 Janire Urrutia1†
Janire Urrutia1† Ane Arrizabalaga-Iriondo1†
Ane Arrizabalaga-Iriondo1† Ana Sanchez-del-Rey2Agustín Martinez-Ibargüen2
Ana Sanchez-del-Rey2Agustín Martinez-Ibargüen2 Mónica Gallego3
Mónica Gallego3 Oscar Casis3
Oscar Casis3 Miren Revuelta1*
Miren Revuelta1*- 1Department of Physiology, Faculty of Medicine and Nursery, University of the Basque Country (UPV/EHU), Bilbao, Spain
- 2Department of Otorhinolaryngology, Faculty of Medicine, University of the Basque Country, Bilbao, Spain
- 3Department of Physiology, Faculty of Pharmacy, University of the Basque Country (UPV/EHU), Vitoria-Gasteiz, Spain
Voltage-gated ion channels are essential for membrane potential maintenance, homeostasis, electrical signal production and controlling the Ca2+ flow through the membrane. Among all ion channels, the key regulators of neuronal excitability are the voltage-gated potassium channels (KV), the largest family of K+ channels. Due to the ROS high levels in the aging brain, K+ channels might be affected by oxidative agents and be key in aging and neurodegeneration processes. This review provides new insight about channelopathies in the most studied neurodegenerative disorders, such as Alzheimer Disease, Parkinson’s Disease, Huntington Disease or Spinocerebellar Ataxia. The main affected KV channels in these neurodegenerative diseases are the KV1, KV2.1, KV3, KV4 and KV7. Moreover, in order to prevent or repair the development of these neurodegenerative diseases, previous KV channel modulators have been proposed as therapeutic targets.
1 Introduction
Ion channels are essential for life as they play a fundamental role in neuronal signaling, muscle contraction or even nutrient transport (Weaver and Wearne, 2008). Moreover, voltage-gated ion channels are responsible for membrane potential maintenance, homeostasis, electrical signal production and controlling the Ca2+ flow through the membrane (Moiseenkova-Bell et al., 2021).
Ion channels are macromolecular pores that control ion flux through the cell membrane and consequently the intracellular ion balance (Eren-Koçak and Dalkara, 2021). The pore opens with mechanical, chemical or electrical stimulus and consequently ion channels allow ions to flow into or out the cell. Voltage-gated ion channels respond to a change in cell membrane potential and are highly selective for a specific ion (Na+, K+, Ca2+ or Cl−) (Trimmer and Rhodes, 2004). Meanwhile, ligand-gated ion channels respond to specific neurotransmitters among other molecules and mechanical-gated ion channels to changes in the mechanical force on the membrane.
Within the group of ion channels, there is a superfamily of K+ ion channels. This family is divided into four main families; the calcium-activated (KCa) family, inward rectifier (Kir) family, two-pore domain family (K2P) and voltage-gated (KV) family (Ocaña et al., 2004; Luo et al., 2021).
The KCa family is formed by 3 members, classified by single-channel conductance. Thereby, KCa1.1 (known as BK channels) shows large conductance, KCa3.1 (IK) intermediate and KCa2.1–3 (SK) small conductance (Sforna et al., 2018). KCa channels are expressed in neurons and other cell types in the central nervous system (CNS). Furthermore, the Kir channels family is formed by 7 subtypes of channels (Kir1–Kir7), and each one has different members. They have an inward-rectification property that permits K+ enter the cell regulating membrane potential. These channels are expressed in different cells and regions of the CNS, and they regulate the hyperpolarization of the membrane potential and excitability (Akyuz et al., 2022). Meanwhile, two-pore domain K+ channels family consists of 15 members (K2P1–K2P7, K2P9–K2P10, K2P12–K2P18). K2P channels are dimers and in the CNS regulates cell excitability and maintains cellular resting potential. Some of the members are implicated in pathological conditions such as stroke, epilepsy, depression or inflammation (Talley et al., 2003).
1.1 KV channels
But among all ion channels, the key regulators of neuronal excitability are the voltage-gated potassium channels (KV), the largest family of K+ channels (Shah and Aizenman, 2014). These KV channels are divided into 12 subfamilies, named as KV1–KV12. They are composed of 4 α-subunits, each one containing 6 α-helical transmembrane domains (S1–S6), voltage sensor (S1–S4) and the ion pore (S5–S6). The N- and C-terminals are intracellular and they have different regulation sites. They differ in biophysical and pharmacological properties and in auxiliary β-subunits too, that modulate their activity, trafficking and location (Kuang et al., 2015). Although some channels regulate neuronal excitability, others participate also in the duration of cardiac action potentials and are involved in cell proliferation or even cancer (Bachmann et al., 2020).
Voltage-gated potassium channels are transmembrane channels responsible for returning the depolarized cell to a resting state after an action potential (Gazulla and Berciano, 2023). Therefore, KV channels are important modulating neuronal excitability in the CNS, but also participate regulating other organs function.
Changes in ion physical function or gaining or depletion of channel function results in channelopathies, several of them associated to neurodegenerative disorders (Orfali et al., 2024). In this review, we will describe the role of some voltage-gated potassium channel in age related neurodegenerative disorders and their modulation for these diseases therapy.
2 Age related neurodegenerative diseases and voltage-gated K+ channel modulation
Age related neurodegenerative diseases have common organ deterioration mechanisms due to ROS production and Ca2+ intracellular accumulation, inflammatory response and apoptosis that results in neuron loss and functional failure. There are some therapeutic options; however, these are limited. Several strategies have been explored during the last decades in order to palliate or reduce the symptoms (Trombetta-Lima et al., 2020). Potassium channels are able to modulate activity patterns, defining their vulnerability to degenerate and their physiological functions (Duda et al., 2016). KV channels regulate cell excitability and homeostasis so they can be considered as therapeutic targets in order to prevent or reduce age related neurodegenerative diseases, since it has been reported that aging itself can affect these channels function. Because ROS levels are highly elevated in the aging brain, K+ channels might be affected by oxidative agents and be key in aging and neurodegeneration processes (reviewed in Sesti et al., 2010). In this condition, molecules involved in the redox balance could modify the channel function (Sahoo et al., 2014).
2.1 KV channels in Alzheimer disease
During Alzheimer disease (AD), the amyloid β-peptide (Aβ) deposition causes synaptic dysfunction and consequently neuronal loss. There have been identified several KV channels that can regulate the firing rate (KV1, KV4 or KV7) or the duration of the action potential (KV2 or KV3) (Li, 2022) (Figure 1). Moreover, the impairment in these KV channels is associated with several pathogenic mechanisms. For instance, KV3.4 expression increase due to Aβ deposition initiates apoptotic processes and KV2.1 contributes to potassium mobilization during neuronal apoptosis, so overexpression of this channel promotes this process (Sun et al., 2022). The formation of KV2.1 oligomers by oxidative agents contribute to neurotoxicity and this phenomenon is aggravated in AD models (Cotella et al., 2012; Wei et al., 2018). This oligomerization triggers integrin signaling, activating Src tyrosine kinases via autophosphorylated FAK (Yu et al., 2019).
