 Eunbeol Lee
Eunbeol Lee- Department of Biological Sciences, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, South Korea
Glial cells are emerging as crucial players that mediate development and homeostasis of the central nervous system (CNS). In particular, glial cells are closely associated with synapses, and control synapse formation, function, plasticity, and elimination during the stages of development and adulthood. Importantly, it is now increasingly evident that abnormal glial function can be an active inducer of the initiation and progression of various neurodegenerative diseases. Here, we discuss recent developments on the physiological roles of glial cells in the brain, and propose that synapse loss, which is a common characteristic of several neurodegenerative diseases, can be initiated by mis-regulation of normal glial function.
Introduction
Glial cells, non-neuronal central nervous system (CNS) cells, are not simply passive support cells for neurons, but active players in neuronal network formation and information processing. The fine processes of astrocytes and microglia closely associate with synapses and affect synapse formation and elimination during development. Even after initial establishment of a neural circuit, glial cells continue to participate in regulation of synapse number and modulation of synaptic function and plasticity. Due to their crucial roles in synapses, it is not surprising that mis-regulation of glial function has recently been revealed to be one of the initiating factors for onset and progression of various neurodegenerative diseases (Phatnani and Maniatis, 2015; Liddelow and Barres, 2017; Li and Barres, 2018).
Synapse loss is one of the most common, and earliest, pathophysiological features of neurodegenerative disease progression. In Alzheimer’s disease (AD), where amyloid β (Aβ) plaques accumulate in the brain, surrounded by reactive glial cells, synapses are weakened and undergo specific loss long before neuronal cell death (Perry and Holmes, 2014; Phatnani and Maniatis, 2015; Ransohoff, 2016). Previously, it was thought that this early synapse loss was mainly due to the direct effects of Aβ oligomers and fibrils on neuronal Aβ receptors or secondary neuroinflammation. However, recent studies have revealed that it is reactive glial cells that actually drive the initial synapse loss in AD (Hong et al., 2016; Shi Q. et al., 2017; Dejanovic et al., 2018; Litvinchuk et al., 2018).
In this review, we summarize the roles of astrocytes and microglia in synapse formation and elimination, and review/propose how mis-regulation of glial function leads to synapse loss in various neurodegenerative diseases (Figure 1).
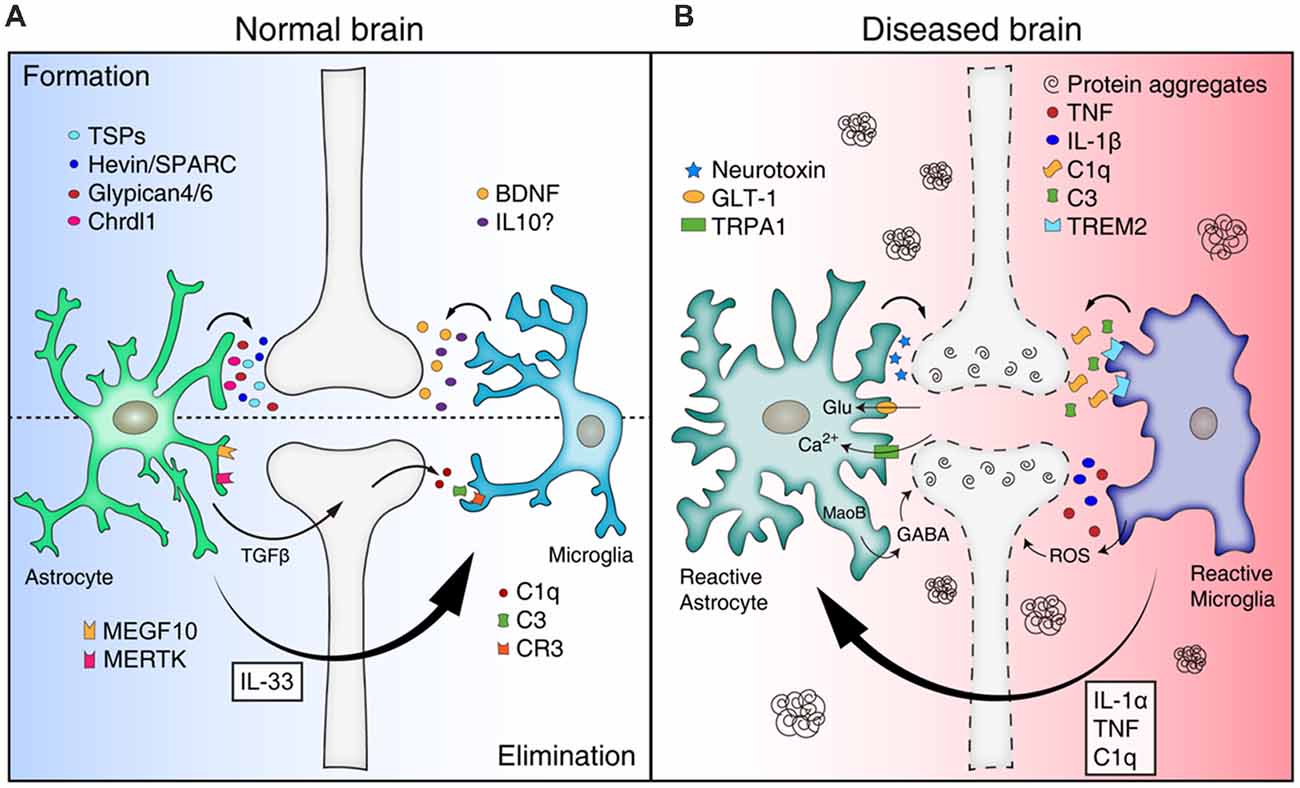
Figure 1. Mis-regulation of glial function can lead to synapse loss in neurodegenerative diseases. (A) Astrocytes and microglia release a number of synaptogenic factors [for example, thrombospondins (TSPs), Hevin/SPARC, Glypican 4/6, Chrdl1 from astrocytes, and brain-derived neurotrophicfactor (BDNF) and interleukin 10 (IL10) from microglia], regulating synapse formation. Astrocytes can mediate synapse pruning through MEGF10, MERTK phagocytic pathways whereas microglia contribute to synapse elimination through complement cascades [C1q, C3, and complement receptor 3 (CR3)]. Astrocytes also can regulate microglia-mediated synapse elimination by secreting transforming growth factor beta (TGFβ) and IL33. (B) In neurodegenerative diseases, reactive astrocytes and microglia are found around protein aggregates [such as amyloid β (Aβ) plaques, α-synuclein aggregates] and mediate synapse loss through various mechanisms. Reactive astrocytes may be induced by cytokines (IL-1α, TNF, and C1q) secreted from reactive microglia in pathological conditions, and produce neurotoxic factors that kill synapses and neurons. In addition, reactive astrocytes in Alzheimer’s disease (AD) show increased expression of glutamate transporter-1 (GLT-1), increased activity of transient receptor potential A1 (TRPA1) channels, and increased release of γ-aminobutyric acid (GABA), leading to aberrant neuronal excitability and synaptic function. Similar to astrocytes, reactive microglia can participate in neuronal damage and synapse loss through inflammatory signaling [such as TNF, IL-1β, and reactive oxygen species (ROS)]. Microglia mediate aberrant synapse loss in AD brains through complement mediators (especially C1q and C3) and triggering receptor expressedon myeloid cells 2 (TREM2).
