 Sanae Ben Mkaddem
Sanae Ben Mkaddem Marc Benhamou
Marc Benhamou Renato C. Monteiro
Renato C. Monteiro
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 April 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00811
This article is part of the Research Topic Roles of Fc Receptors in Disease and Therapy View all 25 articles
Fc receptors (FcRs) belong to the ITAM-associated receptor family. FcRs control the humoral and innate immunity which are essential for appropriate responses to infections and prevention of chronic inflammation or auto-immune diseases. Following their crosslinking by immune complexes, FcRs play various roles such as modulation of the immune response by released cytokines or of phagocytosis. Here, we review FcR involvement in pathologies leading notably to altered intracellular signaling with functionally relevant consequences to the host, and targeting of Fc receptors as therapeutic approaches. Special emphasis will be given to some FcRs, such as the FcαRI, the FcγRIIA and the FcγRIIIA, which behave like the ancient god Janus depending on the ITAM motif to inhibit or activate immune responses depending on their targeting by monomeric/dimeric immunoglobulins or by immune complexes. This ITAM duality has been recently defined as inhibitory or activating ITAM (ITAMi or ITAMa) which are controlled by Src family kinases. Involvement of various ITAM-bearing FcRs observed during infectious or autoimmune diseases is associated with allelic variants, changes in ligand binding ability responsible for host defense perturbation. During auto-immune diseases such as rheumatoid arthritis, lupus or immune thrombocytopenia, the autoantibodies and immune complexes lead to inflammation through FcR aggregation. We will discuss the role of FcRs in autoimmune diseases, and focus on novel approaches to target FcRs for resolution of antibody-mediated autoimmunity. We will finally also discuss the down-regulation of FcR functionality as a therapeutic approach for autoimmune diseases.
Immunoglobulin Fc receptors (FcRs) are membrane molecules expressed by several hematopoietic cells that recognize the Fc region of several immunoglobulin (Ig) classes and subclasses. We distinguish FcR for IgG (FcγRI/CD64, FcγRII/CD32, and FcγRIII/CD16), IgE (FcϵRI), IgA (FcαRI/CD89), IgM (FcμR), and IgA/IgM (Fcα/μR). Several other receptors expressed on different cell types also bind Ig molecules: neonatal FcR for IgG (FcRn) on intestinal epithelium, placenta, and endothelium, low affinity FcϵR (FcϵRII/CD23) on B cells and macrophages, and polymeric Ig receptor (pIgR) on mucosal epithelium (1–3).
The function of antibodies depends on one hand on their ability to recognize antigenic epitopes and, on the other hand, on their dynamic flexibility and their capacity to interact with their cognate FcRs. Engagement of FcRs expressed by leukocytes initiates a number of pro-inflammatory, anti-inflammatory, and immune modulatory functions in the host adaptive immune responses leading to protection but sometimes also to disease.
Several FcRs require the Immunoreceptor Tyrosine-based Activation Motif (ITAM; with the sequence Yxx[L/I]x(6−8)Yxx[L/I]) present in the cytoplasmic tail of the receptor or of associated subunits (FcRγ or FcεRIβ chain) to induce cell signaling. ITAM-mediated functions include phagocytosis, degranulation, antibody-dependent cellular cytotoxicity (ADCC), cytokine, lipid mediator and superoxide production, all of which depend on the cell type and on outside-in signals induced by the ligand. Engagement of the type I FcRs by immune complexes, induces receptor aggregation followed by activation and recruitment of Src family kinases (SFKs), such as Lyn and Fyn (4). The former induces the phosphorylation of the conserved tyrosines in the ITAM motif, followed by activation and recruitment of the tyrosine kinase Syk. This process activates various proteins involved in cell response, such as Phospholipase C gamma 1 (PLCγ), Bruton's tyrosine kinase (Btk), guanine nucleotide exchange factor Vav and phosphoinositide 3-kinase (PI3K). Hydrolysis of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5) P2) by PLCγ generates inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) leading to calcium mobilization and protein kinase C (PKC) activation, respectively. Calcium influx and PKC activation promote cell responses such as degranulation and cytokine production. Vav plays also an important role in actin cytoskeleton remodeling to control phagocytosis and superoxide production by NADPH oxidase. PI3K catalyzes the phosphorylation of PtdIns(4,5)P2 into PtdIns(3,4,5)P3 in the plasma membrane. Pleckstrin homology domains contained in proteins such as PLCγ, GRB2-associated-binding protein 2 (Gab2), protein kinase B (PKB/Akt) and Btk, bind PtdIns(3,4,5)P3 thus recruiting them at the inner leaflet of the plasma membrane promoting their phosphorylation and activation (Figure 1, left).
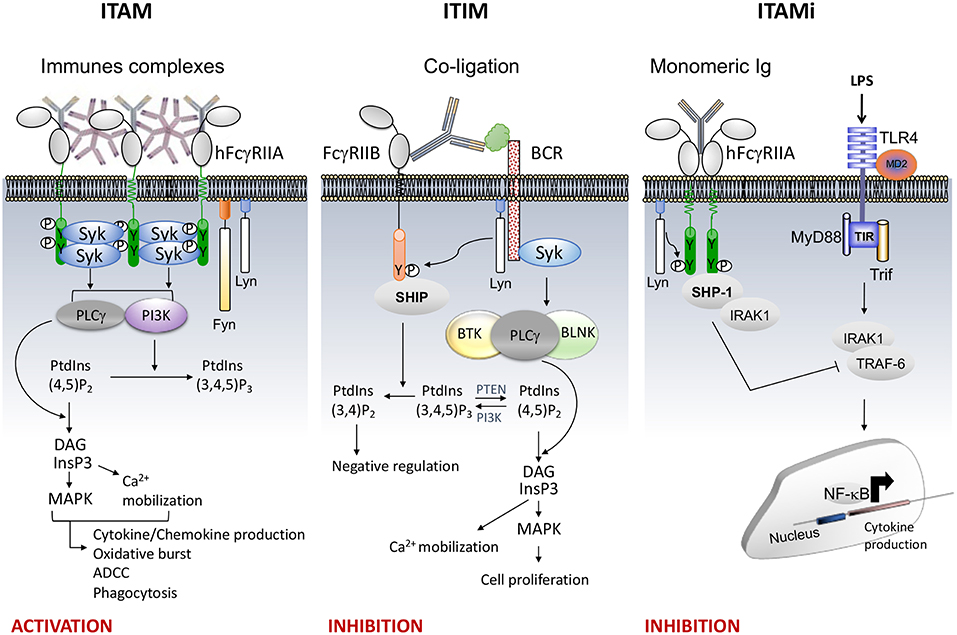
Figure 1. FcR signaling (e.g., FcγRII). (Left), the aggregation by an immune complex of FcR bearing ITAM motif (e.g. FcγRIIA) induces phosphorylation of the two ITAM tyrosine residues by Src kinases Lyn and Fyn responsible for recruitment and phosphorylation of Syk inducing cellular activation through PLCγ and PI3K signaling pathways. The PLCγ converts PI(4,5)P2 into IP3 and DAG. IP3, a soluble inositol phosphate, leads to Ca2+ mobilization while DAG activate MAPK.PI3K converts PI(4,5)P2 to PI(3,4,5)P3 allowing recruitment of signal intermediates through their pleckstrin homology (PH) domain (Middle), co-ligation between an activating heterologous receptor (e.g., the BCR) and the inhibitory FcR (i.e., FcγRIIB) induces phosphorylation of the tyrosine present within the ITIM motif by Lyn (5), leading to the phosphorylation and recruitment of phosphatases (SHIP or SHP). The phosphatases PTEN and SHIP1/2 regulate cellular levels of PI(3,4,5)P3 by hydrolyzing it to PI(4,5)P2 and PI(3,4)P2, respectively. These dephosphorylations inhibit cell proliferation. (Right), monovalent targeting of FcR bearing ITAM motif (e.g., FcγRIIA) induces the phosphorylation of the last tyrosine residue of the ITAM motif by Lyn responsible for transient recruitment of Syk followed by that of SHP-1 which abrogates the activation signal.
The activation of ITAM-bearing immune receptors can be retro-controlled by ITIM-bearing inhibitory FcRs such as the FcγRIIB. The ITIM motif is defined by a single [I/V/L/S]xYxx[L/V] sequence. However, inhibition of cell activation by this motif requires co-ligation between the inhibitory and heterologous activating receptors by immune complexes promoting the recruitment of inositol phosphatases (SHIP-1 and SHIP-2) (6) (Figure 1, Middle). Another inhibitory mechanism has been recently identified that involves ITAM itself. Indeed, following low avidity ligand interactions, ITAM-bearing FcRs induce a sustained inhibitory signal without co-ligation with heterologous receptors. This mechanism was involved in the maintenance of immune homeostasis (7–14). We named this ITAM-mediated inhibitory signal, ITAMi. It has been shown that several low affinity receptors, such as FcαRI, FcγRIIA and FcγRIIIA, can function as such bi-functional receptors to induce either activating or inhibitory signals, a property that can be exploited to reduce the susceptibility to autoimmune and inflammatory diseases (11). Monovalent or divalent targeting of FcRs bearing an ITAM motif induced ITAMi signals that involved activation and recruitment of the Src homology region 2 domain-containing tyrosine phosphatase SHP-1 (Figure 1, Right). It has been demonstrated that other immunoreceptors such as the antigen receptors BCR and TCR can also associate with SHP-1 upon interaction with low avidity ligands (15, 16). Moreover, SHP-1 deficiency in hematopoietic cells favors development of various auto-immunes diseases. For example, the motheaten mice (mev/mev) which express approximately 20% wild type activity of SHP-1, develop severe immune dysregulation and autoantibody production (17).
