 Li Lei1†
Li Lei1† Tao Li
Tao Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Aging Neurosci., 15 July 2021
Sec. Cellular and Molecular Mechanisms of Brain-aging
Volume 13 - 2021 | https://doi.org/10.3389/fnagi.2021.698601
This article is part of the Research TopicMitochondrial Dysfunction in StrokeView all 12 articles
Mitochondrial autophagy is an early defense and protection process that selectively clears dysfunctional or excessive mitochondria through a distinctive mechanism to maintain intracellular homeostasis. Mitochondrial dysfunction during cerebral stroke involves metabolic disbalance, oxidative stress, apoptosis, endoplasmic reticulum stress, and abnormal mitochondrial autophagy. This article reviews the research progress on the mechanism of mitochondrial autophagy in ischemic stroke to provide a theoretical basis for further research on mitochondrial autophagy and the treatment of ischemic stroke.
Stroke is a sudden disease with disturbance of cerebral blood circulation, which has surpassed ischemic heart disease, lung cancer, chronic obstructive pulmonary disease, and liver cancer to become the disease with the highest age-standardized mortality rate in China. According to the latest disease burden report in 2015, stroke has become not only the first cause of disease burden, but also the second leading cause of death and disability in the world (GBD 2015 DALYs and HALE Collaborators, 2016; GBD 2015 Mortality and Causes of Death Collaborators, 2016), and an important cause of premature death. Stroke is primarily divided into two categories: ischemic stroke (accounting for 60% of all strokes) and hemorrhagic stroke (Krishnamurthi et al., 2013). At present, studies have found a substantial link between mitochondrial dysfunction and the occurrence and development of ischemic stroke. Moreover, the abnormal autophagy is profoundly involved in mitochondrial dysfunction.
Autophagy is a process defined by the degradation of abnormal proteins and damaged organelles by bilayer membranes such as exfoliated endoplasmic reticulum or Golgi complex under physiological or pathological circumstances and mediated by autophagy-related proteins. The bilayer structures fuse with lysosomes to form autophagy lysosomes, which degrades the contents of biological macromolecules and organelles, so as to maintain cell homeostasis by meeting the needs of cellular metabolism and organelle renewal (Parzych and Klionsky, 2014; Tagaya and Arasaki, 2017). Autophagy is essential for neuronal survival in the central nervous system. According to the mechanism and the mode of action, autophagy can be divided into three types: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Among them, macroautophagy is the most thoroughly studied. Mitochondrial autophagy, also called mitophagy, is a kind of macrophage autophagy, which selectively degrades damaged mitochondria through autophagy mechanism to sustain the stability of the intracellular environment. Mitochondrial autophagy plays an important role in regulating cellular homeostasis, proliferation, differentiation, genetics, aging, and cell death, as well as in controlling the quantity and quality of mitochondria (Glick et al., 2010).
PTEN Induced Putative Kinase 1 (PINK1)/Parkin-mediated autophagy is the most common type of mitochondrial autophagy in mammalian cells (He et al., 2019). PINK1 is a nuclear-encoded mitochondrial serine (Ser)/threonine kinase (Eiyama and Okamoto, 2015; Quinn et al., 2020), which is divided into N-terminal mitochondrial targeting signal (MTS), α-helical transmembrane domain (TM), serine/threonine kinase domain, and C-terminal mitochondrial outer membrane retention signal peptide sequence (Deas et al., 2011), it can mediate the ubiquitination of substrates, and can regulate protein degradation and signal transduction. Parkin is an E3 ubiquitin ligase encoded by the Park2 gene. Genetic studies have shown that PINK1 plays a role in the upstream of Parkin, and PINK1 and Parkin are in the same pathway to preserve mitochondrial functions (Poole et al., 2008; Whitworth and Pallanck, 2009; Tanaka, 2020). In healthy cells, PINK1 is transported to the mitochondrial inner membrane, where it is cut and degraded rapidly by the ubiquitin-protease system, when the intracellular mitochondrial membrane is damaged, and consequently, the transmembrane potential is disturbed, the transport of PINK1 into the mitochondria will be blocked and PINK1 will accumulate in the outer membrane of the mitochondria, which then mediates the activation of Parkin and ubiquitin phosphorylation (Poole et al., 2008; Jin and Youle, 2013; Durcan and Fon, 2015; Okatsu et al., 2015a; Yamano et al., 2016). PINK1 can simultaneously phosphorylate Ser65 in the N-terminal ubiquitin-like domain of Parkin and produce Ser65 phosphorylated Parkin (Kondapalli et al., 2012; Iguchi et al., 2013; Okatsu et al., 2015b; Yang et al., 2020), phosphorylated Ser65 activates its ubiquitin ligase activity, which is able to connect the polyubiquitin chain to the mitochondrial outer membrane protein (Kazlauskaite et al., 2014), the complex is recognized by LC3, binds to the autophagosome, and then fuses with the lysosome to form the autophagy lysosome that finally degrades the damaged mitochondria. Studies from the Boule laboratory have confirmed that PINK1/Parkin-regulated mitochondrial autophagy plays a key role in the clearance of damaged mitochondria (Lazarou et al., 2015). Mitochondria maintain metabolic balance through continuous fusion and division. Additionally, the fusion/division and the mitochondrial autophagy influence each other; malfunctioning mitochondria are effectively removed by mitochondrial autophagy, and only the mitochondria with normal membrane potential can enter the normal fusion-division cycle (Twig et al., 2008), PINK1 and Parkin are of importance in the process of mitochondrial fusion and division. Related studies have shown that PINK1 and Parkin can promote mitochondrial division and regulate mitochondrial movement by mediating Miro phosphorylation to isolate damaged mitochondria (Poole et al., 2008), maintaining energy balance, and avoiding oxidative stress (Zhuang et al., 2016; Figure 1).
