 Yuan Zhang
Yuan Zhang Yanfang Zhao2
Yanfang Zhao2- 1Institute for Translational Medicine, Qingdao University, Qingdao, China
- 2School for Life Science, Institute of Biomedical Research, Shandong University of Technology, Zibo, China
The pathological features of Alzheimer’s disease (AD) include senile plaques induced by amyloid-β (Aβ) protein deposits, neurofibrillary tangles formed by aggregates of hyperphosphorylated tau proteins and neuronal cell loss in specific position within the brain. Recent observations have suggested the possibility of an association between AD and cellular prion protein (PrPC) levels. PrPC is a high affinity receptor for oligomeric Aβ and is important for Aβ-induced neurotoxicity and thus plays a critical role in AD pathogenesis. The determination of the relationship between PrPC and AD and the characterization of PrPC binding to Aβ will facilitate the development of novel therapies for AD.
Background
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder representing the most common cause of dementia in the elderly. The hallmarks of AD include senile plaques induced by amyloid-β (Aβ) protein deposits, neurofibrillary tangles formed by aggregates of hyperphosphorylated tau proteins and neuronal cell loss in specific position within the brain (Di Lazzaro, 2018). Soluble Aβ and prefibrillar oligomers have been recognized as early and key factors in AD-related synaptic dysfunction. It had initially been suspected that Aβ plaques are directly toxic to neurons, however, studies have revealed that the level of Aβ plaque deposits is not closely associated with the severity of AD (Selkoe, 2011). However, recent studies have shown a strong correlation between the oligomeric forms of Aβ and neurotoxicity and the severity of cognitive impairment in AD (Benilova et al., 2012; Kayed and Lasagna-Reeves, 2013).
The physiological prion protein (PrPC) is an evolutionarily highly conserved protein that is present in all investigated mammals. PrPC is attached to the outer surface of the cell membrane by a glycosylphosphatidylinositol (GPI)-anchor. PrPC is a glycoprotein that expressed in the brain and was first reported to be associated with prion diseases (Singh and Udgaonkar, 2015). Recent observations have indicated the possibility of a connection between prion diseases and AD (Onodera, 2017). PrPC mediates at a certain extent of the toxic effects of Aβ oligomers and thus plays an important role in AD pathogenesis (Salazar and Strittmatter, 2017; Brody and Strittmatter, 2018; Purro et al., 2018). PrPC is a high affinity receptor for oligomeric Aβ, and the expression of PrPC is important for Aβ-induced neurotoxicity, as demonstrated by the loss of long-term potentiation (LTP) and memory impairment in AD mouse models (Gimbel et al., 2010; Kostylev et al., 2015). PrPC deficiency confers resistance to the synaptic toxicity of oligomeric Aβ in mice and in vitro in hippocampal slice cultures (Barry et al., 2011). These findings support the hypothesis that the interaction between PrPC and Aβ is necessary for neurotoxicity and neuronal cell loss in AD.
Changes in PrPC Involved in AD Pathology
Characteristics of Distinct PrPC Isoforms in AD Brains
PrPC is an N-glycosylated GPI-anchored protein usually present in lipid rafts, which is variably glycosylated at two highly conserved sites, namely, the asparagine resides at positions 181 and 197 of the human PrPC (Figure 1). Distinct phenotypes and binding domains are characterized by specific mechanisms during AD progression. N-glycan attachment to these sites in PrPC results in diglycosylated, monoglycosylated and unglycosylated isoforms (Wiseman et al., 2015). The ratios of diglycosylated, monoglycosylated and unglycosylated PrPC have been shown to be variable in AD brains.
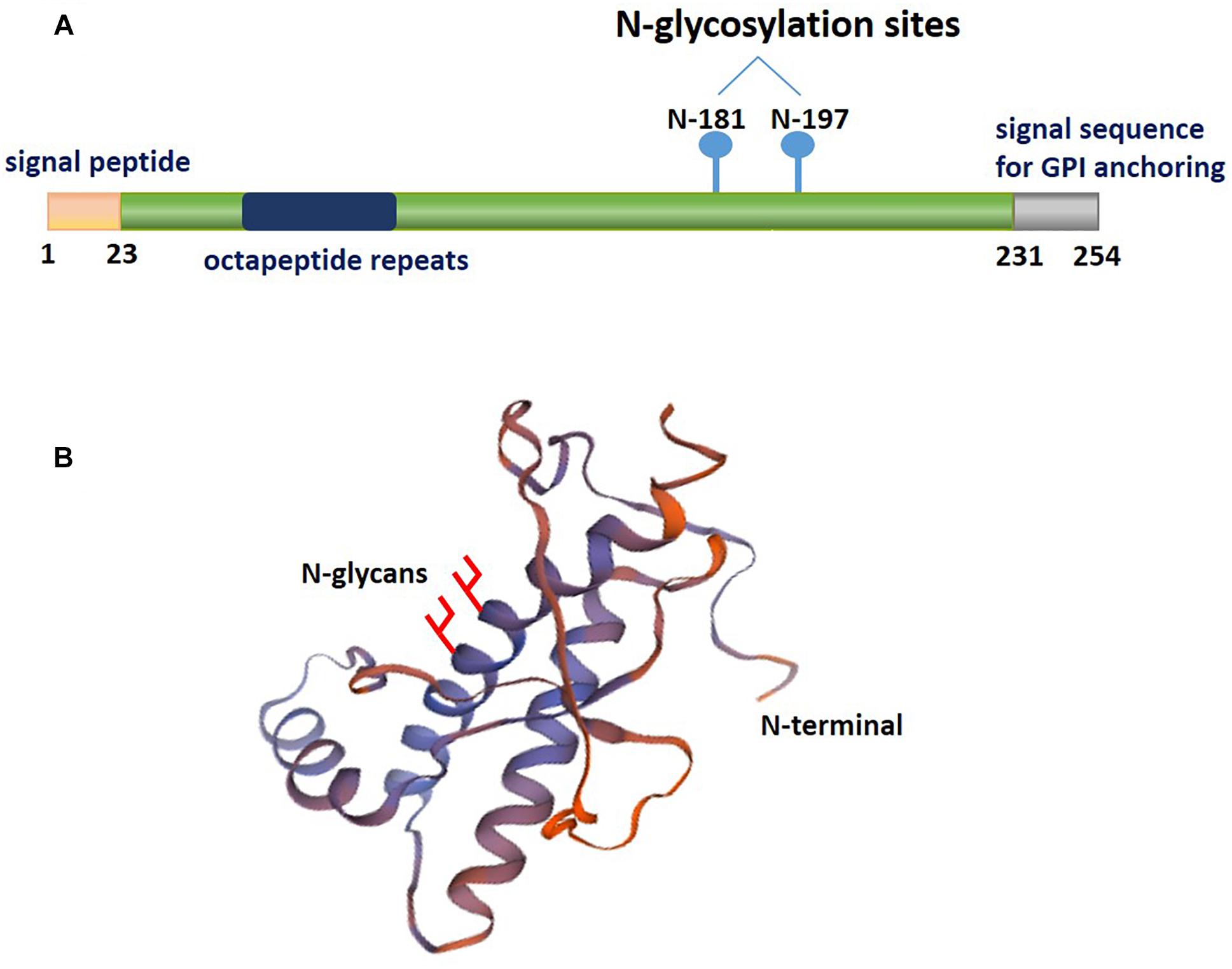
Figure 1. A schematic illustrating the posttranslational modifications of PrPC. The relative size and localization of N-glycans in a schematic representation of the structural domains of PrPC. (A) The structure of PrPC can be divided into two distinct domains: a disordered N-terminal domain and an α-helical C-terminal domain. The N-terminal domain include a positively charged region at the N-terminus that is important for the endocytosis of PrPC, octapeptide repeats that allow PrPC to bind ions, and a hydrophobic tract. The C-terminal domain consists of three α-helices and two short β-strands. This domain is also the site of posttranslational modifications in PrPC; up to two N-glycans are added to the α-helical domain, and a GPI anchor at the C-terminus attaches PrPC to the outer surface of the plasma membrane. (B) The three-dimensional structure of residues 90-31 of recombinant human PrPC (PDB #2lsb.1.A), as determined by NMR spectroscopy.