During ITAMi signaling induced by FcRs, Lyn is essential for the phosphorylation (on tyrosine residue 536) and the activation of SHP-1 (4). It has been reported that Lyn is involved in positive and negative signals induced by antigen receptors (18, 19). Lyn plays an important role in the negative selection of B cells in the bone marrow, since the absence of Lyn was associated with a decreased B cell number in the periphery of mice. In the absence of Lyn, other SFKs, such as Fyn, act as positive regulators of BCR signaling, suggesting a loss of anergy. The opposite roles of Lyn and Fyn were recently demonstrated by in vivo approaches. Lyn deficiency aggravates auto/inflammatory diseases such as nephritis and arthritis, while the absence of Fyn protects against these diseases (4). Additionally, we showed that activation of leukocytes in lupus nephritis patients was associated with Fyn-activated signature, suggesting that the balance between Lyn and Fyn is dysregulated during diseases.
Another FcR that play an essential role in the transcytosis by epithelial cells of dimeric IgA, but also pentameric IgM (notably during IgA deficiencies), is called the polymeric immunoglobulin receptor (pIgR). The pIgR is internalized with its ligands by endocytosis and transcytosed from the basolateral membrane into apical side of the epithelial cell (20). The central role of this receptor is to generate secretory IgA (formed of IgA dimers linked to the extracellular domain of the pIgR, also known as secretory component) in exocrine secretions to establish host-microbiota symbiosis and to mediate the protection of mucosal surfaces against pathogens (20, 21). The Fcα/μR, the Fc receptor for IgA and IgM, may play a role in systemic and mucosal immunity. It has been shown that none of the B cells, T cells, monocyte/macrophages, or NK cells in human blood samples expressed this receptor irrespective of age, ethnic origin or gender. Its expression is restricted to B cells from germinal center, follicular dendritic cells and tonsillar cells. Although, the exact function of the Fcα/μR is not fully clarified, it may play an important role in antigen presentation and B cell selection in the germinal center responses (22).
FcRs are divided into type I and type II on the basis of the conformational state of the Ig Fc domain that interacts with the receptor (1, 23). Type I Fc receptors interact with “open,” but not “closed” Ig Fc conformation (Figure 2). These receptors include FcγRI, FcγRII, FcγRIII, FcεRI, FcαRI, FcμR, and Fcα/μR (25– 29). In contrast, type II FcRs, bind preferentially Ig Fc domains in “closed” conformation. Among these are C-type lectin receptors such as FcεRII (CD23) and DC-SIGN (Figure 3).
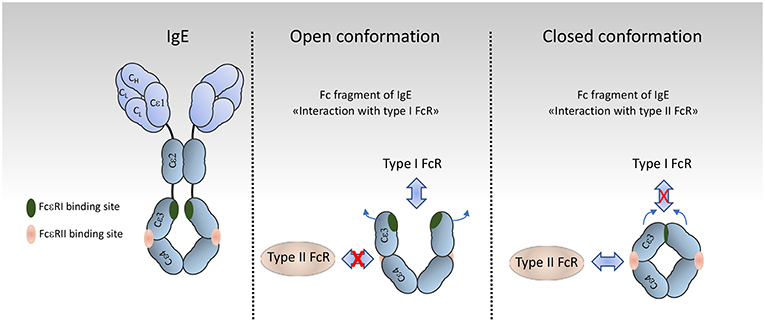
Figure 2. Organization and conformational rearrangements of the IgE Fc. (left), IgE and the binding sites to FcεRI (green) and to CD23 (pink) [adapted from Pennington et al. (24)] (Middle and Right). Representation of the open and closed conformations, respectively, of the IgE Fc Cε3–4 domains, and the mutual allosteric inhibition by FcεRIα (green) and CD23 (pink).
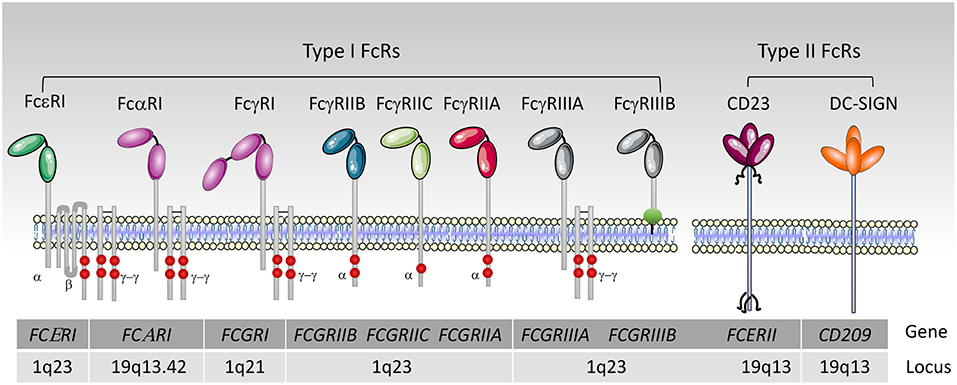
Figure 3. Human type I and II Fc receptors. Schematic representation of human Fc receptors at the cell membrane and their association or not with the FcRγ-chain dimer and the FcεRIβ chain, red circles represent the tyrosine residues. The FcγRIIIB is anchored into the outer leaflet of the plasma membrane by a phosphatidylinositol-glycan (green circle).
For type II Fc receptors, glycosylation of the Fc domain induces a conformational change that occludes the binding site for type I Fc receptors that lies near the hinge region (open conformation) and reveals a binding site at the CH2-CH3 domain interface (closed conformation). These receptors bind antibodies in a two receptors-to-one antibody stoichiometry that may influence signal initiation (1). DC-SIGN and SIGN-R1, for example, bind secretory IgA and play an intriguing role in dendritic cells inducing IL10 and Treg-mediated tolerance (30). Signaling through these receptors, however, is not yet documented as compared to type I FcRs, with the exception of CD23. Crosslinking of CD23 on B cells activates cAMP (31) and intracellular calcium flux (32) which is associated with the activation of the SFK Fyn and of the PI3K pathway (33). These findings are in agreement with our recent data on type I FcRs (4), and indicates that Fyn also plays an activating role in B cells through type II Fc receptors.
Several single-nucleotide polymorphisms (SNPs) have been reported in the genes encoding activating FcγRs (FcγRIIA, FcγRIIIA, and FcγRIIIB). In the gene encoding the inhibitory FcγRIIB, a SNP has been described which is associated with autoimmune diseases such as SLE and rheumatoid arthritis (RA) (34, 35). In addition to SNPs, copy-number variations (CNVs) of FcγR genes are associated with susceptibility to autoimmune disorders (34–40). Most polymorphisms concern the extracellular domains which bind to IgG, affecting the affinity between these receptors and IgG subclasses. However, no polymorphism and CNV have been clearly identified for FcγRI.
The most studied polymorphism is the one in the second Ig-like extracellular domain of the FcγRIIA that results in a point mutation of amino acid at position 131, coding for either arginine (R131) or histidine (H131). FcγRIIA-R131 binds less efficiently IgG2 than FcγRIIA-H131 (34). This Fcgr2a polymorphism has been described as a heritable risk factor for autoimmune and infectious diseases (41, 42). Moreover, genome-wide association studies (GWAS) revealed that FcγRIIA-H131 variant is associated with higher susceptibilities to develop Kawasaki disease and ulcerative colitis (43, 44). This variant is also associated with Guillain–Barré syndrome (45), supporting that immune complexes that include IgG2 auto-antibodies are involved in inflammatory responses. In contrast, the genotype homozygous for the FcγRIIA-R131 variant-encoding gene is associated with SLE, immune thrombocytopenic purpura (ITP) and IgA nephropathy (IgAN), revealing a complex and contrasted picture for the role of IgG2-containing immune complexes in autoimmune diseases (46–48). Regarding infectious diseases, neutrophils homozygous for the gene encoding the H131 variant show a higher capacity for IgG2-mediated phagocytosis than those homozygous for the gene encoding the R131 variant (41). In agreement, patients with the R encoding allele were found to have more severe cases of Severe Acute Respiratory Syndrome infection and were more susceptible to encapsulated microorganism infection, which was attributed to poor IgG2 binding to the R131 variant of FcγRIIA (41, 42). As mice do not express FcγRIIA homologs, transgenic mice expressing the Fcgr2a human gene encoding the R131 variant develop spontaneously autoimmune diseases such as pneumonitis, glomerulonephritis and RA (49, 50). Moreover, the fact that FcγRIIA-R131 expressed on the FcRγ-/- background in mice similarly develop thrombocytopenia (51) and arthritis (11, 52) indicates that pro-autoimmune signals through FcγRIIA-R131 ITAM were sufficient to induce diseases. Biochemical analyses showed that two tyrosines of FcγRIIA ITAM motif were needed to induce inflammatory signals (53). Taken together, these animal models underline the critical involvement of Fcgr2a polymorphism in a number of diseases.