The Role of Glia in Synapse Formation
During CNS development, glial cells participate in regulating synapse number by inducing proper formation and elimination (Ullian et al., 2001). Over the past decades, the molecular mechanisms of glia-mediated synapse formation and elimination have been revealed.
Through in vitro culture of neurons, with or without astrocytes, it was initially found that astrocytes induced synapse formation by secreting several distinct molecules. When retinal ganglion cells (RGCs) were cultured in astrocyte-conditioned media (ACM), the number of synapses were increased both structurally and functionally. Later, thrombospondins (TSPs), especially TSP1 and TSP2, were found to be one of the synaptogenic proteins in the ACM. Despite effects on the formation of structural synapses, TSP1/2-induced synapses are postsynaptically silent due to their lack of functional α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs; Christopherson et al., 2005; Eroglu et al., 2009). Along with TSPs, astrocytes express a number of matricellular proteins, such as hevin and SPARC, which modulate cell-cell and cell-matrix interactions (Eroglu, 2009). Hevin induces structurally normal and postsynaptically silent excitatory synapses, similar to TSP-induced synapses. In contrast, SPARC, a hevin homolog, antagonizes hevin and blocks synapse formation (Kucukdereli et al., 2011). Recently, it was discovered that hevin plays a role in bridging synaptic adhesion molecules neurexin 1α (NRX1α) and neuroligins (NL; Singh et al., 2016), which are localized in pre- and post-synaptic compartments, respectively (Graf et al., 2004). NLs and NRX1α, which alone are interaction-incompatible partners, can associate when transcellularly-linked by hevin. This complex can then recruit more NL1 and NMDAR to synapses (Singh et al., 2016).
So, how do astrocytes increase functional synapses? Through biochemical fractionation of ACM, glypican 4 (Gpc4) and glypican 6 (Gpc6) have been identified as functional synaptogenic molecules that strengthen glutamatergic synapses by recruiting GluA1-containing AMPARs (Allen et al., 2012). Astrocyte-secreted Gpc4 appears to upregulate release of neuronal pentraxins 1 (NP1) through interactions with presynaptic type 2a receptor protein tyrosine phosphatases δ (RPTPδ). Subsequently, NP1 binds postsynaptic AMPARs to recruit GluA1 and induce functional synapse formation (Farhy-Tselnicker et al., 2017). Astrocyte-expressed pentraxin 3 (PTX3) has been also reported to promote functionally-active CNS synapses (Fossati et al., 2019). PTX3, whose activity is regulated by TSP1, increases the surface levels and synaptic clustering of AMPARs through remodeling the perineuronal network, and a β1-integrin/ERK pathway. Chordin-like 1 (Chrdl1) has recently been shown to be another synaptogenic molecule, from astrocytes, that can induce maturation of functional synapses by increasing synaptic GluA2 AMPA receptors. Chrdl1 expression is limited to cortical astrocytes in vivo, and is necessary and sufficient to increase GluA2 clustering, resulting in formation of active synapses in vitro and in vivo (Blanco-Suarez et al., 2018). In addition, astrocyte-derived apolipoprotein E (APOE), which forms lipoprotein particles, with cholesterol and other lipids, has been reported to enhance presynaptic glutamatergic function (Mauch et al., 2001).
Several recent studies have suggested that microglia may also participate in inducing structural synapses. Microglia, the resident macrophages of the CNS, constantly survey and make contacts with synapses in the normal adult brain. Interestingly, when microglia were depleted by diphtheria toxin, synapse formation was disrupted, but synapse elimination rate was unchanged. Removal of brain-derived neurotrophic factor (BDNF), specifically from microglia, recapitulated this phenotype, suggesting that synapse formation is mediated by microglial BDNF (Parkhurst et al., 2013). Additionally, microglial cytokines, such as interleukin 10 (IL-10), have been shown to induce synapse formation (Lim et al., 2013). Using in vivo multiphoton imaging, a recent report found that microglial contact induces neuronal Ca2+ transients and actin accumulation, inducing filopodia formation from the dendritic branches (Miyamoto et al., 2016).
