 Susanna Pucci
Susanna Pucci Michele Zoli
Michele Zoli Francesco Clementi
Francesco Clementi Cecilia Gotti
Cecilia Gotti- 1Institute of Neuroscience, National Research Council (CNR), Milan, Italy
- 2NeuroMi Milan Center for Neuroscience, University of Milano Bicocca, Milan, Italy
- 3Department of Biomedical, Metabolic and Neural Sciences, Center for Neuroscience and Neurotechnology (CfNN), University of Modena and Reggio Emilia, Modena, Italy
- 4Department of Medical Biotechnology and Translational Medicine, Università degli Studi di Milano, Milan, Italy
Neuronal nicotinic acetylcholine receptors containing the α9 or the α9 and α10 subunits are expressed in various extra-neuronal tissues. Moreover, most cancer cells and tissues highly express α9-containing receptors, and a number of studies have shown that they are powerful regulators of responses that stimulate cancer processes such as proliferation, inhibition of apoptosis, and metastasis. It has also emerged that their modulation is a promising target for drug development. The aim of this review is to summarize recent data showing the involvement of these receptors in controlling the downstream signaling cascades involved in the promotion of cancer.
Introduction
There is ample evidence that the risk of developing various types of cancer is greater among smokers than non-smokers (Grando, 2014), although the role of smoking in the etiology of some cancers remains controversial (Li et al., 2016; Shao et al., 2016). Tobacco smoke contains at least 4,000 compounds (nicotine and a number of carcinogenic compounds such as tobacco-specific nitrosamines, polycyclic aromatic hydrocarbons, and aldehydes) that are capable of inducing the DNA damaging response that initiates tumorigenesis and enhances the spread of metastases. The risk and severity of cancer depend on the duration of exposure and the amount of tobacco smoking (Schane et al., 2010).
Nicotine is the most addictive component of tobacco smoke and, although it is unable to initiate tumorigenesis in humans and rodents, it promotes tumor growth and metastasis by inducing cell-cycle progression, migration, epithelial-to-mesenchymal transition, invasion, angiogenesis, and the evasion of apoptosis in a variety of systems (Schaal and Chellappan, 2014; Mucchietto et al., 2016), and ultimately causes multiple cancers to develop resistance to chemotherapeutic drugs (Grando, 2014; Afrashteh Nour et al., 2021). Emerging evidence suggests that there is a pathogenic link between nicotine and an increased susceptibility of cells to malignancies, but the underlying mechanisms are still ill-defined. The primary mechanism underlying the way in which nicotine acts as a tumor promoter is by binding and activating nicotinic acetylcholine receptors (AChRs; Mucchietto et al., 2016), which induce the secretion of the growth factors and cytokines that alter the physiology of various organ systems and can activate a number of intracellular mitogenic signaling pathways to promote cell growth, angiogenesis, and other tissue responses. These receptors represent a highly heterogeneous class of ligand-gated ion channels that are enriched in skeletal muscle (where they transduce nerve-to-muscle communication) and in the central and peripheral nervous systems, where they mediate synaptic transmission (Hurst et al., 2013; Zoli et al., 2015).
Neuronal nicotinic acetylcholine receptors (nAChRs) are a family of subtypes that are mainly heteromeric combinations of nine α (α2-α6) and three β (β2-β4) subunits, or homomeric receptors consisting of α subunits (α7 or α9); these last subunits can also form the heteromeric α7β2 and α9α10 subtypes (Zoli et al., 2015). nAChRs are activated by endogenous ligands such as ACh and choline, and exogenous compounds such as nicotine. The α-bungarotoxin (αBgtx) antagonist binds and blocks only some of the nAChR subtypes, which are therefore divided into two main classes: the αBgtx-sensitive subtypes, which may be homomeric or heteromeric and consist of the α7, α7β2, α9, and α9α10 subtypes, and the αBgtx-insensitive receptors, which are always heteromeric combinations of α (α2-α6) and β (β2-β4) subunits (Gotti et al., 2006; Albuquerque et al., 2009).
Recent studies have clearly shown that nAChRs and their physiological agonist, ACh, are widely expressed in non-neuronal mammalian cells (Grando, 2014; Zoli et al., 2018; Chen et al., 2019) from which ACh is released into the extra-cellular space in an autocrine, paracrine or juxtacrine manner, and that the cholinergic/nicotinic system plays important roles in various aspects of cell biology and homeostasis (Wessler and Kirkpatrick, 2008; Friedman et al., 2019). nAChRs and their endogenous ligands are also expressed by cancer cells and tissues.
Structure, Pharmacology, and Localization of α9 nAChRs
The various combinations of nAChR subunits lead to the formation of a heterogeneous family of pentameric receptor subtypes that have different structural, functional, and pharmacological properties. In heteromeric nAChRs consisting of α and ß subunits, the ACh binding site (also known as the orthosteric site) lies at the interface of the extra-cellular domains of adjacent subunits and consists of a principal component of one α subunit, and a complementary component of the adjacent ß structural subunit.
The α9α10 subtype is a heteromeric receptor that only consists of α subunits (α9 and α10), and it has been proposed that α10 may act as a structural subunit, in much the same way as a β subunit of heteromeric nAChRs providing only complementary components to the agonist binding site (Elgoyhen and Katz, 2012). However, recent studies have shown that α9 and α10 subunits equally contribute to the principal component of the α9α10 nAChR binding site, whereas the contribution of α9 and α10 to the complementary component is non-equivalent (Boffi et al., 2017).
Experiments on the X-ray crystal structures of the extracellular domain of the monomeric state of the human neuronal α9 subunit and its complexes with a αBgtx, methyllycaconitine (MLA; Zouridakis et al., 2014) or the α-conotoxin RgIA (Zouridakis et al., 2019) have shown that, despite the absence of a complementary subunit and transmembrane domain, the α9 extracellular domain has a native folding and can bind MLA, αBgtx or α-conotoxin RgIA through its principal component.
The α9α10 subtype may have two different stoichiometries, (α9)2(α10)3 (Plazas et al., 2005) and (α9)3(α10)2 (Indurthi et al., 2014), which have varying sensitivities to ACh and the Vc1.1 α-conotoxin. In addition to the computationally established α9α10 binding site common to both stoichiometries, the (α9)3(α10)2 stoichiometry has an additional, binding site at the α9/α9 interface of low sensitivity for the α-conotoxin Vc1.1 (Indurthi et al., 2014).
On the basis of their sequence similarity to nAChRs, the α9 and α10 subunits were initially classified as neuronal, but the homomeric α9 and the heteromeric α9α10 receptors are not functionally present in the brain (Morley et al., 2018). The α9α10 subtype is only endogenously expressed in cochlear and vestibular hair cells in the inner ear, and in extra-neuronal tissues. Studies of α9 (Vetter et al., 1999) and α10 knockout (KO) mice (Vetter et al., 2007) have demonstrated the physiological role of this subtype in the medial olivocochlear pathway, in which the targeted disruption of the α9 (Vetter et al., 1999) or α10 subunits (Vetter et al., 2007) abolishes ACh-mediated synaptic currents in both inner and outer hair cells. More recently, Morley et al. (2017) have used constitutive α9 and/or α10 KO mice to show that only the doubly constitutive mice showed abnormal efferent innervation of cochlear hair cells. Moreover, a very recent study (Wang et al., 2021) has demonstrated that, in the developing auditory system, α9 and α10-containing receptors are involved in the spontaneous activity generated in the cochleae that propagates to the CNS to promote circuit formation.
α9 -containing receptors are also expressed at the splanchnic nerve–chromaffin cell synapse, where they are involved in the excitatory neurotransmission. These receptors are upregulated in cold-stressed rats where they become a major determinant of ACh-induced current (Colomer et al., 2010).
α9 subunit is also expressed by the cells of the immune system (Lustig et al., 2001; Hecker et al., 2015) but unlike in neurons and cochlear hair cells, where α9-containing receptors function as ligand-gated ion channels, in immune cells α9 ligand-evoked ion currents have not been observed (Richter et al., 2016).
The functional properties of α9α10 nAChRs vary widely across species (Marcovich et al., 2020). Xenopus oocytes expression of the mammalian α9 subunit forms functional channels, whereas the rat or human α10 subunit only forms functional channels when it is co-expressed with an α9 subunit. The oocytes co-injection of α9 and α10 subunits leads to a subtype that has the same ACh EC50 as the α9 subtype but greater functional expression (Elgoyhen et al., 1994, 2001).
A number of the pharmacological characteristics of α9 and α9α10 nAChRs are particularly interesting: they are activated by ACh but not by the classical agonist nicotine that blocks ACh-gated currents, and the α9α10 nAChR has a mixed nicotinic and muscarinic profile as it is blocked by the nicotinic antagonists d-tubocurarine and αBgtx as well as by the muscarinic antagonist atropine (Elgoyhen et al., 1994, 2001; Sgard et al., 2002).
nAChR-Associated Proteins
All nAChRs are integral membrane proteins that share the same basic topology, and neurotransmitter receptors coupled to an ion-conducting pore. A plethora of proteins and metabolites acting at multiple steps in nAChR biogenesis have recently been discovered (Crespi et al., 2018; Matta et al., 2021): some of these proteins are endoplasmic reticulum-resident chaperone proteins that are necessary for the proper folding and assembly of some nAChR subtypes, whereas others are auxiliary (accessory) subunits that stably associate with receptors and control channel function.
When expressed in transfected cell lines, the α9α10 subtype is not functional. Genome-wide screening has shown that choline acetyltransferase (ChAT), the biosynthetic enzyme for ACh, and the protein TMIE (Gu et al., 2020) are factors enabling the function of α9α10 nAChRs. Functional studies have demonstrated that the assembly and surface expression of α9α10 nAChRs require the ligand binding of an agonist (ACh) or antagonist (αBgtx), and that mutating the α9α10 ACh binding site abolishes receptor surface expression. They have also shown that the presence of TMIE protein, a deafness gene product, is important for the correct functioning of α9α10 nAChRs: TMIE acts as an auxiliary subunit and, by associating with the α9α10 receptors on the cell surface, promotes their function (Gu et al., 2020).
The proteins that stably associate with nAChRs comprise the LY6 prototoxin family, a family of small modulatory proteins that can interact with numerous targets. These proteins have a three- dimensional structure that is similar to that found in snake venom neurotoxins (Miwa, 2021). Some of them are secreted as water-soluble proteins, and others are bound to cell membranes by a glycosylphosphatidylinositol (GPI) anchor. Lynx1, the first member of the LY6 family to be identified, can form a stable complex and regulate the responses of α4β2 and α7 nAChRs. Lynx1 is expressed in normal and neoplastic lung tissue, but its level is lower in lung cancers than in adjacent normal lung tissue (Fu et al., 2015). The recombinant water-soluble variant of Lynx1 inhibits the growth of adenocarcinoma A549 cells by causing cell cycle arrest via the modulation of α7 nAChRs and the activation of various intra-cellular signaling cascades (Bychkov et al., 2019). Although α9α10 nAChRs have not been detected among the targets of Lynx1, it has been shown that a recently synthesized circularized Lynx1-derived peptide binds to the ligand binding domain of α9 nAChRs and inhibits the rat α9α10 subtype expressed in Xenopus oocytes (Kryukova et al., 2019).