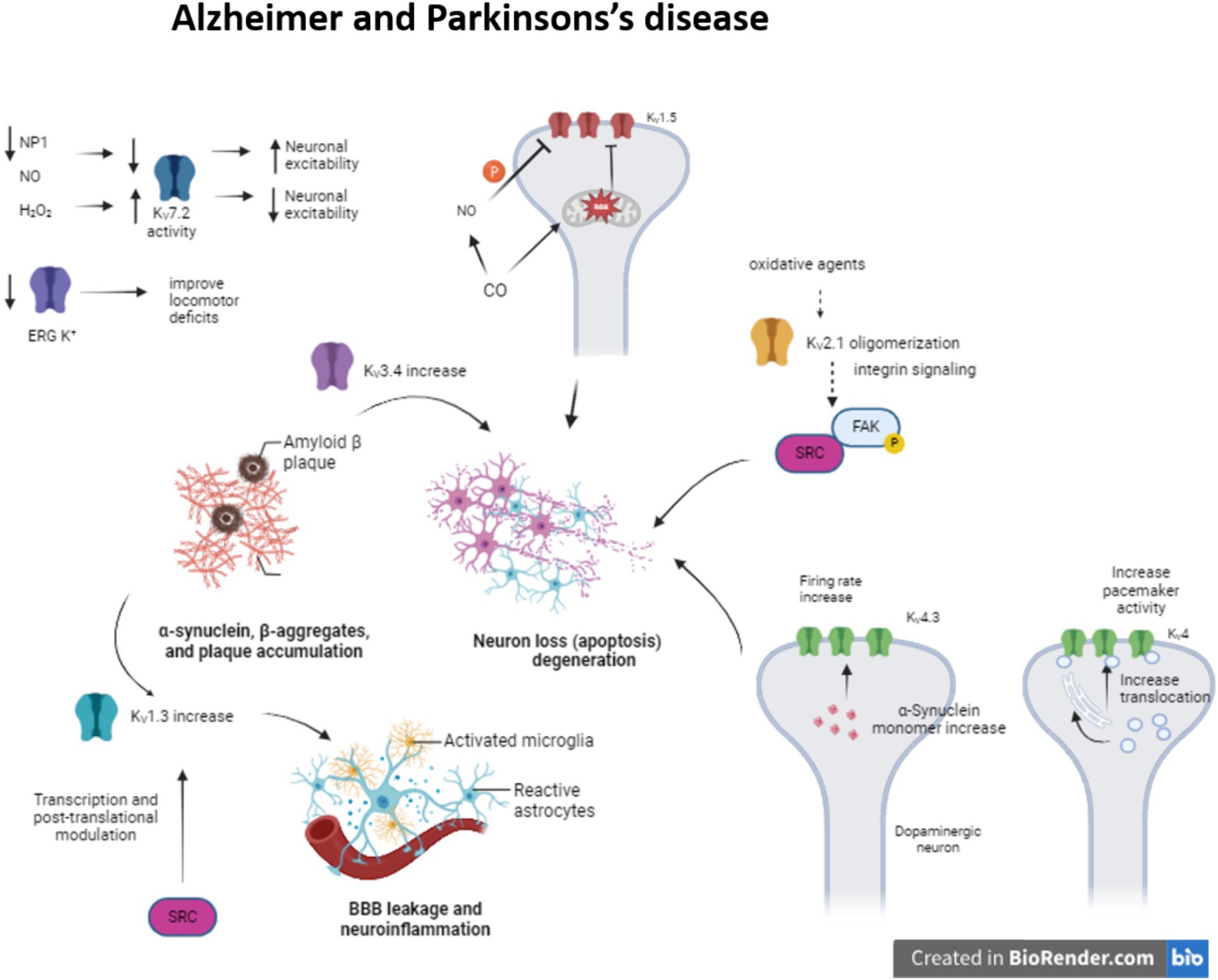
Figure 1. Modulation of KV channels in Alzheimer and Parkinson’s disease. The alteration of some KV channels expression or function modifies neuron excitability arising neuron apoptosis and neuroinflammation in Alzheimer and Parkinson’s disease.
Meanwhile, enhanced KV1.3 expression in microglia, a key regulator of microglia function and related to inflammatory response, after amyloid plaque formation produces proinflammatory cytokine release and apoptotic cascade (reviewed in Revuelta et al., 2022). KV1.5 is mostly studied in the heart, but it is also presented in the brain and its activity is associated with apoptosis. It has been seen that H2O2 increases channel activity (Caouette et al., 2003). In contrast, CO inhibits KV1.5 current by the increase of ROS, which directly regulates the channel. Besides, the increase of NO in response to CO inhibits the channel activity by channel phosphorylation (Al-Owais et al., 2017).
Concerning KV4 channelopathies they have been linked to AD, schizophrenia and epilepsy (Cercós et al., 2021). Particularly in AD, the expression of KChIP3 (KV channel-interacting protein 3 or calsenilin) is increased. This KChIP3 mechanically promote the translocation of KV4 channels to cell membrane, modulating the pacemaker activity (Buxbaum, 2004; Wu et al., 2023). They are associated with presenilins (PS1 and PS2), transmembrane proteins that are related to early-onset familial AD (Bähring, 2018) KChIP3 also modifies the gating of the channel, delaying the kinetic inactivation and accelerates the kinetic recovery from inactivation. Indeed, KV4.3 is involved in transient outward A type potassium current in neurons (Lopez-Hurtado et al., 2019).
The proapoptotic protein pentraxin (NP1) is another protein presented in dystrophic neurites in AD and related to the regulation of synapse density (Ma et al., 2018). This NP1 regulates the surface expression of KV7.2, a channel that controls neuronal excitability. KV7 channels generate M-current, slow voltage dependent outward current that contributes to the maintenance of the resting membrane potential, but can also exert a dampening effect on neuronal excitability. KV7.2 overexpression prevents cells from increased neuronal excitability and synapse, a situation provoked by NP1 downregulation during AD (Figueiro-Silva et al., 2015) (Table 1).