A polymorphism has been found in the inhibitory FcγRIIB-encoding gene that leads to a single I-to-T amino acid substitution in the transmembrane domain (residue 232) (43, 54). Human monocytes expressing the FcγRIIB-T232 failed to inhibit heterologous receptors-mediated cell activation (55). The FcγRIIB-T232 polymorphism is associated with susceptibility to develop auto-immune diseases such as SLE (42, 54, 56). However, there are some discordances concerning the SNPs in the promoter region of Fcgr2b, such as −386G and −120T (haplotype 2B.2), and −386C and −120A (haplotype 2B.4) variants. It has been shown that the 2B.4 SNP promoter haplotype upregulates the expression of FcγRIIB on neutrophils and monocytes that negatively correlates with lupus nephritis (46). This is in agreement with previous reports in mouse SLE-like models and suggests that FcγRIIB expression is protective in SLE (57). However, in striking contrast and in an apparent paradox, the same 2B.4 promoter haplotype was found by the same authors to correlate with SLE (46). This positive (SLE) vs. negative (lupus nephritis) paradoxical association of the 2B.4 promoter haplotype suggests multifaceted impacts of FcγRIIB in SLE that may depend on the affected cell types (e.g., monocytes vs. neutrophils). Alternatively, particular cell types expressing FcγRIIB could have aggravating or protective actions in SLE depending on which affected tissue these cells are recruited to, or on how these cells impact the systemic vs. local aspect of the disease. Thus, further investigation is necessary to elucidate the association of the promoter haplotype in disease development.
The Fcgr3a polymorphism is characterized by a point mutation in the codon for residue 158, encoding valine (V158) or phenylalanine (F158) in the Ig-like domain near the membrane (34–36). The FcγRIIIA-V158 variant has a higher affinity for all human IgGs than the FcγRIIIA-F158 variant (40). The FcγRIIIA-F158 is associated with susceptibility to SLE, Crohn's disease and Behçet's disease (35, 36). Although studies have also explored the association between RA and the V or F 158 variant, their results have been contradictory and this question remains unsettled (58, 59).
Several Fcar1 polymorphisms have been found, including two in the functional promoter region of the FcαRI encoding gene (−114T/C and +56T/C relative to the major transcription start site) (60). The incidence of the −114C/C polymorphism in patients with IgAN was significantly increased compared with other chronic kidney diseases (CKD) and healthy donors (HD) (15.6 vs. 4.0% in other CKD and vs. 2.4% in HD). This Fcar1 polymorphism in the promoter region appears to be associated with susceptibility to IgAN, suggesting the importance of FcαRI expression in this disease. A third Fcar1 polymorphism has been described in the coding region for FcαRI, which changes codon 248 from AGC to GGC leading to G248 instead of S248 in the cytoplasmic domain of the receptor (61). Interestingly, these two different alleles demonstrate significantly different FcαRI-mediated intracellular activating signaling. The proinflammatory FcαRI-G248 variant has been associated with SLE in two ethnic groups (61). However, this Fcar1 polymorphism was not associated with other auto-immune diseases such as systemic sclerosis, RA or IgAN (62, 63). A fourth Fcar1 polymorphism (A/G at nt 324) was also associated with aggressive periodontitis (64). Patients displaying the nt 324 A/A allele presented polymorphonuclear neutrophil dysfunctions with a decreased phagocytosis of periodontopathic bacteria (Porphyromonas gingivalis) as compared to patients expressing the nt 324 G/G (64).
Regarding the pIgR, it has been reported as a susceptibility gene for nasopharyngeal cancer (NPC) associated with Epstein-Barr virus (EBV) (65). This lead to a hypothesis that pIgR could be the nasopharyngeal epithelial receptor for EBV via IgA-EBV complex. Transcytosis failure due to missense C → T mutation on the PIGR1739 nucleotide (resulting in an A-to-V mutation near the endoproteolytic cleavage site of pIgR) could decrease the ability of pIgR to release IgA-EBV complexes, thus increasing susceptibility to develop NPC (65).
The high-affinity FcεRI is expressed by mast cells and basophils and plays an important role in allergic diseases. Several studies have identified two FcεRI polymorphisms associated with allergies. The −66T > C and/or the −315C > T are associated with atopic dermatitis, chronic urticaria, asthma, and high serum IgE levels (66–69). These polymorphisms were also associated with allergic inflammatory diseases such as atopy and nasal allergy (70, 71).
Table 1 summarizes most of described FcR alleles and their expression and functions in physiology and pathology (42–44, 46, 48, 60, 65, 66, 69, 72–83).
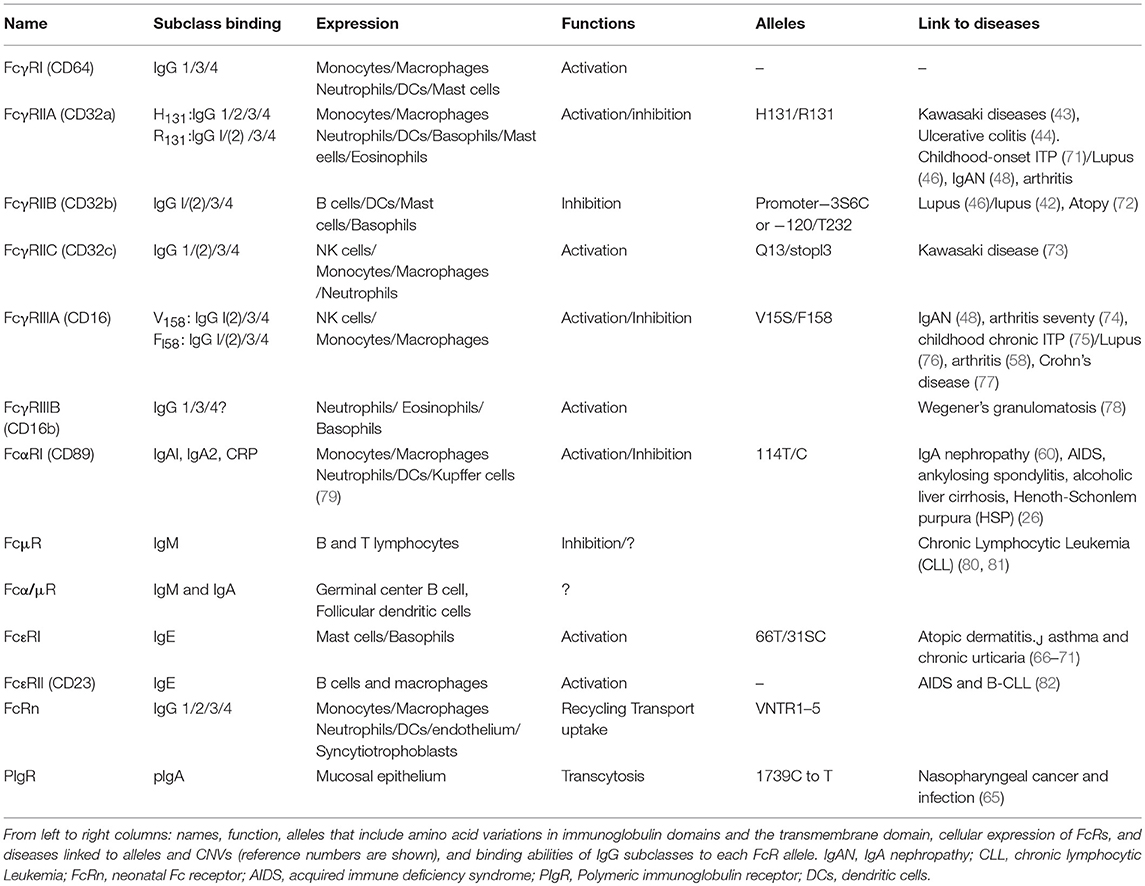
Table 1. Human FcRs: their expression, function and allotypes.
Several studies highlighted altered expression, structure and function of FcγRs in patients. Whereas, CD4+ T cells from healthy donors fail to express significant levels of FcγR, FcγRIIA is expressed in a subpopulation of CD4+ T cells in blood samples from HIV-1-positive patients and is highly enriched in inducible replication-competent proviruses suggestive of an FcγRIIA+ HIV reservoir (84). Yet, in other studies, FcγRIIA expression did not selectively enrich for HIV- or SIV-infected CD4+ T cells in peripheral blood or lymphoid tissue since resting FcγRIIA+ CD4+ T cells have <3% of the total HIV DNA amongst CD4+ T cells (85, 86). Taken together, whereas FcγRIIA expression in CD4+ T cells becomes a marker for HIV infection, the involvement of FcγRIIA+ CD4+ T cells in AIDS remains to be elucidated.
The inhibitory FcγRIIB, in contrast, is down-regulated in autoimmune diseases notably on both memory and plasma B lymphocytes of active SLE patients compared to those from healthy individuals (87). However, this down-regulation was not seen on myeloid-lineage cells. This was also observed in Hashimoto's thyroiditis (88).
High level of FcγRIIIA expression on monocytes together with that of CD14 is associated with proinflammatory cytokine profiles and higher potency in antigen presentation allowing to define monocyte subsets with distinct phenotypes and functions (89).