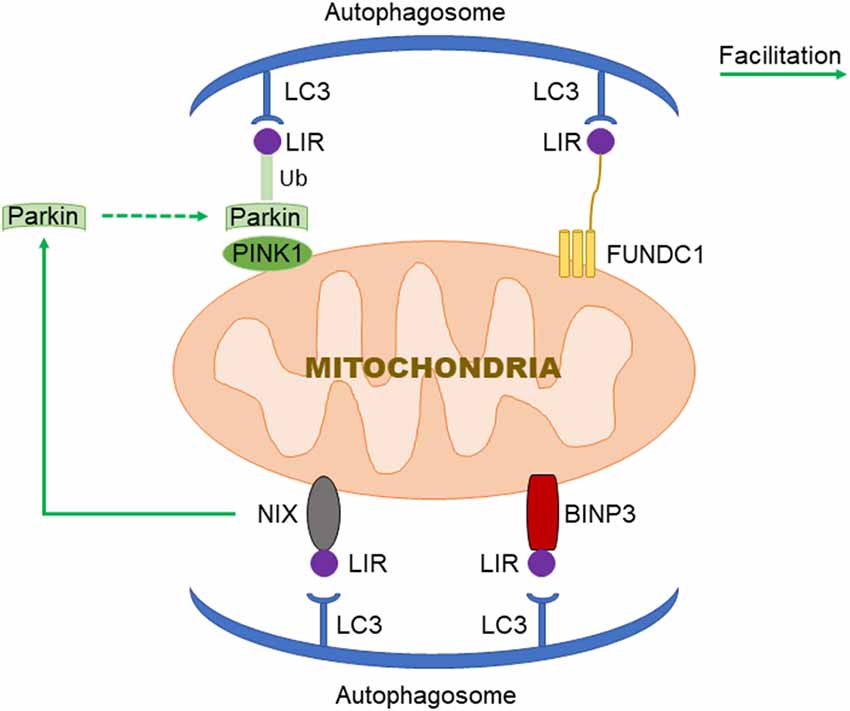
Figure 1. Three signaling pathways regulating mitochondrial autophagy. In PTEN Induced Putative Kinase 1 (PINK1)/Parkin pathway, PINK1 functions as the initiator which recognizes the disturbed outer membrane potential and activates Parkin and ubiquitin phosphorylation, and then the complex is combined by LC3 to autophagosome; FUN14 Domain-Containing 1 (FUNDC1) is a three transmembrane protein having the LC3 interaction region (LIR) motif on the N-terminal which can be recognized by LC3; both BNIP3 and NIX have LIR motif, additionally, NIX can facilitate the localization of Parkin to the damaged mitochondria.
FUN14 Domain-Containing 1 (FUNDC1) is a new type of mitochondrial membrane protein that mediates mitochondrial autophagy in mammalian cells, it mainly acts in controlling mitochondrial autophagy by regulating the phosphorylation level of FUNDC1 on the mitochondrial membrane. FUNDC1 is a protein located in the outer membrane of mitochondria, which contains three transmembrane domains, as well as the N-terminal domain exposed to the cytoplasm and the C-terminal domain inserted into the mitochondrial inner membrane. It has been found that FUNDC1 contains a typical LC3 interaction region (LIR) sequence consisting of amino acid residues at the N-terminal on the cytoplasmic side (Liu et al., 2014; Cai et al., 2021). Under physiological conditions, FUNDC1 inhibits mitochondrial autophagy due to the phosphorylation of Tyr18 in the LIR motif of sarcoma (Src) gene kinase, leading to the inability of FUNDC1 to recruit LC3 and to combine with it, which reduces the interaction between FUNDC1 and LC3 (Liu et al., 2012; Chen et al., 2016; Kuang et al., 2016). Studies have shown that mitochondrial autophagy receptor protein, FUNDC1, is involved in mitochondrial autophagy under the condition of hypoxia in mammalian cells (Wang et al., 2018), when Src kinase and CK2 (formerly known as casein kinase 2) are inactivated during hypoxia or mitochondrial uncoupling, Ser17 on FUNDC1 is dephosphorylated by unc-51-like kinase 1 (ULK1) and Ser13 is dephosphorylated by phosphoglycerol mutase family 5 (PGAM5; Liu et al., 2014; Chen et al., 2016, 2017; Kuang et al., 2016), enhancing FUNDC-LC3 interaction and mitochondrial autophagy (Imai et al., 2010; Liu et al., 2014; Lv et al., 2017). Thus, it can be seen that FUNDC1 phosphorylation or dephosphorylation functions as a specific mitochondrial autophagy receptor in the process of mitochondrial autophagy induced by hypoxia. In addition, FUNDC1 can regulate mitochondrial division-fusion and mitochondrial autophagy by interacting with mitochondrial mitotic protein, dynamin-related protein 1 (Drp1), and optic nerve dystrophin 1 or with Drp1 and calcitonin (Ding and Yin, 2012; Iguchi et al., 2013; Wu et al., 2016a).
BNIP3 (B Lymphoma-2 gene/adenovirus E1B interacting protein 3) and NIX (BNIP3L) are pro-apoptotic mitochondrial proteins located in the outer membrane of mitochondria (Kubli et al., 2007; Liu et al., 2019; Xu et al., 2019), they have 56% homology in amino acid sequence. Under certain conditions, the LIR sequence interacts with the recruited autophagy related protein, LC3, to form an autophagosome to clear the damaged mitochondria (Springer and Macleod, 2016; Šprung et al., 2018; Roperto et al., 2019). It has been reported that BNIP3/NIX mediates mitochondrial autophagy through upstream hypoxia-inducible 1 factor (HIF-1) regulation (Chourasia and Macleod, 2015); the competitive binding of BNIP3 and NIX to the anti-apoptotic protein, Bcl-2, will make Bcl-2-Beclin-1 complex dissociate and release Beclin-1, subsequently activating mitochondrial autophagy and reducing the apoptosis (Zhang et al., 2008). BNIP3 and NIX can promote the enhancement of mitochondrial autophagy by inhibiting the mammalian rapamycin target protein (mTOR) to activate protein Rheb. The upregulation of BNIP3 and NIX expression can be mediated by forkhead box O3 (FOXO3). Some studies have found that BNIP3 and NIX can not only induce cell death, but also increase their expression level under hypoxia, reducing reactive oxygen species (ROS) production and promoting cell survival, and can participate in the regulation of mitochondrial autophagy (de Vries and Przedborski, 2013). BNIP3 can increase the localization of mitochondrial kinesin-associated protein 1, thus promoting the phagocytosis of damaged mitochondria (Liu, 2015). Additionally, BNIP3 promotes the release of cytochrome C. Other studies have found that mitochondrial oxidative phosphorylation uncoupling agent carbonyl cyanide m-chlorophenyl hydrazone (CCCP) treatment on Hela cells can improve the interaction between NIX and LC3, indicating that NIX may also be involved in depolarization-induced mitochondrial autophagy (Novak et al., 2010). NIX mainly regulates mitochondrial autophagy at baseline under physiological conditions and mediates mitochondrial clearance during erythrocyte development by increasing the production of ROS (Novak and Dikic, 2011; Shi et al., 2014). Moreover, NIX can promote the localization of Parkin to the damaged mitochondria, increase the ubiquitination of mitochondrial membrane proteins by Parkin, and eventually induce mitochondrial autophagy through the interaction between p62 and LC3 (Geisler et al., 2010).
Neurological impairment caused by mitochondrial dysfunction is significantly associated with stroke, the main mechanisms include increased adenosine triphosphate (ATP), increased ROS level, and oxidative stress overload. Under mild ischemia or tolerable hypoxic stress, mitochondrial autophagy can benefit cell homeostasis and promote cell survival. On the contrary, vulnerable brain cells in the ischemic core area of persistent ischemia or reperfusion begin to die, which will lead to excessive long–term autophagy hyperactivity, causing cell damage or death (Zhang X. et al., 2013). Therefore, in the course of a stroke, it may be a possible solution of protecting neurons and preventing cell death to regulate the quantity and quality of mitochondria through the process of mitochondrial autophagy (Wong and Cuervo, 2010; Figure 2).