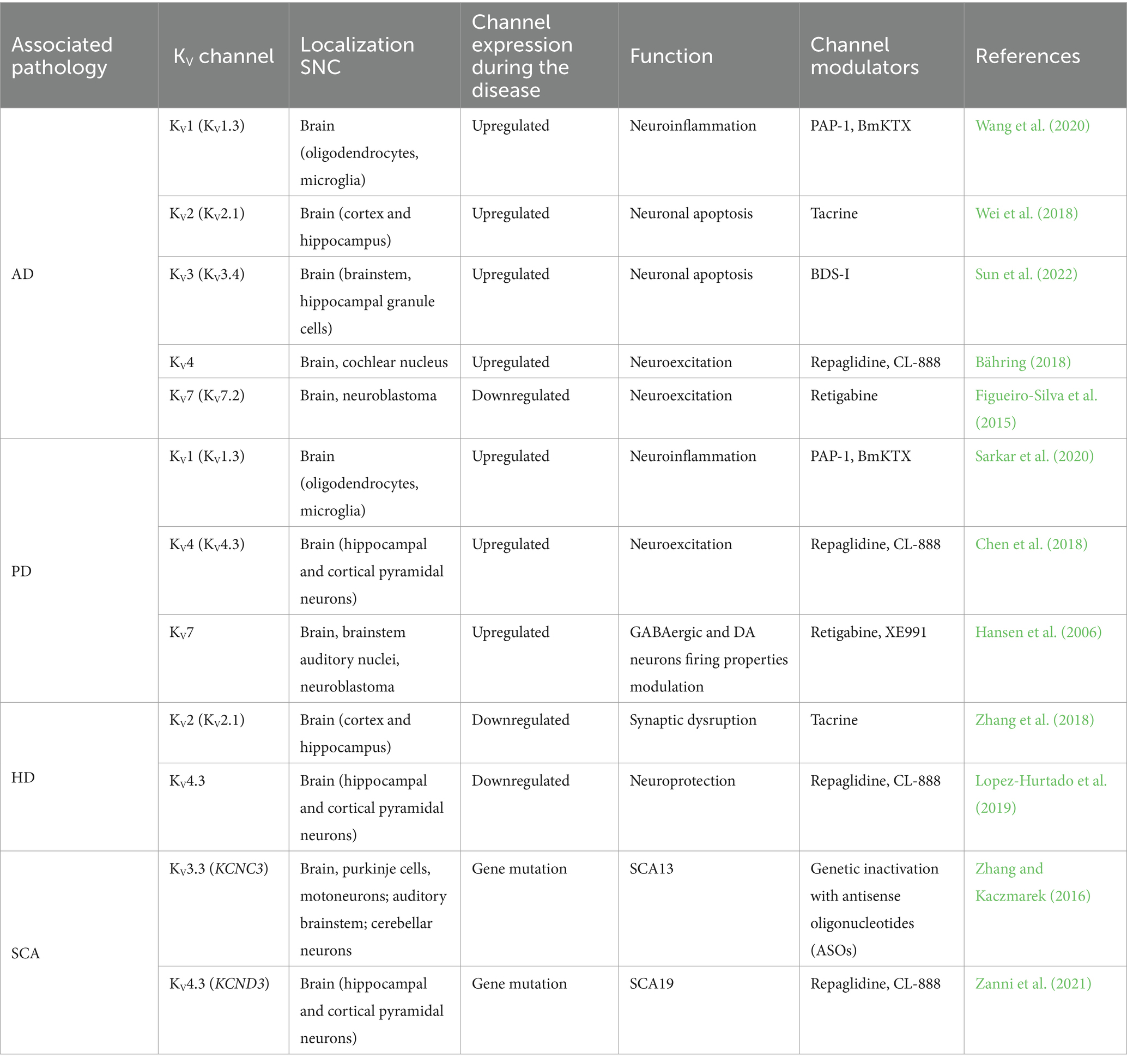
Table 1. Effect of different channelopathies in the neurodegenerative disease and channel modulator.
2.2 KV channels in Parkinson disease
The neuropathological characteristics of Parkinson’s disease (PD) (Figure 1) are the degeneration of dopaminergic neurons in the CNS and the presence of Lewy bodies, α-synuclein-(SNCA)-positive intracytoplasmic inclusions (Poewe et al., 2017). Moreover, in PD pathophysiology there is inflammation due to microgliosis and astrogliosis and it seems that this inflammation is crucial for PD progression (Table 1) (Tansey and Goldberg, 2010).
It has been described that KV1.3 expression is upregulated in some animal models of PD, in vitro experiments, and postmortem human PD brains. Fyn, the Src family kinase that is involved in the microglia activation (Panicker et al., 2015), could regulate the KV1.3 channel expression both transcriptionally and post-translationally modifying its activity and therefore increasing neuroinflammation (Sarkar et al., 2020).
A-type K+ current, generated by KV4.3 and KChip3 interaction, is present in CNS DAergic neurons that contribute to regulating the neuron’s tonic activity (Chen et al., 2018). A53T-SNCA mice mutant which overexpress human α-synuclein with a PD-associated mutation (A53T), showed a oxidative dysfunction of this current induced by the overexpression of the α-synuclein, increasing the firing rate frequency of the dopaminergic substantia nigra neurons (Subramaniam et al., 2014); in both PD animal models and PD patients KV4.3 expression changes have been observed.
KV7 channels are expressed in GABAergic and Dopaminergic neurons in the striatum. Activation of KV7 channels induces hyperpolarization of Dopaminergic neurons and inhibits the excitatory activity (Hansen et al., 2006). Four out of five (KV7.2–KV7.5) M-channels members’ activity is regulated by oxidative and nitrosylation processes in sensory neurons. While oxidation by H2O2 augmented channel activity (Linley et al., 2012), nitrosylation by NO donors inhibited it (Ooi et al., 2013). In oxidative-stress-induced neurodegeneration model, oxidation of the S2–S3 linker of the KV7 enhance the M-current, protecting cells due to neuronal silencing (Gamper et al., 2006; Nuñez et al., 2023).
During PD there is a progressive loss of dopamine (DA) in substantia nigra and consequently in the striatum. Recent studies have proposed the therapeutic role of KCNQ channel blockers as they increase the neuronal bursting pattern in the substantia nigra and enhance DA synthesis in the striatum (Liu et al., 2018).
ERG or Kv11 K+ channels are present in the locus coeruleus (LC) of the brain. This area is related to cognition, learning and memory, among other roles (Uematsu et al., 2017; James et al., 2021; Dahl et al., 2022). This channel prevents increased firing rate and discharge irregularities in those LC neurons (Hasan et al., 2022). In PD, the LC neurons degeneration is present before DAergic neurons degeneration. It has been seen that in Parkinsonian rats the use of ERG K+ channels blockers improves the locomotor deficits, whereas the activators do the opposite, increase burst mode and impaired motor function (Huang et al., 2017). So, this channel dysfunction could be implicated in PD.
2.3 KV channels in Huntington disease
Huntington disease (HD) is a progressive neurodegenerative disease caused by the CAG triples expansion in the Huntington gene (MacDonald et al., 1993). Neurons from striatum and the cerebral cortex are the two main regions affected during HD. Particularly in the medium size spiny neurons (MSNs) from the striatum, K+ channels are necessary to maintain the membrane potential hyperpolarized and the slow firing rate. During HD there is a reduction of KV2.1 channel in MSNs disrupting synaptic integration and consequently information processing (Zhang et al., 2018). At the same time, it is also reported a reduction of M-current, reducing the control of the excitability in striatal output neurons of R6/2 mice (Figure 2). Retigabine, a potential antiepileptic drug, not only restores the hyperactivity network, but also improves motor skills of these mice (Cao et al., 2015).