For FcαRI, its expression is dysregulated in patients with AIDS, ankylosing spondylitis, alcoholic liver cirrhosis, Henoch-Schonlein purpura (HSP) and IgAN (90–93). Some of these studies have shown biochemical abnormalities revealing FcαRI altered protein mobility in SDS-PAGE suggesting altered glycosylation of this receptor (90). Interestingly, mutational studies of FcαRI indicate that the N58 residue of the receptor controls IgA-binding enhancement (94). In parallel, abnormally glycosylated IgA1 molecules (hypogalactosylation and hyposialylation on the hinge region) observed in patients with IgAN and HSP is associated with the shedding of a soluble form of FcαRI (sFcαRI), which participates in the formation of circulating IgA1 complexes (95). These IgA1-sFcαRI complexes were decreased in serum of IgAN patients with severe and progressive disease as compared to non-progressive IgAN patients (96) suggesting a kidney deposition, and hence a possible nephrotoxic action, of such complexes which is further supported by studies in IgAN patients with recurrence of the disease after kidney transplantation (97). In this study, IgA-sFcαRI complexes were decreased in the serum of patients with recurrent IgAN and sFcαRI was detected in the kidney mesangium only in patients with the recurrent disease. Direct evidence for a nephrotoxic role of IgA1-sFcαRI complexes were obtained in humanized animal models. These experimental studies were based on the fact that mouse do not have homologs of IgA1 and FcαRI. Humanized mice expressing human IgA1 and human FcαRI spontaneously develop mesangial deposits of IgA1-sFcαRI complexes (98). In the glomeruli, these complexes are captured by the transferrin receptor 1 (TfR1), which is upregulated on mesangial cells, through interaction with polymeric (p) IgA1 and FcαRI (98, 99). Although the mechanism of TfR1 upregulation remains poorly understood, the crosslinking enzyme transglutaminase 2 has been found to be overexpressed and associated with the receptor controlling mesangial IgA1 complex deposition and renal injury (98). Polymeric IgA1 induce TfR1 expression in vitro on mesangial cells. Polymeric IgA1-TfR1 interaction triggers activating signals through mTOR, PI3K and ERK pathways, and phosphorylated ERK is associated with disease progression (100). Interestingly enough, in physiology TfR1-IgA1 interaction plays a role in erythropoiesis (101). Progression of IgAN to end-stage renal disease may also involve FcαRI activation on tissue macrophages surrounding hypogalactosylated IgA1-mediated mesangial lesions. Indeed, FcαRIR209LTg mice, with an R-to-L substitution at position 209 in the transmembrane region of FcαRI, did not develop macrophage infiltration and proteinuria (102). This mutant receptor cannot associate with the ITAM-bearing FcRγ signaling subunit (103). In agreement, only macrophages expressing wild-type FcαRI, but not those expressing FcαRIR209L, were able to migrate to the kidney after adoptive transfer demonstrating that their chemotaxis depends on the FcRγ subunit. Of note, FcαRI can be found associated and non-associated with FcRγ on the same cells (95). Since mouse IgA and human FcαRI interaction may be sufficient to induce receptor shedding leading to IgA deposits in the kidney, we hypothesized that both receptor types could cooperate to induce disease, the FcRγ-less FcαRI allowing IgA deposits and the FcRγ-associated FcαRI promoting inflammatory cell infiltration and disease progression (102).
For FcμR (IgM receptor), deficiency in the receptor in mice revealed that this receptor plays a crucial role in B cell responses (27, 104). Mice deficient for Fcmr are characterized by the increase in pre-immune serum IgM, dysregulation of humoral immune responses, disturbances in B cell subpopulations, B cell proliferation alteration after BCR ligation, and autoantibody production (104, 105). Accordingly, in chronic lymphocytic leukemia (CLL), the membrane expression and the soluble form of FcμR in serum were increased. The potential mechanism proposed for the up-regulation of FcμR is that the antigen-independent self-ligation of BCR on CLL cells induces activation of Syk thus increasing the cell surface expression of FcμR. Furthermore, the IgM antibodies produced by CLL cells that had differentiated into plasma cells, recognized soluble or lymphocyte membrane self-antigens. IgM/self-antigen immune complexes would then crosslink FcμR and BCR favoring cell survival. An alternative splice variant of the soluble FcμR is increased in CLL patients, but its biological function is unclear (105, 106).
For type II FcRs, an increased expression of FcεRII on monocytes in AIDS patients has been associated with the aberrant activated phenotype of these cells during the immunopathogenesis of AIDS. Interestingly, despite the known ability of IL-10 to downregulate monocyte FcεRII expression, in AIDS the IL-10-enriched environment is not associated with the suppression of FcεRII expression on monocytes (82) indicative of an impairment of this negative regulation in patients. In B-CLL also, patients strongly express FcεRII, which is associated with B cell activation and proliferation. Moreover, altered phosphorylation of FcεRII intracellular tail were reported in B-CLL B lymphocytes (107) further supporting an active role of FcεRII in this disease.
Both murine models and studies in patients suggest a major role of the activating FcRs in initiating and propelling immune complex-mediated inflammatory reactions. For example, human FcγRIIA transgenic mice are hypersensitive to pathogenic antibodies and develop destructive arthritic syndromes. Ex vivo experimentation with circulating monocytes from RA patients suggest that FcγRIIA is responsible for the production of reactive oxygen species (11, 108). Anti-receptor monoclonal antibodies, intact antibodies and antibody fragments as well as a variety of small molecules have been designed to interact with the Ig-binding domains in activating FcRs. Some of these approaches have shown encouraging results when tested in vitro or in vivo for blocking immune complex-mediated cell effects and inflammation. Recently, we have demonstrated that divalent targeting of FcγRIIA by anti-hFcγRII F(ab)′2 fragments ameliorates RA-associated inflammation. This therapeutic effect was mediated by the induction of inhibitory ITAM (ITAMi) signaling through the activation of SHP-1. Moreover, treatment of inflammatory synovial cells from RA patients by F(ab′)2 fragment of hFcγRIIA-specific antibody inhibited production of reactive oxygen species associated with the induction of FcγRIIA-mediated ITAMi signaling. These data suggest that targeting of hFcγRIIA by specific antibody such as clone IV.3 mAb could ameliorate RA-associated inflammation (11). Anti-FcαRI Fab and F(ab)′2 fragments also have demonstrated efficiency on RA (109). Interestingly, in autoimmune blistering skin diseases that involve interaction between IgA autoantibodies and the neutrophil FcαRI, targeting FcαRI by blocking peptides or antibodies prevents neutrophil migration and tissue damage ex-vivo (110, 111).
In allergy, treatment by anti-IgE antibodies has been considered a therapeutic option for a long time. The recombinant anti-IgE humanized monoclonal antibody-E25, named “omalizumab,” is now used in several clinical trials and shows efficacy against IgE-mediated allergic reactions (112, 113) through inhibition of IgE binding to FcεRI on the surface of mast cells and basophils (113).
The above-described upregulation of FcμR expression in CLL cells is of significant clinical interest. It can be easily evaluated by flow cytometry on cells and, additionally, the levels of soluble FcμR may correlate with disease progression. Thus, it may be used as a new biomarker for CLL (106). FcμR is a good target also because it is involved in the pathogenesis of CLL and in the progression of the disease through support of leukemic cell survival (80). Hence, disrupting CLL survival signals might be achieved through FcμR therapeutic targeting. However, a large cohort of CLL patients will be required to validate these two applications (106).
Intravenous immunoglobulins (IVIG) are harvested from the pooled plasma of 3,000 to 100,000 healthy donors. They consist of over 95% IgGs with a subclass distribution corresponding to that found in normal human serum (114). IVIG is used in treatment of several immunodeficiency diseases including idiopathic thrombocytopenic purpura (ITP), Kawasaki disease, and neurologic diseases such as Guillain–Barre syndrome, chronic inflammatory demyelinating polyneuropathy, myasthenia gravis, sclerosis, and autoimmune encephalitis (115). In ITP patients, administration of IVIG can efficiently attenuate platelet clearance from the circulation. The first proposed mechanism was the competitive blockage of the activating FcγRs on myeloid cells by IVIG, which in turn decreases autoantibody-mediated platelet phagocytosis and ADCC against platelets (116). Furthermore, in pediatric ITP patients, intravenous administration of the Fcγ fragments prepared from IVIG resulted in a rapid recovery in platelet counts (117) further indicating the role of FcγRs in IVIG action. Another IVIG anti-inflammatory mechanism involves saturation of FcRn, the IgG recycling receptor (118). FcRn plays an important role in the maintenance of IgG half-life. Therefore, inhibition of autoantibody activity can be induced by the alteration of their interaction with FcRn, impairing their half-life and accelerating their clearing from the circulation. IVIG by competing with autoantibodies for FcRn binding could therefore facilitate their clearing.
A role for the inhibitory FcγRIIB has been proposed to be exclusive in IVIG action to explain their Fc dependent effect (118). This statement was based notably on studies showing a decreased anti-inflammatory effect of IVIG in FcγRIIB-deficient animals. In other studies, a role for FcγRIII in IVIG-mediated inhibition has been reported (119) although the mechanism of action was not clearly established. Recently, we reported that IVIG can control inflammatory responses by ITAMi signaling through FcγRIIA and FcγRIII (10, 11). These data are based on the in vitro targeting of FcγRIIA and FcγRIII by IVIG at the physiological concentration of IgG showing an inhibitory effect on endocytosis. This was confirmed by targeting FcγRIIA or FcγRIII with F(ab′)2 fragments of specific antibodies. These results were further supported in vivo in mice by targeting these receptors with IVIG or with specific antibodies and this inhibitory effect was abolished in receptor-deficient mice (10, 11). Therefore, IVIG could use a combination of non-exclusive mechanisms to promote protection against auto-immune diseases. Although IVIG is well tolerated, some patients develop immediate or delayed adverse effects depending on the time occurrence. The Flu-like symptoms such as fever, fatigue and nausea are the most frequent adverse effects. For the delayed adverse effects, the most frequent are thrombotic events, neurological disorders and renal failure. These delayed adverse effects are rare but dangerous (120). The majority of adverse effects are associated with high doses of immunoglobulins; thus, determining individual dosages to guarantee the efficacy of therapy and minimize adverse effects is an urgent goal.