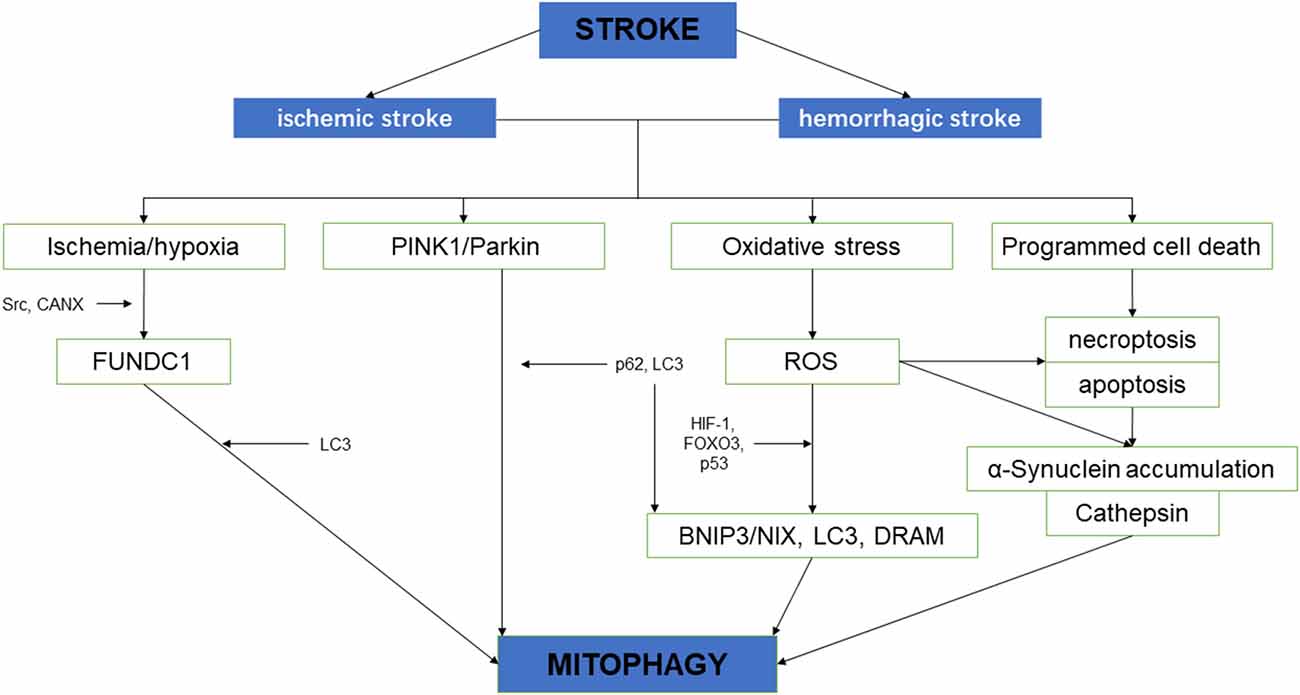
Figure 2. The schematic diagram depicting the relationship between cerebral stroke and mitochondrial autophagy.
Mitochondria are vital organelles that produce energy, regulate cell signals and apoptosis, and are also the major source of ROS (Murphy, 2009). When cerebral ischemia occurs, the decrease of blood flow leads to oxygen and glucose deficiency and changes in mitochondrial structure. Excitatory amino acid toxicity, hypoxia, calcium overload, and other factors cause mitochondrial oxidative phosphorylation disorder (Li et al., 2015; Khandelwal et al., 2016), thus the excessive ROS produced by damaged nerve cells cannot be eliminated by related enzymes. In turn, it results in the destruction of the redox balance of lipids and proteins and extensive cell damage, which further aggravates the mitochondrial damage. Increased level of ROS-enhanced oxidative stress response and excessive generation of ROS can activate hypoxia-inducible factor 1 (HIF-1) and FOXO3, these transcription factors then induce the transcription of BNIP3 and NIX, and LC3 and BNIP3, respectively (Li et al., 2015). HIF-1 is the main factor in the survival response of cells to hypoxia, which can induce the transcription of BNIP3 and NIX genes. Their protein products compete with Beclin-1 to bind Bcl-2 to release Beclin-1 and allow it to induce autophagy (Mahalingaiah and Singh, 2014). It has been found that the increase of intracellular ROS may trigger the FOXO3 signaling pathway, which activates the transcription of ubiquitin-proteasome pathway and autophagy-related genes (such as the genes encoding LC3 and BNIP3), thus inducing autophagy (Aucello et al., 2009). After 3 days of ischemia/reperfusion, superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) in brain tissue decreased significantly. With the intervention using Salvianolic acid, oxygen-free radicals were effectively scavenged in rats, and ischemia-reperfusion injury was significantly suppressed (Xu et al., 2017). Hypoxic preconditioning can activate the HIF-1/Beclin-1 signaling pathway to facilitate autophagy and protect against ischemic and hypoxic injury (Semenza, 2011; Lu et al., 2018). When cells were exposed to hypoxia, the transcriptional level of BNIP3/NIX was directly regulated by HIF-1α and activated (Tracy et al., 2007). Bellot confirmed in a variety of cell types that hypoxia promoted the inhibition of proline hydroxylase (PHD) activity through ROS produced by mitochondria (Bellot et al., 2009), suppressed the degradation of HIF-1α, and up-regulated the expression of autophagy genes such as Beclin-1 and Atg5 to promote mitochondrial autophagy (Jin and Youle, 2012). ROS not only induces mitochondrial autophagy through the HIF-1 signaling pathway (Dias et al., 2013), but also facilitates mitochondrial autophagy by activating the PINK1/Parkin pathway. In addition, ROS can induce autophagy by inhibiting the activity of protein kinase B (Akt) and mTOR in the PI3K/Akt/mTOR signaling pathway (Mellor et al., 2011; van der Vos et al., 2012; Zhang J. et al., 2013; Fiorini et al., 2015; Hambright et al., 2015; Duan et al., 2016).