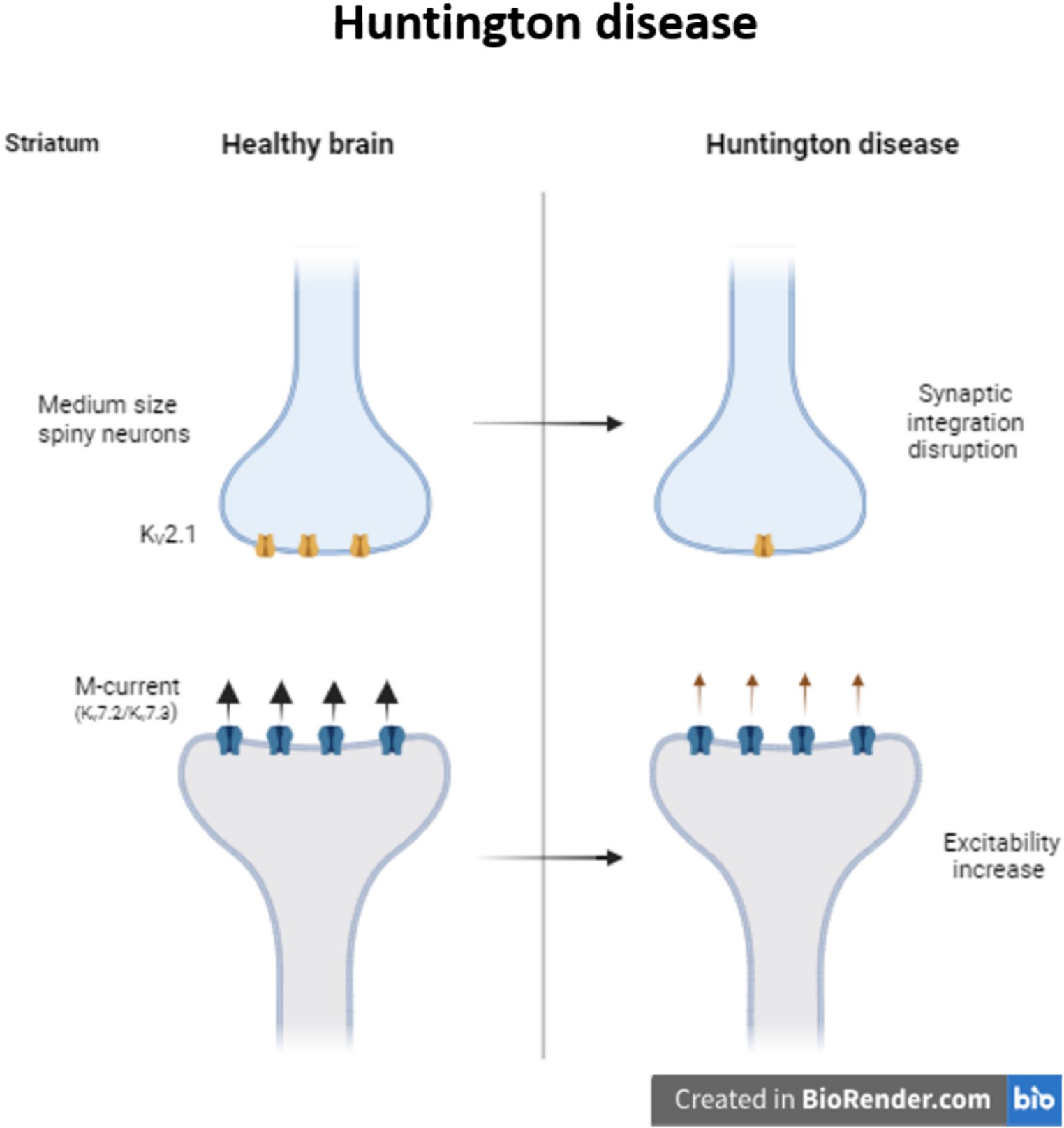
Figure 2. Modulation of KV channels in Huntington disease. Decrease in the expression of KV2.1 provokes synaptic integration disruption in medium size spiny striatum neurons. In Huntington patients, the decrease in the M-current increase the excitability of the striatum neurons.
Moreover, the previously mentioned KChIP3 is downregulated in HD patients and is associated with neuroprotection (An et al., 2000). Several KV4.3/KChIP3 channel complex modulators have been proposed in the last years as therapeutic targets to modulate channel gating and promote neuroprotection during HD (Lopez-Hurtado et al., 2019).
2.4 KV channels in spinocerebellar ataxia
Spinocerebellar ataxia (SCA) is an autosomal dominant neurodegenerative disorder characterized by progressive ataxia with variable symptoms. There are more than 40 distinct genetic SCA (Bhandari et al., 2024). In humans, KV channelopathies are linked to disorders in cell excitability, but only few are the principal responsible for neurodegeneration, mostly the ones that produce spinocerebellar ataxias (SCA) (Zhang and Kaczmarek, 2016). In 2002 there was identified a mutation in the KCND3 gene, that codifies KV4.3 channel that causes SCA19 (Verbeek et al., 2002). Mutations in this gene provoke impairments in the channel traffic from the endoplasmic reticulum to Golgi membrane, reducing the functionality of the channel and consequently provoking the disorder (Duarri et al., 2012; Zanni et al., 2021).
In the mouse model of SCA3, altered KV channel function is associated with Purkinje neuron dysfunction; specifically, the inactivation of KV3 current seems to be the cause (Shakkottai et al., 2011).
The SCA13 is another disorder produced by a mutation in the gene that encodes KV3.3 channel resulting in cerebellar neurodegeneration. The major function of this channel is to drive the repolarization phase of action potential, so mutations in this gene produce disorders of excitability and consequently cerebellar neurodegeneration (Rudy and McBain, 2001; Zhang and Kaczmarek, 2016).
Another KV channel that has been linked to episodic ataxia type 1 (EA1) is the KCNA1 (KV1.1) (Tan et al., 2013). A mutation in KCNA1 gene is the only responsible for the EA1 resulting in episodic ataxia and myokymia. Mutations in this gene can modify the channel current density and consequently channel gating, provoking dysfunctions in the circuits located in several tissues, such as cerebellum, hippocampus or cortex in EA (D’Adamo et al., 2020).
3 Therapeutic approach
Since KV channels are the main regulators of neuronal excitability, their up- and downregulation is linked to enhance several neurodegenerative disorders, such as AD, PD or even provoke ataxias. In order to prevent or repair the development of these neurodegenerative diseases, KV channel modulators have been proposed as therapeutic targets (Table 1).
3.1 KV1 channel modulators
Although certain NaV-blocking anticonvulsant drugs (carbamazepine, phenytoin, and lamotrigine) are used to reduce seizures, they do not work as therapy. In this sense, KV1 (KV1.1 and KV1.3) channelopathies are involved in cell excitability and firing rates in diverse pathological processes, small molecule research and in silico approaches are currently being studied to find modulators of these channels as a target (D’Adamo et al., 2020). KV1.1 dysfunction, for instance, is responsible for episodic ataxia type 1 (EA1). In that regard, some negative modulators of KV1.1 have been found (Wacker et al., 2012), but no molecule able to specifically modulate KV1.1 channels has yet been described. Experimental studies have demonstrated that some resin acids generated by some plants (piramic acid and dehydroabietic acid) are able to open KV channels in vitro by changing voltage-dependent activation towards negative potentials (Ottosson et al., 2015).