Treatment with highly purified serum monomeric IgA (mIgA) decreases cell activation through FcαRI-FcRγ-mediated ITAMi signaling (109). Human mIgA or anti-FcαRI Fab fragments were used to prevent or treat collagen antibody-induced arthritis in FcαRI-transgenic mice. mIgA treatment decreased significantly leukocyte infiltration to the inflamed joints of mice, which was associated with SHP-1 phosphorylation at Y536 residue in joint tissue cells. Moreover, mIgA reversed the activating ITAM to ITAMi signature and the state of inflammation in the synovial fluid isolated from RA patients (109). Of note, protection was also achieved with human serum IgA (4). These findings open new avenues to develop the concept of IVIgA as a new treatment option for inflammatory and auto-immune diseases.
The only FcR containing an inhibitory ITIM motif, FcγRIIB, serves as a critical negative regulator in immune complex driven reactions. In mice lacking FcγRIIB auto-immune symptoms are exacerbated, and a partial restoration of FcγRIIB expression in B cells rescued mice from developing an SLE-like phenotype (57, 121). Several FcγRIIB specific mAbs have now been developed (122, 123), one of which, mAb2B6, has been chimerized and humanized to direct myeloid-cytotoxicity against B cells (123). These antibodies have the potential to serve as novel immune suppressors in auto-immunity either by blocking B cell activation or by targeting their destruction. In addition, they may have an advantage over CD20 antibodies for their ability to target plasma cells (124).
Blocking FcRn-IgG interaction to decrease circulating IgG levels is one strategy to treat auto-immune disease (118). In the absence of interaction with FcRn, IgG would be degraded in lysosomes more quickly instead of being recycled back into circulation. One straightforward method would be to use recombinant soluble human FcRn to compete with membrane FcRn for IgG. Another approach to block IgG-FcRn binding would be through engineered “bait” IgG which occupy FcRn thus preventing binding of endogenous IgG. Such “bait” antibodies have been generated with a much higher affinity for FcRn at both acidic and neutral pH, thereby providing effective occupancy of FcRn, competing with, and resulting in, degradation of endogenous IgG. These antibodies are also called “Abdegs”: antibodies that enhance IgG degradation (125).
An FcRn-specific blocking mAb would also provide interference with FcRn-IgG interaction. One such mAb, 1G3, was examined in rat passive and active models of myasthenia gravis, a prototypical antibody-mediated auto-immune disease (126). Treatment by 1G3 mAb resulted in amelioration of disease symptoms in a dose-dependent manner together with greatly reduced levels of pathogenic antibody in the serum.
Targeting of FcRs by monomeric immunoglobulins or by F(ab′)2 fragments of specific antibodies, induces ITAMi signaling which involved the recruitment of Lyn, but not Fyn. The Src kinase Lyn, leads to partial phosphorylation of the ITAM motif on tyrosine residues (11), and to the conformational change of SHP-1 that allows its recruitment through its SH2 domains to ITAM phosphotyrosine residues (127). This recruitment induces a Lyn-dependent phosphorylation of SHP-1 on Y536 and to SHP-1 phosphatase activity that inhibits the recruitment of various proteins induced by heterologous receptors (52). In contrast, multivalent crosslinking of immunoreceptors by immune complexes induces the recruitment of both Src kinases Lyn and Fyn to the receptor leading to full phosphorylation of the tyrosine residues present in the ITAM motif. This leads to the activation and the recruitment of the kinase Syk. In parallel, Fyn initiates a signaling pathway involving a PI3K-PKCα axis leading the inactivation of SHP-1 through the phosphorylation of its S591 residue barring its recruitment to the plasma membrane (128). Since S591 phosphorylation on SHP-1 keeps the phosphatase in a closed conformation (127), our recent study showed that the phosphorylation of SHP-1 on S591 residue by Fyn axis renders the Y536 residue inaccessible to Lyn. In agreement, the absence of Fyn favors the phosphorylation of SHP-1 on Y536 by Lyn, despite the crosslinking of FcγRIIA (4). These results suggest that the selective absence or inhibition of Fyn may abolish inflammation during auto-immune and proinflammatory diseases. Taken together, inhibition of Fyn or of the molecules which are upstream or downstream this SFK reverses inflammation during auto-immune and inflammatory diseases and thus, could be a new therapeutic strategy to decrease the activating ITAM signaling in these diseases. Along these lines, inhibition of PI3K (a major player of the Fyn-PI3K-PKCα axis (4)) prevents RA and lupus nephritis progression in mouse models (129). However, it should be mentioned that since Fyn is essential for activating ITAM signals (i.e., phagocytosis), the inhibition of this SFK may favor infections. Moreover, Fyn plays also other roles independently of FcRs. It has been shown that the absence of Fyn impaired multipolar-bipolar transition of newly generated neurons and neurite formation during the early phase of migration. Additionally, inhibition of Fyn decreased the branching number of the migrating cortical neurons (130). Another important hurdle is that Lyn and Fyn present a high homology and there are currently no selective inhibitory drugs. Therefore, Fyn does not appear as the best target to treat auto-immune and pro-inflammatory diseases. Identification of new targets which are downstream of Fyn and which are expressed specifically by immune cells involved in auto-immune diseases will permit development of new therapeutic strategies for auto-immune diseases that involve FcR.
Fc receptors may be responsible for diseases when dysregulated in spite of their physiologic protective function. Unraveling all aspects (expression, function, regulation) of FcR biology should help to define approaches to correct the first and to wield the second to restore homeostasis thus representing new hopes for innovative anti-inflammatory strategies. Progress in these two aspects is currently well underway, already proposing new potent therapeutic tools. The future in this field is a promise of scientific excitement.
SB and RM wrote this review. MB has critically read the manuscript.
This work was funded by Inserm, by the Agence Nationale de la Recherche (ANR grants JC -17-CE17-0002-01, ANR PRC -18-CE14-0002-01), and also by LabEx Inflamex (ANR-11-IDEX-0005-02). RM was supported by ≪ Equipe ≫ program of the Fondation pour la recherche médicale (FRM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. (2014) 15:707–16. doi: 10.1038/ni.2939
2. Bruhns P, Jonsson F. Mouse and human FcR effector functions. Immunol Rev. (2015) 268:25–51. doi: 10.1111/imr.12350
3. Kubagawa H, Kubagawa Y, Jones D, Nasti TH, Walter MR, Honjo K. The old but new IgM Fc receptor (FcmuR). Curr Top Microbiol Immunol. (2014) 382:3–28. doi: 10.1007/978-3-319-07911-0_1
4. Mkaddem SB, Murua A, Flament H, Titeca-Beauport D, Bounaix C, Danelli L, et al. Lyn and Fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat Commun. (2017) 8:246. doi: 10.1038/s41467-017-00294-0
5. Malbec O, Fong DC, Turner M, Tybulewicz VL, Cambier JC, Fridman WH, et al. Fc epsilon receptor I-associated lyn-dependent phosphorylation of Fc gamma receptor IIB during negative regulation of mast cell activation. J Immunol. (1998) 160:1647–58.