The PINK1-Parkin pathway is one of the typical signaling pathways in mitochondrial autophagy. Under physiological conditions, PINK1 and Parkin form a strict mitochondrial regulation mechanism and keep a mutual balance, preventing injury of autophagy caused by excessive mitochondrial autophagy. During mitochondrial dysfunction caused by ischemic stroke (Lazarou et al., 2012; Wauer et al., 2015), the PINK1/Parkin pathway mediates the polyubiquitin of the functional or structural proteins in the damaged mitochondria (Riley et al., 2013; Trempe et al., 2013; Kumar et al., 2015; Okatsu et al., 2015b; Yamano et al., 2015). PINK1 can quickly sense the membrane potential abnormality of the damaged mitochondria, recruit Parkin to gather to the damaged ones, and induce mitochondrial autophagy (Lee et al., 2010). In recent years, there has been an increase in the research on this mechanism in stroke, which illustrates to some extent that the PINK1-Parkin pathway is involved in the process of brain injury in stroke. Safarpour’s studies on ischemic stroke using neurons found that knockout of PINK1 or Parkin gene could result in excitatory amino acid toxicity to the neurons, and it was confirmed that either PINK1 or Parkin gene deletion could aggravate neuronal damage in ischemic stroke. Wu et al. (2018) established the ischemic stroke cell model using rat hippocampal neurons in vitro. The results showed that PINK1/Parkin-mediated mitochondrial autophagy could play a neuroprotective role in rat hippocampal neurons. It was also revealed that H2 and rapamycin could increase cell survival, increase the expression of LC3-II, PINK1 and Parkin, reduce the loss of mitochondrial membrane potential, and inhibit the level of ROS and the rate of apoptosis. After autophagy inhibitor 3-methyladeine (3-MA) treatment, mitochondrial autophagy was inhibited, and the cell survival was significantly downscaled, accompanied by a significant increase in the level of ROS and the rate of apoptosis. It was found that in the pathophysiological process of ischemic stroke, PINK1/Parkin signaling pathway exerted a protective effect by mediating mitochondrial autophagy; it was also reported that the expression of LC3B and Beclin-1 was gradually up-regulated and reached the peak 24 h after cerebral ischemia-reperfusion, and the mitochondrial translocation of Parkin and the expression of PINK1 in mitochondrial outer membrane increased significantly at 24 h of reperfusion. When treated with a PINK1 inhibitor, the expression of LC3B and Beclin-1 is decreased, the mitochondrial translocation of Parkin is lessened, and the volume of cerebral infarction is increased significantly. In addition, laboratory studies have proven that in the course of ischemic brain injury, silencing the Parkin gene can interfere with mitochondrial autophagy induced by OGD (oxygen-glucose deprivation) reperfusion model, and further aggravate neuronal injury.
Under normal conditions, the expression of BNIP3/NIX is low in most organs, but under hypoxic-ischemic conditions, its transcriptional level is directly regulated by HIF-1α and activated (Tracy et al., 2007; Semenza, 2011; Guo, 2017; Yuan et al., 2017). Neuronal excitatory injury can induce mitochondrial autophagy during cerebral ischemia (Borsello et al., 2003; Shacka et al., 2007), which might occur via up-regulation of the expression of p53 and damage regulatory autophagy modulator DRAM(an autophagy regulator), and in turn, modulating autophagy by influencing Beclin-1 and LC3 (Rami et al., 2008; Wang et al., 2009; Poluzzi et al., 2014; Maejima et al., 2016). Mitochondrial autophagy can play a neuroprotective role by inhibiting apoptosis in the model of transient ischemia and hypoxia, in which the expression of Beclin-1 protein and the ratio of LC3-II/I is increased, while the expression of p62, TOM20, and HSP60 is decreased, and the progression of cerebral infarction is mild (Huang et al., 2016). In the model of long-term ischemia or permanent middle cerebral artery occlusion (MCAO), the detection of mitochondrial autophagy-related proteins, including Beclin-1, LC3, p62, TOM20, and HSP60 also shows that mitochondrial autophagy is inhibited and that the volume of cerebral infarction increases significantly. However, overactivated autophagy induces neuronal apoptosis and brain tissue injury (Li et al., 2015). Autophagy inhibitor 3-MA exerts the neuroprotective effect by inhibiting the up-regulation of LC3-II and cathepsin B induced by ischemia, preventing the programmed cell death of hippocampal CA1 neurons, and thus reducing the infarct volume and brain edema (Kubota et al., 2010). It is confirmed that the inhibition of Beclin-1 can reduce the cell death caused by MCAO (Xing et al., 2012). Recent research shows that necrostatin-1 treatment suppresses autophagic-associated proteins (LC3-II, Beclin-1) and maintains p62 at a normal level at 24 and 72 h after intracerebral hemorrhage (ICH) in a mouse model; the study also proves that the specific inhibitor, necrostatin-1, inhibits apoptosis and autophagy to exert the neuroprotective effects after ICH (Sekerdag et al., 2018). Zhang et al. (2011) reported that atorvastatin (ATV) weakens the neuroprotective effect of endoplasmic reticulum stress-related apoptosis through the reduction of autophagy in MCAO rats (Yuan et al., 2017), suggesting that compounds that inhibit autophagy may reduce the neuroprotective effect of angiotensin after cerebral ischemia (Adhami et al., 2006). Rapamycin, an inhibitor of mTOR, can activate autophagy after permanent ischemia and ischemia-reperfusion, increase the phosphorylation of Akt and cAMP response element binding protein (CREB), reduce infarction volume, improve stroke prognosis, and participate in protection against cerebral ischemic injury (Sheng et al., 2010). Carloni et al. (2010) used Rapamycin in the neonatal hypoxic-ischemic brain damage model and found that the expression of Beclin-1 was significantly up-regulated and the death of hippocampal and cortical neurons decreased significantly. The study speculated that drugs that appropriately up-regulate autophagy may have a certain neuroprotective effect in patients with large area cerebral infarction or transient ischemic attack (TIA; Carloni et al., 2008). Zheng et al. (2009) applied the ribonucleic acid (RNA) interference technique to the rat model of cerebral ischemia to down-regulate the expression of Beclin-1 in the ischemic brain and inhibit mitochondrial autophagy and noted that it can alleviate cerebral ischemic injury, inhibit neuronal apoptosis in cortex and striatum, and improve the symptoms of neurological impairment. Recent studies have shown that the re-expression of BNIP3L in mouse cortex and striatum transfected with recombinant adeno-associated virus can reverse the loss of BNIP3L after ischemia and promote mitochondrial autophagy. Additionally, this study found that only high expression of wild-type BNIP3L could reverse mitochondrial autophagy and reduce ischemic cerebral infarction volume after cerebral ischemia, whereas high expression monomer mutant BNIP3L could not have such effect. These results suggest that BNIP3L dimer formation is necessary to induce the mitochondrial autophagy to exert a neuroprotective effect, and the decrease of BNIP3L dimer is a key factor for the loss of mitochondrial autophagy in ischemic brain tissue. In ischemia and reperfusion, when the Parkin gene is knockout in vivo and in vitro, BNIP3L is still able to promote mitochondrial autophagy, and the mitochondrial autophagy-related proteins such as Parkin, FUNDC1, Bcl2-L-13, Prohibitin2, and BNIP3L/NIX can be detected. It was suggested that only the protein level of BNIP3L/NIX decreased with the prolonged duration of ischemia time. Recently, a series of studies have reported that tumor suppressor gene p53 and DRAM have an important relationship with the occurrence of autophagy during cerebral ischemia. Nerve excitatory injury can up-regulate the expression of p53 and DRAM, and then induce autophagy by regulating Beclin-1 and LC3 (Wang et al., 2009; Zhang et al., 2009; Sano et al., 2020).
