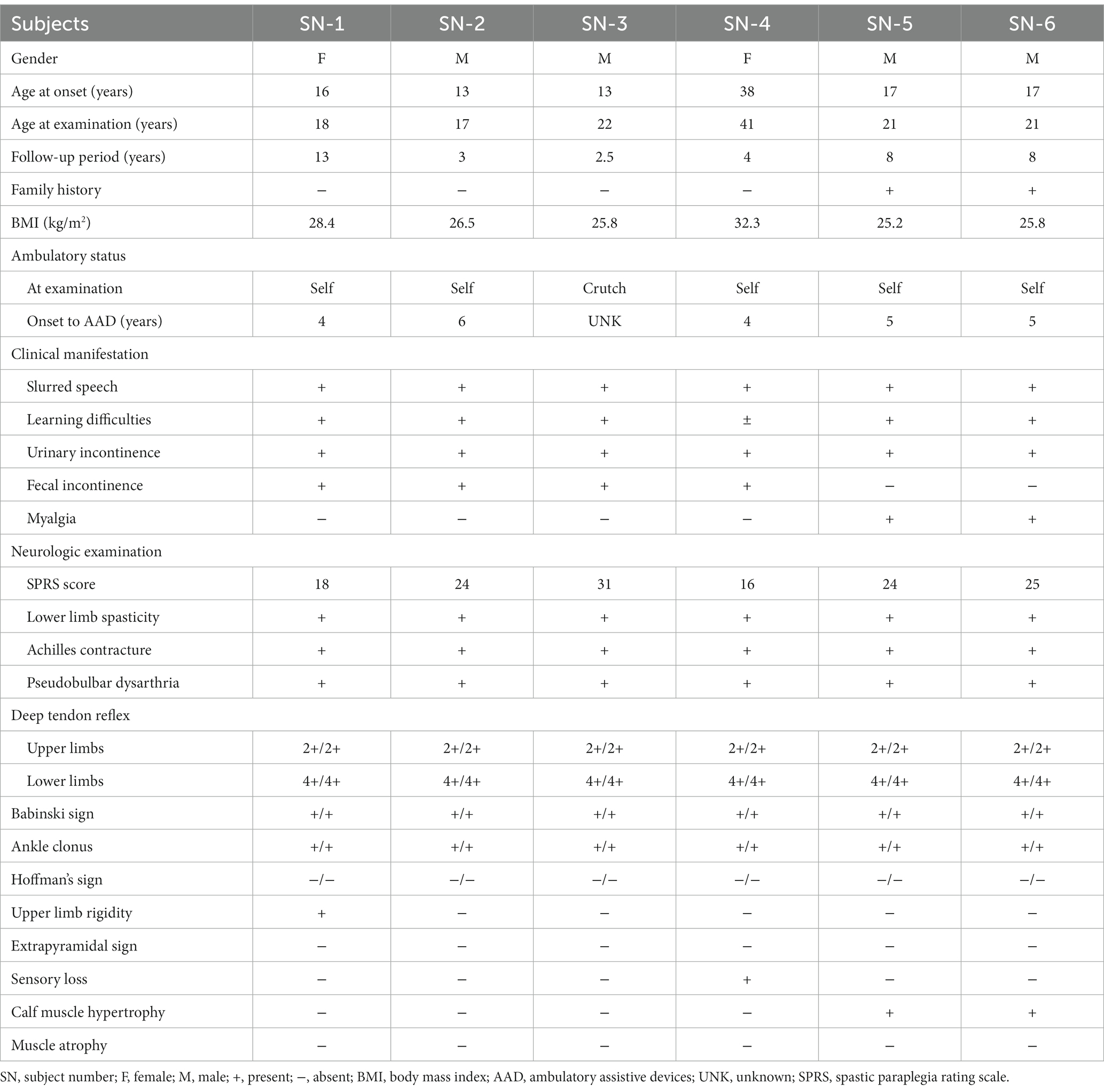
Table 2. Clinical findings of the enrolled patients in this study.