Thus, astrocytes and microglia regulate synapse formation through various mechanisms. How these different molecules engage in crosstalk, and whether neural activity/injury response controls their expression, are important questions for understanding how synapse dynamics are regulated by glial cells in healthy and diseased brains. Aberrant increases in synapse formation during development or after injury may cause hyperactive neural circuits and increased chances of epilepsy (Liuzzi and Lasek, 1987). In contrast, defective glia-mediated synapse formation could impair synaptic turnover and homeostasis, contributing to synapse loss in neurodegenerative diseases, as well as defective synaptic plasticity.
The Role of Glia in Synapse Elimination Through Phagocytosis
To maintain proper synapse numbers, unnecessary synapses need to be eliminated during development and adulthood. Many studies have suggested that excess synapses are eliminated by neuronal activity-dependent competition (Ramiro-Cortés and Israely, 2013; Bian et al., 2015). Surprisingly, glial cells, especially astrocytes and microglia, have been shown to mediate this elimination. Astrocytes express several phagocytic receptors, such as MEGF10 (an ortholog of Drosophila Draper and C. elegans CED-1) and MERTK [a member of the Tyro-Axl-MerTK (TAM) family of receptor tyrosine kinase], and participate in eliminating synapses in the developing brain. RGCs in developing mice deficient in both Megf10 and Mertk pathways show a failure of the normal refinement of connections and retain excess functional synapses with neurons in the dorsal lateral geniculate nucleus (Chung et al., 2013). This finding suggests that astrocytes actively participate in eliminating live synapses rather than simply cleaning up dead synaptic debris. Although microglia have traditionally been thought to be the major glial cells mediating synapse elimination in development, this study shows astrocytes also play a critical role. Astrocytes appear to continuously engulf both excitatory and inhibitory synapses throughout the brain during adulthood as well, suggesting that the synaptic architecture of our brains is constantly being remodeled by astrocytes in response to our experiences.
In addition to synapse elimination via direct phagocytosis, astrocytes also contribute to synapse elimination via inositol 1,4,5-triphosphate receptor type 2 (IP3R2) and P2ry1 dependent signaling (Yang et al., 2016), as well as via microglia-mediated phagocytosis. Microglia mediate synapse pruning through the classical complement cascade (Schafer et al., 2012). C1q, the cascade initiating protein, is expressed by microglia and subsets of neurons (Stephan et al., 2013), and localizes to unwanted synapses for opsonization (Stevens et al., 2007). Synapse-associated C1q, then activates a downstream complement cascade and mediates microglia-dependent synapse elimination through complement receptor 3 (CR3)-mediated phagocytosis (Schafer et al., 2012). Interestingly, C1q expression in neurons is regulated by transforming growth factor (TGF)-β secreted from astrocytes (Bialas and Stevens, 2013). Additionally, it has recently been shown that microglial synapse engulfment during development is regulated by interleukin-33 (IL-33) secreted from astrocytes (Vainchtein et al., 2018), further suggesting a close interplay between astrocytes and microglia in eliminating synapses.
CX3CR1s (fractalkine receptors), expressed by microglia, also play important roles in synapse elimination. Microglia respond to neuronal fractalkine by increasing intracellular calcium transients in vitro and in vivo (Harrison et al., 1998). In Cx3cr1-knockout mice, migration and recruitment of microglia to the brain were impaired, resulting in an increased immature synapse population, which appeared to be responsible for weak functional synaptic connectivity and impaired social interaction in the knockout mice (Fuhrmann et al., 2010; Paolicelli et al., 2011; Hoshiko et al., 2012; Zhan et al., 2014).
Reactive Gliosis in Response to the Injured and Diseased Brain
Reactive gliosis is observed under various conditions, such as infection, ischemia, trauma, and neurodegeneration, and usually involves hypertrophy and proliferation of glial cells and changes in gene expression. Reactive glial cells release various molecules, including chemokines, cytokines, and neurotrophic factors, that can exhibit either neuroprotective or neurotoxic effects (Sofroniew and Vinters, 2010). Although reactive gliosis was previously believed to be a secondary response to neuroinflammation, recent studies suggest that glial cells react differently depending on injury stimulus, and that gliosis can initiate neurodegeneration (Jonsson et al., 2013; Hong et al., 2016; Sekar et al., 2016).
In general, it is believed that reactive microglia can exhibit more than two polarization states, such as M1 and M2. M1-like microglia upregulate inflammatory signaling, such as TNF, IL-1β, and reactive oxygen species (ROS) signaling. In contrast, M2-like microglia express anti-inflammatory molecules, such as TGF-β1, IL-4, and IL-10. By converting their gene expression status, reactive microglia participate not only in neuronal damage and synapse loss, but also regeneration and tissue remodeling through phagocytosis (Block et al., 2007; Boche et al., 2013; Simon et al., 2017). For example, M1-like microglia led to decrease in dendritic spine density in hippocampal neurons in vivo and in vitro by producing extracellular vesicles (EVs) containing a set of miRNAs that regulate the expression of synaptic proteins. Among them, miR-146a-5p controls the expression of synaptotagmin1 and neuroligin1 in EV-receiving neurons. In contrast, M2-like microglia appear to have opposite effects by producing EVs depleted of miR-146a-5p (Prada et al., 2018).