FUNDC1 is a membrane protein located in mitochondria. FUNDC1 binds to LC3 through its LIR domain, causing an autophagic bilayer membrane to envelop mitochondria, inducing mitochondrial autophagy. The resulting mitochondrial autophagy inhibits apoptosis and protects neurons. FUNDC1 is a bridge between tissue-type plasminogen activator (tPA)-regulated apoptosis and mitosis in ischemia-reperfusion injury (IR) damage (Cai et al., 2021; Li et al., 2021). The tPA plays an important role in the treatment of acute cerebral ischemic injury, it has the thrombolytic effect in blood vessels and is also an important neuroprotective agent in the ultra-early ischemic stroke (Jeanneret and Yepes, 2017; Zhou et al., 2018; Yepes, 2019). FUNDC1 is associated with mitosis and is widely involved in the course of IR. The tPA exerts neuroprotective effects by increasing the phosphorylation of AMPK and the expression of FUNDC1, thereby inhibiting apoptosis and improving mitochondrial function. Knocking out the tPA gene significantly aggravated brain injury and increased neuronal apoptosis and mitochondrial damage (Echeverry et al., 2010; Wu et al., 2012; An et al., 2014). Mitochondrial oxidative stress induced by blood re-flow after ischemia can lead to mitochondrial dysfunction and promote the release of cytochrome and Bcl family members (Cai et al., 2021). Recent studies have uncovered that FUNDC1 aggregates on the mitochondrial membrane by mediating the interaction between FUNDC1 and CANX protein on ER under anoxic conditions. Under hypoxic stress, the process of mitochondrial autophagy is activated. The separation of FUNDC1 from CANX, and the recruitment of DNM1L/Drp1 then lead to mitochondrial division (Wu et al., 2016b).
Mitochondria are the key target of cerebral stroke research. As a fundamental organelle, mitochondria play a pivotal role in the fate of neurons suffering an ischemic or hemorrhagic attack, causing neuronal death through a variety of biochemical and molecular processes. In recent years, there are numerous studies with respect to the correlation of cerebral stroke and mitochondrial autophagy, and also some preclinical studies aimed at interfering with autophagy to treat central nervous system diseases (Papadakis et al., 2013; Salminen et al., 2013). However, there is still a shortage of research on mitochondrial autophagy and hemorrhagic stroke, and it is an undeniable fact that the current results show some contradictions. Although the exact role of mitochondrial autophagy in stroke is controversial, it may also become a new hot spot in basic research and potential clinical application. It has been confirmed that drugs such as metformin, resveratrol, ginkgetin, and ezetimibe can activate autophagy to exert an anti-ischemic effect. For example, resveratrol protects the brain from NLRP3 injury by inhibiting the activation of NLRP3 inflammatory bodies through Sirt1-dependent autophagy; ginkgetin can alleviate cerebral ischemia/reperfusion induced autophagy and apoptosis by inhibiting the NF-κB/p53 signaling pathway (He et al., 2017; Yu et al., 2018; Pan et al., 2019). However, due to a lack of studies on drug side effects in experimental animals and the results of clinical trials, the conclusion that if autophagy modulating drugs mediate cytoprotection or cytotoxicity in cerebral stroke remains unavailable. We believe that the in-depth study of the molecular biological mechanism of mitochondrial autophagy will provide new perspectives, treatment strategies, and potential therapeutic targets for stroke and other diseases.
LL, SY, and XL presented the idea and wrote the manuscript. YZ and TL revised the article and are corresponding authors. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (Grant No. 82001278), The Fund for Young Doctors with the First People’s Hospital of Yunnan Province (Grant No. KHBS-2020-014), Yunnan Fundamental Research Projects, and Joint Projects of Yunnan Provincial Science and Technology Department, and Kunming Medical University for Applied Basic Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adhami, F., Liao, G., Morozov, Y. M., Schloemer, A., Schmithorst, V. J., Lorenz, J. N., et al. (2006). Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 169, 566–583. doi: 10.2353/ajpath.2006.051066
An, J., Haile, W. B., Wu, F., Torre, E., and Yepes, M. (2014). Tissue-type plasminogen activator mediates neuroglial coupling in the central nervous system. Neuroscience 257, 41–48. doi: 10.1016/j.neuroscience.2013.10.060
Aucello, M., Dobrowolny, G., and Musarò, A. (2009). Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy 5, 527–529. doi: 10.4161/auto.5.4.7962
Bellot, G., Garcia-Medina, R., Gounon, P., Chiche, J., Roux, D., Pouysségur, J., et al. (2009). Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29, 2570–2581. doi: 10.1128/MCB.00166-09
Borsello, T., Croquelois, K., Hornung, J. P., and Clarke, P. G. (2003). N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur. J. Neurosci. 18, 473–485. doi: 10.1046/j.1460-9568.2003.02757.x
Cai, Y., Yang, E., Yao, X., Zhang, X., Wang, Q., Wang, Y., et al. (2021). FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 38:101792. doi: 10.1016/j.redox.2020.101792
Carloni, S., Buonocore, G., and Balduini, W. (2008). Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 32, 329–339. doi: 10.1016/j.nbd.2008.07.022
Carloni, S., Girelli, S., Scopa, C., Buonocore, G., Longini, M., and Balduini, W. (2010). Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 6, 366–377. doi: 10.4161/auto.6.3.11261
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, Z., Liu, L., Cheng, Q., Li, Y., Wu, H., Zhang, W., et al. (2017). Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 18, 495–509. doi: 10.15252/embr.201643309
Chourasia, A. H., and Macleod, K. F. (2015). Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 11, 1937–1938. doi: 10.1080/15548627.2015.1085136
Deas, E., Plun-Favreau, H., Gandhi, S., Desmond, H., Kjaer, S., Loh, S. H., et al. (2011). PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879. doi: 10.1093/hmg/ddq526