Velayos et al. (2009) found that the unglycosylated isoform predominates in AD patients, unlike in controls, indicating a shift in the profile of PrPC glycosylation in AD pathological progression. Saijo et al. (2011) also observed significantly higher levels of unglycosylated PrPC in the temporal cortex of amnestic mild cognitive impairment (aMCI) patients, but not of mild AD (mAD) patients, compared to that of NCI and AD patients. The higher levels of the unglycosylated isoform compared to the other isoforms in AD patients supports the hypothesis that the presence of certain glycosylation forms has a critical role in AD.
Additionally, Zafar et al. (2017) demonstrated a significant 1.2-fold decrease in the levels of diglycosylated PrPC isoforms in rapidly progressive AD (rpAD) patients compared to controls but significantly increased total PrPC levels in slow progressive AD (spAD) and rpAD patients. This study indicates that posttranslationally modified PrPC isoforms are changed in the different pathological processes of AD, revealing that the different phenotypes of PrPC may be risk factors for the slow or rapid progression of AD pathology.
Extensive structural analysis has shown that the unglycosylated fragment of residues 90–231 PrPC lacks the flexible N-terminal part of the protein (Wuthrich and Riek, 2001). Studies have indicated the energetic stabilization of the structural region of PrPC in residues 127–227 due to the consequence of the presence of an N-glycan at Asn197. In contrast, an N-glycan at Asn181, which is located in a stable secondary structure, does not influence PrPC conformation but may play a functional role (Zuegg and Gready, 2000; Ermonval et al., 2003). Thus, this evidence indicates that distinct PrPC isoforms are involved in the association of altered PrPC interacting proteins with AD pathology. The glycosylation pattern of PrPC, which may become a potential diagnostic biomarker for pathology, is related to the severity of AD.
Altered Levels of PrPC Involved in AD Pathology
PrPC play an important role in the pathogenesis of AD. Some pathological evidence indicates that PrPC deposits often accompany Aβ plaques in AD (Schwarze-Eicker et al., 2005; Takahashi et al., 2011). The importance of the association between PrPC and Aβ is greatly strengthened when it was demonstrated that PrPC was the receptor for the high affinity to Aβ42 oligomers on cells (Purro et al., 2018).
Reported data suggest a regulatory influence of PrPC expression in the pathological process of AD. The altered expression of PrPC in aging and the development of AD are associated with disease progression, and it has been observed that PrPC is decreased in the hippocampus and temporal cortex in aging and sporadic AD but not in familial AD, suggesting that PrPC expression reduced reflects a main mechanism of disease and is not merely a minor consequence of other AD-associated changes (Whitehouse et al., 2010). In a study by Velayos et al. (2009), there was a tendency for a lower expression of PrPC in AD patients than in healthy patients, which indicated that existing PrPC expression may play a protective role in AD.
In addition, other studies have focused on PrPC expression level alterations in advanced stages of AD, mainly stage Braak III to VI, most likely due to neuronal loss. Vergara et al. (2015) demonstrated that, in AD patients with Braak stages I–VI, PrPC protein expression in the brain increases in the early stages of AD and peaks at approximately stage III. Thereafter, PrPC expression decreases until the manifestation of clinical symptoms in both cases (Vergara et al., 2015).
However, there are some conflicting results regarding the elevation of membrane-binding PrPC levels in brain tissue of AD patients compared with that of patients with mild cognitive impairment (MCI) or no cognitive impairment (NCI) (Larson et al., 2012; Beland et al., 2014). There have also been some studies showing that there is no significant diversity in the expression level of PrPC between AD patients and healthy people (Saijo et al., 2011; Dohler et al., 2014; Abu Rumeileh et al., 2017). The discrepancy may due to the lack of specificity of the assay for the prion protein. Diglycosylated fragments overlap with mono- and nonglycosylated forms of full-length PrPC, which may potentially affect the quantification of PrPC levels and explain the discrepancy. As Saijo et al. (2011) further demonstrated, there were no differences in total PrPC levels, however, the glycosylated forms were observed to be significantly changed in AD.
These studies reinforce the hypothesis that changes in PrPC levels are critical for AD pathological development. Clarifying the possible relationship between cognitive decline, PrPC expression and differentially glycosylated PrPC is extremely important for the identification of AD.
PrPC Mediates Neurotoxicity by Aβ Oligomers
Regions of PrPC Involved in Binding to Aβ
The crucial role of PrPC in neurodegeneration, especially in AD, is complex. The distinct functions of the various domains of PrPC have been associated with determining the early trigger of AD pathophysiology. The amino-terminal octapeptide repeat domain of PrPC (resides 60–95) participates in extracellular copper ion binding (Viles et al., 1999; Jackson et al., 2001). The unstructured central domain of PrPC (residues 95–134) contains a charge cluster (residues 95–110) and a segment with hydrophobic character (residues 112–134), which has been implicated in playing a critical role in neurodegenerative activity (Baumann et al., 2007; Li et al., 2007; Lauren et al., 2009).
Several independent studies have shown that the interaction of Aβ oligomers with the N-terminal residues of the PrPC protein region appears critical for neuronal toxicity (Lauren et al., 2009). The PrPC-Aβ42 interaction provides important mechanistic insights into the pathophysiology of AD-related neurodegeneration. Dohler et al. showed that PrPC-Aβ binding always occurs in AD brains and is never detected in nondemented controls and that the binding of Aβ aggregates to PrPC is restricted to the N-terminus of PrPC (Dohler et al., 2014). Antibodies targeting PrPC N-terminal residues can prevent synaptic plasticity deficits induced by Aβ oligomers (Didonna et al., 2015). For instance, Lauren et al. (2009) found that the deletion of residues 32–121 of PrPC abrogates binding to Aβ42 oligomers, indicating that the globular domain alone cannot mediate the role of binding to Aβ. The hydrophobic region of PrPC (residues 105–125) is not an essential determinant for binding activity due to this region binds Aβ42 oligomers in a way that is indistinguishable from its binding to full-length PrPC and further the Δ32–106 variant was similar to the Δ32–121 variant, having no Aβ42-oligomer affinity (Lauren et al., 2009). Pretreatment with an antibody against residues 93–109 of PrPC, 6D11, can prevent neuronal cell death by oligomeric Aβ42 and rescues cognitive deficits in APP/PS1 transgenic mice, while another antibody against residues 144–152 of PrPC, 6H4, fails to block oligomeric Aβ-induced neuronal toxicity (Chung et al., 2010; Kudo et al., 2012). Recently, another antibody against resides 23–111 of PrPC was reported to rescue synapses and cognitive deficits in APP/PS1 mice (Cox et al., 2019).