Similar to microglia, reactive astrocytes isolated from mice with transient ischemia induced by occlusion of the middle cerebral artery (MCAO) or treated with lipopolysaccharide (LPS) differentially expressed certain genes categories depending on injury type. Genes related to metabolic activity, cell-cycle, and transcription factors (e.g., Klf5 and Klf6) were selectively increased in middle cerebral artery (MCAO)-induced reactive astrocytes (A2 astrocytes), whereas genes related to immune response, antigen processing/presentation, and complement pathway were upregulated in lipopolysaccharide (LPS)-induced reactive astrocytes (A1 astrocytes; Zamanian et al., 2012). Subsequent work has revealed that A1 astrocytes can be induced by a combination of cytokines (IL-1α, TNF, and C1q) released by reactive microglia. This study also showed that A1 astrocytes not only lose their ability to induce synapse formation and elimination, but also produce neurotoxic factors that kill neurons and oligodendrocytes (Liddelow et al., 2017). In comparison to signals inducing A1 astrocytes, little is known about the upstream inductive signals that drives A2 astrocytes. Reactive astrocytes after transient ischemic injury can transform into efficient phagocytes, clearing neuronal and synaptic debris through ATP-binding cassette transporter A1 (ABCA1)-dependent mechanisms (Morizawa et al., 2017). Furthermore, astrocytes perform neuroprotective functions through downregulating the P2Y1 purinergic receptor after traumatic brain injury. Downregulation of P2Y1 enhances extension of astrocytic processes and accelerates reactive astrogliosis accompanied by upregulation of glial fibrillary acidic protein (GFAP) and phosphorylation of STAT3. Accelerated activation of astrocytes helps diminish scar formation following injury (Shinozaki et al., 2017). Thus, these astrocytes, which presumably associated with the A2 type, may promote CNS recovery and repair (Bush et al., 1999; Gao et al., 2005; Hayakawa et al., 2014).
Glia-Synapse Interactions in Neurodegenerative Disease
In many neurodegenerative diseases, synapse loss is a common, early sign of disease progression. As we discussed in the previous section, glia play central roles in normal synapse formation and elimination. In this section, we discuss how glial cells also participate in synapse loss in neurodegenerative diseases, especially AD and Parkinson’s disease (PD).
AD is the most common cause of dementia. Its pathology includes extracellular accumulation of Aβ plaques and intracellular, neurofibrillary tangles of hyperphosphorylated tau protein. Aβ, produced by proteolytic cleavage of amyloid precursor protein (APP), self-aggregates to form oligomers, protofibrils, or other fibrils. Soluble Aβ oligomers have been shown to induce synapse loss, tau phosphorylation, and reactive gliosis of astrocytes and microglia (Tomiyama et al., 2010; Mitew et al., 2013; Forny-Germano et al., 2014). Oligomeric Aβ surrounding senile plaques contributes to significant synapse loss in AD mouse models (Koffie et al., 2009). Although the mechanism is not fully understood, it is thought that Aβ oligomers induce overstimulation of NMDA receptors, leading to abnormal redox events and Ca2+ upregulation (Tu et al., 2014). Additionally, Aβ oligomers reduce synaptic adhesion by disrupting neural cell adhesion molecule 2 (NCAM2), leading to synapse loss in the AD hippocampus (Leshchyns’ka et al., 2015).
In AD brains, reactive astrocytes and microglia are found around Aβ plaques and can play beneficial or harmful roles in disease progression (Nagele et al., 2003; Olabarria et al., 2010; Simpson et al., 2010). Reactive astrocytes can internalize Aβ in vitro and in vivo (Wyss-Coray et al., 2003; Pihlaja et al., 2008), although the molecular mechanism and biological impact of this clearance are unclear. In contrast, evidence from aged AD mouse brains shows that astrocytic processes are tightly associated with Aβ plaques, but do not engulf them. Interestingly, reactive astrocytes found in AD brains seem to follow an A1 astrocyte fate, with neuroinflammatory gene expression patterns (Liddelow et al., 2017; Shi Y. et al., 2017). Since A1 astrocytes are less capable of phagocytosing neuronal material, it is possible that reactive astrocytes in AD brains become less able to engulf Aβ plaque during disease progression. Therefore, boosting the phagocytic capacity of astrocytes for Aβ plaques could lower Aβ burden in the CNS. Consistent with this hypothesis, transplantation of wild-type (WT) astrocytes into AD mouse brain results in more efficient removal of Aβ plaques compared to non-transplanted controls (Pihlaja et al., 2011). Moreover, since reactive astrocytes in AD show characteristics of A1 astrocytes, they may produce synaptic and neurotoxic factors, similar to cases of neuroinflammation and brain injury. Identifying such factors may be an important step in preventing reactive astrocyte-induced synapse loss in AD.
In AD, reactive astrocytes also directly affect synaptic function and dynamics. These astrocytes exhibit reduced expression of glutamine synthetase and glutamate transporter-1 (GLT-1; Robinson, 2001; Zumkehr et al., 2015), directly inducing aberrant neuronal excitability and synaptic function, which can lead to synapse loss. It has been suggested that reactive astrocytes can affect synaptic dysfunction through hyper-production and release of inhibitory gliotransmitter γ-aminobutyric acid (GABA). Impaired presynaptic release and spike probability, synaptic plasticity, and learning and memory were rescued by inhibiting the GABA-producing enzyme, MaoB, in AD astrocytes (Jo et al., 2014). Soluble Aβ oligomers also induce astrocytic calcium hyperactivity by activating transient receptor potential A1 (TRPA1) channels, leading to neuronal hyperactivity (Lee et al., 2014; Bosson et al., 2017). Since astrocytic calcium transients may control astrocyte-synapse interaction (Henneberger et al., 2010; Shigetomi et al., 2013; Bazargani and Attwell, 2016), dysregulation of calcium transients in AD astrocytes could be an initiating factor for synapse loss.