de Vries, R. L., and Przedborski, S. (2013). Mitophagy and Parkinson’s disease: be eaten to stay healthy. Mol. Cell. Neurosci. 55, 37–43. doi: 10.1016/j.mcn.2012.07.008
Dias, V., Junn, E., and Mouradian, M. M. (2013). The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 3, 461–491. doi: 10.3233/JPD-130230
Ding, W. X., and Yin, X. M. (2012). Mitophagy: mechanisms, pathophysiological roles and analysis. Biol. Chem. 393, 547–564. doi: 10.1515/hsz-2012-0119
Duan, P., Hu, C., Quan, C., Yu, T., Zhou, W., Yuan, M., et al. (2016). 4-Nonylphenol induces apoptosis, autophagy and necrosis in Sertoli cells: involvement of ROS-mediated AMPK/AKT-mTOR and JNK pathways. Toxicology 341–343, 28–40. doi: 10.1007/s00701-016-2824-2
Durcan, T. M., and Fon, E. A. (2015). The three ‘P’s of mitophagy: PARKIN, PINK1 and post-translational modifications. Genes Dev. 29, 989–999. doi: 10.1101/gad.262758.115
Echeverry, R., Wu, J., Haile, W. B., Guzman, J., and Yepes, M. (2010). Tissue-type plasminogen activator is a neuroprotectant in the mouse hippocampus. J. Clin. Invest. 120, 2194–2205. doi: 10.1172/JCI41722
Eiyama, A., and Okamoto, K. (2015). PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 33, 95–101. doi: 10.1016/j.ceb.2015.01.002
Fiorini, C., Cordani, M., Gotte, G., Picone, D., and Donadelli, M. (2015). Onconase induces autophagy sensitizing pancreatic cancer cells to gemcitabine and activates Akt/mTOR pathway in a ROS-dependent manner. Biochim. Biophys. Acta 1853, 549–560. doi: 10.1016/j.bbamcr.2014.12.016
GBD 2015 DALYs and HALE Collaborators (2016). Global, regional and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388, 1603–1658. doi: 10.1016/S0140-6736(16)31460-X
GBD 2015 Mortality and Causes of Death Collaborators (2016). Global, regional and national life expectancy, all-cause mortality and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388, 1459–1544. doi: 10.1016/S0140-6736(16)31012-1
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Guo, Y. (2017). Role of HIF-1a in regulating autophagic cell survival during cerebral ischemia reperfusion in rats. Oncotarget 8, 98482–98494. doi: 10.18632/oncotarget.21445
Hambright, H. G., Meng, P., Kumar, A. P., and Ghosh, R. (2015). Inhibition of PI3K/AKT/mTOR axis disrupts oxidative stress-mediated survival of melanoma cells. Oncotarget 6, 7195–7208. doi: 10.18632/oncotarget.3131
He, Q., Li, Z., Wang, Y., Hou, Y., Li, L., and Zhao, J. (2017). Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int. Immunopharmacol. 50, 208–215. doi: 10.1016/j.intimp.2017.06.029
He, L., Zhou, Q., Huang, Z., Xu, J., Zhou, H., Lv, D., et al. (2019). PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. J. Cell. Physiol. 234, 8668–8682. doi: 10.1002/jcp.27527
Huang, Y., Chen, H. J., Zhu, J. H., Zhao, F. Y., Qu, Y., and Mu, D. Z. (2016). [Effects of PINK1 gene on cell apoptosis and cell autophagy in neonatal mice with hypoxic-ischemic brain damage]. Zhongguo Dang Dai Er Ke Za Zhi 18, 263–269. doi: 10.7499/j.issn.1008-8830.2016.03.015
Iguchi, M., Kujuro, Y., Okatsu, K., Koyano, F., Kosako, H., Kimura, M., et al. (2013). Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 288, 22019–22032. doi: 10.1074/jbc.M113.467530
Imai, Y., Kanao, T., Sawada, T., Kobayashi, Y., Moriwaki, Y., Ishida, Y., et al. (2010). The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet. 6:e1001229. doi: 10.1371/journal.pgen.1001229
Jeanneret, V., and Yepes, M. (2017). Tissue-type plasminogen activator is a homeostatic regulator of synaptic function in the central nervous system. Neural Regen. Res. 12, 362–365. doi: 10.4103/1673-5374.202924
Jin, S. M., and Youle, R. J. (2012). PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799. doi: 10.1242/jcs.093849
Jin, S. M., and Youle, R. J. (2013). The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757. doi: 10.4161/auto.26122
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/BJ20140334
Khandelwal, P., Yavagal, D. R., and Sacco, R. L. (2016). Acute ischemic stroke intervention. J. Am. Coll. Cardiol. 67, 2631–2644. doi: 10.1016/j.jacc.2016.03.555
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Krishnamurthi, R. V., Feigin, V. L., Forouzanfar, M. H., Mensah, G. A., Connor, M., Bennett, D. A., et al. (2013). Global and regional burden of first-ever ischaemic and haemorrhagic stroke during 1990–2010: findings from the Global Burden of Disease Study 2010. Lancet Glob. Health 1, e259–e281. doi: 10.1016/S2214-109X(13)70089-5
Kuang, Y., Ma, K., Zhou, C., Ding, P., Zhu, Y., Chen, Q., et al. (2016). Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 12, 2363–2373. doi: 10.1080/15548627.2016.1238552
Kubli, D. A., Ycaza, J. E., and Gustafsson, A. B. (2007). Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 405, 407–415. doi: 10.1042/BJ20070319
Kubota, C., Torii, S., Hou, N., Saito, N., Yoshimoto, Y., Imai, H., et al. (2010). Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J. Biol. Chem. 285, 667–674. doi: 10.1074/jbc.M109.053058
Kumar, A., Aguirre, J. D., Condos, T. E., Martinez-Torres, R. J., Chaugule, V. K., Toth, R., et al. (2015). Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 34, 2506–2521. doi: 10.15252/embj.201592337
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, J. Y., Nagano, Y., Taylor, J. P., Lim, K. L., and Yao, T. P. (2010). Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation and HDAC6-dependent mitophagy. J. Cell Biol. 189, 671–679. doi: 10.1083/jcb.201001039
Li, L., Tan, J., Miao, Y., Lei, P., and Zhang, Q. (2015). ROS and autophagy: interactions and molecular regulatory mechanisms. Cell. Mol. Neurobiol. 35, 615–621. doi: 10.1007/s10571-015-0166-x
Li, S., Zhou, Y., Gu, X., Zhang, X., and Jia, Z. (2021). NLRX1/FUNDC1/NIPSNAP1–2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 54:e12986. doi: 10.1111/cpr.12986
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, K. E. and Frazier, W. A. (2015). Phosphorylation of the BNIP3 C-terminus inhibits mitochondrial damage and cell death without blocking autophagy. PLoS One 10:e0129667. doi: 10.1371/journal.pone.0129667