Ionotropic and Metabotropic nAChR Signaling
The ionotropic mechanism of inter-cellular signaling mediated by nAChRs has been studied in neurons (Albuquerque et al., 2009; Corringer et al., 2012), and is due to the opening the ionic channel that is an integral part of the receptor and generating cation fluxes that lead to membrane depolarization. This depolarization opens voltage-gated Ca2+ channels (VGCCs), and leads to a further Ca2+ influx that may induce Ca2+-induced Ca2+ release (CICR) from the endoplasmic reticulum by activating ryanodine receptors (RyRs; King et al., 2015; Kabbani and Nichols, 2018). The nAChR subtypes with the greatest Ca2+ permeability are the homomeric α7 and α9, and the heteromeric α9α10 nAChRs (Fucile et al., 2003, 2006).
In non-excitable cells, the increase in the concentration of free intracellular Ca2+ induced by activation of nAChRs mainly derives from ionic influx through the receptors since VGCCs are poorly expressed.
In astrocytes, whose membrane resting potential is critically lower than that measured in neurons, ionic influx through ligand-gated channels may be largely responsible for the increased intracellular Ca2+ levels that modulates Ca2+-gated channels locally, and activates various Ca2+-dependent signaling mechanisms (Mcneill et al., 2021).
However, over the last decade, it has been shown that α7-, α4-, β2-, and β4-containing nAChRs also have a metabotropic mechanism underlying the neuronal response to nicotinic agonists (Nordman and Kabbani, 2014; King et al., 2015). The metabotropic pathway increases intracellular [Ca2+] via Gαq protein coupled with nAChR signal transduction mediated by phospholipase C (PLC) and inositol-trisphosphate (IP3) receptor-activated Ca2+ release from the endoplasmic reticulum (King et al., 2015; Kabbani and Nichols, 2018).
There are new lines of evidence that, in neurons, agonist stimulation of presynaptic α7 nAChRs mediates Ca2+ signaling via both ionotropic Ca2+ influx through the open-channel state and non-conventional metabotropic channel signaling. Agonist binding to α7 determines sufficient Na+ influx-induced depolarization to activate VGCCs and Ca2+ influx and lead to RyR-mediated CICR from the endoplasmic reticulum. In addition, the transition of α7 nAChRs to a high-affinity ligand-bound desensitized state supports a metabotropic response marked by G protein activation of PLC, IP3 production, and IP3-induced Ca2+ release (Papke and Lindstrom, 2020; Borroni and Barrantes, 2021).
Kabbani’s group (King et al., 2015; King and Kabbani, 2016; Kabbani and Nichols, 2018) has shown that the α7 subunit binds Gα and Gßγ proteins through the intracellular M3-M4 loop, and enables a downstream Ca2+ signaling response that can persist longer than the expected time course of channel activation and leads to the activation of downstream second messenger pathways such as RhoA GTPase and cytoskeletal changes during neurite growth.
In many non-neuronal cell types, metabotropic signaling after ligand binding is a property of α7 receptors and it is present in immune cells where cholinergic signaling plays an important role in regulating cytokine release even though no ACh-dependent currents have been recorded, and despite the fact that α7 receptors are on the plasma membrane (Fujii et al., 2017; Treinin et al., 2017).
It has also been shown that α9 subunit-containing nAChRs have metabotropic functions. Homomeric α9 nAChRs or heteromeric α9α10 nAChRs heterologously expressed in Xenopus oocytes do not generate ion currents when stimulated with phosphocholine, but do so when stimulated by choline, a metabolite of ACh (Richter et al., 2016) that also acts as an agonist of the α7 nAChR.
Various metabotropic functions of α7 and α9 nAChRs have been described in monocytes, in which choline derivatives (choline, phosphocholine, and ACh) inhibit ATP-mediated IL1ß release by blocking purinergic P2X7 receptors (Hecker et al., 2015). The effect of phosphocholine on IL-1ß release takes place via activation of the α9α10 metabotropic pathway. ACh and phosphocholine both inhibit IL-1ß release in monocytes, but only the former evokes ion currents through the receptor. Pre-incubation with phosphocholine reduces ACh-evoked ion flux, thus suggesting that phosphocholine acts as a silent agonist that converts α7 or α9 receptors to a long-lived desensitized state that elicits signal transduction independently of ion channel opening (Richter et al., 2016; Papke and Lindstrom, 2020).
In brief, depending on the type of cell and the nAChR subtypes expressed, the binding of ACh or nicotine can induce conformational changes in nAChRs and/or their associated proteins that can activate various intracellular signaling pathways and regulate gene expression.
Methods Used to Study α9-Containing Receptors
A number of approaches have been used to detect nAChRs in many extra-neuronal tissues. Reverse transcription polymerase chain reaction (RT-PCR), quantitative RT-PCR, and in situ hybridization have all been used to show that they are expressed at variable levels in normal and malignant cells, but interpreting the data obtained is not easy because there is not always a correlation between the levels of mRNA and those of the expressed subunit.
The first evidence of α9 nAChR expression came from rat cochlear hair cells, in which α9 nAChRs are involved in auditory functions (Elgoyhen et al., 1994), but many more locations and biological functions of α9 nAChR have since been identified, including keratinocyte adhesion (Nguyen et al., 2000; Chernyavsky et al., 2007), breast epithelial cancer formation (Lee et al., 2010), endocrine activities (Colomer et al., 2010), immune responses (Hecker et al., 2015), inflammation (Liu et al., 2017, 2019), chronic pain (Romero et al., 2017; Hone et al., 2018; Zheng et al., 2021), the homeostasis of osteocytes, and the regulation of bone mass (Baumann et al., 2019).
Supplementary Table 1 summarizes the studies in which the presence of α9- and α9α10-containing receptors has been shown in cancer cells and tissues.
The most convincing identification of α9 nAChRs in such tissues has been obtained by the use of subtype-specific antagonists (Li et al., 2021) that block many cancer-promoting processes and/or by comparing their effects on the wildtype (WT) nAChR subunit and KO mice. It is important to note that the antagonists αBgtx and MLA not only act on α9 nAChRs but also bind with high affinity to α7 nAChRs (Elgoyhen et al., 1994; Verbitsky et al., 2000). Consequently, if they are used to study the in vivo or in vitro effects of nicotine or ACh on native α9 nAChRs, it needs to be remembered that α7 nAChRs may also be involved. In order to differentiate or analyze the effects mediated only by α9 nAChRs, the further use of α9-selective antagonists such as α-conotoxin Vc1.1 (Indurthi et al., 2014), α-O-conotoxin GeXIVA (Luo et al., 2015), RgIA4 (Romero et al., 2017), and PeIA (Mcintosh et al., 2005; Yu et al., 2018) is crucial. Dimeric constructs of the toxins PeIA, Vc1.1, and RgIA# ([ΔR13]RgIA) have recently been designed, and all three constructs are more potent than their monomeric counterparts in relation to human α9α10 nAChRs (Liang et al., 2020; see Table 1).
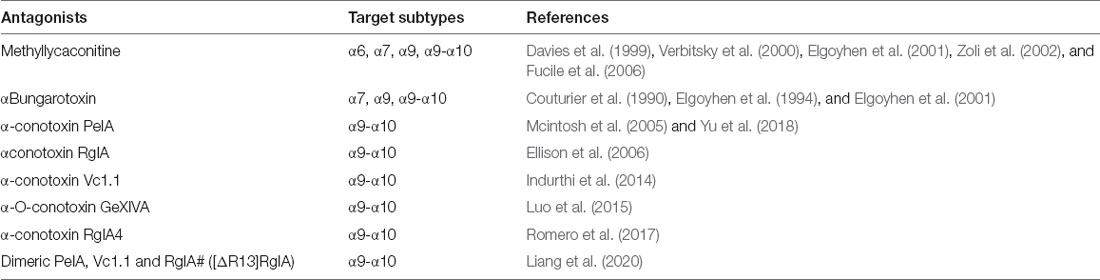
Table 1. Alkaloid and peptidic antagonists targeting α9-containing receptors.
Another successful approach to studying the in vitro and in vivo roles of α9- and α10-containing receptors is to over-express or silence them by RNA interference, a very powerful technique that has given important information concerning the involvement of these receptors in physiological and pathological conditions (Lee et al., 2010).
Extensive work has very recently been done to identify human cancer proteins interacting with membrane proteins and determine their downstream regulated pathways using a systematically integrated method. As α9 is an integral membrane protein, Lin et al. (2019) have tried to identify new α9 nAChR interacting and downstream proteins by means of multiple RNA-seq and microarray data relating to different cancers. Using CHRNA9, they identified 18 new representative candidates that were subsequently verified by means of immunoprecipitation and Western blotting. They also found associated proteins that are important for regulated pathways, such as proteins involved in the cell cycle, adherens junctions, and ErbB signaling. These were related to cell growth and communication in more than six cancer types, thus indicating that α9 plays a role in the formation, progression, and metastatic spread of various tumors (Lin et al., 2019).
As recent exhaustive reviews (Schuller, 2009; Grando, 2014; Schaal and Chellappan, 2014) have already considered the mechanisms by means of which nAChRs are involved in cancer signaling pathways, this review will concentrate on the latest published data concerning the role of α9/α9α10 (α9*) receptors in the physiology and pathology of lung, breast, glioblastoma, and melanoma cancers.
α9-Containing Receptors in Lung Cancer
Lung cancer is the most frequent and lethal cancer in the world, accounting for 1.79 million deaths in 2020 (Ferlay et al., 2021) and, although a subset of patients develop lung cancer without a history of smoking, approximately 70% of all cases of non-small cell lung cancer (NSCLC) and 90% of all cases of small cell lung cancer (SCLC) are caused by smoking (Samet et al., 2009; Pesch et al., 2012; Improgo et al., 2013).
A very recent study (Zhang et al., 2020) has shown that nicotine treatment of human lung epithelial cells can create a pro-tumorigenic environment favoring cell transformation or tumorigenesis in lung epithelium by perturbing intracellular redox status and altering p53 function. Together with a study by Tang et al. (2019) showing that mice exposed to nicotine delivered by means of electronic cigarette smoke develop lung adenocarcinoma, this suggests the need for caution when using nicotine replacement therapies and e-cigarettes.