Similar to astrocytes, microglia are located around Aβ plaques in both human and mouse AD brains (Perlmutter et al., 1990; Bolmont et al., 2008; Grathwohl et al., 2009). It was recently found that the complement cascade that microglia use for pruning unnecessary synapses in the developing brains is responsible for synapse loss in AD brains. In Aβ overproducing AD mouse models, expression of complement mediators, especially C1q and C3, is highly upregulated, mediating aberrant synapse phagocytosis by microglia (Hong et al., 2016; Shi Q. et al., 2017). Blocking complement activation by an antibody blocking C1q function or introduction of C3KO and C3RKO to this AD model mouse background were sufficient to prevent early synapse loss, establishing a direct role of microglial phagocytosis in AD-associated synapse loss. A recent report also showed that in Tau-P301S transgenic mice, a C1q-blocking antibody can also prevent aberrant microglial synapse removal and rescue synaptic density (Dejanovic et al., 2018; Litvinchuk et al., 2018).
Recent studies have shown that triggering receptor expressed on myeloid cells 2 (TREM2) also plays an important role in microglia-mediated synapse elimination. Several phospholipids, including phosphatidylinositol, phosphatidylcholine, and lipoproteins (APOE, LDL, CLU/apoj), as well as Aβ, have been suggested as potential TREM2 ligands. Activation of TREM2 receptors changes microglial gene expression related to inflammatory signals and phagocytosis (Colonna, 2003). In developing brains, TREM2 maintains the phagocytic capacity of microglia and mediates normal synapse pruning (Filipello et al., 2018). Interestingly, TREM2 also mediates Aβ-induced, microglial cytokine expression and Aβ clearance through phagocytosis (Zhao et al., 2018). Determining exact functions of TREM2 in AD would be necessary in understanding the role of microglia in mediating synapse loss in AD.
PD is another common neurodegenerative disease with loss of nigrostriatal dopaminergic neurons. Abnormal intraneuronal and intraneuritic deposits of fibrillary, α-synuclein aggregates are thought to be the main cause of PD. Aggregates of α-synuclein have been found at presynaptic terminals in PD, resulting in synaptic degeneration (Schulz-Schaeffer, 2010). Previous studies demonstrated that α-synuclein induces microglial activation (Austin et al., 2006; Lee E.-J. et al., 2010) and influences microglial phagocytic activity. Park et al. (2008) showed that microglial phagocytosis was enhanced by monomeric α-synuclein, but inhibited by aggregated α-synuclein. It has been also suggested that microglia clear α-synuclein through a C1q-dependent pathway (Depboylu et al., 2011).
In PD brains, α-synuclein also stimulates reactive astrogliosis, and α-synuclein inclusions have been found in astrocytes (Wakabayashi et al., 2000; Lee H.-J. et al., 2010; Braidy et al., 2013). These studies suggest that astrocytes can uptake α-synuclein, although the physiological role of this process is unclear. Recently, it has been shown that inhibiting microglia-mediated conversion of astrocytes into A1 astrocytes significantly alleviate synaptic/neuronal loss in PD mouse models (Yun et al., 2018). Furthermore, microglia in PD show changes in expression of innate immunity genes, such as progranulin, NF-κB, and TDP-43, which may further impact microglial phagocytosis and synapse elimination (Kao et al., 2011; Frakes et al., 2014; Lui et al., 2016; Spiller et al., 2018). Therefore, similar to AD, synapse and neuronal loss in PD may be triggered by reactive microglia and astrocytes. Further studies are necessary to determine whether modulating glial function can prevent α-synuclein accumulation and synapse/neuronal loss in PD.
Conclusions and Perspectives
We here describe recent findings that glial cells, especially astrocytes and microglia, regulate synapse dynamics in normal and neurodegenerative diseases. In the past, neurodegeneration studies have focused mainly on neurons; however, recent studies suggest that abnormal interactions between glia and synapses can initiate many pathophysiological aspects of synapse loss. In diseased brains, glial cells lose their normal functions for proper synapse formation, function, plasticity, and elimination. Furthermore, these cells acquire new, deleterious functions to drive synapse and neuronal loss. In both cases, understanding the exact molecular mechanisms responsible for glia–mediated synapse loss is an important step in developing therapeutic strategies for neurodegenerative diseases. In addition, determining what initiates reactive gliosis in each disease, and whether we can prevent these processes are equally important questions. Astrocytes and microglia may participate in synapse formation and elimination with different degrees in various development and disease conditions. In normal physiological conditions, since major portion of synapses are in direct contact with astrocytic processes, astrocytes may respond more efficiently than microglia in regulating neuronal activity-dependent changes in synaptic density. In contrast, microglia may play dominant roles in eliminating synapses in the diseased conditions due to the significant upregulation of phagocytic machineries after reactive gliosis. How astrocytes and microglia differentially and cooperatively control synapse number in the healthy and diseased brain would be additional fascinating subjects to investigate. Current advances in establishing gene expression databases of disease-affected human brains, as well as in understanding normal glial function in synapse regulation will aid this discovery.
Author Contributions
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP; NRF-2016M3C7A1905391, NRF-2016R1C1B3006969 and NRF-2018R1A4A1020922; W-SC). W-SC is also supported by Cure Alzheimer’s Fund.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all members in Chung’s laboratory for helpful discussion.