Further, hyperactivation of the mTOR pathway is involved in the increased expression and altered distribution of KV1.1 channels in the hippocampus of mice with cortical dysplasia with epilepsy. In those mice, the classical mTOR inhibitor rapamycin normalized the levels of KV1.1, thus proposing that the mTOR pathway may be another possible research target to modulate KV1.1 expression (Nguyen and Anderson, 2018).
On the other hand, the KV1.3 channel is considered a novel therapeutic target to treat neuroinflammatory disorders, such as PD and AD, as it plays a crucial role in microglial cells subsets (Wang et al., 2020). During these neurological disorders, there is an overexpression of KV1.3 channels concluding that KV1.3 specific blockers could mitigate neuroinflammation, and become specific therapeutic candidates during AD or PD (reviewed in Revuelta et al., 2022).
Some studies showed that PAP-1, a KV1.3 blocker, could reduce cerebral Aβ load, diminish neuroinflammation, enhance plasticity of hippocampal neurons and improve behavioral deficits in APP/PS1 transgenic mice (Maezawa et al., 2018). Furthermore, PAP-1 administration reduced neurodegeneration and neuroinflammation in animal models of PD (Sarkar et al., 2020). Moreover, it has been shown that some toxins produced by certain animals can act as a modulator of KV1.3 channels. Specifically, the effects of BmKTX, a scorpion toxin, targeting KV1.3 have been studied as a possible treatment of AD and PD, as it could block microglial activation and thus reduce the neuroinflammation (Wang et al., 2020).
3.2 KV2.1 channel modulators
KV2.1 channel overexpression promotes neurotoxicity and neuronal apoptosis in AD models (Sun et al., 2022), whereas in HD there is a reduction in these channels in medium-sized spiny neurons (MSNs) contributing a synaptic disruption (Zhang et al., 2018).
It has been described that tacrine, a cholinesterase inhibitor, can act on KV channels. It reduces the expression of KV2.1 channels and increases cell proliferation providing neuroprotection during AD (Hu et al., 2020). AD-related mutations can promote increased ROS production leading to KV2.1 channel function loss. Therefore, inhibition of this channel could offer a novel therapeutic approach for AD (Frazzini et al., 2016).
Indeed, several studies relate the activation of KV2.1 channel activators with a better prognosis during HD, since these channels are downregulated in the disease and are associated with the mitochondrial oxidative stress generated in HD (Zhang et al., 2018).
3.3 KV3 channel modulators
KV3.3 channel dysfunction result in the SCA13. A recent study shows that the genetic suppression of KV3.3 channels using antisense oligonucleotides (ASOs) can reverse the SCA13 outcomes (Zhang et al., 2021), meaning that targeting KV3.3 expression may provide a potential therapeutic approach for SCA13.
Concerning the KV3.4 channel, its expression is upregulated during AD due to Aβ deposition, initiating neuronal apoptosis. Recent results suggest that rapid activation/inactivation of these channels could be involved in Aβ-induced neurotoxicity. Therefore, reducing the expression and/or function of KV3.4 in brains with AD could protect Aβ-mediated synaptic alterations (Yeap et al., 2022). Among KV3.4 targets, the BDS-I (blood depressing substance-I), a marine toxin extracted from Anemonia Sulcate, inhibits the channel activity, provoking a reduction of neuronal apoptosis, reducing the expression of certain stress markers, such as active caspase 12; preventing Aβ1-42 induced reactive oxygen species (ROS) production and decreasing the release of pro-inflammatory cytokines (Piccialli et al., 2021).
3.4 KV4 channel modulators
In the hippocampus, KV4 channelopathies are related to epilepsy, schizophrenia, and AD. Therefore, pharmacological modulation of somato-dendritic subthreshold-activating K+ current could function as a therapeutic target for these pathologies (Cercós et al., 2021).
Besides, as previously mentioned, KChIP3 is downregulated in HD patients, promoting neuroprotection. Hence, KV4.3/KChIP3 channel complex inhibitors have been proposed as potential therapeutic targets to promote neuroprotection during HD (Lopez-Hurtado et al., 2019). To date, some molecules, such as repaglidine and CL-888, have been shown to bind and inhibit KV4.3 currents.
3.5 KV7 channel modulators
In addition to their well-known relation with infantile epileptic encephalopathies, KV7 channelopathies are also linked to several age related neurodegenerative diseases (including AD and PD), such as neurotoxicity and alteration of GABAergic and Dopaminergic neuron activation properties. Downregulation of KV7.2 provokes neurotoxicity in AD and therefore, finding activators of these channels could be a therapeutic approach to increase neuronal excitation and synapse. Among other drugs, retigabine, has been described as capable of increasing potassium KV7.2–7.3 channel currents (Czuczwar et al., 2010). Retigabine acts as a positive allosteric modulator, stabilizing the open form of these channels after binding to a hydrophobic pocket near the channel gate (Gunthorpe et al., 2012). Nevertheless, this drug is not in use due to side effects. Even so, this suggests that pharmacological modulation of the M-current could exert beneficial effects on the cognitive deficits involved in the pathophysiology of neurological disorders (Alles and Smith, 2021).
On the other hand, upregulation of KV7 channels causes a modulation of GABAergic and Dopaminergic neuron activation properties. In this sense, XE991 blocks KCNQ channels promoting action potential in DAergic neurons and increasing their excitability. Furthermore, XE991 enhances suprathreshold synaptic responses and promotes depolarization of striatal GABAergic projection neurons (Chen et al., 2018).
3.6 ERG channel modulators
KV11 or ERG channel could be another therapeutic target for PD since the modulation of subthalamic discharge by ERG channel inhibitors attenuates motor dysfunction in PD rats (Huang et al., 2017). The partial block of ERG K+ channels by antipsychotic drugs has also been linked to better dopaminergic neuronal excitability (Shepard et al., 2006).
4 Conclusion
KV channels are essentials for a variety of cell functions. Some of these functions are related with the neuron excitability and it has been probed that the impairment of these channels are implicated in some neurodegeneration diseases. Taking these channels as therapeutic targets and modulating the function of this channel family could be promising to prevent some of the symptoms of these neurodegenerative diseases.