Lung cells express the mRNA of multiple nAChR subunits whose levels are frequently altered in primary NSCLC and SCLC, and their cell lines. It has been found that the levels of α3, α5, ß4, and α9 subunit mRNAs are significantly increased in SCLC (Improgo et al., 2010, 2013), and that the levels of α7, α9, and α10 subunit mRNA are also increased in a number of cell lines derived from SCLC and NSCLC tumors (Improgo et al., 2010).
Various studies have shown that the nAChRs present in lung epithelial and endothelial cells play a direct role in lung cancer. In particular, nAChRs are expressed in airway epithelium cells that synthesize, store, and secrete the physiological ligand ACh, which acts as an autocrine and paracrine growth factor (Spindel, 2016; Friedman et al., 2019). When lung cancer arises from the airway epithelium, cell growth is stimulated by ACh or nicotine, and this may provide endogenous mitogenic signaling without any further activation.
Inhibiting cholinergic signaling causes apoptosis in bronchioalveolar carcinoma cells (Lau et al., 2013), and nicotine protects lung cancer cells against apoptosis induced by chemotherapy (Liang et al., 2020), oxidative stress, and ionising radiation (Friedman et al., 2019).
Case control studies of NSCLC patients (340) and matching controls (435) have shown that an increased risk of developing lung cancer is associated with two single nucleotide polymorphisms (SNPs) in the CHRNA9 gene: rs56159866 and rs6819385 (Chikova et al., 2012).
NSCLC can be subdivided into adenocarcinomas, squamous cell carcinomas, and large-cell lung carcinomas. Adenocarcinomas are the most frequent type of NSCLC and develop from small airway epithelial cells and alveolar type II cells, whereas squamous cell carcinomas arise from large airway epithelial cells (Meza et al., 2015), and α7 and α9 mRNAs have been detected in adenocarcinoma and squamous carcinoma cells, as well as in human tumor samples in which their levels were higher than those observed in normal lung tissue (see Supplementary Table 1).
α9-Mediated Signaling Pathways in Lung Tumors
Studies of A549 adenocarcinoma cells have shown that treatment with a nicotine concentration that is similar to the levels measured in sera of smokers (100 nM) promotes their proliferation (Mucchietto et al., 2018), and this proliferation is blocked by the action of MLA and αBgtx on α7 and α9 receptors, and by subtype-specific α9 (RgIA4) and α7 toxins (AR). The concentration of nicotine increasing cell proliferation was much less than that required to activate α9 or α7 receptors in functional experiments. This suggests that nicotine induced the non-ionic signaling events that regulate the phosphorylation states of the target proteins mediating some of the effects of nicotine on A549 cells (Schaal and Chellappan, 2014). In the same cells, nicotine acting on α9-containing receptors stimulates the Akt and ERK cell signal transduction pathways, and the use of the α9-selective peptidic antagonist RgIA4 prevents nicotine-induced ERK and Akt phosphorylation The involvement of α9-containing nAChRs in nicotine-induced proliferation and intracellular signaling in A549 cells has been further confirmed by the fact that silencing experiments using siRNA directed against α9 mRNA replicated the results obtained using RgIA4. All the effects of nicotine on A549 cell proliferation and signaling activation described above are also blocked by the α7-selective toxin AR and the α7- and α9-unselective antagonists MLA and αBgtx, thus suggesting that both receptors have to be activated in order to reach the cytoplasmic Ca2+ concentration that triggers the biological effects. In addition, the fact that the nicotine-induced proliferation and intracellular signaling of A549 cells is blocked by α7 and α9 peptidic antagonists that are not cell permeable indicates that the effects of nicotine are due to the activation of the nAChRs present on the plasma membrane.
α9-Containing Receptors in Breast Cancer
In 2020, it was estimated that breast cancer accounted for about 2.26 million new cases and 682, 000 deaths, thus making it the most frequently diagnosed cancer worldwide, (Ferlay et al., 2021). It is also the second leading cause of cancer-related deaths among women, mostly of which are due to recurrences and metastases (Ferlay et al., 2021).
Tobacco smoke may contribute to a woman’s risk of developing breast cancer as large-scale cohort studies conducted in the United States and Japan indicate that the risk is associated with both active and passive smoking (Slattery et al., 2008).
One case-control study of 737 breast cancer patients and 719 age-matched healthy controls has studied the association between CHRNA9 SNPs and the risk of breast cancer, and examined the joint effect of cigarette smoke exposure and CHRNA9 SNPs on developing breast cancer. It found that an increased risk of breast cancer is associated with the variant rs73229797 allele on the CHRNA9 gene, and that the risk is greater in both active and passive smokers (Hsieh et al., 2014).
Over the last decade, it has been shown that α9 nAChRs are important in the formation of breast cancer. Lee et al. (2010) have shown that α9-containing nAChRs are expressed in many, breast cancer, epithelial and lung cells most of which also express α10 and α5 subunits. They also found that α9-containing nAChRs are present in both primary tumors and non-malignant breast tissue obtained from patients (see Supplementary Table 1), but their expression is higher in breast cancer cells than in the surrounding normal tissue. Not only did 67.3% of their 276 breast cancer tissue samples express high α9 nAChR levels, this expression was higher in the advanced stage III and IV samples than in the early stage I and II samples, and this occurred more often in smokers than in passive smokers or non-smokers.
α9-Mediated Effects of Nicotine on Breast Cancer Cells
In order to explore the potential carcinogenic effects of the α9 subunit expressed in normal human breast epithelial cells, Lee et al. (2010) established the MCF-10A normal human breast cell line with the tetracycline-regulated (Tet-off) over-expression of α9 nAChRs, and found that it became potentially carcinogenic in vitro and in vivo. Chen et al. (2011) found that the same line xenografted in BALB/c-nu/nu mice induced cyclin D3 over-expression after exposure to nicotine, whereas silencing α9 nAChR expression in breast cancer cells reduced in vitro and in vivo cyclin D3 levels, cell proliferation, and tumorigenic potential (Chen et al., 2011).
It has been shown that 10 μM nicotine stimulates nAChRs (particularly α9-containing nAChRs) to induce proliferation in two breast cancer cell lines (MDA-MB 231 and MCF-7), and induces the growth of tumors xenografted into SCID mice (Lee et al., 2010). In addition, prolonged exposure of normal MCF-10A human breast to 10 μM nicotine or 1 μM 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) triggers pre-cancerous transformation, an effect that was potentiated by increasing the expression of α9 nAChRs (Lee et al., 2010, 2011). It has also been shown that in MCF-7 human breast cancer cells, nicotine and estrogen induce α9 nAChR expression and that this is transcriptionally regulated by estrogen receptors (ERs). The binding of nicotine to α9-containing nAChRs activates the downstream PI3K, Akt or MAPK signaling pathways, thus facilitating the phosphorylation of ERs and their binding to the AP1 transcription factor and the CHRNA9 promoter region.
Nicotine exposure promotes the apoptosis resistance of breast cancer cells by increasing α9 expression, which activates STAT3 nuclear translocation and physical interactions with the promoters of the gene coding for Galectin-3 (LGALS3), an intra-cellular anti-apoptotic α-galactoside-binding lectin (Guha et al., 2014; see Figure 1), and the TWIST1 promoter. Activated STAT3 directly binds these promoters, thus inducing their transcription and up-regulation, and this delays apoptosis and enriches a sub-population of breast cancer cells that have stem cell-like properties.
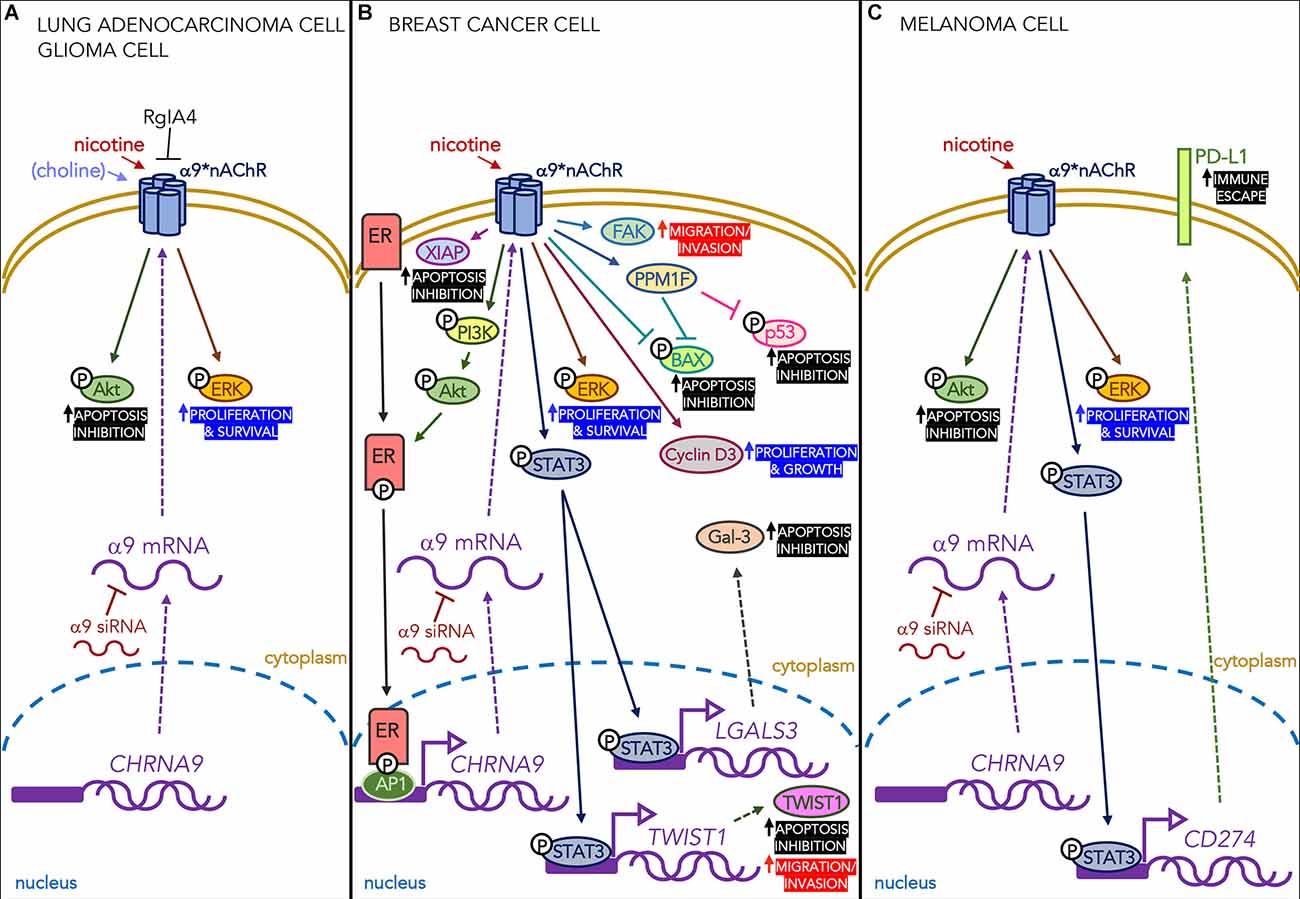
Figure 1. Effects of nicotine acting on α9-containing receptors expressed in different types of cancer cells. (A) Nicotine (in lung adenocarcinoma and glioma cells) and choline (only in glioma cells) mediate proliferative and anti-apoptotic effects through the ERK and Akt signaling pathways. These effects are blocked by the α9-selective antagonist RgIA4 and α9 siRNA. (B) In breast cancer cells, nicotine acting on α9 nAChRs activates the PI3K/Akt pathway that phosphorylates intra-cellular estrogen receptors (ERs) which, in their turn, bind transcription factor AP1 and the CHRNA9 gene promoter region. It also increases the levels of pro-proliferative cyclin D3, the anti-apoptotic protein XIAP, and the PPM1F protein phosphatase that reduces phosphorylated p53 and BAX, thus protecting the cells from apoptosis. Finally, it promotes the phosphorylation of STAT3 and its nuclear translocation, thus increasing the transcription of the gene encoding the anti-apoptotic lectin Gal-3. All of these effects are blocked by α9 siRNA. (C) In melanoma cells, nicotine increases the number of α9 nAChRs and phosphorylates STAT3, which binds the PD-L1 promoter in the nucleus and leads to the higher surface expression of PD-L1, thus promoting the immune escape of cancer cells. The increased expression of PD-L1 protein is down-regulated by α9 siRNA.