References
Allen, N. J., Bennett, M. L., Foo, L. C., Wang, G. X., Chakraborty, C., Smith, S. J., et al. (2012). Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486, 410–414. doi: 10.1038/nature11059
Austin, S. A., Floden, A. M., Murphy, E. J., and Combs, C. K. (2006). α-synuclein expression modulates microglial activation phenotype. J. Neurosci. 26, 10558–10563. doi: 10.1523/JNEUROSCI.1799-06.2006
Bazargani, N., and Attwell, D. (2016). Astrocyte calcium signaling: the third wave. Nat. Neurosci. 19, 182–189. doi: 10.1038/nn.4201
Bialas, A. R., and Stevens, B. (2013). TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 16, 1773–1782. doi: 10.1038/nn.3560
Bian, W. J., Miao, W. Y., He, S. J., Qiu, Z., and Yu, X. (2015). Coordinated spine pruning and maturation mediated by inter-spine competition for cadherin/catenin complexes. Cell 162, 808–822. doi: 10.1016/j.cell.2015.07.018
Blanco-Suarez, E., Liu, T. F., Kopelevich, A., and Allen, N. J. (2018). Astrocyte-secreted chordin-like 1 drives synapse maturation and limits plasticity by increasing synaptic GluA2 AMPA receptors. Neuron 100, 1116.e13–1132.e13. doi: 10.1016/j.neuron.2018.09.043
Block, M. L., Zecca, L., and Hong, J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. doi: 10.1038/nrn2038
Boche, D., Perry, V. H., and Nicoll, J. A. (2013). Review: activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 39, 3–18. doi: 10.1111/nan.12011
Bolmont, T., Haiss, F., Eicke, D., Radde, R., Mathis, C. A., Klunk, W. E., et al. (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008
Bosson, A., Paumier, A., Boisseau, S., Jacquier-Sarlin, M., Buisson, A., and Albrieux, M. (2017). TRPA1 channels promote astrocytic Ca2+ hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-β peptide. Mol. Neurodegener. 12:53. doi: 10.1186/s13024-017-0194-8
Braidy, N., Gai, W. P., Xu, Y. H., Sachdev, P., Guillemin, G. J., Jiang, X. M., et al. (2013). Uptake and mitochondrial dysfunction of α-synuclein in human astrocytes, cortical neurons and fibroblasts. Transl. Neurodegener. 2:20. doi: 10.1186/2047-9158-2-20
Bush, T. G., Puvanachandra, N., Horner, C. H., Polito, A., Ostenfeld, T., Svendsen, C. N., et al. (1999). Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23, 297–308. doi: 10.1016/s0896-6273(00)80781-3
Christopherson, K. S., Ullian, E. M., Stokes, C. C., Mullowney, C. E., Hell, J. W., Agah, A., et al. (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. doi: 10.1016/j.cell.2004.12.020
Chung, W. S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi: 10.1038/nature12776
Colonna, M. (2003). TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453. doi: 10.1038/nri1106
Dejanovic, B., Huntley, M. A., De Mazière, A., Meilandt, W. J., Wu, T., Srinivasan, K., et al. (2018). Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100, 1322.e7–1336.e7. doi: 10.1016/j.neuron.2018.10.014
Depboylu, C., Schäfer, M. K., Arias-Carrión, O., Oertel, W. H., Weihe, E., and Höglinger, G. U. (2011). Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J. Neuropathol. Exp. Neurol. 70, 125–132. doi: 10.1097/nen.0b013e31820805b9
Eroglu, C. (2009). The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J. Cell Commun. Signal. 3, 167–176. doi: 10.1007/s12079-009-0078-y
Eroglu, C., Allen, N. J., Susman, M. W., O’Rourke, N. A., Park, C. Y., Ozkan, E., et al. (2009). Gabapentin receptor α2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. doi: 10.1016/j.cell.2009.09.025
Farhy-Tselnicker, I., van Casteren, A. C. M., Lee, A., Chang, V. T., Aricescu, A. R., and Allen, N. J. (2017). Astrocyte-secreted glypican 4 regulates release of neuronal pentraxin 1 from axons to induce functional synapse formation. Neuron 96, 428.e13–445.e13. doi: 10.1016/j.neuron.2017.09.053
Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48, 979.e8–991.e8. doi: 10.1016/j.immuni.2018.04.016
Forny-Germano, L., Lyra e Silva, N. M., Batista, A. F., Brito-Moreira, J., Gralle, M., Boehnke, S. E., et al. (2014). Alzheimer’s disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J. Neurosci. 34, 13629–13643. doi: 10.1523/JNEUROSCI.1353-14.2014
Fossati, G., Pozzi, D., Canzi, A., Mirabella, F., Valentino, S., Morini, R., et al. (2019). Pentraxin 3 regulates synaptic function by inducing AMPA receptor clustering via ECM remodeling and β1-integrin. EMBO J. 38:e99529. doi: 10.15252/embj.201899529
Frakes, A. E., Ferraiuolo, L., Haidet-Phillips, A. M., Schmelzer, L., Braun, L., Miranda, C. J., et al. (2014). Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 81, 1009–1023. doi: 10.1016/j.neuron.2014.01.013
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Gao, Q., Li, Y., and Chopp, M. (2005). Bone marrow stromal cells increase astrocyte survival via upregulation of phosphoinositide 3-kinase/threonine protein kinase and mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathways and stimulate astrocyte trophic factor gene expression after anaerobic insult. Neuroscience 136, 123–134. doi: 10.1016/j.neuroscience.2005.06.091
Graf, E. R., Zhang, X., Jin, S. X., Linhoff, M. W., and Craig, A. M. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026. doi: 10.1016/j.cell.2004.11.035
Grathwohl, S. A., Kalin, R. E., Bolmont, T., Prokop, S., Winkelmann, G., Kaeser, S. A., et al. (2009). Formation and maintenance of Alzheimer’s disease β-amyloid plaques in the absence of microglia. Nat. Neurosci. 12, 1361–1363. doi: 10.1038/nn.2432