6. Bolland S, Ravetch JV. Inhibitory pathways triggered by ITIM-containing receptors. Adv Immunol. (1999) 72:149–77. doi: 10.1016/S0065-2776(08)60019-X
7. Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, et al. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. (2005) 22:31–42. doi: 10.1016/j.immuni.2004.11.017
8. Pinheiro da Silva F, Aloulou M, Skurnik D, Benhamou M, Andremont A, Velasco IT, et al. CD16 promotes Escherichia coli sepsis through an FcR gamma inhibitory pathway that prevents phagocytosis and facilitates inflammation. Nat Med. (2007) 13:1368–74. doi: 10.1038/nm1665
9. Kanamaru Y, Pfirsch S, Aloulou M, Vrtovsnik F, Essig M, Loirat C, et al. Inhibitory ITAM signaling by Fc alpha RI-FcR gamma chain controls multiple activating responses and prevents renal inflammation. J Immunol. (2008) 180:2669–78. doi: 10.4049/jimmunol.180.4.2669
10. Aloulou M, Ben Mkaddem S, Biarnes-Pelicot M, Boussetta T, Souchet H, Rossato E, et al. IgG1 and IVIg induce inhibitory ITAM signaling through FcgammaRIII controlling inflammatory responses. Blood. (2012) 119:3084–96. doi: 10.1182/blood-2011-08-376046
11. Ben Mkaddem S, Hayem G, Jonsson F, Rossato E, Boedec E, Boussetta T, et al. Shifting FcgammaRIIA-ITAM from activation to inhibitory configuration ameliorates arthritis. J Clin Invest. (2014) 124:3945–59. doi: 10.1172/JCI74572
12. Iborra S, Martinez-Lopez M, Cueto FJ, Conde-Garrosa R, Del Fresno C, Izquierdo HM, et al. Leishmania uses mincle to target an inhibitory ITAM signaling pathway in dendritic cells that dampens adaptive immunity to infection. Immunity. (2016) 45:788–801. doi: 10.1016/j.immuni.2016.09.012
13. Getahun A, Cambier JC. Of ITIMs, ITAMs, and ITAMis: revisiting immunoglobulin Fc receptor signaling. Immunol Rev. (2015) 268:66–73. doi: 10.1111/imr.12336
14. Blank U, Launay P, Benhamou M, Monteiro RC. Inhibitory ITAMs as novel regulators of immunity. Immunol Rev. (2009) 232:59–71. doi: 10.1111/j.1600-065X.2009.00832.x
15. Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. (2003) 4:248–54. doi: 10.1038/ni895
16. Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC. Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. J Exp Med. (2016) 213:751–69. doi: 10.1084/jem.20150537
17. Abram CL, Roberge GL, Pao LI, Neel BG, Lowell CA. Distinct roles for neutrophils and dendritic cells in inflammation and autoimmunity in motheaten mice. Immunity. (2013) 38:489–501. doi: 10.1016/j.immuni.2013.02.018
18. Chan VW, Lowell CA, DeFranco AL. Defective negative regulation of antigen receptor signaling in Lyn-deficient B lymphocytes. Curr Biol. (1998) 8:545–53. doi: 10.1016/S0960-9822(98)70223-4
19. Lowell CA. Src-family kinases: rheostats of immune cell signaling. Mol Immunol. (2004) 41:631–43. doi: 10.1016/j.molimm.2004.04.010
20. Apodaca G, Bomsel M, Arden J, Breitfeld PP, Tang K, Mostov KE. The polymeric immunoglobulin receptor. a model protein to study transcytosis. J Clin Invest. (1991) 87:1877–82. doi: 10.1172/JCI115211
21. Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, et al. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science. (2018) 360:795–800. doi: 10.1126/science.aaq0926
22. Kikuno K, Kang DW, Tahara K, Torii I, Kubagawa HM, Ho KJ, et al. Unusual biochemical features and follicular dendritic cell expression of human Fcalpha/mu receptor. Eur J Immunol. (2007) 37:3540–50. doi: 10.1002/eji.200737655
23. Ahmed AA, Giddens J, Pincetic A, Lomino JV, Ravetch JV, Wang LX, et al. Structural characterization of anti-inflammatory immunoglobulin G Fc proteins. J Mol Biol. (2014) 426:3166–3179. doi: 10.1016/j.jmb.2014.07.006
24. Pennington LF, Tarchevskaya S, Brigger D, Sathiyamoorthy K, Graham MT, Nadeau KC, et al. Structural basis of omalizumab therapy and omalizumab-mediated IgE exchange. Nat Commun. (2016) 7:11610. doi: 10.1038/ncomms11610
25. Hogarth PM. Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr Opin Immunol. (2002) 14:798–802. doi: 10.1016/S0952-7915(02)00409-0
26. Monteiro RC, Van De Winkel JG. IgA Fc receptors. Annu Rev Immunol. (2003) 21:177–204. doi: 10.1146/annurev.immunol.21.120601.141011
27. Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, et al. Identity of the elusive IgM Fc receptor (FcmuR) in humans. J Exp Med. (2009) 206:2779–93. doi: 10.1084/jem.20091107
28. Shibuya A, Sakamoto N, Shimizu Y, Shibuya K, Osawa M, Hiroyama T, et al. Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat Immunol. (2000) 1:441–6. doi: 10.1038/80886
29. Daeron M. Fc receptors as adaptive immunoreceptors. Curr Top Microbiol Immunol. (2014) 382:131–64. doi: 10.1007/978-3-319-07911-0_7
30. Diana J, Moura IC, Vaugier C, Gestin A, Tissandie E, Beaudoin L, et al. Secretory IgA induces tolerogenic dendritic cells through SIGNR1 dampening autoimmunity in mice. J Immunol. (2013) 191:2335–43. doi: 10.4049/jimmunol.1300864
31. Paul-Eugene N, Kolb JP, Abadie A, Gordon J, Delespesse G, Sarfati M, et al. Ligation of CD23 triggers cAMP generation and release of inflammatory mediators in human monocytes. J Immunol. (1992) 149:3066–71.
32. Kolb JP, Abadie A, Paul-Eugene N, Capron M, Sarfati M, Dugas B, et al. Ligation of CD23 triggers cyclic AMP generation in human B lymphocytes. J Immunol. (1993) 150:4798–809.
33. Chan MA, Gigliotti NM, Matangkasombut P, Gauld SB, Cambier JC, Rosenwasser LJ. CD23-mediated cell signaling in human B cells differs from signaling in cells of the monocytic lineage. Clin Immunol. (2010) 137:330–6. doi: 10.1016/j.clim.2010.08.005
34. Hargreaves CE, Rose-Zerilli MJ, Machado LR, Iriyama C, Hollox EJ, Cragg MS, et al. Fcgamma receptors: genetic variation, function, and disease. Immunol Rev. (2015) 268:6–24. doi: 10.1111/imr.12341
35. Gillis C, Gouel-Cheron A, Jonsson F, Bruhns P. Contribution of human fcgammars to disease with evidence from human polymorphisms and transgenic animal studies. Front Immunol. (2014) 5:254. doi: 10.3389/fimmu.2014.00254
36. Li X, Gibson AW, Kimberly RP. Human FcR polymorphism and disease. Curr Top Microbiol Immunol. (2014) 382:275–302 doi: 10.1007/978-3-319-07911-0_13
37. Zhou XJ, Lv JC, Bu DF, Yu L, Yang YR, Zhao J, et al. Copy number variation of FCGR3A rather than FCGR3B and FCGR2B is associated with susceptibility to anti-GBM disease. Int Immunol. (2010) 22:45–51. doi: 10.1093/intimm/dxp113
38. Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. (2007) 39:721–3. doi: 10.1038/ng2046
39. Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. (2002) 2:580–92. doi: 10.1038/nri856
40. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
41. Sanders LA, Feldman RG, Voorhorst-Ogink MM, de Haas M, Rijkers GT, Capel PJ, et al. Human immunoglobulin G (IgG) Fc receptor IIA (CD32) polymorphism and IgG2-mediated bacterial phagocytosis by neutrophils. Infect Immun. (1995) 63:73–81.
42. Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, et al. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. (2002) 46:1242–54. doi: 10.1002/art.10257
43. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. Japan kawasaki disease genome U. S. K. Consortium DG. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
44. Asano K, Matsushita T, Umeno J, Hosono N, Takahashi A, Kawaguchi T, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. (2009) 41:1325–9. doi: 10.1038/ng.482
45. van der Pol WL, van den Berg LH, Scheepers RH, van der Bom JG, van Doorn PA, van Koningsveld R, et al. IgG receptor IIa alleles determine susceptibility and severity of Guillain-Barre syndrome. Neurology. (2000) 54:1661–5. doi: 10.1212/WNL.54.8.1661
46. Tsang ASMW, Nagelkerke SQ, Bultink IE, Geissler J, Tanck MW, Tacke CE, et al. Fc-gamma receptor polymorphisms differentially influence susceptibility to systemic lupus erythematosus and lupus nephritis. Rheumatology (Oxford). (2016) 55:939–48. doi: 10.1093/rheumatology/kev433
47. Qiao J, Al-Tamimi M, Baker RI, Andrews RK, Gardiner EE. The platelet Fc receptor, FcgammaRIIa. Immunol Rev. (2015) 268:241–52. doi: 10.1111/imr.12370
48. Tanaka Y, Suzuki Y, Tsuge T, Kanamaru Y, Horikoshi S, Monteiro RC, et al. FcgammaRIIa-131R allele and FcgammaRIIIa-176V/V genotype are risk factors for progression of IgA nephropathy. Nephrol Dial Transplant. (2005) 20:2439–45. doi: 10.1093/ndt/gfi043
49. McKenzie SE, Taylor SM, Malladi P, Yuhan H, Cassel DL, Chien P, et al. The role of the human Fc receptor Fc gamma RIIA in the immune clearance of platelets: a transgenic mouse model. J Immunol. (1999) 162:4311–8.
50. Tan Sardjono C, Mottram PL, van de Velde NC, Powell MS, Power D, Slocombe RF, et al. Development of spontaneous multisystem autoimmune disease and hypersensitivity to antibody-induced inflammation in Fcgamma receptor IIa-transgenic mice. Arthritis Rheum. (2005) 52:3220–9. doi: 10.1002/art.21344
51. Reilly AF, Norris CF, Surrey S, Bruchak FJ, Rappaport EF, Schwartz E, et al. Genetic diversity in human Fc receptor II for immunoglobulin G: Fc gamma receptor IIA ligand-binding polymorphism. Clin Diagn Lab Immunol. (1994) 1:640–4.