The addition of a synthetic peptide of PrPC residues 98–107 can reduce the neurotoxicity of Aβ oligomers in primary hippocampal cells, whereas the addition of a peptide of PrPC residues 213–230 has no effect on Aβ-induced neurotoxicity (Kudo et al., 2012). Blocking residues 95–105 of PrPC, but not its C-terminal residues, can effectively prevent the inhibition of LTP (Barry et al., 2011; Larson et al., 2012). In addition, Chen et al. indicated that both N-terminal residues 23–27 region and the 92–110 region, which are critically important for PrPC interactions with Aβ42 oligomers because the deletion of either of these regions results in a major loss of binding, are highly flexible and natively unstructured (Chen et al., 2010). Zou et al. demonstrated three Aβ42-specific PrPC peptide regions, including N-terminal residues 47–59, 53–65, and 87–99, as well as three Aβ42-nonspecific peptides, including residues 25–37, 37–49, and 99–111 from the N-terminal domain of PrPC. These Aβ42-specific binding sites are localized in the octapeptide repeat region of the unstructured N-terminal domain. The regions have important implications regarding the pathophysiological consequences of Aβ-PrPC interactions (Zou et al., 2011).
Collectively, these studies strongly suggest that N-terminal residues 23–27 and the 95–110 region of PrPC contain the critical amino acid binding sequence for oligomer Aβ-induced synaptic impairment and neuronal cell death (Figure 2A and Table 1). These soluble recombinant PrP proteins and their fragments are strong inhibitors of the cytotoxic and synaptotoxic effects of Aβ42. The identification of specific huPrP regions that are crucial for the interaction with Aβ may also contribute to the development of therapeutic strategies for targeting this interaction.
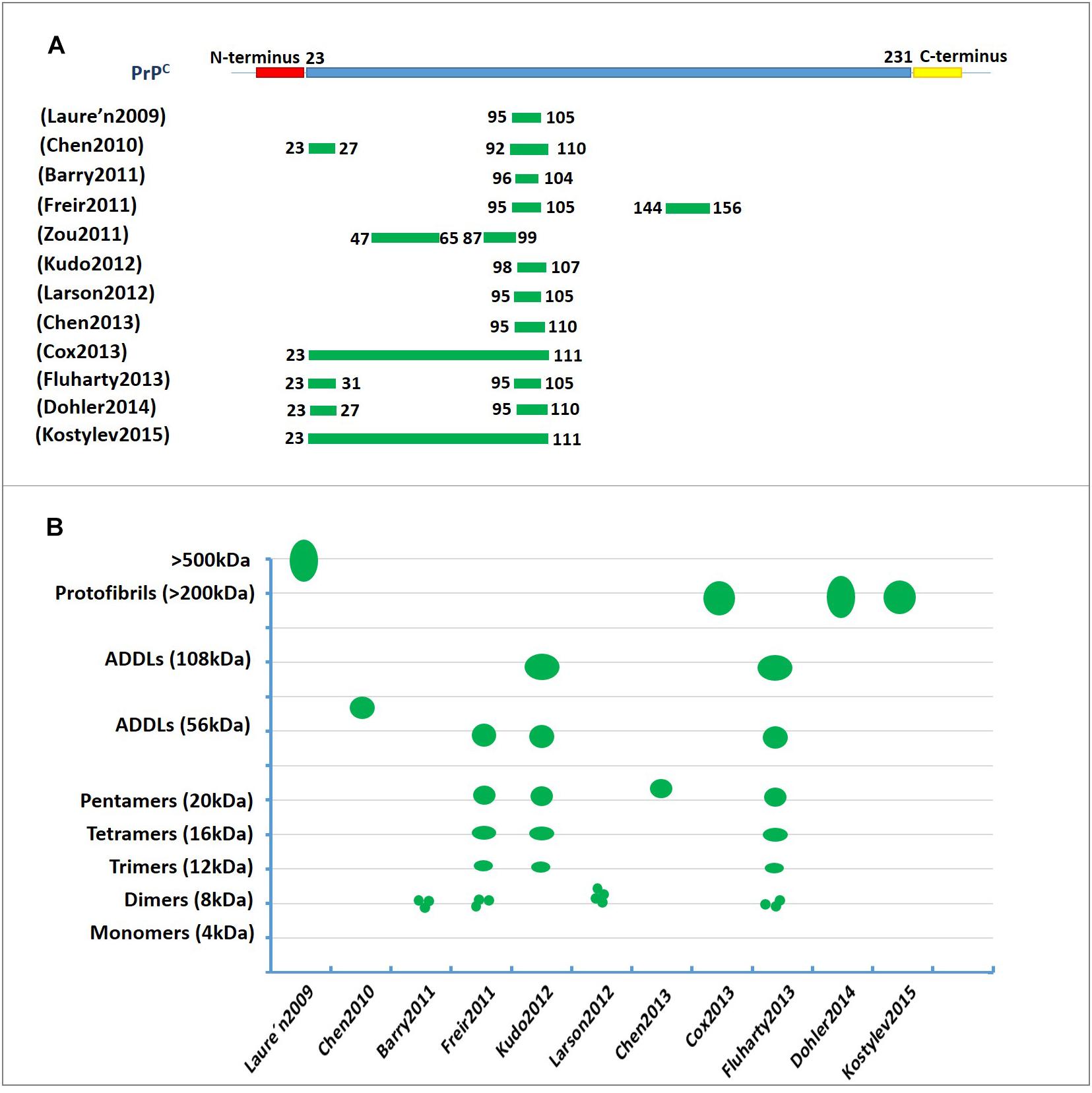
Figure 2. The characterization of PrPC binding to Aβ42. (A) The regions involved in PrPC binding to amyloid-β (Aβ). The green lines represent the amino acid sequence of PrPC involved in binding to Aβ. The residue 95–110 region of PrPC contains the critical amino acid sequence for binding oligomeric Aβ. (B) A diagram depicting the binding of amyloid-β (Aβ) in various aggregation states to PrPC. The green represents the molecular weights of Aβ assemblies bound to PrPC published in previous studies. The Y-axis represents the molecular weight of the various Aβ42 oligomers. ADDL, Aβ-derived diffusible ligand.