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., McNamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U S A 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Hayakawa, K., Pham, L. D., Arai, K., and Lo, E. H. (2014). Reactive astrocytes promote adhesive interactions between brain endothelium and endothelial progenitor cells via HMGB1 and β-2 integrin signaling. Stem Cell Res. 12, 531–538. doi: 10.1016/j.scr.2013.12.008
Henneberger, C., Papouin, T., Oliet, S. H., and Rusakov, D. A. (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–236. doi: 10.1038/nature08673
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoshiko, M., Arnoux, I., Avignone, E., Yamamoto, N., and Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J. Neurosci. 32, 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
Jo, S., Yarishkin, O., Hwang, Y. J., Chun, Y. E., Park, M., Woo, D. H., et al. (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 20, 886–896. doi: 10.1038/nm.3639
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., et al. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U S A 108, 4441–4446. doi: 10.1073/pnas.1100650108
Koffie, R. M., Meyer-Luehmann, M., Hashimoto, T., Adams, K. W., Mielke, M. L., Garcia-Alloza, M., et al. (2009). Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U S A 106, 4012–4017. doi: 10.1073/pnas.0811698106
Kucukdereli, H., Allen, N. J., Lee, A. T., Feng, A., Ozlu, M. I., Conatser, L. M., et al. (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. U S A 108, E440–E449. doi: 10.1073/pnas.1104977108
Lee, L., Kosuri, P., and Arancio, O. (2014). Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients. J. Alzheimers Dis. 38, 49–62. doi: 10.3233/jad-130740
Lee, H.-J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., et al. (2010). Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272. doi: 10.1074/jbc.M109.081125
Lee, E.-J., Woo, M. S., Moon, P. G., Baek, M. C., Choi, I. Y., Kim, W. K., et al. (2010). α-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 185, 615–623. doi: 10.4049/jimmunol.0903480
Leshchyns’ka, I., Liew, H. T., Shepherd, C., Halliday, G. M., Stevens, C. H., Ke, Y. D., et al. (2015). Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease. Nat. Commun. 6:8836. doi: 10.1038/ncomms9836
Li, Q., and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242. doi: 10.1038/nri.2017.125
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lim, S. H., Park, E., You, B., Jung, Y., Park, A. R., Park, S. G., et al. (2013). Neuronal synapse formation induced by microglia and interleukin 10. PLoS One 8:e81218. doi: 10.1371/journal.pone.0081218
Litvinchuk, A., Wan, Y. W., Swartzlander, D. B., Chen, F., Cole, A., Propson, N. E., et al. (2018). Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron 100, 1337.e5–1353.e5. doi: 10.1016/j.neuron.2018.10.031
Liuzzi, F. J., and Lasek, R. J. (1987). Astrocytes block axonal regeneration in mammals by activating the physiological stop pathway. Science 237, 642–645. doi: 10.1126/science.3603044
Lui, H., Zhang, J., Makinson, S. R., Cahill, M. K., Kelley, K. W., Huang, H. Y., et al. (2016). Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 165, 921–935. doi: 10.1016/j.cell.2016.04.001
Mauch, D. H., Nagler, K., Schumacher, S., Göritz, C., Müller, E. C., Otto, A., et al. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357. doi: 10.1126/science.294.5545.1354
Mitew, S., Kirkcaldie, M. T., Dickson, T. C., and Vickers, J. C. (2013). Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 34, 2341–2351. doi: 10.1016/j.neurobiolaging.2013.04.010
Miyamoto, A., Wake, H., Ishikawa, A. W., Eto, K., Shibata, K., Murakoshi, H., et al. (2016). Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 7:12540. doi: 10.1038/ncomms12540
Morizawa, Y. M., Hirayama, Y., Ohno, N., Shibata, S., Shigetomi, E., Sui, Y., et al. (2017). Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 8:28. doi: 10.1038/s41467-017-00037-1
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V., and Wang, H. Y. (2003). Astrocytes accumulate A β 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209. doi: 10.1016/s0006-8993(03)02361-8
Olabarria, M., Noristani, H. N., Verkhratsky, A., and Rodríguez, J. J. (2010). Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58, 831–838. doi: 10.1002/glia.20967
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Park, J. Y., Paik, S. R., Jou, I., and Park, S. M. (2008). Microglial phagocytosis is enhanced by monomeric α-synuclein, not aggregated α-synuclein: implications for Parkinson’s disease. Glia 56, 1215–1223. doi: 10.1002/glia.20691
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R., Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Perlmutter, L. S., Barron, E., and Chui, H. C. (1990). Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci. Lett. 119, 32–36. doi: 10.1016/0304-3940(90)90748-x
Perry, V. H., and Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224. doi: 10.1038/nrneurol.2014.38
Phatnani, H., and Maniatis, T. (2015). Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 7:a020628. doi: 10.1101/cshperspect.a020628
Pihlaja, R., Koistinaho, J., Kauppinen, R., Sandholm, J., Tanila, H., and Koistinaho, M. (2011). Multiple cellular and molecular mechanisms are involved in human Aβ clearance by transplanted adult astrocytes. Glia 59, 1643–1657. doi: 10.1002/glia.21212