As it is still debated whether exposure to second-hand smoke (SHS) affects the risk of breast cancer, Fararjeh et al. (2019) used a model of breast cell carcinogenesis in which immortalized normal human breast epithelial cells exposed to a carcinogen undergo long-term exposure to 1 pM nicotine and/or 100 femtM NNK doses (that can be reached during exposure to SHS). They found that repeated co-exposure to nicotine and NNK disrupted cell growth control and led to the acquisition of cancer cell properties, and increased the levels of a number of α9 nAChR downstream signaling proteins, such as the FAK migration marker and the PI3K/Akt intracellular signaling pathway. They also found the increased expression of XIAP (a protein that may play an important role in breast cancer pathogenesis by inhibiting apoptosis) and a decrease in the expression of the pro-apoptotic protein BAX.
Cell invasion and the spread of metastases are crucial processes in tumor development that are regulated by numerous factors, including growth factors, phosphatases, kinases, and extracellular matrix components. PPM1F is a Ser/Thr protein phosphatase that is overexpressed during breast tumor invasion: the levels of PPM1F protein are higher in invasive MDA-MB-231 breast cancer cells than in non-invasive MCF-7 cells, and PPM1F levels are higher in the breast tumor tissue of smokers than in that of non-smokers. Nicotine induces in vitro PPM1F expression, whereas silencing α9 nAChRs reduces PPM1F protein levels, thus ultimately reducing cell motility and invasiveness. Tu et al. (2016) have found that the overexpression of PPM1F reduces the level of phosphorylated p53 and phosphorylated BAX in nicotine-treated MDA-MB-231 cells. These findings show that PPM1F is a phosphatase that inactivates pro-apoptotic proteins (such as BAX) and proteins involved in cell cycle arrest (such as p53), and may be a downstream mediator of α9 nAChRs.
It has recently been shown that breast cancer cell proliferation in vitro can be inhibited by selectively antagonising α9α10 nAChRs (Sun et al., 2020) using the selective αO-conotoxin GeXIVA.
α9 nAChRs in Triple-Negative Breast Cancer
Breast cancer negative for ERs, progesterone receptors, and excess human epidermal growth factor receptor 2 protein (triple-negative breast cancer, TNBC) is responsible for 15–20% of all breast cancer cases (Lee et al., 2019).
The over-expression of α9 nAChRs is associated with the poor 5-year disease-specific survival of patients diagnosed as having breast cancer as it up-regulates ER expression.
Huang et al. (2017) used a serial spontaneous pulmonary metastasis animal model to generate highly metastatic TNBC cells derived from metastasized lung cells, and found that there was a significant induction of CHRNA9 gene expression and that the level of α9 nAChR expression correlated with the metastatic ability of the tumor. These findings suggest that α9 nAChR expression is required to mediate TNBC metastasis during the development of cancer, and may be a biomarker for targeted therapy.
α9-Containing Receptors in Gliomas and Glioblastomas
Gliomas represent 81% of all primary malignant brain tumors in adults, and are one of the most aggressive human cancers (Jemal et al., 2011). The World Health Organisation (WHO) stratifies gliomas into grades I-IV, the most aggressive of which are grade IV astrocytomas or glioblastomas (GBMs; Louis et al., 2001). The global incidence of GBMs is 2–5 per 100,000 people and has increased over the last decade (Dobes et al., 2011). Median patient survival is approximately 14–17 months (Zhu et al., 2017), and the 5-year survival rate is only 5.1% (Ostrom et al., 2015). GBMs originate in brain neural cells and are highly proliferative and invasive. They may begin in astrocytes, glial precursor cells or neural stem cells, but the pathophysiological and molecular mechanisms underlying their malignant transformation are still unclear. The poor prognosis of GBM is primarily attributable to therapeutic resistance and tumor recurrence after surgical removal, which is due to the presence of a small sub-population of self-renewing and proliferative glioblastoma stem cells (GSCs) similar to the neural stem cells from which they may arise.
The results of numerous retrospective studies have failed to show any causal relationship between smoking and gliomas, which is why the hypothesis of a causative link between smoking and the etiology of malignant gliomas remains controversial (Hou et al., 2016; Li et al., 2016).
Very few articles have described studies of the mechanisms of action of nicotine on glioma or GBM cells and, given the limited amount of material available, this review will thoroughly discuss all of the most recent findings.
Khalil et al. (2013) have shown that nicotine concentrations comparable to those found in the serum of chronic smokers enhance GBM cell migration, proliferation, survival, clonogenicity, and radio-resistance partially by activating EGFRs, ß-adrenergic receptors, and the Akt and ERK signaling pathways, and that these effects can be blocked by pharmacological inhibitors of the same pathways. Nicotine also promotes tumor growth in nude mice xenografted with human malignant glioma cells, thus suggesting that nicotine exposure may enhance the growth of malignant glioma tumors in vivo.
Spina et al. (2016) correlated glioma patient survival with CHRNA gene expression using the Rembrandt portal1, and found that the survival of patients with gliomas or GBMs over-expressing CHRNA1 or CHRNA9 is significantly shorter. They used glial fibrillary acidic protein (GFAP) as a reporter of astrocyte differentiation and isolated multiple clones from three independent GSC lines that over-express CHRNA1 or CHRNA9 and express GFAP. Their results showed that high GFAP expression is associated with reduced clonogenicity in vitro and tumorigenicity in vivo. Their screening of chemical libraries revealed that the neuromuscular AChR blocker atracurium besylate inhibits the clonogenic capacity of patient-derived GSCs and induces astroglial differentiation, whereas a nicotinic receptor agonist prevents atracurium besylate from reducing the self-renewal of GSCs. This study also showed a significant improvement in the survival of mice xenotransplanted with GSCs that were pre-treated with atracurium besylate. Taken together, these findings suggest that α1- and α9-containing nAChRs may be intimately involved in controlling GSC fate by reducing their stemness and/or their population.
Thompson and Sontheimer (2019) have very recently investigated the expression of nAChRs and muscarinic receptors (mAChRs) in GBM datasets and cell lines, and patient-derived xenograft lines. Using the RNA-Seq data relating to the tumor samples taken from 156 GBM patients, they analyzed AChR mRNA expression and found that the expression of the large majority of subunits was low, whereas that of the muscle-type α1 and ß1 subunits, and the neuronal α9 subunit was high. They used the calcium time-lapse technique to show that the activation of mAChRs or nAChRs increases intra-cellular Ca2+ in GBM cells. They also showed that there is a nicotine-mediated increase in intracellular Ca2+ that is due to α7 receptors (and probably α9-containing receptors, which are highly permeable to Ca2+ and show increased mRNA expression in patient-derived xenograft GBM lines). In neurons, the time-course of nicotine and ACh activation of the homomeric α7 receptor is very fast (in the order of milliseconds; Papke and Lindstrom, 2020; Borroni and Barrantes, 2021), whereas in GBM cells nicotine activates Ca2+ influx with a time-course of hundreds of seconds. This Ca2+ signaling response persists longer than channel activation and may be the result of CICR from intra-cellular stores, as has been previously shown in the case of hippocampal astrocytes (Sharma and Vijayaraghavan, 2001). It was also demonstrated that the increase in intracellular Ca2+ induced by ACh has minimal effects on cell proliferation or migration, whereas the stimulation of both mAChRs and nAChRs increases GBM cell invasion due to the increased activity of metalloproteinase-9.
Our group has also been interested in characterizing the nAChRs in glioblastoma cells. We began by examining publicly available data (accession number GSE16805) from a large-scale, multi-dimensional DNA microarray analysis carried out at IRCSS AOU San Martino-IST (Genoa, Italy), which showed that the most frequently expressed nAChR subunit mRNAs were CHRNA5 and CHRNA9 and, to a lesser extent, CHRNA7 and CHRNA10. We then analyzed the in vitro presence and possible role of nAChRs in U87MG human WHO grade IV astrocytoma U87MG cells and the GBM5 cell line derived from CSC-enriched cultures resistant to temozolomide (TMZ) treatment (Banelli et al., 2015), and found that the expression of nicotinic subunits was similar to that recorded in public databases of GBM patients: highly expressed α9 and α5 subunit mRNAs and less expressed α7 and α10 subunit mRNAs.
The results of our study extended the findings of Khalil et al. (2013) as we found that chronic treatment with nM concentrations of choline or nicotine increases the proliferation of U87MG and GBM5 cells, and activates the anti-apoptotic Akt and pro-proliferative ERK pathways. In general, these effects were blocked by silencing the expression of the α7 and α9 subunits by means of siRNAs or by the non-cell-permeable peptide antagonists selective for α7- and α9-containing nAChRs, thus indicating that they required the presence of both subunits. However, the effects of α7 and α9 were not identical as the action of choline on U87MG and GBM5 cell signaling was not significantly reduced after silencing the α7 subunit, whereas nicotine-induced signaling and choline- and nicotine-stimulated cell proliferation were significantly prevented in both cell lines after silencing either of the subunits. This last finding led us to deduce that choline may act mainly through α9-containing nAChRs, at least in activating GBM5 cell signaling. The α9-containing nAChRs are also involved in basal GBM cell proliferation as it decreased after 6 days of treatment with a subtype-specific antagonist (Pucci et al., 2021).