Liu, H., Huang, H., Li, R., Bi, W., Feng, L., E, L., et al. (2019). Mitophagy protects SH-SY5Y neuroblastoma cells against the TNFα-induced inflammatory injury: involvement of microRNA-145 and Bnip3. Biomed. Pharmacother. 109, 957–968. doi: 10.1016/j.biopha.2018.10.123
Liu, L., Sakakibara, K., Chen, Q., and Okamoto, K. (2014). Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 24, 787–795. doi: 10.1038/cr.2014.75
Lu, N., Li, X., Tan, R., An, J., Cai, Z., Hu, X., et al. (2018). HIF-1α/beclin1-mediated autophagy is involved in neuroprotection induced by hypoxic preconditioning. J. Mol. Neurosci. 66, 238–250. doi: 10.1007/s12031-018-1162-7
Lv, M., Wang, C., Li, F., Peng, J., Wen, B., Gong, Q., et al. (2017). Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 8, 25–38. doi: 10.1007/s13238-016-0328-8
Maejima, Y., Isobe, M., and Sadoshima, J. (2016). Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell. Cardiol. 95, 19–25. doi: 10.1016/j.yjmcc.2015.10.032
Mahalingaiah, P. K., and Singh, K. P. (2014). Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS One 9:e87371. doi: 10.1371/journal.pone.0087371
Mellor, K. M., Bell, J. R., Young, M. J., Ritchie, R. H., and Delbridge, L. M. (2011). Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J. Mol. Cell. Cardiol. 50, 1035–1043. doi: 10.1016/j.yjmcc.2011.03.002
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Novak, I., and Dikic, I. (2011). Autophagy receptors in developmental clearance of mitochondria. Autophagy 7, 301–303. doi: 10.4161/auto.7.3.14509
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Okatsu, K., Kimura, M., Oka, T., Tanaka, K., and Matsuda, N. (2015a). Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 128, 964–978. doi: 10.1242/jcs.161000
Okatsu, K., Koyano, F., Kimura, M., Kosako, H., Saeki, Y., Tanaka, K., et al. (2015b). Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 209, 111–128. doi: 10.1083/jcb.201410050
Pan, J., Li, X., Guo, F., Yang, Z., Zhang, L., and Yang, C. (2019). Ginkgetin attenuates cerebral ischemia-reperfusion induced autophagy and cell death via modulation of the NF-κB/p53 signaling pathway. Biosci. Rep. 39:BSR20191452. doi: 10.1042/BSR20191452
Papadakis, M., Hadley, G., Xilouri, M., Hoyte, L. C., Nagel, S., McMenamin, M. M., et al. (2013). Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat. Med. 19, 351–357. doi: 10.1038/nm.3097
Parzych, K. R., and Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism and regulation. Antioxid. Redox Signal. 20, 460–473. doi: 10.1089/ars.2013.5371
Poluzzi, C., Casulli, J., Goyal, A., Mercer, T. J., Neill, T., and Iozzo, R. V. (2014). Endorepellin evokes autophagy in endothelial cells. J. Biol. Chem. 289, 16114–16128. doi: 10.1074/jbc.M114.556530
Poole, A. C., Thomas, R. E., Andrews, L. A., McBride, H. M., Whitworth, A. J., and Pallanck, L. J. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U S A 105, 1638–1643. doi: 10.1073/pnas.0709336105
Quinn, P. M. J., Moreira, P. I., Ambrósio, A. F., and Alves, C. H. (2020). PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 8:189. doi: 10.1186/s40478-020-01062-w
Rami, A., Langhagen, A., and Steiger, S. (2008). Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol. Dis. 29, 132–141. doi: 10.1016/j.nbd.2007.08.005
Riley, B. E., Lougheed, J. C., Callaway, K., Velasquez, M., Brecht, E., Nguyen, L., et al. (2013). Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 4:1982. doi: 10.1038/ncomms2982
Roperto, S., De Falco, F., Perillo, A., Catoi, C., and Roperto, F. (2019). Mitophagy mediated by BNIP3 and BNIP3L/NIX in urothelial cells of the urinary bladder of cattle harbouring bovine papillomavirus infection. Vet. Microbiol. 236:108396. doi: 10.1016/j.vetmic.2019.108396
Salminen, A., Kaarniranta, K., Kauppinen, A., Ojala, J., Haapasalo, A., Soininen, H., et al. (2013). Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 106–107, 33–54. doi: 10.1016/j.pneurobio.2013.06.002
Sano, H., Futamura, M., Gaowa, S., Kamino, H., Nakamura, Y., Yamaguchi, K., et al. (2020). p53/Mieap-regulated mitochondrial quality control plays an important role as a tumor suppressor in gastric and esophageal cancers. Biochem. Biophys. Res. Commun. 529, 582–589. doi: 10.1016/j.bbrc.2020.05.168
Sekerdag, E., Solaroglu, I., and Gursoy-Ozdemir, Y. (2018). Cell death mechanisms in stroke and novel molecular and cellular treatment options. Curr. Neuropharmacol. 16, 1396–1415. doi: 10.2174/1570159X16666180302115544
Semenza, G. L. (2011). Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 1813, 1263–1268. doi: 10.1016/j.bbamcr.2010.08.006
Shacka, J. J., Lu, J., Xie, Z. L., Uchiyama, Y., Roth, K. A., and Zhang, J. (2007). Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci. Lett. 414, 57–60. doi: 10.1016/j.neulet.2006.12.025
Sheng, R., Zhang, L. S., Han, R., Liu, X. Q., Gao, B., and Qin, Z. H. (2010). Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 6, 482–494. doi: 10.4161/auto.6.4.11737
Shi, R. Y., Zhu, S. H., Li, V., Gibson, S. B., Xu, X. S., and Kong, J. M. (2014). BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 20, 1045–1055. doi: 10.1111/cns.12325
Springer, M. Z., and Macleod, K. F. (2016). In brief: mitophagy: mechanisms and role in human disease. J. Pathol. 240, 253–255. doi: 10.1002/path.4774
Šprung, M., Dikic, I., and Novak, I. (2018). Flow cytometer monitoring of bnip3- and bnip3L/nix-dependent mitophagy. Methods Mol. Biol. 1759, 105–110. doi: 10.1007/7651_2017_14
Tagaya, M., and Arasaki, K. (2017). Regulation of mitochondrial dynamics and autophagy by the mitochondria-associated membrane. Adv. Exp. Med. Biol. 997, 33–47. doi: 10.1007/978-981-10-4567-7_3
Tanaka, K. (2020). The PINK1-parkin axis: an overview. Neurosci. Res. 159, 9–15. doi: 10.1016/j.neures.2020.01.006
Tracy, K., Dibling, B. C., Spike, B. T., Knabb, J. R., Schumacker, P., and Macleod, K. F. (2007). BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol. Cell. Biol. 27, 6229–6242. doi: 10.1128/MCB.02246-06
Trempe, J. F., Sauvé, V., Grenier, K., Seirafi, M., Tang, M. Y., Ménade, M., et al. (2013). Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455. doi: 10.1126/science.1237908