Pihlaja, R., Koistinaho, J., Malm, T., Sikkilä, H., Vainio, S., and Koistinaho, M. (2008). Transplanted astrocytes internalize deposited β-amyloid peptides in a transgenic mouse model of Alzheimer’s disease. Glia 56, 154–163. doi: 10.1002/glia.20599
Prada, I., Gabrielli, M., Turola, E., Iorio, A., D’Arrigo, G., Parolisi, R., et al. (2018). Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 135, 529–550. doi: 10.1007/s00401-017-1803-x
Ramiro-Cortés, Y., and Israely, I. (2013). Long lasting protein synthesis- and activity-dependent spine shrinkage and elimination after synaptic depression. PLoS One 8:e71155. doi: 10.1371/journal.pone.0071155
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Robinson, S. R. (2001). Changes in the cellular distribution of glutamine synthetase in Alzheimer’s disease. J. Neurosci. Res. 66, 972–980. doi: 10.1002/jnr.10057
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schulz-Schaeffer, W. J. (2010). The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 120, 131–143. doi: 10.1007/s00401-010-0711-0
Sekar, A., Bialas, A. R., de Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Shi, Q., Chowdhury, S., Ma, R., Le, K. X., Hong, S., Caldarone, B. J., et al. (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Shigetomi, E., Jackson-Weaver, O., Huckstepp, R. T., O’Dell, T. J., and Khakh, B. S. (2013). TRPA1 channels are regulators of astrocyte basal calcium levels and long-term potentiation via constitutive D-serine release. J. Neurosci. 33, 10143–10153. doi: 10.1523/JNEUROSCI.5779-12.2013
Shinozaki, Y., Shibata, K., Yoshida, K., Shigetomi, E., Gachet, C., Ikenaka, K., et al. (2017). Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y1 receptor downregulation. Cell Rep. 19, 1151–1164. doi: 10.1016/j.celrep.2017.04.047
Simon, D. W., McGeachy, M. J., Bayir, H., Clark, R. S. B., Loane, D. J., and Kochanek, P. M. (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 13:572. doi: 10.1038/nrneurol.2017.116
Simpson, J. E., Ince, P. G., Lace, G., Forster, G., Shaw, P. J., Matthews, F., et al. (2010). Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol. Aging 31, 578–590. doi: 10.1016/j.neurobiolaging.2008.05.015
Singh, S. K., Stogsdill, J. A., Pulimood, N. S., Dingsdale, H., Kim, Y. H., Pilaz, L. J., et al. (2016). Astrocytes assemble thalamocortical synapses by bridging NRX1α and NL1 via hevin. Cell 164, 183–196. doi: 10.1016/j.cell.2015.11.034
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Spiller, K. J., Restrepo, C. R., Khan, T., Dominique, M. A., Fang, T. C., Canter, R. G., et al. (2018). Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 21, 329–340. doi: 10.1038/s41593-018-0083-7
Stephan, A. H., Madison, D. V., Mateos, J. M., Fraser, D. A., Lovelett, E. A., Coutellier, L., et al. (2013). A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Tomiyama, T., Matsuyama, S., Iso, H., Umeda, T., Takuma, H., Ohnishi, K., et al. (2010). A mouse model of amyloid β oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 30, 4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 9:48. doi: 10.1186/1750-1326-9-48
Ullian, E. M., Sapperstein, S. K., Christopherson, K. S., and Barres, B. A. (2001). Control of synapse number by glia. Science 291, 657–661. doi: 10.1126/science.291.5504.657
Vainchtein, I. D., Chin, G., Cho, F. S., Kelley, K. W., Miller, J. G., Chien, E. C., et al. (2018). Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273. doi: 10.1126/science.aal3589
Wakabayashi, K., Hayashi, S., Yoshimoto, M., Kudo, H., and Takahashi, H. (2000). NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 99, 14–20. doi: 10.1007/pl00007400
Wyss-Coray, T., Loike, J. D., Brionne, T. C., Lu, E., Anankov, R., Yan, F., et al. (2003). Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 9, 453–457. doi: 10.1038/nm838
Yang, J., Yang, H., Liu, Y., Li, X., Qin, L., Lou, H., et al. (2016). Astrocytes contribute to synapse elimination via type 2 inositol 1,4,5-trisphosphate receptor-dependent release of ATP. Elife 5:e15043. doi: 10.7554/eLife.15043
Yun, S. P., Kam, T. I., Panicker, N., Kim, S., Oh, Y., Park, J. S., et al. (2018). Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 24, 931–938. doi: 10.1038/s41591-018-0051-5
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zhan, Y., Paolicelli, R. C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., et al. (2014). Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406. doi: 10.1038/nn.3641
Zhao, Y., Wu, X., Li, X., Jiang, L. L., Gui, X., Liu, Y., et al. (2018). TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023.e7–1031.e7. doi: 10.1016/j.neuron.2018.01.031
Zumkehr, J., Rodriguez-Ortiz, C. J., Cheng, D., Kieu, Z., Wai, T., Hawkins, C., et al. (2015). Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol. Aging 36, 2260–2271. doi: 10.1016/j.neurobiolaging.2015.04.005
Keywords: astrocytes, microglia, synapse loss, Alzheimer’s disease, neurodegenerative diseases
Citation: Lee E and Chung W-S (2019) Glial Control of Synapse Number in Healthy and Diseased Brain. Front. Cell. Neurosci. 13:42. doi: 10.3389/fncel.2019.00042
Received: 23 November 2018; Accepted: 25 January 2019;
Published: 13 February 2019.
Edited by:
Marie-Eve Tremblay, Laval University, CanadaReviewed by:
Schuichi Koizumi, University of Yamanashi, JapanAnnalisa Buffo, University of Turin, Italy
Copyright © 2019 Lee and Chung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Won-Suk Chung, wonsuk.chung@kaist.ac.kr