The agonist concentrations that increased cell proliferation were much lower than those necessary to activate α7 (nicotine and choline) or α9 nAChRs (choline) in electrophysiological experiments (Elgoyhen et al., 1994; Corradi and Bouzat, 2016). As nicotine is an antagonist of the ionotropic effects of α9 receptors, the occupation of nAChRs in U87MG and GBM5 cells by low concentration of nicotine leads to a conformational change that may alter interactions between nAChR scaffolds and other intracellular signaling molecules or cytoskeletal elements. This may elicit the non-ionic, metabotropic signaling events that regulate the phosphorylation status of target proteins and thus mediates some of the effects of choline and nicotine.
In our experiments, blocking α7 and/or α9 nAChRs by means of the use of selective antagonists or siRNAs substantially abolished choline- and nicotine-induced cell proliferation and decreased the effects of phosphorylation, which again indicates that the presence of receptors containing both subunits is necessary to ensure the effects of nicotinic agonists. This may be because: (1) it is necessary to have both α7- and α9-containing nAChRs in order to ensure sufficient target cell activation; or (2) GBM cells express heteropentameric α7α9-containing nAChRs which are mainly responsible for the effects of nicotine and choline. Given the difference in the levels of α7 and α9 mRNAs (Pucci et al., 2021), the hypothesis of an α7α9-nAChR subtype may seem to be unlikely at first glance, but it cannot be excluded because the turnover of the two mRNAs may be different, and mRNA levels are not always representative of the expressed protein.
As choline is a precursor of ACh, the increase in GBM cell proliferation may have been due to the direct action of either compounds, but the blocking effect of α7/α9 nicotinic antagonists clearly indicates mediation by nAChRs. Thompson and Sontheimer (2019) found that the expression of the mRNAs of the proteins necessary for ACh synthesis and release (many choline transporters, choline acetytransferase, and vescicular acetylcholine transporters) is significantly higher in GBM tissues than in their non-tumoral counterparts, thus suggesting that ACh is the signaling molecule acting on nAChRs.
The α7 and α9α10 nAChRs in GBM cells are pharmacologically similar to those expressed in A549 lung adenocarcinoma cells, whose nicotine-induced activation also increases their proliferation (Mucchietto et al., 2018). This proliferation is blocked in a concentration-dependent manner, by the selective MG624 nicotinic ligand (Gotti et al., 1998). Hybridization of MG624 and a non-nicotinic, resveratrol-derived pro-oxidant mitocan (Bavo et al., 2018) has led to the development of two novel compounds (StN-4 and StN-8) that are more potent than MG624 in reducing the viability of GBM cells, but less potent in reducing the viability of mouse astrocytes (Bavo et al., 2018). Knocking down the α9 subunit in U87MG cells decreases the potency of the effects of both compounds on cell viability, thus clearly indicating that α9-containing nAChRs are involved in sustaining GBM cell growth and survival (Pucci et al., 2022).
α9-Containing Receptors in Melanoma
Melanoma arises from genetic mutations in melanocytes, and is the most aggressive and deadly skin cancer mainly affecting Caucasians. It accounts for less than 5% of all skin cancers diagnosed annually in the United States, but is responsible for more than 75% of skin cancer deaths (Gershenwald and Guy, 2015).
Studies of 2,583 malignant melanoma patients indicate that smoking facilitates the spread of metastases in both men and women (Shaw and Milton, 1981), and a more recent study of a large sample of current smokers has found a close association between smoking and the occurrence of sentinel lymph node metastases (Jones et al., 2017).
Programmed death-ligand 1 (PD-L1) is a type 1 transmembrane protein encoded by the human CD274 gene that is overexpressed in some types of cancer (Salmaninejad et al., 2019), and PD-1 is an inhibitory receptor encoded by the PDCD1 gene that is located on the surface of all T cells (LaFleur et al., 2018). The PD-1 receptors on activated T cells regulates the immune system during various physiological responses, including the responses to cancer and autoimmune diseases (LaFleur et al., 2018): in particular, the PD-L1/PD-1 axis in a tumor microenvironment is hijacked by cancer cells seeking to avoid immune surveillance (Li et al., 2018).
α9-Mediated Signaling Pathways in Melanoma
Nguyen et al. (2019) used RT-PCR and Western blotting to analyze the expression of nAChRs and found that the expression of α9 nAChR mRNA was higher that of the other nAChR subunits in three melanoma cell lines (A375, A2058, and MDA-MB-435) and a primary melanocyte cell line (HEMn-LP), and that it was higher in melanoma cells than in melanocytes. They also found correlations between α9 mRNA expression and the clinical parameters of melanoma patients, and that high levels of α9 expression are significantly associated with lymph node metastases.
The in vitro over-expression of α9 in MDA-MB-435 and A2058 melanoma cells increased cell proliferation and migration, and this was paralleled by the activation of the Akt and ERK signaling pathways, and up-regulated PD-L1 expression. Furthermore, the suppression of α9 expression significantly inhibited melanoma cell growth and migration, decreased Akt and ERK activation, and reduced PD-L1 expression.
The over-expression of α9 nAChRs in A2058 and MDA-MB 435 cells also induced mesenchymal-like melanoma cells with the loss of cell-cell adhesion junctions, whereas α9 knockdown in A2058 cells led to an increase in the number of cell-cell adhesion junctions and epithelial-like melanoma cells.
A number of recent studies have shown positive correlations between smoking and PD-L1 expression: Wang et al. (2019) have shown that tobacco smoke induces PD-L1 expression in lung epithelial cells; and Nguyen et al. (2019) have found that nicotine induces α9 nAChR activity promoting melanoma cell proliferation in a dose- and time-dependent manner by stimulating the α9-mediated Akt, ERK, and STAT3 signaling pathways. In addition, nicotine-induced α9 nAChR activity up-regulated PD-L1 expression by activating the binding of transcription factor STAT3 to the PD-L1 promoter gene, thus suggesting that the functional activation of α9 nAChR by nicotine may regulate cancer-related immunity.
Conclusions and Perspectives
The data described in this review clearly indicate that α9-containing nAChRs play an important role in various cancers. They are expressed in cell types other than neurons and can trigger ion fluxes and/or metabotropic pathways at different time scales. These receptors are higly permeable to Ca2+ and Thompson and Sontheimer (2019) have indirectly shown that α9 activation increases Ca2+ in cancer cells. This suggests that α9 receptors modulate various fundamental processes in signal transduction, which makes them powerful regulators of responses that stimulate a number of cancer processes, including cell proliferation, the spread of metastases, and the inhibition of apoptosis. This has been clearly established by the work of Lin et al. (2019), who used multiple RNA-seq and microarray datasets to show that α9-interacting proteins and signaling pathways are common in various cancers.
Many studies have shown the involvement of α7 nAChRs in tumor-related processes by using the antagonists αBgtx and MLA, which are also active on α9 nAChRs. It is therefore possible that the latter are involved in many more tumor processes than initially hypothesized.
Various clinical studies have shown that the expression of α9 subunits correlates with the unfavorable prognosis of cancer patients, and so α9 nAChRs may act as a novel biomarker when assessing risk and evaluating treatment options.
Greater understanding of the physiological role of α9-containing nAChRs in cells and the molecular mechanisms underlying the way in which they regulate the development of cancer will advance our knowledge of the biology and treatment of different cancers, and recognising their importance in the various stages of tumor progression may allow the design of effective anti-cancer drugs and therefore provide more and better options for cancer patients.
Author Contributions
SP: conceptualization, conducting the research and investigation process, writing—review and editing. MZ: conceptualization, preparation, creation of the published work, writing—review and editing. FC: conceptualization, supervision, and funding acquisition. CG: conceptualization, supervision, statistical analysis, preparation, creation of the published work, writing—review and editing, and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
SP was the recipient of a fellowship from the Fondazione Vollaro (Milan). This work was supported by Fondazione Monzino (Milan) and Fondazione Vollaro (Milan).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
Supplementary Materials
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2021.805123/full#supplementary-material.