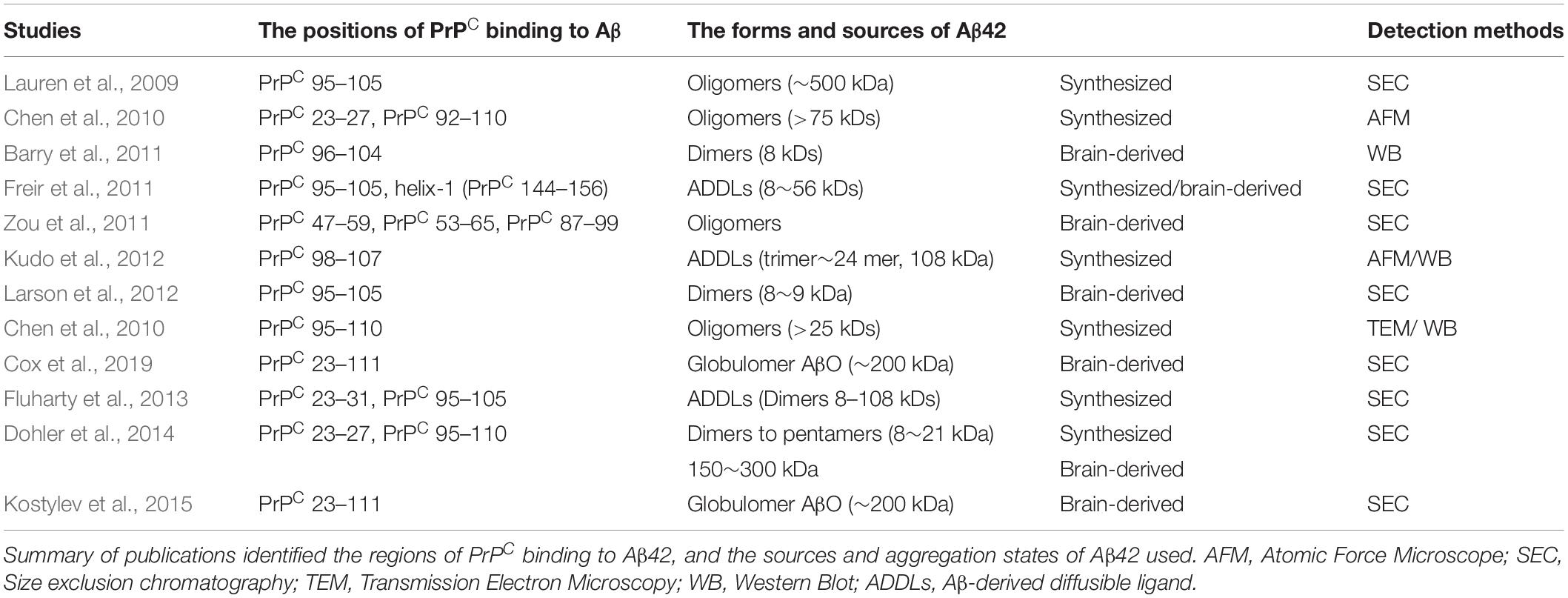
Table 1. The characterization of PrPC binding to Aβ42.
PrPC-Aβ- Binding Assemblies
For decades, research on the molecular mechanism of AD has focused on the compositions of the plaques that are one of the characteristics of the disease (Gouras et al., 2010, 2015). In recent years, it has been found that low molecular weight Aβ aggregates form as Aβ oligomers, and highly structured protofibrils have now emerged as the key neurotoxins in AD (Sakono and Zako, 2010; Brouillette, 2014).
Membrane-binding PrPC, as a receptor for Aβ oligomers, has been demonstrated to be involved in regulating LTP in the hippocampus, which is induced by oligomeric Aβ42. Recently, it was found that PrPC can interact not only with the oligomeric form of Aβ, but also with other forms. For example, synthetic Aβ oligomers, Aβ-derived diffusible ligands (ADDLs) and soluble extracts from AD brains have been reported to interact with PrPC, yet all of these forms of Aβ are unsuccessful at abolishing LTP in PrP-null mice (Freir et al., 2011). Zou et al. (2011) demonstrated that recombinant human PrP (huPrP) also exhibits high affinity and specificity for Aβ42 oligomers from brain extracts of AD patients, and recombinant huPrP may represent an intrinsic molecular spectrum of PrPC in vivo. The inhibition of LTP by human brain extracts containing dimeric amyloid-β is prevented by an antibody fragment (Fab) directed to PrPC in vivo in rats (Barry et al., 2011). Further findings indicate that fragments of the PrPC protein can prevent deficits in synaptic plasticity and neuronal death induced by toxic dimers and trimers of Aβ oligomers species (Scott-McKean et al., 2016). The above studies have revealed that the interaction of certain amyloid species with PrPC leads to neuronal degeneration, and these Aβ species are oligomeric forms with low molecular weights.
Recently, studies have indicated that neurotoxins comprise high molecular weight Aβ assemblies, referred to as ADDLs, which are found to impair synaptic plasticity and memory dysfunction in AD. PrPC has also been shown to bind to ADDLs, which are tightly related with cognitive impairment in multiple mouse models of Alzheimer’s disease (Fluharty et al., 2013; Kostylev et al., 2015). Freir et al. demonstrated that PrPC is a major component for the inhibition of LTP by ADDLs from AD brains, which is consistent with research showing that oligomeric Aβ assemblies bind with strong specificity to PrPC to trigger the disruption of synaptic plasticity in vitro and in vivo (Freir et al., 2011; Kostylev et al., 2015).
However, a separate study indicated that PrPC shows strong binding to high molecular mass assemblies of Aβ (158–300 kDa) derived from the brains of Alzheimer’s disease patients, but not to small synthetic oligomeric Aβ42 (Dohler et al., 2014). It is possible Aβ protofibrils or high molecular mass assemblies correlate better than either globular oligomers or amyloid fibrils with PrPC binding. High molecular mass assemblies are not simply chains of globular oligomers, but instead contain a defined triple helical nanotube structure that may relate to their specific PrPC-dependent toxicity (Nicoll et al., 2013; Purro et al., 2018). It is possible that the connections of Aβ42 and PrPC are not exclusive, and other sterically similar β-sheet rich assemblies might interact with and signal through PrPC (Resenberger et al., 2011).
Thus, PrPC appears to be important to mediate the plasticity impairments induced by certain Aβ species or conformations that must be clarified in the future. Figure 2B and Table 1 show the molecular weights and aggregation states of Aβ42 binding to PrPC currently. Dimers, ∼108 kDa oligomers and ADDLs display a much higher ability to interact with PrPC.
Characterization of Soluble and Insoluble Aβ in AD
The major components of Aβ aggregates that form in AD brain are neuritic plaques, diffuse amyloid, and vascular amyloid. A variety of other assemblies including Aβ protofibrils and soluble oligomers of various sizes have also been identified (Caughey and Lansbury, 2003; Haass and Selkoe, 2007). Evidence suggests that Aβ oligomers are soluble and may spread throughout the brain, yet amyloid fibrils are larger and insoluble and may assemble into amyloid plaques (Ferreira et al., 2015). However, the present findings strongly support that soluble Aβ oligomers are more detrimental to synaptic plasticity.