Radiologically, TCC was observed in all subjects. Three subjects (SN-1, SN-5, and SN-6) showed focal thinning limited to region 3, and another (SN-2) showed thinning in regions 3 and 4. The other two (SN-3 and SN-4) showed diffuse thinning involving the entire or anterior half of the corpus callosum, respectively (Figure 1). The FLAIR hyperintensities at the forceps minor, called ears of the lynx sign, were found in all subjects, PVWM signal abnormalities at the forceps major were seen in three (SN-2, SN-3, and SN-4), and the widening of the lateral ventricles was found in one (SN-3; Figure 2). A follow-up brain MRI was performed after 12 years in one subject (SN-1) and 3 years in the other (SN-4). In SN-1, the thinning of region 3 had further progressed and the corpus callosum had become diffusely thinned. More pronounced FLAIR signal abnormalities at the forceps minor and major were accompanied by the enlargement of the lateral ventricles (Figures 3A–D). In SN-4, TCC did not significantly worsen, although PVWM signal abnormalities accompanied by ventricular widening became prominent (Figures 3E–H). The spine MRI performed on three subjects was unremarkable. In the electrodiagnostic studies, MEP to the lower limbs showed an absent response in all subjects, and MEP to the upper limbs was absent in three subjects (SN-1, SN-2, and SN-3). In the follow-up MEP tests performed 2 years later, upper limb CMCT was absent in one subject (SN-4) and prolonged in two subjects (SN-5 and SN-6). SEP revealed unremarkable results in both the upper and lower limbs. NCS was unremarkable, except for one subject (SN-4) who had distal symmetric axonal sensory polyneuropathies in the legs. Needle EMG showed myotonic discharges typical of non-dystrophic myotonia in the twins. Concerning neuropsychologic studies, the median K-MMSE score was 27 (range, 6–28), all of which were deducted in the serial sevens task. Cognitive ability was examined using WAIS-III in two (SN-1 and SN-2) or WAIS-IV in the others, and the median full-scale IQ was 48 (range, 42–76). The detailed information on the radiological, electrodiagnostic, and neuropsychologic findings is summarized in Table 3.
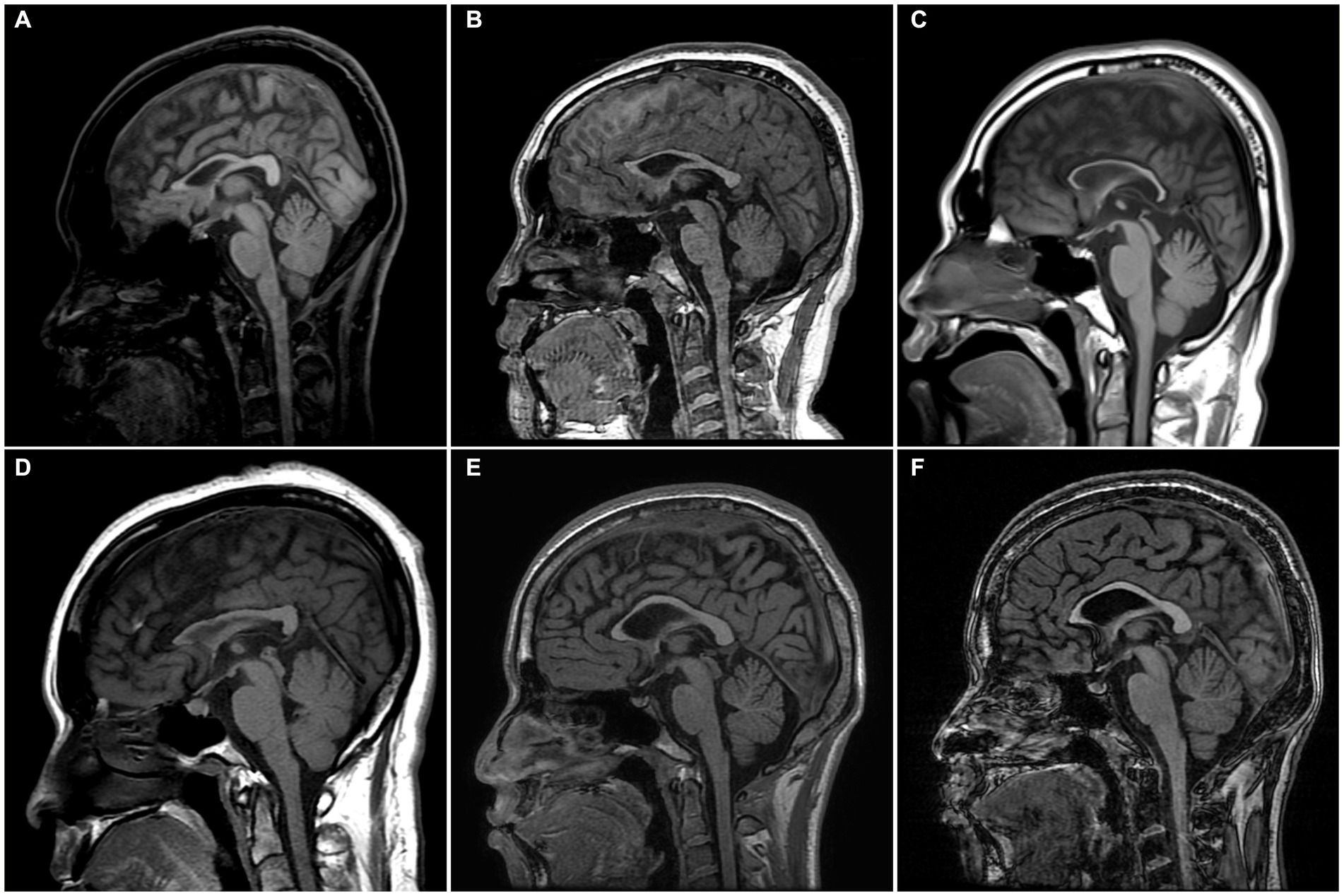
Figure 1. Sagittal T1-weight MR images of the enrolled patients. (A) SN-1, 18 years of age, has focal thinning of region 3 in the corpus callosum. (B) SN-2, 17 years of age, has thinning of regions 3 and 4. (C) SN-3, 22 years of age, has thinning of the entire corpus callosum involving regions 1–7. (D) SN-4, 41 years of age, has thinning of the anterior half of the corpus callosum involving regions 1–4. (E,F) SN-5 and SN-6, 21-year-old identical twins, have focal thinning of region 3. The corpus callosum was divided into seven regions according to Witelson’s guidelines. SN, subject number.