References
Afrashteh Nour, M., Hajiasgharzadeh, K., Kheradmand, F., Asadzadeh, Z., Bolandi, N., and Baradaran, B. (2021). Nicotinic acetylcholine receptors in chemotherapeutic drugs resistance: an emerging targeting candidate. Life Sci. 278:119557. doi: 10.1016/j.lfs.2021.119557
Albuquerque, E. X., Pereira, E. F., Alkondon, M., and Rogers, S. W. (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120. doi: 10.1152/physrev.00015.2008
Banelli, B., Carra, E., Barbieri, F., Wurth, R., Parodi, F., Pattarozzi, A., et al. (2015). The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle 14, 3418–3429. doi: 10.1080/15384101.2015.1090063
Baumann, L., Kauschke, V., Vikman, A., Durselen, L., Krasteva-Christ, G., Kampschulte, M., et al. (2019). Deletion of nicotinic acetylcholine receptor alpha9 in mice resulted in altered bone structure. Bone 120, 285–296. doi: 10.1016/j.bone.2018.11.003
Bavo, F., Pucci, S., Fasoli, F., Lammi, C., Moretti, M., Mucchietto, V., et al. (2018). Potent antiglioblastoma agents by hybridizing the onium-alkyloxy-stilbene based structures of an alpha7-nAChR, alpha9-nAChR antagonist and of a pro-oxidant mitocan. J. Med. Chem. 61, 10531–10544. doi: 10.1021/acs.jmedchem.8b01052
Boffi, J. C., Marcovich, I., Gill-Thind, J. K., Corradi, J., Collins, T., Lipovsek, M. M., et al. (2017). Differential contribution of subunit interfaces to alpha9alpha10 nicotinic acetylcholine receptor function. Mol. Pharmacol. 91, 250–262. doi: 10.1124/mol.116.107482
Borroni, V., and Barrantes, F. J. (2021). Homomeric and heteromeric alpha7 nicotinic acetylcholine receptors in health and some central nervous system diseases. Membranes (Basel) 11:664. doi: 10.3390/membranes11090664
Bychkov, M., Shenkarev, Z., Shulepko, M., Shlepova, O., Kirpichnikov, M., and Lyukmanova, E. (2019). Water-soluble variant of human Lynx1 induces cell cycle arrest and apoptosis in lung cancer cells via modulation of alpha7 nicotinic acetylcholine receptors. PLoS One 14:e0217339. doi: 10.1371/journal.pone.0217339
Chen, J., Cheuk, I. W. Y., Shin, V. Y., and Kwong, A. (2019). Acetylcholine receptors: Key players in cancer development. Surg. Oncol. 31, 46–53. doi: 10.1016/j.suronc.2019.09.003
Chen, C. S., Lee, C. H., Hsieh, C. D., Ho, C. T., Pan, M. H., Huang, C. S., et al. (2011). Nicotine-induced human breast cancer cell proliferation attenuated by garcinol through down-regulation of the nicotinic receptor and cyclin D3 proteins. Breast Cancer Res. Treat. 125, 73–87. doi: 10.1007/s10549-010-0821-3
Chernyavsky, A. I., Arredondo, J., Vetter, D. E., and Grando, S. A. (2007). Central role of alpha9 acetylcholine receptor in coordinating keratinocyte adhesion and motility at the initiation of epithelialization. Exp. Cell Res. 313, 3542–3555. doi: 10.1016/j.yexcr.2007.07.011
Chikova, A., Bernard, H. U., Shchepotin, I. B., and Grando, S. A. (2012). New associations of the genetic polymorphisms in nicotinic receptor genes with the risk of lung cancer. Life Sci. 91, 1103–1108. doi: 10.1016/j.lfs.2011.12.023
Colomer, C., Olivos-Ore, L. A., Vincent, A., Mcintosh, J. M., Artalejo, A. R., and Guerineau, N. C. (2010). Functional characterization of alpha9-containing cholinergic nicotinic receptors in the rat adrenal medulla: implication in stress-induced functional plasticity. J. Neurosci. 30, 6732–6742. doi: 10.1523/JNEUROSCI.4997-09.2010
Corradi, J., and Bouzat, C. (2016). Understanding the bases of function and modulation of alpha7 nicotinic receptors: implications for drug discovery. Mol. Pharmacol. 90, 288–299. doi: 10.1124/mol.116.104240
Corringer, P. J., Poitevin, F., Prevost, M. S., Sauguet, L., Delarue, M., and Changeux, J. P. (2012). Structure and pharmacology of pentameric receptor channels: from bacteria to brain. Structure 20, 941–956. doi: 10.1016/j.str.2012.05.003
Couturier, S., Bertrand, D., Matter, J. M., Hernandez, M. C., Bertrand, S., Millar, N., et al. (1990). A neuronal nicotinic acetylcholine receptor subunit (alpha 7) is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron 5, 847–856. doi: 10.1016/0896-6273(90)90344-f
Crespi, A., Colombo, S. F., and Gotti, C. (2018). Proteins and chemical chaperones involved in neuronal nicotinic receptor expression and function: an update. Br. J. Pharmacol. 175, 1869–1879. doi: 10.1111/bph.13777
Davies, A. R., Hardick, D. J., Blagbrough, I. S., Potter, B. V., Wolstenholme, A. J., and Wonnacott, S. (1999). Characterisation of the binding of [3H]methyllycaconitine: a new radioligand for labelling alpha 7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology 38, 679–690. doi: 10.1016/s0028-3908(98)00221-4
Dobes, M., Khurana, V. G., Shadbolt, B., Jain, S., Smith, S. F., Smee, R., et al. (2011). Increasing incidence of glioblastoma multiforme and meningioma and decreasing incidence of schwannoma (2000–2008): findings of a multicenter australian study. Surg. Neurol. Int. 2:176. doi: 10.4103/2152-7806.90696
Elgoyhen, A. B., Johnson, D. S., Boulter, J., Vetter, D. E., and Heinemann, S. (1994). Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79, 705–715. doi: 10.1016/0092-8674(94)90555-x
Elgoyhen, A. B., and Katz, E. (2012). The efferent medial olivocochlear-hair cell synapse. J. Physiol. Paris 106, 47–56. doi: 10.1016/j.jphysparis.2011.06.001
Elgoyhen, A. B., Vetter, D. E., Katz, E., Rothlin, C. V., Heinemann, S. F., and Boulter, J. (2001). alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc. Natl. Acad. Sci. U S A 98, 3501–3506. doi: 10.1073/pnas.051622798
Ellison, M., Haberlandt, C., Gomez-Casati, M. E., Watkins, M., Elgoyhen, A. B., Mcintosh, J. M., et al. (2006). Alpha-RgIA: a novel conotoxin that specifically and potently blocks the alpha9alpha10 nAChR. Biochemistry 45, 1511–1517. doi: 10.1021/bi0520129
Fararjeh, A. S., Tu, S. H., Chen, L. C., Cheng, T. C., Liu, Y. R., Chang, H. L., et al. (2019). Long-term exposure to extremely low-dose of nicotine and 4-(methylnitrosamino)-1–(3-pyridyl)-1-butanone (NNK) induce non-malignant breast epithelial cell transformation through activation of the a9-nicotinic acetylcholine receptor-mediated signaling pathway. Environ. Toxicol. 34, 73–82. doi: 10.1002/tox.22659
Ferlay, J., Colombet, M., Soerjomataram, I., Parkin, D. M., Pineros, M., Znaor, A., et al. (2021). Cancer statistics for the year 2020: an overview. Int. J. Cancer [Online ahead of print]. doi: 10.1002/ijc.33588
Friedman, J. R., Richbart, S. D., Merritt, J. C., Brown, K. C., Nolan, N. A., Akers, A. T., et al. (2019). Acetylcholine signaling system in progression of lung cancers. Pharmacol. Ther. 194, 222–254. doi: 10.1016/j.pharmthera.2018.10.002
Fu, X. W., Song, P. F., and Spindel, E. R. (2015). Role of Lynx1 and related Ly6 proteins as modulators of cholinergic signaling in normal and neoplastic bronchial epithelium. Int. Immunopharmacol. 29, 93–98. doi: 10.1016/j.intimp.2015.05.022
Fucile, S., Renzi, M., Lax, P., and Eusebi, F. (2003). Fractional Ca(2+) current through human neuronal alpha7 nicotinic acetylcholine receptors. Cell Calcium 34, 205–209. doi: 10.1016/s0143-4160(03)00071-x
Fucile, S., Sucapane, A., and Eusebi, F. (2006). Ca2+ permeability through rat cloned alpha9-containing nicotinic acetylcholine receptors. Cell Calcium 39, 349–355. doi: 10.1016/j.ceca.2005.12.002
Fujii, T., Mashimo, M., Moriwaki, Y., Misawa, H., Ono, S., Horiguchi, K., et al. (2017). Expression and function of the cholinergic system in immune cells. Front. Immunol. 8:1085. doi: 10.3389/fimmu.2017.01085
Gershenwald, J. E., and Guy, G. P., Jr. (2015). Stemming the rising incidence of melanoma: calling prevention to action. J. Natl. Cancer Inst. 108:djv381. doi: 10.1093/jnci/djv381
Gotti, C., Balestra, B., Moretti, M., Rovati, G. E., Maggi, L., Rossoni, G., et al. (1998). 4-Oxystilbene compounds are selective ligands for neuronal nicotinic alphaBungarotoxin receptors. Br. J. Pharmacol. 124, 1197–1206. doi: 10.1038/sj.bjp.0701957
Gotti, C., Zoli, M., and Clementi, F. (2006). Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol. Sci. 27, 482–491. doi: 10.1016/j.tips.2006.07.004
Grando, S. A. (2014). Connections of nicotine to cancer. Nat. Rev. Cancer 14, 419–429. doi: 10.1038/nrc3725
Gu, S., Knowland, D., Matta, J. A., O’carroll, M. L., Davini, W. B., Dhara, M., et al. (2020). Hair cell alpha9alpha10 nicotinic acetylcholine receptor functional expression regulated by ligand binding and deafness gene products. Proc. Natl. Acad. Sci. U S A 117, 24534–24544. doi: 10.1073/pnas.2013762117
Guha, P., Bandyopadhyaya, G., Polumuri, S. K., Chumsri, S., Gade, P., Kalvakolanu, D. V., et al. (2014). Nicotine promotes apoptosis resistance of breast cancer cells and enrichment of side population cells with cancer stem cell-like properties via a signaling cascade involving galectin-3, alpha9 nicotinic acetylcholine receptor and STAT3. Breast Cancer Res. Treat. 145, 5–22. doi: 10.1007/s10549-014-2912-z
Hecker, A., Kullmar, M., Wilker, S., Richter, K., Zakrzewicz, A., Atanasova, S., et al. (2015). Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit ATP-induced IL-1beta release. J. Immunol. 195, 2325–2334. doi: 10.4049/jimmunol.1400974
Hone, A. J., Servent, D., and Mcintosh, J. M. (2018). alpha9-containing nicotinic acetylcholine receptors and the modulation of pain. Br. J. Pharmacol. 175, 1915–1927. doi: 10.1111/bph.13931
Hou, L., Jiang, J., Liu, B., Han, W., Wu, Y., Zou, X., et al. (2016). Smoking and adult glioma: a population-based case-control study in China. Neuro Oncol. 18, 105–113. doi: 10.1093/neuonc/nov146
Hsieh, Y. C., Lee, C. H., Tu, S. H., Wu, C. H., Hung, C. S., Hsieh, M. C., et al. (2014). CHRNA9 polymorphisms and smoking exposure synergize to increase the risk of breast cancer in Taiwan. Carcinogenesis 35, 2520–2525. doi: 10.1093/carcin/bgu179