Aβ oligomers are categorized by molecular weight as low-molecular-weight (LMW) oligomers ranging from dimers-tetramers and high-molecular-weight (HMW) oligomers ranging from ∼50 to 150 kDa (Figueiredo et al., 2013). A recent study indicated that LMW and HMW oligomers have differential binding affinities for neurons and neurotoxicity. LMW oligomers acutely impair synaptic plasticity, whereas HMW oligomers induce neuronal oxidative stress via activation of NMDA receptors (Wisniewski et al., 2011).
However, other studies have shown that both soluble and insoluble fractions of brain homogenates bind to PrPC in transgenic mouse models of AD (Zou et al., 2011; Larson et al., 2012). Dohler et al. (2014) demonstrated that optimal binding to PrPC occurs in the insoluble fraction of Aβ. Their data show that Aβ is present as insoluble oligomers in all tested high molecular weight fractions. Another study using AD and healthy brains also showed the preferential binding of high molecular weight Aβ42 assemblies to PrPC, which occurs mainly in the insoluble fraction of Aβ, in AD (Zou et al., 2011).
In fact, the composition, concentration and purity of Aβ samples from AD brains are always different. For synthetic Aβ42, small oligomeric species show prominent binding to PrPC, whereas in AD brains larger protein assemblies containing Aβ42 bind efficiently to PrPC (Dohler et al., 2014). Furthermore, the natural separation of different Aβ aggregates present in AD brain samples is complex because preparation methods can disrupt and alter the conformation of Aβ assemblies (Stine et al., 2011). Synthetic Aβ is generally soluble, while the Aβ extracted from AD brains may contain both soluble and insoluble fractions (Table 1). Some soluble oligomers may bind to other macromolecules or to cell membranes and can therefore become insoluble (Dohler et al., 2014).
In addition, Wildburger et al. (2017) recently found 26 unique proteoforms in soluble and more insoluble Aβ aggregates of AD brain samples, including 73% N-terminal truncations and 30% C-terminal truncations of the total Aβ proteoforms. The Aβ proteoforms segregated between the soluble and more insoluble aggregates, with N-terminal truncations predominating in the insoluble material and C-terminal truncations segregating into the soluble aggregates. This result suggests that the Aβ aggregates in AD are heterogeneous and offers much new evidence for investigation into the pathological mechanisms of AD.
Molecular Consequences of the PrPC/AβO Interaction in AD
The molecular and cellular consequences of the PrPC-Aβ oligomer interaction are dependent on raft-based complexes. The integrity of cholesterol-rich lipid rafts is critical for the interaction between Aβ42 with PrPC. It has been demonstrated that the PrPC-mediated toxicity of Aβ oligomers and the activation of downstream pathways require lipid rafts (Rushworth et al., 2013). GPI-anchored PrPC is localized to the cholesterol-rich lipid raft microdomains of the plasma membrane (Taylor and Hooper, 2006). Cholesterol depletion disrupts these rafts with PrPC being redistributed into nonraft regions of the membrane (Taylor et al., 2005). A study revealed that the disruption of the rafts causes a significant reduction in Aβ oligomer binding to cells and prevents the activation of Fyn kinase (Rushworth et al., 2013).
A growing body of evidence suggests that PrPC mediates downstream intracellular processes through many different receptors, including the metabotropic glutamate receptors mGluR1 and mGluR5, the α7 nicotinic acetylcholine receptor, the kainite receptor GluR6/7, and AMPA receptor subunits GluA1 and GluA2 (Zhao et al., 2010; Carulla et al., 2011; Jeong and Park, 2015; Haas and Strittmatter, 2016). These studies indicate that PrPC functions as an extracellular scaffolding protein that is able to organize multiprotein complexes that mediate intracellular signal transduction at the cell surface.
A separate study implicated group I mGluR signaling in the regulation of Aβ42 toxicity in neurons and in AD mouse models, and Aβ42 oligomers interact with PrPC to increase metabotropic glutamate receptor 5 (mGluR5)-dependent long-term depression (LTD) and LTP (Hu et al., 2014). Another study showed that the role of the Aβ/PrPC complex in AD pathophysiology; the complex has been demonstrated to induce intracellular Fyn activation, leading to the further phosphorylation of the NR2B subunit of the N-methyl-D-aspartate (NMDA) receptor and to the destabilization of dendritic spines (Um et al., 2012; Brody and Strittmatter, 2018). In addition, Pyk2 has been reported to be a downstream effector of PrPC-Aβ signaling. This study revealed Pyk2 as a direct tyrosine kinase of tau that is active downstream of Fyn (Li and Gotz, 2018).
Recently, the Aβ-PrPC complex was reported to be internalized into endosomes via low-density lipoprotein 1 (LRP1) in AD. Rushworth et al. (2013) revealed that PrPC-mediated binding and toxicity and the subsequent PrPC-mediated internalization of Aβ oligomers are dependent upon LRP1. Research has indicated that LRP1 functions as a transmembrane coreceptor that is involved in the PrPC-mediated binding of Aβ oligomers (Rushworth et al., 2013).
The laminin receptor (LRP/LR) has been demonstrated to play a significant role in the interaction between Aβ and PrPC, given that the laminin receptor binds and internalizes PrPC. The blockade of LRP/LR ameliorates the detrimental effect of PrPC overexpression on cell viability upon exposure to exogenous Aβ (Pinnock et al., 2016). The binding of Aβ to PrPC leads to the induction of apoptosis by interacting with LRP/LR, which, as a transmembrane receptor, may influence the activity of the JNK signaling pathway. However, the proapoptotic signals pathways may not be directly transduced by PrPC, as PrPC is not a transmembrane protein and therefore must be transduced through other receptors binding to PrPC, such as LRP/LR (Da Costa Dias et al., 2014).
Thus, further investigations have been made to expound these complex and their downstream pathways to prevent neurotoxic consequences (Figure 3).