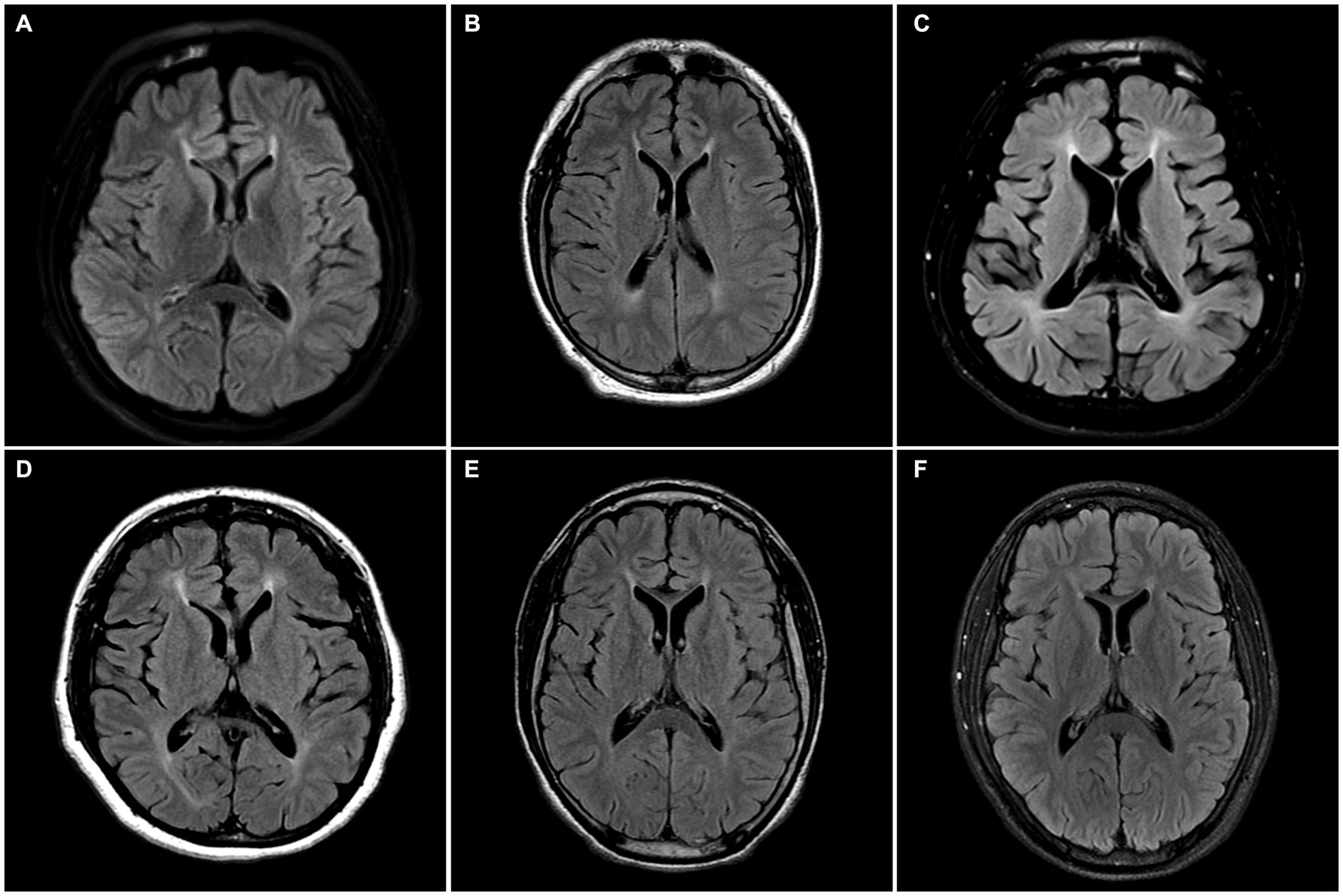
Figure 2. Axial FLAIR MR images of the enrolled patients. (A–F) All subjects have hyperintensities at the forceps minor region called the ears of the lynx sign. (B–D) SN-2, SN-3, and SN-4 have forceps major region signal abnormality. (C) SN-3 has the widening of the lateral ventricle.