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
van der Vos, K. E., Eliasson, P., Proikas-Cezanne, T., Vervoort, S. J., van Boxtel, R., Putker, M., et al. (2012). Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat. Cell Biol. 14, 829–837. doi: 10.1038/ncb2536
Wang, Y., Dong, X. X., Cao, Y., Liang, Z. Q., Han, R., Wu, J. C., et al. (2009). p53 induction contributes to excitotoxic neuronal death in rat striatum through apoptotic and autophagic mechanisms. Eur. J. Neurosci. 30, 2258–2270. doi: 10.1111/j.1460-9568.2009.07025.x
Wang, L., Wang, P., Dong, H., Wang, S., Chu, H., Yan, W., et al. (2018). Ulk1/FUNDC1 prevents nerve cells from hypoxia-induced apoptosis by promoting cell autophagy. Neurochem. Res. 43, 1539–1548. doi: 10.1007/s11064-018-2568-x
Wauer, T., Simicek, M., Schubert, A., and Komander, D. (2015). Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374. doi: 10.1038/nature14879
Whitworth, A. J., and Pallanck, L. J. (2009). The PINK1/Parkin pathway: a mitochondrial quality control system? J. Bioenerg. Biomembr. 41, 499–503. doi: 10.1007/s10863-009-9253-3
Wong, E., and Cuervo, A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811. doi: 10.1038/nn.2575
Wu, W., Li, W., Chen, H., Jiang, L., Zhu, R., and Feng, D. (2016a). FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 12, 1675–1676. doi: 10.1080/15548627.2016.1193656
Wu, W., Lin, C., Wu, K., Jiang, L., Wang, X., Li, W., et al. (2016b). FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 35, 1368–1384. doi: 10.15252/embj.201593102
Wu, X., Li, X., Liu, Y., Yuan, N., Li, C., Kang, Z., et al. (2018). Hydrogen exerts neuroprotective effects on OGD/R damaged neurons in rat hippocampal by protecting mitochondrial function via regulating mitophagy mediated by PINK1/Parkin signaling pathway. Brain Res. 1698, 89–98. doi: 10.1016/j.brainres.2018.06.028
Wu, F., Wu, J., Nicholson, A. D., Echeverry, R., Haile, W. B., Catano, M., et al. (2012). Tissue-type plasminogen activator regulates the neuronal uptake of glucose in the ischemic brain. J. Neurosci. 32, 9848–9858. doi: 10.1523/JNEUROSCI.1241-12.2012
Xing, S., Zhang, Y., Li, J., Zhang, J., Li, Y., Dang, C., et al. (2012). Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 8, 63–76. doi: 10.4161/auto.8.1.18217
Xu, D., Chen, P., Wang, B., Wang, Y., Miao, N., Yin, F., et al. (2019). NIX-mediated mitophagy protects against proteinuria-induced tubular cell apoptosis and renal injury. Am. J. Physiol. Renal Physiol. 316, F382–F395. doi: 10.1152/ajprenal.00360.2018
Xu, S., Zhong, A., Ma, H., Li, D., Hu, Y., Xu, Y., et al. (2017). Neuroprotective effect of salvianolic acid B against cerebral ischemic injury in rats via the CD40/NF-κB pathway associated with suppression of platelets activation and neuroinflammation. Brain Res. 1661, 37–48. doi: 10.1016/j.brainres.2017.02.011
Yamano, K., Matsuda, N., and Tanaka, K. (2016). The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 17, 300–316. doi: 10.15252/embr.201541486
Yamano, K., Queliconi, B. B., Koyano, F., Saeki, Y., Hirokawa, T., Tanaka, K., et al. (2015). Site-specific interaction mapping of phosphorylated ubiquitin to uncover parkin activation. J. Biol. Chem. 290, 25199–25211. doi: 10.1074/jbc.M115.671446
Yang, X., Pan, W., Xu, G., and Chen, L. (2020). Mitophagy: a crucial modulator in the pathogenesis of chronic diseases. Clin. Chim. Acta 502, 245–254. doi: 10.1016/j.cca.2019.11.008
Yepes, M. (2019). The plasminogen activation system promotes neurorepair in the ischemic brain. Curr. Drug Targets 20, 953–959. doi: 10.2174/1389450120666181211144550
Yu, J., Li, X., Matei, N., McBride, D., Tang, J., Yan, M., et al. (2018). Ezetimibe, a NPC1L1 inhibitor, attenuates neuronal apoptosis through AMPK dependent autophagy activation after MCAO in rats. Exp. Neurol. 307, 12–23. doi: 10.1016/j.expneurol.2018.05.022
Yuan, Y., Zheng, Y., Zhang, X., Chen, Y., Wu, X., Wu, J., et al. (2017). BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 13, 1754–1766. doi: 10.1080/15548627.2017.1357792
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. doi: 10.1074/jbc.M800102200
Zhang, X., Deguchi, S., Deguchi, K., Ohta, Y., Yamashita, T., Shang, J., et al. (2011). Amlodipine and atorvastatin exert protective and additive effects via antiapoptotic and antiautophagic mechanisms after transient middle cerebral artery occlusion in Zucker metabolic syndrome rats. J. Neurosci. Res. 89, 1228–1234. doi: 10.1002/jnr.22633
Zhang, J., Kim, J., Alexander, A., Cai, S., Tripathi, D. N., Dere, R., et al. (2013). A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 15, 1186–1196. doi: 10.1038/ncb2822
Zhang, X. D., Wang, Y., Wang, Y., Zhang, X., Han, R., Wu, J. C., et al. (2009). p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 5, 339–350. doi: 10.4161/auto.5.3.8174
Zhang, X., Yan, H., Yuan, Y., Gao, J., Shen, Z., Cheng, Y., et al. (2013). Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 9, 1321–1333. doi: 10.4161/auto.25132
Zheng, Y. Q., Liu, J. X., Li, X. Z., Xu, L., and Xu, Y. G. (2009). RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol. Sin. 30, 919–927. doi: 10.1038/aps.2009.79
Zhou, H., Zhu, P., Wang, J., Zhu, H., Ren, J., and Chen, Y. (2018). Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 25, 1080–1093. doi: 10.1038/s41418-018-0086-7
Keywords: mitochondrial autophagy, PINK1/Parkin, FUNDC1, NIX/BNIP3, cerebral stroke
Citation: Lei L, Yang S, Lu X, Zhang Y and Li T (2021) Research Progress on the Mechanism of Mitochondrial Autophagy in Cerebral Stroke. Front. Aging Neurosci. 13:698601. doi: 10.3389/fnagi.2021.698601
Received: 21 April 2021; Accepted: 02 June 2021;
Published: 15 July 2021.
Edited by:
Lin Wang, Zhejiang University, ChinaReviewed by:
Cameron Lenahan, Burrell College of Osteopathic Medicine, United StatesCopyright © 2021 Lei, Yang, Lu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongfa Zhang, MTM4ODgxNjgwOThAMTYzLmNvbQ==; Tao Li, dGFvbGlfa2hAMTYzLmNvbQ==
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.