52. Pfirsch-Maisonnas S, Aloulou M, Xu T, Claver J, Kanamaru Y, Tiwari M, et al. Inhibitory ITAM signaling traps activating receptors with the phosphatase SHP-1 to form polarized “inhibisome” clusters. Sci Signal. (2011) 4:ra24. doi: 10.1126/scisignal.2001309
53. Pietersz GA, Mottram PL, van de Velde NC, Sardjono CT, Esparon S, Ramsland PA, et al. Inhibition of destructive autoimmune arthritis in FcgammaRIIa transgenic mice by small chemical entities. Immunol Cell Biol. (2009) 87:3–12. doi: 10.1038/icb.2008.82
54. White AL, Beers SA, Cragg MS. FcgammaRIIB as a key determinant of agonistic antibody efficacy. Curr Top Microbiol Immunol. (2014) 382:355–72. doi: 10.1007/978-3-319-07911-0_16
55. Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, et al. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nat Med. (2005) 11:1056–8. doi: 10.1038/nm1288
56. Espeli M, Smith KG, Clatworthy MR. FcgammaRIIB and autoimmunity. Immunol Rev. (2016) 269:194–211. doi: 10.1111/imr.12368
57. McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. (2005) 307:590–3. doi: 10.1126/science.1105160
58. Morgan AW, Griffiths B, Ponchel F, Montague BM, Ali M, Gardner PP, et al. Fcgamma receptor type IIIA is associated with rheumatoid arthritis in two distinct ethnic groups. Arthritis Rheum. (2000) 43:2328–34. doi: 10.1002/1529-0131(200010)43:10<2328::AID-ANR21>3.0.CO;2-Z
59. Nieto A, Pascual M, Caliz R, Mataran L, Martin J. Association of Fcgamma receptor IIIA polymorphism with rheumatoid arthritis: comment on the article by Morgan et al. Arthritis Rheum. (2002) 46:556–9. doi: 10.1002/art.10122
60. Tsuge T, Shimokawa T, Horikoshi S, Tomino Y, Ra C. Polymorphism in promoter region of Fcalpha receptor gene in patients with IgA nephropathy. Hum Genet. (2001) 108:128–33. doi: 10.1007/s004390100458
61. Wu J, Ji C, Xie F, Langefeld CD, Qian K, Gibson AW, et al. FcalphaRI (CD89) alleles determine the proinflammatory potential of serum IgA. J Immunol. (2007) 178:3973–82. doi: 10.4049/jimmunol.178.6.3973
62. Broen JC, Coenen MJ, Rueda B, Witte T, Padyukov L, Klareskog L, et al. The functional polymorphism 844 A>G in FcalphaRI (CD89) does not contribute to systemic sclerosis or rheumatoid arthritis susceptibility. J Rheumatol. (2011) 38:446–9. doi: 10.3899/jrheum.100427
63. Maillard N, Thibaudin L, Abadja F, Masson I, Garraud O, Berthoux F, et al. Single nucleotidic polymorphism 844 A->G of FCAR is not associated with IgA nephropathy in Caucasians. Nephrol Dial Transplant. (2012) 27:656–60. doi: 10.1093/ndt/gfr246
64. Kaneko S, Kobayashi T, Yamamoto K, Jansen MD, van de Winkel JG, Yoshie H. A novel polymorphism of FcalphaRI (CD89) associated with aggressive periodontitis. Tissue Antigens. (2004) 63:572–7. doi: 10.1111/j.0001-2815.2004.0228.x
65. Hirunsatit R, Kongruttanachok N, Shotelersuk K, Supiyaphun P, Voravud N, Sakuntabhai A, et al. Polymeric immunoglobulin receptor polymorphisms and risk of nasopharyngeal cancer. BMC Genet. (2003) 4:3. doi: 10.1186/1471-2156-4-3
66. Hasegawa M, Nishiyama C, Nishiyama M, Akizawa Y, Mitsuishi K, Ito T, et al. A novel−66T/C polymorphism in Fc epsilon RI alpha-chain promoter affecting the transcription activity: possible relationship to allergic diseases. J Immunol. (2003) 171:1927–33. doi: 10.4049/jimmunol.171.4.1927
67. Potaczek DP, Sanak M, Mastalerz L, Setkowicz M, Kaczor M, Nizankowska E, et al. The alpha-chain of high-affinity receptor for IgE (FcepsilonRIalpha) gene polymorphisms and serum IgE levels. Allergy. (2006) 61:1230–3. doi: 10.1111/j.1398-9995.2006.01195.x
68. Kim SH, Ye YM, Lee SK, Park HS. Genetic mechanism of aspirin-induced urticaria/angioedema. Curr Opin Allergy Clin Immunol. (2006) 6:266–70. doi: 10.1097/01.all.0000235899.57182.d4
69. Zhou J, Zhou Y, Lin LH, Wang J, Peng X, Li J, et al. Association of polymorphisms in the promoter region of FCER1A gene with atopic dermatitis, chronic uticaria, asthma, and serum immunoglobulin E levels in a Han Chinese population. Hum Immunol. (2012) 73:301–5. doi: 10.1016/j.humimm.2011.12.001
70. Zhang X, Zhang W, Qiu D, Sandford A, Tan WC. The E237G polymorphism of the high-affinity IgE receptor beta chain and asthma. Ann Allergy Asthma Immunol. (2004) 93:499–503. doi: 10.1016/S1081-1206(10)61419-6
71. Yang HJ, Zheng L, Zhang XF, Yang M, Huang X. Association of the MS4A2 gene promoter C-109T or the 7th exon E237G polymorphisms with asthma risk: a meta-analysis. Clin Biochem. (2014) 47:605–11. doi: 10.1016/j.clinbiochem.2014.01.022
72. Wu J, Lin R, Huang J, Guan W, Oetting WS, Sriramarao P, et al. Functional Fcgamma receptor polymorphisms are associated with human allergy. PLoS ONE. (2014) 9:e89196. doi: 10.1371/journal.pone.0089196
73. van der Heijden J, Breunis WB, Geissler J, de Boer M, van den Berg TK, Kuijpers TW. Phenotypic variation in IgG receptors by nonclassical FCGR2C alleles. J Immunol. (2012) 188:1318–24. doi: 10.4049/jimmunol.1003945
74. Lee YH, Ji JD, Song GG. Associations between FCGR3A polymorphisms and susceptibility to rheumatoid arthritis: a metaanalysis. J Rheumatol. (2008) 35:2129–35. doi: 10.3899/jrheum.080186
75. Carcao MD, Blanchette VS, Wakefield CD, Stephens D, Ellis J, Matheson K, et al. Fcgamma receptor IIa and IIIa polymorphisms in childhood immune thrombocytopenic purpura. Br J Haematol. (2003) 120:135–41. doi: 10.1046/j.1365-2141.2003.04033.x
76. Koene HR, Kleijer M, Swaak AJ, Sullivan KE, Bijl M, Petri MA, et al. The Fc gammaRIIIA-158F allele is a risk factor for systemic lupus erythematosus. Arthritis Rheum. (1998) 41:1813–8. doi: 10.1002/1529-0131(199810)41:10<1813::AID-ART13>3.0.CO;2-6
77. Moroi R, Endo K, Kinouchi Y, Shiga H, Kakuta Y, Kuroha M, et al. FCGR3A-158 polymorphism influences the biological response to infliximab in Crohn's disease through affecting the ADCC activity. Immunogenetics. (2013) 65:265–71. doi: 10.1007/s00251-013-0679-8
78. Kelley JM, Monach PA, Ji C, Zhou Y, Wu J, Tanaka S, et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci U.S.A. (2011) 108:20736–41. doi: 10.1073/pnas.1109227109
79. van Egmond M, van Garderen E, van Spriel AB, Damen CA, van Amersfoort ES, van Zandbergen G, et al. FcalphaRI-positive liver Kupffer cells: reappraisal of the function of immunoglobulin A in immunity. Nat Med. (2000) 6:680–5. doi: 10.1038/76261
80. Pallasch CP, Schulz A, Kutsch N, Schwamb J, Hagist S, Kashkar H, et al. Overexpression of TOSO in CLL is triggered by B-cell receptor signaling and associated with progressive disease. Blood. (2008) 112:4213–9. doi: 10.1182/blood-2008-05-157255
81. Hitoshi Y, Lorens J, Kitada SI, Fisher J, LaBarge M, Ring HZ, et al. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity. (1998) 8:461–71. doi: 10.1016/S1074-7613(00)80551-8
82. Miller LS, Atabai K, Nowakowski M, Chan A, Bluth MH, Minkoff H, et al. Increased expression of CD23 (Fc(epsilon) receptor II) by peripheral blood monocytes of aids patients. AIDS Res Hum Retroviruses. (2001) 17:443–52. doi: 10.1089/088922201750102544
83. Morgan AW, Keyte VH, Babbage SJ, Robinson JI, Ponchel F, Barrett JH, et al. FcgammaRIIIA-158V and rheumatoid arthritis: a confirmation study. Rheumatology (Oxford). (2003) 42:528–33. doi: 10.1093/rheumatology/keg169
84. Descours B, Petitjean G, Lopez-Zaragoza JL, Bruel T, Raffel R, Psomas C, et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature. (2017) 543:564–567. doi: 10.1038/nature21710
85. Abdel-Mohsen M, Kuri-Cervantes L, Grau-Exposito J, Spivak AM, Nell RA, Tomescu C, et al. CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells. Sci Transl Med. (2018) 10:eaar6759. doi: 10.1126/scitranslmed.aar6759.
86. Badia R, Ballana E, Castellvi M, Garcia-Vidal E, Pujantell M, Clotet B, et al. CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir. Nat Commun. (2018) 9:2739. doi: 10.1038/s41467-018-05157-w
87. Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, et al. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. (2007) 178:3272–80. doi: 10.4049/jimmunol.178.5.3272
88. Liu Y, Gong Y, Qu C, Zhang Y, You R, Yu N, et al. CD32b expression is down-regulated on double-negative memory B cells in patients with Hashimoto's thyroiditis. Mol Cell Endocrinol. (2017) 440:1–7. doi: 10.1016/j.mce.2016.11.004
89. Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. (2012) 53:41–57. doi: 10.1007/s12026-012-8297-3
90. Grossetete B, Launay P, Lehuen A, Jungers P, Bach JF, Monteiro RC. Down-regulation of Fc alpha receptors on blood cells of IgA nephropathy patients: evidence for a negative regulatory role of serum IgA. Kidney Int. (1998) 53:1321–35.