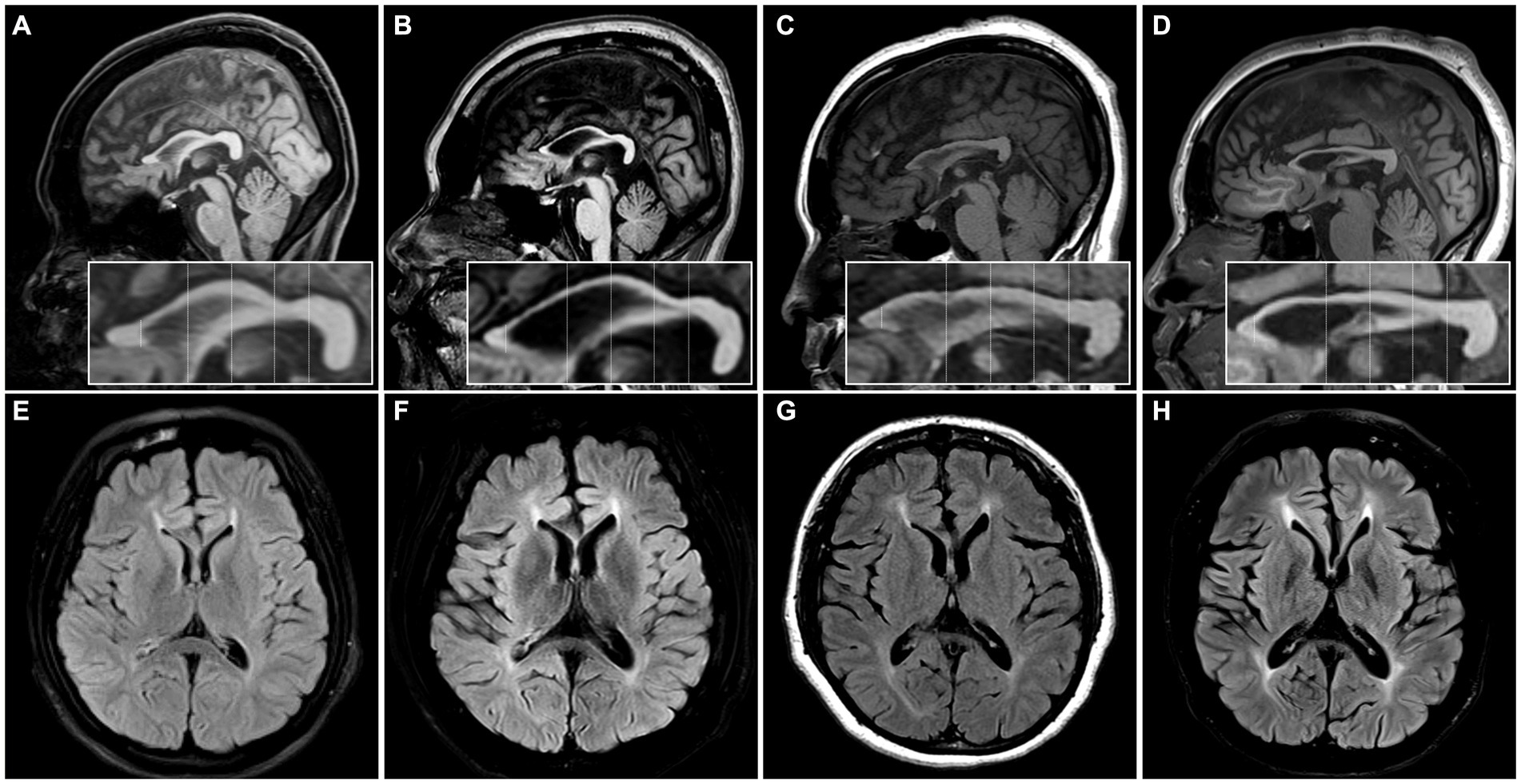
Figure 3. The initial and follow-up images of two patients. (A–D) MR images of SN-1. (A,B) The focal thinning of region 3 in the initial image was further thinned and other regions of the corpus callosum were also slenderized on the MRI performed 12 years later. (C,D) The forceps minor region signal abnormality became more distinct in the subsequent image, and the forceps major region signal abnormality with ventricular widening was observed in the follow-up image. (E–H) MR images of the SN-4. (E,F) The thinning in the anterior half of the corpus callosum was still observed on the MRI performed 3 years later, although there was no significant change. (G,H) The forceps minor and major regions signal abnormalities with ventricular widening were more distinct in the follow-up image.