Huang, L. C., Lin, C. L., Qiu, J. Z., Lin, C. Y., Hsu, K. W., Tam, K. W., et al. (2017). Nicotinic acetylcholine receptor subtype Alpha-9 mediates triple-negative breast cancers based on a spontaneous pulmonary metastasis mouse model. Front. Cell Neurosci. 11:336. doi: 10.3389/fncel.2017.00336
Hurst, R., Rollema, H., and Bertrand, D. (2013). Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol. Ther. 137, 22–54. doi: 10.1016/j.pharmthera.2012.08.012
Improgo, M. R., Schlichting, N. A., Cortes, R. Y., Zhao-Shea, R., Tapper, A. R., and Gardner, P. D. (2010). ASCL1 regulates the expression of the CHRNA5/A3/B4 lung cancer susceptibility locus. Mol. Cancer Res. 8, 194–203. doi: 10.1158/1541-7786.MCR-09-0185
Improgo, M. R., Soll, L. G., Tapper, A. R., and Gardner, P. D. (2013). Nicotinic acetylcholine receptors mediate lung cancer growth. Front. Physiol. 4:251. doi: 10.3389/fphys.2013.00251
Indurthi, D. C., Pera, E., Kim, H. L., Chu, C., Mcleod, M. D., Mcintosh, J. M., et al. (2014). Presence of multiple binding sites on alpha9alpha10 nAChR receptors alludes to stoichiometric-dependent action of the alpha-conotoxin, Vc1.1. Biochem. Pharmacol. 89, 131–140. doi: 10.1016/j.bcp.2014.02.002
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., and Forman, D. (2011). Global cancer statistics. CA Cancer J. Clin. 61, 69–90. doi: 10.3322/caac.20107
Jones, M. S., Jones, P. C., Stern, S. L., Elashoff, D., Hoon, D. S. B., Thompson, J., et al. (2017). The impact of smoking on sentinel node metastasis of primary cutaneous melanoma. Ann. Surg. Oncol. 24, 2089–2094. doi: 10.1245/s10434-017-5775-9
Kabbani, N., and Nichols, R. A. (2018). Beyond the channel: metabotropic signaling by nicotinic receptors. Trends Pharmacol. Sci. 39, 354–366. doi: 10.1016/j.tips.2018.01.002
Khalil, A. A., Jameson, M. J., Broaddus, W. C., Lin, P. S., and Chung, T. D. (2013). Nicotine enhances proliferation, migration and radioresistance of human malignant glioma cells through EGFR activation. Brain Tumor Pathol. 30, 73–83. doi: 10.1007/s10014-012-0101-5
King, J. R., and Kabbani, N. (2016). Alpha 7 nicotinic receptor coupling to heterotrimeric G proteins modulates RhoA activation, cytoskeletal motility and structural growth. J. Neurochem. 138, 532–545. doi: 10.1111/jnc.13660
King, J. R., Nordman, J. C., Bridges, S. P., Lin, M. K., and Kabbani, N. (2015). Identification and characterization of a G protein-binding cluster in alpha7 nicotinic acetylcholine receptors. J. Biol. Chem. 290, 20060–20070. doi: 10.1074/jbc.M115.647040
Kryukova, E. V., Egorova, N. S., Kudryavtsev, D. S., Lebedev, D. S., Spirova, E. N., Zhmak, M. N., et al. (2019). From synthetic fragments of endogenous three-finger proteins to potential drugs. Front. Pharmacol. 10:748. doi: 10.3389/fphar.2019.00748
LaFleur, M. W., Muroyama, Y., Drake, C. G., and Sharpe, A. H. (2018). Inhibitors of the PD-1 pathway in tumor therapy. J. Immunol. 200, 375–383. doi: 10.4049/jimmunol.1701044
Lau, J. K., Brown, K. C., Thornhill, B. A., Crabtree, C. M., Dom, A. M., Witte, T. R., et al. (2013). Inhibition of cholinergic signaling causes apoptosis in human bronchioalveolar carcinoma. Cancer Res. 73, 1328–1339. doi: 10.1158/0008-5472.CAN-12-3190
Lee, C. H., Chang, Y. C., Chen, C. S., Tu, S. H., Wang, Y. J., Chen, L. C., et al. (2011). Crosstalk between nicotine and estrogen-induced estrogen receptor activation induces alpha9-nicotinic acetylcholine receptor expression in human breast cancer cells. Breast Cancer Res. Treat. 129, 331–345. doi: 10.1007/s10549-010-1209-0
Lee, C. H., Huang, C. S., Chen, C. S., Tu, S. H., Wang, Y. J., Chang, Y. J., et al. (2010). Overexpression and activation of the alpha9-nicotinic receptor during tumorigenesis in human breast epithelial cells. J. Natl. Cancer Inst. 102, 1322–1335. doi: 10.1093/jnci/djq300
Lee, K. L., Kuo, Y. C., Ho, Y. S., and Huang, Y. H. (2019). Triple-negative breast cancer: current understanding and future therapeutic breakthrough targeting cancer stemness. Cancers (Basel) 11:1334. doi: 10.3390/cancers11091334
Li, H. X., Peng, X. X., Zong, Q., Zhang, K., Wang, M. X., Liu, Y. Z., et al. (2016). Cigarette smoking and risk of adult glioma: a meta-analysis of 24 observational studies involving more than 2.3 million individuals. Onco Targets Ther. 9, 3511–3523. doi: 10.2147/OTT.S99713
Li, X., Shao, C., Shi, Y., and Han, W. (2018). Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 11:31. doi: 10.1186/s13045-018-0578-4
Li, X., Tae, H. S., Chu, Y., Jiang, T., Adams, D. J., and Yu, R. (2021). Medicinal chemistry, pharmacology and therapeutic potential of alpha-conotoxins antagonizing the alpha9alpha10 nicotinic acetylcholine receptor. Pharmacol. Ther. 222:107792. doi: 10.1016/j.pharmthera.2020.107792
Liang, J., Tae, H.-S., Xu, X., Jiang, T., Adams, D. J., and Yu, R. (2020). Dimerization of alpha-conotoxins as a strategy to enhance the inhibition of the human α7 and α9α10 nicotinic acetylcholine receptors. J. Med. Chem. 63, 2974–2985. doi: 10.1021/acs.jmedchem.9b01536
Lin, C. Y., Lee, C. H., Chuang, Y. H., Lee, J. Y., Chiu, Y. Y., Wu Lee, Y. H., et al. (2019). Membrane protein-regulated networks across human cancers. Nat. Commun. 10:3131. doi: 10.1038/s41467-019-10920-8
Liu, Q., Li, M., Whiteaker, P., Shi, F. D., Morley, B. J., and Lukas, R. J. (2019). Attenuation in nicotinic acetylcholine receptor alpha9 and alpha10 subunit double knock-out mice of experimental autoimmune encephalomyelitis. Biomolecules 9:827. doi: 10.3390/biom9120827
Liu, Q., Whiteaker, P., Morley, B. J., Shi, F. D., and Lukas, R. J. (2017). Distinctive roles for alpha7*- and alpha9*-nicotinic acetylcholine receptors in inflammatory and autoimmune responses in the murine experimental autoimmune encephalomyelitis model of multiple sclerosis. Front. Cell Neurosci. 11:287. doi: 10.3389/fncel.2017.00287
Louis, D. N., Holland, E. C., and Cairncross, J. G. (2001). Glioma classification: a molecular reappraisal. Am. J. Pathol. 159, 779–786. doi: 10.1016/S0002-9440(10)61750-6
Luo, S., Zhangsun, D., Harvey, P. J., Kaas, Q., Wu, Y., Zhu, X., et al. (2015). Cloning, synthesis and characterization of alphaO-conotoxin GeXIVA, a potent alpha9alpha10 nicotinic acetylcholine receptor antagonist. Proc. Natl. Acad. Sci. U S A 112, E4026–E4035. doi: 10.1073/pnas.1503617112
Lustig, L. R., Peng, H., Hiel, H., Yamamoto, T., and And Fuchs, P. A. (2001). Molecular cloning and mapping of the human nicotinic acetylcholine receptor alpha10 (CHRNA10). Genomics 73, 272–283. doi: 10.1006/geno.2000.6503
Marcovich, I., Moglie, M. J., Carpaneto Freixas, A. E., Trigila, A. P., Franchini, L. F., Plazas, P. V., et al. (2020). Distinct evolutionary trajectories of neuronal and hair cell nicotinic acetylcholine receptors. Mol. Biol. Evol. 37, 1070–1089. doi: 10.1093/molbev/msz290
Matta, J. A., Gu, S., Davini, W. B., and Bredt, D. S. (2021). Nicotinic acetylcholine receptor redux: discovery of accessories opens therapeutic vistas. Science 373:eabg6539. doi: 10.1126/science.abg6539
Mcintosh, J. M., Plazas, P. V., Watkins, M., Gomez-Casati, M. E., Olivera, B. M., and Elgoyhen, A. B. (2005). A novel alpha-conotoxin, PeIA, cloned from conus pergrandis, discriminates between rat alpha9alpha10 and alpha7 nicotinic cholinergic receptors. J. Biol. Chem. 280, 30107–30112. doi: 10.1074/jbc.M504102200
Mcneill, J., Rudyk, C., Hildebrand, M. E., and Salmaso, N. (2021). Ion channels and electrophysiological properties of astrocytes: implications for emergent stimulation technologies. Front. Cell Neurosci. 15:644126. doi: 10.3389/fncel.2021.644126
Meza, R., Meernik, C., Jeon, J., and Cote, M. L. (2015). Lung cancer incidence trends by gender, race and histology in the united states, 1973–2010. PLoS One 10:e0121323. doi: 10.1371/journal.pone.0121323
Miwa, J. M. (2021). Lynx1 prototoxins: critical accessory proteins of neuronal nicotinic acetylcholine receptors. Curr. Opin. Pharmacol. 56, 46–51. doi: 10.1016/j.coph.2020.09.016
Morley, B. J., Dolan, D. F., Ohlemiller, K. K., and Simmons, D. D. (2017). Generation and characterization of alpha9 and alpha10 nicotinic acetylcholine receptor subunit knockout mice on a C57BL/6J background. Front. Neurosci. 11:516. doi: 10.3389/fnins.2017.00516
Morley, B. J., Whiteaker, P., and Elgoyhen, A. B. (2018). Commentary: nicotinic acetylcholine receptor alpha9 and alpha10 subunits are expressed in the brain of mice. Front. Cell Neurosci. 12:104. doi: 10.3389/fncel.2018.00104
Mucchietto, V., Crespi, A., Fasoli, F., Clementi, F., and Gotti, C. (2016). Neuronal acetylcholine nicotinic receptors as new targets for lung cancer treatment. Curr. Pharm. Des. 22, 2160–2169. doi: 10.2174/1381612822666160203144114
Mucchietto, V., Fasoli, F., Pucci, S., Moretti, M., Benfante, R., Maroli, A., et al. (2018). alpha9- and alpha7-containing receptors mediate the pro-proliferative effects of nicotine in the A549 adenocarcinoma cell line. Br. J. Pharmacol. 175, 1957–1972. doi: 10.1111/bph.13954
Nguyen, H. D., Liao, Y. C., Ho, Y. S., Chen, L. C., Chang, H. W., Cheng, T. C., et al. (2019). The alpha9 nicotinic acetylcholine receptor mediates nicotine-induced PD-L1 expression and regulates melanoma cell proliferation and migration. Cancers (Basel) 11:1991. doi: 10.3390/cancers11121991
Nguyen, V. T., Ndoye, A., and Grando, S. A. (2000). Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am. J. Pathol. 157, 1377–1391. doi: 10.1016/s0002-9440(10)64651-2
Nordman, J. C., and Kabbani, N. (2014). Microtubule dynamics at the growth cone are mediated by alpha7 nicotinic receptor activation of a Galphaq and IP3 receptor pathway. FASEB J. 28, 2995–3006. doi: 10.1096/fj.14-251439
Ostrom, Q. T., Gittleman, H., Fulop, J., Liu, M., Blanda, R., Kromer, C., et al. (2015). CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the united states in 2008–2012. Neuro Oncol. 17, iv1–iv62. doi: 10.1093/neuonc/nov189