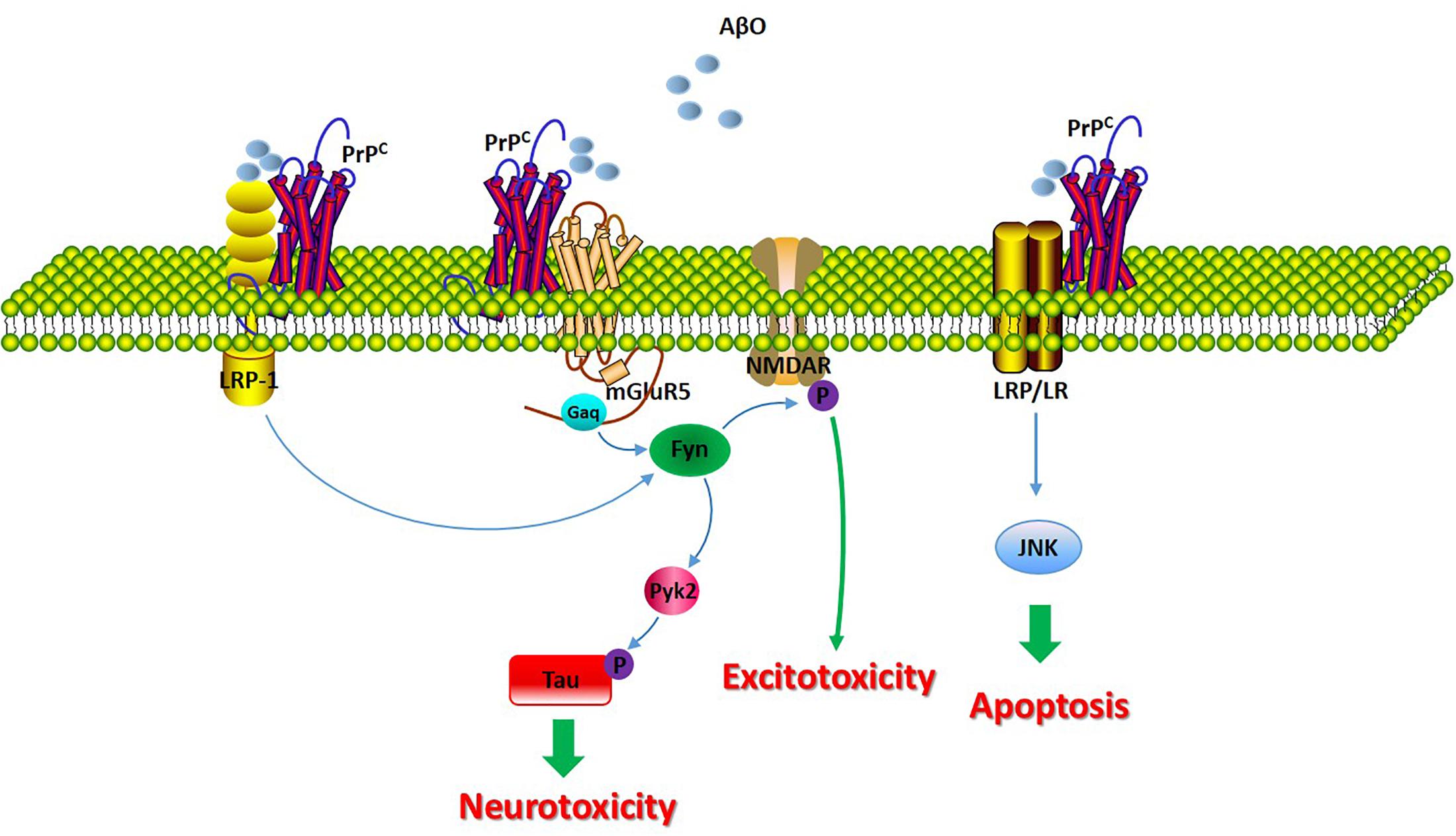
Figure 3. The molecular consequences of the PrPC/Aβ interaction in AD. The signaling pathway involved in the PrPC/Aβ interaction in AD. The PrPC/Aβ complex can interact with mGluR5, LRP1, and LRP/LR. The PrPC/Aβ complex induces Fyn activation, leading to the further phosphorylation of the NR2B subunit of the NMDA receptor and to the destabilization of dendritic spines. Additionally, Fyn activation of Pyk2 leads to the further phosphorylation of tau. LRP1 functions as a transmembrane coreceptor that is involved in PrPC/AβO-mediated Fyn activation. LRP/LR is a transmembrane receptor involved in the apoptotic signaling pathway through interactions with PrPC-Aβ.
Conclusion
Currently most researches show that PrPC may play an important role in the pathogenesis of AD. There is a connection between AD and PrPC levels. PrPC, as a high affinity receptor for oligomeric Aβ and ADDLs (8∼108 kDa), is essential for Aβ-induced synaptic toxicity. N-terminal residues 23–27 and the 95–110 region of PrPC contain the critical amino acid sequence for oligomer Aβ-induced synaptic impairment and neuronal cell death.
However, there is some controversy about the neurotoxicity of PrPC. Some literature reports that PrPC deletion does not inhibit the toxic effects of Aβ oligomers. For example, Balducci et al. (2010) demonstrated that Aβ oligomers induced in vivo memory impairment and bound PrPC with high affinity but found that PrPC is not responsible for the recognition impairment in AD induced by Aβ. However, others have reported that PrPC is a key receptor mediating toxic effects, but these assumption have not been confirmed (Larson et al., 2012). Perhaps PrPC is not the unique receptor that mediates synaptic damage induced by Aβ oligomers. PrPC has been involved in neurotoxic signaling with high-affinity binding to Aβ oligomers, suggesting that the interaction of PrPC with Aβ is part of a common molecular pathway. The most recent investigations indicated the existence of a strong interaction between Aβ oligomers and PrPC and suggest that this interaction may impact synaptic plasticity functions. Further investigations will be necessary to clarify the involvement of PrPC in the neuropathology of AD. The identification of specific huPrP regions that are crucial for the interaction with Aβ may also contribute to the development of therapeutic strategies that target this interaction.
Author Contributions
YuZ conceived and designed the manuscript. YuZ, YaZ, and LZ analyzed and collected the literature. WY, YW, and WC collected the literature. YuZ wrote the review. All authors revised the manuscript.