Table 3. Laboratory findings of the enrolled patients in this study.
Discussion
In addition to spastic weakness in the legs, a core feature of HSP, the major additional symptoms of SPG11-HSP were learning difficulties, pseudobulbar dysarthria, bladder dysfunction, and being overweight. A few subjects complained of rigidity in their upper limbs or neuropathic pain, which are known as possible accompanying manifestations (4, 5, 17, 18). The twins presented myotonic muscle stiffness, which has been reported in a family with autosomal recessive HSP (19). However, it could be due to an incidental coexistence of myotonia congenita rather than an additional symptom of SPG11-HSP, given that a pathogenic CLCN1 mutation was identified. Intriguingly, all subjects had abnormal BMI scores of 25 kg/m2 or higher, indicating they were overweight according to the WHO classification, yet implying obesity when applying the refined cut-off for Asian-Pacific populations (20, 21). Although metabolic aspects have not been well implicated in SPG11-HSP, recent studies have suggested a relationship between bodyweight changes and hypothalamic dysfunction in this disease (22, 23). Therefore, weight gain may not be simply because of physical inactivity following gait disturbance but an additional manifestation of SPG11-HSP (22). Furthermore, since body composition and the related metabolic profile of the Asian population can differ from Western society (24), further investigations into changes in bodyweight among SPG11-HSP patients by race may be warranted.
The distinct radiologic finding in this study was TCC. We divided the corpus callosum into subregions to identify the thinning pattern since the callosal fibers of the corpus callosum are thought to have a topographic organization in accordance with projecting cortical areas (25, 26). The most commonly affected area was region 3 or 4, which contains commissural fibers to the premotor and motor cortex (12, 27). Considering that a patient’s serial MRI images spaced over 10 years showed aggravation of TCC, the thinning may extend from the rostral or anterior midbody to the entirety of the corpus callosum with disease progression (5, 18, 28, 29). In addition, all subjects had PVWM signal abnormalities in common, even those who had mild and focal TCC. Notably, these signal changes worsened without further thinning of the corpus callosum on the follow-up MRI of SN-4. Thus, PVWM signal abnormality, including the ears of the lynx sign, seemed to parallel or precede the worsening of TCC (30). The enlargement of lateral ventricles observed in three subjects has been rarely described before (31, 32), and is presumed to be related to the decreased white and grey matter volume (31).
Peripheral neuropathy has been infrequently reported in SPG11-HSP (5). Its clinical symptoms are usually mild, but it has been reported that severe axonopathy, neuropathic pain, or autonomic dysfunction may be accompanied in the late stage of the disease (17). In this study, NCS was abnormal in the only patient with neuropathic pain, which is inconsistent with a large cohort study of 25 SPG11-HSP subjects, in which 96% subjects showed non-length dependent motor neuropathy (33). Interestingly, all subjects showed absent lower limb CMCT regardless of SPRS scores or disease duration, similar to the many studies on the neurophysiologic features of HSP that have reported abnormal lower limb MEP (34). Meanwhile, less attention has been paid to MEP in the upper limbs, and investigations specific to SPG11-HSP are still lacking, even though it is the most common subtype of autosomal recessive HSP. In this study, the frequency of abnormal findings in the upper limb MEP was relatively high compared to an abnormal rate of 26% in a systemic review covering HSP patients with variable genotypes (34). MEP is thought to reflect the neuronal integrity of the upper motor neurons and has been proposed as a disease severity marker (34–36). Therefore, rapid conversion of the upper limb CMCT abnormalities may reflect progressive neurodegeneration of SPG11-HSP, and its longitudinal evaluation of the MEP test could provide a clue to assess the clinical course of this disease.