Papke, R. L., and Lindstrom, J. M. (2020). Nicotinic acetylcholine receptors: conventional and unconventional ligands and signaling. Neuropharmacology 168:108021. doi: 10.1016/j.neuropharm.2020.108021
Pesch, B., Kendzia, B., Gustavsson, P., Jockel, K. H., Johnen, G., Pohlabeln, H., et al. (2012). Cigarette smoking and lung cancer–relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int. J. Cancer 131, 1210–1219. doi: 10.1002/ijc.27339
Plazas, P. V., Katz, E., Gomez-Casati, M. E., Bouzat, C., and Elgoyhen, A. B. (2005). Stoichiometry of the alpha9alpha10 nicotinic cholinergic receptor. J. Neurosci. 25, 10905–10912. doi: 10.1523/JNEUROSCI.3805-05.2005
Pucci, S., Bolchi, C., Bavo, F., Pallavicini, M., De Palma, C., Renzi, M., et al. (2022). Evidence of a dual mechanism of action underlying the anti-proliferative and cytotoxic effects of ammonium-alkyloxy-stilbene-based α7- and α9- nicotinic ligands on glioblastoma cells. Pharmacol. Res. 105959. doi: 10.1016/j.phrs.2021.105959
Pucci, S., Fasoli, F., Moretti, M., Benfante, R., Di Lascio, S., Viani, P., et al. (2021). Choline and nicotine increase glioblastoma cell proliferation by binding and activating alpha7- and alpha9- containing nicotinic receptors. Pharmacol. Res. 163:105336. doi: 10.1016/j.phrs.2020.105336
Richter, K., Mathes, V., Fronius, M., Althaus, M., Hecker, A., Krasteva-Christ, G., et al. (2016). Phosphocholine - an agonist of metabotropic but not of ionotropic functions of alpha9-containing nicotinic acetylcholine receptors. Sci. Rep. 6:28660. doi: 10.1038/srep28660
Romero, H. K., Christensen, S. B., Di Cesare Mannelli, L., Gajewiak, J., Ramachandra, R., Elmslie, K. S., et al. (2017). Inhibition of alpha9alpha10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. U S A 114, E1825–E1832. doi: 10.1073/pnas.1621433114
Salmaninejad, A., Valilou, S. F., Shabgah, A. G., Aslani, S., Alimardani, M., Pasdar, A., et al. (2019). PD-1/PD-L1 pathway: basic biology and role in cancer immunotherapy. J. Cell Physiol. 234, 16824–16837. doi: 10.1002/jcp.28358
Samet, J. M., Avila-Tang, E., Boffetta, P., Hannan, L. M., Olivo-Marston, S., Thun, M. J., et al. (2009). Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin. Cancer Res. 15, 5626–5645. doi: 10.1158/1078-0432.CCR-09-0376
Schaal, C., and Chellappan, S. P. (2014). Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 12, 14–23. doi: 10.1158/1541-7786.MCR-13-0541
Schane, R. E., Ling, P. M., and Glantz, S. A. (2010). Health effects of light and intermittent smoking: a review. Circulation 121, 1518–1522. doi: 10.1161/CIRCULATIONAHA.109.904235
Schuller, H. M. (2009). Is cancer triggered by altered signalling of nicotinic acetylcholine receptors. Nat. Rev. Cancer 9, 195–205. doi: 10.1038/nrc2590
Sgard, F., Charpantier, E., Bertrand, S., Walker, N., Caput, D., Graham, D., et al. (2002). A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9-subunit. Mol. Pharmacol. 61, 150–159. doi: 10.1124/mol.61.1.150
Shao, C., Zhao, W., Qi, Z., and He, J. (2016). Smoking and glioma risk: evidence from a meta-analysis of 25 observational studies. Medicine (Baltimore) 95:e2447. doi: 10.1097/MD.0000000000002447
Sharma, G., and Vijayaraghavan, S. (2001). Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. U S A 98, 4148–4153. doi: 10.1073/pnas.071540198
Shaw, H. M., and Milton, G. W. (1981). Smoking and the development of metastases from malignant melanoma. Int. J. Cancer 28, 153–156. doi: 10.1002/ijc.2910280207
Slattery, M. L., Curtin, K., Giuliano, A. R., Sweeney, C., Baumgartner, R., Edwards, S., et al. (2008). Active and passive smoking, IL6, ESR1 and breast cancer risk. Breast Cancer Res. Treat. 109, 101–111. doi: 10.1007/s10549-007-9629-1
Spina, R., Voss, D. M., Asnaghi, L., Sloan, A., and Bar, E. E. (2016). Atracurium Besylate and other neuromuscular blocking agents promote astroglial differentiation and deplete glioblastoma stem cells. Oncotarget 7, 459–472. doi: 10.18632/oncotarget.6314
Spindel, E. R. (2016). Cholinergic targets in lung cancer. Curr. Pharm. Des. 22, 2152–2159. doi: 10.2174/1381612822666160127114237
Sun, Z., Bao, J., Zhangsun, M., Dong, S., Zhangsun, D., and Luo, S. (2020). alphaO-conotoxin GeXIVA inhibits the growth of breast cancer cells via interaction with alpha9 nicotine acetylcholine receptors. Mar. Drugs 18:195. doi: 10.3390/md18040195
Tang, M. S., Wu, X. R., Lee, H. W., Xia, Y., Deng, F. M., Moreira, A. L., et al. (2019). Electronic-cigarette smoke induces lung adenocarcinoma and bladder urothelial hyperplasia in mice. Proc. Natl. Acad. Sci. U S A 116, 21727–21731. doi: 10.1073/pnas.1911321116
Thompson, E. G., and Sontheimer, H. (2019). Acetylcholine receptor activation as a modulator of glioblastoma invasion. Cells 8:1203. doi: 10.3390/cells8101203
Treinin, M., Papke, R. L., Nizri, E., Ben-David, Y., Mizrachi, T., and Brenner, T. (2017). Role of the alpha7 nicotinic acetylcholine receptor and RIC-3 in the cholinergic anti-inflammatory pathway. Cent. Nerv. Syst. Agents Med. Chem. 17, 90–99. doi: 10.2174/1871524916666160829114533
Tu, S. H., Lin, Y. C., Huang, C. C., Yang, P. S., Chang, H. W., Chang, C. H., et al. (2016). Protein phosphatase Mg2+/Mn2+ dependent 1F promotes smoking-induced breast cancer by inactivating phosphorylated-p53-induced signals. Oncotarget 7, 77516–77531. doi: 10.18632/oncotarget.12717
Verbitsky, M., Rothlin, C. V., Katz, E., and Elgoyhen, A. B. (2000). Mixed nicotinic-muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology 39, 2515–2524. doi: 10.1016/s0028-3908(00)00124-6
Vetter, D. E., Katz, E., Maison, S. F., Taranda, J., Turcan, S., Ballestero, J., et al. (2007). The alpha10 nicotinic acetylcholine receptor subunit is required for normal synaptic function and integrity of the olivocochlear system. Proc. Natl. Acad. Sci. U S A 104, 20594–20599. doi: 10.1073/pnas.0708545105
Vetter, D. E., Liberman, M. C., Mann, J., Barhanin, J., Boulter, J., Brown, M. C., et al. (1999). Role of alpha9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron 23, 93–103. doi: 10.1016/s0896-6273(00)80756-4
Wang, Y., Sanghvi, M., Gribizis, A., Zhang, Y., Song, L., Morley, B., et al. (2021). Efferent feedback controls bilateral auditory spontaneous activity. Nat. Commun. 12:2449. doi: 10.1038/s41467-021-22796-8
Wang, G. Z., Zhang, L., Zhao, X. C., Gao, S. H., Qu, L. W., Yu, H., et al. (2019). The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 10:1125. doi: 10.1038/s41467-019-08887-7
Wessler, I., and Kirkpatrick, C. J. (2008). Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br. J. Pharmacol. 154, 1558–1571. doi: 10.1038/bjp.2008.185
Yu, R., Tae, H. S., Tabassum, N., Shi, J., Jiang, T., and Adams, D. J. (2018). Molecular determinants conferring the stoichiometric-dependent activity of alpha-conotoxins at the human alpha9alpha10 nicotinic acetylcholine receptor subtype. J. Med. Chem. 61, 4628–4634. doi: 10.1021/acs.jmedchem.8b00115
Zhang, Q., Ganapathy, S., Avraham, H., Nishioka, T., and Chen, C. (2020). Nicotine exposure potentiates lung tumorigenesis by perturbing cellular surveillance. Br. J. Cancer 122, 904–911. doi: 10.1038/s41416-020-0730-0
Zheng, N., Christensen, S. B., Dowell, C., Purushottam, L., Skalicky, J. J., Mcintosh, J. M., et al. (2021). Discovery of methylene thioacetal-incorporated alpha-RgIA analogues as potent and stable antagonists of the human alpha9alpha10 nicotinic acetylcholine receptor for the treatment of neuropathic pain. J. Med. Chem. 64, 9513–9524. doi: 10.1021/acs.jmedchem.1c00802
Zhu, P., Du, X. L., Lu, G., and Zhu, J. J. (2017). Survival benefit of glioblastoma patients after FDA approval of temozolomide concomitant with radiation and bevacizumab: a population-based study. Oncotarget 8, 44015–44031. doi: 10.18632/oncotarget.17054
Zoli, M., Moretti, M., Zanardi, A., Mcintosh, J. M., Clementi, F., and Gotti, C. (2002). Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J. Neurosci. 22, 8785–8789. doi: 10.1523/JNEUROSCI.22-20-08785.2002
Zoli, M., Pistillo, F., and Gotti, C. (2015). Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96, 302–311. doi: 10.1016/j.neuropharm.2014.11.003
Zoli, M., Pucci, S., Vilella, A., and Gotti, C. (2018). Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr. Neuropharmacol. 16, 338–349. doi: 10.2174/1570159X15666170912110450
Zouridakis, M., Giastas, P., Zarkadas, E., Chroni-Tzartou, D., Bregestovski, P., and Tzartos, S. J. (2014). Crystal structures of free and antagonist-bound states of human alpha9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 21, 976–980. doi: 10.1038/nsmb.2900
Zouridakis, M., Papakyriakou, A., Ivanov, I. A., Kasheverov, I. E., Tsetlin, V., Tzartos, S., et al. (2019). Crystal structure of the monomeric extracellular domain of alpha9 nicotinic receptor subunit in complex with alpha-conotoxin RgIA: molecular dynamics insights into RgIA binding to alpha9alpha10 nicotinic receptors. Front. Pharmacol. 10:474. doi: 10.3389/fphar.2019.00474
Keywords: nicotinic acethylcholine receptor (nAChR), cancer, lung, breast, glioblastoma, melanoma
Citation: Pucci S, Zoli M, Clementi F and Gotti C (2022) α9-Containing Nicotinic Receptors in Cancer. Front. Cell. Neurosci. 15:805123. doi: 10.3389/fncel.2021.805123
Received: 29 October 2021; Accepted: 29 December 2021;
Published: 21 January 2022.
Edited by:
Barbara Jane Morley, Boys Town National Research Hospital, United StatesReviewed by:
Yuan Soon Ho, Taipei Medical University, TaiwanUrsula H. Winzer-Serhan, Texas A&M Health Science Center, United States
Copyright © 2022 Pucci, Zoli, Clementi and Gotti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Gotti, Y2VjaWxhLmdvdHRpQGluLmNuci5pdA==