Funding
This work was supported by funding from the Qingdao Applied Basic Research Project (No.18-2-2-48-jch), Shandong Provincial Natural Science Foundation of China (No. ZR2016CQ23), and National Natural Science Foundation of China (No. 81603357).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abu Rumeileh, S., Lattanzio, F., Stanzani Maserati, M., Rizzi, R., Capellari, S., and Parchi, P. (2017). Diagnostic Accuracy of a Combined Analysis of Cerebrospinal Fluid t-PrP, t-tau, p-tau, and Abeta42 in the Differential Diagnosis of Creutzfeldt-Jakob Disease from Alzheimer’s Disease with Emphasis on Atypical Disease Variants. J. Alzheimer’s Dis. 55, 1471–1480. doi: 10.3233/JAD-160740
Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., et al. (2010). Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci.U.S.A. 107, 2295–2300. doi: 10.1073/pnas.0911829107
Barry, A. E., Klyubin, I., Mc Donald, J. M., Mably, A. J., Farrell, M. A., Scott, M., et al. (2011). Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011
Baumann, F., Tolnay, M., Brabeck, C., Pahnke, J., Kloz, U., Niemann, H. H., et al. (2007). Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 26, 538–547. doi: 10.1038/sj.emboj.7601510
Beland, M., Bedard, M., Tremblay, G., Lavigne, P., and Roucou, X. (2014). Abeta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol. Aging. 35, 1537–1548. doi: 10.1016/j.neurobiolaging.2014.02.001
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Brody, A. H., and Strittmatter, S. M. (2018). Synaptotoxic signaling by amyloid beta oligomers in Alzheimer’s disease through prion protein and mGluR5. Adv. Pharmacol. 82, 293–323. doi: 10.1016/bs.apha.2017.09.007
Brouillette, J. (2014). The effects of soluble Abeta oligomers on neurodegeneration in Alzheimer’s disease. Curr. Pharm. Design. 20, 2506–2519. doi: 10.2174/13816128113199990498
Carulla, P., Bribian, A., Rangel, A., Gavin, R., Ferrer, I., Caelles, C., et al. (2011). Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell. 22, 3041–3054. doi: 10.1091/mbc.E11-04-0321
Caughey, B., and Lansbury, P. T. (2003). Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. doi: 10.1146/annurev.neuro.26.010302.081142
Chen, S., Yadav, S. P., and Surewicz, W. K. (2010). Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J. Biol. Chem. 285, 26377–26383. doi: 10.1074/jbc.M110.145516
Chung, E., Ji, Y., Sun, Y., Kascsak, R. J., Kascsak, R. B., Mehta, P. D., et al. (2010). Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer’s disease model mouse. BMC Neurosci. 11:130. doi: 10.1186/1471-2202-11-130
Cox, T. O., Gunther, E. C., Brody, A. H., Chiasseu, M. T., Stoner, A., Smith, L. M., et al. (2019). Anti-PrP(C) antibody rescues cognition and synapses in transgenic alzheimer mice. Ann. Clin. Transl. Neurol. 6, 554–574. doi: 10.1002/acn3.730
Da Costa Dias, B., Jovanovic, K., Gonsalves, D., Moodley, K., Reusch, U., Knackmuss, S., et al. (2014). The 37kDa/67kDa laminin receptor acts as a receptor for Abeta42 internalization. Sci. Rep. 4:5556. doi: 10.1038/srep05556
Di Lazzaro, V. (2018). Emergence of neurophysiological biomarkers of Alzheimer disease. J. Neurol.Neurosurg.Psychiatry. 89:1235. doi: 10.1136/jnnp-2018-318867
Didonna, A., Venturini, A. C., Hartman, K., Vranac, T., Curin Serbec, V., and Legname, G. (2015). Characterization of four new monoclonal antibodies against the distal N-terminal region of PrP(c). PeerJ. 3:e811. doi: 10.7717/peerj.811
Dohler, F., Sepulveda-Falla, D., Krasemann, S., Altmeppen, H., Schluter, H., Hildebrand, D., et al. (2014). High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer’s disease. Brain J. Neurol. 137, 873–886. doi: 10.1093/brain/awt375
Ermonval, M., Mouillet-Richard, S., Codogno, P., Kellermann, O., and Botti, J. (2003). Evolving views in prion glycosylation: functional and pathological implications. Biochimie. 85, 33–45. doi: 10.1016/s0300-9084(03)00040-3
Ferreira, S. T., Lourenco, M. V., Oliveira, M. M., and De Felice, F. G. (2015). Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 9:191. doi: 10.3389/fncel.2015.00191
Figueiredo, C. P., Clarke, J. R., Ledo, J. H., Ribeiro, F. C., Costa, C. V., Melo, H. M., et al. (2013). Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 33, 9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013
Fluharty, B. R., Biasini, E., Stravalaci, M., Sclip, A., Diomede, L., Balducci, C., et al. (2013). An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866. doi: 10.1074/jbc.M112.423954
Freir, D. B., Nicoll, A. J., Klyubin, I., Panico, S., Mc Donald, J. M., Risse, E., et al. (2011). Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2:336. doi: 10.1038/ncomms1341
Gimbel, D. A., Nygaard, H. B., Coffey, E. E., Gunther, E. C., Lauren, J., Gimbel, Z. A., et al. (2010). Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010
Gouras, G. K., Olsson, T. T., and Hansson, O. (2015). beta-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 12, 3–11.
Gouras, G. K., Tampellini, D., Takahashi, R. H., and Capetillo-Zarate, E. (2010). Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 119, 523–541. doi: 10.1007/s00401-010-0679-9
Haas, L. T., and Strittmatter, S. M. (2016). Oligomers of Amyloid beta Prevent Physiological Activation of the Cellular Prion Protein-Metabotropic Glutamate Receptor 5 Complex by Glutamate in Alzheimer Disease. J. Biol. Chem. 291, 17112–17121. doi: 10.1074/jbc.M116.720664
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hu, N. W., Nicoll, A. J., Zhang, D., Mably, A. J., O’Malley, T., Purro, S. A., et al. (2014). mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat. Commun. 5:3374. doi: 10.1038/ncomms4374
Jackson, G. S., Murray, I., Hosszu, L. L., Gibbs, N., Waltho, J. P., Clarke, A. R., et al. (2001). Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 98, 8531–8535. doi: 10.1073/pnas.151038498
Jeong, J. K., and Park, S. Y. (2015). Neuroprotective effect of cellular prion protein (PrPC) is related with activation of alpha7 nicotinic acetylcholine receptor (alpha7nAchR)-mediated autophagy flux. Oncotarget 6, 24660–24674. doi: 10.18632/oncotarget.4953
Kayed, R., and Lasagna-Reeves, C. A. (2013). Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimer’s Dis. 33(Suppl. 1), S67–S78.
Kostylev, M. A., Kaufman, A. C., Nygaard, H. B., Patel, P., Haas, L. T., Gunther, E. C., et al. (2015). Prion-Protein-interacting Amyloid-beta Oligomers of High Molecular Weight Are Tightly Correlated with Memory Impairment in Multiple Alzheimer Mouse Models. J. Biol. Chem. 290, 17415–17438. doi: 10.1074/jbc.M115.643577
Kudo, W., Lee, H. P., Zou, W. Q., Wang, X., Perry, G., Zhu, X., et al. (2012). Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum. Mol. Genetics. 21, 1138–1144. doi: 10.1093/hmg/ddr542
Larson, M., Sherman, M. A., Amar, F., Nuvolone, M., Schneider, J. A., Bennett, D. A., et al. (2012). The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857a–16871a. doi: 10.1523/JNEUROSCI.1858-12.2012
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 457, 1128–1132. doi: 10.1038/nature07761