91. Grossetete B, Viard JP, Lehuen A, Bach JF, Monteiro RC. Impaired Fc alpha receptor expression is linked to increased immunoglobulin A levels and disease progression in HIV-1-infected patients. AIDS. (1995) 9:229–34. doi: 10.1097/00002030-199509030-00003
92. Silvain C, Patry C, Launay P, Lehuen A, Monteiro RC. Altered expression of monocyte IgA Fc receptors is associated with defective endocytosis in patients with alcoholic cirrhosis. Potential role for IFN-gamma. J Immunol. (1995) 155:1606–18.
93. Berthelot L, Jamin A, Viglietti D, Chemouny JM, Ayari H, Pierre M, et al. Value of biomarkers for predicting immunoglobulin A vasculitis nephritis outcome in an adult prospective cohort. Nephrol Dial Transplant. (2017) 33:1579–90. doi: 10.1093/ndt/gfx300
94. Xue J, Zhao Q, Zhu L, Zhang W. Deglycosylation of FcalphaR at N58 increases its binding to IgA. Glycobiology. (2010) 20:905–15. doi: 10.1093/glycob/cwq048
95. Monteiro RC. Recent advances in the physiopathology of IgA nephropathy. Nephrol Ther. (2018) 14(Suppl 1):S1–8. doi: 10.1016/j.nephro.2018.02.004
96. Vuong MT, Hahn-Zoric M, Lundberg S, Gunnarsson I, van Kooten C, Wramner L, et al. Association of soluble CD89 levels with disease progression but not susceptibility in IgA nephropathy. Kidney Int. (2010) 78:1281–7. doi: 10.1038/ki.2010.314
97. Berthelot L, Robert T, Vuiblet V, Tabary T, Braconnier A, Drame M, et al. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. (2015) 88:815–22. doi: 10.1038/ki.2015.158
98. Berthelot L, Papista C, Maciel TT, Biarnes-Pelicot M, Tissandie E, Wang PH, et al. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J Exp Med. (2012) 209:793–806. doi: 10.1084/jem.20112005
99. Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, et al. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med. (2001) 194:417–25. doi: 10.1084/jem.194.4.417
100. Tamouza H, Chemouny JM, Raskova Kafkova L, Berthelot L, Flamant M, Demion M, et al. The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int. (2012) 82:1284–96. doi: 10.1038/ki.2012.192
101. Coulon S, Dussiot M, Grapton D, Maciel TT, Wang PH, Callens C, et al. Polymeric IgA1 controls erythroblast proliferation and accelerates erythropoiesis recovery in anemia. Nat Med. (2011) 17:1456–65. doi: 10.1038/nm.2462
102. Kanamaru Y, Arcos-Fajardo M, Moura IC, Tsuge T, Cohen H, Essig M, et al. Fc alpha receptor I activation induces leukocyte recruitment and promotes aggravation of glomerulonephritis through the FcR gamma adaptor. Eur J Immunol. (2007) 37:1116–28. doi: 10.1002/eji.200636826
103. Launay P, Patry C, Lehuen A, Pasquier B, Blank U, Monteiro RC. Alternative endocytic pathway for immunoglobulin A Fc receptors (CD89) depends on the lack of FcRgamma association and protects against degradation of bound ligand. J Biol Chem. (1999) 274:7216–25. doi: 10.1074/jbc.274.11.7216
104. Honjo K, Kubagawa Y, Suzuki Y, Takagi M, Ohno H, Bucy RP, et al. Enhanced auto-antibody production and Mott cell formation in FcmuR-deficient autoimmune mice. Int Immunol. (2014) 26:659–72. doi: 10.1093/intimm/dxu070
105. Ouchida R, Mori H, Hase K, Takatsu H, Kurosaki T, Tokuhisa T, et al. Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc Natl Acad Sci USA. (2012) 109:E2699–706. doi: 10.1073/pnas.1210706109
106. Li FJ, Kubagawa Y, McCollum MK, Wilson L, Motohashi T, Bertoli LF, et al. Enhanced levels of both the membrane-bound and soluble forms of IgM Fc receptor (FcmuR) in patients with chronic lymphocytic leukemia. Blood. (2011) 118:4902–9. doi: 10.1182/blood-2011-04-350793
107. Madarova M, Mucha R, Hresko S, Makarova Z, Gdovinova Z, Szilasiova J, et al. Identification of new phosphorylation sites of CD23 in B-cells of patients with chronic lymphocytic leukemia. Leuk Res. (2018) 70:25–33. doi: 10.1016/j.leukres.2018.05.002
108. Tsuboi N, Asano K, Lauterbach M, Mayadas TN. Human neutrophil Fcgamma receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. (2008) 28:833–46. doi: 10.1016/j.immuni.2008.04.013
109. Rossato E, Ben Mkaddem S, Kanamaru Y, Hurtado-Nedelec M, Hayem G, Descatoire V, et al. Reversal of arthritis by human monomeric IgA through the receptor-mediated SH2 domain-containing phosphatase 1 Inhibitory pathway. Arthritis Rheumatol. (2015) 67:1766–77. doi: 10.1002/art.39142
110. van der Steen LP, Bakema JE, Sesarman A, Florea F, Tuk CW, Kirtschig G, et al. Blocking Fcalpha receptor I on granulocytes prevents tissue damage induced by IgA autoantibodies. J Immunol. (2012) 189:1594–601. doi: 10.4049/jimmunol.1101763
111. Heineke MH, van der Steen LPE, Korthouwer RM, Hage JJ, Langedijk JP M, Benschop JJ, et al. Peptide mimetics of immunoglobulin A (IgA) and FcalphaRI block IgA-induced human neutrophil activation and migration. Eur J Immunol. (2017) 47:1835–45. doi: 10.1002/eji.201646782
112. Jardieu PM, Fick RB Jr. IgE inhibition as a therapy for allergic disease. Int Arch Allergy Immunol. (1999) 118:112–5 (1999) doi: 10.1159/000024043
113. Milgrom H, Fick RB Jr, Su JQ, Reimann JD, Bush RK, Watrous ML, et al. Treatment of allergic asthma with monoclonal anti-IgE antibody. rhuMAb-E25 Study Group. N Engl J Med. (1999) 341:1966–73. doi: 10.1056/NEJM199912233412603
114. Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. (2001) 345:747–55. doi: 10.1056/NEJMra993360
115. Pecoraro A, Crescenzi L, Granata F, Genovese A, Spadaro G. Immunoglobulin replacement therapy in primary and secondary antibody deficiency: the correct clinical approach. Int Immunopharmacol. (2017). 52:136–42. doi: 10.1016/j.intimp.2017.09.005
116. Jin F, Balthasar JP. Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Hum Immunol. (2005) 66:403–10. doi: 10.1016/j.humimm.2005.01.029
117. Debre M, Bonnet MC, Fridman WH, Carosella E, Philippe N, Reinert P, et al. Infusion of Fc gamma fragments for treatment of children with acute immune thrombocytopenic purpura. Lancet. (1993) 342:945–9. doi: 10.1016/0140-6736(93)92000-J
118. Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. (2008) 26:513–33. doi: 10.1146/annurev.immunol.26.021607.090232
119. Siragam V, Crow AR, Brinc D, Song S, Freedman J, Lazarus AH. Intravenous immunoglobulin ameliorates ITP via activating Fc gamma receptors on dendritic cells. Nat Med. (2006) 12:688–92. doi: 10.1038/nm1416
120. Epstein JS, Zoon KC. Important drug warning: immune globulin intravenous (human) (IGIV) products. Neonatal Netw. (2000) 19:60–2.
121. Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. (2000) 13:277–85. doi: 10.1016/S1074-7613(00)00027-3
122. van Mirre E, van Royen A, Hack CE. IVIg-mediated amelioration of murine ITP via FcgammaRIIb is not necessarily independent of SHIP-1 and SHP-1 activity. Blood. (2004) 103:1973. doi: 10.1182/blood-2003-11-3933
123. Veri MC, Gorlatov S, Li H, Burke S, Johnson S, Stavenhagen J, et al. Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology. (2007) 121:392–404. doi: 10.1111/j.1365-2567.2007.02588.x
124. Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, et al. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol. (2007) 8:419–29. doi: 10.1038/ni1440
125. Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. (2005) 23:1283–8. doi: 10.1038/nbt1143
126. Liu L, Garcia AM, Santoro H, Zhang Y, McDonnell K, Dumont J, et al. Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal FcR blockade. J Immunol. (2007) 178:5390–8. doi: 10.4049/jimmunol.178.8.5390
127. Zhang Z, Shen K, Lu W, Cole PA. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J Biol Chem. (2003) 278:4668–74. doi: 10.1074/jbc.M210028200
128. Jones ML, Craik JD, Gibbins JM, Poole AW. Regulation of SHP-1 tyrosine phosphatase in human platelets by serine phosphorylation at its C terminus. J Biol Chem. (2004) 279:40475–83. doi: 10.1074/jbc.M402970200
129. Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. (2005) 11:936–43. doi: 10.1038/nm1284
Keywords: immunoglobilins, Fc receptor, antibody treatment, signaling/signaling pathways, inflammatory diseases
Citation: Ben Mkaddem S, Benhamou M and Monteiro RC (2019) Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front. Immunol. 10:811. doi: 10.3389/fimmu.2019.00811
Received: 29 December 2018; Accepted: 27 March 2019;
Published: 12 April 2019.
Edited by:
Mark S. Cragg, University of Southampton, United KingdomReviewed by:
Carlos Rosales, National Autonomous University of Mexico, MexicoCopyright © 2019 Ben Mkaddem, Benhamou and Monteiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renato C. Monteiro, cmVuYXRvLm1vbnRlaXJvQGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.