Author contributions
JU: Data curation, Funding acquisition, Resources, Validation, Visualization, Writing – original draft, Writing – review & editing. AA-I: Conceptualization, Methodology, Resources, Writing – original draft. AS-d-R: Conceptualization, Validation, Writing – review & editing. AM-I: Conceptualization, Data curation, Supervision, Writing – review & editing. MG: Supervision, Writing – review & editing. OC: Supervision, Writing – review & editing. MR: Data curation, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work has been financed by the University of the Basque Country UPV/EHU (GIU22-015), Basque Government (IT1707-22) and Ministerio de Ciencia e Innovación MICINN (PID2020-118814RB-I00).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akyuz, E., Koklu, B., Uner, A., Angelopoulou, E., and Paudel, Y. N. (2022). Envisioning the role of inwardly rectifying potassium (Kir) channel in epilepsy. J. Neurosci. Res. 100, 413–443. doi: 10.1002/jnr.24985
Alles, S. R. A., and Smith, P. A. (2021). Peripheral voltage-gated cation channels in neuropathic pain and their potential as therapeutic targets. Front. Pain Res. 2:750583. doi: 10.3389/fpain.2021.750583
Al-Owais, M. M., Hettiarachchi, N. T., Boyle, J. P., Scragg, J. L., Elies, J., Dallas, M. L., et al. (2017). Multiple mechanisms mediating carbon monoxide inhibition of the voltage-gated K+ channel KV1.5. Cell Death Dis. 8:e3163. doi: 10.1038/cddis.2017.568
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., et al. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. doi: 10.1038/35000592
Bachmann, M., Li, W., Edwards, M. J., Ahmad, S. A., Patel, S., Szabo, I., et al. (2020). Voltage-gated potassium channels as regulators of cell death. Front. Cell Dev. Biol. 8:611853. doi: 10.3389/fcell.2020.611853
Bähring, R. (2018). KV channel-interacting proteins as neuronal and non-neuronal calcium sensors. Channels 12, 187–200. doi: 10.1080/19336950.2018.1491243
Bhandari, J., Thada, P. K., and Samanta, D. (2024). “Spinocerebellar ataxia” in StatPearls (Treasure Island, FL: StatPearls).
Buxbaum, J. D. (2004). A role for calsenilin and related proteins in multiple aspects of neuronal function. Biochem. Biophys. Res. Commun. 322, 1140–1144. doi: 10.1016/j.bbrc.2004.08.001
Cao, Y., Bartolomé-Martín, D., Rotem, N., Rozas, C., Dellal, S. S., Chacon, M. A., et al. (2015). Rescue of homeostatic regulation of striatal excitability and locomotor activity in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 112, 2239–2244. doi: 10.1073/pnas.1405748112
Caouette, D., Dongmo, C., Bérubé, J., Fournier, D., and Daleau, P. (2003). Hydrogen peroxide modulates the KV1.5 channel expressed in a mammalian cell line. Naunyn Schmiedeberg’s Arch. Pharmacol. 368, 479–486. doi: 10.1007/s00210-003-0834-0
Cercós, P., Peraza, D. A., Benito-Bueno, A., Socuéllamos, P. G., Aziz-Nignan, A., Arrechaga-Estévez, D., et al. (2021). Pharmacological approaches for the modulation of the potassium channel KV4.x and KChIPs. Int. J. Mol. Sci. 22:1419. doi: 10.3390/ijms22031419
Chen, X., Xue, B., Wang, J., Liu, H., Shi, L., and Xie, J. (2018). Potassium channels: a potential therapeutic target for Parkinson’s disease. Neurosci. Bull. 34, 341–348. doi: 10.1007/s12264-017-0177-3
Cotella, D., Hernandez-Enriquez, B., Wu, X., Li, R., Pan, Z., Leveille, J., et al. (2012). Toxic role of K+ channel oxidation in mammalian brain. J. Neurosci. 32, 4133–4144. doi: 10.1523/JNEUROSCI.6153-11.2012
Czuczwar, P., Wojtak, A., Cioczek-Czuczwar, A., Parada-Turska, J., Maciejewski, R., and Czuczwar, S. J. (2010). Retigabine: the newer potential antiepileptic drug. Pharmacol. Rep. 62, 211–219. doi: 10.1016/S1734-1140(10)70260-7
D’Adamo, M. C., Liantonio, A., Rolland, J. F., Pessia, M., and Imbrici, P. (2020). KV1.1 channelopathies: pathophysiological mechanisms and therapeutic approaches. Int. J. Mol. Sci. 21:2935. doi: 10.3390/ijms21082935
Dahl, M. J., Mather, M., and Werkle-Bergner, M. (2022). Noradrenergic modulation of rhythmic neural activity shapes selective attention. Trends Cogn. Sci. 26, 38–52. doi: 10.1016/j.tics.2021.10.009
Duarri, A., Jezierska, J., Fokkens, M., Meijer, M., Schelhaas, H. J., den Dunnen, W. F. A., et al. (2012). Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann. Neurol. 72, 870–880. doi: 10.1002/ana.23700
Duda, J., Pötschke, C., and Liss, B. (2016). Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J. Neurochem. 139, 156–178. doi: 10.1111/jnc.13572
Eren-Koçak, E., and Dalkara, T. (2021). Ion channel dysfunction and neuroinflammation in migraine and depression. Front. Pharmacol. 12:777607. doi: 10.3389/fphar.2021.777607
Figueiro-Silva, J., Gruart, A., Clayton, K. B., Podlesniy, P., Abad, M. A., Gasull, X., et al. (2015). Neuronal pentraxin 1 negatively regulates excitatory synapse density and synaptic plasticity. J. Neurosci. 35, 5504–5521. doi: 10.1523/JNEUROSCI.2548-14.2015
Frazzini, V., Guarnieri, S., Bomba, M., Navarra, R., Morabito, C., Mariggiò, M. A., et al. (2016). Altered KV2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis. 7:e2100. doi: 10.1038/cddis.2016.18
Gamper, N., Zaika, O., Li, Y., Martin, P., Hernandez, C. C., Perez, M. R., et al. (2006). Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 25, 4996–5004. doi: 10.1038/sj.emboj.7601374
Gazulla, J., and Berciano, J. (2023). Potential benefit of channel activators in loss-of-function primary potassium channelopathies causing heredoataxia. Cerebellum 23, 833–837. doi: 10.1007/s12311-023-01584-8
Gunthorpe, M. J., Large, C. H., and Sankar, R. (2012). The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 53, 412–424. doi: 10.1111/j.1528-1167.2011.03365.x
Hansen, H. H., Ebbesen, C., Mathiesen, C., Weikop, P., Rønn, L. C., Waroux, O., et al. (2006). The KCNQ channel opener retigabine inhibits the activity of mesencephalic dopaminergic systems of the rat. J. Pharmacol. Exp. Ther. 318, 1006–1019. doi: 10.1124/jpet.106.106757