Li, A., Christensen, H. M., Stewart, L. R., Roth, K. A., Chiesa, R., and Harris, D. A. (2007). Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J. 26, 548–558. doi: 10.1038/sj.emboj.7601507
Li, C., and Gotz, J. (2018). Pyk2 is a novel tau tyrosine kinase that is regulated by the tyrosine kinase fyn. J. Alzheimer’s Dis. 64, 205–221. doi: 10.3233/JAD-180054
Nicoll, A. J., Panico, S., Freir, D. B., Wright, D., Terry, C., Risse, E., et al. (2013). Amyloid-beta nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 4:2416. doi: 10.1038/ncomms3416
Onodera, T. (2017). Dual role of cellular prion protein in normal host and Alzheimer’s disease. Proc. Japan Acad. Ser. BPhys. Biol. Sci. 93, 155–173. doi: 10.2183/pjab.93.010
Pinnock, E. C., Jovanovic, K., Pinto, M. G., Ferreira, E., Dias Bda, C., Penny, C., et al. (2016). LRP/LR antibody mediated rescuing of amyloid-beta-induced cytotoxicity is dependent on PrPc in Alzheimer’s disease. J. Alzheimer’s Dis. 49, 645–657. doi: 10.3233/JAD-150482
Purro, S. A., Nicoll, A. J., and Collinge, J. (2018). Prion protein as a toxic acceptor of amyloid-beta oligomers. Biol. psychiatry. 83, 358–368. doi: 10.1016/j.biopsych.2017.11.020
Resenberger, U. K., Harmeier, A., Woerner, A. C., Goodman, J. L., Muller, V., Krishnan, R., et al. (2011). The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070. doi: 10.1038/emboj.2011.86
Rushworth, J. V., Griffiths, H. H., Watt, N. T., and Hooper, N. M. (2013). Prion protein-mediated toxicity of amyloid-beta oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 288, 8935–8951. doi: 10.1074/jbc.M112.400358
Saijo, E., Scheff, S. W., and Telling, G. C. (2011). Unaltered prion protein expression in Alzheimer disease patients. Prion. 5, 109–116. doi: 10.4161/pri.5.2.16355
Sakono, M., and Zako, T. (2010). Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 277, 1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x
Salazar, S. V., and Strittmatter, S. M. (2017). Cellular prion protein as a receptor for amyloid-beta oligomers in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 483, 1143–1147. doi: 10.1016/j.bbrc.2016.09.062
Schwarze-Eicker, K., Keyvani, K., Gortz, N., Westaway, D., Sachser, N., and Paulus, W. (2005). Prion protein (PrPc) promotes beta-amyloid plaque formation. Neurobiol. Aging. 26, 1177–1182. doi: 10.1016/j.neurobiolaging.2004.10.004
Scott-McKean, J. J., Surewicz, K., Choi, J. K., Ruffin, V. A., Salameh, A. I., Nieznanski, K., et al. (2016). Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Abeta oligomers: implications for novel therapeutic strategy in Alzheimer’s disease. Neurobiol. Dis. 91, 124–131. doi: 10.1016/j.nbd.2016.03.001
Selkoe, D. J. (2011). Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 3:a004457. doi: 10.1101/cshperspect.a004457
Singh, J., and Udgaonkar, J. B. (2015). Molecular mechanism of the misfolding and oligomerization of the prion protein: current understanding and its implications. Biochemistry 54, 4431–4442. doi: 10.1021/acs.biochem.5b00605
Stine, W. B., Jungbauer, L., Yu, C., and LaDu, M. J. (2011). Preparing synthetic Abeta in different aggregation states. Methods Mol. Biol. 670, 13–32. doi: 10.1007/978-1-60761-744-0_2
Takahashi, R. H., Tobiume, M., Sato, Y., Sata, T., Gouras, G. K., and Takahashi, H. (2011). Accumulation of cellular prion protein within dystrophic neurites of amyloid plaques in the Alzheimer’s disease brain. Neuropathology 31, 208–214. doi: 10.1111/j.1440-1789.2010.01158.x
Taylor, D. R., and Hooper, N. M. (2006). The prion protein and lipid rafts. Mol. Membr. Biol. 23, 89–99. doi: 10.1080/09687860500449994
Taylor, D. R., Watt, N. T., Perera, W. S., and Hooper, N. M. (2005). Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 118, 5141–5153. doi: 10.1242/jcs.02627
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
Velayos, J. L., Irujo, A., Cuadrado-Tejedor, M., Paternain, B., Moleres, F. J., and Ferrer, V. (2009). The cellular prion protein and its role in Alzheimer disease. Prion 3, 110–117. doi: 10.4161/pri.3.2.9135
Vergara, C., Ordonez-Gutierrez, L., Wandosell, F., Ferrer, I., del Rio, J. A., and Gavin, R. (2015). Role of PrP(C) expression in tau protein levels and phosphorylation in Alzheimer’s disease evolution. Mol. Neurobiol. 51, 1206–1220. doi: 10.1007/s12035-014-8793-7
Viles, J. H., Cohen, F. E., Prusiner, S. B., Goodin, D. B., Wright, P. E., and Dyson, H. J. (1999). Copper binding to the prion protein: structural implications of four identical cooperative binding sites. Proc Natl. Acad. Sci.U.S.A. 96, 2042–2047. doi: 10.1073/pnas.96.5.2042
Whitehouse, I. J., Jackson, C., Turner, A. J., and Hooper, N. M. (2010). Prion protein is reduced in aging and in sporadic but not in familial Alzheimer’s disease. J. Alzheimer’s Dis. 22, 1023–1031. doi: 10.3233/JAD-2010-101071
Wildburger, N. C., Esparza, T. J., LeDuc, R. D., Fellers, R. T., Thomas, P. M., Cairns, N. J., et al. (2017). Diversity of amyloid-beta proteoforms in the Alzheimer’s disease brain. Sci. Rep. 7:9520. doi: 10.1038/s41598-017-10422-x
Wiseman, F. K., Cancellotti, E., Piccardo, P., Iremonger, K., Boyle, A., Brown, D., et al. (2015). The glycosylation status of PrPC is a key factor in determining transmissible spongiform encephalopathy transmission between species. J. Virol. 89, 4738–4747. doi: 10.1128/JVI.02296-14
Wisniewski, M. L., Hwang, J., and Bahr, B. A. (2011). Submicromolar Abeta42 reduces hippocampal glutamate receptors and presynaptic markers in an aggregation-dependent manner. Biochim. Biophys. Acta. 1812, 1664–1674. doi: 10.1016/j.bbadis.2011.09.011
Wuthrich, K., and Riek, R. (2001). Three-dimensional structures of prion proteins. Adv. Protein Chem. 57, 55–82. doi: 10.1016/s0065-3233(01)57018-7
Zafar, S., Shafiq, M., Younas, N., Schmitz, M., Ferrer, I., and Zerr, I. (2017). Prion protein interactome: identifying novel targets in slowly and rapidly progressive forms of Alzheimer’s disease. J. Alzheimer’s Dis. 59, 265–275. doi: 10.3233/JAD-170237
Zhao, W. Q., Santini, F., Breese, R., Ross, D., Zhang, X. D., Stone, D. J., et al. (2010). Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J. Biol. Chem. 285, 7619–7632. doi: 10.1074/jbc.M109.057182
Zou, W. Q., Xiao, X., Yuan, J., Puoti, G., Fujioka, H., Wang, X., et al. (2011). Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J. Biol. Chem. 286, 15095–15105. doi: 10.1074/jbc.M110.199356
Keywords: cellular prion protein, amyloid-β, oligomers, receptor, Alzheimer’s disease
Citation: Zhang Y, Zhao Y, Zhang L, Yu W, Wang Y and Chang W (2019) Cellular Prion Protein as a Receptor of Toxic Amyloid-β42 Oligomers Is Important for Alzheimer’s Disease. Front. Cell. Neurosci. 13:339. doi: 10.3389/fncel.2019.00339
Received: 21 April 2019; Accepted: 10 July 2019;
Published: 30 July 2019.
Edited by:
Antonio Gambardella, Università degli Studi Magna Græcia di Catanzaro, ItalyReviewed by:
Diana Laura Castillo-Carranza, Hampton University, United StatesHelmut Kessels, Netherlands Institute for Neuroscience (KNAW), Netherlands
Copyright © 2019 Zhang, Zhao, Zhang, Yu, Wang and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Zhang, eXVhbl96aGFuZzg0QDEyNi5jb20=