Intellectual disability is known as one of the non-motor symptoms of SPG11-HSP (37). The MMSE scores of the subjects were slightly lower for age and education, which is in line with the fact that intellectual disability is a frequent feature of SPG11-HSP (38). The MMSE is a 30-point questionnaire composed of several domains, although few papers on HSP have described it in detail by domain. Notably, all subjects exclusively showed impairment in the attention/calculation domain, which evaluates working memory. Contrarily, IQ was conspicuously lower than the age-matched population in five patients, and one showed borderline intellectual functioning. A similar discrepancy between the Montreal Cognitive Assessment and MMSE was observed in a study of four patients with SPG11-HSP (39). Thus, additional comprehensive assessments of intellectual function would be needed, even if the MMSE score seems satisfactory. Nonetheless, since the MMSE is the most widely used cognitive screening tool in clinical settings, the serial sevens task may be a straightforward and useful method to identify cognitive impairment in those suspected of complicated HSP.
This study has several limitations. First, small number of patients were studied. Second, this study was designed retrospectively. Third, copy number variants are known to account for about 19% of pathogenic SPG11 alleles (40), but no further genetic tests, such as multiplex ligation-dependent probe amplification and segregation analysis, were performed. Nevertheless, the subjects had clinicoradiological features consistent with SPG11-HSP, and they only carried one or two pathogenic variants in the SPG11 gene without clinically or genetically significant variants in other HSP-related genes. Additional prospective studies with larger sample sizes are necessary to comprehensively understand the clinical features of patients with SPG11-HSP.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board at Chonnam National University Hospital. The patients/participants provided their written informed consent to participate in this study.
Author contributions
Y-RK: data curation and writing-original draft. T-SN: conceptualization, writing-review and editing, and supervision. J-MK, KK, and BK: data curation and formal analysis. S-MC and S-HL: formal analysis. M-KK: formal analysis and supervision. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Health Fellowship Foundation and a grant from the Chonnam National University Hospital Biomedical Research Institute (BCRI 21025).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2023.1198728/full#supplementary-material
References
1. Salinas, S, Proukakis, C, Crosby, A, and Warner, TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. (2008) 7:1127–38. doi: 10.1016/S1474-4422(08)70258-8
2. Kim, TH, Lee, JH, Park, YE, Shin, JH, Nam, TS, Kim, HS, et al. Mutation analysis of SPAST, ATL1, and REEP1 in Korean patients with hereditary spastic paraplegia. J Clin Neurol. (2014) 10:257–61. doi: 10.3988/jcn.2014.10.3.257
3. McDermott, C, White, K, Bushby, K, and Shaw, P. Hereditary spastic paraparesis: a review of new developments. J Neurol Neurosurg Psychiatry. (2000) 69:150–60. doi: 10.1136/jnnp.69.2.150
4. Pozner, T, Regensburger, M, Engelhorn, T, Winkler, J, and Winner, B. Janus-faced spatacsin (SPG11): involvement in neurodevelopment and multisystem neurodegeneration. Brain. (2020) 143:2369–79. doi: 10.1093/brain/awaa099
5. Stevanin, G, Azzedine, H, Denora, P, Boukhris, A, Tazir, M, Lossos, A, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. (2008) 131:772–84. doi: 10.1093/brain/awm293
6. Vander Stichele, G, Durr, A, Yoon, G, Schüle, R, Blackstone, C, Esposito, G, et al. An integrated modelling methodology for estimating global incidence and prevalence of hereditary spastic paraplegia subtypes SPG4, SPG7, SPG11, and SPG15. BMC Neurol. (2022) 22:115. doi: 10.1186/s12883-022-02595-4
7. Anheim, M, Lagier-Tourenne, C, Stevanin, G, Fleury, M, Durr, A, Namer, IJ, et al. SPG11 spastic paraplegia. A new cause of juvenile parkinsonism. J Neurol. (2009) 256:104–8. doi: 10.1007/s00415-009-0083-3
8. Pensato, V, Castellotti, B, Gellera, C, Pareyson, D, Ciano, C, Nanetti, L, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain. (2014) 137:1907–20. doi: 10.1093/brain/awu121
9. da Graça, FF, de Rezende, TJR, Vasconcellos, LFR, Pedroso, JL, Barsottini, OGP, and França, MC. Neuroimaging in hereditary spastic paraplegias: current use and future perspectives. Front Neurol. (2018) 9:1117. doi: 10.3389/fneur.2018.01117
10. Crimella, C, Arnoldi, A, Crippa, F, Mostacciuolo, ML, Boaretto, F, Sironi, M, et al. Point mutations and a large intragenic deletion in SPG11 in complicated spastic paraplegia without thin corpus callosum. J Med Genet. (2009) 46:345–51. doi: 10.1136/jmg.2008.063321
11. Meyer-Kleine, C, Steinmeyer, K, Ricker, K, Jentsch, TJ, and Koch, MC. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am J Hum Genet. (1995) 57:1325–34.