Hasan, S., Delicata, F., Guasti, L., Duranti, C., Haidar, F. M., Arcangeli, A., et al. (2022). Locus coeruleus neurons’ firing pattern is regulated by ERG voltage-gated K+ channels. Int. J. Mol. Sci. 23:15334. doi: 10.3390/ijms232315334
Hu, X.-M., Ren, S., Li, K., and Li, X. T. (2020). Tacrine modulates KV2.1 channel gene expression and cell proliferation. Int. J. Neurosci. 130, 781–787. doi: 10.1080/00207454.2019.1705811
Huang, C.-S., Wang, G. H., Tai, C. H., Hu, C. C., and Yang, Y. C. (2017). Antiarrhythmics cure brain arrhythmia: the imperativeness of subthalamic ERG K+ channels in parkinsonian discharges. Sci. Adv. 3:e1602272. doi: 10.1126/sciadv.1602272
James, T., Kula, B., Choi, S., Khan, S. S., Bekar, L. K., and Smith, N. A. (2021). Locus coeruleus in memory formation and Alzheimer’s disease. Eur. J. Neurosci. 54, 6948–6959. doi: 10.1111/ejn.15045
Kuang, Q., Purhonen, P., and Hebert, H. (2015). Structure of potassium channels. Cell. Mol. Life Sci. 72, 3677–3693. doi: 10.1007/s00018-015-1948-5
Li, X.-T. (2022). Alzheimer’s disease therapy based on acetylcholinesterase inhibitor/blocker effects on voltage-gated potassium channels. Metab. Brain Dis. 37, 581–587. doi: 10.1007/s11011-022-00921-w
Linley, J. E., Ooi, L., Pettinger, L., Kirton, H., Boyle, J. P., Peers, C., et al. (2012). Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc. Natl. Acad. Sci. U.S.A. 109, E1578–E1586. doi: 10.1073/pnas.1201544109
Liu, H., Jia, L., Chen, X., Shi, L., and Xie, J. (2018). The KV7/KCNQ channel blocker XE991 protects nigral dopaminergic neurons in the 6-hydroxydopamine rat model of Parkinson’s disease. Brain Res. Bull. 137, 132–139. doi: 10.1016/j.brainresbull.2017.11.011
Lopez-Hurtado, A., Peraza, D. A., Cercos, P., Lagartera, L., Gonzalez, P., Dopazo, X. M., et al. (2019). Targeting the neuronal calcium sensor DREAM with small-molecules for Huntington’s disease treatment. Sci. Rep. 9:7260. doi: 10.1038/s41598-019-43677-7
Luo, L., Song, S., Ezenwukwa, C. C., Jalali, S., Sun, B., and Sun, D. (2021). Ion channels and transporters in microglial function in physiology and brain diseases. Neurochem. Int. 142:104925. doi: 10.1016/j.neuint.2020.104925
Ma, Q.-L., Teng, E., Zuo, X., Jones, M., Teter, B., Zhao, E. Y., et al. (2018). Neuronal pentraxin 1: a synaptic-derived plasma biomarker in Alzheimer’s disease. Neurobiol. Dis. 114, 120–128. doi: 10.1016/j.nbd.2018.02.014
MacDonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Maezawa, I., Nguyen, H. M., Di Lucente, J., Jenkins, D. P., Singh, V., Hilt, S., et al. (2018). KV1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: preclinical proof of concept. Brain 141, 596–612. doi: 10.1093/brain/awx346
Moiseenkova-Bell, V., Delemotte, L., and Minor, D. L. (2021). Ion channels: intersection of structure, function, and pharmacology. J. Mol. Biol. 433:167102. doi: 10.1016/j.jmb.2021.167102
Nguyen, L. H., and Anderson, A. E. (2018). mTOR-dependent alterations of KV1.1 subunit expression in the neuronal subset-specific Pten knockout mouse model of cortical dysplasia with epilepsy. Sci. Rep. 8:3568. doi: 10.1038/s41598-018-21656-8
Nuñez, E., Jones, F., Muguruza-Montero, A., Urrutia, J., Aguado, A., Malo, C., et al. (2023). Redox regulation of KV7 channels through EF3 hand of calmodulin. eLife 12:e81961. doi: 10.7554/eLife.81961
Ocaña, M., Cendán, C. M., Cobos, E. J., Entrena, J. M., and Baeyens, J. M. (2004). Potassium channels and pain: present realities and future opportunities. Eur. J. Pharmacol. 500, 203–219. doi: 10.1016/j.ejphar.2004.07.026
Ooi, L., Gigout, S., Pettinger, L., and Gamper, N. (2013). Triple cysteine module within M-type K + channels mediates reciprocal channel modulation by nitric oxide and reactive oxygen species. J. Neurosci. 33, 6041–6046. doi: 10.1523/JNEUROSCI.4275-12.2013
Orfali, R., Alwatban, A. Z., Orfali, R. S., Lau, L., Chea, N., Alotaibi, A. M., et al. (2024). Oxidative stress and ion channels in neurodegenerative diseases. Front. Physiol. 15:1320086. doi: 10.3389/fphys.2024.1320086
Ottosson, N. E., Wu, X., Nolting, A., Karlsson, U., Lund, P. E., Ruda, K., et al. (2015). Resin-acid derivatives as potent electrostatic openers of voltage-gated K channels and suppressors of neuronal excitability. Sci. Rep. 5:13278. doi: 10.1038/srep13278
Panicker, N., Saminathan, H., Jin, H., Neal, M., Harischandra, D. S., Gordon, R., et al. (2015). Fyn kinase regulates microglial neuroinflammatory responses in cell culture and animal models of Parkinson’s disease. J. Neurosci. 35, 10058–10077. doi: 10.1523/JNEUROSCI.0302-15.2015
Piccialli, I., Ciccone, R., Secondo, A., Boscia, F., Tedeschi, V., de Rosa, V., et al. (2021). The Na+/Ca2+ exchanger 3 is functionally coupled with the NaV1.6 voltage-gated channel and promotes an endoplasmic reticulum Ca2+ refilling in a transgenic model of Alzheimer’s disease. Front. Pharmacol. 12:775271. doi: 10.3389/fphar.2021.775271
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3:17013. doi: 10.1038/nrdp.2017.13
Revuelta, M., Urrutia, J., Villarroel, A., and Casis, O. (2022). Microglia-mediated inflammation and neural stem cell differentiation in Alzheimer’s disease: possible therapeutic role of KV1.3 channel blockade. Front. Cell. Neurosci. 16:868842. doi: 10.3389/fncel.2022.868842
Rudy, B., and McBain, C. J. (2001). KV3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 24, 517–526. doi: 10.1016/S0166-2236(00)01892-0
Sahoo, N., Hoshi, T., and Heinemann, S. H. (2014). Oxidative modulation of voltage-gated potassium channels. Antioxid. Redox Signal. 21, 933–952. doi: 10.1089/ars.2013.5614
Sarkar, S., Nguyen, H. M., Malovic, E., Luo, J., Langley, M., Palanisamy, B. N., et al. (2020). KV1.3 modulates neuroinflammation and neurodegeneration in Parkinson’s disease. J. Clin. Invest. 130, 4195–4212. doi: 10.1172/JCI136174
Sesti, F., Liu, S., and Cai, S.-Q. (2010). Oxidation of potassium channels by ROS: a general mechanism of aging and neurodegeneration? Trends Cell Biol. 20, 45–51. doi: 10.1016/j.tcb.2009.09.008
Sforna, L., Megaro, A., Pessia, M., Franciolini, F., and Catacuzzeno, L. (2018). Structure, gating and basic functions of the Ca2+-activated K channel of intermediate conductance. Curr. Neuropharmacol. 16, 608–617. doi: 10.2174/1570159X15666170830122402
Shah, N. H., and Aizenman, E. (2014). Voltage-gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Transl. Stroke Res. 5, 38–58. doi: 10.1007/s12975-013-0297-7
Shakkottai, V. G., do Carmo Costa, M., Dell’Orco, J. M., Sankaranarayanan, A., Wulff, H., and Paulson, H. L. (2011). Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J. Neurosci. 31, 13002–13014. doi: 10.1523/JNEUROSCI.2789-11.2011
Shepard, P. D., Canavier, C. C., and Levitan, E. S. (2006). Ether-a-go-go related gene potassium channels: what’s all the buzz about? Schizophr. Bull. 33, 1263–1269. doi: 10.1093/schbul/sbm106
Subramaniam, M., Althof, D., Gispert, S., Schwenk, J., Auburger, G., Kulik, A., et al. (2014). Mutant α-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J. Neurosci. 34, 13586–13599. doi: 10.1523/JNEUROSCI.5069-13.2014
Sun, Q., Liu, F., Zhao, J., Wang, P., and Sun, X. (2022). Cleavage of KV2.1 by BACE1 decreases potassium current and reduces neuronal apoptosis. Neurochem. Int. 155:105310. doi: 10.1016/j.neuint.2022.105310
Talley, E. M., Sirois, J. E., Lei, Q., and Bayliss, D. A. (2003). Two-pore-domain (KCNK) potassium channels: dynamic roles in neuronal function. Neuroscientist 9, 46–56. doi: 10.1177/1073858402239590
Tan, S. V., Wraige, E., Lascelles, K., and Bostock, H. (2013). Episodic ataxia type 1 without episodic ataxia: the diagnostic utility of nerve excitability studies in individuals with KCNA1 mutations. Dev. Med. Child. Neurol. 55, 959–962. doi: 10.1111/dmcn.12236
Tansey, M. G., and Goldberg, M. S. (2010). Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 37, 510–518. doi: 10.1016/j.nbd.2009.11.004
Trimmer, J. S., and Rhodes, K. J. (2004). Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol. 66, 477–519. doi: 10.1146/annurev.physiol.66.032102.113328
Trombetta-Lima, M., Krabbendam, I. E., and Dolga, A. M. (2020). Calcium-activated potassium channels: implications for aging and age-related neurodegeneration. Int. J. Biochem. Cell Biol. 123:105748. doi: 10.1016/j.biocel.2020.105748
Uematsu, A., Tan, B. Z., Ycu, E. A., Cuevas, J. S., Koivumaa, J., Junyent, F., et al. (2017). Modular organization of the brainstem noradrenaline system coordinates opposing learning states. Nat. Neurosci. 20, 1602–1611. doi: 10.1038/nn.4642
Verbeek, D., Schelhaas, J., Ippel, E., Beemer, F., Pearson, P., and Sinke, R. (2002). Identification of a novel SCA locus (SCA19) in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21-q21. Hum. Genet. 111, 388–393. doi: 10.1007/s00439-002-0782-7
Wacker, S. J., Jurkowski, W., Simmons, K. J., Fishwick, C. W. G., Johnson, A. P., Madge, D., et al. (2012). Identification of selective inhibitors of the potassium channel KV1.1–1.2(3) by high-throughput virtual screening and automated patch clamp. ChemMedChem 7, 1775–1783. doi: 10.1002/cmdc.201100600
Wang, X., Li, G., Guo, J., Zhang, Z., Zhang, S., Zhu, Y., et al. (2020). Corrigendum: KV1.3 channel as a key therapeutic target for neuroinflammatory diseases: state of the art and beyond. Front. Neurosci. 14:163. doi: 10.3389/fnins.2020.00163
Weaver, C. M., and Wearne, S. L. (2008). Neuronal firing sensitivity to morphologic and active membrane parameters. PLoS Comput. Biol. 4:e11. doi: 10.1371/journal.pcbi.0040011
Wei, Y., Shin, M. R., and Sesti, F. (2018). Oxidation of KCNB1 channels in the human brain and in mouse model of Alzheimer’s disease. Cell Death Dis. 9:820. doi: 10.1038/s41419-018-0886-1
Wu, L.-Y., Song, Y. J., Zhang, C. L., and Liu, J. (2023). KV channel-interacting proteins in the neurological and cardiovascular systems: an updated review. Cells 12:1894. doi: 10.3390/cells12141894
Yeap, J., Sathyaprakash, C., Toombs, J., Tulloch, J., Scutariu, C., Rose, J., et al. (2022). Reducing voltage-dependent potassium channel KV3.4 levels ameliorates synapse loss in a mouse model of Alzheimer’s disease. Brain. Neurosci. Adv. 6:239821282210864. doi: 10.1177/23982128221086464
Yu, W., Gowda, M., Sharad, Y., Singh, S. A., and Sesti, F. (2019). Correction: oxidation of KCNB1 potassium channels triggers apoptotic integrin signaling in the brain. Cell Death Dis. 10:756. doi: 10.1038/s41419-019-1982-6
Zanni, G., Hsiao, C. T., Fu, S. J., Tang, C. Y., Capuano, A., Bosco, L., et al. (2021). Novel KCND3 variant underlying nonprogressive congenital ataxia or SCA19/22 disrupt KV4.3 protein expression and K+ currents with variable effects on channel properties. Int. J. Mol. Sci. 22:4986. doi: 10.3390/ijms22094986
Zhang, Y., and Kaczmarek, L. K. (2016). KV3.3 potassium channels and spinocerebellar ataxia. J. Physiol. 594, 4677–4684. doi: 10.1113/JP271343
Zhang, Y., Quraishi, I. H., McClure, H., Williams, L. A., Cheng, Y. C., Kale, S., et al. (2021). Suppression of KV3.3 channels by antisense oligonucleotides reverses biochemical effects and motor impairment in spinocerebellar ataxia type 13 mice. FASEB J. 35:e22053. doi: 10.1096/fj.202101356R
Keywords: ion channel, Alzheimer, Parkinson, spinocerebellar ataxia, Huntington, channelopathies
Citation: Urrutia J, Arrizabalaga-Iriondo A, Sanchez-del-Rey A, Martinez-Ibargüen A, Gallego M, Casis O and Revuelta M (2024) Therapeutic role of voltage-gated potassium channels in age-related neurodegenerative diseases. Front. Cell. Neurosci. 18:1406709. doi: 10.3389/fncel.2024.1406709
Edited by:
Alessandro Tozzi, University of Perugia, ItalyReviewed by:
Paola Imbrici, University of Bari Aldo Moro, ItalyEnes Akyuz, University of Wisconsin-Madison, United States
Copyright © 2024 Urrutia, Arrizabalaga-Iriondo, Sanchez-del-Rey, Martinez-Ibargüen, Gallego, Casis and Revuelta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miren Revuelta, bWlyZW4ucmV2dWVsdGFAZWh1LmV1cw==
†These authors have contributed equally to this work and share first authorship