12. Esteban, J, Neumeyer, AM, McKenna-Yasek, D, and Brown, RH. Identification of two mutations and a polymorphism in the chloride channel CLCN-1 in patients with Becker's generalized myotonia. Neurogenetics. (1998) 1:185–8. doi: 10.1007/s100480050027
13. Schule, R, Holland-Letz, T, Klimpe, S, Kassubek, J, Klopstock, T, Mall, V, et al. The spastic paraplegia rating scale (SPRS): a reliable and valid measure of disease severity. Neurology. (2006) 67:430–4. doi: 10.1212/01.wnl.0000228242.53336.90
14. Witelson, SF. Hand and sex differences in the isthmus and genu of the human corpus callosum. A postmortem morphological study. Brain. (1989) 112 (Pt 3):799–835. doi: 10.1093/brain/112.3.799
15. Witelson, SF, and Goldsmith, CH. The relationship of hand preference to anatomy of the corpus callosum in men. Brain Res. (1991) 545:175–82. doi: 10.1016/0006-8993(91)91284-8
16. MANO, Y, NAKAMURO, T, IKOMA, K, SUGATA, T, MORIMOTO, S, TAKAYANAGI, T, et al. Central motor conductivity in aged people. Intern Med. (1992) 31:1084–7. doi: 10.2169/internalmedicine.31.1084
17. Manole, A, Chelban, V, Haridy, NA, Hamed, SA, Berardo, A, Reilly, MM, et al. Severe axonal neuropathy is a late manifestation of SPG11. J Neurol. (2016) 263:2278–86. doi: 10.1007/s00415-016-8254-5
18. Stevanin, G, Santorelli, FM, Azzedine, H, Coutinho, P, Chomilier, J, Denora, PS, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. (2007) 39:366–72. doi: 10.1038/ng1980
19. Wessel, K, Kessler, C, Rosengart, A, and Kömpf, D. Myotonia congenita with familial spastic paraparesis. Nervenarzt. (1988) 59:675–8.
20. WHO Expert Consultation. Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies. Lancet. (2004) 363:157–63. doi: 10.1016/S0140-6736(03)15268-3
21. World Health Organization, International Association for the Study of Obesity, International Obesity Task Force. The Asia-Pacific perspective: Redefining obesity and its treatment. Sydney, Australia: WHO/IASO/IOTF (2000), p.15–21.
22. Cardozo-Hernández, ALC, Rezende, TJR, and França, MC. Hereditary spastic paraplegia type 11 (SPG11) is associated with obesity and hypothalamic damage. J Neurol Sci. (2020) 416:116982. doi: 10.1016/j.jns.2020.116982
23. Regensburger, M, Krumm, L, Schmidt, MA, Schmid, A, Spatz, IT, Marterstock, DC, et al. Neurometabolic dysfunction in SPG11 hereditary spastic paraplegia. Nutrients. (2022) 14:4803. doi: 10.3390/nu14224803
24. Wulan, SN, Westerterp, KR, and Plasqui, G. Ethnic differences in body composition and the associated metabolic profile: a comparative study between Asians and Caucasians. Maturitas. (2010) 65:315–9. doi: 10.1016/j.maturitas.2009.12.012
25. Zarei, M, Johansen-Berg, H, Smith, S, Ciccarelli, O, Thompson, AJ, and Matthews, PM. Functional anatomy of interhemispheric cortical connections in the human brain. J Anat. (2006) 209:311–20. doi: 10.1111/j.1469-7580.2006.00615.x
26. Friedrich, P, Forkel, SJ, and Thiebaut de Schotten, M. Mapping the principal gradient onto the corpus callosum. NeuroImage. (2020) 223:117317. doi: 10.1016/j.neuroimage.2020.117317
27. Scepkowski, LA, and Cronin-Golomb, A. The alien hand: cases, categorizations, and anatomical correlates. Behav Cogn Neurosci Rev. (2003) 2:261–77. doi: 10.1177/1534582303260119
28. Denora, PS, Schlesinger, D, Casali, C, Kok, F, Tessa, A, Boukhris, A, et al. Screening of ARHSP-TCC patients expands the spectrum of SPG11 mutations and includes a large scale gene deletion. Hum Mutat. (2009) 30:E500–19. doi: 10.1002/humu.20945
29. Pan, MK, Huang, SC, Lo, YC, Yang, CC, Cheng, TW, Yang, CC, et al. Microstructural integrity of cerebral fiber tracts in hereditary spastic paraparesis with SPG11 mutation. AJNR Am J Neuroradiol. (2013) 34:s991:990–6. doi: 10.3174/ajnr.A3330
30. Riverol, M, Samaranch, L, Pascual, B, Pastor, P, Irigoyen, J, Pastor, MA, et al. Forceps minor region signal abnormality "ears of the lynx": an early MRI finding in spastic paraparesis with thin corpus callosum and mutations in the spatacsin gene (SPG11) on chromosome 15. J Neuroimaging. (2009) 19:52–60. doi: 10.1111/j.1552-6569.2008.00327.x
31. Hayakawa, M, Matsubara, T, Mochizuki, Y, Takeuchi, C, Minamitani, M, Imai, M, et al. An autopsied case report of spastic paraplegia with thin corpus callosum carrying a novel mutation in the SPG11 gene: widespread degeneration with eosinophilic inclusions. BMC Neurol. (2022) 22:2. doi: 10.1186/s12883-021-02514-z
32. Utz, KS, Kohl, Z, Marterstock, DC, Doerfler, A, Winkler, J, Schmidt, M, et al. Neuropsychology and MRI correlates of neurodegeneration in SPG11 hereditary spastic paraplegia. Orphanet J Rare Dis. (2022) 17:301. doi: 10.1186/s13023-022-02451-1
33. Faber, I, Martinez, ARM, de Rezende, TJR, Martins, CR Jr, Martins, MP, Lourenço, CM, et al. SPG11 mutations cause widespread white matter and basal ganglia abnormalities, but restricted cortical damage. Neuroimage Clin. (2018) 19:848–57. doi: 10.1016/j.nicl.2018.05.031
34. Siow, SF, Cameron Smail, R, Ng, K, Kumar, KR, and Sue, CM. Motor evoked potentials in hereditary spastic paraplegia-a systematic review. Front Neurol. (2019) 10:967. doi: 10.3389/fneur.2019.00967
35. Groppa, S, Oliviero, A, Eisen, A, Quartarone, A, Cohen, LG, Mall, V, et al. A practical guide to diagnostic transcranial magnetic stimulation: report of an IFCN committee. Clin Neurophysiol. (2012) 123:858–82. doi: 10.1016/j.clinph.2012.01.010
36. Karle, KN, Schüle, R, Klebe, S, Otto, S, Frischholz, C, Liepelt-Scarfone, I, et al. Electrophysiological characterisation of motor and sensory tracts in patients with hereditary spastic paraplegia (HSP). Orphanet J Rare Dis. (2013) 8:158. doi: 10.1186/1750-1172-8-158
37. Winner, B, Uyanik, G, Gross, C, Lange, M, Schulte-Mattler, W, Schuierer, G, et al. Clinical progression and genetic analysis in hereditary spastic paraplegia with thin corpus callosum in spastic gait gene 11 (SPG11). Arch Neurol. (2004) 61:117–21. doi: 10.1001/archneur.61.1.117
38. Crum, RM, Anthony, JC, Bassett, SS, and Folstein, MF. Population-based norms for the mini-mental state examination by age and educational level. JAMA. (1993) 269:2386–91. doi: 10.1001/jama.1993.03500180078038
39. Li, C, Yan, Q, Duan, FJ, Zhao, C, Zhang, Z, du, Y, et al. Mild cognitive impairment in novel SPG11 mutation-related sporadic hereditary spastic paraplegia with thin corpus callosum: case series. BMC Neurol. (2021) 21:12. doi: 10.1186/s12883-020-02040-4
Keywords: SPG11 , spastic paraplegia, corpus callosum, magnetic resonance imaging, evoked potential (EP), intellectual disabilities
Citation: Kang Y-R, Nam T-S, Kim J-M, Kang KW, Choi S-M, Lee S-H, Kim BC and Kim M-K (2023) Clinical analysis in patients with SPG11 hereditary spastic paraplegia. Front. Neurol. 14:1198728. doi: 10.3389/fneur.2023.1198728
Edited by:
Marcondes C. França, State University of Campinas, BrazilReviewed by:
Gabriele Siciliano, University of Pisa, ItalySun Ah Choi, Ewha Womans Medical Center, Republic of Korea
Copyright © 2023 Kang, Nam, Kim, Kang, Choi, Lee, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tai-Seung Nam, bnRzMDAyMkBoYW5tYWlsLm5ldA==
†ORCID: You-Ri Kang https://orcid.org/0000-0001-5189-1323
Tai-Seung Nam https://orcid.org/0000-0003-2771-8728
Jae-Myung Kim https://orcid.org/0000-0003-0483-4179
Kyung Wook Kang https://orcid.org/0000-0001-9362-8670
Seong-Min Choi https://orcid.org/0000-0003-3138-1881
Seung-Han Lee https://orcid.org/0000-0002-4410-646X
Byeong C. Kim https://orcid.org/0000-0001-6827-6730
Myeong-Kyu Kim https://orcid.org/0000-0001-8673-7561